

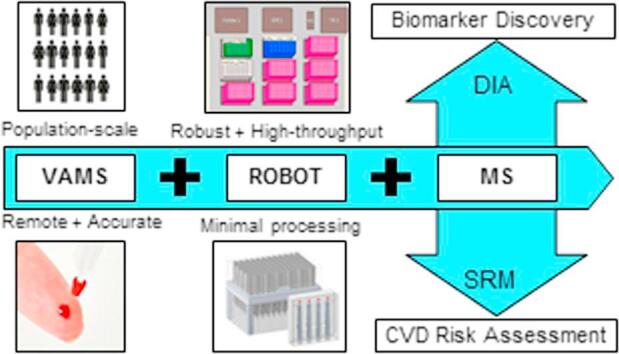
Graphical abstract
Keywords: Remote monitoring, Dried blood, Selected reaction monitoring, Data-independent acquisition, Mass spectrometry, Apolipoproteins
Highlights
-
•
Volumetric absorptive microsampling (VAMS) collects a fixed volume (10 µL) of blood.
-
•
VAMS can enable remote biomarker monitoring and is not biased by hematocrit.
-
•
We integrated VAMS into an automated platform for protein extraction and digestion.
-
•
The workflow’s analytical performance is shown for clinically relevant proteins.
-
•
The workflow’s broad applicability is shown by data-independent acquisition MS.
Abstract
Volumetric absorptive micro sampling (VAMS™) allows accurate sampling of 10 µL of blood from a minimally invasive finger prick and could enable remote personalized health monitoring. Moreover, VAMS overcomes effects from hematocrit and sample heterogeneity associated with dried blood spots (DBS). We describe the first application of VAMS with the Mitra® microsampling device for the quantification of protein biomarkers using an automated, high-throughput sample preparation method coupled with mass spectrometric (MS) detection.
The analytical performance of the developed workflow was evaluated for 10 peptides from six clinically relevant proteins: apolipoproteins A-I, B, C-I, C-III, E, and human serum albumin (HSA). Extraction recovery from blood with three different levels of hematocrit varied between 100% and 111% for all proteins. Within-day and total assay reproducibility (i.e., 5 replicates on 5 days) ranged between 3.2–10.4% and 3.4–12.6%, respectively. In addition, after 22 weeks of storage of the Mitra microsampling devices at −80 °C, all peptide responses were within ±15% deviation from the initial response. Application to data-independent acquisition (DIA) MS further demonstrated the potential for broad applicability and the general robustness of the automated workflow by reproducible detection of 1661 peptides from 423 proteins (average 15.7%CV (n = 3) in peptide abundance), correlating to peptide abundances in corresponding plasma (R = 0.8383).
In conclusion, we have developed an automated workflow for efficient extraction, digestion, and MS analysis of a variety of proteins in a fixed small volume of dried blood (i.e., 10 µL). This robust and high-throughput workflow will create manifold opportunities for the application of remote, personalized disease biomarker monitoring.
1. Introduction
Dried blood can be collected in small amounts from a minimally invasive finger prick, which, in contrast to serum or plasma specimens, eliminates the need for trained phlebotomists, hospital visits, elaborate sample processing, and controlled transportation. The collection of dried blood has the potential to enable sample collection in remote locations, which would increase patient participation and sample access. Dried blood collection, primarily performed using dried blood spots (DBS), involves the placement of small volumes of capillary blood onto special paper cards, and is particularly well-established for the analysis of small molecule biomarkers, e.g., in newborn screening [3]. However, DBS suffer from sample heterogeneity and hematocrit issues that cause non-homogeneous spreading of blood and analyte onto the filter paper and complicate analysis of a standardized volume [4]. Volumetric absorptive microsampling (VAMS™) offers accurate sampling of a fixed small volume of blood (e.g., 10 µL), while overcoming the negative effects associated with DBS [5], [6]. Numerous studies have successfully applied VAMS with the Mitra® microsampling device to the bioanalysis of small molecules and have reported better accuracy and correlation to venous blood than that found with DBS [7], [8], [9], [10].
Remote blood sampling to quantitate protein biomarkers is, however, less well-established. Proteins are major drivers of many disease pathways, and remote or longitudinal sample collection would offer great opportunities in population-based or personalized health monitoring [1], [2]. In recent years, targeted quantification of protein biomarkers in DBS has been increasingly explored by taking advantage of the specificity and multiplexing capacity of mass spectrometry (MS) [11], [12], [13], [14], [15], [16], [17]. The application of VAMS with the Mitra® microsampling device for quantification of protein biomarkers has, nonetheless, not yet been reported.
MS-based analysis of protein biomarkers typically involves digestion of the protein by a proteolytic enzyme and detection of representative peptides by: (i) selected reaction monitoring (SRM) or multiple reaction monitoring (MRM) for targeted quantification, (ii) data-dependent acquisition (DDA) for protein identification, or (iii) data-independent acquisition (DIA) for protein identification and SRM-like quantification. SRM has been introduced into the clinical laboratory as a more specific alternative for antibody-based clinical protein assays [18], additionally enabling the quantification of specific protein variants [19], [20] or the quantification of multiplexed biomarker panels [21], [22]. Important considerations in the clinical application of SRM-based laboratory-developed tests for protein biomarkers include: (i) reduction of the sources of analytical errors [23], [24], (ii) automation or on-line coupling of sample preparation [21], [25], [26], [27], and (iii) metrological traceability of measured protein concentrations [28]. In line with the improvements in robustness, precision, and trueness of clinical MS for protein quantification, DIA is rapidly evolving to provide precise, unbiased (i.e., without a priori selection) quantification of large numbers of proteins by targeted extraction of continuously acquired MS/MS spectra [32], [33]. Whereas DDA provides high resolution MS/MS spectra and, hence, high specificity of identification for any compound that exceeds a detection threshold, DIA provides SRM-like MS/MS peak profiles that can be (also retrospectively) integrated for more-precise quantification of, theoretically, any compound as long as the compound is defined by its specific mass transitions and stored in a spectral library [34].
DBS are reported to have been applied to SRM [11], [12], [13], [14], [15], [16], [17] and DDA [35], but not to global quantification by DIA. In this manuscript, we describe the development of an automated platform for efficient extraction and digestion of proteins from the Mitra microsampling device with applicability to both SRM and DIA MS. The combination of accurate volumetric sampling and automated sample preparation follow the current trends to: (i) improve the accuracy of targeted and discovery-based MS [23], [24], and (ii) explore the potential of remote blood collection for longitudinal monitoring of multiplexed biomarker panels [1].
