


Abstract
A general approach is described for controlling the RNA-cleaving activity of nucleic acid enzymes (ribozymes and DNAzymes) via the use of oligonucleotide effectors (regulators). In contrast to the previously developed approaches of allosteric and facilitator-mediated regulation of such enzymes, this approach, called ‘expansive’ regulation, requires that the regulator bind simultaneously to both enzyme and substrate to form a branched three-way complex. Such three-way enzyme–substrate–regulator complexes are catalytically competent relative to the structurally unstable enzyme–substrate complexes. Using the 8-17 and bipartite DNAzymes and the hammerhead ribozyme as model systems, 20- to 30-fold rate enhancements were achieved in the presence of regulators of engineered variants of the above three enzymes, even under unoptimized conditions. Broadly, using this approach ribozyme and DNAzyme variants that are amenable to regulation by oligonucleotide effectors can be designed even in the absence of any knowledge of the folded structure of the relevant ribozyme or DNAzyme. Expansive regulation therefore represents a new and potentially useful technology for both the regulation of nucleic acid enzymes and the detection of specific RNA transcripts.
INTRODUCTION
The discovery of catalytic RNA (1,2) and the subsequent development of in vitro selection and evolution (reviewed in 3–6) have revolutionized conceptions about biological catalysis and enzyme function. All naturally occurring ribozymes (with the notable exception of the ribosome; 7) and many of the in vitro selected nucleic acid enzymes catalyze chemical reactions on the phosphodiester backbone of nucleic acids. Examples of such reactions include the cleavage (8–18) and/or ligation (19–24) of RNA and DNA. With a few exceptions [such as tertiary structure-mediated substrate recognition by the Neurospora VS ribozyme (25) and the triplex interaction required by a Cu2+-dependent DNAzyme (26)], the target site specificity of these nucleic acid enzymes is achieved exclusively through Watson–Crick base pairing between sequences flanking the target phosphodiester bond and those flanking the catalytic core of the enzyme. Therefore, the catalytic cycle of these enzymatic reactions (see Fig. 1A, upper) can be simply described as a series of three sequential steps: (i) binding of enzyme to substrate(s) (defined by a forward rate constant k1 and back constant k–1); (ii) chemical modification of the substrate(s) (defined by k2); (iii) release of the product(s) (defined by k3). Since both the enzyme and substrate(s) are nucleic acids, the rates of substrate binding and product release follow the general rules of nucleic acid hybridization. Factors such as the number of base pairs formed between enzyme and substrate(s) and the GC content can greatly influence the overall rate of catalysis. For optimal enzyme activity, the interaction between the enzyme substrate recognition arms and the substrate(s) should be stable enough to facilitate chemistry, yet loose enough to allow rapid dissociation of the product(s) (27,28).
Figure 1.

The concept of expansive regulation. (A) The kinetic framework for expansively regulated nucleic acid enzymes. In the absence of the regulator oligonucleotide (top pathway), formation of the enzyme–substrate complex is unfavorable (k–1 >> k1) and, therefore, catalysis does not proceed efficiently. However, in the presence of the regulator (bottom pathway), formation of an enzyme–substrate–regulator complex becomes favorable (k1 >> k–1) and allows catalysis to proceed efficiently. (B) Optimal design of an expansively regulated 10-23 DNAzyme (46). While one substrate-binding arm of the 10-23 DNAzyme remains unchanged (forms 6 bp with the substrate, stem A), the other substrate-binding arm is modified such that it is only able to form 3 bp with the substrate (stem B). The regulator oligonucleotide (either DNA or RNA) is complementary to both the substrate (forming 6 bp, stem C) and the DNAzyme (forming 7 bp, stem D). At the three-way junction a bulge composed of two adenosine bases was introduced in the DNAzyme strand to enhance stability of the junction. The arrow identifies the site of RNA cleavage.
One feature of the above mentioned nucleic acid enzymes is that they are relatively simple catalytic systems. Catalysis occurs when the enzyme and substrate associate through complementary base pair hybridization and therefore is not amenable to regulation in the manner of protein enzymes found in living systems (29). Recently, however, ribozymes with more sophisticated kinetic characteristics have been created using molecular engineering strategies such as modular rational design and in vitro selection (reviewed in 30,31). Both of these approaches exploit a considerable understanding of the secondary and/or tertiary folding of the relevant ribozyme such that an aptamer element for effector binding (or, alternatively, a random sequence element from which an effector-binding site may evolve) and the ribozyme entities are joined in such a way that conformational stabilization of the aptamer upon ligand binding also leads to stabilization and catalytic activation of the ribozyme. To date, a number of allosteric ribozymes have been created and/or selected to be responsive to a variety of effector molecules, including ATP (32), flavin mononucleotide (FMN) (33–37), theophylline (35–37), the antibiotics doxycycline (38) and pefloxacin (39), the second messengers cAMP and cGMP (40), oligonucleotides (23,41–43) and proteins (44,45).
We have recently reported a distinct strategy for constructing the first DNA enzyme (the 10-23 DNAzyme) whose catalytic activity can be specifically controlled by the binding of effector oligonucleotides (46). Unlike the generation of allosteric ribozymes, our strategy, termed ‘expansive regulation’, requires no prior knowledge of the secondary or tertiary folding of the enzyme. Whereas most allosteric ribozymes are regulated at the chemical step (defined by k2), expansive regulation modulates catalysis mainly at the substrate-binding step (defined by k1 and k–1) (46). In this present set of studies we wished to investigate the generality of expansive regulation of ribozymes and DNAzymes utilizing oligonucleotide effectors. Expansive regulation is attempted herein with two further RNA-cleaving DNAzymes [8-17 (14) and bipartite (18)] as well as with the hammerhead ribozyme (whose folded structure is known). To our knowledge, the design strategy described herein is the only general method to be successfully applied to both RNA and DNA enzymes, including those whose folding is understood and those whose folding is not yet understood. Our approach therefore promises a considerable versatility of effector-mediated control of a variety of catalytic nucleic acids (including the larger ribozymes), provided that recognition between such enzymes and their substrates is based substantially on Watson–Crick base pairing.
