

Abstract
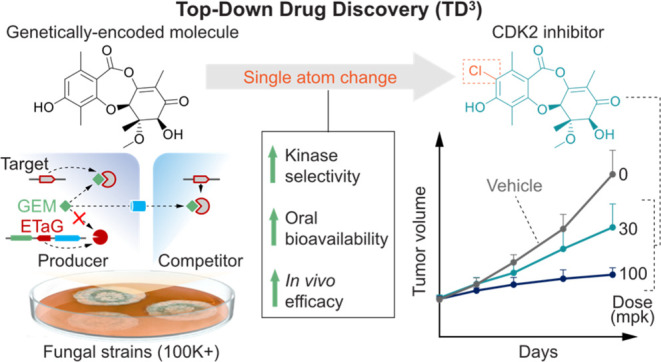
Fungi have historically been the source of numerous important medicinal compounds, but full exploitation of their genetic potential for drug development has been hampered in traditional discovery paradigms. Here we describe a radically different approach, top-down drug discovery (TD3), starting with a massive digital search through a database of over 100,000 fully genomicized fungi to identify loci encoding molecules with a predetermined human target. We exemplify TD3 by the selection of cyclin-dependent kinases (CDKs) as targets and the discovery of two molecules, 1 and 2, which inhibit therapeutically important human CDKs. 1 and 2 exhibit a remarkable mechanism, forming a site-selective covalent bond to the CDK active site Lys. We explored the structure–activity relationship via semi- and total synthesis, generating an analog, 43, with improved kinase selectivity, bioavailability, and efficacy. This work highlights the power of TD3 to identify mechanistically and structurally novel molecules for the development of new medicines.
Introduction
Several of the most impactful medicines known to humankind are the products of small molecule evolution in fungi. Lovastatin was the first drug approved by the FDA to lower cholesterol by blocking sterol biogenesis,1 and is the direct progenitor to the entire family of statin drugs that are among the most widely prescribed medicines in the US, with over 800 million prescriptions filled annually to 40 million patients.2 Cyclosporin enabled routine and widespread organ transplantation in humans by suppressing T cell mediated rejection of the donor-derived organs.3 Cyclosporin also served as an invaluable tool for elucidation of many fundamental aspects of T cell biology, while revealing a powerful new modality for drug action, launching the field of molecules known as molecular glues.4 Lovastatin and cyclosporin exemplify the potential of genetically encoded small molecules (GEMs), otherwise known as natural products, to provide mechanistically and structurally novel medicines that have saved and extended millions of lives.
In the predominant paradigm for small molecule drug discovery, a clinical molecule is elaborated through an iterative optimization process that typically requires the synthesis and characterization of thousands of fully synthetic compounds. In contrast to this bottom-up approach, lovastatin and cyclosporin represent an appealing alternative paradigm, which we term top-down drug discovery (TD3), where a new lead molecule emerges fully fledged from a discovery program as a clinical candidate. Both lovastatin and cyclosporin achieved FDA approval with no further elaboration; importantly, they also served as an advanced starting point for focused semisynthetic optimization that subsequently yielded next-gen FDA-approved drugs with improved clinical properties.5 Penicillin, a drug that has saved millions of lives, is the most famous example of a fungal GEM discovered in the TD3 paradigm that was life-cycle-managed, in this case to overcome emergent bacterial drug resistance.6 Mycophenolic acid is yet another prominent example of a clinically administered drug discovered from fungi by this approach, and subsequently life-cycle-managed by semisynthesis (CellCept).7
Notwithstanding the obvious appeal of TD3 from fungi, broader deployment of this strategy has been hampered by the numerous difficulties encountered during activity-guided isolation of fungal GEMs.8 These molecules are often potent and target-specific, but are typically present in vanishingly small quantities and in complex mixtures comprising hundreds to thousands of contaminating GEMs, many of which exhibit diverse biological activities that confound cell-based screens.9 Having successfully carried out a large-scale genome-mining operation in bacteria that led to the discovery of new members of the FK506/rapamycin class of GEMs,10 it occurred to us that exploitation of advances in fungal genomics, bioengineering, and multiomics might also accelerate, simplify and render more predictable the scalable discovery of GEMs from fungi. In both bacteria and fungi, the genes encoding GEM biosynthesis are often colocated within the genome in biosynthetic gene clusters (BGCs), each comprising a cassette of coordinately regulated genes that evolve and act together to perform context-dependent production of their self-encoded GEM.11 Many fungal GEMs have evolved to provide a competitive advantage over other fungi, and this creates the need for self-protection of fungi from their own GEMs.12 Such protective genes typically reside within the BGC, as resistance mechanisms are coevolved with, and mechanistically tailored to, individual GEMs. Many BGCs, for example, contain one or more genes encoding GEM-specific transmembrane pumps that export their cognate GEM from the producing fungus.13 However, most relevant to the field of drug discovery, a small but important subset of BGCs contain a gene embedded within their evolutionarily conserved boundaries that encodes a copy of the molecular target of the encoded GEM, which we refer to as an embedded target gene (ETaG). For example, colocalized within the lovastatin BGC can be found a copy of the HMG-CoA reductase gene, the target of lovastatin that, when inhibited in humans, is responsible for its cholesterol-lowering effect.14 Typically, fungi hosting BGCs using this method of self-resistance encode two copies of the target gene: an ancestral copy that remains susceptible to inhibition, and a second ETaG copy that has acquired GEM-resistant mutations while maintaining the capacity for functional complementation; for example, the protein encoded by the HMG-CoA reductase ETaG is active enzymatically but resistant to inhibition by lovastatin.
Fungi and humans share many genes that can be considered evolutionarily conserved, and often perform the same or a related function in both organisms.15 These orthologs typically exhibit moderate overall sequence identity, but active site amino acid residues can be conserved almost identically.16 However, it is exactly these functionally important and highly conserved regions that comprise important drug binding sites. While fungal GEMs can be assumed to have evolved to compete with organisms encountered in their native environments, such as other fungi, bacteria, or plants, there is considerable potential for cross-kingdom binding to homologous human proteins at conserved active sites. In this context, an ETaG can be viewed as an avatar for a relevant human therapeutic target, enabling a digital search through genomic space to identify BGCs that encode novel GEMs with important clinical properties.17 These concepts suggested to us the possibility of broadly enabling an ETaG-based platform for more rapid, scalable, and targeted implementation of TD3 in fungi than has previously been available, by (1) assembling a massive database of phylogenetically diverse, comprehensively annotated fungal genomes; (2) selecting a disease-relevant human protein; (3) performing genomic search to query the database for BGCs having an ETaG avatar of the preselected human target; (4) using multiomic methods to monitor BGC-related RNA expression and metabolite production under a variety of conditions; (5) identifying the fully elaborated GEM product of the BGC; then (6) performing minor structural optimization if necessary to develop the GEM as a drug.
By applying our integrated TD3 platform we report the use of ETaG-based search to identify a BGC that encodes two inhibitors of human cyclin-dependent kinases (CDKs): compounds 1 and 2, respectively roseopurpurins D and C. The roseopurpurins were originally isolated from the fungi Aspergillus spp.(18,19) and Penicillium roseopurpureum(20) and shown to exhibit selective cytotoxicity toward cancer cell lines,20 though the target responsible for the activity of these GEMs was not identified in that earlier work. We recently disclosed that 1 and 2 are novel CDK-inhibitors, with potential utility in the treatment of CDK-dependent cancers.21 Subsequent to our disclosure, the target identification of 1 and 2 was replicated independently in a publication that describes the CDK-active properties of the roseopurpurins and delineates the encoding BGC, designated as rosA–rosK.22 Here we provide a fully detailed account of our genomic deorphanization of these kinase-inhibitor GEMs, comprehensive elucidation of the biosynthetic pathway, characterization of their extraordinary covalent kinase engagement mechanism, an initial structure–activity exploration by semisynthetic and synthetic modification, and preliminary in cellulo and in vivo characterization.
Results and Discussion
Computational Discovery of a BGC Family with a CDK ETaG
With a goal to discover novel CDK inhibitors, we applied our TD3 platform to identify CDK ETaGs in our proprietary database of over 100,000 annotated fungal genomes (LifeBase). After gathering all candidate BGCs having a CDK ETaG, we performed comparative genome analyses to identify the consensus gene set comprising each family of BGCs, and established that the candidate ETaG was embedded within the biosynthetic cluster.
We identified a candidate BGC family with a putative CDK ETaG and a polyketide synthase core (Supporting Table 1), consisting of 900 members in 173 species distributed across four taxonomic classes of ascomycete fungi, representing close to 1% of all fungi within in our collection (Figure 1). During the preparation of this manuscript, two alleles of this same BGC family (ros) were described in another publication.22 Of the 900 ros BGC alleles identified in our database, 98.9% contain the candidate ETaG. Furthermore, we observed 70 distinct BGC variants, each having a unique gene order and orientation. The presence of the putative ETaG in most members of this BGC family, despite the substantial degree of gene rearrangement, strongly suggested functional association of the ETaG with the ros BGC.
Figure 1.

Gene content and taxonomic distribution of a representative ros BGC. (A) The ros BGC has 11 genes, including a polyketide synthase core (rosJ) and a putative ETaG homologous to human CDKs (rosG). (B) Shown are 4 of the 70 distinct alleles of the ros BGC discovered in diverse taxonomic classes (indicated on the left) within LifeBase. Though the order and orientation of genes differ, the consensus genes are maintained, including the ETaG (red), consistent with functional essentiality.
A commonly observed characteristic of ETaGs is that they represent an extra copy of a housekeeping gene.13 Indeed, phylogenetic analysis comparing a CDK ETaG from a LifeBase strain of Preussia flanaganii with CDK proteins from Saccharomyces cerevisiae (yeast), and Homo sapiens (human) revealed that the ETaG is most closely related to fungal housekeeping Pho85 protein, which in turn is most closely related to human CDK5 (Figure 2). An important feature of CDKs is that they are central to cellular processes in both fungi and humans, with conserved evolutionary, structural, and functional relationships between subgroups. The CDK family is greatly expanded (radiated) in humans, which presents an opportunity to optimize a single fungal GEM scaffold to inhibit evolutionarily divergent human kinases. We anticipated that identifying the GEM encoded by this BGC might provide a starting point for the development of novel inhibitors of human CDK2, an attractive target for drug development23 and a close sequence homologue of CDK5.
Figure 2.
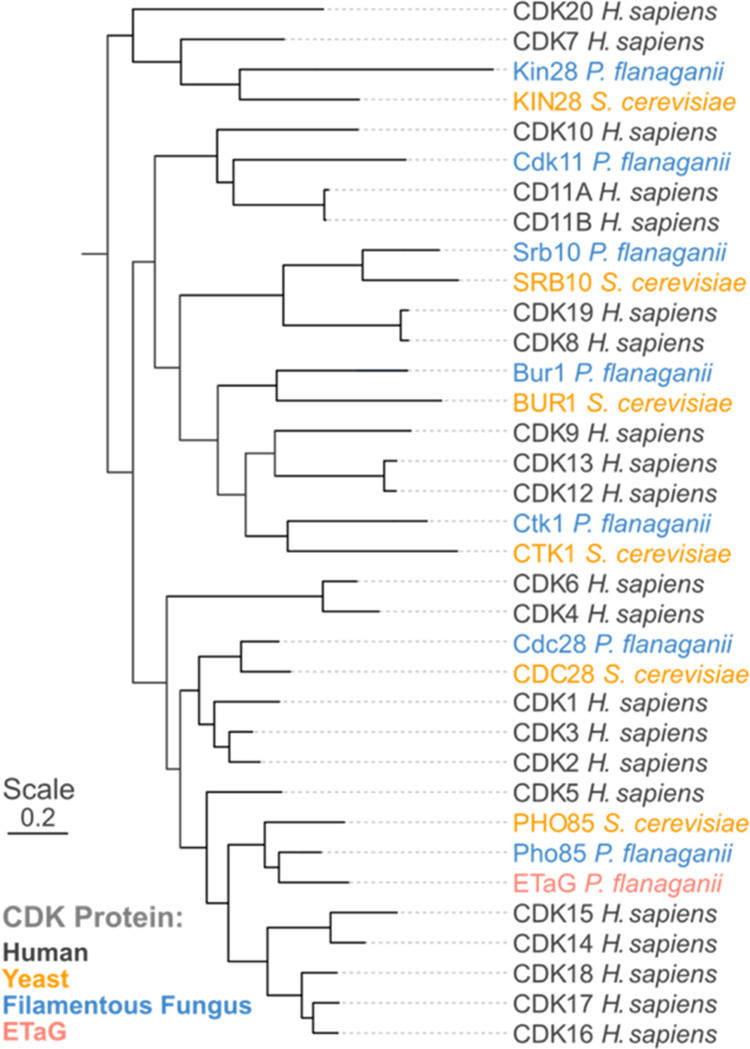
Phylogenetic comparison of the candidate ros BGC ETaG protein (red) from P. flanaganii with yeast and human CDK protein isoforms. Note that the ETaG (red) is a second copy of and is most closely related to the corresponding housekeeping Pho85 (blue).
ETaGs may provide self-resistance via the acquisition of mutations that reduce or abolish the binding of an inhibitor, but that maintain normal enzymatic function.12 We thus performed residue conservation analysis comparing 100 diverse housekeeping Pho85 proteins (selected as having a single pho85 gene copy and lacking the ros BGC) and the sequences of 38 diverse Pho85 ETaGs, to identify statistically significant sequence variations. Using this approach, we identified 8 residues that consistently differed between the housekeeping protein set and the ETaG protein set, and where the most abundant residue was present in more than 50% of each set of sequences (Table 1). Three of these potential resistance mutations were colocalized within the ATP binding pocket, as judged from the structure of the yeast Pho85-Pho80 CDK-Cyclin complex (PDB ID 2PMI, Figure 3). As resistance mutations are most likely to interfere directly with drug binding, these observations suggest that the GEM encoded by the ros BGC is an active site (orthosteric) inhibitor that competes directly with ATP. One of the putative resistance mutations occurs at the so-called gatekeeper residue; gatekeeper mutations in human kinases are a known, frequent escape mechanism that confers clinical resistance to FDA-approved kinase inhibitors.24 Here, the gatekeeper Phe in Pho85 is replaced by Ser in the ETaG. In the ATP binding pocket, human CDK2 has identical residues with the fungal housekeeping Pho85 in two out of three mutation sites, including the gatekeeper Phe, which led us to hypothesize that human CDK2, similarly to fungal housekeeping Pho85, would be susceptible to the GEM produced by this BGC.
Table 1. Conserved ETaG Mutant Residuesa.
| predominant
residue (abundance) |
||||
|---|---|---|---|---|
| positionb | Pho85 | ETaG | distance to ATP-γ-S/Å | human CDK2c |
| 66 | Val (92%) | Leu (93%) | 4.9 | 64 Val |
| 69 | His (66%) | Tyr (53%) | 8.8 | 67 Leu |
| 79 | Met (57%) | Ile (56%) | 12.4 | 77 Tyr |
| 82 | Phe (87%) | Ser (88%) | 4.4 | 80 Phe |
| 123 | Phe (54%) | Tyr (52%) | 17.3 | 117 Phe |
| 129 | Val (58%) | Ile (55%) | 14.6 | 123 Val |
| 150 | Gly (58%) | Ala (58%) | 7.9 | 144 Ala |
| 176 | Ala (64%) | Pro (57%) | 17.8 | 170 Ala |
Residue conservation analysis between housekeeping Pho85 and ETaG proteins identified 8 candidate resistance mutations that differ between housekeeping (Pho85) and ETaG sets, and are more than 50% conserved in each set. Three of these residues (bold) are within 8 Å of the active site (PDB ID 2PMI)25 and are proposed here to be determinants of GEM resistance in the ETaG.
Positions are in reference to yeast Pho85 (Uniprot ID P17157).
Human CDK2 (Uniprot ID P24941).
Figure 3.
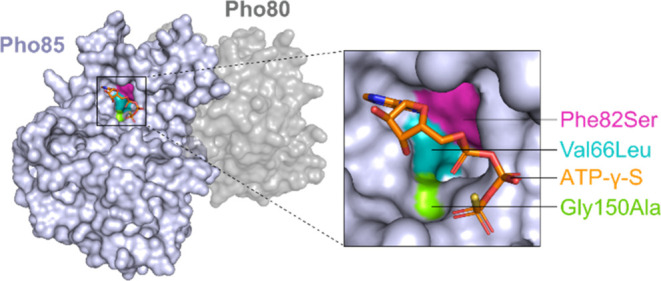
Residue conservation analysis identified three putative resistance mutations in the active site of the CDK ETaG (Table 1), suggesting that the product of the ros BGC is an active site, ATP-competitive inhibitor of CDKs. Mutations are mapped onto the structure of the yeast Pho85-Pho80 CDK-Cyclin complex (PDB ID 2PMI).25
Identification of GEMs 1 and 2 as CDK Inhibitors
To induce high-level expression of the GEM encoded by the ros BGC, we replaced the promoter of the rosH transcription factor with a strong constitutive promoter in a LifeBase strain Mycophilomyces sp., with the expectation that overproduction of this transcription factor would coordinately up-regulate all the biosynthetic genes in the ros BGC. By screening extract fractions from these engineered overproducer strains in an enzyme assay utilizing fungal Pho85 and human CDK5/p25, we identified and purified to homogeneity two CDK-inhibitory products, 1 and 2, of the candidate BGC. Detailed nuclear magnetic resonance (NMR) analysis (Supporting Tables 2 and 3) identified 1 and 2 as a diastereomeric pair of tricyclic polyketides containing a vinylogous anhydride, previously isolated and structurally characterized as roseopurpurins D and C, respectively.
To confirm our functional analysis of rosG as a cluster-specific ETaG, and our genomic prediction of the resistance-determining residues of the ETaG, we expressed and purified the wild-type Pho85 protein ortholog of Mycophilomyces sp. alongside a chimeric variant incorporating the putative resistance-determining residues (Val66Leu, Phe82Ser, and Gly150Ala) in Escherichia coli. In vitro functional assays of both proteins, wild-type and chimeric variant, in the presence of 2 revealed that the three conserved active site residues from the ETaG were sufficient to confer resistance to Pho85 against 2 at greater than 50 mM concentration (Supporting Figure 1). This result validated our TD3 platform as a means to identify both GEMs and their molecular target through genomic search, as well as provided key mechanistic insights into binding mode and resistance in silico.
Having established the product of the ros gene cluster in an engineered strain of Mycophilomyces sp., we returned to the LifeBase strain collection to see whether we could identify, by targeted metabolite screening, a high-producing wild-type strain. We selected a set of 83 fungal strains containing the ros cluster, so as to reasonably represent the full phylogenetic diversity of the 900 total strains in which this cluster is present. Each strain was fermented on a panel of seven media conditions and harvested at day 7 and 14. Samples were subjected to gene expression analysis by RNAseq and detection of roseopurpurin production by targeted liquid chromatography with tandem mass spectrometry (LC-MS/MS) (Figure 4). Fifteen strains were identified with increased cluster gene expression and accumulation of 1 and 2, as compared to the promoter-engineered strain of Mycophilomyces sp. In general, the highest expressing and producing strains were observed within two phylogenetic groups, the genus Penicillium, and the family Lasiosphaeriaceae. We selected strains of Penicillium sp. (sect. Excilicaulis, Penicillium restrictum clade, Supporting Figure 2)26 for GEM production, on the basis of their favorable properties for growth and handling.
Figure 4.
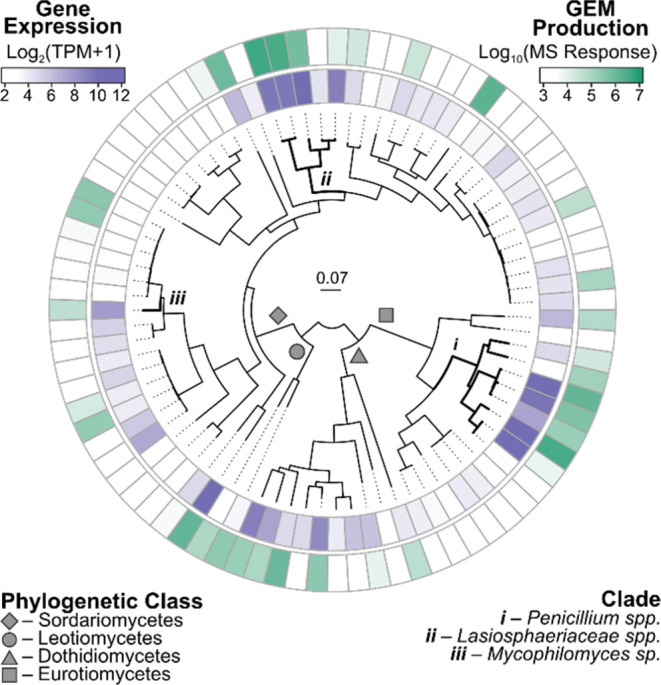
Heatmap of the sum of LC-MS/MS response from targeted detection of 1 and 2 (green) and the average expression of ros cluster genes (purple), as determined by quantitative RNAseq, in fermentations of LifeBase strains containing the ros BGC. Plotted values are the maximum observed in fermentations on 7 different media conditions at days 7 and 14. The heatmap is organized by multilocus phylogenetic analysis of 100 fungal housekeeping proteins.
Biosynthetic Pathway of 1 and 2
To fully understand the biosynthetic pathway of 1 and 2, single-gene knockout experiments were conducted on each of the 11 genes of the ros BGC in the wild-type production strain (Penicillium sp.) discussed above. Biosynthetic intermediates that uniquely accumulated in each knockout strain were characterized by LC-MS/MS analysis (Scheme 1). Seven of the ros genes (rosABCDFJK) were found to be essential for the biosynthesis of 2, while deletions of the four remaining ros genes (rosEGHI) led to similar or reduced production levels of 2, suggesting that they play a supporting role. Disruption of rosE significantly reduced both molecule production and BGC transcription, effects similar to those seen upon targeted deletion of the transcription factor rosH, suggesting that rosE also serves a role in transcriptional activation, despite its lack of sequence or structural similarity to any known transcription factor (Supporting Figure 3).
Scheme 1. Biosynthetic Pathway of 1 and 2.