The developed analytical pipeline offers opportunities to integrate accurate remote fingerprick blood collection with the Mitra microsampling device into population-scale biomarker discovery studies, as well as longitudinal clinical risk assessment based on targeted protein panels. The analytical performance of the developed workflow has been carefully evaluated for 10 peptides from six clinically relevant proteins, including the cardiovascular risk-associated apolipoproteins (i.e., apoA-I, apoB, apoC-I, apoC-III, and apoE), as well as human serum albumin (HSA), using an SRM assay that includes stable-isotope labeled peptide analogs for each targeted peptide and exogenous beta-galactosidase (BGAL) for process quality control. To evaluate the broader applicability and global robustness of the workflow, global protein quantification of dried blood was performed using a DIA assay optimized for plasma biomarker discovery.
2. Experimental section
2.1. Loading of Mitra tips
Six pools of human whole blood (with K2EDTA preservative) were obtained from Bioreclamation I VT (Chestertown, MD, US) over a period of 11 months. All blood pools were stored at 4 °C and used within seven days of collection. Mitra® microsampling devices (Neoteryx, Torrance, CA, US) with 10 µL volumes were loaded with blood by dipping the tips into a 1.5 mL Eppendorf tube filled with blood. Care was taken that the Mitra tips only touched the liquid surface and were not submerged. The tips were held at the surface until fully colored red, and then for an additional 3 s to ensure a complete fill. The filled Mitra tips were allowed to dry for at least 16 h at room temperature and stored until further use in a closed container at room temperature, unless otherwise specified.
2.2. Preparation of combined heavy peptide working solution
For each targeted peptide, a stable-isotope labeled (heavy) peptide analog was included. The 13 stable-isotope labeled peptides, with 13C615N4 labeled arginine or 13C615N2 labeled lysine as C-terminal amino acid residues (New England Peptides, Gardner, MA, US), were obtained as lyophilized powder and reconstituted in 20% (v/v) acetonitrile, 0.1% (v/v) formic acid in water. The individual heavy peptide solutions were combined to make a heavy peptide working solution that provided, as based on amino acid analysis, 1250 pmol DDNPNLPR and LVNEVTEFAK (HSA), 100 pmol WVGYGQDSR, IDPNAWVER, and GDFQFNISR (BGAL), 50 pmol THLAPYSDELR (apoA-I), 10 pmol FPEVDVLTK and GFEPTLEALFGK (apoB), 10 pmol TPDVSSALDK (apoC-I), 10 pmol GWVTDGFSSLK (apoC-III), and 5 pmol SELEEQLTPVAEETR, LGPLVEQGR, and AATVGSLAGQPLQER (apoE) per sample. The combined heavy peptide working solution was aliquoted into single-use portions and stored at
−80 °C.
2.3. Preparation of reagents for automated extraction and digestion
20% (w/v) octyl-beta-glucopyranoside (OGS, Sigma-Aldrich) in water, 50 mMol/L Tris (2-carboxyethyl) phosphine (TCEP, Thermo Scientific, Waltham, MA, US) in water, 200 mMol/L methyl methane thiosulfate (MMTS, Thermo Scientific) in isopropanol, and 5 mg/mL BGAL from E. Coli (Sigma-Aldrich) were prepared as single-use aliquots and stored at -80 °C. A stock solution of 10% (v/v) formic acid was prepared by dilution of a 1 mL ampule of formic acid (Optima LC-MS, Fisher Scientific) in 9 mL water. The stock solution was stored in the dark at room temperature. On each day of analysis, the above-described reagents for denaturation, reduction, and alkylation were thawed and placed on-deck on the robotic liquid handling platform (Biomek NXP Span-8 Workstation, Beckman Coulter Life Sciences, Indianapolis, IN, US). In addition, the buffer for extraction and digestion was freshly prepared by dissolving Tris pre-set crystals (Sigma-Aldrich), in 4 mMol/L CaCl2 (Sigma-Aldrich) to make a 100 mMol/L Tris, 4 mmol/L CaCl2 buffer with pH 8.5. The combined heavy peptide working solution was diluted with buffer resulting in 16.1 µL of the combined heavy peptide working solution per 127.5 µL buffer. Lastly, N-tosyl-L-phenylalanine chloromethyl ketone (TPCK)-treated trypsin (Sciex, Framingham, MA, US) was dissolved in 0.1% (v/v) formic acid at a concentration of 2 µg/µL and stored on-deck until use.
2.4. Automated extraction and digestion workflow
The automated protocol for extraction and digestion was adopted from a previously optimized workflow for trypsin digestion of plasma on the same Biomek NXP Workstation[25]. First, a 150 µL mixture composed of 15 µL 20% w/v OGS, 25 µL 50 mmol/L TCEP, and 110 µL 100 mmol/L Tris, 4 mmol/L CaCl2 buffer pH 8.5 was added to each well of a 1 mL deep well titer plate (Beckman Coulter), hereafter named as sample plate. Mitra tips were removed manually from the Mitra microsampler body by gently pushing with the long side of a needle while holding the tip to the edge of the destination well. The sample plate was then placed on the shaking peltier ALP (Inheco, Martinsried, Germany) for a one hour incubation with rigorous shaking (i.e., 1200 RPM) at 60 °C to simultaneously effect extraction, denaturation, and reduction; 12.5 µL 200 mmol/L MMTS was then added and the sample plate was incubated for five minutes at 25 °C and 1000 RPM to allow alkylation. Subsequently, 127.5 µL buffer, containing 16.1 µL combined heavy peptide working solution, as well as 10 μg BGAL, was added, followed by addition of 10 µL trypsin (at 2 µg/µL), and incubation of the samples for six hours at 37 °C at 1000 RPM. Trypsin digestion was blocked by addition of 15 µL 10% (v/v) formic acid and the sample plate was centrifuged for 30 min at 2800 RCF (Sorvall Legend™ RT, Thermo Scientific). After centrifugation, 8 µL of the supernatant was diluted with 72 µL 10% (v/v) acetonitrile 0.1% (v/v) formic acid in water in a 96-well PCR plate (Biorad, Irvine, CA, US). Five µL of the diluted digest was injected onto the LC-MS/MS.