MATERIALS AND METHODS
DNA and RNA oligomers: synthesis and purification
Synthetic DNA oligomers were purchased from Sigma-Genosys (Woodlands, TX) or were synthesized on an Applied Biosystems 392 DNA/RNA synthesizer using standard phosphoramidite methodology. Synthetic RNA substrates were purchased from Dharmacon Research (Boulder, CO). All oligonucleotides were purified by denaturing (8 M urea) PAGE. Purified RNA substrates were radiolabeled with T4 polynucleotide kinase (Gibco BRL) and [γ-32P]ATP (NEN) using standard kinasing protocols and then re-purified by 20% PAGE. Purified DNA/RNA was isolated from the gel by crush-soaking in 10 mM Tris–HCl (pH 7.5 at 25°C), 300 mM NaOAc and 1 mM EDTA and were precipitated from solution by the addition of 2.5 vol of 100% ethanol.
Hammerhead ribozyme variants were generated by in vitro transcription of the appropriate DNA templates made double stranded by extension using Taq polymerase (45). Transcription reactions (200 µl) containing ∼500 pmol template DNA, 40 mM Tris–HCl (pH 8.0 at 25°C), 20 mM MgCl2, 5 mM DTT, 1 mM spermidine, 20 mg/ml PEG-8000, 3 mM each NTP and 0.2 mg/ml T7 RNA polymerase were incubated at 37°C for 3 h. The resulting transcription reactions were phenol:cholorform extracted and the RNA products were ethanol precipitated, followed by purification by denaturing 10% PAGE and recovery as described above.
Single turnover assays and kinetic analyses
Kinetic analyses of all engineered DNAzyme and ribozyme constructs were carried out under single turnover conditions, as previously described (46), with enzyme (500 nM) in excess over trace concentrations (∼2 nM) of γ-32P-labeled substrate RNA, at 23°C. The above concentration of enzyme is sufficient to saturate the substrate present, since doubling or tripling the concentration of substrate had no discernable effect on the cleavage rates. The final concentration of regulator oligonucleotides, if used, was 10 µM. The combined enzyme/regulator and substrate/regulator solutions in water were heated, separately, for 1 min at 90°C to disrupt folded structures that may have formed during storage. Following cooling to 23°C, the enzyme/regulator and substrate/regulator solutions were made up to 50 mM Tris–HCl (pH 7.5 at 25°C), 20 mM MgCl2, 100 mM NaCl and 0.01% SDS, for reactions involving the DNAzymes, or to 50 mM Tris–HCl (pH 7.5 at 25°C) and 20 mM MgCl2, for reactions involving hammerhead ribozymes. The enzyme/regulator and substrate/regulator solutions were further incubated for 10 min at the assay temperature to prevent anomalous initial rates that might result from a slow adoption of Mg2+-dependent folded structures. Cleavage reactions were initiated by combining the enzyme/regulator and substrate/regulator solutions. Aliquots were removed at appropriate time intervals and quenched with stop buffer (10 mM Tris–HCl pH 8.0 at 25°C, 40 mM EDTA, 95% formamide, 0.01% bromophenol blue, 0.005% xylene cyanol). These aliquoted samples were analyzed by 20% denaturing PAGE and quantified using a Molecular Dynamics PhosphorImager.
Cleavage rates were found to be first order through the first three half-lives of the reaction. Rate constants for reactions were determined from the negative slopes of the natural log of fractions of uncleaved substrate plotted against time (and normalized to the final extent of cleavage by the unmodified enzyme; 32). Each rate constant was obtained as an average of at least two independent experiments.
RESULTS
The expansive regulation strategy
The general concept of expansive regulation is illustrated in Figure 1A. The design strategy can be divided into two components. First, to ensure that the catalytic activity of the enzyme is severely hindered in the absence of the oligonucleotide effector, the enzyme is constructed to have a sub-optimal number of potential base pairs with the substrate. This effectively hampers catalysis by lowering the stability of the enzyme–substrate complex (Fig. 1A, top pathway). The second aspect of the design is restoration of efficient enzyme–substrate association by providing additional base pairing between enzyme and substrate via the introduction of a third, ‘regulator’ oligonucleotide. This regulator oligonucleotide is in part complementary to the enzyme and in part complementary to the substrate; it is therefore able to base pair to both simultaneously via the formation of a three-way junction (Fig. 1A, bottom pathway).
This design strategy was previously applied successfully (46) to the in vitro selected 10-23 DNAzyme (14). The optimized design of such a 10-23 variant is illustrated in Figure 1B. This variant was designed to form 6 and 3 bp (stems A and B, the two substrate-binding arms flanking the catalytic core) with the substrate in the absence of regulator. In the presence of regulator, additional 6 bp interactions were provided between the enzyme and the substrate (stem C) and 7 bp to mediate the interaction between the enzyme and the regulator (stem D). In addition to these, the design incorporated two unpaired adenosines (adenosine bulge) in the enzyme strand, located at the three-way junction, to provide further stabilization of the enzyme–substrate–regulator complex by permitting two of the three stems to stack. With this optimized design, the variant of the 10-23 enzyme shown in Figure 1B had kobs values ∼50-fold and ∼250-fold higher in the presence of DNA and RNA regulators, respectively (relative to kobs values measured in the absence of any regulator), under single turnover conditions. Multiple turnover analysis of this 10-23 variant revealed that presence of the regulator improved both the kcat and Km of the catalytic system (46).
