

Abstract
Mitochondria lie at the crossroads of neuronal survival and cell death. They play important roles in cellular bioenergetics, control intracellular Ca2+ homeostasis, and participate in key metabolic pathways. Mutations in genes involved in mitochondrial quality control cause a myriad of neurodegenerative diseases. Mitochondria have evolved strategies to kill cells when they are not able to continue their vital functions. This review provides an overview of the role of mitochondria in neurologic disease and the cell death pathways that are mediated through mitochondria, including their role in accidental cell death, the regulated cell death pathways of apoptosis and parthanatos, and programmed cell death. It details the current state of parthanatic cell death and discusses potential therapeutic strategies targeting initiators and effectors of mitochondrial-mediated cell death in neurologic disorders.
Keywords: apoptosis, apoptosis-inducing factor, neurodegeneration, parthanatos, poly (ADP-ribose) polymerase, stroke
INTRODUCTION
Neurologic diseases are a leading cause of disability and death in the developed world. The economic costs are correspondingly high. Alzheimer’s Disease International indicates that “for 2010 the global cost of dementia … [was] $604 billion – about 1% of world gross domestic product” (1). Other brain disorders have similarly high economic costs. In the United States in 2008, the cost of stroke was estimated to be $65.5 billion (2), and the cost of Parkinson’s disease (PD) was estimated to be $14.4 billion (3). Thus, the global burden of neurologic diseases is huge. Accordingly, preventing and treating neurologic disorders is of paramount importance, as it will improve the lives of many people in a myriad of ways.
Many neurologic disorders including stroke, Alzheimer’s disease (AD), PD, multiple sclerosis (MS), and others are characterized by the death of neurons. Understanding and interfering with these neuronal cell death pathways will have a huge impact on the quality of life of patients who suffer from these disorders. Unfortunately, the underlying mechanisms of neuronal damage that occur in neurodegenerative diseases, stroke, and other neurologic disorders are not fully understood. Mitochondria orchestrate neuronal cell death and survival. This review provides an overview of mitochondrial-mediated mechanisms of neuronal cell death and strategies for treatment of acute and chronic neurologic diseases.
MITOCHONDRIA
Mitochondria are intracellular organelles composed of two bilayers containing lipid and protein: the external outer mitochondrial membrane and the inner mitochondrial membrane. These two membranes enclose the intermembrane space and matrix. Mitochondria play critical roles as the energy generators of the cell through oxidative phosphorylation and in the electron transfer in the electron transport chain through a series of electron carriers in the inner mitochondrial membrane leading to the production of ATP (4) (Figure 1). In addition to their important role in cellular bioenergetics, mitochondria participate in the synthesis of iron-sulfur clusters (5, 6), and they control intracellular Ca2+ homeostasis. Mitochondria also participate in the Krebs cycle and fatty acid metabolism via enzymes in the matrix (7). Reactive oxygen species (ROS) including the hydroxyl radical (•OH) and the superoxide anion (O2•−) are produced via mitochondrial bioenergetics. Mitochondrial-generated O2•− is detoxified by manganese superoxide dismutase and generates hydrogen peroxide (H2O2) (Figure 1). Glutathione peroxidase detoxifies H2O2. Failure to detoxify ROS can cause cellular injury.
Figure 1.
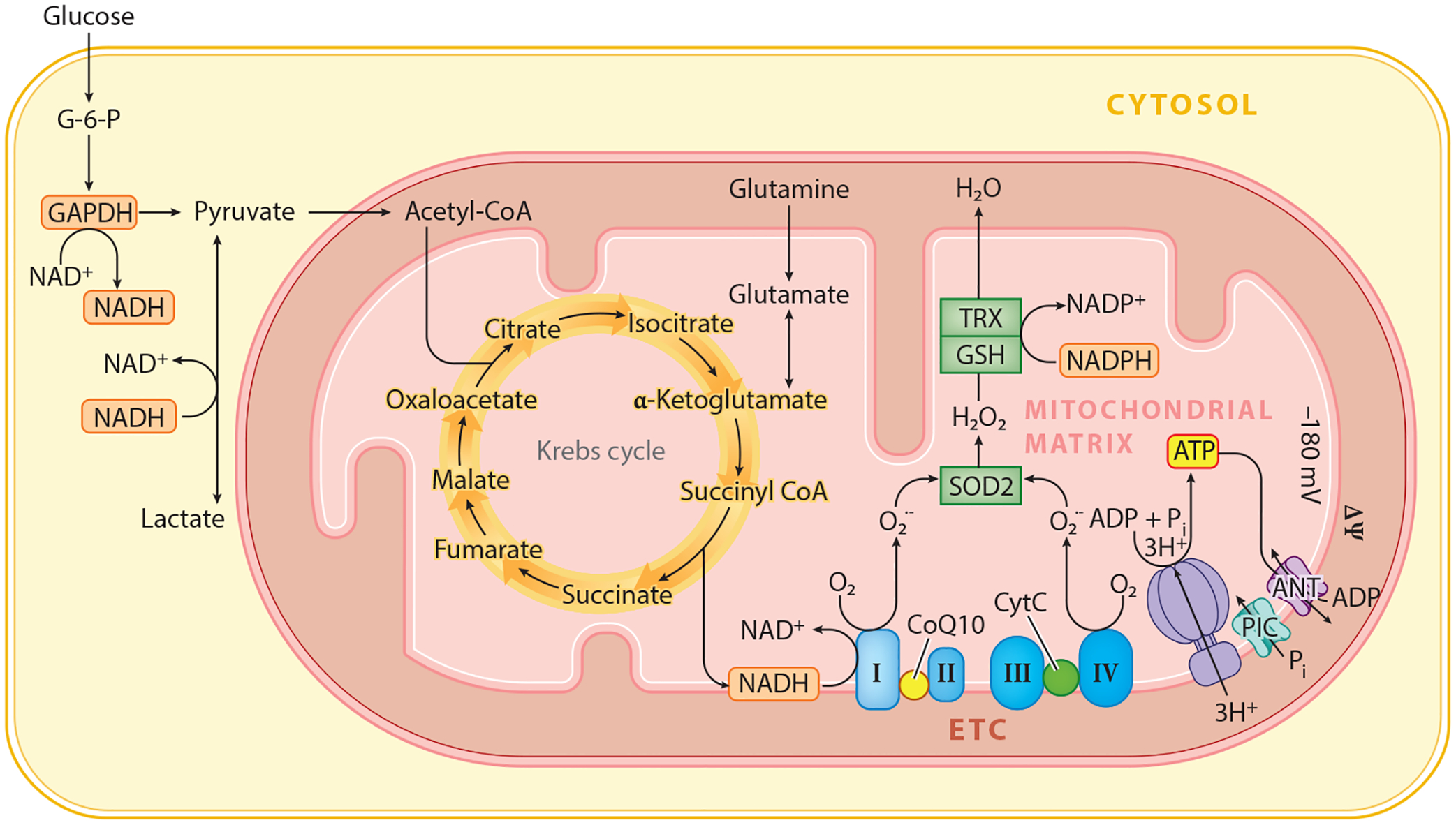
Mitochondria play vital cellular functions including oxidative phosphorylation through the Krebs cycle and the electron transport chain. Abbreviations: ADP, adenosine diphosphate; ANT, adenine nucleotide transporter; ATP, adenosine triphosphate; CoQ10, coenzyme Q 10; CytC, cytochrome C; G-6-P, glucose-6-phosphate; ETC, electron transport chain; GSH, glutathione; H2O2, hydrogen peroxide; mV, millivolt; ΔΨ, mitochondrial membrane potential; NAD+, nicotinamide adenine dinucleotide; NADP+, nicotinamide adenine dinucleotide phosphate; O2, oxygen; Pi, phosphate; PIC, phosphate channel; O2•−, superoxide anion; SOD2, superoxide dismutase 2; TRX, thioredoxin; I, complex I; II, complex II; III, complex III; IV, complex IV.
Defects in mitochondrial respiration have been implicated in many neurodegenerative diseases (8). Although defects in oxidative phosphorylation and the accompanying ROS generation contribute to neuronal cell death and neurodegenerative diseases, these defects may not be the primary driver of pathology. Instead, defects in oxidative phosphorylation and the accompanying ROS are downstream consequences of primary defects that set in motion mitochondrial dysfunction that ultimately contributes to neurodegeneration.
Mitochondrial Quality Control
To maintain oxidative phosphorylation and other important mitochondrial functions, quality control systems maintain the integrity of mitochondria (9). Mitochondria are in a constant state of flux to meet cellular metabolic demands. Fusion and fission dictate the size of mitochondria. Damaged mitochondria must be identified, segregated, and eliminated, and new mitochondria must be synthesized (Figure 2). In the nervous system, owing to long cellular processes, active axonal and dendritic transport mechanisms also participate in the quality control (Figure 2). Damaged mitochondria are segregated through dynamin-related protein (DRP1)-mediated fission and eliminated through autophagy (mitophagy). Mitochondria also combine through fusion to maintain their intrinsic functions. A fastidious quality control system is important because defective mitochondria would lead to calcium dysregulation, loss of energy, oxidative damage, and cell death from activation of intrinsic mitochondrial cell death machinery (9).