2.5. LC-MS/MS settings for selected reaction monitoring
The LC system was a Prominence 20AD (Shimadzu, Columbia, MD, US) consisting of a SIL-20ACXR autosampler, a CTO-20AC controller, a CBM-20A Lite column oven, and two LC-20ADXR pumps. The analytical column was an XBridge® Peptide BEH C18 (100 mm, 2.1 mm i.d., 3.5 µm particle size), protected by a VanGuard XBridge® BEH C18 guard column (5 mm, 2.1 mm i.d., 3.5 µm particle size), both from Waters (Milford, MA, US). The column temperature was set at 36 °C. Mobile phase A consisted of 2% (v/v) acetonitrile and 0.1% (v/v) formic acid in water, whereas mobile phase B consisted of 5% water (v/v) and 0.1% (v/v) formic acid in acetonitrile. At a constant flow rate of 0.250 mL/min, for the first five minutes the gradient increased from 2 to 5% mobile phase B, then increased over 10 min to 20% mobile phase B, and then over four minutes to 45% mobile phase B, and finally to 90% mobile phase B over 0.5 min. Mobile phase B was then maintained at 90% for three minutes, after which the column was re-equilibrated for three minutes at 2% mobile phase B. The total LC run time was 26 min. During the first eight and the last five minutes the flow was diverted directly to the waste.
SRM was performed on a QTRAP 6500 mass spectrometer (Sciex) in the low mass resolution configuration using positive electrospray ionization. Ion spray voltage and temperature were set at 5 kV and 500 °C, respectively. Curtain gas, GS1, and GS2 were set at 35, 60, and 55 arbitrary units, respectively. A scheduled SRM transition list was created with a 90 min window, a one second cycle time, and unit resolution for both Q1 and Q3. A detailed list of SRM settings is provided in Table 1. Data acquisition and analysis for SRM was performed with Analyst® 1.6.2 and MultiQuant™ 3.0 software (both Sciex), respectively.
Table 1.
Optimized SRM parameters for the selected proteins and peptides.
| Protein | Peptide | Rt (min) | Precursor ion m/z (charge) |
Fragment ion m/z (type) |
DP (V) | CE (V) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| light | heavy | light | heavy | |||||||
| apoA-I | THLAPYSDELR | 12.4 | 651.3 | 656.3 | +2 | 950.5 | 960.5 | y8+ | 70 | 31 |
| 879.4 | 889.4 | y7+ | 70 | 31 | ||||||
| 440.2 | 445.2 | y72+ | 70 | 34 | ||||||
| apoB | FPEVDVLTK | 17.1 | 524.3 | 528.3 | +2 | 674.4 | 682.4 | y6+ | 60 | 28 |
| 803.5 | 811.5 | y7+ | 60 | 24 | ||||||
| 450.8 | 454.8 | y82+ | 60 | 24 | ||||||
| GFEPTLEALFGK | 19.0 | 654.8 | 658.8 | +2 | 664.4 | 672.4 | y6+ | 60 | 29 | |
| 975.6 | 983.6 | y9+ | 60 | 22 | ||||||
| 488.3 | 492.3 | y92+ | 60 | 24 | ||||||
| apoC-I | TPDVSSALDK | 11.5 | 516.8 | 520.8 | +2 | 620.3 | 628.3 | y6+ | 27 | 45 |
| 466.2 | 842.4 | y8+ | 24 | 45 | ||||||
| 834.4 | 470.3 | y92+ | 27 | 45 | ||||||
| apoC-III | GWVTDGFSSLK | 17.7 | 598.8 | 602.8 | +2 | 953.5 | 961.5 | y9+ | 23 | 40 |
| 854.4 | 862.4 | y8+ | 23 | 40 | ||||||
| 244.1 | 244.1 | b2+ | 26 | 50 | ||||||
| apoE | SELEEQLTPVAEETR | 15.1 | 865.9 | 870.9 | +2 | 902.5 | 912.5 | y8+ | 41 | 70 |
| 801.4 | 811.4 | y7+ | 41 | 70 | ||||||
| 605.3 | 615.3 | y5+ | 41 | 70 | ||||||
| LGPLVEQGR | 12.4 | 484.8 | 489.8 | +2 | 588.3 | 598.3 | y5+ | 50 | 29 | |
| 489.2 | 499.3 | y4+ | 50 | 26 | ||||||
| 399.7 | 404.7 | y72+ | 50 | 23 | ||||||
| AATVGSLAGQPLQER | 13.7 | 749.4 | 754.4 | +2 | 898.5 | 908.5 | y8+ | 70 | 36 | |
| 827.4 | 837.5 | y7+ | 90 | 33 | ||||||
| 642.4 | 652.4 | y5+ | 70 | 33 | ||||||
| HSA | DDNPNLPR | 9.3 | 470.8 | 475.8 | +2 | 272.0 | 282.0 | y2+ | 40 | 19 |
| 596.2 | 606.2 | y5+ | 40 | 20 | ||||||
| 298.7 | 303.7 | y52+ | 50 | 25 | ||||||
| LVNEVTEFAK | 14.8 | 575.3 | 579.3 | +2 | 694.2 | 702.2 | y6+ | 50 | 27 | |
| 823.4 | 831.4 | y7+ | 50 | 26 | ||||||
| 937.3 | 945.3 | y8+ | 40 | 25 | ||||||
| BGAL | WVGYGQDSR | 11.0 | 534.3 | 539.3 | +2 | 286.1 | 286.1 | b2+ | 40 | 22 |
| 782.1 | 792.1 | y7+ | 40 | 26 | ||||||
| 562.1 | 572.1 | y5+ | 40 | 26 | ||||||
| IDPNAWVER | 14.7 | 550.3 | 555.3 | +2 | 774.2 | 784.2 | y6+ | 50 | 31 | |
| 871.2 | 881.2 | y7+ | 50 | 25 | ||||||
| 436.1 | 441.1 | y72+ | 50 | 22 | ||||||
| GDFQFNISR | 16.0 | 542.3 | 547.3 | +2 | 262.1 | 272.1 | y2+ | 50 | 25 | |
| 636.0 | 646.0 | y5+ | 50 | 28 | ||||||
| 764.2 | 774.2 | y6+ | 50 | 28 | ||||||
Rt: Retention time; CE: Collision energy; DP: Declustering potential.
The transitions used for quantification are listed in bold. The stable isotope labeled (heavy) amino acid residues are underlined.
2.6. Optimization of automated extraction and digestion protocol
Plasma was prepared by centrifugation of fresh whole blood (K2EDTA preservative) for 12 min at 1200 RCF. The supernatant (i.e., approximately 50% of the initial volume of whole blood) was used to perform eight individual digestions of 5 µL plasma simultaneously with eight individual digestions of 10 µL of the original whole blood. For each of these samples, a digestion time curve was prepared, according to our optimized automated digestion protocol [25] that was adjusted to accommodate a starting volume of 150 µL during reduction and alkylation. Stable-isotope labeled peptides were added before digestion. Triplicate 50 µL aliquots were taken at eight different time points (i.e., 0.5, 1, 2, 3, 4, 6, 20, and 24 h) and quenched with 2.5 µL 10% (v/v) formic acid. The average light-to-heavy response ratio was plotted versus digestion time.