Design and characterization of expansively controlled 8-17 DNAzyme variants
Considering the simplicity of the expansive regulation design used for the 10-23 DNAzyme, we speculated that various other RNA-cleaving DNAzymes as well as ribozymes could be engineered to be comparably or even more responsive to oligonucleotide effectors. We initiated our investigation of possible generality on the 8-17 DNAzyme, another small and sequence-specific RNA-cleaving DNA enzyme (14). Interestingly, the catalytic core of 8-17 has also evolved from another, independent, in vitro selection that used a different starting library and also different selection conditions (16). The 8-17 catalytic core is simple and slightly smaller than the 10-23 catalytic core (13 versus 15 nt, respectively). Figure 2A shows that the conserved core of 8-17 consists of a 3 bp internal stem–loop followed by an unpaired region of 4 nt. In addition, the 8-17 DNAzyme requires a rG·dT wobble base pair immediately downstream of the scissile phosphodiester bond for full catalytic activity (14).
Figure 2.
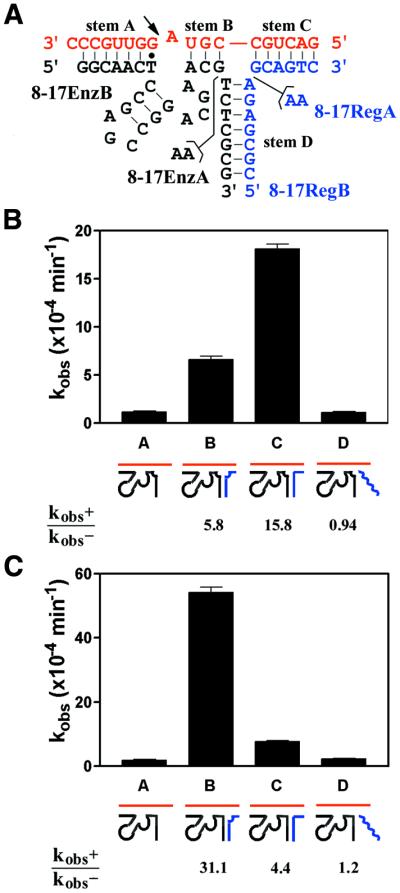
Design of and kinetic modulation by expansively regulated 8-17 DNAzyme variants. (A) Sequence and secondary structure of the 8-17 variants (black) complexed with RNA substrate (red) and regulator oligonucleotide (blue). The four stem elements present in the structure are indicated and the site of RNA cleavage is marked by an arrow. The observed rate constants (kobs) were measured for 8-17EnzA (B) and 8-17EnzB (C) under single turnover conditions [500 nM enzyme, ∼2 nM substrate, 10 µM regulator (if used), 50 mM Tris–HCl (pH 7.5 at 25°C), 20 mM MgCl2, 100 mM NaCl and 0.01% SDS]. Lane A, no regulator; lane B, with 8-17RegA; lane C, with 8-17RegB; lane D, with control oligonucleotide (BpRegA). Catalytic modulation (kobs+/kobs–) for each construct is indicated.
At 20 mM Mg2+ the unmodified 8-17 DNAzyme, with 6 and 9 bp on the substrate-binding arms, yielded kobs of 0.0107 ± 0.0003 min–1 and addition of the regulator oligonucleotides had no effect on the catalytic rate. We then constructed a series of regulator-dependent 8-17 variants (Fig. 2A) based on the optimized 10-23 construct (Fig. 1B), in all of which 6 bp (in addition to the rG·dT wobble base pair) for stem A and 3 bp for stem B flanked the catalytic 8-17 motif. The number of base pairs between the regulator and the substrate (stem C) and between the regulator and the enzyme (stem D) were also maintained at 6 and 7 bp, respectively (Fig. 2A).
Our experiments with the 10-23 DNAzyme had demonstrated that the presence of a two-adenosine bulge at the three-way junction site could greatly enhance regulator-dependent catalytic activity by contributing to a more stable assembly of the enzyme–substrate–regulator complex (presumably utilizing a coaxial stacking interaction between two of the three stems; 46). However, depending on the location of the adenosine bulge (i.e. whether it was located on the enzyme strand or the regulator strand), alternative conformers were possible. Such conformers in DNA three-way junctions are stereochemically non-equivalent and may have significant differences in stability (47,48). In our constructs it was also possible that given conformers could sterically interfere with the enzyme core and inhibit catalysis (46). Because no tertiary structural information on the 8-17 motif is available and the spatial orientation of helices in given three-way junctions could not be simply predicted, two constructs of the 8-17 enzyme strand were created, one containing a two-adenosine bulge (8-17EnzA) and the other lacking such a bulge (8-17EnzB). Likewise, two regulator sequences were constructed (8-17RegA, with an adenosine bulge, and 8-17RegB, without an adenosine bulge) (Fig. 2A). The catalytic properties of the two 8-17 DNAzyme variants were then investigated in the absence and presence of the two different regulator oligonucleotides. Figure 2B and C presents the results of the investigation for 8-17EnzA and 8-17EnzB, respectively.
Overall, the presence of regulators (irrespective of the location of the adenosine bulge) enhanced catalysis (Fig. 2B and C, lanes B and C) while control DNA oligonucleotides (that did not base pair to either the enzyme or the substrate) used in place of regulators did not influence catalysis (Fig. 2B and C, lane D). The location of the adenosine bulge did appear to have a major influence on the degree of catalytic activation (kobs+/kobs–, defined as the observed rate constant in the presence of a regulator divided by the rate constant in the absence of any regulator). In marked contrast to the 10-23 variant, where the preferred location for the adenosine bulge was on the enzyme strand (46), the most profitable location for the adenosine bulge on the 8-17 variant was on the regulator strand (Fig. 2C, lane B, showing 31-fold activation).
Design and characterization of expansively regulated variants of the bipartite DNAzyme
We also investigated oligonucleotide-mediated control of yet another general RNA-cleaving DNAzyme, the ‘bipartite’ DNAzyme (18). The tertiary folding of this DNAzyme, like those of 10-23 and 8-17, remains to be elucidated. The catalytic core of the bipartite DNAzyme is slightly larger than that of the 10-23 motif (22 versus 15 nt, respectively) and has a purine-rich 5′ segment of the catalytic core and a pyrimidine-rich 3′ segment (Fig. 3A). At 20 mM Mg2+ and 23°C the unmodified bipartite DNAzyme, with 8 and 9 nt substrate-binding arms, yielded a kobs value of 0.754 ± 0.025 min–1. The addition of regulator oligonucleotides did not affect the catalytic rate of this ‘wild-type’ construct. The modified versions of the bipartite DNAzyme are shown in Figure 3A. In the absence of regulator such modified enzymes bind substrate by forming 8 + 3 bp (the number of base pairs in stem A was increased in these constructs to compensate for the lower GC content of this substrate-binding arm relative to that of the optimized 10-23 variant) (Fig. 1B). Stems C and D, which mediated the substrate–regulator and enzyme–regulator interactions, respectively, were maintained at 6 and 7 bp.
