

Abstract
Gene delivery from biomaterial scaffolds has been employed to induce the expression of tissue inductive factors for applications in regenerative medicine. The delivery of viral vectors has been described as reflecting a balance between vector retention and release. Herein, we investigated the design of hydrogels in order to retain the vector at the material in order to enhance transgene expression. Poly(ethylene-glycol) (PEG) hydrogels were modified with poly-L-lysine (PLL) to non-covalently bind lentivirus. For cells cultured on the hydrogels, increasing the PLL molecular weight from 1 to 70 kDa led to increased transgene expression. The incubation time of the virus with the hydrogel and the PLL concentration modulated the extent of virus adsorption, and adsorbed virus had a 20% increase in the half-life at 37°C. Alternatives to high molecular weight PLL were identified through phage display technology, with peptide sequences specific for the VSV-G ectodomain, an envelope protein pseudotyped on the virus. These affinity peptides could easily be incorporated into the hydrogel, and expression was increased 20-fold relative to control peptide, and comparable to levels observed with the high molecular weight PLL. The modification of hydrogels with affinity proteins or peptides to bind lentivirus can be a powerful strategy to enhance and localized transgene expression.
Keywords: localized gene delivery, lentivirus, polylysine, phage display
Introduction
The delivery of gene therapy vectors represents a versatile method to promote the sustained expression of inductive factors in numerous applications of regenerative medicine. The delivery of gene therapy vectors is considered versatile as the nucleotide sequence can readily be exchanged to express one or more factors using a single delivery system (Shen et al., 2000; Thomas et al., 2014). Both non-viral and viral vectors can promote prolonged transgene expression, with non-viral vectors generally considered to be safer as they generally do not integrate into the host chromosome, yet have significantly lower levels of transgene expression than viral vectors. Viral vectors have evolved mechanisms for effectively transducing target cells and thus provide the greatest levels of expression. In particular, lentiviral vectors have recently received approval in a Phase 2/3 clinical trial (Bluebird Bio, 2013), and have been able to promote long-term transgene expression (Lentz et al., 2012). Lentiviral tropism has been greatly expanded by pseudotyping its envelope with the glycoprotein of the vesicular stomatitis virus (VSV-G), which allows the virus to infect a wide range of cell types and deliver genes to both dividing and non-dividing cells (Bukrinsky et al., 1993; Lewis and Emerman, 1993; Lewis et al., 1992; Naldini et al., 1996). Additionally, their ease of production, relatively low immunogenic profile (Abordo-Adesida et al., 2005), and relatively large genetic cargo make them excellent candidates as gene delivery vectors.
Biomaterial scaffolds serve a central role in regenerative medicine by creating a space for tissue growth and a support for cell interactions, and they have also served as a vehicle for gene delivery vectors as a means to localize expression of tissue inductive factors. Localized expression of tissue inductive factors enables some control over the local microenvironment that has been effective in multiple models such as spinal cord injury and bone regeneration (Luk et al., 2003; Thomas et al., 2014). The delivery of viral vectors from scaffolds has been attempted through a variety of approaches, such as non-specific adsorption of the vector (Jen et al., 2013), entrapment (Raut et al., 2010), modifying the vector to bind to a material (Padmashali and Andreadis, 2011), or modifying the material to interact with the vector. Material modifications have included modification of surfaces with hydroxyapatite (Boehler et al., 2013; Shin and Shea, 2010), or phosphatidylserine that is known to have specific interactions with the vector (Shin et al., 2010). Proteins and peptides are regularly employed to functionalize biomaterials, and are emerging as a tool for providing binding sites for vectors on material surfaces. Poly-L-lysine (PLL) with a molecular weight of 70–150 kDa has been used for delivery of viral vectors (Phillips et al., 2008). Identifying the design requirements for peptides or proteins for promoting efficient gene delivery would be invaluable for developing biomaterials for use in regenerative medicine.
In this report, we investigate the design parameters for peptides and proteins to enhance delivery of lentiviral vectors from biomaterial scaffolds. Poly(ethylene glycol)-based hydrogels were employed as the biomaterial scaffold in these studies as they are widely used in vitro and in vivo for studies with cell culture, encapsulation, and transplantation and ultimately tissue formation, and provide a relatively low amount of non-specific binding for lentiviral vectors (Raeber et al., 2005; Shepard et al., 2012). Four-arm PEG-acrylate hydrogels were functionalized with peptides containing a cysteine to provide quick and straightforward functionalization via Michael-type addition. The design of proteins and peptides for gene delivery initially employed PLL at a range of molecular weights. These studies investigated the mechanism by which PLL enhances gene delivery through characterizing virus binding and stability. Although PLL can produce efficient delivery, the relatively high molecular weight and polydispersity of PLL may be a challenge for translation. We thus sought to identify peptides using a phage display technology, which has had success with identifying ligands for multiple cell types (Chamarthy et al., 2004; Hardy et al., 2007; Mummert et al., 2000; Romanczuk et al., 1999; Samoylova and Smith, 1999), proteins (Binetruy-Tournaire et al., 2000; Birkenmeier et al., 1997; Dore et al., 1998), and small molecules (Miura et al., 2004; Rodi et al., 1999; Rozinov and Nolan, 1998). The peptides can provide a high affinity and specific binding interactions with the viral vector, and we investigated multiple strategies for their presentation from the hydrogel. Modifying biomaterials with peptides offers great potential to enhance and modulate virus localization and promote transgene expression for numerous regenerative medicine applications.
Materials and Methods
Virus Production
Lentivirus was produced by co-transfecting HEK-293T cells with lentiviral packaging vectors (pMDL-GagPol, pRSV-Rev, pIVS-VSV-G), previously described by Dull et al. (1998), and the gene of interest (pLenti-CMV-GFP and pLenti-CMV-GLuc) using jetPRIME (Polyplus Transfection, Illkirch, France). After 17 h, supernatant was replaced with fresh media containing 4 mM caffeine. After an additional 31 h, supernatant was collected and cell debris was spun down and removed. Virus particles were concentrated using PEG-it (Systems Biosciences, Mountain View, CA) and re-suspended in PBS. Lentivirus titers were determined by qRT-PCR lentivirus titration kit (ABM, Inc., Richmond, Canada). Virus was further purified for SDS–PAGE gel analysis via a Lenti-X Maxi Purification Kit (Clonetech, Mountain View, CA) and desalted using a PD-10 column (GE Healthcare, Buckinghamshire, England).