Figure 2.
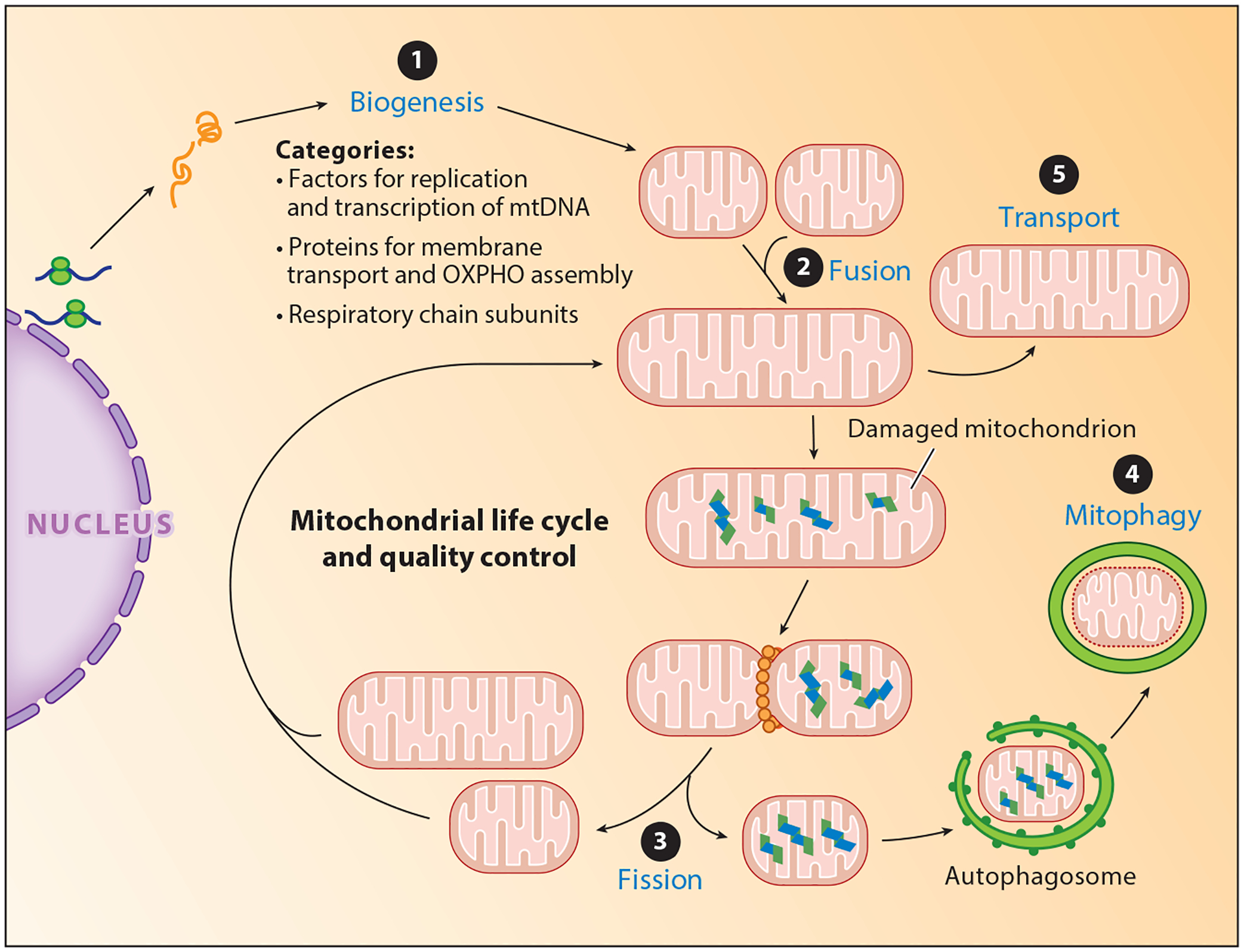
The life cycle of mitochondria. Mitochondrial quality control is a tightly controlled, multistep process to meet the metabolic demands of the cell. ❶ Biogenesis coordinates the synthesis of new mitochondria. ❷ Fusion leads to joining of mitochondria ❸ whereas fission functions in part to segregate damaged mitochondria, which are engulfed by the autophagosome and degraded via ❹ mitophagy. In the nervous system, mitochondria are ❺ transported to the distal ends of axons and dendrites. Abbreviations: mtDNA, mitochondrial DNA; OXPHO, oxidative phosphorylation.
Mutations in key regulators of mitochondrial quality controls cause neurodegenerative diseases. Mutations in PTEN-induced putative kinase 1 (PINK1) and parkin, which play roles in all aspects of mitochondrial control, cause autosomal recessive PD (10–12). Failure to eliminate damaged, ROS-generating mitochondria through mitophagy causes inflammasome activation, which can be further exacerbated by defects in mitochondrial biogenesis (9, 13, 14). Mutations in the mitochondrial DNA polymerase γ, which lead to defects in mitochondrial DNA replication, can contribute to some cases of PD. Charcot-Marie-Tooth disease type 2A is caused by mutations in mitofusin 2 (MFN2), and mutations in optic atrophy 1 and 3 (OPA1 and 3) cause optic atrophy and are linked to defects in mitochondrial fusion (15). MFN1/2 and OPA1 are dynamin-like GTPases localized to the outer and inner mitochondrial membranes, where they coordinate fusion. Mutations in DRP1, another dynamin-like GTPase, cause severe infantile neurodegeneration (15). Other mutations in mitochondrial genes or regulators of mitochondrial function are linked to neurologic disorders (for reviews, see 8, 15).
Mitochondria and Cell Death
Owing to the essential functions of mitochondria in energy bioenergetics and other important and critical metabolic functions, mitochondria have evolved strategies to kill cells that are not able to continue their vital functions. They use different effectors to kill cells. Several cell death pathways have been identified and are segregated into accidental, regulated, and programmed cell death (16). Accidental cell death, as described by Galluzzi et al. (16), is uncontrolled cell death due to extreme physical, chemical or mechanical stimuli and manifests morphologically with necrotic features. Regulated cell death can be influenced, at least to some extent, by specific pharmacologic or genetic interventions and exhibits both necrotic and apoptotic features. Programmed cell death is a regulated cell death program that can occur as part of a developmental program or to preserve physiologic adult tissue homeostasis and is apoptotic morphologically (16). Mitochondria play roles in all three forms of cell death. In accidental cell death, intracellular calcium levels increase owing to complete disruption of mitochondrial function and energy failure. Ultimately, calcium-dependent catabolic enzyme activation is the coup de grâce.
In regulated and programmed cell death, mitochondria contribute to these processes by the release of proteins that activate intrinsic cell death programs. The morphologic classification of cell death (apoptosis versus necrosis) has been replaced by quantifiable biochemical assessments (17). Mitochondria play important roles in intrinsic apoptosis and parthanatos and less important roles in other forms of regulated cell death (16). For instance, mitochondria are not required for necroptosis (18). The final step in all forms of cell death is the inability to meet the energy and metabolic demands of the cell. For further insight into nonmitochondrial-mediated and/or-initiated forms of cell death, the reader is referred to Reference 16.
Intrinsic apoptosis is initiated by a variety of stressors that lead to activation of the initiator caspase 9 followed by proteolytic maturation of the executioner caspase 3 (Figure 3). This process is set in motion by mitochondrial outer membrane permeability (MOMP) in a B cell lymphoma 2 (Bcl-2)-associated X protein (BAX)- and Bcl-2 homologous antagonist/killer (BAK)-dependent manner, which releases the intrinsic intermembrane space mitochondrial protein cytochrome C (CytC). In combination with the apoptotic peptidase-activating factor 1 and deoxy-ATP, CytC forms the apoptosome that activates the initiator caspase 9 (19–21) followed by activation of caspase 3 and downstream processes such as activation of DNAases and substrate proteins that orchestrate the apoptotic cell death program (16) (Figure 3). Activation of apoptosis stimulating fragment or tumor necrosis factor receptors or other death receptors can lead to mitochondrial-mediated cell death through cleavage of caspase 8 and activation of BID [BH3 (Bcl-2 homology 3) interacting-domain death agonist], which causes MOMP (22). The cellular signals regulating both intrinsic and extrinsic apoptosis have been reviewed extensively (see 23–25).
Figure 3.