Figure 3.
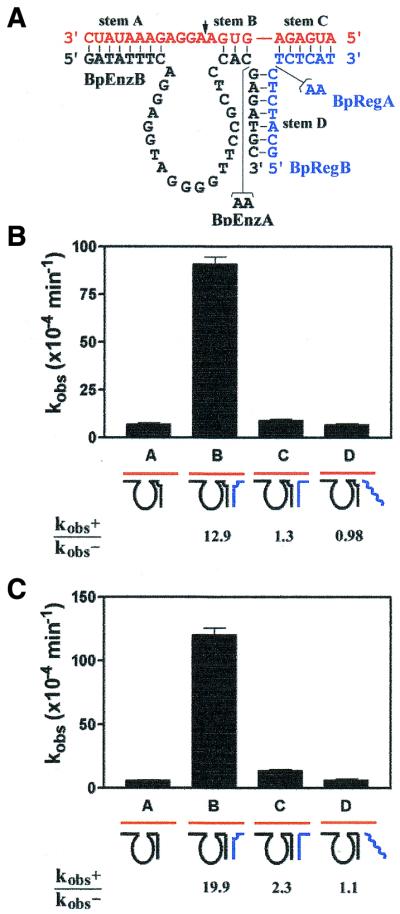
Design of and kinetic modulation by expansively regulated bipartite DNAzyme variants. (A) Sequence and secondary structure of the bipartite variants (black) complexed with RNA substrate (red) and regulator oligonucleotide (blue). The four stem elements present in the structure are indicated and the site of RNA cleavage is marked by an arrow. The observed rate constants (kobs) were measured for BpEnzA (B) and BpEnzB (C) under single turnover conditions (see Fig. 2). Lane A, no regulator; lane B, with BpRegA; lane C, with BpRegB; lane D, with control oligonucleotide (8-17RegA). Catalytic modulation (kobs+/kobs–) for each construct is indicated.
Comparable to the design of distinct variants for the 8-17 DNAzyme, two variant constructs of the bipartite DNAzyme were created (BpEnzA, with a two-adenosine bulge, and BpEnzB, lacking such an adenosine bulge) (Fig. 3A). Their catalytic properties were likewise investigated in both the absence and presence of two regulator sequences (BpRegA, containing an adenosine bulge, and BpRegB, lacking such a bulge). Figure 3B and C presents the results for BpEnzA and BpEnzB, respectively. Similar to the case of the 8-17 DNAzyme, but different from that of the 10-23 DNAzyme, the most optimal location for the adenosine bulge for the bipartite DNAzyme appeared to be on the regulator strand. Such a bulge yielded a 20-fold catalytic activation (Fig. 3C, lane B). However, unlike either the 10-23 (46) or the 8-17 variants, where the addition of any regulator (irrespective of the location of the adenosine bulge) gave rise to a minimum of a 4-fold catalytic activation, the bipartite variants having no adenosine bulge (Fig. 3C, lane C) or only having an adenosine bulge located on the enzyme strand (Fig. 3B, lane C) showed minimal catalytic activation (2.3- and 1.3-fold, respectively). The precise steric causes for the above observation remain to be elucidated, although a few possibilities will be discussed below.
Investigation of expansively regulated variants of the hammerhead ribozyme
We extended our investigation of the utility of oligonucleotide-mediated expansive regulation to an RNA enzyme, the hammerhead ribozyme. Currently there exists no regulatory strategy that can be applied in generic fashion to both in vitro selected DNAzymes and ribozymes. The hammerhead ribozyme is a logical choice for this investigation owing to certain structural similarities to the above mentioned DNAzymes (i.e. having two substrate-binding arms flanking a conserved catalytic core) as well as its well-characterized kinetics (27). In addition, the three-dimensional structure of the hammerhead ribozyme has been elucidated by X-ray crystallography (reviewed in 49) and this structural information provided us with a starting point for the creation of expansively controlled variants of this ribozyme.
Our decision to shorten stem III of the hammerhead ribozyme to create potentially regulatable variants is based on published reports that as few as 3 bp in the other substrate-binding stem (stem I) was sufficient for full catalytic activity (50). In other words, it would not necessarily prove fruitful to shorten stem I in an attempt to destabilize the enzyme–substrate interaction. The first generation of modified constructs based on the hammerhead ribozyme are shown in Figure 4A and B. Stem I (seven Watson–Crick and one wobble base pairs) in our constructs were identical to those in the well-characterized HH15 hammerhead ribozyme (27). We examined two initial classes of constructs, the first with two potential base pairs forming in stem III, in the absence of regulator, between the enzyme and the substrate (Class I, Fig. 4A), while in the second class three such potential base pairs would be formed (Class II, Fig. 4B). Within these two classes two further variants of the hammerhead ribozyme were created, one with a two-adenosine bulge (I-HamEnzA in Fig. 4A and II-HamEnzA in Fig. 4B) and the other lacking such a bulge (I-HamEnzB in Fig. 4A and II-HamEnzB in Fig. 4B). The catalytic properties of the above four constructs were investigated in the absence as well as presence of two regulator sequences (I-HamRegA and II-HamRegA, both with a two-adenosine bulge, and I-HamRegB and II-HamRegB, both lacking such a bulge).
Figure 4.