PLL Functionalization With Cysteine
A low molecular weight PLL (10 kDa, Alamanda Polymers, Huntsville, AL) and a high molecular weight PLL (30–70 kDa, Sigma–Aldrich, St. Louis, MO) were functionalized with a cysteine using EDC/NHS chemistry to facilitate incorporation into the PEG-acrylate hydrogels. A solution of 2 mM EDC and 5 mM NHS was added to a 0.1 M MES buffer (pH 5.0) and the peptide was added to make a concentration of 1 mg/mL. After 15 min of incubation, the buffer was exchanged with centrifugal filtration (10 kDa, Amicon Ultra-0.5) to 0.1 M PBS (pH 8) containing 30 mg/mL of oxidized cysteine. After allowing the solution to react for 2 h, excess cysteine was removed via dialysis and any oxidized thiol groups were reduced with 50 mg/mL of dithiothreitol. The PLL was quantified with a fluorescamine assay and the cysteines were quantified via Ellman’s test.
Peptide Synthesis
Peptides were synthesized at Northwestern University’s Peptide Synthesis Core Facility of the Institute for BioNanotechnology in Medicine. To facilitate peptide incorporation into the PEG-acrylate hydrogels via Michael-type addition, a cysteine was added to the C-terminus of the synthesized phage display peptide.
Biotinylated VSV-G
The VSV glycoprotein ectodomain (kindly gifted by Yves Gaudin, CNRS, Unité Mixte de Recherche) was biotinylated using sulfo-NHS-LC-biotin (Thermo Scientific, Rockford, IL) according to the manufacturer’s recommended methods. Briefly, VSV-G was diluted in a 0.1 M NaHCO3 buffer and the biotinylation reagent was added. The reaction was incubated on ice for 2 h then excess reagent was removed using centrifugal filtration (10 kDa, Amicon Ultra-0.5). The degree of biotinylation on the VSV-G proteins was assayed using a fluorescence biotin quanititation kit (Thermo Scientific).
Solution Phase Phage Display
Peptides that bind to the biotinylated VSV-G protein were identified using a 12-mer phage display library (New England Biolabs, Ipswich, MA) using the suggested methods for solution phase panning. The phage library and biotinylated protein were combined in a TBS buffer with Tween-20 and allowed to interact. The mixture was then added to a streptavidin-coated 96-well plate blocked with BSA and incubated for 10 min. Biotin (0.5 μL, 10 mM) was added to displace any phages bound to the streptavidin and the plates were washed 15 times with TBST to remove non-binding phages. Bound phages were eluted from the immobilized protein by incubating in an acidic glycine elution buffer (pH 2.2) for 30 min followed by neutralization with 1 M Tris buffer (pH 9.1). Phages were then amplified in Escherichia coli and purified using a PEG solution (20% w/v).
A total of three rounds of panning were completed, with each round introducing more rigorous conditions to select for stronger binding candidates. Tween-20 concentration was increased 0.1, 0.5, and 1%, and NaCl concentration was increased 150, 300, and 750 mM. Time the phages spent incubating with the target protein was reduced to 60, 45, and 30 min. After the third round of panning, individual phage clones were randomly sampled and their DNA was extracted then purified. DNA was sequenced at Northwestern University’s Genomics Core Facility.
Hydrogel Preparation
Hydrogels were formed by dissolving 4-arm poly(ethylene glycol) acrylate (20 kDa) (Laysan Bio, Inc., Arab, AL) in 8.5 mM HEPES buffer (pH 8.0) at a concentration of 100 mg/mL. In hydrogels containing PLL, cys-functionalized PLL was added for a total concentration of 0.45 mg/mL, unless otherwise noted, in additionto 2.5 mM of the cell adhesion peptide, RGD (Ac-CGRGDS-NH3; Celtek Peptides). The RGD control gels contained 5 mM of the peptide. The PEG precursor solution was incubated at 37°C for 30 min to facilitate the Michael-type addition between the acrylate and thiol. To initiate the free-radical polymerization of the acrylate groups, Irgacure 2959 dissolved in N-vinylpyrrolidinone (600 mg/mL) was added to the PEG for a final concentration of 1% (wt/vol). Gel precursor was added to non-adhesive silicon molds (diameter = 4.5 mm, height = 0.8 mm), cross linked with UV light for 90 s, then washed with PBS to remove unbound peptide and unreacted photoinitiator. To determine if virus binding to the walls of the polystyrene plate affected the results, wells of a tissue culture treated 96-well plate were blocked with bovine serum albumin (5 mg/mL in 0.1 M NaHCO3 buffer, pH = 8.6). No significant binding was observed.
Hydrogels functionalized with peptides from the phage display panning used a 5 kDa PEG linker (acrylate—PEG—maleimide; Creative PEG Works, Winston Salem, NC). The PEG linker was added to the PEG precursor solution at a 2.5 mM concentration then photopolymerized using UV light. The peptides were incubated with the virus for 3 h and then incubated with the hydrogels for 15 min, with hydrogels subsequently washed 2× with PBS.
Transgene Expression
Lentivirus (1 × 107 particles) was added to the gels and allowed to incubate for 3 h at room temperature (unless otherwise noted). Virus solution was then removed and gels were washed twice with 150 μL of PBS to remove unbound virus. HT1080 cells were added (104 cells/well) and incubated with Dulbecco’s modified Eagle’s medium plus 10% fetal bovine serum at 37°C, and 5% CO2. Cells expressing GFP were imaged 72 h later, unless noted otherwise. The supernatant of cells expressing GLuc was gathered after 72 h of incubation and measured using a Gaussia luciferase assay kit (New England BioLabs) with a luminometer (Turner Design, Sunnyvale, CA).