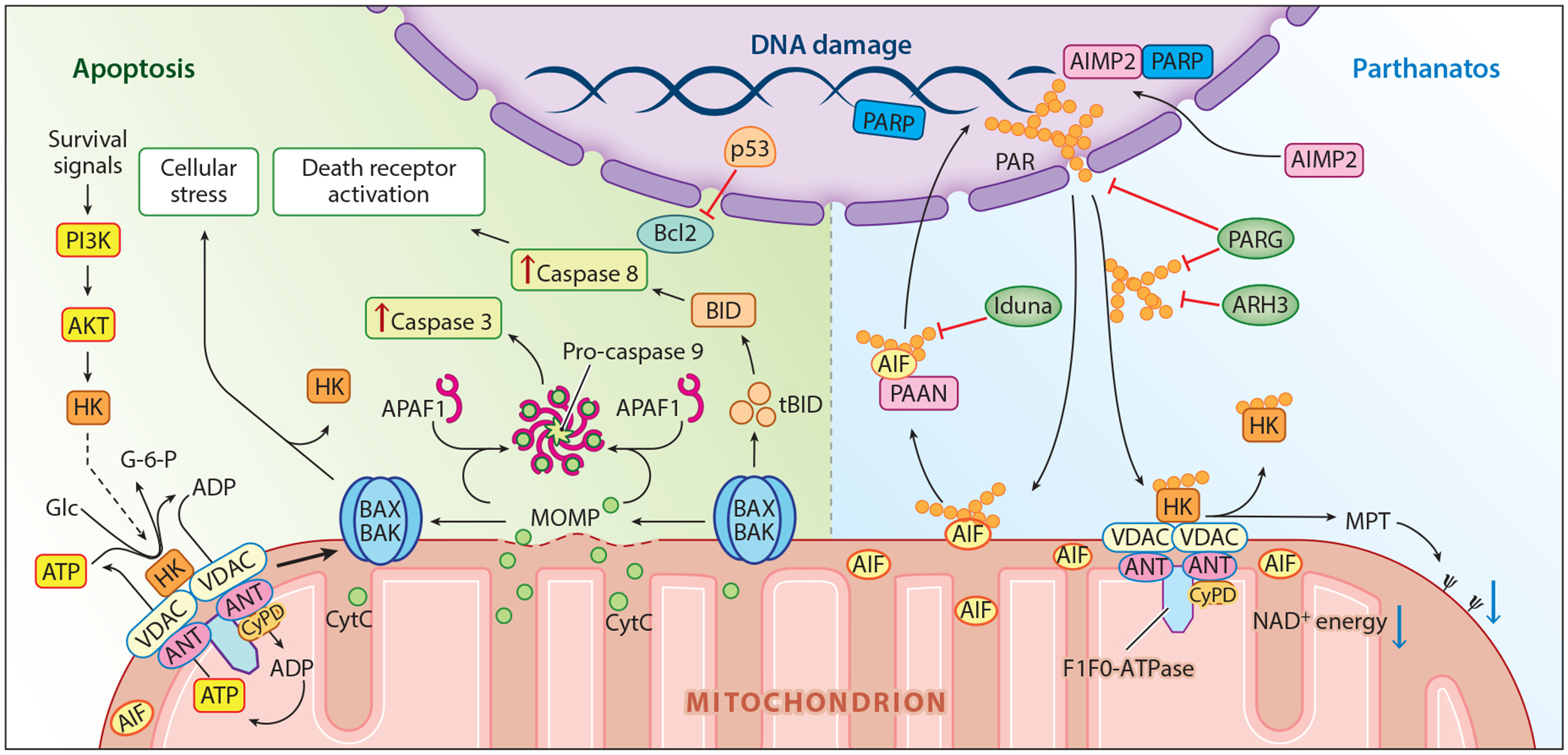
Mitochondrial-regulated cell death. Key players in apoptosis (left) and parthanatos (right) are highlighted. Abbreviations: AIF, apoptosis-inducing factor; AIMP2, aminoacyl-tRNA synthetase complex interacting multifunctional protein-2; AKT, protein kinase B; ANT, adenine nucleotide transporter; APAF1, apoptotic peptidase-activating factor 1; ARH3, ADP-ribosyl-acceptor hydrolase 3; ATP, adenosine triphosphate; BAK, Bcl-2 homologous antagonist/killer; BAX, Bcl-2-associated X; Bcl-2, B cell lymphoma 2; BID, BH3 (Bcl-2 homology 3) interacting-domain death agonist; CyPD, cyclophilin D; CytC, cytochrome C; Glc, glucose; G-6-P, glucose-6-phosphate; HK, hexokinase; Ψ, mitochondrial membrane potential; MOMP, mitochondrial outer membrane permeability; MPT, mitochondrial permeability transition; NAD+, nicotinamide adenine dinucleotide; PAAN, parthanatos AIF-associated nuclease; PAR, poly (ADP-ribose); PARG, PAR glycohydrolase; PARP, PAR polymerase; PI3K, phosphatidylinositol-3-kinase; p53, tumor suppressor p53; tBID, truncated BID.
Oxidative stress– or calcium overload–mediated (26) mitochondrial membrane permeabilization leads to permeability transition pore complex (PTPC) opening, which causes mitochondrial permeability transition (MPT). Mitochondria can accumulate large amounts of calcium through the calcium uniporter under pathologic conditions in which intracellular calcium concentrations increase (27). Osmotic swelling and rupture of the mitochondrial membranes and release of mitochondrial intermembrane proteins such as CytC, apoptosis-inducing factor (AIF), and the matrix protein endonuclease G occur after complete MPT (26). Cell death induced by MPT can occur during oxidative stress or ischemia-reperfusion injury, and unlike MOMP, MPT killing of cells does not require caspase activation (28). MPT can switch from low- to high-conductance states (29), and prolonged high conductance leads to mitochondrial collapse (30). Gradations of MPT can occur because mild MPT can be reversed by inhibitors of cyclophilin D (CyPD) (31). When a small fraction of mitochondria undergo MPT, they can be eliminated by mitophagy.
Although the exact molecular composition of the PTPC is not known, researchers do know it is a supramolecular multiprotein complex spanning the outer and inner mitochondrial membranes (Figure 3). It is composed, in part, of the integral protein of the inner mitochondrial membrane, the adenine nucleotide translocase (ANT), the outer mitochondrial membrane protein, the voltage-dependent anion channel (VDAC), and the matrix protein CyPD. The mitochondrial ATP synthase, another multiprotein complex, also known as F1F0-ATP synthase, that is responsible for the synthesis of ATP from dissipation of the chemiosmotic gradient (32), is emerging as a key component of the PTPC (33, 34). In addition, the PTPC also contains hexokinase (HK), creatine kinase, and several other modulatory enzymes (35). The rate-limiting step of glycolysis is controlled by HK, which in an ATP-dependent manner converts glucose to glucose-6-phosphate. HK’s tight association with VDAC provides ready access to mitochondrial-generated ATP. Disruption of the interaction of HK with VDAC can induce MPT (36). Only CyPD seems to be critical for MPT-mediated cell death, as loss of CyPD attenuates MPT-induced cell death (28), whereas ANT and VDAC are dispensable for MPT-induced cell death (26). Ultimately, complete MPT leads to cell death through energy collapse.
STROKE, NEURODEGENERATION, AND NEUROLOGIC DISORDERS
Disease-modifying therapies for neurodegenerative disorders do not exist or have very minimal benefits. Currently, no therapeutic strategies are available for treating neurologic injury due to stroke and other neurologic disorders. Thus, a clearer understanding of the pathologic processes that contribute to cell death in stroke, neurodegenerative diseases such as AD and PD, and neurologic disorders such as MS is essential. Inroads into developing effective disease-modifying therapies for neurologic disorders rest on gaining a detailed molecular understanding of the mechanisms of neuronal cell death. Having laid the foundation for why mitochondria play an important regulatory and contributory role to cell death, the remainder of this review highlights how neuronal cell death is orchestrated by this organelle. Cell death due to stroke is highlighted, as stroke is the leading neurologic cause of death and disability, and the basic molecular mechanisms of cell death that play important roles in neurologic injury due to stroke are likely to extrapolate to other neurologic disorders
Stroke
Ischemic neuronal injury that occurs following loss of oxygen and glucose to the brain leads initially to accidental cell death. Secondary pathologic processes follow the initial insult (Figure 4). Although prevention is paramount to treating stroke, once it occurs, both the control of these secondary processes and the restoration of blood supply are essential to limit ischemic neuronal damage. Advances in restoration of blood flow following stroke, including tissue plasminogen activator therapy (37) and mechanical thrombectomy (38), have led to effective therapies to restore blood flow, limiting neuronal and glial damage, which has changed medical practice. Despite the advances in restoring blood flow, substantial neuronal, glial, and neurovascular damage still occurs, particularly due to reperfusion injury of the penumbra (compromised but not necrotic brain tissue).
Figure 4.