Initial design of expansively regulated hammerhead ribozyme variants. Two classes of hammerhead variants (black) are shown. Class I variants (A) contain 2 nt in stem III for base pairing to the substrate RNA (red) while Class II variants (B) contain 3 nt. Stem I for both classes form 8 bp with the substrate. The regulator oligonucleotides are shown in blue. The three stem elements present in the structure are indicated and the site of RNA cleavage is marked by an arrow. The observed rate constants (kobs) were measured for Class I variants (C) and Class II variants (D) under single turnover conditions [500 nM enzyme, ∼2 nM substrate, 10 µM regulator (if used), 50 mM Tris–HCl (pH 7.5 at 25°C) and 20 mM MgCl2]. Open bars indicate the kobs measured in the absence of regulator. Solid bars for lanes A and D, regulator containing the bulge; lanes B and E, regulator lacking the bulge; lanes C and F, control oligonucleotide (BpRegA).
Unexpectedly, addition of either regulator oligomer to the Class I variants significantly inhibited their catalysis (Fig. 4C, lanes A, B, D and E), while control oligonucleotides used in place of the regulators did not impact on catalysis (Fig. 4C, lanes C and F). The most detrimental construct (shown in lane E) contained no adenosine bulge at its junction site; it slowed the catalytic rate by ∼115-fold in the presence of regulator. The most likely explanation for this inhibition may be structural: a steric clash between the catalytic core and the newly formed stem between the enzyme and the regulator oligonucleotide.
Interestingly, the regulator-induced inhibition effect observed with the Class I hammerhead variants was not observed with the Class II hammerhead variants. It is conceivable that the one extra base pair in stem III relieved a steric clash (a 1 bp movement along an A-type helix would rotate the projection of a stem by 30–36°). Nevertheless, even with the Class II constructs, only a small catalytic activation (<2-fold) was observed with two of the constructs (Fig. 4D, lanes A and D).
One factor that had not been previously investigated in a systematic way in characterizations of expansive regulation was the effect of the number of base pairs formed by the ‘unaltered’ substrate-binding arm (stem A in the DNAzyme constructs and stem I in the hammerhead constructs). Presumably, in order to achieve a high degree of effector-induced catalytic activation, the number of base pairs formed by this unaltered arm should be kept to a minimum (such that the association between the enzyme and the substrate is severely compromised). It is conceivable that the hammerhead variants described above were sub-optimal in their design, in that they incorporated a robust 8 bp in stem I formed by the enzyme and substrate. Such a stable stem I may not have necessitated additional base pairing supplied by the regulator oligonucleotide in order to carry out efficient catalysis. Therefore, in an attempt to improve the catalytic activation observed in the Class II hammerhead variants, we generated two additional enzyme constructs (II-HamEnzC, containing the two-adenosine bulge, and II-HamEnzD, lacking such a bulge), each with the ability to form only 4 bp in stem I (three Watson–Crick plus one G·U wobble base pairs) (Fig. 5A). Figure 5B and C presents data on the catalytic properties of II-HamEnzC and II-HamEnzD, respectively. As predicted, destabilization of the stem I element significantly lowered the rate of ‘background’ catalysis of the hammerhead variants, i.e. in the absence of regulator oligonucleotides. For example, the background catalytic rate (in the absence of any regulator) of II-HamEnzD was ∼20-fold lower than that of II-HamEnzB. As a consequence, the additional base pairing supplied by the regulator sequence played a proportionately much more significant role in the enhancement of catalysis in the constructs shown in Figure 5C. In lane B, for instance, a catalytic activation of 22-fold was achieved with II-HamEnzD.
Figure 5.
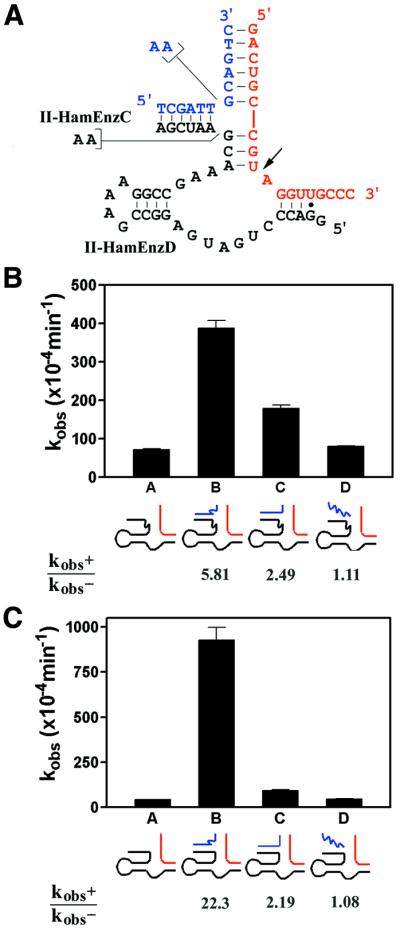
Class II hammerhead variants, with a reduced stem I element. (A) Sequence and secondary structure of the hammerhead variants (black) complexed with the RNA substrate (red) and the regulator oligonucleotide (blue). The number of base pairs formed with substrate in the stem I element is here reduced from 8 to 4. The observed rate constants (kobs) were measured for II-HamEnzC (B) and II-HamEnzD (C) under single turnover conditions (see Fig. 4). Lane A, no regulator; lane B, with II-HamRegA; lane C, with II-HamRegB; lane D, with control oligonucleotide (BpRegA). Catalytic modulation (kobs+/kobs–) for each construct is indicated.
DISCUSSION
In this report we describe an alternative strategy to that of classic allosteric regulation for the design of effector-controlled nucleic acid enzymes. Here we have reported single turnover data on the expansive control of different RNA and DNA enzymes. Earlier, multiple turnover analysis of an expansively regulated 10-23 DNAzyme revealed that presence of the regulator improved both the kcat and Km of the catalytic system (46). Future work will determine the multiple turnover parameters for the additional enzymes described in this paper.