For optimization of protein extraction from Mitra tips, we investigated the advantage of in-tip proteolytic digestion by analysis of eight replicate Mitra tips with the adjusted digestion procedure and a one hour incubation at 60 °C for reduction and denaturation. Four out of the eight Mitra tips were removed after denaturation and reduction, whereas the other four Mitra tips remained in solution during the digestion. The peptide response ratios in the samples with removed tips were expressed as the fraction of the response ratios in the samples with in-tip digestion. In addition, we evaluated the effect of sonication and/or prolonged incubation at 60 °C on peptide response. To summarize, we analyzed two Mitra tips that followed the general procedure (i.e., one hour at 60 °C), three tips that were incubated for two hours at 60 °C, and three tips that were incubated for two hours at 60 °C preceded by a one hour water bath sonication.
2.7. Analytical performance Evaluation
Extraction recovery was assessed by comparison of the relative response ratios in 10 μL blood that was either loaded onto the Mitra microsamplers or pipetted directly into the sample plate. Three different levels of hematocrit were created by transferring 1 mL blood (defined HCTnormal) to two separate tubes. Both tubes were centrifuged for 12 min at 1200 RCF. 100 μL of the supernatant was then transferred from one tube to the other, to create 1.1 ∗ HCTnormal and 0.9 ∗ HCTnormal hematocrit levels, respectively. Mitra tips were loaded with HCTnormal, 0.9 ∗ HCTnormal and 1.1 ∗ HCTnormal (3 Mitra tips each) and dried for 16 h before analysis. Likewise, 10 μL whole blood from all three hematocrit levels was transferred into the sample plate and stored for 16 h at 4 °C.
Intra- and inter-day precision of the entire workflow were assessed by analysis of 25 Mitra microsamplers loaded with the same source of blood on five different days (n = 5 each day). These Mitra microsamplers had been stored for 9–13 weeks at room temperature before analysis. The precision of microsampling by different, untrained, individuals was assessed by sampling 10 µL of blood from a 1.5 mL tube by five individuals (n = 6 for each operator). All 30 Mitra microsamplers were analyzed on the same day, following 13 weeks of storage at room temperature.
Storage stability of the peptides and proteins in dried blood was evaluated by analysis of Mitra microsamplers loaded with the same source of blood after 0, 8, and 22 weeks of storage at −80 °C in a closed container (n = 3 at each time point). All light-to-heavy response ratios were expressed as percentage of the response ratio on the first day of measurement.
2.8. Sample preparation for DIA
For DIA analysis, the 200 mmol/L MMTS alkylation reagent was replaced by a freshly prepared 200 mmol/L iodoacetamide (Sigma Aldrich) solution (for compatibility with the carbamidomethylated-cysteine containing spectral library) and the sample plate was covered with aluminum foil during the 10 min alkylation reaction. After quenching of the digestion, the sample plate was removed from the Biomek NXP Workstation and 250 µL of each sample (i.e., plasma and blood) was transferred to Vivacon 500 centrifugation tubes with 10,000 MW cut-off filters (Sartorius, Göttingen, Germany) and centrifuged for 15 min at 14,000 RCF at room temperature. From the filtrate, 225 µL was mixed with 275 µL 0.1% (v/v) formic acid and 500 µL 4% (v/v) phosphoric acid for subsequent solid-phase extraction (SPE). SPE was performed in a 96-well format using Oasis hydrophilic-lipohilic balanced (HLB) SPE plates (Waters, 30 mg per well) and an extraction plate vacuum manifold (Waters). The HLB sorbent was conditioned with 1 mL methanol and equilibrated with three 1 mL portions of 0.1% (v/v) formic acid. The diluted sample (1 mL) was loaded at an approximate rate of two drops per second. After loading the samples, the sorbent was washed with three 1 mL portions of 0.1% (v/v) formic acid before elution with 1 mL 80% acetonitrile (v/v) in 0.1% (v/v) formic acid. The eluates were dried down using a SPD2010 SpeedVac™ system (Thermo Scientific) and reconstituted in 0.1% (v/v) formic acid in water (Optima LC-MS grade, Fisher Scientific) to an approximate total protein concentration of 2 µg/µL. Five µL of the reconstituted peptides were combined with 5 µL indexed retention time standard peptides (iRTs, Biognosys, Zürich, Switzerland) diluted to 0.5 injection equivalents/μL in 0.1% (v/v) formic acid in water.
2.9. Data acquisition and analysis for DIA
Four µL (∼4 µg protein) was injected onto an Eksigent NanoLC™ 415 System coupled to a TripleTOF® 6600 mass spectrometer (both Sciex). LC separation was performed on a ChromXP™ C18 column (150 x 0.3 mm, Sciex) at a flow rate of 5 µL/min, using 0.1% (v/v) formic acid in water as mobile phase A and 0.1% (v/v) formic acid in acetonitrile as mobile phase B (both Optima LC-MS grade). Mobile phase B was increased from 3% to 35% over 60 min, then from 35% to 85% over three minutes, and held at 85% for five minutes before re-equilibration at 3% mobile phase B for seven minutes (total LC run time was 75 min).
DIA was performed using sequential windowed acquisition of all theoretical fragment ion mass spectra (SWATH®) consisting of 100 Q1 variable isolation windows across a mass range from 400-1250 Da (30 ms dwell time per window). The variable sized Q1 windows were optimized based on precursor density and further increased specificity while ensuring broad mass range coverage [36]. A full MS1 scan (250 ms dwell time) was acquired before each SWATH cycle. Ion spray voltage was set at 5500 V and ion spray temperature at 100 °C. Curtain gas was set as 25 AU, GS1 at 5 AU, and GS2 at 20 AU. The declustering potential was 80 V and rolling collision energy was enabled for each SWATH isolation window.
Data-independent acquisition was controlled by Analyst TF software version 1.7.1 (Sciex). SWATH-MS.wiff files were converted to profile mzML using ProteoWizard v.3.0.6002 [37]. SWATH-targeted data analysis was carried out on an internal computing cluster using OpenSWATH v.2.0.0 [33], employing a target-decoy scoring system (PyProphet v.0.13.3) to estimate the false discovery rate (FDR) and using the Twin Study Library [38] to identify peak groups at a 1% peptide FDR. Peak groups were aligned based on the clustering behaviors of retention time in each run with a non-linear alignment algorithm [39], and realigned to each other using LOcally WEighted Scatterplot Smoothing and peak group clustering with ‘LocalMST’. Specifically, only those peak groups within 3 standard deviations of the retention time were considered for alignment. Peptides that are shared between multiple different proteins were discarded from further analysis.