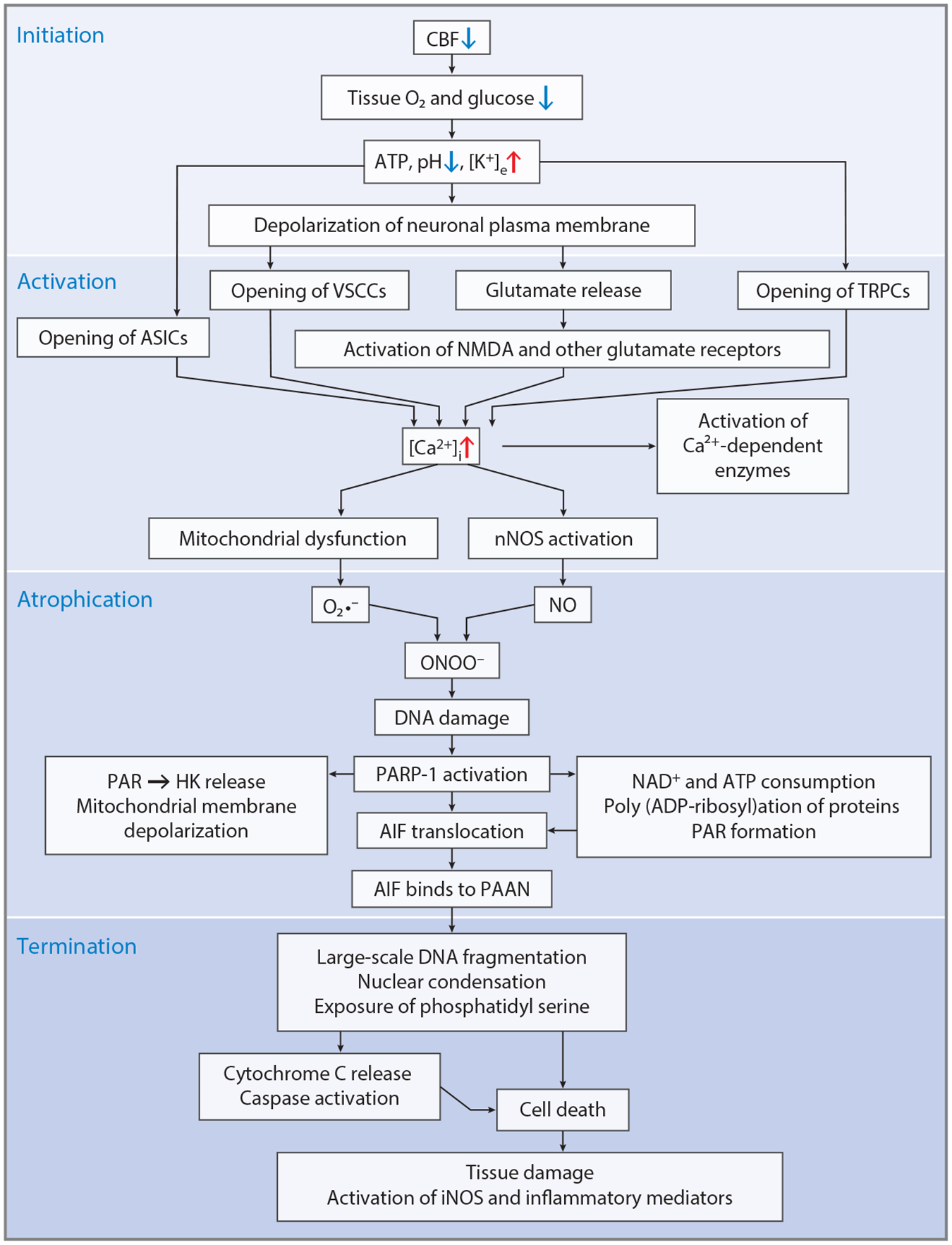
Mechanisms of neuronal cell death during stroke. Neuronal cell death occurs in a four-step process involving initiation due to loss of cerebral blood flow; activation due to opening of a variety of calcium channels; atrophication, which involves the activation of calcium-dependent enzymes and oxidative and nitrosative stress, which leads to parthanatos; and, ultimately, death through large-scale DNA fragmentation and caspase activation. Tissue damage continues and is amplified through inflammatory mediators. Other causes of neuronal cell death may occur through similar mechanisms. Abbreviations: AIF, apoptosis-inducing factor; ASIC, acid-sensitive ion channel; ATP, adenosine triphosphate; [Ca2+]i, intracellular calcium; CBF, cerebral blood flow; DNA, deoxyribonucleic acid; HK, hexokinase; iNOS, inducible nitric oxide synthase; [K+]e, extracellular potassium; NAD+, nicotinamide adenine dinucleotide; NMDA, N-methyl-d-aspartate; NO, nitric oxide; nNOS, neuronal NO synthase; O2, oxygen; O2•−, superoxide anion; ONOO−, peroxynitrite; PAAN, parthanatos AIF-associated nuclease; PAR, poly (ADP-ribose); PARP-1, PAR polymerase-1; TRPC, transient receptor potential channel; VSCC, voltage-sensitive cation channel.
Researchers have proposed many pathways that may contribute to cell death in the penumbra following stroke, but the more extensively investigated mediators include glutamate excitotoxicity, which drives the increases in intracellular calcium, production of oxygen free radicals, and nitric oxide (NO) that set cellular death mechanisms in motion (39–42). In addition, other routes of calcium entry through acid-sensing channels or transient receptor potential channels play important roles in neuronal injury (for reviews, see 43–45). Inappropriate activation of other transmitter second-messenger pathways owing to the initial ischemic insult may also contribute to the neuronal injury (39–42, 46). Accompanying the neuronal damage is activation of neuroinflammatory pathways, which contribute to damage and late regenerative processes (47).
Excitotoxicity
Glutamate excitotoxicity plays a role in ischemia/stroke, seizures, Huntington’s disease (HD), PD, AD, amyotrophic lateral sclerosis, hepatic encephalopathy, MS, traumatic brain injury, and metabolic disorders of the brain (39–41, 48–51). Thus, understanding the molecular mechanisms by which glutamate excitotoxicity kills neurons has relevance to many disorders of the nervous system.
Glutamate binds to four major types of receptors: N-methyl-d-aspartate (NMDA) receptors, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, metabotropic receptors, and kainate receptors, all of which transduce glutamate’s action in the nervous system (52). Ischemia-induced glutamate excitotoxicity and neuronal damage is mediated in large part through NMDA receptor activation (39, 41, 50). Evidence in support of targeting glutamate excitotoxicity in stroke comes from recent work with postsynaptic density-95 (PSD-95) inhibitors, which uncouple the NMDA receptor from its signaling cascade. PSD-95 inhibitors show unequivocal protection in rodent brains (53–56), high-order brains of old-world primates (57, 58), and humans (59). These recent exciting results have reinvigorated the search for novel neuroprotectants to treat acute ischemic stroke in humans (60–62). In addition, there is tremendous interest in interfering with nonglutamate receptor–mediated calcium entry through blockade of acid-sensing channels (63) and transient receptor potential channels (64), particularly because these channels seem to play a role in the later stages of calcium entry following ischemic injury in the brain.
Nitric Oxide and Poly (ADP-ribose) Polymerase-1
Glutamate excitotoxicity is mediated and initiated predominantly by glutamate activation of NMDA receptors, intracellular calcium flux, NO, and free radicals (Figure 4) (65–67). Neuronal cell death due to a short pulse (5 min) of glutamate receptor activation causes cell death 24 h later (68). This form of cell death, called delayed or rapidly triggered neurotoxicity, is dependent on NMDA receptor activation, calcium influx, and NO (65–67). Extrasynaptic NMDA receptors account for most of the toxicity associated with NMDA receptor activation, whereas synaptic NMDA receptor activation leads to activation of cell survival pathways (69).
Treatment with NO synthase (NOS) inhibitors, disruption of the interaction between the NMDA receptor and neuronal NOS (nNOS), or gene deletion of nNOS prevent cell death in cell culture, slice models, and in vivo models of stroke (53, 66, 67, 70–74). Overproduction of NO generated by NOS leads to the generation of peroxynitrite (ONOO−) (75) through the reaction of mitochondrial-generated O2•− with NO (76). ONOO− modification of DNA and the subsequent DNA nicks trigger the next step in the excitotoxic death program by activating the DNA damage-sensing enzyme poly (ADP-ribose) (PAR) polymerase-1 (PARP-1) (77–79). Cell demise from PARP-1 overactivation has been attributed to depletion of cellular energy; release of the mitochondrial death effector AIF from mitochondria; and production of excess PAR polymer, a novel death signal (46, 80–88).
Apoptosis
Glutamate excitotoxicity can induce apoptosis by increasing intracellular calcium and mitochondrial calcium overload and triggering MOMP or MPT (89). Oxidative stress can also lead to apoptosis through MOMP or MPT. MOMP leads to CytC release and activation of intrinsic apoptosis. Inflammation can induce both intrinsic and extrinsic apoptosis (90). Many studies in experimental animal and culture models of neurodegenerative and neurologic disorders, as well as human postmortem tissue examination, have revealed caspase activation and markers of apoptosis, raising the possibility that interference with both intrinsic and extrinsic apoptotic cascades could be neuroprotective in neurologic disorders (for reviews, see 91–93). Numerous other stressors or signals can lead to apoptosis in neurons. For instance, in response to DNA damage, tumor suppressor p53 inhibits the antiapoptotic protein Bcl-2 (94). The lack of neurotrophic support, which promotes cell survival through kinase signaling cascades such as the phosphatidylinositol-3-kinase and protein kinase B pathways, is the classic neuronal trigger of apoptosis, which plays important roles during neuronal development (93). Lack of neurotrophic support may also play a role in some neurodegenerative and neurologic disorders. Although apoptotic mechanisms have dominated the discourse on cell death, most of the cell death that occurs during stroke is nonapoptotic (95).