It is important to note that the engineered nucleic acid enzymes described in this study have not been subjected to any efforts at optimization, to achieve the highest possible regulation response. Our goal here was to demonstrate, through preliminary profiling, the generality and the adaptability of the oligonucleotide-dependent expansive regulation design strategy. In our earlier work on the design of an expansively controlled 10-23 DNAzyme, several design parameters emerged as being important (46), including: the number of base pairs mediating formation of the three-way junction (stems B–D in the DNAzyme constructs); the presence and location of a two-adenosine bulge; the concentration dependence of the regulator sequence. In this report, in which we have extended the utility of expansive regulation to a ribozyme and two other DNA enzymes, the critical importance of the adenosine bulge and its location are emphasized. However, since no empirical formalism has been established to date to precisely predict the stacking interactions in three-way junctions, systematic trials were required to identify the preferred location(s) of the adenosine bulge. The necessity, moreover, of starting out with unstable base pairing interactions in the ‘unmodified’ substrate-binding arm (stem I in the case of the hammerhead variants) was highlighted in this study. This is logical since expansive regulation relies on the inability of enzymes modified in this way to bind substrate satisfactorily. The function of the regulator oligonucleotide is, then, to enhance binding between enzyme and substrate via the formation of a three-way enzyme–regulator–substrate complex. The above general observation suggests that the relatively low regulation responses observed with a number of the bipartite variants may be improved by simply decreasing the number of base pairs in stem A (Fig. 3A).
A number of oligonucleotide-dependent allosteric ribozymes have been previously designed and reported (23,41–43). In some of these the regulation of catalytic activity was achieved by modulating antisense interactions. These ribozymes in such cases carried an additional, ‘inhibitory’ domain that normally inactivated ribozyme function by base pairing directly with the ribozyme catalytic and/or substrate-binding elements. The addition of an oligonucleotide complementary to the inhibitory sequence prevented formation of the inactive ribozyme conformation and stimulated catalytic activity (23,41). In some respects the regulation of these above mentioned allosteric ribozymes was similar to our expansively regulated DNAzymes and ribozyme because regulation occurred at the substrate-binding step. However, in the allosteric cases the regulator sequence bound only to the ribozyme, whereas in expansive regulation the regulator sequence binds both enzyme and substrate. The use of ‘oligonucleotide facilitators’ is another approach to modulation of the catalytic activity of nucleic acid enzymes (51). These facilitators are designed to bind exclusively to the substrate sequences adjacent to the ribozyme-binding site. The presence of facilitators enhances catalysis via two distinct mechanisms: (i) by increasing the accessibility of the enzymes to the substrates by preventing the formation of a stable secondary structure near the site of RNA cleavage (52); (ii) by increasing the stability of the enzyme–substrate complex by coaxial stacking of the facilitator with the enzyme (53). Whereas allosteric effectors only bind to the enzyme, facilitators bind only to the substrate; both of these paradigms are therefore different from expansive regulation via the formation of a three-way junction, as we have described.
Nucleic acids whose function can be regulated by the binding of other nucleic acids are already known to exist in nature, where functional modulation serves important roles in cellular operations. In many respects the role of the regulator oligonucleotides described here mirrors the roles of naturally occurring regulatory RNAs. These regulatory RNAs (or riboregulators) use antisense interactions to modulate the functions of other RNAs to provide precise temporal and spatial cellular control, in both prokaryotes and eukaryotes (54,55). Conceivably, the expansively regulated DNAzymes or ribozyme described here may be utilized as ‘intelligent’ gene silencing agents due to their ability to be activated by the presence of external signals (e.g. specific mRNA transcripts that function as effector, distinct from those that function as substrate). Possible applications of this approach include the temporal activation of ribozymes (sensing specific mRNAs expressed at particular developmental stages) and spatial activation (sensing specific mRNAs present in particular cellular locations). In addition, expansive regulation of ribozymes may be of utility in therapeutic applications, to ablate cellular mRNAs essential for cellular viability (by sensing viral/oncogenic mRNA transcripts), rather than in directly targeting virally coded or oncogenic mRNAs (46).
Single-stranded nucleic acids possess the potential for formation of diverse folded structures (4), for diverse catalytic functions (5,56) and for molecular recognition properties (57). It is now apparent that all of these features of nucleic acids can be combined to generate allosteric ribozymes that are regulated by a variety of effector molecules. The same features can also be utilized to generate expansively regulated DNAzymes and ribozymes that are responsive to effector molecules other than oligonucleotides. For example, we have developed DNAzymes responsive to adenosines and ribozymes responsive to ATP and FMN (58). Recently, Breaker and colleagues showed that allosteric ribozymes can be arrayed and used to detect analytes as diverse as specific metal ions, nucleotides, cofactors and drugs, all within a complex mixture (59). Comparable applications may also be anticipated for expansively regulated DNAzymes, to detect specific ligands, given that DNAzymes could be mounted in arrays using technologies similar to those used to create DNA chips. DNA is significantly less costly to synthesize and is more resistant to hydrolytic degradation than RNA. Thus, DNAzymes would provide an excellent alternative to the use of allosteric ribozymes under more extreme reaction conditions.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Dr P. Unrau for setting up the DNA synthesizing facility and members of the Sen laboratory for helpful discussions. This work is supported by the Medical Research Council of Canada (MRC)/Canadian Institutes of Health Research (CIHR). D.S. is a Senior Scholar of the Michael Smith Foundation for Health Research.