Statistics
One-way ANOVA followed by Tukey’s post-test for multiple comparisons and two-tailed Student’s t-test, where appropriate, was performed using GraphPad version 5.04 for Windows (La Jolla, CA). Statistical significance was set at P ≤ 0.05 unless noted. Values shown represent the mean ± SEM.
Results
PLL Length
PEG hydrogels were functionalized with PLL of three molecular weights and subsequently investigated for their ability to localize lentiviral vectors to the substrate and promote gene transfer. PLL was modified with cysteine for attachment to the acrylate groups on the PEG hydrogel. Studies were performed with varying molecular weights of PLL, which were incorporated at equal masses (Table I). Hydrogels functionalized with 1, 10, or 30–70 kDa PLL were incubated for 3 h with lentivirus encoding for GFP. Fluorescence images demonstrated that the high molecular weight PLL (30–70 kDa) provided the greatest number of GFP positive cells (Fig. 1C), whereas the 1 kDa PLL had almost no cells expressing the transgene (Fig. 1A). The high molecular weight PLL resulted in transduction of 25% of the cells, whereas the lowest molecular weight PLL transduced <0.1% of the cells (Fig. 1D). Based on its ability to promote the greatest extent of transduction relative to the other PLL’s, the 30–70 kDa was selected for further analysis. To demonstrate this system’s broad potential to transduce other cell types, bone marrow derived macrophages were isolated from mice and incubated with hydrogels functionalized with 30–70 kDa PLL (Supplementary Information). The extent of transgene expression for the virus immobilized to the hydrogel was subsequently characterized. The control condition for this study involved hydrogels without PLL, yet had the cell-adhesion peptide RGD, which is necessary for cell adhesion and provides minimal interactions with the virus. The assay displays effectively zero background signal in the absence of virus, indicating that any measured signal is produced via transgene expression (Fig. 2). Luciferase activity was more than 10-fold greater on the PLL-containing gels relative to the control hydrogels, and the control gels were not statistically different from gels without virus.
Table I.
Equal masses of the different poly-l-lysines were added to provide an identical net charge for each condition.
| PLL MW (kDa) | Lysine units |
|---|---|
|
| |
| 1 | 8 |
| 10 | 78 |
| 30–70 | 230–550 |
Figure 1.

Influence of PLL molecular weight on virus localization. PEG hydrogels containing 2.5 mM RGD were functionalized with 0.45 mg/mL of 1 kDa (n = 3) (A), 10 kDa (n = 3) (B), and 30–70 kDa (n = 5) (C) PLL and incubated with GFP-encoding lentivirus. Hydrogels were washed to remove non-binding virus then seeded with HT1080 cells for 72 h. (D) Significantly more cells were transduced with 30–70 kDa PLL than the shorter PLL’s (* P ≤ 0.05; *** P ≤ 0.001).
Figure 2.
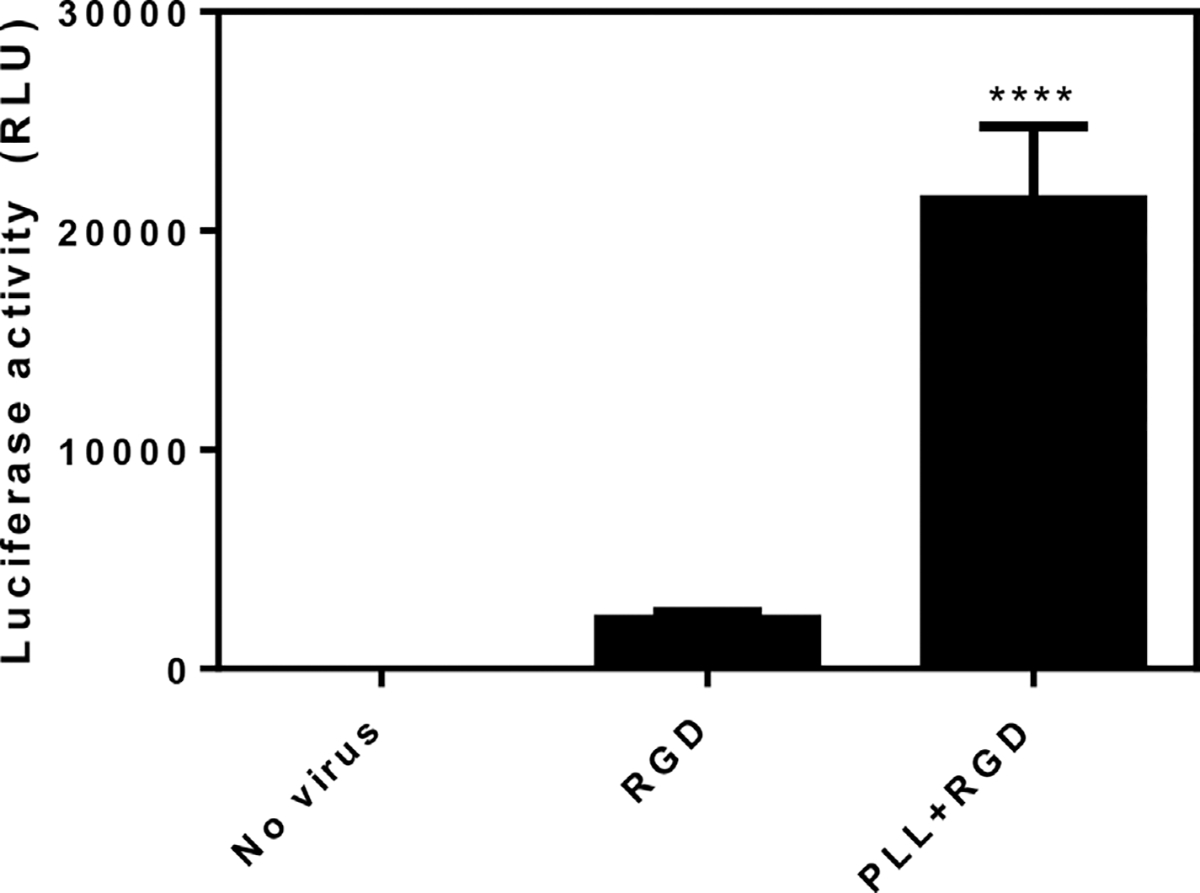
PLL enhances virus localization on PEG hydrogels. PEG hydrogels containing 2.5 mM RGD were functionalized with either 30–70 kDa PLL (0.45 mg/mL) or additional RGD (2.5 mM; n = 3). The “No virus” condition contains PLL but was incubated with PBS instead of lentivirus. Lentivirus encoding for GLuc was incubated with the hydrogels then washed with PBS to remove non-binding virus. HT1080 cells were incubated with the hydrogels for 3 days and then luciferase activity was assayed. Significant difference compared to No virus and RGD (**** P ≤ 0.0001).