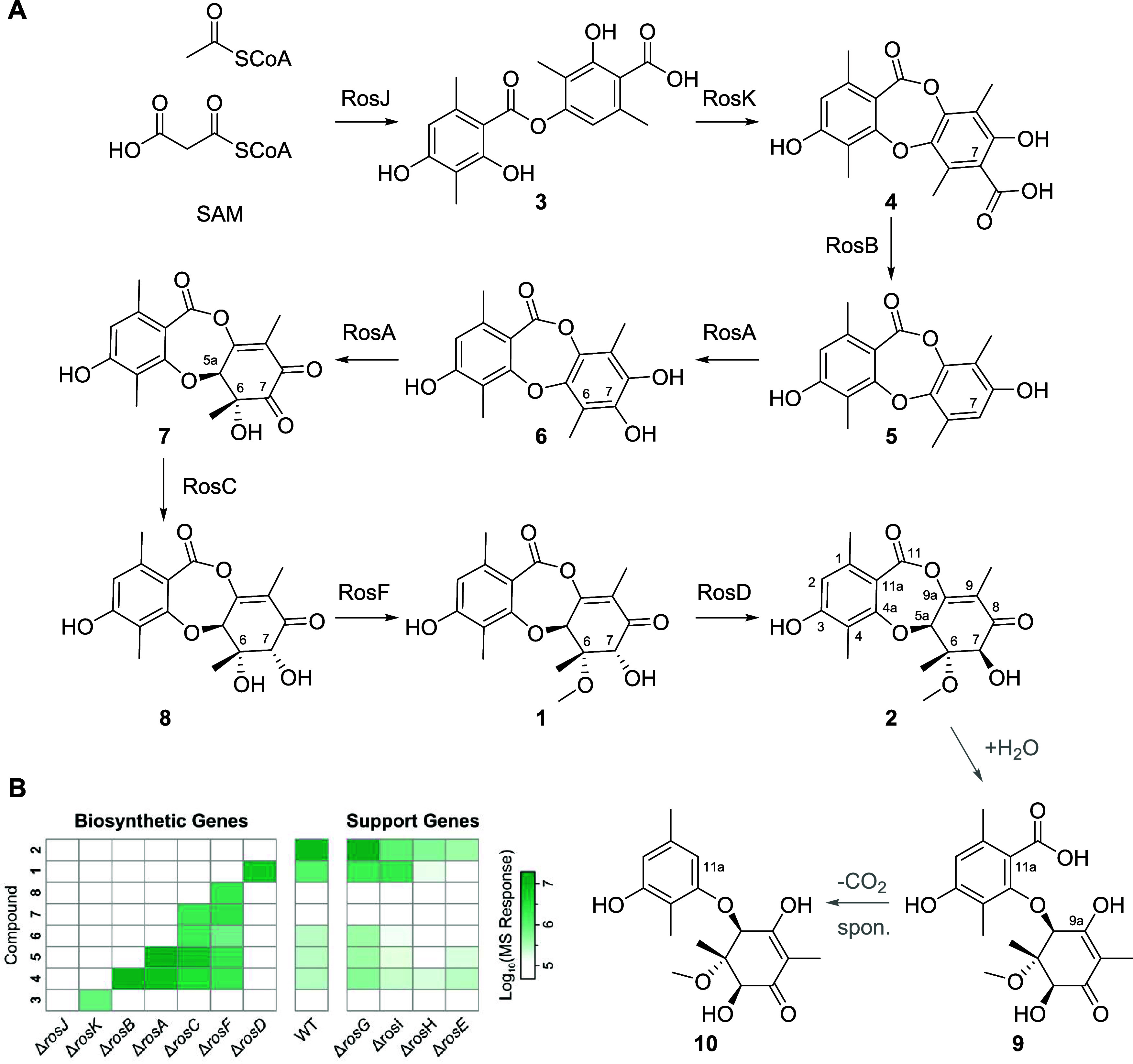
(A) Biosynthetic pathway of 1 and 2, showing nonenzymatic degradation pathway of 2 to 9 by hydrolysis and spontaneous decarboxylation at C11a to form 10 (gray arrows). (B) Heatmap of LC-MS/MS response for biosynthetic intermediates of 2 accumulated in single-gene KO strains of Penicillium sp.
The structures of intermediates 3 and 5 (Scheme 1) were confirmed by comparison to reference standards prepared by chemical synthesis (Supporting Figure 4). The early steps of the biosynthetic pathway of 2, up to formation of 5, are similar to other recently characterized fungal depsidone biosynthetic systems as well as previous proposals for the biosynthesis of 2.27 However, our observations have more clearly delineated several late-stage steps in the pathway, involving three genes responsible for oxidation, reduction and methylation that are proposed to convert 5 to 1, via roseopurpurin G 6. The hydroxylation of 5 by RosA was confirmed after heterologous expression of the encoding gene in S. cerevisiae, however we did not observe further oxidation to putative intermediate 7 in this system (Supporting Figure 5). Disruption of rosF, encoding an O-methyltransferase, led to accumulation of 8, which was isolated and its structure was confirmed by NMR. While previous studies have proposed a different biosynthetic pathway, one in which 6 is formed by elimination of water from 8,22 we did not observe such an elimination reaction during purification. In our extensively characterized pathway, 6 is not a shunt artifact but a bona fide biosynthetic intermediate.
The final gene in the biosynthesis encodes RosD, which is a unique protein in fungal biosynthesis having no significant sequence identity to any functionally characterized enzyme (Supporting Table 1). RosD does however have a conserved domain belonging to the vicinal oxygen chelating (VOC) superfamily, which has been associated with highly diverse functions in other proteins.28 RosD has been suggested to catalyze the hydrolysis of 2 to aculeatusquinone C as a detoxification mechanism,22 but the comprehensive analysis here demonstrates unambiguously that rosD encodes a C7 hydroxyl epimerase that interconverts 1 and 2 to the equilibrium mixture of ∼30/70% 1/2. Consistent with this role, disruption of rosD resulted in accumulation of 1, without production of 2. The purified RosD protein, generated by heterologous expression in E. coli, enabled reconstitution of the epimerization reaction in vitro, affording rapid conversion of 1 to the equilibrium 1/2 mixture (Supporting Figure 5). In these reconstitution reactions, we also observed two new products by LC-MS/MS, these being consistent with hydrolytic ring-opening of the vinylogous anhydride in 2 to form the known compound roseopurpurin A 9, which undergoes spontaneous decarboxylation to form aculeatusquinone C 10 (Scheme 1). Interestingly, the corresponding products of hydrolytic ring-opening and decarboxylation from 1 were not observed in significant quantities under reaction conditions, consistent with our general observation that the vinylogous anhydride in 2 is more reactive than that in 1. Degradants 9 and 10 were formed at the same rate under conditions in which the RosD enzyme was omitted, with all other conditions being held constant. To further establish the nonenzymatic nature of this pathway, 2 was dissolved in a mixture of acetonitrile and water with a mild base (NaHCO3), resulting in formation of 9 and 10. Because intermediates such as 6 in the biosynthetic pathway can also undergo nonenzymatic hydrolysis and decarboxylation, or methanolysis to produce methyl esters, we propose that a number of the aculeatusquinones and roseopurpurins derived from intermediates in the ros biosynthetic pathway similarly result from shunt pathways and are not evolved products of the ros BGC (Supporting Figure 6).
Characterization of Mechanism of Action of 1 and 2 on Human CDK2
Our initial characterization of the interaction of 1 with the Aspergillus nidulans CDK Pho85 (PhoA) revealed the site-selective formation of a covalent 1:1 adduct, with the inhibitor being linked to the active site Lys residue that engages the α- and β-phosphate moieties of ATP. Peptide mapping by mass spectrometry identified a single 348 Da mass shift of the Lys38-containing peptide, along with a missed tryptic cleavage site following this residue, consistent with site-specific modification by 1 (Supporting Figure 7). This residue is highly conserved in the vast majority of enzymatically active protein kinases, including all CDKs, and mutation to any other residue is known to abolish kinase activity.29 The kinase activity of CDK enzymes is strongly stimulated in vitro and in vivo by complexation with a cognate cyclin, and this is fundamental to the biological role of CDK/cyclin complexes exerting exquisite regulatory control over cell cycle progression. The preliminary observations of covalent inhibition by the fungal CDK employed the enzyme in the absence of its cyclin, and yet robust covalent conjugation of 1 and 2 was observed. In our mechanistic and structural studies of the human CDKs, we deemed it essential to use only the physiologically active form of the enzyme, namely its fully phosphorylated complex with the cognate cyclin.
In enzyme activity assays with human CDK2/cyclin E, we observed time-dependence in the IC50 for inhibition by 2, behavior that can be ascribed to covalent inhibition. We therefore employed a full battery of kinetic assays (Figure 5) to characterize the potency of 2 with respect to the relevant covalent inactivation parameters,30 the reversible equilibrium dissociation constant (KI) and the maximal rate of inactivation (kinact). In these assays, 2 exhibited rapid enzyme inactivation, with a kinact of 0.04 s–1. The preinactivation binding equilibrium was of moderate strength, with a KI of 1.9 μM, while the ratio (kinact/ KI), generally used as a gauge of potency for covalent inhibitors, was determined to be 23, which is comparable to FDA-approved covalent kinase inhibitors such as afatinib and dacomitinib.31 To further elucidate the mechanism of action, we performed intact mass spectrometry analysis of CDK2/cyclin E incubated with 2, confirming formation of a 1:1 covalent adduct (Supporting Figure 7).
Figure 5.
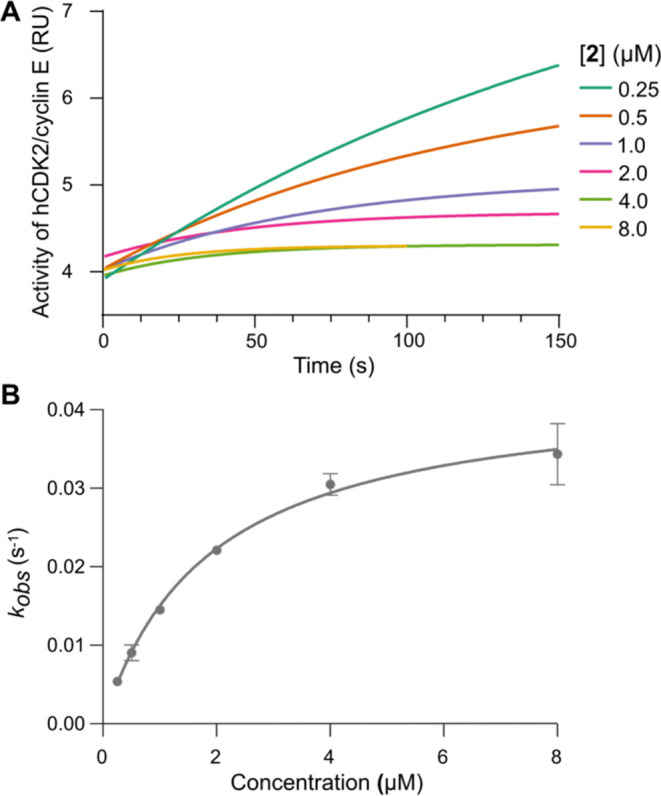
Kinase activity assays utilizing the CDK2/cyclin E kinase complex to characterize covalent binding interactions with 2. (A) Enzyme inhibition by 2 exhibited time-dependence of the IC50, here plotted in relative units (RU). (B) CDK2/cyclin E activity as a function of preincubation time was fit to determine the kobs rate constants which are plotted as a function of 2 concentration. Mean kobs reported ± SEM. These data were fit to determine KI (the apparent inhibitor affinity), kinact (the maximal inactivation rate), and the ratio kinact/KI (potency).
The binding interactions and inhibitory mechanism of 2 with the CDK2/cyclin E protein complex were elucidated at the molecular level by X-ray crystallography (Figure 6A). As expected, the reaction product of 2 with CDK2/cyclin E was located in the ATP binding site, with the phenolic ring engaging the kinase active site hinge region in a prototypical manner; specifically, the C3 phenol forms a hydrogen bond with the backbone of hinge residues Glu81 and Leu83. The central 7-membered ring has undergone ring-opening, and electron density clearly shows that the catalytic lysine (Lys33) is within covalent bonding distance to the carbonyl moiety of the original vinylogous anhydride (Figure 6B). The nonaromatic ring is rotated away, with the C8 carbonyl forming a hydrogen bond to Gln131 on the periphery of the active site.
Figure 6.

(A) Co-crystal structure of CDK2/cyclin E in complex with 2 (PDB ID 9BJB). (B) Active site of CDK2 with bound 2, showing H-bonding interactions from C3-OH to the kinase hinge; electron density map of 2 overlaid in mesh (2fo-fc contoured at 1 sigma) demonstrates covalent attachment to Lys33. (C) Binding of amide analog XC208 51 (PDB ID 9BJC) without covalent interaction, demonstrating a prereaction complex. (D) 2D map of binding site interactions between CDK2 and 2, showing covalent (solid line) and H-bonding (dotted lines) positions. (E) Structure of CDK2/cyclin A with ATP bound showing similar interactions to 2 and 51 at the hinge and catalytic Lys33 (PDB ID 4EOQ).
The structure depicted in Figure 6B clearly represents that of a postreaction complex; in order to determine the binding mode of the compound prior to the covalent reaction, we used the known isostructural amide derivative 51(18) (XC208) as a nonelectrophilic crystallographic probe. In enzyme activity assays, compound 51 showed reversible binding to CDK2/cyclin E, with an IC50 of 3 μM. The cocrystal structure of 51 bound to CDK2/cyclin E (Figure 6C) revealed similar contacts at the kinase hinge region compared to the postreaction complex of 2, but importantly no covalent bond was evident to Lys33, confirming that this structure approximates the prereaction binding mode for this series of compounds.
H-bonding interactions are observed between residues Asp86, Lys89, Gln131 and substituents at C8 and the chiral position C7 (Figure 6D), highlighting the importance of the chiral groups distant from the covalent reaction center in defining the optimal binding pose for nucleophilic attack (Supporting Figure 8). The binding mode of 2 is very similar to that observed for the kinase substrate ATP (Figure 6E), which forms interactions from the hinge region to the nucleobase and from the catalytic Lys33 to the α and β phosphates, suggesting that the structure and binding mode of 2 is primed for potent inhibition of CDK2.
To evaluate the biological activity of 2 in cellulo, we explored whether a durable irreversible response could be achieved in cells with just a short exposure for 4 h followed by washout (Figure 7). A CCNE1 amplified ovarian cancer cell line (OVCAR-3), reported as a CDK2 dependent cell line by DepMap,32 was treated in culture with a range of concentrations of 2 for 4 h and subsequently washed out and assessed at 120 h. After the transient 4 h exposure, 2 maintained full potency for 120 h (IC50 = 0.64 μM) comparable to conditions when cells were treated constantly with inhibitor for the entire 120 h, IC50 = 1 μM.
Figure 7.
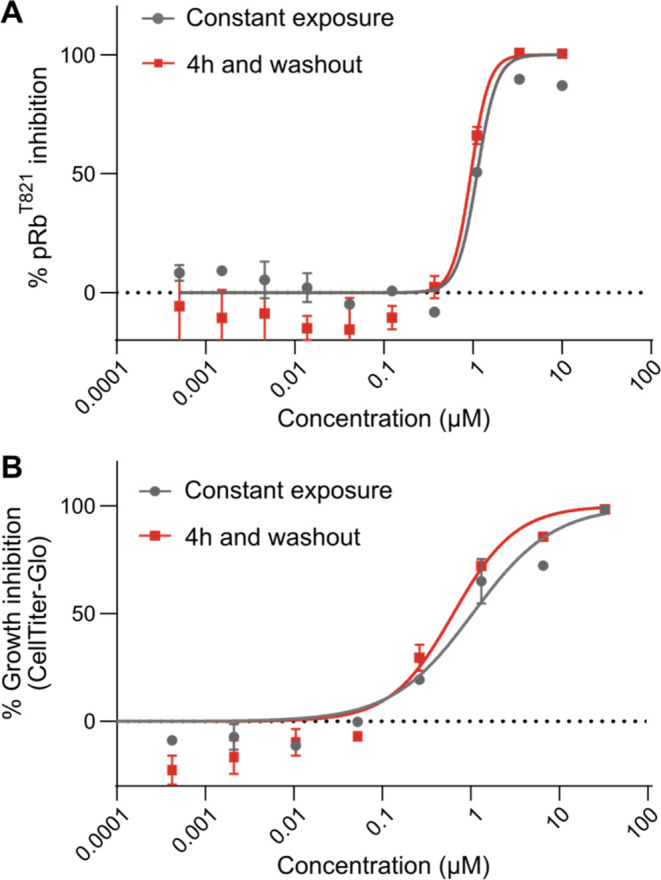
(A) % Inhibition of pRbT821 dose response of 2 in OVCAR-3 cells. Potency of 2 with transient 4 h exposure was similar to constant treatment. Mean IC50 reported ± SEM (B) % growth inhibition dose response measured by Cell Titer-Glo in OVCAR-3 cells. Potency of 2 with transient 4h exposure was similar to constant treatment. Mean IC50 reported ± SEM.
In a parallel experiment we evaluated the cellular impact of treatment with 2 on an extensively characterized direct CDK2 substrate, Retinoblastoma protein (Rb), wherein CDK2 inhibition is known to result in reduced phosphorylation of Rb (pRb) and consequential antigrowth activity in OVCAR-3 cells.33 Treatment with 2 resulted in inhibition of pRb (IC50 = 1.1 μM) with equipotency to the antigrowth activity IC50 = 1.0 μM (Figure 7). Furthermore, pRb inhibition was sustained with only a short exposure of 4 h and washout (IC50 = 0.95 μM) with equipotency to that of cells treated constantly for 18 h. In summary, the cellular mechanistic and phenotypic responses in living cells are fully sustained after just transient exposure and washout, corroborating with the irreversible nature of the catalytic inhibition.
Taken together, the time-dependent inhibition, intact mass spectrometry, crystallographic evidence and cellular washout clearly demonstrated a covalent mechanism of action via the key catalytic lysine residue of human CDK2.
Synthetic Chemistry
Compounds 1 and 2 have been evolved to target fungal CDKs for covalent inactivation, while our primary interest lies in selective inhibition of human CDK2. With a Ki in the low micromolar range, and as described below, low selectivity within the CDK family and modest oral bioavailability (12% in rats), it became clear that optimization by medicinal chemistry would be desirable. Our TD3 lead-optimization strategy focused on making structure–activity explorations using three synthetic approaches executed in parallel: (1) a total synthesis campaign to execute the first total synthesis of 1 and 2, gaining broad control over chemical matter in this series; (2) a campaign to a relevant series of analogs probing various aspects of structure–activity relationships; and (3) semisynthetic modification starting from 2 produced by fungal fermentation. With respect to fully synthetic approaches, the compactness of this scaffold (MW = 348 Da) is attractive, but the structure poses a formidable synthetic challenge, due to the three contiguous chiral centers (C5a, C6 and C7), a quaternary chiral center, a reactive and water-sensitive vinylogous anhydride warhead, and an oxidatively dearomatized moiety with the potential to rearomatize.
The hydrolysis of 2 (Scheme 1) served as an inspiration for our synthetic strategy, featuring a late-stage dehydration to form the central 7-member ring. Disconnecting into a suitably protected orsellinic acid derivative and a nonaromatic ring bearing the stereochemical complexity, we reasoned that an epoxide coupling approach would set the desired relative stereochemistry. Our initial experiments coupling known compound 14 with the racemic epoxide of a protected benzoquinone delivered a trans intermediate 15, which was methylated to form 16 (Scheme 2). Relative stereochemistry was confirmed by NOE NMR experiments. Epoxidation to 17, followed by substrate-controlled stereoselective reduction afforded intermediate 18 with the desired relative stereochemistry at C7 (highlighted blue). We had planned to induce the rearrangement of the epoxide to the vinylogous carboxylic acid in preparation for dehydrative ring closure to form 2 but were unable to effect the transformation. Instead, we opted for a stepwise approach by first reducing the epoxide to intermediate 19 then reoxidation; this was successful, but overoxidized to intermediate 20 ablating the C7 stereocenter. Regioselective reduction was achieved using zinc with ammonium chloride, however this delivered exclusively the undesired stereochemistry at C7 (21, highlighted red). Debenzylation to 22 occurred smoothly, and our key dehydrative ring closure was achieved with TFAA to afford racemic mixture 23, which was resolved by chiral supercritical fluid chromatography (SFC) to afford each enantiomer (1 and 24) in 99% e.e. The desired enantiomer was identified by comparison of chiral analytical SFC retention times to fermented samples of 1.
Scheme 2.

To complete the synthesis of 2, we leveraged our unique insight into the biosynthetic pathway (Scheme 1), specifically that the enzyme RosD would catalyze the transformation of 1 to 2 by epimerization of the C7 hydroxyl. Synthetic 1 was treated with the RosD epimerase which resulted in a 70% conversion, but only 11% isolated yield of 2 due to the small scale and the requirement for HPLC to separate the unreacted starting material. Under the same conditions, the racemic mixture 23 shows diminished conversion (30%), and the chirally purified diastereomer 24 showed no conversion to 25. These results provide further evidence that epimerization of the C7 hydroxyl is an enzymatically driven process catalyzed by RosD, and that the enzyme is stereospecific for (S)-stereochemistry at C5a. The analytical data for synthetic 1 and 2 matched the data for samples prepared by fermentation, which confirmed that we have completed the total synthesis of two GEMs.
During the synthetic sequence above we observed that ring opening of the racemic epoxide afforded an unexpected 2:1 mixture of trans (15a and 15b) and cis (15c and 15d) products (Scheme 3). We deduced that the cis product was formed by epimerization of the stereocenter α to the carbonyl of the initially formed trans product under the basic reaction conditions (i.e., 15a → 15c and 15b → 15d), and furthermore that 15d had the configuration corresponding to the epimerized C6 quaternary center (5aS,6R; highlighted blue). The cis product was isolated as a racemic mixture of 15c and 15d and using the same synthetic sequence as above elaborated to racemic mixture 26 and then resolved by chiral SFC. Leveraging the equilibrating nature of the epimerase the (5aS,6R,7S)-enantiomer 27 was treated with RosD, which resulted in 35% conversion by LC–MS and 6% isolated yield of the C7 epimer 28, requiring SFC purification to remove the starting material. The relative stereochemistry at C5a, C6 and C7 of 27 and 28 were confirmed by NOESY data (see Supporting Information). The (5aR,6S,7R)-enantiomer 29 was isolated but showed no conversion with the RosD epimerase.
Scheme 3.

In parallel to the total synthesis approach, we undertook a systematic simplification of the molecule, especially with respect to stereochemistry. We used the same disconnection strategy, but substituted into the synthesis a simplified nonaromatic ring, which expedited the synthesis and avoided the late-stage redox challenges discussed above. The β-diketone 30 was methylated to form 31 and protected with a benzoyl group in 32, then brominated with NBS. The resulting α-bromo ketone 33 was alkylated with an orsellinic acid derivative 34 (Scheme 4). The resulting intermediate 35 was cyclized using TFAA to afford racemic 36. The unsubstituted intermediate 36a (R6 = H) was resolved to 37 using chiral SFC then deprotected to afford 38. The simplified intermediate 36b (R6 = Me) was oxidized using Davis reagent to install the C7 OH, then deprotected and resolved using chiral SFC to afford 39 and subsequent RosD-catalyzed epimerization afforded 40.