3. Results and discussion
3.1. Optimization and quality control of protein digestion in blood
The efficiency of trypsin digestion in blood was evaluated by comparison to a previously optimized automated proteomics sample preparation workflow for plasma [25]. Although trypsin digestion generally occurred slightly slower in the blood samples (10 μL) than in the plasma samples (5 μL), complete cleavage was readily achieved with response ratios that were almost identical between plasma and blood samples (Fig. 1). For one peptide (GFEPTLEALFGK from apoB, Fig. 1C), a lower maximum response ratio was observed in blood samples than in plasma. This discrepancy in released peptide could be a result of binding, precipitation, or other interactions of the apoB molecule with components in the blood matrix that hinder release of peptides in certain regions of the molecule. All digestion profiles, nonetheless, were consistent and reached a plateau within six hours. A six-hour incubation time was, therefore, selected to allow sample preparation and analysis within the same day.
Fig. 1.

Evaluation of trypsin digestion efficiency in blood (red lines) as compared to plasma (black lines) for peptides from apoA-I (a), apoB (b-c), apoC-I (d), apoC-III (e), apoE (f-h), and HSA (i-j). The data points indicate the average response ratio, i.e., the endogenous peptide signal relative to the stable-isotope labeled peptide (added before digestion) signal, of three analytical replicates. Error bars indicate ± one standard deviation.
To monitor the quality of trypsin digestion, we evaluated the addition of an exogenous protein, beta-galactosidase (BGAL), and three equimolar stable-isotope labeled peptides (transitions listed in Table 1). The digestion of BGAL was reproducible and consistent among five different blood pools in tips stored up to seven months at room temperature (Fig. 2), suggesting the absence of matrix or storage effects on the digestion efficiency of BGAL in Mitra microsamplers. Nonetheless, whereas digestion was complete for all peptides when BGAL was added after extraction (as indicated by the 1-to-1 relative response ratio of the recombinant protein and the stable-isotope labeled peptides), incomplete digestion was observed in blood samples when beta-galactosidase was added before extraction (Fig. 2). The incomplete cleavage of BGAL in blood was confirmed by the digestion time profiles (Fig. 2) and suggests interaction of beta-galactosidase with blood matrix components during the one hour incubation at 60 °C, limiting cleavability by trypsin. Although, for quantitative analysis, consistency is more important than completeness of protein cleavage, for quality control of digestion conditions, completeness of protein cleavage was considered a prerequisite. Therefore, we decided to add BGAL after extraction to serve as a specific control for digestion conditions. Quality control of extraction efficiency would only be possible if a standard protein were present in the tip before loading with blood.
Fig. 2.

Optimization of the use of the exogenous protein beta-galactosidase (BGAL) as quality control for digestion efficiency among five blood specimens (indicated by different data symbols) at various time points (ordered chronologically with at least three analytical replicates at each time point). When the recombinant protein was spiked into the digestion buffer (after extraction, denaturation, reduction, and alkylation), the relative responses for three different peptides were consistently monitored around a 1:1 stoichiometry (a). When the recombinant protein was spiked into the extraction buffer (before extraction, denaturation, reduction, and alkylation), the relative peptide responses could still be consistently monitored, but did not provide an optimal 1:1 stoichiometry (b). Evaluation of the trypsin digestion efficiency of BGAL, which was spiked into plasma or blood before extraction, revealed a lower recovery of all three peptides in blood, compared to plasma (n = 3 at each time point). Of the three peptides, IDPNAWVER (c) showed the most-complete digestion, whereas WVGYGQDSR (d) and GDFQFNISR (e) did not reach more than 25% of the peptide reponse obtained after digestion in plasma.
3.2. Optimization and automation of protein extraction from Mitra tips
To enable the capacity for high-throughput, protein extraction was evaluated with specific regard to its amenability to be incorporated within the automated digestion workflow. Although leaving the tips on the Mitra microsampler body allowed automated tip transfer to and from the extraction solvent, removal of the tips from the sampler body improved extraction recovery via improved liquid flow and increased the effect of vigorous shaking. In addition, removal of the tips from the sampler body allowed the tips to remain in solution during the entire process of denaturation and reduction (i.e., one hour at 60 °C and 1200 RPM), alkylation, and digestion (i.e., six hours at 37 °C and 1000 RPM). Integration of the extraction with the entire digestion procedure had two major advantages: (i) in-tip proteolytic digestion yielded better recovery in comparison to tips removed before digestion, and (ii) digestion eliminated the need for off-line sonication, as is recommended for the extraction of small molecules from Mitra microsamplers [10] (Fig. 3).
Fig. 3.

The benefits of integrating protein extraction with protein digestion. The final protocol for Mitra microsamplers with dried blood includes denaturation and reduction for one hour at 60 °C and 1200 RPM and digestion with trypsin for six hours at 37 °C and 1000 RPM and (1) improves recovery, as compared to VAMS samplers that are removed after denaturation and reduction (orange bars, n = 4) and (2) eliminates the need for longer or more rigorous incubation (grey bars, include two hours instead of one hour denaturation and reduction, n = 3) or sonication (blue bars, include one hour sonication and two hours denaturation and reduction, n = 3). Error bars indicate the standard deviation. All relative response ratios are normalized to the relative response ratio of the final protocol, indicated by the dashed line at y = 1.0. The inlets below the legend show the final color of the Mitra tips after the different protocols.
3.3. Analytical performance Evaluation
3.3.1. Extraction recovery
The extraction recovery was determined by comparison of the relative peptide responses in Mitra microsamplers with 10 μL blood volumetrically measured by a pipet (n = 3 at three hematocrit levels). With the integrated extraction and digestion procedure, average extraction recoveries (n = 9) ranged between 100.3% and 111.8% (Table 2), indicating complete recovery of all peptides from the Mitra microsamplers. Moreover, the extraction recoveries were independent of hematocrit levels, suggesting absence of matrix effects on protein extraction.
Table 2.