Binding Dynamics, Release, and Stability
We subsequently investigated the duration over which virus was incubated with hydrogels and the density of functionalization, as both have been previously reported to influence the binding of non-viral vectors and the extent of transgene expression. Hydrogels were functionalized with PLL and incubated with virus for times ranging from 15 to 270 min. A significant increase in luciferase activity was observed for virus incubated for 90 min with the hydrogels, with longer incubations of 270 min having no significant effect on transgene expression (Fig. 3A). The PLL concentration similarly influenced transgene expression, with increasing transgene expression observed between 0.15 and 1.35 mg/mL, and subsequent increases to 4.05 mg/mL not significantly affecting transgene expression (Fig. 3B). These observed trends in incubation time and PLL concentration are consistent with those reported for non-viral vectors, and are likely due to the quantities of the lentivirus associated with the substrate.
Figure 3.
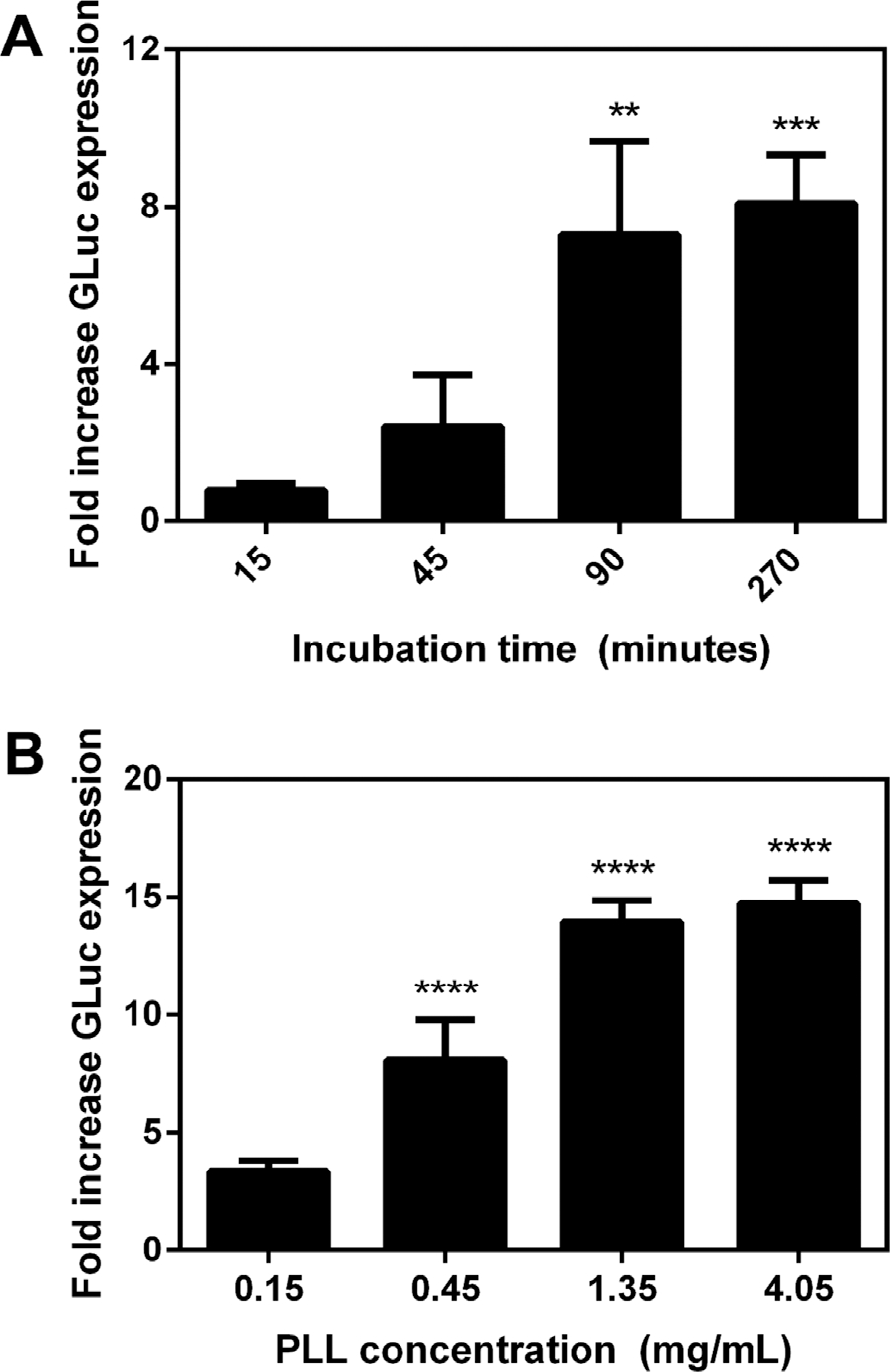
Transgene expression can be modulated via virus-hydrogel incubation time and PLL concentration. (A) PEG hydrogels functionalized with 2.5 mM RGD and 0.45 mg/mL of PLL were incubated with virus for varying times (n = 3). Fold increase in GLuc expression relative to RGD control. Significant difference compared to RGD control (**P ≤ 0.01; ***P ≤ 0.001). (B) Hydrogels were functionalized with varied concentrations of PLL and incubated with virus for 3 h (n = 3). Significant difference compared to RGD control (****P ≤ 0.0001).