Scheme 4.

Utilizing high-producing strains of Penicillium sp., identified from our fungal collection (Figure 4), we performed medium-scale static flask fermentations at up to 45 L batch size to isolate 2, which formed the basis for a semisynthesis campaign. Derivatives 41–50 were prepared in 1–4 steps (Scheme 5). Direct halogenation was effective, but silyl protection of the phenol was employed to enable palladium-catalyzed coupling and derivatization of the C7 OH. The structures of 2 and the chlorinated derivative 43 were confirmed by single crystal X-ray analysis (see Supporting Information).
Scheme 5.

Lead Optimization and SAR
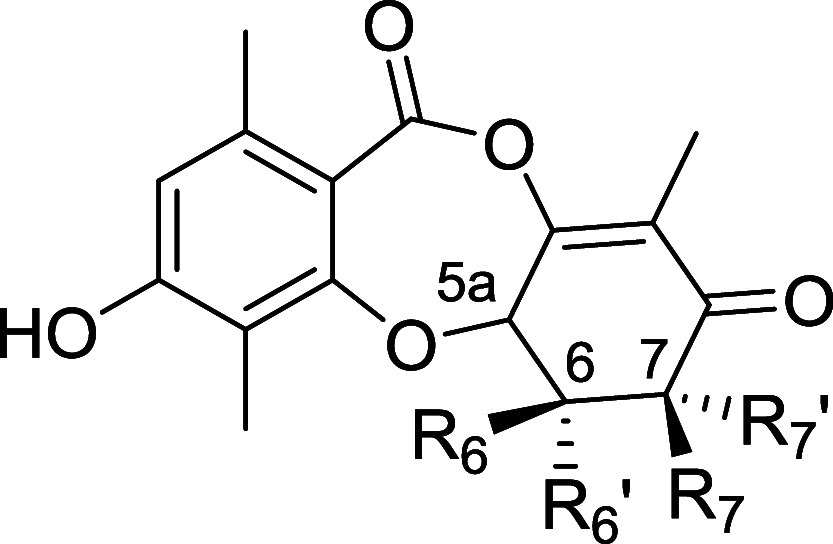
We were intrigued by the observation that 2 shows 25-fold enhanced biochemical inhibition of CDK2/cyclin E relative to the coisolated GEM 1, despite only a single difference between the two compounds, specifically the configuration at the C7 stereocenter. To ascertain the impact of the stereochemical configuration on the activity, we characterized our panel of stereoisomer analogs (Table 2). To confirm biological activity we tested compounds in parallel in live cells employing NanoBRET alongside in vitro enzyme assays; these data corroborated each other well. Compound 2 showed 21-fold greater biochemical inhibition than its C6 epimer 28, and for the epimerized scaffold the C7 diastereomer 28 was superior to 27, showing the same trend as 2 and 1. Compounds 24 and 29 (enantiomers of 1 and 27, respectively) lost significant activity against CDK2/cyclin E, and displayed noncovalent binding properties. The C6/C7-simplified analog set was also profiled, however complete removal of substituents at C6 and C7 (compound 38) led to substantial loss of activity on CDK2/cyclin E, which was partially restored upon reintroduction of both a methyl group at C6 and hydroxyl at C7 (39). Epimerization at C7 in 40 provided a further boost to activity, achieving a similar level of cellular inhibition to 1, but still 6-fold lower than 2.
Table 2. SAR of the Stereoisomers of 2 and Simplified Analogsa.
| enzyme
assay (kinact/KI × 10–3/s–1 μM–1) |
NanoBRET
(EC50, μM) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| compound | 5a | R6 | R6′ | R7 | R7′ | CDK2 | CDK1 | CDK7 | CDK2 | CDK1 | CDK7 |
| 1 (5aS,6S,7R) | (S) | Me | OMe | H | OH | 0.92 | 0.018 | 0.18 | 0.90 | 2.89 | 5.36 |
| 2 (5aS,6S,7S) | (S) | Me | OMe | OH | H | 23 | 1.1 | 9 | 0.14 | 0.61 | 0.30 |
| 24 (5aR,6R,7S) | (R) | OMe | Me | OH | H | rev (70 μM) | rev (>100 μM) | rev | >33 | >33 | >33 |
| 27 (5aS,6R,7S) | (S) | OMe | Me | OH | H | 1.1 | 0.11 | 0.08 | 0.33 | 1.6 | 4.0 |
| 28 (5aS,6R,7R) | (S) | OMe | Me | H | OH | 0.16 | 0.012 | 0.007 | 3.0 | 8.0 | 11.6 |
| 29 (5aR,6S,7R) | (R) | Me | OMe | H | OH | rev (9 μM) | rev (19 μM) | rev | 22 | >33 | >33 |
| 38 | (S) | H | H | H | H | 0.0002 | 26.6 | ||||
| 39 | (S) | H | Me | H | OH | 0.14 | 0.01 | 1.62 | 16.35 | 30.2 | |
| 40 | (S) | H | Me | OH | H | 0.10 | 0.0057 | 0.01 | 0.87 | 35 | 55.5 |
Stereochemistry indicated for stereoisomers of 2. Rev = Reversible. Enzyme and NanoBRET activity assays were assessed on the active CDK/cyclin holoenzyme as follows: CDK2/cyclin E1, CDK1/cyclin B1, CDK7/cyclin H/MAT1. Mean values calculated from at least two independent replicates for enzyme assays and NanoBRET; exception: NanoBRET compounds 24 and 38 were run n = 1 and inactive.
In aggregate, these observations suggest that both the presence of substituents and the natural stereochemical configuration are crucial for the covalent inhibition of human CDK2/cyclin E, likely determining the proximity and orientation of the vinylogous anhydride to the catalytic lysine. Furthermore, the range of activities across the chemical series demonstrates that this unusual motif is not an indiscriminate electrophile, but rather a tunable warhead.
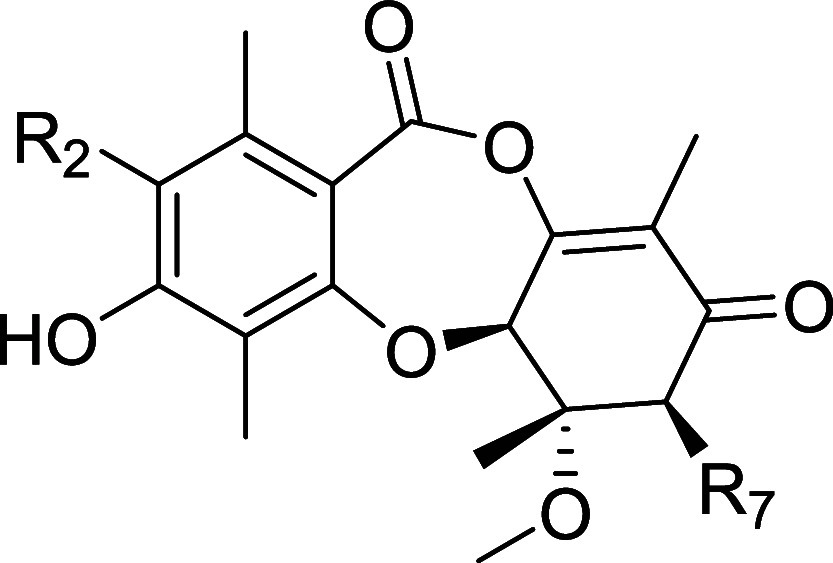
The CDK inhibitory profiles on the semisynthetic derivatives of 2 are shown in Table 3. The C7 OH tolerated diverse functionality, such as acetyl, glycine, methylcarbamate and a linked biotin moiety, but there was a universal loss of activity. The R2 position was also synthetically tractable, and we were able to establish an SAR trend according to size across compounds 41–45. The smaller R2 substituents are more active: biochemically H > F, Me, Cl > Br ≫ Et; and in the NanoBRET cellular context H, F > Cl > Me > Br ≫ Et. It is also worthy of note that larger substituents R2 = Cl (43) and Br (44) show increased selectivity for CDK2 over CDK7 relative to smaller substituents H and F (2 and 41, respectively).
Table 3. SAR of Semisynthetic Analogsa.
| enzyme
assay (kinact/KI × 10–3/s–1μM–1) |
NanoBRET
(EC50, μM) |
|||||||
|---|---|---|---|---|---|---|---|---|
| compound | R2 | R7 | CDK2 | CDK1 | CDK7 | CDK2 | CDK1 | CDK7 |
| 2 | H | OH | 23 | 1.1 | 9 | 0.14 | 0.61 | 0.3 |
| 41 | F | OH | 5.9 | 0.044 | 0.71 | 0.13 | 0.57 | 0.35 |
| 42 | Me | OH | 2.7 | 0.23 | 0.3 | 0.99 | 5.25 | 5.76 |
| 43 (XC219) | Cl | OH | 5.8 | 0.35 | 0.21 | 0.23 | 1.0 | 3.4 |
| 44 | Br | OH | 1.0 | 0.14 | 0.05 | 0.7 | 4.9 | 12.2 |
| 45 | Et | OH | 0.025 | 0.012 | 0.005 | >33 | >33 | >33 |
| 47 | H | OAc | 3.18 | 0.17 | 1.1 | 1.5 | 4.3 | 1.6 |
| 48 | H | OC(O)OMe | 0.5 | 0.059 | 0.31 | 6.1 | 17.7 | 6.3 |
| 49 | H | OGly | 3.9 | 0.28 | 5.6 | 1.4 | 6.5 | 1.8 |
| 50 | H | OC(O)(CH2)2–PEG4-biotin | 0.28 | 0.077 | ||||
| 51 (XC208) | H | OH | rev (3 μM) | rev (7 μM) | rev (9 μM) | >33 | >33 | >33 |
Rev = Reversible. Enzyme and NanoBRET activity assays were assessed on the active CDK/cyclin holoenzyme as follows: CDK2/cyclin E1, CDK1/cyclin B1, CDK7/cyclin H/MAT1. Mean values calculated from at least two independent replicates for enzyme assays and NanoBRET.
The chlorinated analog 43 (LifeMine code XC219) was selected for additional profiling as it retained potent biochemical and cellular inhibition of CDK2 while demonstrating improved selectivity against CDK7. We acquired an X-ray crystal structure of 43 in complex with CDK2/cyclin E, which showed that its binding mode matches that of 2, with the C3 phenol binding the hinge and formation of a covalent bond from C11 to Lys33 (Figure 8). The R2 vector projects toward Val64 of CDK2, which rotates to accommodate the Cl atom of 43. There is a point-change of the corresponding residue to Ile75 in CDK7, and we propose that the larger residue cannot accommodate the R2 = Cl, affording selectivity for CDK2 over CDK7.
Figure 8.

(A) Cocrystal structure of CDK2/cyclin E with 2. (B) Cocrystal structure of CDK2/cyclin E with XC219 43 (PDB ID 9BJD) showing the same binding pose as 2 with rotation of Val64 to accommodate the larger R2 = Cl substituent.
Selectivity across the Kinome and Proteome
The vinylogous anhydride of 2 reacts with the active site lysine of CDK2 (Lys33) to form a covalent bond. This residue is highly conserved within the protein kinase family, which raises the question of the selectivity of 2 not only for CDKs but for the entire kinome.34 Furthermore, the vinylogous anhydride of 2 clearly shows conditional reactivity with CDKs, but its electrophilic nature could give rise to off-target reactivity. To assess the selectivity of 2 across the human kinome, we tested 2 in a competitive Kinobead pull-down assay.35 The assay revealed remarkably high overall selectivity, with the primary human targets of 2 being the CMGC family of proline-directed serine/threonine kinases (Figure 9A), which contains 21 CDK family members. A small number of additional kinase targets were identified in the CK1, CAMK, and TKL classes. A broader competitive chemoproteomic screen using the biotinylated probe 50 in lysates treated with 25 μM of 2 detected no additional nonkinase targets (Supporting Figure 9).
Figure 9.

Kinome binding profile of 2 and analogs using the Kinobead assay. (A) Profile of GEM 2 (2 μM) compared to (B) chlorinated analog 43 (2 μM) with improved CDK2 selectivity; (C) acetylated analog 47 (2 μM) with alternate selectivity in the CAMK group; (D) analog 38 (50 μM) with alternate specificity in the CK1 group. Kinases with engagement >50% are labeled, kinase classes that show differentiation between analogs of 2 are highlighted.
We also performed an analogous Kinobead experiment in S. cerevisiae lysate to profile the fungal kinase targets of 2, revealing potent (EC50 = 31 nM) and selective engagement of yeast PHO85, consistent with the genomic target prediction based on the fungal Pho85 ETaG in the ros BGC. Compound 2 engaged yeast PHO85 as the top ranking target out of 81 detected kinases in the cell lysate, demonstrating >100-fold higher potency relative to nearly all other detected kinases, including fungal CDK orthologs CDC28, KIN28, and SGV1 (Supporting Figure 10).
We profiled a diverse selection of synthetic analogs in the human Kinobead assay (Figure 9B–D). The improved selectivity of 43 versus 2 was recapitulated in the Kinobead assay, and extended to the larger CDK family CDK5, 7, 12, and 13, and importantly the kinome in general. The transcriptional regulators CDK7, 12, and 13 are known to have a narrow therapeutic window, while reduced inhibition of CDK5 offers more favorable neurological safety.36 Beyond the CDK family we also noted improved selectivity on kinases such as MAP3K11 and CSKN1E. The C7 acetylated analog 47 led to a slight shift of activity favoring the CMGC kinase CLK2, and the CAMK kinase AURKC. The simplified analog 38 showed barely detectable CDK2 activity but was highly selective for a therapeutically valued target, CK1 kinase CSNK1A1,37 albeit with weak activity.
Pharmacokinetics
The in vivo pharmacokinetic profiles of 2 and 43 are reported in Table 4. 2 shows high clearance and low oral bioavailability in mouse and rat. In contrast, 43 shows low clearance and good oral bioavailability in both rodent species. Here, a single atom change afforded a significant improvement in the PK profile, and we attribute this to the improved microsomal stability observed in vitro.
Table 4. Pharmacokinetic and Select In Vitro ADME Parameters of Compounds 2 and 43.
| Cpd 2 | Cpd 43 | |
|---|---|---|
| Mouse PK | ||
| IV dose (mg/kg) | 0.94 | 0.81 |
| AUC (ng·h/mL) | 93 | 1263 |
| Cl (mL/min/kg) | 176 | 13.1 |
| Vss (L/kg) | 6.3 | 1.6 |
| t1/2 (h) | 0.6 | 5.2 |
| oral dose (mg/kg) | 2.79 | 2.42 |
| AUC (ng·h/mL) | 42 | 2058 |
| F (%) | 12 | 54 |
| Rat PK | ||
| IV dose (mg/kg) | 0.89 | 0.83 |
| AUC (ng·h/mL) | 229 | 1288 |
| Cl (mL/min/kg) | 68 | 11 |
| Vss (L/kg) | 29.5 | 2.0 |
| t1/2 (h) | 9.0 | 3.1 |
| oral dose (mg/kg) | 2.73 | 2.55 |
| AUC (ng·h/mL) | 227 | 2261 |
| F (%) | 25 | 58 |
| Microsomal Stability | ||
| mouse t1/2 (min) | 18.2 | 65.2 |
| rat t1/2 (min) | 41.0 | 148 |
Brain and plasma exposures were evaluated over a 24 h period in mice administered with a single dose of either 2 or 43, at 30 mpk. Interestingly, 2 showed exceptionally high levels of brain exposure (AUC blood-to-plasma ratio of 0.68), and 43 significantly reduced brain exposures (AUC b/p ratio = 0.0003).
High-dose intraperitoneal (i.p.) mouse PK was conducted to determine tolerability. Although animals treated with a single 50 mpk dose of 2 presented with acute body weight loss within 3 days, animals treated with 43 tolerated 7 days of daily dosing at 100 mpk with no adverse observations noted. We attributed the improved tolerability to the improved selectivity profile.
In Vivo Efficacy
Given the favorable selectivity, PK and tolerability profiles, 43 was advanced to an efficacy study to evaluate this novel mechanism of CDK2 inhibition in a gastric patient derived xenotransplant (PDX) model. We selected a rapidly proliferating PDX, CRT00292 gastric mouse model harboring a high CCNE1 amplification (CN = 12), which is a predictive biomarker of response to CDK2 inhibition. Animals with established subcutaneous tumor fragments were dosed daily i.p. for 14 days, resulting in tumor growth inhibition (TGI) measured relative to the vehicle control over the duration of the study (Figure 10). At 14 days 43 showed 44 and 80% TGI at 30 and 100 mpk, respectively, demonstrating a dose-dependent response and a remarkable level of TGI at the high dose in this aggressive PDX model.
Figure 10.
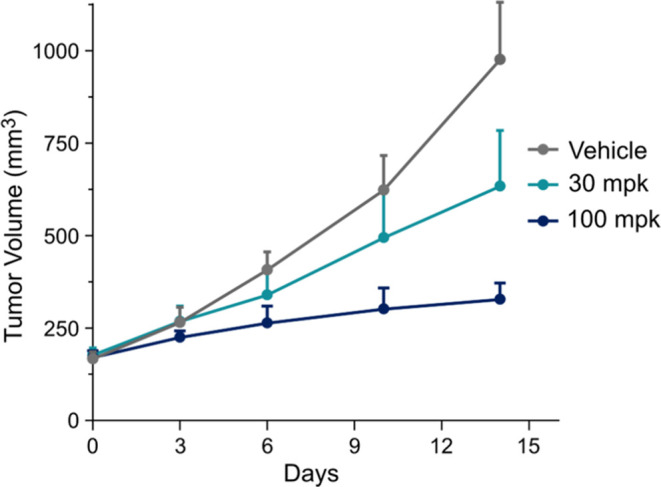
Antitumor growth activity in a CCNE amplified (CN = 12) gastric PDX model CRT00292. NOG mice were inoculated subcutaneously with tumor fragments and randomized (n = 7/group) when tumors reached an average of 150–200 mm3. Animals were dosed daily with XC219 43 (30 and 100 mpk × 14d, i.p.). In the highest dose group 3 animals were moribund or withdrawn from treatment. One-way ANOVA, Dunnett’s multiple comparison test was performed reporting statistical significance of 100 mpk group, p value = 0.0079.
Conclusions
In this work, we have employed ETaG-based genomic search across a comprehensively sequenced and functionally annotated collection of over 100,000 diverse wild-type fungi (LifeBase) to identify an inhibitor of a preselected human target family, cyclin-dependent kinases. To our knowledge, this represents the first example in which a fungal GEM inhibiting a human protein of therapeutic interest was discovered first by a targeted search in genomic space.21 In other words, our TD3 discovery paradigm starts with a human target, progresses to the discovery of genes encoding a fungal GEM predicted to engage that human target, and then progresses further to the isolation and characterization of the GEM. This is in reversed order compared to the historical paradigm of “natural product” drug discovery, in which a GEM was first isolated and in some cases its target identified, but only in relatively rare instances was the producing organism sequenced and the genetic origins of the GEM characterized. The historical emphasis on searching through chemical space has led to a situation in which many GEMs in the literature have been isolated, structurally characterized and reported to possess biological activity in human cells, but the target(s) responsible for that activity has not been elucidated. Genomic search for BGCs using the presence of a colocated ETaG offers a powerful means to deorphanize the targets of such GEMs. The present work exemplifies the use of genomic search for GEM deorphanization—1 and 2 had been isolated decades ago and shown to possess antiproliferative activity in cultured human cells, but their molecular target(s) had not been identified. Here we found that, as predicted by the presence of the CDK-homologous rosG ETaG in the ros BGC, 1 and 2 indeed encode inhibitors of both fungal and human CDK enzymes. In work to be reported elsewhere, we have undertaken a systematic deorphanization of known GEMs and are developing AI-based methods to correlate BGC and GEM structure.
The ability to search in genomic space for therapeutics that engage a predetermined human target offers substantial advantages over searching for GEMs through chemical space using activity-based fractionation. As demonstrated in this study, up-front knowledge of the genes encoding a GEM enables the transcriptional activity of the cluster to be monitored as a function of growth conditions, allowing for GEM production to be maximized prior to initiation of isolation. The functions of the genes within the BGC can provide important insights into the chemical structure of the GEM that aid in isolation based on MS/MS fragmentation, for example analysis of the ros BGC suggested a heavily oxidized orsellinic acid derivative with a methylated heteroatom. Furthermore, enzymes encoded within the BGC can be used as reagents in a hybrid bio/chemo-catalytic synthesis to aid downstream preparation of the GEM and analogs for SAR. Of the roughly 100,000 genomicized strains in LifeBase, 900 bore a colocated assemblage of the ros genes, though the order of genes within the cluster was not conserved. Targeted profiling of a subset of these strains for the transcriptional activity of the ros genes under various growth conditions enabled us efficiently to identify a wild-type fungal strain that natively overproduces 1 and 2.
Fungal proteins exhibit a high degree of structural relatedness to their functionally related human counterparts, with sequence and structural conservation being especially high in enzyme active sites and surfaces that engage in protein–protein interactions. This relatedness leads to species crossover, whereby a GEM evolved in fungi to target fungal proteins engages the corresponding human protein. Given the high relatedness of the fungal CDK Pho85 to human CDKs, it comes as no surprise that 1 and 2 engage human CDKs. Species crossover is however complicated by homologue radiation; whereas there are 6 CDKs in fungi, the need for more specialized orchestration of cell cycle related mechanisms in humans versus fungi has resulted in radiation to 21 CDK homologues in humans. For radiated human targets, sequence comparisons alone are unlikely to be predictive of human homologue selectivity for a fungal-derived GEM, because interactions with a drug are determined by a handful of amino acid residues on the target, whereas phylogenetic analysis encompasses the entire protein sequence. In the case of 1 and 2, human homologue selectivity is further complicated by the covalent inhibition mechanism of these GEMs, with contributions from both noncovalent binding (KI) and the kinetics of covalent bond formation (kinact). Thus, while 1 and 2 show marked selectivity for human CDKs over other protein kinases, selectivity among the human CDK homologues is poorly predicted by phylogenetic distance (compare Figures 9A and 2). To emphasize this point for proteins of therapeutic importance, 1 and 2 show little activity on CDK4 and CDK6 but have robust activity on CDK1 and CDK2, though the former are only marginally more distant from Pho85 than the latter. However, in the context of fungal kinases 1 and 2 show exquisite selectivity for Pho85 versus the other 5 endogenous CDK homologues.