Extraction recoveries at different hematocrit levels.
| 0.9 * normal HCT |
normal HCT |
1.1 * normal HCT |
|||||
|---|---|---|---|---|---|---|---|
| Protein | Peptide | avg | sd | avg | sd | avg | sd |
| apoA-I | THLAPYSDELR | 108.6 | 10.3 | 111.8 | 7.1 | 112.6 | 10.5 |
| apoB | FPEVDVLTK | 108.8 | 9.8 | 111.2 | 7.3 | 110.4 | 9.7 |
| GFEPTLEALFGK | 91.6 | 18.6 | 115.8 | 30.5 | 112.3 | 17.3 | |
| apoC-I | TPDVSSALDK | 104.2 | 6.9 | 106.5 | 7.8 | 103.3 | 5.2 |
| apoC-III | GWVTDGFSSLK | 97.1 | 2.0 | 102.2 | 1.9 | 101.6 | 2.2 |
| apoE | SELEEQLTPVAEETR | 104.8 | 17.3 | 97.3 | 10.6 | 107.7 | 18.8 |
| AATVGSLAGQPLQER | 111.8 | 18.9 | 104.2 | 11.1 | 105.6 | 11.1 | |
| LGPLVEQGR | 113.2 | 15.4 | 112.5 | 9.4 | 109.6 | 11.5 | |
| HSA | DDNPNLPR | 107.8 | 8.9 | 113.2 | 6.4 | 109.0 | 8.0 |
| LVNEVTEFAK | 106.7 | 10.4 | 113.0 | 4.9 | 109.6 | 9.2 | |
% Recovery is calculated as the average (avg) response ratio after extraction of blood from VAMS samplers (n = 3) compared to the average response ratio from 10 µL blood pipetted to the 96-well plate (n = 3) for blood samples at three different hematocrit (HCT) levels. Standard deviation (sd) is calculated as (sd2VAMS + sd2pipet)0.5.
3.3.2. Intra- and inter-day reproducibility
The within- and between-day reproducibility of the entire workflow, including sampling, automated extraction and digestion, and SRM analysis, was determined by the preparation of five Mitra microsamplers on five different days. All Mitra microsamplers were simultaneously loaded from the same blood source, stored at room temperature, and prepared within 20 days of storage. The average within-day reproducibility ranged between 3.2 and 10.4%CV, whereas the total workflow reproducibility, including between-day variability, was between 3.4 and 12.6%CV (Table 3). The intra- and inter day reproducibility meet the requirements for clinical use [40] and demonstrate the robustness of our automated workflow for extraction and digestion.
Table 3.
The intra- and inter-operator reproducibility of loading the Mitra devices with blood and the intra- and inter-day reproducibility of extraction, digestion, and LC-MS/MS measurement.
| Protein | Peptide | Operator reproducibility |
Workflow reproducibility |
||||
|---|---|---|---|---|---|---|---|
| %CV intra | %CV inter | %CV total | %CV intra | %CV inter | %CV total | ||
| n = 6 | o = 5 | n = 5 | d = 5 | ||||
| apoA-I | THLAPYSDELR | 5.2 | 8.2 | 9.7 | 6.0 | 0.0 | 6.0 |
| apoB | FPEVDVLTK | – | – | – | 3.8 | 0.0 | 3.8 |
| GFEPTLEALFGK | – | – | – | 8.1 | 9.7 | 12.6 | |
| apoC-I | TPDVSSALDK | 4.9 | 7.8 | 9.2 | 6.6 | 2.9 | 7.2 |
| apoC-III | GWVTDGFSSLK | 4.7 | 6.2 | 7.8 | 6.2 | 5.8 | 8.5 |
| apoE | SELEEQLTPVAEETR | 6.5 | 6.9 | 9.5 | 10.4 | 0.0 | 10.4 |
| AATVGSLAGQPLQER | 4.2 | 8.0 | 9.0 | 6.1 | 3.2 | 6.9 | |
| LGPLVEQGR | 5.7 | 8.4 | 10.2 | 4.6 | 2.7 | 5.4 | |
| HSA | DDNPNLPR | – | – | – | 3.6 | 2.4 | 4.3 |
| LVNEVTEFAK | 4.5 | 8.8 | 9.9 | 3.2 | 1.0 | 3.4 | |
Within-operator and within-day%CV are expressed as the average%CV of n replicates over o operators or d days, respectively. The total%CV (%CVintra+inter) is calculated as (CVintra2 + CVinter2)1/2, where CVinter is calculated as the%CV of the individual or daily mean (B) corrected for%CVintra (CVinter = (B2 − CVintra2/n)1/2.
3.3.3. Intra- and inter-operator reproducibility
To enable remote, at-home, blood collection, it is important that blood sampling be non-invasive, simple, and easy to perform precisely without training. When blood was sampled by five untrained individuals (six replicates each), the average within-individual precision was between 4.2 and 9.3%CV, which is comparable to our within-run precision and indicates that each individual could accurately sample blood from a tube (Table 3). We did observe a minor systematic bias in the measured relative response ratios between operators, possibly as a result of sampling bias from immersing the Mitra tip too deeply in the blood pool [5]; while noteworthy, this is less likely to occur when blood is loaded from a drop on the finger.
3.3.4. Long-term storage stability
Long-term storage stability of Mitra tips in the laboratory was evaluated by analysis of three analytical replicates after 0, 8, and 22 weeks of storage at −80 °C (Table 4). The response ratios for all peptides were within ±15% deviation from the response ratio of the initial measurement that was performed one day after loading, indicating the suitability of long term storage at −80 °C for quality control, standard, and patient Mitra tips up to at least five months. For application to remote monitoring, it will, nonetheless, be essential to assess the stability of the target protein and peptides under the potential conditions between time of sampling and arrival in the laboratory. Therefore, stability assessment using sealed packages with desiccants, as provided in remote sampling kits, needs to be performed; preferably for a broader panel of peptides and proteins to evaluate general peptide stability.
Table 4.
Stability of the peptides and proteins in Mitra tips stored at −80 °C.
| Protein | Peptide | 8 weeks |
22 weeks |
||
|---|---|---|---|---|---|
| % of initial | SD | % of initial | SD | ||
| apoA-I | THLAPYSDELR | 105.1 | 2.5 | 95.6 | 5.3 |
| apoB | FPEVDVLTK | 103.6 | 1.5 | 96.3 | 7.3 |
| GFEPTLEALFGK | 107.8 | 5.1 | 110.4 | 8.6 | |
| apoC-I | TPDVSSALDK | 108.9 | 6.2 | 106.3 | 10.9 |
| apoC-III | GWVTDGFSSLK | 108.3 | 1.4 | 90.4 | 7.4 |
| apoE | SELEEQLTPVAEETR | 92.8 | 3.6 | 94.5 | 2.1 |
| AATVGSLAGQPLQER | 96.6 | 5.4 | 99.0 | 7.7 | |
| LGPLVEQGR | 104.4 | 2.0 | 101.5 | 5.7 | |
| HSA | DDNPNLPR | 106.2 | 0.6 | 99.5 | 7.7 |
| LVNEVTEFAK | 109.3 | 1.1 | 94.6 | 6.7 | |
Stability is expressed as the% response ratio after 8 and 22 weeks of storage at −80 °C compared to the response ratio obtained at the initial measurement (1 day after loading).