The release of lentivirus from PLL-functionalized PEG gels was investigated, as retention of the vector at the material can localize gene delivery. After virus incubation with the hydrogel and subsequent washing, released virus in the supernatant was collected and quantified with RT-qPCR (Fig. 4A and B). After washing, the PLL modified hydrogel did not have detectable levels of virus in the supernatant (Fig. 4A). In contrast, the RGD control had a steady release of virus for the duration of the study (Fig. 4B).
Figure 4.

Retention of lentivirus on PLL-functionalized PEG hydrogels. (A) PEG hydrogels containing 2.5 mM RGD were functionalized with either 30–70 kDa PLL or additional RGD (0.45 mg/mL; n = 4). Virus was incubated with the hydrogels for three hours at 37°C then washed with PBS to remove non-binding virus. Unbound virus in the supernatant was collected and assayed via qPCR at different time points. Following the two washes, no detectable level of virus was found in the PLL-functionalized hydrogels. (B) The fraction of lentivirus released from the hydrogel was calculated by dividing the eluted virus particles by the initial virus loading. Significant differences between corresponding RGD and PLL conditions are denoted by an asterisk (**P ≤ 0.01; ****P ≤ 0.0001).
The stability of the immobilized virus, another factor affecting transduction, was subsequently investigated by incubating hydrogels with immobilized virus at 37°C for varying amounts of time. Following incubation, cells were seeded onto the hydrogels and luciferase expression was assayed at 72 h. Increasing times of incubation led to decreased levels of transgene expression for both the PLL condition and the RGD control, and this decline in activity was used to determine a half-life of activity. The half-life of lentivirus on the control hydrogel was 8.3 h, consistent with previous reports (Higashikawa and Chang, 2001; Tolmachov et al., 2011; Zhang et al., 2004), whereas the PLL-functionalized hydrogels demonstrated a half-life of 10 h, a 20% increase in half-life relative to control (P ≤ 0.05; Fig. 5).
Figure 5.
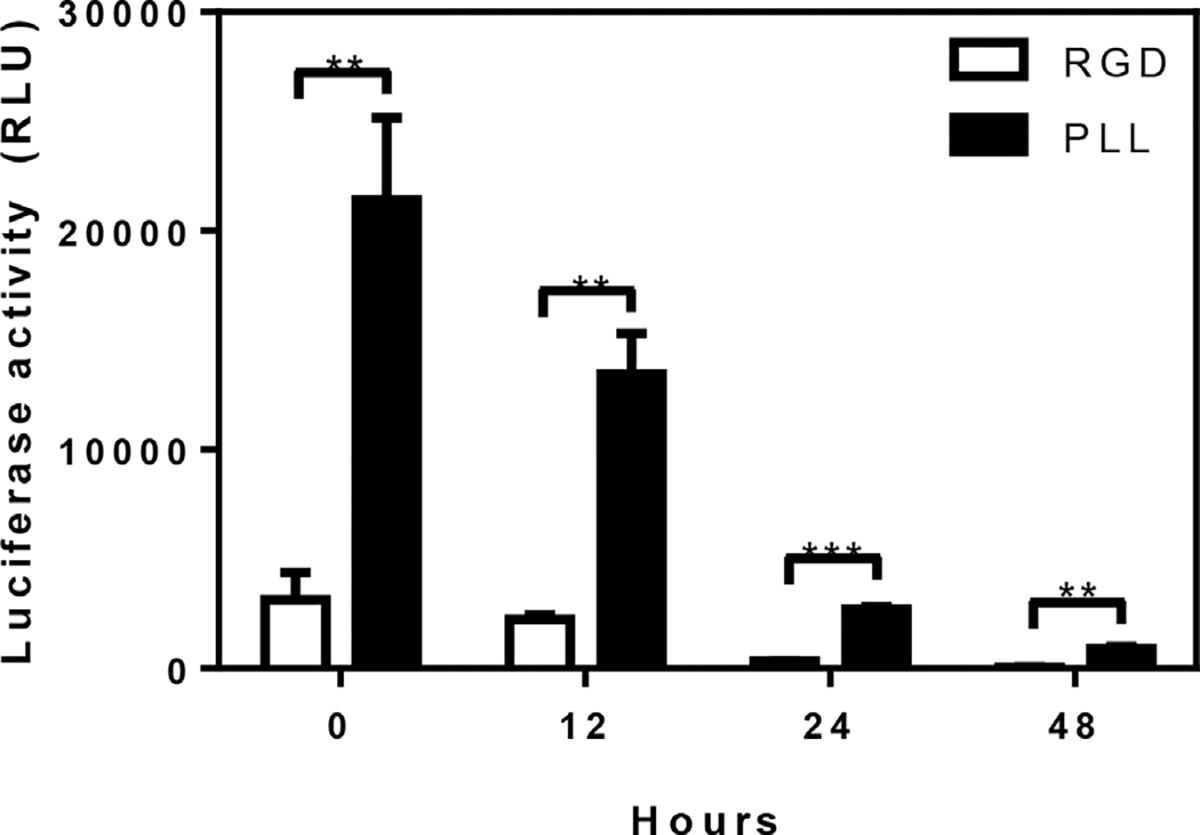
Increased viral stability in PLL-functionalized hydrogels at 37°C. PEG hydrogels with 2.5 mM RGD and either 30–70 kDa PLL or additional RGD (0.45 mg/mL) were incubated with virus at 37°C for 3 h (n = 3). All hydrogels were washed and incubated with PBS. At each time point, the PBS was replaced with HT1080 cells and subsequent luciferase expression was assayed 72 h later (**P ≤ 0.01; ***P ≤ 0.001).