The structural features in kinase inhibitors that give rise to selectivity can be subtle and difficult to predict. That is indeed so in the case of 1 and 2, for which the kinetics of covalent bond formation make a major contribution to selectivity. The aromatic ring of 1 and 2 slots into the active site in a manner consistent with other orthosteric kinase inhibitors, with the phenolic hydroxyl engaging the hinge region (Figure 6B). We were able to gain insight into the binding interaction that precedes covalent bond formation using the analog 51, in which the vinylogous anhydride of 2 is replaced by an amide-NH, rendering the analog inert. The structure of CDK2/cyclin E bound to 51 (Figure 6C) shows the molecule optimally positioned in the active site for nucleophilic attack by Lys33. GEMs 1 and 2 are selective by many orders of magnitude for Lys33 over other lysines in CDK2 as well as lysines in other proteins, which in combination with the optimal binding pose demonstrates that covalent bond formation with Lys33 is enzyme-activated, most likely by a combination of orbital steering and enzyme-assisted lysine desolvation. Nearly all protein kinases possess a lysine residue in the same position as Lys33 in CDK2, and yet 1 and 2 are far from promiscuous kinase inhibitors.
The origins of kinase selectivity in 1 and 2 are likely to derive from the substituents and stereochemistry of the GEMs, especially at the junction with the 7-membered ring (C5a) and the adjacent C6. Both of these critical positions are, interestingly, remote from the hinge-interacting region. The stereocenter at C7 may be less important for selectivity, as 1 and 2 are stereoisomeric at this position, yet exhibit nearly identical kinase selectivity. Our SAR studies provided further insights into the role of these positions in determining selectivity. For example, at equal concentrations the enantiomer of 1 is devoid of kinase inhibitory activity when 1 and 2 show nearly complete inhibition. Analog 38, stripped of stereochemistry and substituents at C6 and C7, was drastically reduced in CDK-inhibitory activity but had undergone reprogramming to target a completely different kinase, CSNK1A1 (Figure 9D). This observation suggests that a systematic SAR exploration of the tricyclic scaffold in 1 and 2 is warranted, as it is likely to yield a family of novel Lys-targeting covalent kinase inhibitors, possibly with high brain penetration. Finally, semisynthetic chloro-substitution at C2 provided an analog with improved selectivity for CDK2 compared to 1 and 2, retaining good CDK2 activity but reducing activity against several other CDKs (e.g., CDK7) and against other protein kinases (e.g., MAP3K11) (compare Figure 9A,B).
A recent report confirming our discovery that 2 targets CDKs failed to observe a covalent binding mode, despite presentation of a ligand-protein cocrystal structure (PDB ID 8OY2).22 In our previous disclosure and as further elaborated here we provide multiple lines of evidence, from intact mass spectrometry to rigorous enzymological characterization and high-resolution structural information, all confirming that 1 and 2 are irreversible covalent inhibitors that modify the active site Lys residue (Lys33 in CDK2) of their kinase targets. The discrepancy is most likely attributable to the fact that noncovalent binding was reported from CDK2 in the absence of its obligate cyclin partner, whereas this study utilized the relevant physiologic form of CDK2 in complex with its cognate cyclin E. Cyclins are essential binding partners required to activate CDKs both in vitro and in vivo and, since ATP interacts differently with these enzymes in the presence and absence of the cyclin,38 it is straightforward to imagine that the CDK would interact differently with inhibitors that also bind the ATP site.
The molecule series reported here has many interesting and promising properties that differentiate them radically from existing orthosteric kinase inhibitors. First, they have a rigid tricyclic ring system devoid of nitrogen atoms, as opposed to the nitrogen-rich beads-on-a-string structure of most human-designed kinase inhibitors. This difference may underlie the high brain exposure seen with 2, as brain penetration in small molecule drugs is typically counter-associated with rotatable bonds.39 Brain penetration is a highly desirable feature of cancer drugs in general but even more so in those being used to treat breast cancer, as metastasis to the brain is commonly observed and is often aggressively lethal in patients taking nonbrain-penetrant precision medicines.40 Second, this family of molecules possess highly conditional enzyme-activated Lys-specific warheads, an attribute that humans have found difficult to design but fungi have produced via evolutionary selection. In cancer drugs that covalently attach to Cys residues, mutational escape by genetic alteration of the Cys residue is common, because there is strong selective pressure favoring such mutation and little selective pressure against it.41 Similar mutational escape would be ineffective for molecules of the 1/2 scaffold class, because the Lys residue is essential for the catalytic activity of the kinase targets, hence there is strong selective pressure against its loss. While the most promising molecules presented here will require further optimization to increase potency and to improve the modest ∼5-fold CDK2 versus CDK1 selectivity, the work detailed here provides substantial enablement for that optimization campaign. Beyond CDKs, we expect that this pharmacologically privileged scaffold will find broader utility in the important and burgeoning field of kinase inhibition.
In a future publication, we will report the results of a comprehensive genomic search detailing all BGCs in the LifeBase collection of over 100k fungal genomes having an ETaG predictive of the fungal and human target of the encoded GEMs. Given the speed of retrieval and the structural and mechanistic novelty of fungal GEMs, we expect that the TD3 paradigm of drug discovery will make an increasingly visible contribution to the transformative medicines of the future.
Experimental Section
CDK Activity Assay
Kinase inhibition experiments were performed using recombinant and commercial-available (Carna Biosciences, ProQinase) cyclin-dependent kinases utilizing previously published fluorescence-based kinase probes.42 First, a 12-point dose curve of each compound was serially diluted in DMSO in 96-well V-bottom plates (Costar #3357) from a 10 mM DMSO stock, and 1 μL transferred to 384 well black flat bottom plates (Corning #3575). For each CDK, a 5× solution of ATP and the corresponding fluorogenic peptide substrate (AssayQuant Technologies, Inc.) were prepared in Kinase Assay Buffer (50 mM HEPES, 10 mM MgCl2, 0.01% n-octyl β-d glucopyranoside (NOG), 50 μM TCEP, pH 7.5) and 5 μL added to each well so the final concentrations of ATP and peptide substrate were 1× Km and 10 μM, respectively. Finally, a 1.2× solution of CDK enzyme in Kinase Assay Buffer was prepared and added using the instrument syringe of the ClarioStar 2 (BMG LabTech) to a final volume of 25 μL. Final concentrations of enzyme and ATP were 2 nM and 25 μM for CDK2/cyclin E1, 4 nM and 25 μM for CDK1/cyclin B1 and 20 nM and 50 μM for CDK7/cyclin H/MAT1. Once added, the fluorescent signal (Ex 360 nm, Em 485 nm, Gain 1800) was measured continuously over a 60 min period to produce a time course curve.
For compounds with fast kinact kinetic assays were performed by monitoring phosphorylation of a fluorescence-based kinase probe (AQT0297, AssayQuant) by CDK2/cyclin E1 (Carna Biosciences) using a stopped flow fluorescence spectrometer (SX-20, Applied Photophysics). Two solutions were prepared for the runs: Solution A: ATP (50 μM) and AQT0297 (10 μM) with varied concentrations of inhibitor in a modified Kinase Assay Buffer (50 mM HEPES pH 7.5, 10 mM MgCl2, 0.01% n-octyl β-d glucopyranoside, 50 μM TCEP, 4% DMSO), and Solution B: CDK2/cyclin E1 (35 nM) in the modified Kinase Assay Buffer were prepared. Each solution was used to fill one syringe of the stopped flow system. Each analyte concentration was flushed 5 times using the drive function followed by three acquisitions with 0.5 s intervals (excitation 360 nm, PMT filter 435 nm, voltage 520 V), and data was exported from the instrument for fitting.
For time-dependent inhibitors, the time course was fitted using the following equation to determine the pseudo-first-order rate constant, kobs for each inhibitor concentration
where V is the initial rate of fluorescence change; kobs is the rate constant for the progression of the rate from V to zero; t is time; FI is fluorescence intensity and BF is background fluorescence. Fitted kobs were plotted against compound concentrations [I] to determine if covalent inactivation shows either single-step or two-step inactivation kinetics. For single-step inactivation kinetics, a linear fit of the plot determines the kinact/KI value for the tested compound. For two-step inactivation, the data was fitted using the following equation to determine KI and kinact for each compound against the tested CDK enzyme, and kinact/KI was calculated from those determined values.
For reversible inhibitors, the slope of the time course was determined for each well through the ClarioStar analysis software (MARS, BMG Labtech). Slopes values (S) were normalized against positive (P, 100% inhibited) and negative (N, 0% inhibited) control wells using the following equation, plotted against compound concentrations, and fitted to a four-parameter nonlinear curve model shown below to determine the IC50 value.
NanoLuc Bioluminescence Resonance Energy Transfer (NanoBRET) Assay for Target Engagement
HEK293 cells were cotransfected with CDK2-NanoLuc Fusion Vector (CDK2: NV2781) and CCNE1 Expression Vector (CCNE1: NV2781); CDK1-NanoLuc Fusion Vector (CDK1: NV2701) and CCNB1 Expression Vector (CCNB1: NV2601); or CDK7-NanoLuc Fusion Vector (CDK7: NV2851) (Promega, Madison, WI) and Expression Vectors CCNH1 and MNAT1 (GenScript, Piscataway, NJ). Cells were seeded at 20,000 cells/well in 96-well plates in DMEM media supplemented with 10% FBS. The compounds were serially diluted from a 10 mM DMSO stock and tested at a top concentration of 33 μM with 3-fold dilution in a 10-point dose curve. Cells were incubated for 2 h at 37 °C 5% CO2 and performed per manufacturer’s specifications (Promega, Madison, WI). Tracer #9 (K-9; N2631) and Tracer #10 (K-10; N2641) were added in appropriate wells. NanoBRET NanoGlo Substrate and Extracellular NanoLuc Inhibitor were added, and donor (450 nm) and acceptor (610 nm) emission were measured within 10 min on a ClarioStar 2. Mean corrected milliBRET units (mBU) was calculated as follows
Cell Proliferation Assay
Ovarian cancer cell line, OVCAR-3 cells were seeded at 3000 cells/well in 96-well plates in RPMI media supplemented with 20% FBS + 10 μg/mL insulin for 24 h at 37 °C 5% CO2. The compounds were serially diluted from a 10 mM DMSO stock and tested at a top concentration of 33 μM with 3-fold dilution in an 8-point dose curve. Cells were incubated for 5 days at 37 °C 5% CO2. For the washout conditions, the same method was applied except media containing DMSO or compound was removed after 4 h of treatment, then washed 2× with 1× PBS and replaced with complete media without compound.
To determine the relative growth upon treatment, CellTiter-Glo 2.0 Viability Assay (Promega, Madison, WI) was performed per manufacturer’s specifications and luminescence was read on the ClarioSTAR1 with a gain of 3100. Mean GI50 values were determined using a four-parameter fit determined by GraphPad Prism software.
pRb T821/826 AlphaLISA Assay
Phosphorylation of RbT821/826 was measured in OVCAR-3 cells using the Multiplex Surefire Ultra Phospho Thr821/826/Total Rb Assay Kit MPSU-PTRB-N500 according to manufacturer’s specifications (Revvity, Waltham, MA). Cells were seeded at 25,000 cells/well in a 96 well plate in serum-free DMEM for 24–30 h at 37 °C 5% CO2. Media was replaced the following day with complete DMEM containing 20% FBS+ 10 μg/mL insulin and treated for 18 h with either DMSO or compounds. The compounds were serially diluted from a 10 mM DMSO stock and tested at a top concentration of 10 μM with 3-fold dilution, to obtain a 9-pt dose curve for pRb levels on an EnVision plate reader (PerkinElmer). For the washout conditions, the same method was applied except media containing DMSO or compound was removed after 4 h of treatment, then washed 2× with 1× PBS and replaced with complete media without compound. Data were processed with GraphPad Prism using a four-parameter fit to calculate mean IC50.
In Vivo Pharmacology and Pharmacokinetics
Female NOG mice were purchased from Taconic and implanted at 10–16 weeks old. Animals were housed for a minimum 3-day acclimation period for all nonanesthesia-based procedures. Animals were housed in individual HEPA ventilated cages (Innocage IVC, Innovive). Fluorescent lighting was maintained on a 12-h cycle. Temperature and humidity were monitored, recorded daily, and maintained at 68–74 °F (20–23 °C) and 30–70% humidity, respectively. 2920X.10 18% soy irradiated rodent feed (Teklad) was provided and available ad libitum. Acidified water (pH 2.5–3) (Aquavive, Innovive) was provided and available ad libitum. female NOG mice were implanted with CRT_STAD_00292 tumor fragments subcutaneously into the right rear flank. The mice were anesthetized using isoflurane induction. Animals were shaved and surgically prepped using 70% isopropyl alcohol. A small incision was made to the lower right rear flank of the mouse. A subcutaneous space was created using gentle blunt dissection. A tumor fragment was inserted into the subcutaneous pocket and a wound clip was applied to seal the incision. Analgesic was administered. Tumor volumes were recorded twice weekly using digital calipers. The length (L) and the width (W) of the tumors were measured and tumor volumes were automatically calculated using the following formula: 0.5 × L × W2. When tumors reached an average of 170 mm3, ranging from 111–262 mm3, 35 animals were randomized into the respective treatment groups and dosed within 24 h. Body weights were measured 2 times weekly following randomization and throughout treatment. Animals were dosed at 30 or 100 mpk daily, q.d. for 14 days and monitored until animals reached a tumor volume of 1500 mm3, or humane end point, whichever occurred first. BW of the mouse Body weight changes were calculated based on the first day of treatment.
Percent inhibition was calculated as follows
Tt = mean tumor volume of treated at time t, T0 = mean tumor volume of treated at time 0, Ct = mean tumor volume of control at time t and C0 = mean tumor volume of control at time 0. One-way ANOVA, Dunnett’s multiple comparison test was performed.
Pharmacokinetic Assessment was conducted in Laboratory Animal studies following Institutional Animal Care and Use Committee Care, Welfare for laboratory animals used including male Sprague–Dawley rat, male CD-1 mice following single intravenous and/or oral administration. Blood samples were collected in EDTA collection tubes for plasma protein precipitation followed by LC/MS/MS analysis. PK profile parameters were analyzed by noncompartmental methods (WinNonlin Professional, version 4.1).
Chemistry
General Methods
All commercially available materials were used as received without further purification. Air or moisture sensitive reactions were carried out under an atmosphere of nitrogen. Flash chromatography was conducted on Biotage instruments with the silica gel cartridges and elution gradients indicated. Chiral SFC was conducted on a Waters UPC2 or Agilent 1260 instrument using column and elution system indicated. Reverse phase HPLC was conducted on an ISCO instrument with Sunfire column unless otherwise specified. NMR spectra were recorded on a Bruker 400 or 500 MHz spectrometer. Chemical shifts (δ) are quoted in parts per million (ppm) referenced to the solvent peak. Coupling constants (J) are reported in hertz (Hz). LC–MS were recorded on Agilent instrument with C18 column eluting with acetonitrile/water gradient with 0.1% formic acid. All tested compounds were determined to be ≥95% pure by LC–MS or analytical HPLC.
General Procedure for the Epimerase Reaction
To a 40 mL vial was added substrate (10 mg) as a solution in DMSO (0.29 mL), aqueous epimerase enzyme solution (0.29 mL 400 μM) and TRIS buffer (28.1 mL, 50 mM, pH 9.0). The vial was shaken for 90 min at 30 °C then 30 mL acetonitrile was added and the reaction filtered. The resulting mixture was concentrated in vacuo and purified by reverse phase HPLC to afford epimerized material.
6,9-Dimethyl-1,4-dioxaspiro[4.5]deca-6,9-dien-8-one (12)
To a solution of PhI(OAc)2 (131.8 g, 409.3 mmol, 4 equiv) in hexane (500 mL) was added 2,5-dimethylphenol (12.5 g, 102 mmol, 1 equiv) and ethylene glycol (82.6 g, 1.33 mol, 74.4 mL, 13 equiv) at 0 °C. The mixture was stirred at 25 °C for 16 h. The reaction mixture was diluted with H2O (500 mL) and extracted with EtOAc (3 × 500 mL). The combined organic phase was washed with brine (3 × 500 mL), dried with anhydrous Na2SO4, filtered and concentrated in vacuum. The residue was purified by flash chromatography (0–10% EtOAc in petroleum ether, 220 g column) to afford 6,9-dimethyl-1,4-dioxaspiro[4.5]deca-6,9-dien-8-one (12) (18 g, 47% yield) as a yellow oil. LC–MS m/z = 181.0 [M + H]+.
1,4-Dimethyl-7-oxaspiro[bicyclo[4.1.0]heptane-2,2′-[1,3]dioxolan]-3-en-5-one (13)
To a solution of 6,9-dimethyl-1,4-dioxaspiro[4.5]deca-6,9-dien-8-one (12) (10 g, 55.5 mmol, 1 equiv) in MeOH (200 mL) was added H2O2 (30% w/w, 23.6 g, 208 mmol, 20 mL, 3.75 equiv) and Na2CO3 (1 M, 111 mL, 2 equiv) at 0 °C. The mixture was degassed, purged with N2 3 times and stirred at 25 °C for 5 h. The reaction mixture was quenched by the addition of aqueous Na2SO3 (30 mL) at 0 °C, and then the reaction mixture was diluted with H2O (500 mL) and extracted with EtOAc (3 × 500 mL). The combined organic phase was washed with brine (3 × 200 mL), dried with anhydrous Na2SO4, filtered and concentrated in vacuum. The residue was purified by flash chromatography (0–5% EtOAc in petroleum ether, 80 g column) to afford 1,4-dimethyl-7-oxaspiro[bicyclo[4.1.0]heptane-2,2′-[1,3]dioxolan]-3-en-5-one (13) (2.3 g, 10% yield) as a yellow oil. LC–MS m/z = 196.9 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 6.07 (ddd, J = 1.5, 3.6, 5.2 Hz, 1 H), 4.22–4.14 (m, 1 H), 4.08 (brdd, J = 2.8, 6.5 Hz, 2 H), 4.03–3.96 (m, 1 H), 3.28–3.20 (m, 1 H), 1.76–1.70 (m, 3 H), 1.45–1.38 (m, 3 H).
rac-Benzyl 2-(((6R,7R)-6-Hydroxy-6,9-dimethyl-8-oxo-1,4-dioxaspiro[4.5]dec-9-en-7-yl)oxy)-4-((4-methoxybenzyl)oxy)-3,6-dimethylbenzoate (15)
A mixture of 1,4-dimethyl-7-oxaspiro[bicyclo[4.1.0]heptane-2,2′-[1,3]dioxolan]-3-en-5-one (13) (4.2 g, 1 equiv, 21 mmol) and benzyl 2-hydroxy-4-((4-methoxybenzyl)oxy)-3,6-dimethylbenzoate (14) (25 g, 3 equiv, 64 mmol), NaOH (2.6 g, 3 equiv, 64 mmol) in ACN (400 mL) was degassed and purged with N2 3 times, and then the mixture was stirred at 65 °C for 72 h under N2 atmosphere. The reaction mixture was filtered and concentrated under reduced pressure to give a residue. The residue was purified by flash chromatography (1–25% EtOAc in petroleum ether) to afford rac-benzyl 2-(((6R,7R)-6-hydroxy-6,9-dimethyl-8-oxo-1,4-dioxaspiro[4.5]dec-9-en-7-yl)oxy)-4-((4-methoxybenzyl)oxy)-3,6-dimethylbenzoate (15) (2.5 g, 20%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.47–7.40 (m, 2H), 7.40–7.31 (m, 5H), 6.92 (d, J = 8.8 Hz, 2H), 6.47 (s, 1H), 6.31 (d, J = 1.3 Hz, 1H), 5.41–5.35 (m, 1H), 5.31–5.25 (m, 1H), 5.03 (s, 1H), 4.97 (s, 2H), 4.91 (s, 1H), 4.31–4.26 (m, 1H), 4.25–4.16 (m, 1H), 4.12–4.01 (m, 2H), 3.83 (s, 3H), 2.17 (s, 3H), 2.11 (s, 3H), 1.86 (d, J = 1.5 Hz, 3H), 1.31 (s, 3H).
rac-Benzyl 2-(((6S,7R)-6-Hydroxy-6,9-dimethyl-8-oxo-1,4-dioxaspiro[4.5]dec-9-en-7-yl)oxy)-4-((4-methoxybenzyl)oxy)-3,6-dimethylbenzoate (15c and 15d)
Also isolated from the above purification was the title compound (1.5 g, 12%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.47–7.41 (m, 2H), 7.39–7.29 (m, 5H), 6.92 (d, J = 8.5 Hz, 2H), 6.52 (s, 1H), 6.38 (s, 1H), 5.38–5.27 (m, 2H), 5.02–4.89 (m, 2H), 4.39 (s, 1H), 4.21–4.02 (m, 4H), 3.83 (s, 3H), 3.64 (s, 1H), 2.23 (s, 3H), 2.02 (s, 3H), 1.84 (s, 3H), 1.29 (s, 3H).
rac-Benzyl 4-Hydroxy-2-(((1R,6R)-6-methoxy-3,6-dimethyl-2,5-dioxocyclohex-3-en-1-yl)oxy)-3,6-dimethylbenzoate (16)
Twelve batches of the following experiment were conducted then combined before purification. To a solution of rac-benzyl 2-(((6R,7R)-6-hydroxy-6,9-dimethyl-8-oxo-1,4-dioxaspiro[4.5]dec-9-en-7-yl)oxy)-4-((4-methoxybenzyl)oxy)-3,6-dimethylbenzoate (15) (500 mg, 1 equiv, 0.85 mmol) in THF (18 mL) was added NaH (60% dispersion in mineral oil, 0.17 g, 5 equiv, 4.25 mmol) at 0 °C for 50 min under N2 atmosphere, then added methyl trifluoromethanesulfonate (348 mg, 2.5 equiv, 2.12 mmol). The mixture was stirred at 0 °C for 1 h under N2 atmosphere. The combined reaction mixture was poured into water (18 mL) and then extracted with EtOAc (3 × 25 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by flash chromatography (1–25% EtOAc in petroleum ether) to afford rac-benzyl 2-(((6R,7R)-6-methoxy-6,9-dimethyl-8-oxo-1,4-dioxaspiro[4.5]dec-9-en-7-yl)oxy)-4-((4-methoxybenzyl)oxy)-3,6-dimethylbenzoate (3.6 g, 58% yield) as a white solid. LC–MS m/z = 603.3 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.49–7.40 (m, 2H), 7.39–7.29 (m, 5H), 6.92 (d, J = 8.6 Hz, 2H), 6.47 (s, 1H), 6.22 (d, J = 1.3 Hz, 1H), 5.39–5.32 (m, 1H), 5.30–5.21 (m, 2H), 4.98 (s, 2H), 4.26–4.19 (m, 1H), 4.17–4.07 (m, 2H), 4.05–3.97 (m, 1H), 3.88–3.78 (m, 3H), 3.55–3.48 (m, 3H), 2.27 (s, 3H), 2.19 (s, 3H), 1.77 (d, J = 1.4 Hz, 3H), 1.57 (s, 2H), 1.55 (s, 3H).