3.3.5. General applicability to biomarker Research
To evaluate the applicability of VAMS to a larger number of proteins, we applied the developed workflow for extraction and digestion to global, unbiased quantification by DIA. DIA does not require, in contrast to SRM, a priori knowledge of the target proteins, but extracts ion chromatograms for all transitions from peptides for which MS1 and MS2 spectra have been previously acquired and archived in a spectral library. In total, 2265 peptides corresponding to a total number of 549 proteins were identified in three analytical replicates of Mitra microsamplers. Of these, 1661 peptides from 423 proteins were detected in all three analytical replicates (73% and 77%, respectively) and a total of 785 peptides (corresponding to 229 proteins) overlapped with the identifications in plasma from the same source (Fig. 4). The average within-day reproducibility was 15.7%CV for the most-precise transitions of 1661 peptides in three analytical replicates measured on one day. Although higher than the average within-day reproducibility of the SRM assay (5.9%CV for 10 peptides from 6 proteins), application of VAMS to DIA demonstrated that (i) the reproducibility of the analysis of dried blood samples is comparable to the analysis of more-established plasma samples, and (ii) an increase in proteome coverage can be achieved with an acceptable decrease in accuracy for application to biomarker discovery. Moreover, the correlation between peptide abundances in dried blood and plasma (R = 0.8383, Fig. 4) indicate global consistency in extraction and digestion efficiency, as well as global peptide stability and the potential for transferability between data obtained from dried blood and plasma.
Fig. 4.

The correlation between the average peptide abundance (n = 3) in plasma and dried blood (a). The correlation (R) is calculated based on all transitions, except the transitions from hemoglobins (green circles). Pink circles represent HSA, the highest abundance plasma protein. The Venn diagrams indicate the overlap between the total number of peptides (top) or proteins (bottom) identified in all analytical replicates (n = 3) from blood (red circles) or plasma (blue circles) (b).
4. Conclusion
We have developed a robust, high-throughput protocol for extraction and digestion of proteins from Mitra microsamplers that allows mass spectrometric quantification by SRM and DIA. The developed workflow has been successfully applied to targeted measurement of six high abundance proteins with varying size and molecular weight, as well as global quantification of thousands of peptides by DIA. Further optimization of the evaluated targeted protein panel, including five cardiovascular disease (CVD) associated apolipoproteins, could advance clinical CVD risk assessment and/or treatment by enabling remote monitoring of apolipoprotein concentrations via home-based collection of blood using Mitra tips followed by delivery to a clinical laboratory for analysis by mass spectrometry. Peptide and protein selection requires further optimization to guarantee the use of at least two robust proteotypic peptides per protein. In addition, pre-analytical factors such as at-home sampling from a finger prick, stability of peptides and proteins during storage and shipment prior to arrival in the laboratory, and effects of, for example, humidity, should be carefully assessed. Furthermore, more accurate quantification requires the incorporation of appropriate reference standards and the generation of daily calibration curves. The performance of VAMS for quantification of protein biomarkers in capillary blood should then be compared to quantitative results obtained in venous blood, plasma, and/or DBS. The measurement of lower-abundance CVD-risk associated proteins would require additional sample clean-up, incorporating the use of stable-isotope standards and capture with anti-peptide antibodies (SISCAPA®), as has reportedly been integrated on a platform for automated DBS sample analysis [17]. In conclusion, this workflow will support the integration of remote blood collection, based on VAMS, into personalized health monitoring of protein biomarkers by mass spectrometry.
Conflict of interest statement
Jennifer E. Van Eyk has a sponsored research agreement with Neoteryx; the research agreement has no overlap with the research described in this manuscript. Stuart Kushon is employed through Neoteryx; he has no competing financial interest. Michael Kowalski is employed through Beckman-Coulter Life Sciences; he has no competing financial interest. All other authors have no conflict of interest to disclose.
Acknowledgments
Jennifer E. van Eyk is supported by funding from the National Institutes of Health (Grant 1R01HL111362-01A1).
References
- 1.Anderson L., Razavi M., Skates S., Anderson N.G., Pearson T.W. Bioanalysis. 2016 doi: 10.4155/bio-2016-0088. [DOI] [PubMed] [Google Scholar]
- 2.McDade T.W., Williams S., Snodgrass J.J. Demography. 2007;44:899–925. doi: 10.1353/dem.2007.0038. [DOI] [PubMed] [Google Scholar]
- 3.Keevil B.G. Clin. Biochem. 2011;44:110–118. doi: 10.1016/j.clinbiochem.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 4.Wagner M., Tonoli D., Varesio E., Hopfgartner G. Mass Spectrom. Rev. 2016;35:361–438. doi: 10.1002/mas.21441. [DOI] [PubMed] [Google Scholar]
- 5.Denniff P., Spooner N. Anal. Chem. 2014;86:8489–8495. doi: 10.1021/ac5022562. [DOI] [PubMed] [Google Scholar]
- 6.Spooner N., Denniff P., Michielsen L., De Vries R., Ji Q.C., Arnold M.E., Woods K., Woolf E.J., Xu Y., Boutet V., Zane P., Kushon S., Rudge J.B. Bioanalysis. 2015;7:653–659. doi: 10.4155/bio.14.310. [DOI] [PubMed] [Google Scholar]
- 7.De Kesel P.M., Lambert W.E., Stove C.P. Anal. Chim. Acta. 2015;881:65–73. doi: 10.1016/j.aca.2015.04.056. [DOI] [PubMed] [Google Scholar]
