

Abstract
Fatty acid binding protein 5 (FABP5) is a highly promising target for the development of analgesics as its inhibition is devoid of CB1R-dependent side-effects. The design and discovery of highly potent and FABP5-selective truxillic acid (TA) monoesters (TAMEs) is the primary aim of the present study. On the basis of molecular docking analysis, ca. 2,000 TAMEs were designed and screened in silico, to funnel down to 55 new TAMEs, which were synthesized and assayed for their affinity (Ki) to FABP5, 3 and 7. The SAR study revealed that the introduction of H-bond acceptors to the far end of the 1,1′-biphenyl-3-yl and 1,1′-biphenyl-2-yl ester moieties improved the affinity of α-TAMEs to FABP5. Compound γ-3 is the first γ-TAME, demonstrating a high affinity to FABP5 and competing with α-TAMEs. We identified the best 20 TAMEs based on the FABP5/3 selectivity index. The clear front runner is α-16, bearing a 2‑indanyl ester moiety. In sharp contrast, no ε-TAMEs made the top 20 in this list. However, α-19 and ε-202, have been identified as potent FABP3-selective inhibitors for applications related to their possible use in the protection of cardiac myocytes and the reduction of α-synuclein accumulation in Parkinson’s disease. Among the best 20 TAMEs selected based on the affinity to FABP7, 13 out of 20 TAMEs were found to be FABP7-selective, with α-21 as the most selective. This study identified several TAMEs as FABP7-selective inhibitors, which would have potentially beneficial therapeutic effects in diseases such as Down’s syndrome, schizophrenia, breast cancer, and astrocytoma. We successfully introduced the α-TA monosilyl ester (TAMSE)-mediated protocol to dramatically improve the overall yields of α-TAMEs. α-TAMSEs with TBDPS as the silyl group is isolated in good yields and unreacted α-TA/ α-MeO-TA, as well as disilyl esters (α-TADSEs) are fully recycled. Molecular docking analysis provided rational explanations for the observed binding affinity and selectivity of the FABP3, 5 and 7 inhibitors, including their α, γ and ε isomers, in this study.
Keywords: Fatty acid binding protein, FABP inhibitor, Truxillic acid, Antinociceptive agent, SAR, Molecular docking, Computer-aided drug design
1. Introduction
Fatty acid binding proteins (FABPs) are a family of intracellular inflammatory proteins that transport various lipophilic ligands, including fatty acids, eicosanoids, cannabinoids, and N-acylethanolamines (NAEs) [1–5]. The transport of NAEs, particularly endocannabinoids, plays a critical role in signaling pathways.[6–8] Modulation of FABP function holds therapeutic potential for the treatment of many disorders, including atherosclerosis, diet-induced obesity, inflammation, cancer, and pain [9–11]. FABPs exhibit heterogeneous expression patterns in mammalian tissues, e.g., the central and peripheral nervous systems express FABP3, FABP5, and FABP7 [12].
There are two well-characterized endocannabinoids, i.e., anandamide (AEA) and 2-arachidonoylglycerol (2-AG), which serve as endogenous ligands for the type-1 and type-2 cannabinoid receptors (CB1R and CB2R), respectively, whose activation produces a variety of beneficial effects, including antinociception [13,14]. Efforts to develop therapeutics targeting CB1R have been hampered by the observation that global CB1R activation by exogenous agonists such as Δ-9-tetrahydrocannabinol is accompanied by undesirable side effects including catalepsy and hypolocomotion [15,16]. In contrast, the elevation of endocannabinoid levels produces CB1R and CB2R mediated beneficial effects while bypassing the adverse effects of direct CB1R agonists [16].