To a solution of rac-benzyl 2-(((6R,7R)-6-methoxy-6,9-dimethyl-8-oxo-1,4-dioxaspiro[4.5]dec-9-en-7-yl)oxy)-4-((4-methoxybenzyl)oxy)-3,6-dimethylbenzoate (4 g, 1 equiv, 7 mmol) in 1,3-dimethoxybenzene (18.34 g, 20 equiv, 0.13 mol) was added dropwise a mixture of 1,3-dimethoxybenzene (18.34 g, 20 equiv, 0.13 mol) and formic acid (305 mg, 4 mL, 1 equiv, 7 mmol). The mixture was stirred at 25 °C for 16 h. The residue was purified by flash chromatography (0–10% EtOAc in petroleum ether, 40 g column) to afford rac-benzyl 4-hydroxy-2-(((1R,6R)-6-methoxy-3,6-dimethyl-2,5-dioxocyclohex-3-en-1-yl)oxy)-3,6-dimethylbenzoate (16) (5.65 g, 62% yield) as a yellow solid. LC–MS m/z = 439.0 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 8.64–8.55 (m, 1H), 7.32–7.25 (m, 2H), 7.22–7.10 (m, 3H), 6.47 (d, J = 1.5 Hz, 1H), 6.23 (s, 1H), 5.13–5.07 (m, 1H), 5.02–4.96 (m, 1H), 4.60 (s, 1H), 2.94 (s, 3H), 1.96 (s, 3H), 1.87 (s, 3H), 1.84–1.82 (m, 3H), 1.61 (br s, 14H), 1.37–1.29 (m, 4H).
rac-Benzyl 4-Hydroxy-2-(((3R,4R)-4-methoxy-1,4-dimethyl-2,5-dioxo-7-oxabicyclo[4.1.0]heptan-3-yl)oxy)-3,6-dimethylbenzoate (17)
To a solution of rac-benzyl 4-hydroxy-2-(((1R,6R)-6-methoxy-3,6-dimethyl-2,5-dioxocyclohex-3-en-1-yl)oxy)-3,6-dimethylbenzoate (16) (1.5 g, 3.42 mmol, 1 equiv) in H2O2 (30% w/w, 776 mg, 6.84 mmol, 0.65 mL, 2 equiv) and MeOH (40 mL) was added Na2CO3 (1 M, 13.68 mL, 4 equiv). The mixture was stirred at 25 °C for 10 min. The reaction mixture was quenched by the addition of Na2SO3 (30 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by flash chromatography (1–50% EtOAc in petroleum ether) to afford rac-benzyl 4-hydroxy-2-(((3R,4R)-4-methoxy-1,4-dimethyl-2,5-dioxo-7-oxabicyclo[4.1.0]heptan-3-yl)oxy)-3,6-dimethylbenzoate (17) (600 mg, 33% yield) as a white solid. LC–MS m/z = 455.1 [M + H]+.
rac-Benzyl 4-Hydroxy-2-(((3R,4S,5R)-5-hydroxy-4-methoxy-1,4-dimethyl-2-oxo-7-oxabicyclo[4.1.0]heptan-3-yl)oxy)-3,6-dimethylbenzoate (18)
To a solution of rac-benzyl 4-hydroxy-2-(((3R,4R)-4-methoxy-1,4-dimethyl-2,5-dioxo-7-oxabicyclo[4.1.0]heptan-3-yl)oxy)-3,6-dimethylbenzoate (17) (0.6 g, 1.32 mmol, 1 equiv) in toluene (20 mL) was added Ti(i-PrO)4 (1.13 g, 3.96 mmol, 1.17 mL, 3.0 equiv) and 2-trimethylsilylethanol (0.63 g, 5.28 mmol, 4.0 equiv). The mixture was stirred at 110 °C for 10 h. The reaction mixture was quenched by the addition of H2O (25 mL), diluted with EtOAc (2 × 35 mL). The combined organic layers were concentrated under reduced pressure to give a residue. The residue was purified by flash chromatography (1–50% EtOAc in petroleum ether) to afford rac-benzyl 4-hydroxy-2-(((3R,4S,5R)-5-hydroxy-4-methoxy-1,4-dimethyl-2-oxo-7-oxabicyclo[4.1.0]heptan-3-yl)oxy)-3,6-dimethylbenzoate (18) (300 mg, 50% yield) as a white solid. LC–MS m/z = 457.0 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 9.72 (s, 1H), 7.50–7.34 (m, 5H), 6.40 (s, 1H), 5.89 (d, J = 5.7 Hz, 1H), 5.24–5.04 (m, 3H), 4.26 (d, J = 5.7 Hz, 1H), 3.01 (s, 3H), 2.06 (s, 3H), 1.99 (s, 3H), 1.39 (s, 3H), 1.02 (s, 3H).
rac-Benzyl 2-(((1R,2S,3R)-3,4-Dihydroxy-2-methoxy-2,5-dimethyl-6-oxocyclohexyl)oxy)-4-hydroxy-3,6-dimethylbenzoate (19)
To a solution of rac-benzyl 4-hydroxy-2-(((3R,4S,5R)-5-hydroxy-4-methoxy-1,4-dimethyl-2-oxo-7-oxabicyclo[4.1.0]heptan-3-yl)oxy)-3,6-dimethylbenzoate (18) (0.3 g, 0.65 mol, 1 equiv) in EtOH (6 mL) was added hydrazine hydrate (85% in water, 1.2 g, 20 mol, 0.38 mL, 30 equiv). The mixture was stirred at 25 °C for 2 h. The reaction mixture was quenched by the addition of HCl (2 M, 15 mL), and then extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine (2 × 30 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by flash chromatography (1–50% EtOAc in petroleum ether) to afford rac-benzyl 2-(((1R,2S,3R)-3,4-dihydroxy-2-methoxy-2,5-dimethyl-6-oxocyclohexyl)oxy)-4-hydroxy-3,6-dimethylbenzoate (19) (0.2 g, 66% yield) as a white solid. LC–MS m/z = 459.2 [M + H]+.
rac-Benzyl 4-Hydroxy-2-(((1S,6R)-2-hydroxy-6-methoxy-3,6-dimethyl-4,5-dioxocyclohex-2-en-1-yl)oxy)-3,6-dimethylbenzoate (20)
To a solution of rac-benzyl 2-(((1R,2S,3R)-3,4-dihydroxy-2-methoxy-2,5-dimethyl-6-oxocyclohexyl)oxy)-4-hydroxy-3,6-dimethylbenzoate (19) (200 mg, 436 μmol, 1 equiv) in acetone (25 mL) was added Jones reagent (2.5 M, 349 μL, 2 equiv) at 0 °C. The mixture was stirred at 0 °C for 0.2 h. The residue was diluted with H2O (20 mL) and extracted with EtOAc (3 × 30 mL). The combined organic layers were washed with brine (2 × 20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by prep-TLC (50% EtOAc in petroleum ether) to afford rac-benzyl 4-hydroxy-2-(((1S,6R)-2-hydroxy-6-methoxy-3,6-dimethyl-4,5-dioxocyclohex-2-en-1-yl)oxy)-3,6-dimethylbenzoate (20) (40 mg, 20% yield) as a white solid. LC–MS m/z = 455.1 [M + H]+.
rac-Benzyl 2-(((1S,5R,6S)-2,5-Dihydroxy-6-methoxy-3,6-dimethyl-4-oxocyclohex-2-en-1-yl)oxy)-4-hydroxy-3,6-dimethylbenzoate (21)
To a solution of rac-benzyl 4-hydroxy-2-(((1S,6R)-2-hydroxy-6-methoxy-3,6-dimethyl-4,5-dioxocyclohex-2-en-1-yl)oxy)-3,6-dimethylbenzoate (20) (150 mg, 330 μmol, 1 equiv) in THF (9 mL) was added Zn (432 mg, 6.6 mmol, 20 equiv) and saturated aqueous NH4Cl (9 mL, 30 equiv). The mixture was stirred at 0 °C for 15 min. The reaction mixture was quenched by the addition of HCl (2 M, 5 mL), and then diluted with H2O (15 mL) and extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine (2 × 30 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by prep-TLC (1–25% EtOAc in petroleum ether) to afford rac-benzyl 2-(((1S,5R,6S)-2,5-dihydroxy-6-methoxy-3,6-dimethyl-4-oxocyclohex-2-en-1-yl)oxy)-4-hydroxy-3,6-dimethylbenzoate (21) (65 mg, 43% yield) as a white solid. LC–MS m/z = 457.1 [M + H]+.
rac-2-(((1S,5R,6S)-2,5-Dihydroxy-6-methoxy-3,6-dimethyl-4-oxocyclohex-2-en-1-yl)oxy)-4-hydroxy-3,6-dimethylbenzoic Acid (22)
To a solution of rac-benzyl 2-(((1S,5R,6S)-2,5-dihydroxy-6-methoxy-3,6-dimethyl-4-oxocyclohex-2-en-1-yl)oxy)-4-hydroxy-3,6-dimethylbenzoate (21) (65 mg, 0.14 mmol, 1 equiv) in THF (3 mL) was added Pd/C (10 wt %, 25 mg). The suspension was degassed under vacuum and purged with H2. The mixture was stirred under H2 (15 psi) at 25 °C for 1 h. The reaction mixture was concentrated under reduced pressure to afford rac-2-(((1S,5R,6S)-2,5-dihydroxy-6-methoxy-3,6-dimethyl-4-oxocyclohex-2-en-1-yl)oxy)-4-hydroxy-3,6-dimethylbenzoic acid (22) (45 mg, 86% yield) was obtained as a white solid which was used into the next step without further purification. LC–MS m/z = 366.9 [M + H]+.
rac-(5aS,6S,7R)-3,7-Dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (23)
To a solution of 4-hydroxy-2-(((1S,2R,3R)-3-hydroxy-2-methoxy-2,5-dimethyl-4,6-dioxocyclohexyl)oxy)-3,6-dimethylbenzoic acid (22) (45 mg, 1 equiv, 0.12 mmol) in DCM (5 mL) was added TFAA (0.10 g, 69 μL, 4 equiv, 0.49 mmol) at 25 °C. The mixture was stirred at 25 °C for 30 min. The reaction was diluted with H2O (25 mL), and then extracted with EtOAc (3 × 15 mL). The combined organic layers were dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by prep-TLC (25% EtOAc in petroleum ether) to afford rac-(5aS,6S,7R)-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (18 mg, 42% yield) as a white solid. LC–MS m/z = 349.0 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 9.12 (s, 1H), 6.33 (s, 1H), 4.79 (s, 1H), 4.30 (d, J = 4.0 Hz, 1H), 4.25 (d, J = 4.1 Hz, 1H), 3.22 (s, 3H), 2.25 (s, 3H), 1.97 (s, 3H), 1.55 (s, 3H).
(5aS,6S,7R)-3,7-Dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (1)
The racemate (23) (18 mg) was purified by chiral SFC chromatography (Chiralpak AS (250 mm × 30 mm, 10 μm); 25% EtOH in CO2) to afford the title compound (1) (5.46 mg, 30% yield) as a white solid. LC–MS m/z = 349.2 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 9.11 (s, 1H), 6.42 (s, 1H), 4.89 (s, 1H), 4.36 (s, 1H), 3.33 (s, 3H), 2.36 (s, 3H), 2.08 (s, 3H), 1.66 (s, 3H).
(5aR,6R,7S)-3,7-Dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (24)
Also isolated from the above resolution was the title compound (24) (5.57 mg, 31% yield) as a white solid. LC–MS m/z = 349.2 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 9.11 (s, 1H), 6.42 (s, 1H), 4.89 (s, 1H), 4.36 (s, 1H), 3.33 (s, 3H), 2.36 (s, 3H), 2.08 (s, 3H), 1.66 (s, 3H).
(5aS,6S,7S)-3,7-Dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (2)
Using the general procedure for the epimerization (0.46 mg, 1.3 μmol, 11%) as a white solid. LC–MS m/z = 346.8 [M – H]−. 1H NMR (500 MHz, THF-d8) δ 9.24 (s, 1H), 6.50 (s, 1H), 5.04 (q, J = 2.0 Hz, 1H), 4.54 (d, J = 3.2 Hz, 1H), 4.23 (d, J = 2.9 Hz, 1H), 3.44 (s, 3H), 2.44 (s, 3H), 2.18 (s, 3H), 1.86 (d, J = 1.9 Hz, 3H), 1.15 (s, 3H).
rac-(5aS,6R,7S)-3,7-Dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (26)
To a solution of rac-benzyl 2-(((6S,7R)-6-hydroxy-6,9-dimethyl-8-oxo-1,4-dioxaspiro[4.5]dec-9-en-7-yl)oxy)-4-((4-methoxybenzyl)oxy)-3,6-dimethylbenzoate (15c and 15d) (1 g, 1 equiv, 2 mmol) and MeI (5 g, 2 mL, 20 equiv, 0.03 mol) in THF (40 mL) was added NaH (60% dispersion in mineral oil, 0.3 g, 3 equiv, 8 mmol) at 0 °C. The mixture was stirred at 0 °C for 1 h. The mixture was diluted with H2O (20 mL) and extracted with EtOAc (3 × 40 mL). The combined organic layers were washed with brine (50 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by flash chromatography (25% EtOAc in petroleum ether) to afford rac-benzyl 2-(((6S,7R)-6-methoxy-6,9-dimethyl-8-oxo-1,4-dioxaspiro[4.5]dec-9-en-7-yl)oxy)-4-((4-methoxybenzyl)oxy)-3,6-dimethylbenzoate (600 mg, 60% yield) was obtained as yellow oil. LC–MS m/z = 603.3 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.46 (br d, J = 7.0 Hz, 2H), 7.38–7.30 (m, 5H), 6.92 (br d, J = 8.3 Hz, 2H), 6.89–6.89 (m, 1H), 6.48 (s, 1H), 6.26 (s, 1H), 5.31 (s, 2H), 5.12 (s, 1H), 4.97 (s, 2H), 4.19–4.04 (m, 4H), 3.83 (s, 3H), 3.41 (s, 3H), 2.24–2.19 (m, 3H), 2.18 (s, 3H), 1.85–1.75 (m, 3H), 1.39 (s, 3H).
To a solution of rac-benzyl 2-(((6S,7R)-6-methoxy-6,9-dimethyl-8-oxo-1,4-dioxaspiro[4.5]dec-9-en-7-yl)oxy)-4-((4-methoxybenzyl)oxy)-3,6-dimethylbenzoate (6.4 g, 1 equiv, 11 mmol) in 1,3-dimethoxybenzene (29 g, 20 equiv, 0.21 mol) was added formic acid (20 g, 40 equiv, 0.42 mol). The mixture was stirred at 50 °C for 16 h. The mixture was diluted with H2O (20 mL) and extracted with EtOAc (3 × 40 mL). The combined organic layers were washed with brine (50 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by flash chromatography (25% EtOAc in petroleum ether) to afford rac-benzyl 4-hydroxy-2-(((1R,6S)-6-methoxy-3,6-dimethyl-2,5-dioxocyclohex-3-en-1-yl)oxy)-3,6-dimethylbenzoate (3.4 g, 73% yield) as brown oil. LC–MS m/z = 439.3 [M + H]+.
To a solution of rac-benzyl 4-hydroxy-2-(((1R,6S)-6-methoxy-3,6-dimethyl-2,5-dioxocyclohex-3-en-1-yl)oxy)-3,6-dimethylbenzoate (1.2 g, 1 equiv, 2.7 mmol) and H2O2 (30% w/w, 0.34 g, 0.31 mL, 1.1 equiv, 3.0 mmol) in MeOH (30 mL)was added Na2CO3 (0.87 g, 8.2 mL, 1 M, 3 equiv, 8.2 mmol) at 0 °C. The mixture was stirred at 25 °C for 16 h. The mixture was quenched with saturated aqueous NaS2O3 and adjusted to pH 6–7 (1 N HCl(aq)). The mixture was diluted with H2O (5 mL), extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by flash chromatography (0–30% EtOAc in petroleum ether, 40 g column) to give rac-benzyl 4-hydroxy-2-(((3R,4S)-4-methoxy-1,4-dimethyl-2,5-dioxo-7-oxabicyclo[4.1.0]heptan-3-yl)oxy)-3,6-dimethylbenzoate (980 mg, 79% yield) as yellow oil. LC–MS m/z = 455.1 [M + H]+.
To a solution of rac-benzyl 4-hydroxy-2-(((3R,4S)-4-methoxy-1,4-dimethyl-2,5-dioxo-7-oxabicyclo[4.1.0]heptan-3-yl)oxy)-3,6-dimethylbenzoate (4.6 g, 1 equiv, 10 mmol) in toluene (140 mL) were added titanium(IV) isopropoxide (8.6 g, 9.2 mL, 3 equiv, 30 mmol) and 2-(trimethylsilyl)ethan-1-ol (4.8 g, 4 equiv, 40 mmol). The mixture was stirred at 110 °C for 10 h. The mixture was quenched with H2O (100 mL) and extracted with EtOAc (3 × 200 mL). The combined organic layers were washed with brine (200 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by flash chromatography (0–20% EtOAc in petroleum ether, 80 g column) to afford rac-benzyl 4-hydroxy-2-(((3R,4R,5R)-5-hydroxy-4-methoxy-1,4-dimethyl-2-oxo-7-oxabicyclo[4.1.0]heptan-3-yl)oxy)-3,6-dimethylbenzoate (2.6 g, 5.7 mmol, 56%) as yellow oil. LC–MS m/z = 457.2 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.49 (s, 2H), 7.43–7.33 (m, 3H), 6.35 (s, 1H), 5.32 (d, J = 17.1 Hz, 2H), 5.24 (br s, 1H), 5.19 (s, 1H), 3.78–3.70 (m, 1H), 3.50 (s, 3H), 3.31 (s, 1H), 2.93 (d, J = 11.5 Hz, 1H), 2.25 (s, 3H), 2.14 (s, 3H), 1.49 (s, 3H), 1.39 (s, 3H).
To a solution of rac-benzyl 4-hydroxy-2-(((3R,4R,5R)-5-hydroxy-4-methoxy-1,4-dimethyl-2-oxo-7-oxabicyclo[4.1.0]heptan-3-yl)oxy)-3,6-dimethylbenzoate (700 mg, 1 equiv, 1.53 mmol) in EtOH (4.2 mL) was added hydrazine hydrate (85% solution in water, 4.33 g, 4.20 mL, 48 equiv, 73.6 mmol). The mixture was stirred at 25 °C for 16 h. The mixture was diluted with H2O (20 mL), extracted with EA (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The mixtures were purified by flash chromatography (0–100% EtOAc in petroleum ether, 20 g column) to give rac-benzyl 2-(((1R,2R,3R)-3,4-dihydroxy-2-methoxy-2,5-dimethyl-6-oxocyclohexyl)oxy)-4-hydroxy-3,6-dimethylbenzoate (350 mg, 763 μmol, 50%) as a yellow solid. LC–MS m/z = 459.3 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.52–7.46 (m, 2 H), 7.45–7.35 (m, 3 H), 6.37 (s, 1 H), 5.44 (d, J = 12.0 Hz, 1 H), 5.34–5.26 (m, 1 H), 5.19 (d, J = 12.0 Hz, 1 H), 4.60 (d, J = 1.0 Hz, 1 H), 3.52–3.49 (m, 3 H), 3.44–3.35 (m, 1 H), 3.04 (br d, J = 8.9 Hz, 1 H), 2.28 (s, 3 H), 2.18–2.13 (m, 3 H), 1.53–1.49 (m, 3 H), 1.08 (d, J = 6.4 Hz, 3 H).
To a solution of rac-benzyl 2-(((1R,2R,3R)-3,4-dihydroxy-2-methoxy-2,5-dimethyl-6-oxocyclohexyl)oxy)-4-hydroxy-3,6-dimethylbenzoate (980 mg, 1 equiv, 2.14 mmol) in acetone (64 mL) was added Jones reagent (847 mg, 0.65 mL, 2 equiv, 4.27 mmol) at −10 °C. The mixture was stirred at 25 °C for 1 h. The mixture was quenched with H2O (30 mL), extracted with DCM (3 × 60 mL). The combined organic layers were washed with brine (50 mL), dried over Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by reverse phase HPLC (36–76% acetonitrile in water, 0.1% HCl) to afford rac-benzyl 4-hydroxy-2-(((1S,6S)-2-hydroxy-6-methoxy-3,6-dimethyl-4,5-dioxocyclohex-2-en-1-yl)oxy)-3,6-dimethylbenzoate (190 mg, 418 μmol, 20%) as yellow solid. LC–MS m/z = 455.1 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.42–7.37 (m, 3 H), 7.34–7.30 (m, 2 H), 6.83 (s, 1 H), 6.40 (s, 1 H), 5.31 (d, J = 12.0 Hz, 1 H), 5.23–5.16 (m, 1 H), 5.05–4.88 (m, 1 H), 4.69 (s, 1 H), 3.30 (s, 3 H), 2.18 (s, 3 H), 2.15 (s, 3 H), 1.91 (s, 3 H), 1.44 (s, 3 H).
To a solution of rac-benzyl 4-hydroxy-2-(((1S,6S)-2-hydroxy-6-methoxy-3,6-dimethyl-4,5-dioxocyclohex-2-en-1-yl)oxy)-3,6-dimethylbenzoate (170 mg, 1 equiv, 0.37 mmol) in THF (20 mL) and saturated aqueous NH4Cl (4 mL) was added zinc (489 mg, 20 equiv, 7.48 mmol) at 0 °C. The mixture was stirred at 0 °C for 10 min, then filtered. The filtrate was concentrated under reduced pressure to give a residue. The residue was purified by prep-TLC (50% EtOAc in petroleum ether) to give rac-benzyl 2-(((1S,5S,6R)-2,5-dihydroxy-6-methoxy-3,6-dimethyl-4-oxocyclohex-2-en-1-yl)oxy)-4-hydroxy-3,6-dimethylbenzoate (100 mg, 0.22 mmol, 59%) as yellow oil. LC–MS m/z = 457.0 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.65 (s, 1 H), 7.51 (br d, J = 6.5 Hz, 1 H), 7.44 (br d, J = 6.5 Hz, 2 H), 7.41–7.35 (m, 2 H), 6.48 (s, 1 H), 5.68 (d, J = 11.4 Hz, 1 H), 5.23–4.91 (m, 3 H), 3.91 (d, J = 1.7 Hz, 1 H), 3.37–3.34 (m, 3 H), 3.23 (br d, J = 11.7 Hz, 1 H), 2.36 (s, 3 H), 2.22 (s, 3 H), 1.84 (d, J = 1.3 Hz, 3 H), 1.08 (s, 3 H).