Parthanatos
Because apoptosis may not be the major form of cell death in stroke, what other mechanisms might be at play in the death of neurons due to stroke? The term parthanatos was coined from Thanatos, the personification of death in Greek mythology, to describe cell death initiated by the PAR polymer (Figure 2) (16, 17, 46, 80, 84, 88). Parthanatos is distinct biochemically from apoptosis and necrosis. Biochemically, it is characterized by rapid PARP-1 activation followed by PAR accumulation, early NAD+ depletion, and mitochondrial AIF translocation to the nucleus. Ultimately, late caspase activation and loss of cellular ATP occur (46, 80, 81, 84–88). In stroke, both PARP-1 and PARP-2 contribute to injury via AIF (96).
DNA damage is the traditional activator of PARP-1. DNA breaks are detected by PARP-1 via its zinc-finger domain and are followed by synthesis of PAR, which acts as a signaling scaffold and participates in the orchestration of the DNA repair response (97, 98). Other PARPs, including PARP-2 and PARP-3, contribute to DNA repair (97, 98). Excessive activation of the PARPs leads to parthanatos. Mitochondria play a key role in parthanatos, as mitochondrial-generated ROS due to mitochondrial calcium uptake are required for PARP-1 activation (99) and mitochondria house the death effector AIF (100). Bursts of O2•− production that occur during transient MPTs (101) could contribute to PARP-1 activation through formation of ONOO− (102). Other mechanisms of activation of PARP-1 may not require DNA damage and expand the ways in which parthanatos can be initiated. For instance, in PD, aminoacyl-tRNA synthetase complex interacting multifunctional protein-2 activates PARP-1 directly and contributes to the degeneration of dopamine neurons (103). Glyceraldehyde-3-phosphate dehydrogenase can promote PARP-1 activation in the setting of oxidative and nitrosative stress (104).
Shrunken and condensed nuclei are the hallmark morphologic features of parthanatos. Shortly after activation of PARP-1 in parthanatos, cells become propidium iodide positive. Caspase inhibitors cannot prevent parthanatos (46, 81, 85, 87, 88). Experimental studies indicate that over-activity of parthanatos plays a prominent role in neuronal injury in stroke (105–107). Parthanatos also plays an important role in cell damage following trauma, ischemia-reperfusion of the retina, PD, AD, HD, and the experimental autoimmune encephalomyelitis model of MS (for reviews, see 42, 46, 83, 108).
Once AIF enters the nucleus, chromatinolysis and large-scale DNA degradation happens, followed by cell death. The identity of the AIF-associated nuclease has remained elusive. Recently the macrophage migration inhibitory factor (MIF) was identified as a PARP-1–dependent AIF-associated nuclease (PAAN) (109). Molecular modeling indicates that the MIF trimer possesses the same topologic structure as the PD-D/E(X)K nuclease superfamily, in which it has both 3′ exonuclease and endonuclease activity (110, 111). MIF was shown to be a Mg2+/Ca2+-dependent nuclease that is carried into the nucleus by AIF after PARP-1 activation. Prevention of the translocation of MIF from the cytosol to the nucleus by disruption of the AIF and MIF protein-protein interaction protects against glutamate excitotoxicity and stroke. Moreover, knockout of MIF and a nuclease–deficient MIF prevents glutamate excitotoxicity and stroke to an extent similar to that afforded by deletion or inhibition of PARP-1. Identification of MIF as the elusive PAAN opens up a new therapeutic avenue to explore as a potential disease-modifying therapy in neurologic disorders. Inhibition of MIF’s nuclease activity may have substantial advantages over inhibition of PARP, particularly in chronic neurodegenerative diseases, as it would avoid impairment of the DNA repair response by inhibition of PARP.
AIF binding to PAR is required for parthanatos both in vitro and in vivo (85). AIF’s interaction with the outer mitochondrial membrane is disrupted by PAR binding, which leads to AIF release from mitochondria. The PAR binding site on AIF is distinct from its DNA binding site. This PAR-dependent release of AIF occurs in a calpain-independent manner (101). An AIF PAR binding mutant (R588A, K589A, R592A) cannot be released from mitochondria and prevents parthanatos despite being fully capable of AIF’s other functions, including FAD and DNA binding, NADH oxidase activity, and AIF-mediated nuclear condensation. Thus, PAR polymer binding to AIF is required for execution and provides a mechanism for AIF-mediated cell death. The PAR-dependent releasable pool of AIF is on the outer membrane of mitochondria (112). This mitochondrial outer membrane localization enables AIF to bind PAR and accounts for AIF’s release prior to the mitochondrial membrane depolarization (85), which explains the later release of CytC in parthanatic cell death after MPT occurs (88). Calpain- and BAX-dependent mechanisms of mitochondrial AIF release have also been described (113).
One of the remaining mysteries in parthanatos is how the highly negatively charged PAR leaves the nucleus to translocate to mitochondria to induce the release of AIF. Multiple routes of delivery of PAR from the nucleus to mitochondria likely exist, including PAR-binding proteins such as histone 1.2, which is known to translocate from the nucleus to mitochondria following cellular injury (114). In addition, free PAR polymer, which is released from PARylated proteins by the action of PAR glycohydrolase (PARG), contributes to mitochondrial AIF release (115). This may be the main route of cytosolic PAR, as ADP-ribosyl-acceptor hydrolase 3 cleavage of PAR protects against parthanatos by lowering PAR levels in the cytoplasm that are created through the action of PARG (115). Future studies will be required to determine whether a PAR binding protein or PARylated protein such as histone 1.2 plays a role in carrying PAR out of the nucleus to mitochondria.
Like in other cell death pathways, there are endogenous inhibitors of parthanatos. PARG is an enzyme that removes PAR from PARylated proteins, terminating the actions of PARP modification of proteins in much the same way that phosphatases terminate the action of kinases (116). Overexpressing PARG reduces cell death, and lowering PARG levels exacerbates cell death induced by activators of PARP-1, including glutamate excitotoxicity and stroke, consistent with the notion that PAR is a cell death messenger (81). In addition, failure to degrade PAR leads to early embryonic lethality and increased sensitivity to cytotoxicity (117). The majority of Drosophila lacking PARG die in the larval stage, but 25% of Drosophila survive, accumulate PAR, and develop a progressive neurodegenerative disorder (118). This PAR-dependent cell death induced by the lowering of PARG levels seems to occur via AIF, as knockdown of AIF by RNA interference reduces the enhanced, chemotherapy-induced cell death due to lowering of PARG (119). Mutations of c6orf130, which interacts with terminal ADP-ribose protein glycohydrolase, cause severe neurodegeneration through impairment of the turnover and recycling of PAR (120).
Iduna (RNF146) is another endogenous inhibitor of parthanatos. Iduna protects against stroke and NMDA receptor–mediated excitotoxicity both in vitro and in vivo by interfering with PAR-dependent cell death (121). It is a PAR-dependent ubiquitin E3 ligase targeting proteins that are PARylated or bind PAR (122–124). Iduna is activated by binding PAR, which serves as an allosteric activation signal (125). It also protects against DNA damage, facilitates DNA repair, regulates cell survival in a PAR-dependent fashion (123), and regulates Wnt signaling (122, 124). Interestingly, Iduna is a gene that is induced by preconditioning (121). Considering the importance of this inhibitor in survival and DNA repair, it will be important to understand how its expression is regulated as well as how its E3 ligase PAR-dependent substrates are involved in cell death.
The mechanism of energetic collapse following PARP-1 activation has been clarified recently. Consumption of cellular NAD+ owing to PARylation was thought to be the underlying mechanism of PARP-1–induced energetic collapse (126, 127). Instead, PAR-dependent inhibition of HK leads to defects in glycolysis that account for the bioenergetics collapse (128, 129). PAR binds to HK and inhibits HK’s activity, which leads to inhibition of glycolysis and subsequent reductions in cellular NAD+ and ATP. Consistent with notion that PAR binding to HK causes defects in glycolysis, these PAR-dependent defects can be rescued by the mitochondrial substrates pyruvate and glutamine. Thus, the reduction in cellular NAD+ is not due to excessive PARylation, as researchers proposed originally (126, 127). These results are consistent with the idea that PAR is a death-signaling molecule (46, 81, 87) and suggest that direct interference of PAR polymer signaling may provide unique opportunities for preventing cell death following activation of PARP-1.
A recent report by Xu et al. (130, p. 333ra48, 1) stated aptly that “translating neuroprotective treatment from discovery in cell and animal models to the clinic has proven challenging.” Recent advances in deriving human neurons from embryonic stem cells or inducible pluripotent stem cells provide an opportunity to validate processes identified in simpler model systems in human neurons (131–133). Human cortical neurons possessing a balanced excitatory and inhibitor network die in a NO- and parthanatos-dependent fashion when exposed to glutamate excitotoxicity or oxygen glucose deprivation. Moreover, inhibitors of PARP that are currently in clinical trials prevent neuronal cell death, providing further support for the notion that inhibition of the parthanatic death cascade may offer therapies that are disease modifying in human neurologic disorders (130).