FABPs facilitate the intracellular trafficking of endocannabinoids to their catabolic enzyme, fatty acid amide hydrolase (FAAH), for hydrolysis and inactivation [2,17]. The inhibition of FAAH produces therapeutically beneficial effects but it is unfortunately accompanied by adverse effects including the emergence of insulin resistance [18]. Therefore, targeting FABPs may offer a distinct advantage as compared to the inhibition of FAAH. We reported previously that the augmentation of AEA levels through FABP5 inhibition produced CB1R-mediated antinociceptive effects in a variety of preclinical pain models [4,19–22]. Consequently, FABP5 would serve as a better target for the development of analgesics as its inhibition is devoid of CB1R-dependent side effects. Furthermore, FABP5 inhibitors would neither produce reinforcing effects nor motor and cognitive impairment [23].
In addition to FABP5 inhibitors, those selectively targeting FABP3 or FABP7 may also have therapeutic value. FABP3 is abundantly expressed in cardiac, skeletal muscle, and neurons, and FABP3 deficiency protects cardiac myocytes from apoptosis after myocardial infarction and reduces α-synuclein accumulation in a Parkinson’s disease model [24–26]. FABP7 is highly expressed in radial glial cells of the developing brain, and the fetal brain expresses more FABP7 than the adult brain [27]. Overexpression of FABP7 is observed in many diseases, including Down’s syndrome, schizophrenia, breast cancer, and astrocytoma [28–31]. FABP7-selective inhibitors could help reveal the potentially beneficial therapeutic effects of FABP7 inhibition in these diseases.
Employing a structure-based drug discovery approach by means of in silico screening of a large library of compounds followed by in vitro affinity assays with FABP5, we identified α-truxillic acid 1‑naphthyl ester (SB-FI-26, L1) as a promising hit/lead compound for FABP5 inhibition, which exhibited potent antinociceptive and anti-inflammatory effects in vivo in rodent models [32]. Our first systematic optimization of L1 was performed based on the SAR study of a library of L1 analogs, based on the successfully determined X-ray crystal structures of FABP5-L1 and FABP7-L1 complexes in high resolution [33], and in vitro affinity assays with FABP3, 5 and 7, as well as in vivo efficacy assay [34]. This first optimization study identified two new lead compounds, SB-FI-102 (L2) and SB-FI-103 (L3), wherein L2 is a highly potent non-selective inhibitor for FABP3, 5 and 7, while L3 is less potent, but selective FABP5 inhibitor (Fig. 1) [34]. The in vivo efficacy assays of potent compounds selected based on Ki values revealed that those with very high ClogP values (>7) did not show efficacy even though the Ki values were better or comparable [34].
Fig. 1.

Structures of the hit (L1) and current lead (L2 and L3) compounds.
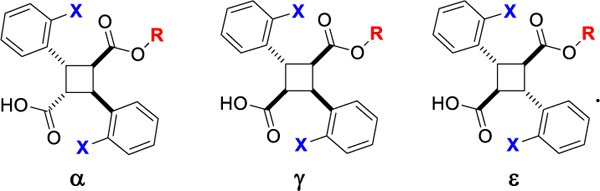
Accordingly, we launched the second optimization study (this work) by taking into account all the findings obtained in the first optimization study on mostly α-truxillic acid monoesters (α-TAMEs) and also expanding the structures of TAMEs not only within the α-isomers, but also to the γ and ε isomers, i.e., γ-TAMEs and ε-TAMEs, respectively (Fig. 2).
Fig. 2.

Structures of α-, γ- and ε-TAMEs.
In this study, we continued our structure-based computer-aided drug design, molecular docking analysis and SAR based on in vitro affinity (Ki) assays to identify highly selective and potent FABP5 inhibitors, as well as FABP3 and FABP7 inhibitors, and promising lead compounds were examined for their in vivo efficacy on antinociceptive activity. It is worthy of note that the results described here include the very first SAR study on selective FABP3 and FABP7 inhibitors.