VSV-G Protein Solution-Phase Panning
We subsequently sought to replace the high molecular weight PLL with a peptide, which are routinely used to functionalize biomaterials. The low molecular weight PLL has an insufficient affinity for lentivirus binding, and subsequently applied phage display to identify peptides with a high affinity for the lentivirus. The production of lentivirus resulted in the presence of contaminating proteins at sufficient quantities that prohibited solution-phase phage display, and this technique was thus applied to the envelope protein of the lentivirus, VSV-G. VSV-G was biotinylated at a ratio of 0.8 mol of biotin per 1 mol of VSV-G protein. Three rounds of panning with the VSV-G protein were performed to enrich the phage pool for VSV-G binding sequences. The binding specificity of these sequences was assessed using a fourth round of panning with and without the VSV-G protein, with 105 pfu/μL in the presence of the VSV-G protein, and 101 pfu/μL in the absence of the protein. In the absence of the target protein, the titer of the eluted phages drops 10,000-fold, supporting the hypothesis that the phages are specifically interacting with VSV-G. From solutions with the target protein, 108 phage clones were sequenced, with 32 displaying the sequence STQHHHHSKQSR. Additionally, three other sequences appeared multiple times. These sequences were selected for further analysis (Table II).
Table II.
Phage display clones that appeared multiple times were selected for further analysis.
| Peptide | Occurrence |
|---|---|
|
| |
| HLKHTHNTHYKT | 4 |
| HWKPHSNLHLSR | 8 |
| STQHHHHSKQSR | 32 |
| WPGHHNHSMKHK | 6 |
The potential of these sequences to bind lentivirus and enhance gene delivery was subsequently investigated. Peptides were synthesized with a terminal thiol group, and initial studies incorporated these peptides through Michael type addition, consistent with the mechanism of attachment for PLL and RGD peptides. The peptide density and incubation time of the peptide with virus were those identified to maximize expression with PLL (Fig 3). Peptides that were directly incorporated into the PEG hydrogel failed to promote transgene expression. Subsequent studies involved attaching a 5 kDa PEG linker to the peptide, which was hypothesized to provide greater flexibility for lentivirus binding. PEG hydrogels functionalized with linker-modified peptides promoted substantial transgene expression (Fig. 6). The four peptides identified by phage display provided a 6- to 20-fold increase in luciferase activity relative to RGD modified hydrogels (with RGD presented on a linker), and had expression levels that were comparable to the 30–70 kDa PLL. The use of the linker to connect the peptide to the hydrogel enhanced expression by the 30–70 kDa PLL relative to the absence of the linker (Fig 2). Interestingly, a 1 kDa PLL peptide immobilized on a linker did not promote significant gene transfer above that produced with RGD. Taken together, these results indicate that peptides can promote substantial transgene expression by immobilization to hydrogels, though their presentation on a linker is necessary.
Figure 6.

Phage display identified peptides specific to the VSV-G protein on the lentivirus. Specific binding of lentivirus to phage display peptides. PEG hydrogels were functionalized with 2.5 mM RGD and 2.5 mM of the synthesized phage display peptides conjugated with a 5 kDa PEG linker (n= 4). HLKHTHNTHYKTCG (“HLK”), HWKPHSNLHLSRCG (“HWK”), STQHHHHSKQSRCG (“STQ”), and WPGHHNHSMKHKCG (“WPG”). RGD and 30–70 kDa PLL (0.45mg/mL) were also functionalized to the PEG linker to serve as negative and positive controls, respectively. Lentivirus was incubated with the hydrogels for 3 h followed by washing to remove non-binding virus. HT1080cellswere seeded on the hydrogels and luciferase activity was assayed 72h later. Fold increase in GLuc expression relative to RGD control. Significant differences between RGD and PLL are denoted by an asterisk (P < 0.05).
Discussion
This report investigated the design of peptides for immobilization of lentivirus to hydrogels and subsequently promote transgene expression. PLL is a cationic polypeptide that has been previously employed in non-viral gene delivery and has also been used to modify biomaterials to promote virus association. PLL has been proposed to associate with the lentiviral vector through non-specific interactions. Our studies, consistent with previous reports (Davis et al., 2004), demonstrated that relatively high molecular weight PLL led to greater transgene expression. The enhanced transgene expression likely results from the retention of virus at the surface, which would overcome mass transport limitations by localizing the virus to the substrate to which cells were adhered (Bengali et al., 2009; Luo and Saltzman, 2000). Immobilization also served to increase the stability by approximately 20%, and the extent of immobilization increased through concentrations of 1.35 mg/mL and subsequently reached a plateau. Hydrogels functionalized with PLL concentrations higher than 4.05 mg/mL were generally associated with decreased cell viability. Gene expression could theoretically be further manipulated by modifying PEG concentration, the molecular weight of PEG, and the amount of loaded virus. The collection of positive charges presented by the PLL provide sufficient avidity to effectively act to bind, yet the interaction between the virion and cell is sufficient to disrupt the PLL-lentivirus binding.
PEG hydrogels were functionalized with PLL at multiple molecular weights (1, 10, and 30–70 kDa). These studies were performed for a similar extent of surface modification to isolate the effect of chain length. These studies demonstrated a significant effect of the chain length, with the shortest not supporting binding, the intermediate providing low level transduction, and the longest providing the greatest levels of expression. The peptide RGD served as a control for these studies and small quantities of virus was associated with this condition; however, this low association may be influenced by non-specific interaction with the hydrogel.
We sought to identify shorter peptides for virus immobilization, as large peptides can be difficult to work with, relatively expensive, and have potential for cytotoxicity. The results with 1 kDa PLL indicated that the peptide would need greater affinity, and thus phage displayed was used. Phage display requires a highly pure target, which was not achievable with the existing lentivirus purification kits (LentiX). The crystal structure for the pre-fusion form of the VSV-G protein had recently been determined (Roche et al., 2007), and the purified VSV-G ectodomain was generously provided by the Gaudin group. Using the 12-mer library, we identified 108 clones, and the sequences that were most commonly observed were investigated for lentivirus binding. The STQ sequence was obtained with the greatest frequency after sequencing the phage clones. Several of the non-STQ sequences displayed a preference for four histidines at amino acid positions 4, 5, 6, and 7. A high abundance of aromatic amino acids could signify that the phages are interacting with the plastic instead of the target protein (Adey et al., 1995; Menendez and Scott, 2005). In a fourth round of panning, the phages showed a 10,000-fold higher binding affinity for wells coated with VSV-G, suggesting that our experiments did not identify proteins based on non-specific binding to the plastic.