To a solution of rac-benzyl 2-(((1S,5S,6R)-2,5-dihydroxy-6-methoxy-3,6-dimethyl-4-oxocyclohex-2-en-1-yl)oxy)-4-hydroxy-3,6-dimethylbenzoate (50 mg, 1 equiv, 0.11 mmol) in THF (1 mL) was added Pd/C (10 wt %, 12 mg). The mixture was degassed under vacuum and purged with H2. The mixture was stirred under H2 (15 psi) at 25 °C for 2 h. The mixture was filtered and the filtrate was concentrated under reduced pressure to afford rac-2-(((1S,5S,6R)-2,5-dihydroxy-6-methoxy-3,6-dimethyl-4-oxocyclohex-2-en-1-yl)oxy)-4-hydroxy-3,6-dimethylbenzoic acid (30 mg, 75% yield) as yellow oil which was used without purification. LC–MS m/z = 389.0 [M + Na]+.
Two batches of the following reaction were conducted. To a solution of rac-2-(((1S,5S,6R)-2,5-dihydroxy-6-methoxy-3,6-dimethyl-4-oxocyclohex-2-en-1-yl)oxy)-4-hydroxy-3,6-dimethylbenzoic acid (35 mg, 1 equiv, 96 μmol) in DCM (7 mL) was added TFAA (80 mg, 4 equiv, 0.38 mmol). The mixture was stirred at 25 °C for 30 min. The combined reaction mixtures were diluted with H2O (10 mL) and extracted with DCM (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried with anhydrous Na2SO4, filtered and concentrated under reduced pressure to give a residue. The residue was purified by prep-TLC (33% EtOAc in petroleum ether) to afford rac-(5aS,6R,7R)-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (26) (26 mg, 39% yield) as a yellow solid. LC–MS m/z = 349.2 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 9.05 (br s, 1 H), 6.40 (s, 1 H), 5.20 (d, J = 2.3 Hz, 1 H), 4.64 (br s, 1 H), 4.12 (s, 1 H), 3.16 (s, 3 H), 2.30 (s, 3 H), 2.11 (s, 3 H), 1.78 (d, J = 2.1 Hz, 3 H), 1.76 (s, 3 H).
(5aS,6R,7S)-3,7-Dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (27)
The racemate (26) (24 mg) was purified by chiral SFC chromatography (Chiralcel OJ, 250 mm × 30 mm, 10 μm, 20% EtOH in CO2) to afford the title compound (27) (10.8 mg, 31.0 μmol, 32%) as a white solid. LC–MS m/z = 349.0 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 8.94 (s, 1H), 6.38 (s, 1H), 5.20 (q, J = 2.1 Hz, 1H), 4.62 (s, 1H), 4.11 (br s, 1H), 3.16 (s, 3H), 2.30 (s, 3H), 2.11 (s, 3H), 1.78 (d, J = 2.1 Hz, 3H), 1.76 (s, 3H).
(5aR,6S,7R)-3,7-Dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (29)
Also isolated from the above resolution was the title compound (29) (10.6 mg, 30.4 μmol, 32%) as a white solid. LC–MS m/z = 349.0 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 8.94 (s, 1H), 6.38 (s, 1H), 5.20 (q, J = 2.2 Hz, 1H), 4.62 (br s, 1H), 4.11 (s, 1H), 3.16 (s, 3H), 2.30 (s, 3H), 2.11 (s, 3H), 1.78 (d, J = 2.2 Hz, 3H), 1.76 (s, 3H).
(5aS,6R,7R)-3,7-Dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (28)
Using the general procedure for the epimerization, (0.62 mg, 1.8 μmol, 6%) as a white solid. LC–MS m/z = 346.8 [M – H]−. 1H NMR (500 MHz, THF-d8) δ 9.02 (s, 1H), 6.41 (s, 1H), 5.12 (t, J = 1.3 Hz, 1H), 4.85 (s, 1H), 4.25 (s, 1H), 3.29 (s, 3H), 2.35 (s, 3H), 2.13 (s, 3H), 1.38 (s, 3H), 1.30 (s, 3H).
2-Methyl-3-oxocyclohex-1-en-1-yl benzoate (32a)
2-Methyl-1,3-cyclohexadione (2.5 g, 1 equiv, 20 mmol), DMAP (300 mg, 0.12 equiv, 2.5 mmol) and triethylamine (3.5 mL, 1.25 equiv, 25 mmol) were placed in a vial with DCM (15 mL). Benzoyl chloride (3 mL, 1.25 equiv, 25 mmol) was added portion wise. The reaction was stirred at room temperature overnight. The reaction was then quenched with saturated aqueous sodium bicarbonate solution. The organics were washed with brine and concentrated. The residue was purified by flash chromatography (0–20% EtOAc in hexanes) to afford 2-methyl-3-oxocyclohex-1-en-1-yl benzoate (32a) (4.1 g, 17.8 mmol, 90%). LC–MS m/z = 230.8 [M + H]+. 1H NMR (500 MHz, CDCl3) δ 8.14–8.08 (m, 2H), 7.64 (td, J = 7.3, 1.4 Hz, 1H), 7.50 (t, J = 7.8 Hz, 2H), 2.67 (tq, J = 6.1, 1.9 Hz, 2H), 2.50 (dd, J = 7.5, 6.0 Hz, 2H), 2.08 (p, J = 6.4 Hz, 2H), 1.72 (t, J = 1.9 Hz, 3H).
4-Bromo-2-methyl-3-oxocyclohex-1-en-1-yl benzoate (33a)
A mixture of 4-methylbenzenesulfonic acid (149.5 mg, 0.86 mmol, 0.1 equiv), NBS (1.93 g, 10.86 mmol, 1.25 equiv) in DCM (10 mL), and then 2-methyl-3-oxocyclohex-1-en-1-yl benzoate (32a) (2 g, 8.69 mmol, 1 equiv) was added. The mixture was stirred at 55 °C for 2 h under microwave tube. The mixture was quenched with H2O (50 mL) and extracted with DCM (3 × 100 mL). The combined organic layers were washed with brine (100 mL) dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (0–1% EtOAc in petroleum ether, 40 g column) to afford 4-bromo-2-methyl-3-oxocyclohex-1-en-1-yl benzoate (33a) (10 g, 32.4 mmol, 74% yield) as yellow oil. LC–MS m/z = 308.8 [M + H]+.
Methyl 4-(Benzyloxy)-2-hydroxy-3,6-dimethylbenzoate (34)
To a solution of methyl 2,4-dihydroxy-3,6-dimethylbenzoate (25 g, 127.42 mmol, 1 equiv) in MeCN (30 mL) was added K2CO3 (35.22 g, 254.8 mmol, 2 equiv) and BnBr (26.15 g, 152.9 mmol, 18.16 mL, 1.2 equiv) at 0 °C. The mixture was stirred at 25 °C for 16 h. The liquor was quenched with H2O (400 mL) and extracted with EtOAc (3 × 500 mL). The combined organic layers were washed with brine (400 mL) dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was triturated with petroleum ether in MTBE (1:1, 100 mL) and filtered to give methyl methyl 4-(benzyloxy)-2-hydroxy-3,6-dimethylbenzoate (34) (114 g, 78% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ 11.28 (s, 1H), 7.49–7.43 (m, 2H), 7.43–7.37 (m, 2H), 7.36–7.30 (m, 1H), 6.59 (s, 1H), 5.17 (s, 2H), 3.86 (s, 3H), 2.43 (s, 3H), 2.02 (s, 3H).
rac-4-Benzyloxy-3,6-dimethyl-2-(3-methyl-2,4-dioxo-cyclohexoxy)benzoic Acid (35a)
To a solution of methyl 4-(benzyloxy)-2-hydroxy-3,6-dimethylbenzoate (34) (10.19 g, 35.58 mmol, 1.1 equiv) in THF (50 mL) was added NaH (60% dispersion in mineral oil, 2.20 g, 55.0 mmol, 1.7 equiv) at 25 °C. The mixture was stirred at 25 °C for 30 min. The mixture was added (4-bromo-2-methyl-3-oxo-cyclohexen-1-yl) benzoate (10 g, 32.35 mmol, 1 equiv) in THF (20 mL) at 25 °C. The mixture was stirred at 25 °C for 4 h. The mixture was quenched with H2O (40 mL), and HCl (2 N, 15 mL) added to give pH ∼ 2, the mixture was extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine (100 mL) dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was triturated with EtOAc (15 mL) and filtered to afford rac-methyl 4-(benzyloxy)-3,6-dimethyl-2-((3-methyl-2,4-dioxocyclohexyl)oxy)benzoate (5.6 g, 13.6 mmol, 42% yield) as a white solid. LC–MS m/z = 411.0 [M + H]+.
To a solution of rac-methyl 4-(benzyloxy)-3,6-dimethyl-2-((3-methyl-2,4-dioxocyclohexyl)oxy)benzoate (7 g, 17.05 mmol, 1 equiv) in EtOH (140 mL) was added KOH (31.58 g, 563 mmol, 33 equiv). The mixture was stirred at 80 °C for 4 h. The mixture was quenched with H2O (20 mL), and HCl (2 N) was added to give pH ∼ 2, the mixture quenched with H2O (20 mL), and extracted with EA (3 × 300 mL). The combined organic layers was washed with brine (10 mL) dried over Na2SO4, filtered and concentrated under reduced pressure to afford crude rac-4-(benzyloxy)-2-((2-hydroxy-3-methyl-4-oxocyclohex-2-en-1-yl)oxy)-3,6-dimethylbenzoic acid (35a) (7 g, crude) as a brown solid. LC–MS m/z = 397.0 [M + H]+.
rac-3-(Benzyloxy)-1,4,9-trimethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (36a)
To a solution of rac-4-(benzyloxy)-2-((2-hydroxy-3-methyl-4-oxocyclohex-2-en-1-yl)oxy)-3,6-dimethylbenzoic acid (35a) (7 g, 17.7 mmol, 1 equiv) in DCM (70 mL) was added TFAA (122.38 g, 582.70 mmol, 81.05 mL, 33 equiv). The mixture was stirred at 46 °C for 1 h. The mixture was filtered and concentrated under reduced pressure. The mixture was quenched with H2O (20 mL) and extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with brine (50 mL) dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was triturated with a mixture of petroleum ether and EtOAc (20:1, 20 mL) and filtered to give a solid. The filtrate was concentrated and the residue was purified by flash chromatography (0–20% EtOAc in petroleum ether, 20 g column), and combined with the solid above to afford rac-3-(benzyloxy)-1,4,9-trimethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (36a). (5.7 g, 84% yield) as a white solid. LC–MS m/z = 379.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.30–7.48 (m, 5H), 6.95 (s, 1H), 5.56 (ddd, J = 10.76, 5.00, 2.13 Hz, 1H), 5.17 (s, 2H), 2.52–2.57 (m, 1H), 2.37–2.43 (m, 2H), 2.35 (s, 3H), 2.05 (s, 3H), 1.71–1.83 (m, 1H), 1.62 (d, J = 2.00 Hz, 3H).
(R)-3-(Benzyloxy)-1,4,9-trimethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (37)
rac-3-(Benzyloxy)-1,4,9-trimethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (36a) (5 g, 13.2 mmol, 1 equiv) was purified by chiral SFC (Daicel Chiralpak IC 250 × 50 mm2, 10 μm, 40% EtOH in water, 0.1% NH3) to afford (R)-3-(benzyloxy)-1,4,9-trimethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (37) (2.3 g, 45% yield) as a white solid. LC–MS m/z = 379.1 [M + H]+. 1H NMR (400 MHz, DMSO-d6) δ 7.47–7.33 (m, 5H), 6.95 (s, 1H), 5.56 (ddd, J = 2.1, 5.0, 10.7 Hz, 1H), 5.17 (s, 2H), 2.56–2.51 (m, 1H), 2.42–2.36 (m, 2H), 2.35 (s, 3H), 2.05 (s, 3H), 1.85–1.70 (m, 1H), 1.62 (d, J = 2.0 Hz, 3H).
(R)-3-Hydroxy-1,4,9-trimethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (38)
To a solution of (R)-3-(benzyloxy)-1,4,9-trimethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (37) (100.0 mg, 0.26 mmol, 1 equiv) in THF (6 mL) was added Pd/C (10 wt %, 12 mg). The mixture was stirred at 25 °C for 4 h under H2. The reaction mixture was filtered and the filtrate was concentrated. The residue was purified by flash chromatography (0–20% EtOAc in petroleum ether, 12 g column), then purified by reverse phase HPLC (Welch Xtimate C18 100 × 40 mm2 × 3 μm, 30–60% acetonitrile in water, 0.1% HCl) to afford (R)-3-hydroxy-1,4,9-trimethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (38) (15.6 mg, 31% yield) as a gray solid. LC–MS m/z = 289.1 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 6.49 (s, 1H), 5.47 (s, 1H), 5.35–5.26 (m, 1H), 2.63–2.52 (m, 1H), 2.47–2.37 (m, 2H), 2.35 (s, 3H), 2.09 (s, 3H), 2.03–1.88 (m, 1H), 1.74 (d, J = 2.0 Hz, 3H).
2,5-Dimethylcyclohexane-1,3-dione (31b)
To a solution of 5-methylcyclohexane-1,3-dione (100 g, 1 equiv, 793 mmol) and NaOH (29 g, 0.91 equiv, 721 mmol) in H2O (250 mL) was added MeI (225 g, 99.1 mL, 2 equiv, 1.59 mol) at 0 °C. The mixture was stirred at 100 °C for 16 h. Additional MeI (225 g, 99.1 mL, 2 equiv, 1.59 mol) was added at 0 °C and the mixture was stirred at 100 °C for 16 h. Additional MeI (113 g, 49.6 mL, 1 equiv, 793 mmol) was added at 0 °C and the mixture was stirred at 100 °C for 16 h. The mixture was cooled to 0 °C and stirred for 30 min. The mixture was filtrated and afforded crude compound 2,5-dimethylcyclohexane-1,3-dione (31b) (76 g, 0.54 mol, 68% yield) as a brown solid which was used without purification.
2,5-Dimethyl-3-oxocyclohex-1-en-1-yl Benzoate (32b)
To a solution of 2,5-dimethylcyclohexane-1,3-dione (31b) (99 g, 1 equiv, 0.71 mol) in DCM (990 mL) was added TEA (140 g, 0.20 L, 2 equiv, 1.4 mol) and DMAP (8.6 g, 0.1 equiv, 71 mmol). Then the mixture was added benzoyl chloride (120 g, 98 mL, 1.2 equiv, 0.85 mol) at 0 °C. The mixture was stirred at 0 °C for 2 h. The mixture was quenched with H2O (100 mL) and extracted with DCM (3 × 300 mL). The combined organic layers were washed with brine (100 mL) dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (0–3% EtOAc in petroleum ether, 220 g column) to give 2,5-dimethyl-3-oxocyclohex-1-en-1-yl benzoate (32b) (85 g, 49% yield, 93% purity) as an oil. LC–MS m/z = 245.1 [M + H]+.
4-Bromo-2,5-dimethyl-3-oxocyclohex-1-en-1-yl benzoate (33b)
Twenty batches of the following reaction were conducted. A mixture of NBS (1.6 g, 1.1 equiv, 9.00 mmol), 4-methylbenzenesulfonic acid (0.14 g, 0.1 equiv, 0.82 mmol) in DCE (10 mL), and then 2,5-dimethyl-3-oxocyclohex-1-en-1-yl benzoate (32b) (2.0 g, 1 equiv, 8.2 mmol) was added. The mixture was stirred at 100 °C for 5 min under microwave tube. The batches were combined and the mixture was quenched with H2O (100 mL) and extracted with EtOAc (3 × 200 mL). The combined organic layers were washed with brine (200 mL) dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (0–1% EtOAc in petroleum ether, 220 g column) to give 4-bromo-2,5-dimethyl-3-oxocyclohex-1-en-1-yl benzoate (33b) (37 g, 70% yield, 92% purity) as yellow oil. LC–MS m/z = 325.0 [M + H]+.
rac-4-(Benzyloxy)-2-(((1R,6S)-2-hydroxy-3,6-dimethyl-4-oxocyclohex-2-en-1-yl)oxy)-3,6-dimethylbenzoic Acid (35b)
To a solution of methyl 4-(benzyloxy)-2-hydroxy-3,6-dimethylbenzoate (34) (29 g, 1.1 equiv, 0.10 mol) in THF (300 mL) was added NaH (60% dispersion in mineral oil, 6.3 g, 1.7 equiv, 0.16 mol) at 25 °C for 30 min. To the mixture was added a solution of 4-bromo-2,5-dimethyl-3-oxocyclohex-1-en-1-yl benzoate (33b) (30 g, 1 equiv, 93 mmol) in THF (120 mL) at 25 °C. The mixture was stirred at 25 °C for 4 h. The mixture was quenched with H2O (100 mL) and extracted with EtOAc (3 × 200 mL). The combined organic layers were washed with brine (100 mL) dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was triturated with EtOAc (50 mL) and filtered to give rac-methyl 4-(benzyloxy)-2-(((1R,6S)-3,6-dimethyl-2,4-dioxocyclohexyl)oxy)-3,6-dimethylbenzoate (10.4 g, 26% yield) as a white solid. LC–MS m/z = 425.2[M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.66 (s, 1H), 7.45–7.34 (m, 5H), 6.59 (s, 1H), 5.10 (d, J = 1.5 Hz, 2H), 4.37 (dt, J = 6.9, 1.4 Hz, 1H), 3.93 (s, 3H), 2.63 (dd, J = 16.4, 4.7 Hz, 1H), 2.45 (dtd, J = 9.3, 6.7, 4.7 Hz, 1H), 2.38 (s, 3H), 2.21 (s, 3H), 2.11 (dd, J = 16.5, 9.3 Hz, 1H), 1.80 (d, J = 1.3 Hz, 3H), 0.84 (d, J = 6.7 Hz, 3H).
Three batches of the following reaction were conducted. To a solution of rac-methyl 4-(benzyloxy)-2-(((1R,6S)-3,6-dimethyl-2,4-dioxocyclohexyl)oxy)-3,6-dimethylbenzoate (17 g, 1 equiv, 40 mmol) in EtOH (500 mL) was added KOH (74 g, 33 equiv, 1.3 mol). The mixture was stirred at 80 °C for 4 h. The mixture was acidified with HCl (2 N) to give pH ∼ 2. The batches were combined and the mixture was filtered and the filtrate was concentrated to give crude compound rac-4-(benzyloxy)-2-(((1R,6S)-3,6-dimethyl-2,4-dioxocyclohexyl)oxy)-3,6-dimethylbenzoic acid (35b) (45.4 g, 93% yield) as a brown solid. LC–MS m/z = 411.0 [M + H]+.
rac-(5aR,6S)-3-(Benzyloxy)-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (36b)
Two batches of the following reaction were conducted. To solution of rac-4-(benzyloxy)-2-(((1R,6S)-3,6-dimethyl-2,4-dioxocyclohexyl)oxy)-3,6-dimethylbenzoic acid (35b) (22.7 g, 1 equiv, 55.3 mmol) in DCM (227 mL) was added TFAA (383 g, 258 mL, 33 equiv, 1.82 mol). The mixture was stirred at 46 °C for 1 h. The mixture was concentrated under reduced pressure and quenched with H2O (100 mL) and extracted with EA (3 × 200 mL). The combined organic layers were washed with brine (100 mL) dried over Na2SO4, filtered and concentrated under reduced pressure. The batches were combined and the residue was purified by flash chromatography (0–20% EtOAc in petroleum ether, 120 g column) to afford rac-(5aR,6S)-3-(benzyloxy)-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (36b) (23.3 g, 53% yield, 88% purity) as a white solid. LC–MS m/z = 393.2 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 7.46–7.33 (m, 5H), 6.63 (s, 1H), 5.10 (d, J = 1.8 Hz, 2H), 4.93 (dd, J = 1.8, 8.8 Hz, 1H), 2.59–2.51 (m, 1H), 2.42 (s, 3H), 2.31–2.19 (m, 2H), 2.18 (s, 3H), 1.75 (d, J = 2.0 Hz, 3H), 1.40 (d, J = 6.0 Hz, 3H).
(5aR,6S,7R)-3,7-Dihydroxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (39)
To a solution of rac-(5aR,6S)-3-(benzyloxy)-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (36b) (4.2 g, 1 equiv, 11 mmol) in THF (42 mL) was added LiHMDS (2.1 g, 13 mL, 1 M, 1.2 equiv, 13 mmol) at −78 °C for 0.5 h. Then the mixture was added 3-phenyl-2-(phenylsulfonyl)-1,2-oxaziridine (4.2 g, 1.5 equiv, 16 mmol) in THF (10.5 mL) at −78 °C. The mixture was stirred at −78 °C for 1 h. The mixture was quenched with saturated aqueous NH4Cl (50 mL), then the mixture was concentrated under reduced pressure. The mixture was extracted with EtOAc (3 × 100 mL). The combined organic layers were washed with brine (100 mL) dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified by flash chromatography (0–20% EtOAc in petroleum ether, 40 g column) to afford rac-(5aR,6S,7R)-3-(benzyloxy)-7-hydroxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (1.65 g, 38% yield) as a white solid. LC–MS m/z = 409.0 [M + H]+.
To a solution of rac-(5aR,6S,7R)-3-(benzyloxy)-7-hydroxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (2 g, 1 equiv, 5 mmol) in THF (120 mL) was added Pd/C (10 wt %, 480 mg) under N2 atmosphere. The suspension was degassed and purged with H2 3 times. The mixture was stirred under H2 (15 Psi) at 25 °C for 1 h. The mixture was filtered and the filtrate was concentrated. The residue was purified by flash chromatography (0–10% EtOAc in petroleum ether, 12 g column) to afford rac-(5aR,6S,7R)-3,7-dihydroxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (1.3 g, 80% yield) as a white solid. LC–MS m/z = 319.0 [M + H]+. 1H NMR (500 MHz, THF-d8) δ 9.11 (s, 1H), 6.43 (s, 1H), 4.99 (dd, J = 6.2, 1.6 Hz, 1H), 4.92 (d, J = 3.6 Hz, 1H), 4.06 (t, J = 3.6 Hz, 1H), 2.40–2.34 (m, 1H), 2.32 (s, 3H), 2.10 (s, 3H), 1.69 (d, J = 1.5 Hz, 3H), 1.28 (d, J = 6.9 Hz, 3H).
rac-(5aR,6S,7R)-3,7-dihydroxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (1.3 g) was resolved by reverse phase chiral HPLC (Daicel Chiralpak AD, 250 × 50 mm2,10 μm, EtOH in water, 0.1% NH3) to afford (5aR,6S,7R)-3,7-dihydroxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (39) (571 mg, 44% yield) as a white solid. LC–MS m/z = 319.2 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 8.98 (br s, 1H), 6.32 (s, 1H), 4.96–4.64 (m, 2H), 3.95 (t, J = 3.4 Hz, 1H), 2.26 (ddd, J = 3.3, 6.6, 9.8 Hz, 1H), 2.21 (s, 3H), 1.99 (s, 3H), 1.58 (d, J = 1.4 Hz, 3H), 1.17 (d, J = 6.9 Hz, 3H).