PERSPECTIVE AND THERAPIES
Based on the central role mitochondria play in neuronal cell death, strategies that target mitochondrial-mediated cell death pathways have particular promise as neuroprotective therapies. Prevention of neurologic injury is likely to have the greatest impact, such as in trauma, stroke, and the restoration of blood flow in stroke. As advances in genomic manipulation such as CRISPR-Cas9 (clustered regularly interspaced short palindromic repeats-CRISPR associated 9) technology enable the correction of genetic defects, it may be possible to prevent neurodegenerative diseases that are due to mutations in causal genes (134). Owing to the daunting barrier or inefficiency of human brain gene transduction, including brain size and the blood-brain barrier, genetic correction may be limited to regions or areas that are readily accessible, such as the retina or small areas such as the substantia nigra that would be amenable to current viral and nanoparticle transduction approaches. Wide application of genetic correction in neurologic disease would require concomitant advances in gene transduction approaches.
Fortunately, as discussed here, many other approaches besides prevention and correction of genetic mutations could alter the course of neurologic disorders. Inhibitors of MPT and MOMP have been touted as cytoprotective agents (35). Targeting mitochondrial dynamics and mitophagy has received attention as a strategy to treat mitochondrial disorders and is the subject of recent reviews (9, 135). Areas of focus include efforts to enhance mitochondrial biogenesis, increase mitophagy, and control of the balance between fission and fusion.
Methods to interfere with mitochondrial-initiated regulated cell pathways include inhibition of caspases or PARP. Although evidence exists to implicate both caspases and PARP in neurologic disorders, the identification of PARP and caspase activity does not indicate causality or a beneficial effect through PARP or caspase inhibition (46, 136). Because multiple cell death pathways are initiated by mitochondrial dysfunction, simple inhibition of one path to cell death may not be sufficient. Moreover, inhibition of caspases or PARP, particularly in chronic neurologic disorders, could have long-term off-target consequences. Caspase or PARP inhibition in acute disorders such as stroke may avoid potential long-term complications. Inhibition of PARP has been particularly attractive since the first PARP inhibitor, olaparib, was approved by the US Food and Drug Administration for ovarian cancer (137). Caspase inhibitors have yet to meet this threshold. Several other PARP inhibitors are also in clinical trials for the treatment of different cancers (138, 139). It will be important to identify brain-penetrant PARP inhibitors with good safety profiles to repurpose and evaluate in neurologic disorders.
Recent advances in elucidating the molecular mechanisms of parthanatos also offer additional therapeutic targets, including interference with PAR signaling and inhibition of PAAN. Both targets are amenable to high-throughput screening, and agents targeting both could potentially avoid the long-term consequences of PARP inhibition.
In summary, research to improve our understanding of mitochondrial function and the cell death pathways orchestrated by mitochondria offers several interventions that hold the potential for disease-modifying therapies to treat neurologic disorders. Substantial work remains before we can identify safe and potent inhibitors of mitochondrial cell death pathways as well as agents that improve mitochondrial function that can be translated for use in humans.
ACKNOWLEDGMENTS
This work was supported by US National Institutes of Health grants NS38377, NS67525, and DA00266 and by the JPB Foundation. The authors acknowledge the joint participation by the Adrienne Helis Malvin Medical Research Foundation and the Diana Helis Henry Medical Research Foundation through its direct engagement in the continuous active conduct of medical research in conjunction with the Johns Hopkins Hospital and the Johns Hopkins University School of Medicine and the Foundation’s Parkinson’s Disease Programs M-1, M-2, M-2013, and H-2014. The authors thank I-Hsun Wu for assistance with the illustrations. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases.
Footnotes
DISCLOSURE STATEMENT
T.M.D. and V.L.D. are founders of Valted, LLC and hold an ownership equity interest in the company. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies.
LITERATURE CITED
- 1.McGovern Inst. Brain Res. MIT. 2014. Brain Disorders: By the Numbers. Cambridge, MA: MIT. https://mcgovern.mit.edu/brain-disorders/by-the-numbers [Google Scholar]
- 2.Di Carlo A 2009. Human and economic burden of stroke. Age Ageing 38:4–5 [DOI] [PubMed] [Google Scholar]
- 3.Kowal SL, Dall TM, Chakrabarti R, Storm MV, Jain A. 2013. The current and projected economic burden of Parkinson’s disease in the United States. Mov. Disord 28:311–18 [DOI] [PubMed] [Google Scholar]
- 4.Nath S, Villadsen J. 2015. Oxidative phosphorylation revisited. Biotechnol. Bioeng 112:429–37 [DOI] [PubMed] [Google Scholar]
- 5.Paul VD, Lill R. 2015. Biogenesis of cytosolic and nuclear iron-sulfur proteins and their role in genome stability. Biochim. Biophys. Acta 1853:1528–39 [DOI] [PubMed] [Google Scholar]
- 6.Maio N, Rouault TA. 2015. Iron-sulfur cluster biogenesis in mammalian cells: new insights into the molecular mechanisms of cluster delivery. Biochim. Biophys. Acta 1853:1493–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan DC. 2007. Mitochondrial dynamics in disease. N. Engl. J. Med 356:1707–9 [DOI] [PubMed] [Google Scholar]
- 8.Schon EA, Przedborski S. 2011. Mitochondria: the next (neurode)generation. Neuron 70:1033–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suliman HB, Piantadosi CA. 2016. Mitochondrial quality control as a therapeutic target. Pharmacol. Rev 68:20–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickrell AM, Youle RJ. 2015. The roles of PINK1, Parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85:257–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scarffe LA, Stevens DA, Dawson VL, Dawson TM. 2014. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 37:315–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Winklhofer KF. 2014. Parkin and mitochondrial quality control: toward assembling the puzzle. Trends Cell Biol. 24:332–41 [DOI] [PubMed] [Google Scholar]
- 13.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, et al. 2011. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol 12:222–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou R, Yazdi AS, Menu P, Tschopp J. 2011. A role for mitochondria in NLRP3 inflammasome activation. Nature 469:221–25 [DOI] [PubMed] [Google Scholar]
- 15.Burte F, Carelli V, Chinnery PF, Yu-Wai-Man P. 2015. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol 11:11–24 [DOI] [PubMed] [Google Scholar]
- 16.Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, et al. 2015. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ. 22:58–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, et al. 2012. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19:107–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tait SW, Oberst A, Quarato G, Milasta S, Haller M, et al. 2013. Widespread mitochondrial depletion via mitophagy does not compromise necroptosis. Cell Rep. 5:878–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, et al. 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91:479–89 [DOI] [PubMed] [Google Scholar]
- 20.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. 1996. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86:147–57 [DOI] [PubMed] [Google Scholar]
- 21.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. 1997. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c–dependent activation of caspase-3. Cell 90:405–13 [DOI] [PubMed] [Google Scholar]
- 22.Yin XM, Wang K, Gross A, Zhao Y, Zinkel S, et al. 1999. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400:886–91 [DOI] [PubMed] [Google Scholar]
- 23.Gillies LA, Kuwana T. 2014. Apoptosis regulation at the mitochondrial outer membrane. J. Cell. Biochem 115:632–40 [DOI] [PubMed] [Google Scholar]
- 24.Kroemer G, Galluzzi L, Brenner C. 2007. Mitochondrial membrane permeabilization in cell death. Physiol. Rev 87:99–163 [DOI] [PubMed] [Google Scholar]
- 25.Tait SW, Green DR. 2010. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell. Biol 11:621–32 [DOI] [PubMed] [Google Scholar]
- 26.Orrenius S, Gogvadze V, Zhivotovsky B. 2015. Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun 460:72–81 [DOI] [PubMed] [Google Scholar]
- 27.Foskett JK, Philipson B. 2015. The mitochondrial Ca2+ uniporter complex. J. Mol. Cell. Cardiol 78:3–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, et al. 2005. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434:658–62 [DOI] [PubMed] [Google Scholar]
- 29.Ichas F, Mazat JP. 1998. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim. Biophys. Acta 1366:33–50 [DOI] [PubMed] [Google Scholar]
- 30.Bernardi P 1999. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev 79:1127–55 [DOI] [PubMed] [Google Scholar]
- 31.Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, et al. 2005. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434:652–58 [DOI] [PubMed] [Google Scholar]
- 32.Mitchell P 1961. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 191:144–48 [DOI] [PubMed] [Google Scholar]