REFERENCES
- 1.Kruger K., Grabowski,P., Zaug,A., Sands,J., Gottschling,D. and Cech,T. (1982) Autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell, 31, 147–157. [DOI] [PubMed] [Google Scholar]
- 2.Guerrier-Takada C., Gardiner,K., Marsh,T., Pace,N. and Altman,S. (1983) The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell, 35, 849–857. [DOI] [PubMed] [Google Scholar]
- 3.Joyce G.F. (1994) In vitro evolution of nucleic acids. Curr. Opin. Struct. Biol., 4, 331–336. [DOI] [PubMed] [Google Scholar]
- 4.Gold L., Polsky,B., Uhlenbeck,O.C. and Yarus,M. (1995) Diversity of oligonucleotide functions. Annu. Rev. Biochem., 64, 763–797. [DOI] [PubMed] [Google Scholar]
- 5.Breaker R.R. (1997) In vitro selection of catalytic polynucleotides. Chem. Rev., 97, 371–390. [DOI] [PubMed] [Google Scholar]
- 6.Wilson D. and Szostak,J.W. (1999) In vitro selection of functional nucleic acids. Annu. Rev. Biochem., 68, 611–647. [DOI] [PubMed] [Google Scholar]
- 7.Nissen P., Hansen,J., Ban,N., Moore,P.B. and Steitz,T.A. (2000) The structural basis of ribosome activity in peptide bond synthesis. Science, 289, 920–930. [DOI] [PubMed] [Google Scholar]
- 8.Robertson D.L. and Joyce,G.F. (1990) Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature, 344, 467–468. [DOI] [PubMed] [Google Scholar]
- 9.Breaker R.R. and Joyce,G.F. (1994) A DNA enzyme that cleaves RNA. Chem. Biol., 1, 223–229. [DOI] [PubMed] [Google Scholar]
- 10.Breaker R.R. and Joyce,G.F. (1995) A DNA enzyme with Mg2+-dependent RNA phosphoesterase activity. Chem. Biol., 2, 655–660. [DOI] [PubMed] [Google Scholar]
- 11.Carmi N., Shultz,L.A. and Breaker,R.R. (1996) In vitro selection of self-cleaving DNAs. Chem. Biol., 3, 1039–1046. [DOI] [PubMed] [Google Scholar]
- 12.Faulhammer D. and Famulok,M. (1997) Characterization and divalent metal-ion dependence of in vitro selected deoxyribozymes which cleave DNA/RNA chimeric oligonucleotide. Angew. Chem. Int. Ed. Engl., 35, 2837–2841. [DOI] [PubMed] [Google Scholar]
- 13.Geyer C.R. and Sen,D. (1997) Evidence for the metal-cofactor independence of an RNA phosphodiester-cleaving DNA enzyme. Chem. Biol., 4, 579–593. [DOI] [PubMed] [Google Scholar]
- 14.Santoro S.W. and Joyce,G.F. (1997) A general purpose RNA-cleaving DNA enzyme. Proc. Natl Acad. Sci. USA, 94, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roth A. and Breaker,R.R. (1998) An amino acid as a cofactor for a catalytic polynucleotide. Proc. Natl Acad. Sci. USA, 95, 6027–6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J., Zeng,W., Kwon,A.H. and Lu,Y. (2000) In vitro selection and characterization of a highly efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res., 28, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang J. and Breaker,R.R. (2000) Structural diversity of self-cleaving ribozymes. Proc. Natl Acad. Sci. USA, 97, 5784–5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feldman A.R. and Sen,D. (2001) A new and efficient DNA enzyme for the sequence-specific cleavage of RNA. J. Mol. Biol., 313, 283–294. [DOI] [PubMed] [Google Scholar]
- 19.Bartel D.P. and Szostak,J.W. (1993) Isolation of new ribozymes from a large pool of random sequences. Science, 261, 1411–1418. [DOI] [PubMed] [Google Scholar]
- 20.Cuenoud B. and Szostak,J.W. (1995) A DNA metalloenzyme with ligase activity. Nature, 375, 611–614. [DOI] [PubMed] [Google Scholar]
- 21.Hager A.J. and Szostak,J.W. (1997) Isolation of novel ribozymes that ligate AMP-activated RNA substrate. Chem. Biol., 4, 607–617. [DOI] [PubMed] [Google Scholar]
- 22.Jaeger L., Wright,M.C. and Joyce,G.F. (1999) A complex ligase ribozyme evolved in vitro from a group I ribozyme domain. Proc. Natl Acad. Sci. USA, 96, 14712–14717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robertson M.P. and Ellington,A.D. (1999) In vitro selection of an allosteric ribozyme that transduces analytes to amplicons. Nature Biotechnol., 17, 62–66. [DOI] [PubMed] [Google Scholar]
- 24.Rogers J. and Joyce,G.F. (2000) A ribozyme that lacks cytidine. Nature, 402, 323–325. [DOI] [PubMed] [Google Scholar]
- 25.Guo H.C.T. and Collins,R.A. (1995) Efficient trans-cleavage of a stem-loop RNA substrate by a ribozyme derived from Neurospora VS RNA. EMBO J., 14, 368–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carmi N. Balkhi,S.R. and Breaker,R.R. (1998) Cleaving DNA with DNA. Proc. Natl Acad. Sci. USA, 95, 2233–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fedor M.J. and Uhlenbeck,O.C. (1992) Kinetics of intermolecular cleavage by hammerhead ribozymes. Biochemistry, 31, 12042–12054. [DOI] [PubMed] [Google Scholar]
- 28.Hertel K.J., Herschlag,D. and Uhlenbeck,O.C. (1994) A kinetic and thermodynamic framework for the hammerhead ribozyme. Biochemistry, 33, 3374–3385. [DOI] [PubMed] [Google Scholar]
- 29.Fersht A. (1985) Enzyme Structure and Mechanism, 2nd Edn. W.H. Freeman and Co., New York, NY.