Peptides identified through phage display were able to support transduction if a linker was employed for peptide immobilization. Initial studies with direct peptide attachment to the PEG hydrogel had minimal levels of transgene expression. Linkers have been used by others to conjugate antibodies to epidermal growth factor (EGF), which were hypothesized to reduce steric hindrance and thereby improve interactions between the target (Deguchi et al., 1998). Peptide immobilization with a 5 kDa PEG linker led to significant transgene expression, with the STQ and WPG peptides providing the greatest levels of expression. Interestingly, the transgene expression levels obtained with the immobilized peptide were comparable to the levels obtained with the relatively high molecular weight PLL. A linker applied to the 30–70 kDa PLL increased transgene expression by approximately 80%, yet was required for the peptides. Finally, we note that the affinity provided by the peptides identified by phage display was necessary for transduction, as a 1 kDa PLL did not support gene transfer.
Conclusions
Viral gene delivery represents a versatile tool to modify the microenvironment of damaged or diseased tissue and promote regeneration by converting the transduced cells into bioreactors to produce therapeutic proteins or downregulate undesired genes. Hydrogels are employed as a substrate that creates a space to promote regeneration, possess mechanical properties similar to native extracellular matrices, and can be readily functionalized. Hydrogels functionalized with proteins or peptides capable of binding lentivirus retained the virus at the material, enhanced the virus stability, and ultimately promoted gene transfer. High molecular weight proteins that non-specifically bind the lentivirus were directly attached to support binding and gene transfer. Alternatively, short peptides that specifically bind the lentivirus had to be immobilized onto biomaterials through linkers in order to promote binding and gene transfer, yet offered comparable gene expression levels. Taken together, affinity peptides or proteins can be attached to biomatetrials to promote the binding of gene therapy vectors and subsequent gene transfer, with the efficiency a function of the peptide length and binding affinity.
Supplementary Material
Acknowledgments
We would like to acknowledge funding from the NIH grants EB003806 and EB005678. Additionally, we would like to thank the Gaudin group (CNRS, Unite Mixte de Recherche) for their generous donation of the VSV-G protein.
Contract grant sponsor: NIH
Contract grant numbers: EB003806; EB005678
Footnotes
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
References
- Abordo-Adesida E, Follenzi A, Barcia C, Sciascia S, Castro MG, Naldini L, Lowenstein PR. 2005. Stability of lentiviral vector-mediated transgene expression in the brain in the presence of systemic antivector immune responses. Hum Gene Ther 16(6):741–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adey NB, Mataragnon AH, Rider JE, Carter JM, Kay BK. 1995. Characterization of phage that bind plastic from phage-displayed random peptide libraries. Gene 156:27–31. [DOI] [PubMed] [Google Scholar]
- Bengali Z, Rea JC, Gibly RF, Shea LD. 2009. Efficacy of immobilized polyplexes and lipoplexes for substrate-mediated gene delivery. Biotechnol Bioeng 102(6):1679–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binetruy-Tournaire R, Demangel C, Malavaud B, Vassy R, Rouyre S, Kraemer M, Plouet J, Derbin C, Perret G, Mazie JC. 2000. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J 19(7):1525–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenmeier G, Osman AA, Kopperschl€ager G, Thomas M. 1997. Epitope mapping by screening of phage display libraries of a monoclonal antibody directed against the receptor binding domain of human a2-macroglobulin. FEBS Lett 416(2):193–196. [DOI] [PubMed] [Google Scholar]
- Bluebird Bio. 2013–2018. A phase 2/3 study of the efficacy and safety of hematopoietic stem cells transduced with Lenti-D lentiviral vector for the treatment of Childhood Cerebral Adrenoleukodystrophy (CCALD). Clinical-Trials.gov. NLM Identifier: NCT01896102. [Google Scholar]
- Boehler RM, Shin S, Fast AG, Gower RM, Shea LD. 2013. A PLG/HAp composite scaffold for lentivirus delivery. Biomaterials 34(21):5431–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukrinsky MI, Haggerty S, Dempsey MP, Sharova N, Adzhubei A, Spitz L, Lewis P, Goldfarb D, Emerman M, Stevenson M. 1993. A nuclear localization signal within HIV-1 matrix protein that governs infection of non-dividing cells. Nature 365(14):666–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamarthy SP, Jia L, Kovacs JR, Anderson KR, Shen H, Firestine SM, Meng WS. 2004. Gene delivery to dendritic cells facilitated by a tumor necrosis factor alpha-competing peptide. Mol Immunol 41(8):741–749. [DOI] [PubMed] [Google Scholar]
- Davis HE, Rosinski M, Morgan JR, Yarmush ML. 2004. Charged polymers modulate retrovirus transduction via membrane charge neutralization and virus aggregation. Biophys J 86:1234–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguchi Y, Kurihara A, Pardridge WM. 1998. Rentention of biologic activity of human epidermal growth factor following conjugation to a blood–brain barrier druge delivery vector via an extended poly(ethylene glycol) linker. Bioconjug Chem 10:32–37. [DOI] [PubMed] [Google Scholar]
- Dore JM, Morard F, Vita N, Wijdenes J. 1998. Identification and location on syndecan-1 core protein of the epitopes of B-B2 and B-B4 monoclonal antibodies. FEBS Lett 426:67–70. [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. 1998. A third-generation lentivirus vector with a conditional packaging system. J Virol 72(11):8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy B, Raiter A, Weiss C, Kaplan B, Tenenbaum A, Battler A. 2007. Angiogenesis induced by novel peptides selected from a phage display library by screening human vascular endothelial cells under different physiological conditions. Peptides 28(3):691–701. [DOI] [PubMed] [Google Scholar]