- 33.Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, et al. 2014. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. PNAS 111:10580–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, et al. 2013. Dimers of mitochondrial ATP synthase form the permeability transition pore. PNAS 110:5887–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonora M, Wieckowski MR, Chinopoulos C, Kepp O, Kroemer G, et al. 2015. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 34:1475–86 [DOI] [PubMed] [Google Scholar]
- 36.Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, et al. 2008. Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLOS ONE 3:e1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Adams HP Jr., del Zoppo G, Alberts MJ, Bhatt DL, Brass L, et al. 2007. Guidelines for the early management of adults with ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council, Clinical Cardiology Council, Cardiovascular Radiology and Intervention Council, and the Atherosclerotic Peripheral Vascular Disease and Quality of Care Outcomes in Research Interdisciplinary Working Groups. Stroke 38:1655–711 [DOI] [PubMed] [Google Scholar]
- 38.Berkhemer OA, Fransen PS, Beumer D, van den Berg LA, Lingsma HF, et al. 2015. A randomized trial of intraarterial treatment for acute ischemic stroke. N. Engl. J. Med 372:11–20 [DOI] [PubMed] [Google Scholar]
- 39.Lai TW, Zhang S, Wang YT. 2014. Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog. Neurobiol 115:157–88 [DOI] [PubMed] [Google Scholar]
- 40.Mehta A, Prabhakar M, Kumar P, Deshmukh R, Sharma PL. 2013. Excitotoxicity: bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol 698:6–18 [DOI] [PubMed] [Google Scholar]
- 41.Arundine M, Tymianski M. 2004. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell. Mol. Life Sci 61:657–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu SW, Wang H, Dawson TM, Dawson VL. 2003. Poly(ADP-ribose) polymerase-1 and apoptosis inducing factor in neurotoxicity. Neurobiol. Dis 14:303–17 [DOI] [PubMed] [Google Scholar]
- 43.Besancon E, Guo S, Lok J, Tymianski M, Lo EH. 2008. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol. Sci 29:268–75 [DOI] [PubMed] [Google Scholar]
- 44.MacDonald JF, Jackson MF. 2007. Transient receptor potential channels of the melastatin family and ischemic responses of central neurons. Stroke 38:665–69 [DOI] [PubMed] [Google Scholar]
- 45.Simard JM, Tarasov KV, Gerzanich V. 2007. Non-selective cation channels, transient receptor potential channels and ischemic stroke. Biochim. Biophys. Acta 1772:947–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fatokun AA, Dawson VL, Dawson TM. 2014. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol 171:2000–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Iadecola C, Anrather J. 2011. The immunology of stroke: from mechanisms to translation. Nat. Med 17:796–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kostic M, Zivkovic N, Stojanovic I. 2013. Multiple sclerosis and glutamate excitotoxicity. Rev. Neurosci 24:71–88 [DOI] [PubMed] [Google Scholar]
- 49.Meldrum B 1990. Protection against ischaemic neuronal damage by drugs acting on excitatory neurotransmission. Cerebrovasc. Brain Metab. Rev 2:27–57 [PubMed] [Google Scholar]
- 50.Meldrum BS. 1992. Excitatory amino acid receptors and disease. Curr. Opin. Neurol. Neurosurg 5:508–13 [PubMed] [Google Scholar]
- 51.Sepers MD, Raymond LA. 2014. Mechanisms of synaptic dysfunction and excitotoxicity in Huntington’s disease. Drug Discov. Today 19:990–96 [DOI] [PubMed] [Google Scholar]
- 52.Nakanishi S 1992. Molecular diversity of glutamate receptors and implications for brain function. Science 258:597–603 [DOI] [PubMed] [Google Scholar]
- 53.Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, et al. 2002. Treatment of ischemic brain damage by perturbing NMDA receptor–PSD-95 protein interactions. Science 298:846–50 [DOI] [PubMed] [Google Scholar]
- 54.Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. 1999. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 284:1845–48 [DOI] [PubMed] [Google Scholar]
- 55.Soriano FX, Martel MA, Papadia S, Vaslin A, Baxter P, et al. 2008. Specific targeting of pro-death NMDA receptor signals with differing reliance on the NR2B PDZ ligand. J. Neurosci 28:10696–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun HS, Doucette TA, Liu Y, Fang Y, Teves L, et al. 2008. Effectiveness of PSD95 inhibitors in permanent and transient focal ischemia in the rat. Stroke 39:2544–53 [DOI] [PubMed] [Google Scholar]
- 57.Cook DJ, Teves L, Tymianski M. 2012. A translational paradigm for the preclinical evaluation of the stroke neuroprotectant Tat-NR2B9c in gyrencephalic nonhuman primates. Sci. Transl. Med 4:154ra33. [DOI] [PubMed] [Google Scholar]
- 58.Cook DJ, Teves L, Tymianski M. 2012. Treatment of stroke with a PSD-95 inhibitor in the gyrencephalic primate brain. Nature 483:213–17 [DOI] [PubMed] [Google Scholar]
- 59.Hill MD, Martin RH, Mikulis D, Wong JH, Silver FL, et al. 2012. Safety and efficacy of NA-1 in patients with iatrogenic stroke after endovascular aneurysm repair (ENACT): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 11:942–50 [DOI] [PubMed] [Google Scholar]
- 60.Neuhaus AA, Rabie T, Sutherland BA, Papadakis M, Hadley G, et al. 2014. Importance of preclinical research in the development of neuroprotective strategies for ischemic stroke. JAMA Neurol. 71:634–39 [DOI] [PubMed] [Google Scholar]
- 61.Tymianski M 2014. Stroke in 2013: disappointments and advances in acute stroke intervention. Nat. Rev. Neurol 10:66–68 [DOI] [PubMed] [Google Scholar]
- 62.Tymianski M 2015. Neuroprotective therapies: preclinical reproducibility is only part of the problem. Sci. Transl. Med 7:299fs32. [DOI] [PubMed] [Google Scholar]
- 63.O’Bryant Z, Vann KT, Xiong ZG. 2014. Translational strategies for neuroprotection in ischemic stroke—focusing on acid-sensing ion channel 1a. Transl. Stroke Res 5:59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moran MM, McAlexander MA, Biro T, Szallasi A. 2011. Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discov 10:601–20 [DOI] [PubMed] [Google Scholar]
- 65.Dawson VL, Dawson TM. 1998. Nitric oxide in neurodegeneration. Prog. Brain Res 118:215–29 [DOI] [PubMed] [Google Scholar]
- 66.Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH. 1993. Mechanisms of nitric oxide–mediated neurotoxicity in primary brain cultures. J. Neurosci 13:2651–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dawson VL, Dawson TM, London ED, Bredt DS, Snyder SH. 1991. Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. PNAS 88:6368–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choi DW, Rothman SM. 1990. The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci 13:171–82 [DOI] [PubMed] [Google Scholar]
- 69.Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB. 2009. Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. PNAS 106:9854–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cazevieille C, Muller A, Meynier F, Bonne C. 1993. Superoxide and nitric oxide cooperation in hypoxia/reoxygenation-induced neuron injury. Free Radic. Biol. Med 14:389–95 [DOI] [PubMed] [Google Scholar]
- 71.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. 1994. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science 265:1883–85 [DOI] [PubMed] [Google Scholar]
- 72.Kollegger H, McBean GJ, Tipton KF. 1993. Reduction of striatal N-methyl-d-aspartate toxicity by inhibition of nitric oxide synthase. Biochem. Pharmacol 45:260–64 [DOI] [PubMed] [Google Scholar]
- 73.Lustig HS, von Brauchitsch KL, Chan J, Greenberg DA. 1992. Cyclic GMP modulators and excitotoxic injury in cerebral cortical cultures. Brain Res. 577:343–46 [DOI] [PubMed] [Google Scholar]
- 74.Moncada C, Lekieffre D, Arvin B, Meldrum B. 1992. Effect of NO synthase inhibition on NMDA- and ischaemia-induced hippocampal lesions. NeuroReport 3:530–32 [PubMed] [Google Scholar]
- 75.Beckman JS, Koppenol WH. 1996. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol 271:C1424–37 [DOI] [PubMed] [Google Scholar]
- 76.Xia Y, Dawson VL, Dawson TM, Snyder SH, Zweier JL. 1996. Nitric oxide synthase generates superoxide and nitric oxide in arginine-depleted cells leading to peroxynitrite-mediated cellular injury. PNAS 93:6770–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.de Murcia G, Ménissier de Murcia J. 1994. Poly(ADP-ribose) polymerase: a molecular nick-sensor. Trends Biochem. Sci 19:172–76 [DOI] [PubMed] [Google Scholar]
- 78.de Murcia G, Schreiber V, Molinete M, Saulier B, Poch O, et al. 1994. Structure and function of poly(ADP-ribose) polymerase. Mol. Cell. Biochem 138:15–24 [DOI] [PubMed] [Google Scholar]
- 79.Szabo C, Dawson VL. 1998. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol. Sci 19:287–98 [DOI] [PubMed] [Google Scholar]
- 80.Andrabi SA, Dawson TM, Dawson VL. 2008. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann. N. Y. Acad. Sci 1147:233–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Andrabi SA, Kim NS, Yu SW, Wang H, Koh DW, et al. 2006. Poly(ADP-ribose) (PAR) polymer is a death signal. PNAS 103:18308–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Koh DW, Dawson TM, Dawson VL. 2005. Poly(ADP-ribosyl)ation regulation of life and death in the nervous system. Cell. Mol. Life Sci 62:760–68 [DOI] [PubMed] [Google Scholar]
- 83.Virag L, Szabo C. 2002. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol. Rev 54:375–429 [DOI] [PubMed] [Google Scholar]