- 30.Soukup G.A. and Breaker,R.R. (1999) Nucleic acid molecular switches. Trends Biotechnol., 17, 469–476. [DOI] [PubMed] [Google Scholar]
- 31.Soukup G.A. and Breaker,R.R. (2000) Allosteric nucleic acid catalysts. Curr. Opin. Struct. Biol., 10, 318–325. [DOI] [PubMed] [Google Scholar]
- 32.Tang J. and Breaker,R.R. (1997) Rational design of allosteric ribozymes. Chem. Biol., 4, 453–459. [DOI] [PubMed] [Google Scholar]
- 33.Araki M., Okuno,Y., Hara,Y. and Sugiura,Y. (1998) Allosteric regulation of a ribozyme activity through ligand-induced conformational change. Nucleic Acids Res., 26, 3379–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soukup G.A. and Breaker,R.R. (1999) Engineering precision RNA molecular switches. Proc. Natl Acad. Sci. USA, 96, 3584–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soukup G.A. and Breaker,R.R. (1999) Design of allosteric hammerhead ribozymes activated by ligand-induced structure stabilization. Structure, 7, 783–791. [DOI] [PubMed] [Google Scholar]
- 36.Robertson M.P. and Ellington,A.D. (2000) Design and optimization of effector-activated ribozyme ligases. Nucleic Acids Res., 28, 1751–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jose A.M., Soukup,G.A. and Breaker,R.R. (2001) Cooperative binding of effectors by an allosteric ribozyme. Nucleic Acids Res., 29, 1631–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Piganeau N., Jenne,A., Thuillier,V. and Famulok,M. (2000) An allosteric ribozyme regulated by doxycyline. Angew. Chem. Int. Ed. Engl., 39, 4369–4373. [DOI] [PubMed] [Google Scholar]
- 39.Piganeau N., Thuillier,V. and Famulok,M. (2000) In vitro selection of allosteric ribozymes: theory and experimental validation. J. Mol. Biol., 312, 1177–1190. [DOI] [PubMed] [Google Scholar]
- 40.Kozumi M., Soukup,G.A., Kerr,J.N. and Breaker,R.R. (1999) Allosteric selection of ribozymes that respond to second messengers cGMP and cAMP. Nature Struct. Biol., 6, 1062–1071. [DOI] [PubMed] [Google Scholar]
- 41.Porta H. and Lizardi,P.M. (1995) An allosteric hammerhead ribozyme. Biotechnology, 13, 161–164. [DOI] [PubMed] [Google Scholar]
- 42.Kuwabara T., Warashina,M., Tanabe,T., Tani,K., Asano,S. and Taira,K. (1998) A novel allosterically trans-activated ribozyme, the maxizyme, with exceptional specificity in vitro and in vivo. Mol. Cell, 2, 617–627. [DOI] [PubMed] [Google Scholar]
- 43.Komatsu Y., Yamashita,S., Kazama,N., Nobuoka,K. and Ohtsuka,E. (2000) Construction of new ribozymes requiring short regulator oligonucleotides as a cofactor. J. Mol. Biol., 299, 1231–1243. [DOI] [PubMed] [Google Scholar]
- 44.Robertson M.P. and Ellington,A.D. (2001) In vitro selection of nucleoprotein enzymes. Nature Biotechnol., 19, 650–655. [DOI] [PubMed] [Google Scholar]
- 45.Wang D.Y. and Sen,D. (2002) Rationally designed allosteric variants of hammerhead ribozymes responsive to the HIV-1 Tat protein. Comb. Chem. High Throughput Screening, in press. [DOI] [PubMed] [Google Scholar]
- 46.Wang D.Y. and Sen,D. (2001) A novel mode of regulation of an RNA-cleaving DNAzyme by effectors that bind to both enzyme and substrate. J. Mol. Biol., 310, 723–734. [DOI] [PubMed] [Google Scholar]
- 47.Zhong M., Rashes,M.S., Leontis,N.B. and Kallenbach,N.R. (1994) Effects of unpaired bases on the conformation and stability of three-arm DNA junctions. Biochemistry, 33, 3660–3667. [DOI] [PubMed] [Google Scholar]
- 48.Welch J.B., Duckett,D.R. and Lilley,D.M.J. (1995) Two inequivalent folding isomers of the three-way junction with unpaired bases: sequence-dependence of the folded conformation. J. Mol. Biol., 251, 507–519. [DOI] [PubMed] [Google Scholar]
- 49.Scott W.G. (1999) Biophysical and biochemical investigations of RNA catalysis in the hammerhead ribozyme. Q. Rev. Biophys., 32, 241–284. [DOI] [PubMed] [Google Scholar]
- 50.Tabler M., Homann,M., Tzortzakaki,S. and Sczakiel,G. (1994) A three-nucleotide helix I is sufficient for full activity of a hammerhead ribozyme: advantages of an asymmetric design. Nucleic Acids Res., 22, 3958–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goodchild J. (1992) Enhancement of ribozyme catalytic activity by a contiguous oligonucleotide (facilitator) and by 2′-O-methylation. Nucleic Acids Res., 20, 4607–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jankowsky E. and Schwenzer,B. (1996) Efficient improvement of hammerhead ribozyme mediated cleavage of long substrates by oligonucleotide facilitators. Biochemistry, 35, 15313–15321. [DOI] [PubMed] [Google Scholar]
- 53.Perkins T.A., Wolf,D.E. and Goodchild,J. (1996) Fluorescence resonance energy transfer analysis of ribozyme kinetics reveals the mode of action of a facilitator oligonucleotide. Biochemistry, 35, 16370–16377. [DOI] [PubMed] [Google Scholar]
- 54.Wassarman K.M., Zhang,A. and Storz,G. (1999) Small RNAs in Escherichia coli. Trends Microbiol., 7, 37–45. [DOI] [PubMed] [Google Scholar]
- 55.Altuvia S. and Wagner,E.G.H. (2000) Switching on and off with RNA. Proc. Natl Acad. Sci. USA, 97, 9824–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Geyer C.R. and Sen,D. (1998) DNA enzymes. Curr. Opin. Chem. Biol., 2, 680–687. [DOI] [PubMed] [Google Scholar]
- 57.Hermann T. and Patel,D.J. (2000) Adaptive recognition by nucleic acid aptamers. Science, 287, 820–825. [DOI] [PubMed] [Google Scholar]
- 58.Wang D.Y., Lai,B.H.Y. and Sen,D. (2002) J. Mol. Biol., in press. [Google Scholar]
- 59.Seetharaman S., Zivarts,M., Sudarsan,N. and Breaker,R.R. (2001) Immobilized RNA switches for the analysis of complex chemical and biological mixtures. Nature Biotechnol., 19, 336–341. [DOI] [PubMed] [Google Scholar]