(5aR,6S,7S)-3,7-Dihydroxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (40)
Using the general procedure for the epimerization, (27.0 mg, 84.8 μmol, 54%) as a white solid. LC–MS m/z = 316.8 [M – H]−. 1H NMR (500 MHz, THF-d8) δ 9.07 (s, 1H), 6.44 (s, 1H), 4.99 (dd, J = 9.5, 2.1 Hz, 1H), 4.40 (s, 1H), 3.78 (d, J = 11.9 Hz, 1H), 2.30 (s, 3H), 2.08 (s, 3H), 1.85 (ddq, J = 12.2, 9.5, 6.2 Hz, 1H), 1.71 (d, J = 2.0 Hz, 3H), 1.53 (d, J = 6.2 Hz, 3H).
(5aS,6S,7S)-2-Fluoro-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (41)
(5aS,6S,7S)-3,7-Dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (2) (50 mg, 1 equiv, 0.14 mmol) and selectfluor (61 mg, 1.2 equiv, 0.17 mmol) were added to a vial under air. Acetonitrile (1.4 mL) was added and the reaction was stirred at room temperature for 18 h. The reaction mixture was concentrated and purified by reverse phase HPLC (ISCO, Sunfire column, 10–100% acetonitrile in water with 0.1% formic acid) to give (5aS,6S,7S)-2-fluoro-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (41) (2.1 mg, 5.8 μmol, 4%) as a yellow solid. LC–MS m/z = 364.8 [M – H]−. 1H NMR (400 MHz, THF-d8) δ 5.76 (s, 1H), 5.09 (q, J = 2.0 Hz, 1H), 4.62 (d, J = 3.3 Hz, 1H), 4.23 (d, J = 2.8 Hz, 1H), 3.44 (s, 3H), 2.38 (d, J = 2.9 Hz, 3H), 2.24 (s, 3H), 1.86 (d, J = 2.0 Hz, 3H), 1.10 (s, 3H). 19F NMR (376 MHz, THF-d8) δ −146.64 (q, J = 2.9 Hz).
(5aS,6S,7S)-3,7-Dihydroxy-6-methoxy-1,2,4,6,9-pentamethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (42)
(5aS,6S,7S)-2-bromo-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (44) (90 mg, 1 equiv, 0.21 mmol) and 1H-imidazole (22 mg, 1.5 equiv, 0.32 mmol) were added to a vial under air. Chloroform (2 mL) was added, followed by chlorotrimethylsilane (27 mg, 32 μL, 1.2 equiv, 0.25 mmol), and the mixture was heated to 50 °C and stirred overnight. Additional portions of chlorotrimethylsilane (27 mg, 32 μL, 1.2 equiv, 0.25 mmol) and 1H-imidazole (22 mg, 1.5 equiv, 0.32 mmol) were added, and the mixture was stirred at 50 °C for an additional 6 h. The mixture was quenched with saturated aqueous NH4Cl and extracted with DCM, and the organic layer was dried over MgSO4 and concentrated. Purification over silica gel (0–25% EtOAc in hexanes) afforded (5aS,6S,7S)-2-bromo-7-hydroxy-6-methoxy-1,4,6,9-tetramethyl-3-((trimethylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (52 mg, 0.10 mmol, 49% yield) as a light yellow solid. LC–MS m/z = 500.0 [M + H]+. 1H NMR (400 MHz, CDCl3) δ 6.19 (s, 1H), 4.97 (q, J = 1.9 Hz, 1H), 4.09 (s, 1H), 3.45 (s, 3H), 2.54 (d, J = 0.7 Hz, 3H), 2.28 (d, J = 0.7 Hz, 3H), 1.88 (d, J = 2.0 Hz, 3H), 1.26 (s, 3H), 0.13 (s, 9H).
(5aS,6S,7S)-2-Bromo-7-hydroxy-6-methoxy-1,4,6,9-tetramethyl-3-((trimethylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (25 mg, 1 equiv, 50 μmol), potassium ethyltrifluoroborate (18 mg, 3 equiv, 0.15 mmol), potassium phosphate (32 mg, 3 equiv, 0.15 mmol), and P(t-Bu)3 Pd G4 (2.9 mg, 0.1 equiv, 5.0 μmol) were added to a microwave vial, and the vial was evacuated and refilled with N2 3 times. Toluene (1 mL) and water (0.1 mL) that had been degassed were added, and the mixture was heated to 120 °C in a microwave reactor for 4 h. The mixture was diluted in DCM and washed with saturated aqueous NH4Cl, and the organic layer was dried over MgSO4 and concentrated. The residue was purified by flash chromatography (20–50% EtOAc in hexanes) afforded (5aS,6S,7S)-7-hydroxy-6-methoxy-1,2,4,6,9-pentamethyl-3-((trimethylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (7 mg, 0.02 mmol, 30% yield) as a yellow oil. LC–MS m/z = 435.0 [M + H]+.
(5aS,6S,7S)-7-hydroxy-6-methoxy-1,2,4,6,9-pentamethyl-3-((trimethylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (7 mg, 1 equiv, 0.02 mmol) was dissolved in THF (1 mL) in a vial under air. Pyridine hydrofluoride (4 mg, 3 μL, 2 equiv, 0.04 mmol) was added, and the mixture was stirred at room temperature for 4 h. Additional portions of pyridine hydrofluoride (8 mg, 6 μL, 4 equiv, 0.08 mmol) were added, and the mixture was stirred at room temperature overnight. The mixture was concentrated and the residue was purified by reverse phase HPLC (10–70% acetonitrile in water, 0.1% formic acid) afforded (5aS,6S,7S)-3,7-dihydroxy-6-methoxy-1,2,4,6,9-pentamethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (42) (1.2 mg, 3.3 μmol, 20% yield) as a white solid. LC–MS m/z = 363.0 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 8.25 (s, 1H), 5.17 (q, J = 2.1 Hz, 1H), 4.57 (d, J = 3.3 Hz, 1H), 4.25 (d, J = 3.4 Hz, 1H), 3.45 (s, 3H), 2.33 (s, 3H), 2.25 (s, 3H), 2.15 (s, 3H), 1.84 (d, J = 2.1 Hz, 3H), 1.00 (s, 3H).
(5aS,6S,7S)-2-Chloro-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (43)
(5aS,6S,7S)-3,7-Dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (2) (1 g, 1 equiv, 3 mmol) was dissolved in THF (60 mL) and transferred to an oven-dried, 2-neck round-bottom flask under N2, and the solution was cooled to 0 °C in an ice bath. Sulfuryl dichloride (1 M in DCM, 0.9 g, 6 mL, 2.2 equiv, 6 mmol) was added dropwise via addition funnel. The mixture immediately turned a pale yellow. The mixture was diluted in DCM and washed with saturated NH4Cl, and the organic layer was dried over MgSO4 and concentrated. The residue was purified by flash chromatography (20–50% EtOAc in hexanes) to afford (5aS,6S,7S)-2-chloro-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (43) (837 mg, 2.19 mmol, 76% yield) as a yellow solid. LC–MS m/z = 383.8 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 9.11 (s, 1H), 5.21 (q, J = 2.1 Hz, 1H), 4.66 (dd, J = 3.4, 0.8 Hz, 1H), 4.29–4.21 (m, 1H), 3.46 (s, 3H), 2.28 (s, 3H), 1.85 (d, J = 2.1 Hz, 3H), 1.04 (s, 3H).
(5aS,6S,7S)-2-Bromo-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (44)
(5aS,6S,7S)-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (2) (100 mg, 1 equiv, 287 μmol) was dissolved in ACN (3 mL) in a vial under air. NBS (56.2 mg, 1.1 equiv, 316 μmol) was added, and the mixture was stirred at room temperature for 1 h. The mixture was concentrated without workup. The residue was purified by flash chromatography (20–50% EtOAc in hexanes) to afford (5aS,6S,7S)-2-bromo-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (44) (87 mg, 0.20 mmol, 71% yield) as a yellow solid. LC–MS m/z = 428.8 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 8.80 (s, 1H), 5.23 (q, J = 2.1 Hz, 1H), 4.66 (d, J = 3.4 Hz, 1H), 4.25 (d, J = 3.5 Hz, 1H), 3.46 (s, 3H), 2.52 (s, 3H), 2.30 (s, 3H), 1.85 (d, J = 2.1 Hz, 3H), 1.02 (s, 3H).
(5aS,6S,7S)-2-Ethyl-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (45)
(5aS,6S,7S)-2-Bromo-7-hydroxy-6-methoxy-1,4,6,9-tetramethyl-3-((trimethylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (50 mg, 1 equiv, 0.10 mmol), potassium ethyltrifluoroborate (41 mg, 3 equiv, 0.30 mmol), potassium phosphate (64 mg, 3 equiv, 0.30 mmol), and P(t-Bu)3 Pd G4 (5.9 mg, 0.1 equiv, 10 μmol) were added to a microwave vial under air. The vial was sealed, then evacuated and refilled with N2 3 times. Degassed toluene (1 mL) and water (0.1 mL) were added, and the mixture was heated to 120 °C in a microwave vial for 4 h. The mixture was diluted in DCM (10 mL) and washed with NH4Cl (2 × 10 mL). The organic layer was dried over MgSO4 and concentrated. The residue was purified by flash chromatography (0–100% EtOAc in hexanes) to afford (5aS,6S,7S)-2-ethyl-7-hydroxy-6-methoxy-1,4,6,9-tetramethyl-3-((trimethylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (14.4 mg, 32.1 μmol, 32%) as a yellow oil. LC–MS m/z = 449.0 [M + H]+.
(5aS,6S,7S)-2-Ethyl-7-hydroxy-6-methoxy-1,4,6,9-tetramethyl-3-((trimethylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (14.4 mg, 1 equiv, 32.1 μmol) was dissolved in THF (1 mL), pyridine hydrofluoride (6.36 mg, 5.8 μL, 2 equiv, 64.2 μmol) was added, then the mixture was stirred at room temperature for 30 min. At this time, additional pyridine hydrofluoride (6.36 mg, 5.8 μL, 2 equiv, 64.2 μmol) was added, and the mixture was stirred at room temperature for 18 h. The mixture was concentrated and the residue was purified by reverse phase HPLC (10–100% acetonitrile in water, 0.1% formic acid) to afford (5aS,6S,7S)-2-ethyl-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (45) (4.8 mg, 13 μmol, 40%) as a white solid. LC–MS m/z = 377.0 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 8.28 (s, 1H), 5.16 (d, J = 2.2 Hz, 1H), 4.58 (d, J = 3.2 Hz, 1H), 4.24 (d, J = 3.2 Hz, 1H), 3.46 (s, 3H), 2.70 (qd, J = 7.5, 5.7 Hz, 2H), 2.37 (s, 3H), 2.25 (s, 3H), 1.85 (d, J = 2.2 Hz, 3H), 1.08 (t, J = 7.5 Hz, 3H), 1.01 (s, 3H).
(5aS,6S,7S)-7-Hydroxy-6-methoxy-1,4,6,9-tetramethyl-3-((triisopropylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (46)
A vial was charged sequentially with (5aS,6S,7S)-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (2) (200 mg, 1 equiv, 574 μmol), THF (5 mL), followed by 1H-imidazole (46.9 mg, 1.2 equiv, 689 μmol). Finally, chlorotriisopropylsilane (133 mg, 147 μL, 1.2 equiv, 689 μmol) was added, and the mixture was stirred at room temperature overnight. Additional portions of chlorotriisopropylsilane (133 mg, 147 μL, 1.2 equiv, 689 μmol) and 1H-imidazole (46.9 mg, 1.2 equiv, 689 μmol) were added, and the reaction was stirred an additional 6 h. The mixture was diluted in DCM and washed with saturated aqueous NH4Cl, and the organic layer was dried over MgSO4 and concentrated. The residue was purified by flash chromatography (0–25% EtOAc in hexanes) to afford (5aS,6S,7S)-7-hydroxy-6-methoxy-1,4,6,9-tetramethyl-3-((triisopropylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (46) (196 mg, 388 μmol, 68% yield) as a colorless oil. LC–MS m/z = 505.2 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 6.61 (s, 1H), 5.12 (q, J = 2.0 Hz, 1H), 4.60 (d, J = 3.3 Hz, 1H), 4.28–4.19 (m, 1H), 3.44 (s, 3H), 2.45 (s, 3H), 2.26 (s, 3H), 1.86 (d, J = 2.0 Hz, 3H), 1.45–1.30 (m, 3H), 1.15 (dd, J = 7.5, 3.5 Hz, 18H), 1.12 (s, 3H).
(5aS,6R,7S)-6-Methoxy-1,4,6,9-tetramethyl-8,11-dioxo-3-((triisopropylsilyl)oxy)-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl Acetate (47)
(5aS,6S,7S)-7-Hydroxy-6-methoxy-1,4,6,9-tetramethyl-3-((triisopropylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (46) (50 mg, 1 equiv, 99 μmol) was dissolved in DCM (2.0 mL). Pyridine (78 mg, 90 μL, 10 equiv, 0.99 mmol) was added, followed by acetic anhydride (0.10 g, 94 μL, 10 equiv, 0.99 mmol), and the mixture was stirred at room temperature for 1 h, then the mixture was heated to 45 °C and stirred overnight. An additional portion of acetic anhydride (0.10 g, 94 μL, 10 equiv, 0.99 mmol) was added, and the mixture was stirred overnight at room temperature. The mixture was diluted in DCM and washed with saturated aqueous NH4Cl, and the organic layer was dried over MgSO4 and concentrated. The residue was purified by flash chromatography (0–25% EtOAc in hexanes) to afford (5aS,6R,7S)-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-3-((triisopropylsilyl)oxy)-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl acetate (46 mg, 99 μmol, 85% yield) as a colorless oil. LC–MS m/z = 547.2 [M + H]+.
(5aS,6R,7S)-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-3-((triisopropylsilyl)oxy)-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl acetate (46 mg, 1 equiv, 84 μmol) was dissolved in THF (3 mL) and transferred to a vial under air. Pyridine hydrofluoride (17 mg, 15 μL, 2 equiv, 0.17 mmol) was added, and the mixture was stirred at room temperature for 1 h. The mixture was concentrated without workup. The residue was purified by flash chromatography (0–40% EtOAc in hexanes) to afford (5aS,6R,7S)-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-3-((triisopropylsilyl)oxy)-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl acetate (47) (24 mg, 61 μmol, 73% yield) as a white solid. LC–MS m/z = 391.0 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 9.31 (s, 1H), 6.52 (s, 1H), 5.61 (s, 1H), 5.33 (q, J = 2.0 Hz, 1H), 3.27 (s, 3H), 2.45 (s, 3H), 2.18 (s, 3H), 2.14 (s, 3H), 1.84 (d, J = 1.9 Hz, 3H), 1.27 (s, 3H).
(5aS,6R,7S)-3-Hydroxy-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl methyl Carbonate (48)
(5aS,6S,7S)-7-Hydroxy-6-methoxy-1,4,6,9-tetramethyl-3-((triisopropylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (46) (20 mg, 1 equiv, 40 μmol) was dissolved in THF (1 mL). Dry sodium hydride (95%, 1.9 mg, 2 equiv, 79 μmol) was added, and the mixture was stirred at 25 °C for 3 min. Then, methyl chloroformate (7.5 mg, 6.1 μL, 2 equiv, 79 μmol) was added, and the mixture was allowed to stir at 25 °C for 16 h. The mixture was then diluted with EtOAc (20 mL), washed with saturated aqueous NH4Cl (2 × 20 mL), and the organic layer was dried over Na2SO4 and concentrated. The residue was purified by flash chromatography (0–100% EtOAc in hexanes) to afford (5aS,6R,7S)-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-3-((triisopropylsilyl)oxy)-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl methyl carbonate (5.0 mg, 8.9 μmol, 22% yield) as a yellow oil. LC–MS m/z = 563.2 [M + H]+.
To a stirred solution of (5aS,6R,7S)-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-3-((triisopropylsilyl)oxy)-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl methyl carbonate (5.0 mg, 1 equiv, 8.9 μmol) in THF (1 mL) was added pyridine hydrofluoride (2.6 mg, 2.4 μL, 3 equiv, 27 μmol). The mixture was allowed to stir at 25 °C for 1 h then concentrated and the residue was purified by reverse phase HPLC (10–100% acetonitrile in water, 0.1% formic acid) to afford (5aS,6R,7S)-3-hydroxy-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl methyl carbonate (48) (0.60 mg, 1.5 μmol, 17% yield) as a white solid. LC–MS m/z = 404.8 [M – H]−. 1H NMR (400 MHz, THF-d8) δ 9.37 (s, 1H), 6.53 (s, 1H), 5.41 (s, 1H), 5.31 (q, J = 1.9 Hz, 1H), 3.78 (s, 3H), 3.31 (s, 3H), 2.45 (s, 3H), 2.18 (s, 3H), 1.85 (d, J = 1.9 Hz, 3H), 1.26 (s, 3H).
(5aS,6R,7S)-3-Hydroxy-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl Glycinate (49)
(5aS,6S,7S)-7-Hydroxy-6-methoxy-1,4,6,9-tetramethyl-3-((triisopropylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (46) (35 mg, 1 equiv, 69 μmol) was dissolved in THF (2 mL). Sodium hydride (3.3 mg, 2 equiv, 0.14 mmol) was added, and the mixture was stirred at 25 °C for 3 min. Then, Boc-Gly-OSu (38 mg, 2 equiv, 0.14 mmol) was added, and the mixture was allowed to stir at 25 °C for 16 h. The mixture was diluted in DCM and quenched with saturated aqueous NH4Cl, and the organic layer was dried over MgSO4 and concentrated. The residue was purified by flash chromatography (0–50% EtOAc in hexanes) afforded (5aS,6R,7S)-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-3-((triisopropylsilyl)oxy)-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl (tert-butoxycarbonyl)glycinate (18 mg, 27 μmol, 39% yield) as a colorless oil. LC–MS m/z = 662.2 [M + H]+.
(5aS,6R,7S)-6-Methoxy-1,4,6,9-tetramethyl-8,11-dioxo-3-((triisopropylsilyl)oxy)-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl (tert-butoxycarbonyl)glycinate (18 mg, 1 equiv, 27 μmol) was dissolved in THF (2 mL) in a vial under air. Pyridine hydrofluoride (13 mg, 12 μL, 5 equiv, 0.14 mmol) was added, and the mixture was stirred at 25 °C for 1 h. The mixture was washed with NH4Cl and extracted with DCM and the organic layer was dried over MgSO4 and concentrated. The residue was purified by flash chromatography (0–60% EtOAc in hexanes) to afford (5aS,6R,7S)-3-hydroxy-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl (tert-butoxycarbonyl)glycinate (8 mg, 0.02 mmol, 60% yield) as a colorless oil. LC–MS m/z = 504.0 [M – H]−.
(5aS,6R,7S)-3-Hydroxy-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl (tert-butoxycarbonyl)glycinate (8 mg, 1 equiv, 0.02 mmol) was dissolved in DCM (1 mL) and transferred to a vial under air. 2,2,2-trifluoroacetic acid (10 μL, 10 equiv, 0.2 mmol) was added, and the mixture was stirred at 25 °C for 1 h. The mixture was concentrated to an oil and then lyophilized to afford (5aS,6R,7S)-3-hydroxy-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl glycinate (49) (6 mg, 0.01 mmol, 90%) as a white solid. LC–MS m/z = 406.0 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 6.54 (s, 1H), 5.72 (s, 1H), 5.31 (q, J = 2.0 Hz, 1H), 4.16 (d, J = 17.7 Hz, 1H), 4.05 (d, J = 17.6 Hz, 1H), 3.32 (s, 3H), 2.46 (s, 3H), 2.18 (s, 3H), 1.85 (d, J = 1.9 Hz, 3H), 1.32 (s, 3H).
(5aS,6R,7S)-3-Hydroxy-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl 17-oxo-21-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)-4,7,10,13-tetraoxa-16-azahenicosanoate (50)
To a vial under nitrogen containing (5aS,6S,7S)-7-hydroxy-6-methoxy-1,4,6,9-tetramethyl-3-((triisopropylsilyl)oxy)-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (46) (47.4 mg, 1 equiv, 93.9 μmol) and dry sodium hydride (95%, 6.8 mg, 3 equiv, 282 μmol) was added anhydrous THF (1 mL). The mixture was stirred at room temperature for 5 min. NHS-PEG4-biotin (175 mg, 3 equiv, 282 μmol) and DMF (0.2 mL) were added, and the mixture was stirred for 3 h. An additional portion of dry sodium hydride (95%, 4.5 mg, 2 equiv, 188 μmol) and DMF (0.2 mL) were added, and the mixture was stirred overnight at room temperature. The mixture was diluted in EtOAc and washed with saturated aqueous NH4Cl, and the aqueous layer was extracted with EtOAc. The combined organic layer was dried over Na2SO4 and concentrated. The residue was purified by reverse phase chromatography (RediSep C18 column, 10–100% acetonitrile in water with 0.1% formic acid) to afford (5aS,6R,7S)-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-3-((triisopropylsilyl)oxy)-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl 17-oxo-21-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)-4,7,10,13-tetraoxa-16-azahenicosanoate (24 mg, 24 μmol, 26% yield) as a white solid. LC–MS m/z = 979.4 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 7.05 (s, 1H), 6.63 (s, 1H), 5.80 (s, 1H), 5.67 (s, 1H), 5.58 (s, 1H), 5.43 (d, J = 2.1 Hz, 1H), 4.35 (t, J = 6.5 Hz, 1H), 4.19 (t, J = 6.3 Hz, 1H), 3.77 (t, J = 6.5 Hz, 2H), 3.56 (s, 3H), 3.55–3.50 (m, 1H), 3.45 (t, J = 5.5 Hz, 2H), 3.30 (s, 3H), 3.28 (d, J = 5.2 Hz, 2H), 3.16–3.09 (m, 1H), 2.85 (dd, J = 12.3, 5.1 Hz, 1H), 2.71 (t, J = 6.4 Hz, 2H), 2.61 (d, J = 12.5 Hz, 1H), 2.48 (d, J = 10.8 Hz, 8H), 2.26 (s, 3H), 2.10 (t, J = 7.3 Hz, 2H), 1.85 (d, J = 2.0 Hz, 3H), 1.62 (q, J = 7.7 Hz, 2H), 1.47–1.33 (m, 3H), 1.24 (s, 3H), 1.15 (dd, J = 7.5, 3.7 Hz, 18H).