- 84.Wang Y, Dawson VL, Dawson TM. 2009. Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp. Neurol 218:193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang Y, Kim NS, Haince JF, Kang HC, David KK, et al. 2011. Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is critical for PAR polymerase-1-dependent cell death (parthanatos). Sci. Signal 4:ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang Y, Kim NS, Li X, Greer PA, Koehler RC, et al. 2009. Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (parthanatos). J. Neurochem 110:687–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu SW, Andrabi SA, Wang H, Kim NS, Poirier GG, et al. 2006. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. PNAS 103:18314–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, et al. 2002. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297:259–63 [DOI] [PubMed] [Google Scholar]
- 89.Nicholls DG. 2004. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr. Mol. Med 4:149–77 [DOI] [PubMed] [Google Scholar]
- 90.Inohara N, Nunez G. 2003. NODs: intracellular proteins involved in inflammation and apoptosis. Nat. Rev. Immunol 3:371–82 [DOI] [PubMed] [Google Scholar]
- 91.Friedlander RM. 2003. Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med 348:1365–75 [DOI] [PubMed] [Google Scholar]
- 92.Martin LJ. 2010. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals 3:839–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mattson MP. 2000. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell. Biol 1:120–29 [DOI] [PubMed] [Google Scholar]
- 94.Hemann MT, Lowe SW. 2006. The p53–Bcl-2 connection. Cell Death Differ. 13:1256–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lipton P 1999. Ischemic cell death in brain neurons. Physiol. Rev 79:1431–568 [DOI] [PubMed] [Google Scholar]
- 96.Li X, Klaus JA, Zhang J, Xu Z, Kibler KK, et al. 2010. Contributions of poly(ADP-ribose) polymerase-1 and −2 to nuclear translocation of apoptosis-inducing factor and injury from focal cerebral ischemia. J. Neurochem 113:1012–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dantzer F, Amé JC, Schreiber V, Nakamura J, Ménissier-de Murcia J, de Murcia G. 2006. Poly(ADP-ribose) polymerase-1 activation during DNA damage and repair. Methods Enzymol. 409:493–510 [DOI] [PubMed] [Google Scholar]
- 98.Krietsch J, Rouleau M, Pic E, Ethier C, Dawson TM, et al. 2013. Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Mol. Aspects Med 34:1066–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Duan Y, Gross RA, Sheu SS. 2007. Ca2+-dependent generation of mitochondrial reactive oxygen species serves as a signal for poly(ADP-ribose) polymerase-1 activation during glutamate excitotoxicity. J. Physiol 585:741–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, et al. 1999. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397:441–46 [DOI] [PubMed] [Google Scholar]
- 101.Wang W, Fang H, Groom L, Cheng A, Zhang W, et al. 2008. Superoxide flashes in single mitochondria. Cell 134:279–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wang H, Yu SW, Koh DW, Lew J, Coombs C, et al. 2004. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J. Neurosci 24:10963–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee Y, Karuppagounder SS, Shin JH, Lee YI, Ko HS, et al. 2013. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat. Neurosci 16:1392–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nakajima H, Kubo T, Ihara H, Hikida T, Danjo T, et al. 2015. Nuclear-translocated glyceraldehyde-3-phosphate dehydrogenase promotes poly(ADP-ribose) polymerase-1 activation during oxidative/nitrosative stress in stroke. J. Biol. Chem 290:14493–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, et al. 1997. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med 3:1089–95 [DOI] [PubMed] [Google Scholar]
- 106.Moroni F 2008. Poly(ADP-ribose)polymerase 1 (PARP-1) and postischemic brain damage. Curr. Opin. Pharmacol 8:96–103 [DOI] [PubMed] [Google Scholar]
- 107.Pacher P, Szabo C. 2008. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am. J. Pathol 173:2–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Martire S, Mosca L, d’Erme M. 2015. PARP-1 involvement in neurodegeneration: a focus on Alzheimer’s and Parkinson’s diseases. Mech. Ageing Dev 146–148:53–64 [DOI] [PubMed] [Google Scholar]
- 109.Wang Y, An R, Umanah GK, Park H, Nambiar K, et al. 2016. A nuclease that mediates cell death induced by DNA damage and poly(ADP-ribose) (PAR) polymerase-1. Science. In press. 10.1126/science.aad6872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Laganeckas M, Margelevičius M, Venclovas C. 2011. Identification of new homologs of PD-(D/E)XK nucleases by support vector machines trained on data derived from profile–profile alignments. Nucleic Acids Res. 39:1187–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Steczkiewicz K, Muszewska A, Knizewski L, Rychlewski L, Ginalski K. 2012. Sequence, structure and functional diversity of PD-(D/E)XK phosphodiesterase superfamily. Nucleic Acids Res. 40:7016–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yu SW, Wang Y, Frydenlund DS, Ottersen OP, Dawson VL, Dawson TM. 2009. Outer mitochondrial membrane localization of apoptosis-inducing factor: mechanistic implications for release. ASN Neuro 1:AN20090046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Moubarak RS, Yuste VJ, Artus C, Bouharrour A, Greer PA, et al. 2007. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol. Cell. Biol 27:4844–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Konishi A, Shimizu S, Hirota J, Takao T, Fan Y, et al. 2003. Involvement of histone H1.2 in apoptosis induced by DNA double-strand breaks. Cell 114:673–88 [DOI] [PubMed] [Google Scholar]
- 115.Mashimo M, Kato J, Moss J. 2013. ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. PNAS 110:18964–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Feng X, Koh DW. 2013. Roles of poly(ADP-ribose) glycohydrolase in DNA damage and apoptosis. Int. Rev. Cell Mol. Biol 304:227–81 [DOI] [PubMed] [Google Scholar]
- 117.Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, et al. 2004. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. PNAS 101:17699–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hanai S, Kanai M, Ohashi S, Okamoto K, Yamada M, et al. 2004. Loss of poly(ADP-ribose) glycohydrolase causes progressive neurodegeneration in Drosophila melanogaster. PNAS 101:82–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Feng X, Zhou Y, Proctor AM, Hopkins MM, Liu M, Koh DW. 2012. Silencing of Apoptosis-Inducing factor and poly(ADP-ribose) glycohydrolase reveals novel roles in breast cancer cell death after chemotherapy. Mol. Cancer 11:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sharifi R, Morra R, Appel CD, Tallis M, Chioza B, et al. 2013. Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J. 32:1225–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Andrabi SA, Kang HC, Haince JF, Lee YI, Zhang J, et al. 2011. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat. Med 17:692–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Callow MG, Tran H, Phu L, Lau T, Lee J, et al. 2011. Ubiquitin ligase RNF146 regulates tankyrase and Axin to promote Wnt signaling. PLOS ONE 6:e22595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kang HC, Lee YI, Shin JH, Andrabi SA, Chi Z, et al. 2011. Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. PNAS 108:14103–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, et al. 2011. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat. Cell Biol 13:623–29 [DOI] [PubMed] [Google Scholar]
- 125.DaRosa PA, Wang Z, Jiang X, Pruneda JN, Cong F, et al. 2015. Allosteric activation of the RNF146 ubiquitin ligase by a poly(ADP-ribosyl)ation signal. Nature 517:223–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. 2010. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci 30:2967–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ha HC, Snyder SH. 1999. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. PNAS 96:13978–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, et al. 2014. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. PNAS 111:10209–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fouquerel E, Goellner EM, Yu Z, Gagné JP, Barbi de Moura M, et al. 2014. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 8:1819–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Xu JC, Fan J, Wang X, Eacker SM, Kam TI, et al. 2016. Cultured networks of excitatory projection neurons and inhibitory interneurons for studying human cortical neurotoxicity. Sci. Transl. Med 8:333ra48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. 2009. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol 27:275–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, et al. 2008. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3:519–32 [DOI] [PubMed] [Google Scholar]
- 133.Gaspard N, Bouschet T, Hourez R, Dimidschstein J, Naeije G, et al. 2008. An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature 455:351–57 [DOI] [PubMed] [Google Scholar]
- 134.Doudna JA, Charpentier E. 2014. The new frontier of genome engineering with CRISPR-Cas9. Science 346:1258096. [DOI] [PubMed] [Google Scholar]
- 135.Whitaker RM, Corum D, Beeson CC, Schnellmann RG. 2016. Mitochondrial biogenesis as a pharmacological target: a new approach to acute and chronic diseases. Annu. Rev. Pharmacol. Toxicol 56:229–49 [DOI] [PubMed] [Google Scholar]
- 136.Fischer U, Schulze-Osthoff K. 2005. New approaches and therapeutics targeting apoptosis in disease. Pharmacol. Rev 57:187–215 [DOI] [PubMed] [Google Scholar]
- 137.Deeks ED. 2015. Olaparib: first global approval. Drugs 75:231–40 [DOI] [PubMed] [Google Scholar]
- 138.Ricks TK, Chiu HJ, Ison G, Kim G, McKee AE, et al. 2015. Successes and challenges of PARP inhibitors in cancer therapy. Front. Oncol 5:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Vinayak S, Ford JM. 2010. PARP inhibitors for the treatment and prevention of breast cancer. Curr. Breast Cancer Rep 2:190–97 [DOI] [PMC free article] [PubMed] [Google Scholar]