- Higashikawa F, Chang L. 2001. Kinetic analyses of stability of simple and complex retroviral vectors. Virology 280(1):124–131. [DOI] [PubMed] [Google Scholar]
- Jen MC, Baler K, Hood AR, Shin S, Shea LD, Ameer GA. 2013. Sustained, localized transgene expression mediated from lentivirus-loaded biodegradable polyester elastomers. J Biomed Mater Res A 101(5):1328–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz TB, Gray SJ, Samulski RJ. 2012. Viral vectors for gene delivery to the central nervous system. Neurobiol Dis 48(2):179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis P, Hensel M, Emerman M. 1992. Human immunodeficiency virus infection of cells arrested in the cell cycle. EMBO J 11(8):3053–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis PF, Emerman M. 1993. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J Virol 68(1):510–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KDK, Chen Y, Cheung KMC, Kung H-F, Lu WW, Leong JCY. 2003. Adeno-associated virus-mediated bone morphogenetic protein-4 gene therapy for in vivo bone formation. Biochem Biophys Res Commun 308(3):636–645. [DOI] [PubMed] [Google Scholar]
- Luo D, Saltzman MW. 2000. Enhancement of transfection by physical concentration of DNA at the cell surface. Nat Biotechnol 18:893–895. [DOI] [PubMed] [Google Scholar]
- Menendez A, Scott JK. 2005. The nature of target-unrelated peptides recovered in the screening of phage-displayed random peptide libraries with antibodies. Anal Biochem 336(2):145–157. [DOI] [PubMed] [Google Scholar]
- Miura Y, Sasao Y, Kamihira M, Sakaki A, Iijima S, Kobayashi K. 2004. Peptides binding to a Gb3 mimic selected from a phage library. Biochim Biophys Acta 1673(3):131–138. [DOI] [PubMed] [Google Scholar]
- Mummert ME, Mohamadzadeh M, Mummert D, Mizumoto N. 2000. Development of a peptide inhibitor of hyaluronan-mediated leukocyte trafficking. J Exp Med 192(6):769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gage FH, Trono D, Verma IM. 1996. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci USA 93:11382–11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmashali RM, Andreadis ST. 2011. Engineering fibrinogen-binding VSV-G envelope for spatially- and cell-controlled lentivirus delivery through fibrin hydrogels. Biomaterials 32(12):3330–3339. [DOI] [PubMed] [Google Scholar]
- Phillips JE, Burns KL, Le Doux JM, Guldberg RE, Garcia AJ. 2008. Engineering graded tissue interfaces. Proc Natl Acad Sci USA 105(34):12170–12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raeber GP, Lutolf MP, Hubbell JA. 2005. Molecularly engineered PEG hydrogels: A novel model system for proteolytically mediated cell migration. Biophys J 89(2):1374–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raut SD, Lei P, Padmashali RM, Andreadis ST. 2010. Fibrin-mediated lentivirus gene transfer: Implications for lentivirus microarrays. J Control Release 144(2):213–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche S, Rey FA, Gaudin Y, Bressanelli S. 2007. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315(5813):843–848. [DOI] [PubMed] [Google Scholar]
- Rodi DJ, Janes RW, Sanganee HJ, Holton RA, Wallace BA, Makowski L. 1999. Screening of a library of phage-displayed peptides identifies human Bcl-2 as a taxol-binding protein. J Mol Biol 285:197–203. [DOI] [PubMed] [Google Scholar]
- Romanczuk H, Galer CE, Zabner J, Barsomian G, Wadsworth SC, O’Riordan CR. 1999. Modification of an adenoviral vector with biologically selected peptides: a novel strategy for gene delivery to cells of choice. Hum Gene Ther 10:2615–2626. [DOI] [PubMed] [Google Scholar]
- Rozinov MN, Nolan GP. 1998. Evolution of peptides that modulate the spectral qualities of bound, small-molecule fluorophores. Chem Biol 5:713–728. [DOI] [PubMed] [Google Scholar]
- Samoylova TI, Smith BF. 1999. Elucidation of muscle-binding peptides by phage display screening. Muscle Nerve 22(4):460–466. [DOI] [PubMed] [Google Scholar]
- Shen Y, Muramatsu S-I, Ikeguchi K, Fujimoto K-I, Fan D-S, Ogawa M, Mizukami H, Urabe M, Kume A, Nagatsu I, Urano F, Suzuki T, Ichinose H, Nagatsu T, Monahan J, Nakano I, Ozawa K. 2000. Triple transduction with adeno-associated virus vectors expressing tyrosine hydroxylase, aromatic-L-amino-acid decarboxylase, and GTP cyclohydrolase I for gene therapy of Parkinson’s disease. Hum Gene Ther 11:1509–1519. [DOI] [PubMed] [Google Scholar]
- Shepard JA, Virani FR, Goodman AG, Gossett TD, Shin S, Shea LD. 2012. Hydrogel macroporosity and the prolongation of transgene expression and the enhancement of angiogenesis. Biomaterials 33(30):7412–7421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Shea LD. 2010. Lentivirus immobilization to nanoparticles for enhanced and localized delivery from hydrogels. Mol Ther 18(4):700–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S, Tuinstra HM, Salvay DM, Shea LD. 2010. Phosphatidylserine immobilization of lentivirus for localized gene transfer. Biomaterials 31(15):4353–4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AM, Seidlits SK, Goodman AG, Kukushliev TV, Hassani DM, Cummings BJ, Anderson AJ, Shea LD. 2014. Sonic hedgehog and neurotrophin-3 increase oligodendrocyte numbers and myelination after spinal cord injury. Integr Biol (Camb) 6(7):694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolmachov OE, Tolmachova T, Al-Allaf FA. 2011. Designing lentiviral gene vectors. Designing gene vectors. Rijeka, Croatia: InTech. p 263–284. [Google Scholar]
- Zhang B, Metharom P, Jullie H, Ellem KA, Cleghorn G, West MJ, Wei MQ. 2004. The significance of controlled conditions in lentiviral vector titration and in the use of multiplicity of infection (MOI) for predicting gene transfer events. Genet Vaccines Ther 2(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Goncalves R, Mosser D. 2008. The isolation and characterization of murine macrophages. Curr Protoc Immunol 1:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.