To a vial under nitrogen containing (5aS,6R,7S)-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-3-((triisopropylsilyl)oxy)-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl 17-oxo-21-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)-4,7,10,13-tetraoxa-16-azahenicosanoate (9.4 mg, 1 equiv, 9.6 μmol) and TBAT (7.8 mg, 1.5 equiv, 14 μmol) was added anhydrous THF (0.4 mL). The mixture was stirred at room temperature for 10 min then concentrated. The residue was purified by reverse phase chromatography (RediSep C18 column, 10–100% acetonitrile in water with 0.1% formic acid) afforded (5aS,6R,7S)-3-hydroxy-6-methoxy-1,4,6,9-tetramethyl-8,11-dioxo-5a,6,7,8-tetrahydro-11H-dibenzo[b,e][1,4]dioxepin-7-yl 17-oxo-21-((3aS,4S,6aR)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)-4,7,10,13-tetraoxa-16-azahenicosanoate (50) (4.2 mg, 5.1 μmol, 53% yield) as a white solid. LC–MS m/z = 822.2 [M + H]+. 1H NMR (400 MHz, THF-d8) δ 7.10 (s, 1H), 6.59 (s, 1H), 5.97 (s, 1H), 5.79 (s, 1H), 5.70 (s, 1H), 5.36 (d, J = 2.2 Hz, 1H), 4.42–4.37 (m, 1H), 4.25–4.19 (m, 1H), 3.77 (t, J = 6.2 Hz, 2H), 3.56–3.50 (m, 6H), 3.44 (t, J = 5.4 Hz, 2H), 3.27 (d, J = 2.5 Hz, 5H), 3.16–3.10 (m, 1H), 2.87 (dd, J = 12.5, 5.0 Hz, 1H), 2.70 (td, J = 6.3, 2.1 Hz, 2H), 2.64 (d, J = 12.5 Hz, 1H), 2.46 (s, 3H), 2.18 (s, 3H), 2.08 (t, J = 7.3 Hz, 2H), 1.84 (d, J = 1.9 Hz, 3H), 1.57 (h, J = 6.4 Hz, 3H), 1.41 (td, J = 14.4, 6.5 Hz, 2H), 1.29 (s, 3H).
(5aS,6S,7S)-3,7-Dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydrodibenzo[b,f][1,4]oxazepine-8,11(5aH,10H)-dione (51)
(5aS,6S,7S)-3,7-Dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydro-11H-dibenzo[b,e][1,4]dioxepine-8,11(5aH)-dione (60.0 mg, 1 equiv, 172 μmol), and DMF (0.3 mL) were added to a microwave vial under air. Ammonia solution (0.4 M in THF, 2.15 mL, 5 equiv, 861 μmol) was added, then the mixture was stirred at room temperature for 4 h. The reaction was concentrated then toluene (5.0 mL) and PPTS (17.3 mg, 0.4 equiv, 68.9 μmol) were added to the reaction mixture and the mixture was heated to 110 °C for 1 h. Additional PPTS (17.3 mg, 0.4 equiv, 68.9 μmol) was added and the reaction was heated to 115 °C in the microwave reactor for 1 h. The reaction was heated to 120 °C in the microwave reactor for 3 h. The reaction was concentrated and the residue was purified by flash chromatography (0–50% EtOAc in hexane, 10 g column) then further purified by reverse phase HPLC (10–100% acetonitrile in water, 0.1% formic acid) to give (5aS,6S,7S)-3,7-dihydroxy-6-methoxy-1,4,6,9-tetramethyl-6,7-dihydrodibenzo[b,f][1,4]oxazepine-8,11(5aH,10H)-dione (51) (26 mg, 75 μmol, 43% yield) as a white solid. LC–MS m/z = 346.0 [M – H]−. 1H NMR (400 MHz, THF-d8) δ 8.96 (s, 1H), 8.44 (s, 1H), 6.48 (s, 1H), 4.77 (d, J = 1.9 Hz, 1H), 4.38 (d, J = 2.6 Hz, 1H), 4.15 (d, J = 2.5 Hz, 1H), 3.42 (s, 3H), 2.43 (s, 3H), 2.21 (s, 3H), 1.90 (d, J = 1.8 Hz, 3H), 1.22 (s, 3H).
Acknowledgments
Thanks to John Bacsa and the Emory X-ray Crystallography Center for single crystal X-ray crystallographic analysis, Eric Smith for graphic design, and several contract research organizations for experimental support—WuXi AppTec (Shanghai, China), WuXi AppTec (New Jersey), Viva Biotech (Shanghai, China), CreLux (Martinsried, Germany), Certis Oncology Solutions, Inc. (San Diego, CA), Promega (Madison, WI), XenoBiotic Laboratory (Cranbury, NJ).
Glossary
Abbreviations Used
- CDK
cyclin-dependent kinase
- GEM
genetically encoded small molecule
- ETaG
embedded target gene
- TD3
top-down drug discovery
- BGC
biosynthetic gene cluster
- PDX
patient-derived xenotransplant
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c01095.
Pho85:ETaG chimera activity; phylogenetic analysis of production strain; transcriptional and metabolomic effects of BGC gene KOs; in vitro assays with BGC enzymes, proposed biosynthesis of known roseopurpurins and aculeatusquinones; intact MS and peptide mapping of CDKs bound to 2; chemoproteomics of 2 in human cell lysate; BGC function; NMR assignments; X-ray crystallography; nucleic acids used; UniProt IDs for sequence data used; MS and NMR spectra of compounds isolated from fungi; MS and NMR spectra of synthetic compounds; X-ray crystallography reports for compounds 2 and 43; additional genomics (ETaG search, resistance mutation analysis, phylogenetic reconstruction, RNA sequencing and analysis); synthetic biology (filamentous fungal engineering, heterologous expression and assay of RosA and RosD, fungal Pho85 and human CDKs); microbiology (fungal fermentation and GEM extraction); and biochemistry (protein intact mass spectrometry, X-ray crystallography, chemoproteomics) methods (PDF)
SMILES strings for all compounds in this study (CSV)
Raw data for phylogenetic trees (ZIP)
Author Present Address
§ Mount Sinai South Nassau, One Healthy Way, Oceanside, New York 11572, United States
Author Present Address
∥ Matchpoint Therapeutics, 99 Coolidge Ave, Watertown, Massachusetts 02472, United States
Author Present Address
⊥ Sail Biomedicines, 140 First St, Suite 600, Cambridge, Massachusetts 02141, United States
Author Present Address
# Moderna Inc., 1 Upland Rd, Norwood, Massachusetts 02062, United States
Author Present Address
∇ Bactobio Ltd., 1–45 Durham St, London SE11 5JH, U.K.
Author Present Address
○ JN Analytical Solutions LLC, 2 Oneida Place, Hudson, Massachusetts 01749, United States
Author Present Address
◆ Sensorium Therapeutics, 40 Guest Street, Boston, Massachusetts 02135, United States
Author Present Address
¶ Center for Cystic Fibrosis and Airways Disease Research (CF-AIR), Department of Pediatrics, Emory University School of Medicine, 2015 Uppergate Dr NE, Atlanta, Georgia 30322, United States
Author Present Address
†† University of Iowa. 230 N Madison St, Iowa City, Iowa 52240, United States
Author Present Address
‡‡ Atavistik Bio Inc., 101 Cambridgepark Drive, Cambridge, Massachusetts 02140, United States
Author Present Address
§§ Entact Bio, 65 Grove St, Watertown, Massachusetts 02472, United States
Author Present Address
∥∥ Servier Pharmaceuticals, 200 Pier 4 Blvd., Boston, Massachusetts 02210, United States
Author Present Address
⊥⊥ Empress Therapeutics, 99 Coolidge Ave, Watertown, Massachusetts 02472, United States
Author Present Address
## Fungal Biotech Solutions, 20 fourth St, Medford, Massachusetts 02155, United States
Author Present Address
∇∇ Revolution Medicines, Inc., 700 Saginaw Dr., Redwood City, California 94063, United States
Author Contributions
○○ J.D., M.H., S.P.R., and Y.J.C. contributed equally to this work.
The authors declare the following competing financial interest(s): All authors are current or former employees of LifeMine Therapeutics.
Special Issue
Published as part of Journal of Medicinal Chemistryvirtual special issue “Natural Products Driven Medicinal Chemistry”.
Supplementary Material
References
- Tobert J. A. Lovastatin and beyond: The History of the HMG-CoA Reductase Inhibitors. Nat. Rev. Drug Discovery 2003, 2 (7), 517–526. 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- Matyori A.; Brown C. P.; Ali A.; Sherbeny F. Statins Utilization Trends and Expenditures in the U.S. before and after the Implementation of the 2013 ACC/AHA Guidelines. Saudi Pharm. J. 2023, 31 (6), 795–800. 10.1016/j.jsps.2023.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzi J. R.; Sayegh M. H.; Mallat S. G. Calcineurin Inhibitors: 40 Years Later, Can’t Live Without···. J. Immunol 2013, 191 (12), 5785–5791. 10.4049/jimmunol.1390055. [DOI] [PubMed] [Google Scholar]
- Schreiber S. L. The Rise of Molecular Glues. Cell 2021, 184 (1), 3–9. 10.1016/j.cell.2020.12.020. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83 (3), 770–803. 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- Rolinson G. N.; Geddes A. M. The 50th Anniversary of the Discovery of 6-Aminopenicillanic Acid (6-APA). Int. J. Antimicrob. Agents 2007, 29 (1), 3–8. 10.1016/j.ijantimicag.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Bentley R. Mycophenolic Acid: A One Hundred Year Odyssey from Antibiotic to Immunosuppressant. Chem. Rev. 2000, 100 (10), 3801–3826. 10.1021/cr990097b. [DOI] [PubMed] [Google Scholar]
- Beutler J. A. Natural Products as a Foundation for Drug Discovery. Curr. Protoc. Pharmacol. 2019, 86 (1), e67 10.1002/cpph.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasov A. G.; Zotchev S. B.; Dirsch V. M.; Orhan I. E.; Banach M.; Rollinger J. M.; Barreca D.; Weckwerth W.; Bauer R.; Bayer E. A.; Majeed M.; Bishayee A.; Bochkov V.; Bonn G. K.; Braidy N.; Bucar F.; Cifuentes A.; D’Onofrio G.; Bodkin M.; Diederich M.; Dinkova-Kostova A. T.; Efferth T.; Bairi K. E.; Arkells N.; Fan T.-P.; Fiebich B. L.; Freissmuth M.; Georgiev M. I.; Gibbons S.; Godfrey K. M.; Gruber C. W.; Heer J.; Huber L. A.; Ibanez E.; Kijjoa A.; Kiss A. K.; Lu A.; Macias F. A.; Miller M. J. S.; Mocan A.; Müller R.; Nicoletti F.; Perry G.; Pittalà V.; Rastrelli L.; Ristow M.; Russo G. L.; Silva A. S.; Schuster D.; Sheridan H.; Skalicka-Woźniak K.; Skaltsounis L.; Sobarzo-Sánchez E.; Bredt D. S.; Stuppner H.; Sureda A.; Tzvetkov N. T.; Vacca R. A.; Aggarwal B. B.; Battino M.; Giampieri F.; Wink M.; Wolfender J.-L.; Xiao J.; Yeung A. W. K.; Lizard G.; Popp M. A.; Heinrich M.; Berindan-Neagoe I.; Stadler M.; Daglia M.; Verpoorte R.; Supuran C. T. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discovery 2021, 20, 200–216. 10.1038/s41573-020-00114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigdel U. K.; Lee S.-J.; Sowa M. E.; Bowman B. R.; Robison K.; Zhou M.; Pua K. H.; Stiles D. T.; Blodgett J. A. V.; Udwary D. W.; Rajczewski A. T.; Mann A. S.; Mostafavi S.; Hardy T.; Arya S.; Weng Z.; Stewart M.; Kenyon K.; Morgenstern J. P.; Pan E.; Gray D. C.; Pollock R. M.; Fry A. M.; Klausner R. D.; Townson S. A.; Verdine G. L. Genomic Discovery of an Evolutionarily Programmed Modality for Small-Molecule Targeting of an Intractable Protein Surface. Proc. Natl. Acad. Sci. U.S.A. 2020, 117 (29), 17195–17203. 10.1073/pnas.2006560117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osbourn A. Secondary Metabolic Gene Clusters: Evolutionary Toolkits for Chemical Innovation. Trends Genet. 2010, 26 (10), 449–457. 10.1016/j.tig.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Cundliffe E.; Demain A. L. Avoidance of Suicide in Antibiotic-Producing Microbes. J. Ind. Microbiol. Biotechnol. 2010, 37 (7), 643–672. 10.1007/s10295-010-0721-x. [DOI] [PubMed] [Google Scholar]
- Almabruk K. H.; Dinh L. K.; Philmus B. Self-Resistance of Natural Product Producers: Past, Present, and Future Focusing on Self-Resistant Protein Variants. ACS Chem. Biol. 2018, 13 (6), 1426–1437. 10.1021/acschembio.8b00173. [DOI] [PubMed] [Google Scholar]
- Hutchinson C. R.; Kennedy J.; Park C.; Kendrew S.; Auclair K.; Vederas J. Aspects of the Biosynthesis of Non-Aromatic Fungal Polyketides by Iterative Polyketide Synthases. Antonie van Leeuwenhoek 2000, 78 (3–4), 287–295. 10.1023/A:1010294330190. [DOI] [PubMed] [Google Scholar]
- Kachroo A. H.; Laurent J. M.; Yellman C. M.; Meyer A. G.; Wilke C. O.; Marcotte E. M. Systematic Humanization of Yeast Genes Reveals Conserved Functions and Genetic Modularity. Science 2015, 348 (6237), 921–925. 10.1126/science.aaa0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedoya-Cardona J. E.; Rubio-Carrasquilla M.; Ramírez-Velásquez I. M.; Valdés-Tresanco M. S.; Moreno E. Identifying Potential Molecular Targets in Fungi Based on (Dis)Similarities in Binding Site Architecture with Proteins of the Human Pharmacolome. Molecules 2023, 28 (2), 692 10.3390/molecules28020692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.; Liu N.; Tang Y. Recent Developments in Self-Resistance Gene Directed Natural Product Discovery. Nat. Prod. Rep. 2020, 37 (7), 879–892. 10.1039/C9NP00050J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooda D. U.; Haruyoshi S.; Yuki T.; Michael W. P.; Nami Y.. Dibenzo-Oxazepine and -Dioxepine Derivatives and Their Use as Anti-Tumor Agents. WO Patent WO1997047611A1, 1997.
- Chen L.; Zhang W.; Zheng Q.; Liu Q.; Zhong P.; Hu X.; Fang Z.; Zhang Q. Aculeatusquinones A—D, Novel Metabolites from the Marine-Derived Fungus Aspergillus Aculeatus. Heterocycles 2013, 87 (4), 861–867. 10.3987/COM-13-12667. [DOI] [Google Scholar]
- Shang Z.; Khalil Z.; Li L.; Salim A. A.; Quezada M.; Kalansuriya P.; Capon R. J. Roseopurpurins: Chemical Diversity Enhanced by Convergent Biosynthesis and Forward and Reverse Michael Additions. Org. Lett. 2016, 18 (17), 4340–4343. 10.1021/acs.orglett.6b02099. [DOI] [PubMed] [Google Scholar]
- Verdine G. L.; Cumming J. N.; Saalau S. M.; Castaldi P. M.; Choi Y.. CDK Inhibitor Compounds for Use in Methods of Treatment. WO Patent WO2022187611A1, 2022.
- Dunbar K. L.; Perlatti B.; Liu N.; Cornelius A.; Mummau D.; Chiang Y.-M.; Hon L.; Nimavat M.; Pallas J.; Kordes S.; Ng H. L.; Harvey C. J. B. Resistance Gene–Guided Genome Mining Reveals the Roseopurpurins as Inhibitors of Cyclin-Dependent Kinases. Proc. Natl. Acad. Sci. U.S.A. 2023, 120 (48), e2310522120 10.1073/pnas.2310522120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadesse S.; Anshabo A. T.; Portman N.; Lim E.; Tilley W.; Caldon C. E.; Wang S. Targeting CDK2 in Cancer: Challenges and Opportunities for Therapy. Drug Discovery Today 2020, 25 (2), 406–413. 10.1016/j.drudis.2019.12.001. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. Targeting Cancer with Small Molecule Kinase Inhibitors. Nat. Rev. Cancer 2009, 9 (1), 28–39. 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- Huang K.; Ferrin-O’Connell I.; Zhang W.; Leonard G. A.; O’Shea E. K.; Quiocho F. A. Structure of the Pho85-Pho80 CDK-Cyclin Complex of the Phosphate-Responsive Signal Transduction Pathway. Mol. Cell 2007, 28 (4), 614–623. 10.1016/j.molcel.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visagie C. M.; Seifert K. A.; Houbraken J.; Samson R. A.; Jacobs K. A Phylogenetic Revision of Penicillium Sect. Exilicaulis, Including Nine New Species from Fynbos in South Africa. IMA Fungus 2016, 7 (1), 75–117. 10.5598/imafungus.2016.07.01.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X.; Chen Y.; Long T.; Liu Z.; Zhang Q.; Zhang H.; Yan Y.; Zhang C.; Zhu Y. Genome Mining and Biosynthetic Reconstitution of Fungal Depsidone Mollicellins Reveal a Dual Functional Cytochrome P450 for Ether Formation. J. Nat. Prod. 2023, 86 (8), 2046–2053. 10.1021/acs.jnatprod.3c00609. [DOI] [PubMed] [Google Scholar]
- Armstrong R. N. Mechanistic Diversity in a Metalloenzyme Superfamily. Biochemistry 2000, 39 (45), 13625–13632. 10.1021/bi001814v. [DOI] [PubMed] [Google Scholar]
- Endicott J. A.; Noble M. E. M.; Johnson L. N. The Structural Basis for Control of Eukaryotic Protein Kinases. Annu. Rev. Biochem. 2012, 81 (1), 587–613. 10.1146/annurev-biochem-052410-090317. [DOI] [PubMed] [Google Scholar]
- Copeland R. A.; Luo L.; Auger K. R.; Huang P. S. A Method for Determining Intracellular Concentrations of Enzyme Substrates from a Combination of Competitive Inhibition and Mutagenesis Studies. Anal. Biochem. 2005, 337 (2), 351–353. 10.1016/j.ab.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Schwartz P. A.; Kuzmic P.; Solowiej J.; Bergqvist S.; Bolanos B.; Almaden C.; Nagata A.; Ryan K.; Feng J.; Dalvie D.; Kath J. C.; Xu M.; Wani R.; Murray B. W. Covalent EGFR Inhibitor Analysis Reveals Importance of Reversible Interactions to Potency and Mechanisms of Drug Resistance. Proc. Natl. Acad. Sci. U.S.A. 2014, 111 (1), 173–178. 10.1073/pnas.1313733111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsherniak A.; Vazquez F.; Montgomery P. G.; Weir B. A.; Kryukov G.; Cowley G. S.; Gill S.; Harrington W. F.; Pantel S.; Krill-Burger J. M.; Meyers R. M.; Ali L.; Goodale A.; Lee Y.; Jiang G.; Hsiao J.; Gerath W. F. J.; Howell S.; Merkel E.; Ghandi M.; Garraway L. A.; Root D. E.; Golub T. R.; Boehm J. S.; Hahn W. C. Defining a Cancer Dependency Map. Cell 2017, 170 (3), 564–576.e16. 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarkowska T.; Mittnacht S. Differential Phosphorylation of the Retinoblastoma Protein by G1/S Cyclin-Dependent Kinases*. J. Biol. Chem. 1997, 272 (19), 12738–12746. 10.1074/jbc.272.19.12738. [DOI] [PubMed] [Google Scholar]
- Scheeff E. D.; Bourne P. E. Structural Evolution of the Protein Kinase–Like Superfamily. PLoS Comput. Biol. 2005, 1 (5), e49 10.1371/journal.pcbi.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Médard G.; Pachl F.; Ruprecht B.; Klaeger S.; Heinzlmeir S.; Helm D.; Qiao H.; Ku X.; Wilhelm M.; Kuehne T.; Wu Z.; Dittmann A.; Hopf C.; Kramer K.; Kuster B. Optimized Chemical Proteomics Assay for Kinase Inhibitor Profiling. J. Proteome Res. 2015, 14 (3), 1574–1586. 10.1021/pr5012608. [DOI] [PubMed] [Google Scholar]
- Malumbres M.; Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 2009, 9 (3), 153–166. 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Zhang M.; Sun J.; Yang X. Casein kinase 1α: biological mechanisms and theranostic potential. Cell Commun. Signaling 2018, 16, 23 10.1186/s12964-018-0236-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A.; Burban D. J.; Muretta J. M.; Thompson A. R.; Engel T. A.; Rasmussen D. M.; Subrahmanian M. V.; Veglia G.; Thomas D. D.; Levinson N. M. Allostery governs Cdk2 activation and differential recognition of CDK inhibitors. Nat. Chem. Biol. 2021, 17, 456–464. 10.1038/s41589-020-00725-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeson P. D.; Davis A. M. Time-Related Differences in the Physical Property Profiles of Oral Drugs. J. Med. Chem. 2004, 47 (25), 6338–6348. 10.1021/jm049717d. [DOI] [PubMed] [Google Scholar]
- Lin N. U.; Amiri-Kordestani L.; Palmieri D.; Liewehr D. J.; Steeg P. S. CNS Metastases in Breast Cancer: Old Challenge, New Frontiers. Clin. Cancer Res. 2013, 19 (23), 6404–6418. 10.1158/1078-0432.CCR-13-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P.; Cross D.; Jänne P. A. Kinase Drug Discovery 20 Years after Imatinib: Progress and Future Directions. Nat. Rev. Drug Discovery 2021, 20, 551–569. 10.1038/s41573-021-00195-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shults M. D.; Imperiali B. Versatile Fluorescence Probes of Protein Kinase Activity. J. Am. Chem. Soc. 2003, 125 (47), 14248–14249. 10.1021/ja0380502. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.