- 8.Denniff P., Parry S., Dopson W., Spooner N. J. Pharm. Biomed. Anal. 2015;108:61–69. doi: 10.1016/j.jpba.2015.01.052. [DOI] [PubMed] [Google Scholar]
- 9.Miao Z., Farnham J.G., Hanson G., Podoll T., Reid M.J. Bioanalysis. 2015;7:2071–2083. doi: 10.4155/bio.15.125. [DOI] [PubMed] [Google Scholar]
- 10.Mano Y., Kita K., Kusano K. Bioanalysis. 2015;7:1821–1829. doi: 10.4155/bio.15.111. [DOI] [PubMed] [Google Scholar]
- 11.deWilde A., Sadilkova K., Sadilek M., Vasta V., Hahn S.H. Clin. Chem. 2008;54:1961–1968. doi: 10.1373/clinchem.2008.111989. [DOI] [PubMed] [Google Scholar]
- 12.Boemer F., Ketelslegers O., Minon J.M., Bours V., Schoos R. Clin. Chem. 2008;54:2036–2041. doi: 10.1373/clinchem.2008.106369. [DOI] [PubMed] [Google Scholar]
- 13.Daniel Y.A., Turner C., Haynes R.M., Hunt B.J., Dalton R.N. Clin. Chem. 2007;53:1448–1454. doi: 10.1373/clinchem.2007.088682. [DOI] [PubMed] [Google Scholar]
- 14.Edwards R.L., Creese A.J., Baumert M., Griffiths P., Bunch J., Cooper H.J. Anal. Chem. 2011;83:2265–2270. doi: 10.1021/ac1030804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chambers A.G., Percy A.J., Yang J., Borchers C.H. Mol. Cell Proteomics. 2015;14:3094–3104. doi: 10.1074/mcp.O115.049957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chambers A.G., Percy A.J., Yang J., Camenzind A.G., Borchers C.H. Mol. Cell Proteomics. 2013;12:781–791. doi: 10.1074/mcp.M112.022442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Razavi M., Anderson N.L., Yip R., Pope M.E., Pearson T.W. Bioanalysis. 2016:8. doi: 10.4155/bio-2016-0059. [DOI] [PubMed] [Google Scholar]
- 18.Hoofnagle A.N., Wener M.H. J. Immunol. Methods. 2009;347:3–11. doi: 10.1016/j.jim.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y., Snyder M.R., Zhu Y., Tostrud L.J., Benson L.M., Katzmann J.A., Bergen H.R., 3rd. Clin. Chem. 2011;57:1161–1168. doi: 10.1373/clinchem.2011.163006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ladwig P.M., Barnidge D.R., Snyder M.R., Katzmann J.A., Murray D.L. Clin. Chem. 2014;60:1080–1088. doi: 10.1373/clinchem.2014.222208. [DOI] [PubMed] [Google Scholar]
- 21.van den Broek I., Romijn F.P., Nouta J., van der Laarse A., Drijfhout J.W., Smit N.P., van der Burgt Y.E., Cobbaert C.M. Clin. Chem. 2016;62:188–197. doi: 10.1373/clinchem.2015.246702. [DOI] [PubMed] [Google Scholar]
- 22.Agger S.A., Marney L.C., Hoofnagle A.N. Clin. Chem. 2010;56:1804–1813. doi: 10.1373/clinchem.2010.152264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van den Broek I., Romijn F.P., Smit N.P., van der Laarse A., Drijfhout J.W., van der Burgt Y.E., Cobbaert C.M. J Proteome Res. 2015;14:928–942. doi: 10.1021/pr5011179. [DOI] [PubMed] [Google Scholar]
- 24.Addona T.A., Abbatiello S.E., Schilling B., Skates S.J., Mani D.R., Bunk D.M., Spiegelman C.H., Zimmerman L.J., Ham A.J., Keshishian H., Hall S.C., Allen S., Blackman R.K., Borchers C.H., Buck C., Cardasis H.L., Cusack M.P., Dodder N.G., Gibson B.W., Held J.M., Hiltke T., Jackson A., Johansen E.B., Kinsinger C.R., Li J., Mesri M., Neubert T.A., Niles R.K., Pulsipher T.C., Ransohoff D., Rodriguez H., Rudnick P.A., Smith D., Tabb D.L., Tegeler T.J., Variyath A.M., Vega-Montoto L.J., Wahlander A., Waldemarson S., Wang M., Whiteaker J.R., Zhao L., Anderson N.L., Fisher S.J., Liebler D.C., Paulovich A.G., Regnier F.E., Tempst P., Carr S.A. Nat. Biotechnol. 2009;27:633–641. doi: 10.1038/nbt.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu, Q.; Kowalski, M. P.; Mastali, M.; R, H.; Venkatraman, V.; van den Broek, I.; Ackerman, A. L.; Anger, J. T.; Hunter, C. L.; Van Eyk, J. E. Unpublished work, Cedars-Sinai Medical Center (2016) 2016.
- 26.Zhang Y.V., Rockwood A. Clin. Chim. Acta. 2015;450:298–303. doi: 10.1016/j.cca.2015.08.027. [DOI] [PubMed] [Google Scholar]
- 27.Toth C.A., Kuklenyik Z., Jones J.I., Parks B.A., Gardner M.S., Schieltz D.M., Rees J.C., Andrews M.L., McWilliams L.G., Pirkle J.L., Barr J.R. J. Proteomics. 2016;150:258–267. doi: 10.1016/j.jprot.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smit N.P., Romijn F.P., van den Broek I., Drijfhout J.W., Haex M., van der Laarse A., van der Burgt Y.E., Cobbaert C.M. J. Proteomics. 2014;109:143–161. doi: 10.1016/j.jprot.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 32.Gillet L.C., Navarro P., Tate S., Rost H., Selevsek N., Reiter L., Bonner R., Aebersold R. Mol Cell Proteomics. 2012;11(O111):016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rost H.L., Rosenberger G., Navarro P., Gillet L., Miladinovic S.M., Schubert O.T., Wolski W., Collins B.C., Malmstrom J., Malmstrom L., Aebersold R. Nat. Biotechnol. 2014;32:219–223. doi: 10.1038/nbt.2841. [DOI] [PubMed] [Google Scholar]
- 34.Parker S.J., Venkatraman V., Van Eyk J.E. Proteomics. 2016;16:2221–2237. doi: 10.1002/pmic.201600007. [DOI] [PubMed] [Google Scholar]
- 35.Martin N.J., Bunch J., Cooper H.J. J. Am. Soc. Mass Spectrom. 2013;24:1242–1249. doi: 10.1007/s13361-013-0658-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holewinski R.J., Parker S.J., Matlock A.D., Venkatraman V., Van Eyk J.E. Methods Mol Biol. 2016;1410:265–279. doi: 10.1007/978-1-4939-3524-6_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kessner D., Chambers M., Burke R., Agus D., Mallick P. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y., Buil A., Collins B.C., Gillet L.C., Blum L.C., Cheng L.Y., Vitek O., Mouritsen J., Lachance G., Spector T.D., Dermitzakis E.T., Aebersold R. Mol. Syst. Biol. 2015;11:786. doi: 10.15252/msb.20145728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weisser H., Nahnsen S., Grossmann J., Nilse L., Quandt A., Brauer H., Sturm M., Kenar E., Kohlbacher O., Aebersold R., Malmstrom L. J. Proteome Res. 2013;12:1628–1644. doi: 10.1021/pr300992u. [DOI] [PubMed] [Google Scholar]
- 40.Percy A.J., Byrns S., Pennington S.R., Holmes D.T., Anderson N.L., Agreste T.M., Duffy M.A. Expert Rev. Proteomics. 2016;13:673–684. doi: 10.1080/14789450.2016.1205950. [DOI] [PubMed] [Google Scholar]