2. Results and discussion
2.1. Computer-aided drug design of TAME-based FABP inhibitors
AutoDock 4.2 was used to assist the structure-based design and scoring of new TAMEs. Co-crystal structures of FABP5 and FABP7 with α-TAME L1 (PDB: 5UR9 and 5URA, respectively) provided the structural basis for computational binding scoring and docking pose analysis. Although racemic L1 was used in the crystallization process, only its (S, S,S,S)-enantiomer was found at the canonical binding site [33]. Consequently, we carried out the optical resolution of (S,S,S,S)- and (R,R,R,R)-enantiomers of L1 and their Ki values determined, which showed practically the equivalent affinity within experimental errors (Ki for FABP5: (S,S,S,S) 0.80 ± 0.14 μM; (R,R,R,R) 0.78 ± 0.14 μM) [33]. Also, the molecular docking analysis with molecular dynamics (MD) simulations revealed that the (R,R,R,R)-enantiomer also bound with virtually the same docking energy as that of the (S,S,S,S)-enantiomer within statistical errors (~−40 kcal/mol for both enantiomers) [33].
One of the most critical criteria for our validation of the molecular docking solutions was whether the key interaction of the carboxylate group of TAMEs with Arg129 and Tyr131 in FABP5 was conserved or not. Well-docked poses were generated more consistently for (S,S,S,S)-enantiomers compared to those for (R,R,R,R)-enantiomers, using the 5UR9 and 5URA crystal structures. This is easily understandable since the crystal structure of FABP-L1 should be the result of the induced fit of the FABP protein to accommodate (S,S,S,S)-L1. Since (R,R,R,R)-L1 was found to possess the same Ki and docking score as those of (S,S,S,S)-L1 as mentioned above [33], we decided to run α-TAME-based drug design only using (S,S,S,S)-enantiomers. It should be noted that there is only a factor of 2 difference even when the (R,R,R,R)-isomer of a TAME is totally inactive, i.e., not binding at all, and it is very likely that (R,R,R, R)-TAMEs have comparable Ki values to their (S,S,S,S)-counterparts. It should be noted that γ-TAMEs have two enantiomers, i.e., (1S,2R,3S,4R)- and (1R,2S,3R,4S)-isomers, while ε-TAMEs are achiral due to two planes of symmetry in a molecule. It should also be noted that the cyclobutane moiety of α-TAMEs can take quasi-axial and quasi-equatorial conformations, due to the strained puckering of the cyclobutene ring (Figure S1, Table S1, Supporting Information). In the X-ray crystal structures of FABP5-L1 (5UR9) and FABP7-L1 (5URA), the ester moiety of L1 is taking a quasi-equatorial conformation, which has turned out to be the lowest molecular energy conformation for all α-TAMEs. Since no co-crystal structure of FABPs with a γ- or ε-TAME is available at present, we have chosen to use the lowest molecular energy conformation of each of all γ- and ε-TAMEs to initiate the molecular docking and docking pose analysis. The lowest energy structures of γ- and ε-TAMEs also take a quasi-equatorial conformation for the ester group. In both γ- and ε-isomers, the two carboxyl groups are syn to each other which exacerbates the steric strain in the quasi-axial conformation. Calculated values of quasi-axial and quasi-equatorial conformations of α-, γ- and ε-L1, using the MMFF94 force field indicates the energy difference of 5 kcal/mol for α-L1 and 11 kcal/mol for γ-L1. Unlike the other two isomers, the quasi-axial conformer of ε-L1 was not able to reach a local minimum, due to the immense steric strain of all four substituents in the quasi-axial conformation (Table S1, Supporting Information).
We reported previously that α-TAMEs with (1S,2R)- and (1R,2S)-2-phenylcyclohex-1-yl ester groups have high affinities to all three FABPs, especially to FABP5 (Ki 0.18–0.21 μM), in vitro.[34] However, these excellent Ki values were not reflected in the efficacy against hyper-algesia in vivo, which is very likely to be attributed to their very high hydrophobicity (ClogP 7.17) [34]. As an achiral isostere of the 2‑phenylcyclohexyl group, we introduced a 1,1′-biphenyl-2-yl group to α -TAME (L4), which exhibited very good Ki values (0.35–0.71 μM) for FABP3, 5 and 7, with improved ClogP (6.12) (Fig. 3) [34]. We also examined two other biphenyl regioisomers and found that the 1,1′-biphenyl-3-yl isomer (L5) (Fig. 3) exhibited only slightly reduced affinities to FABP5 and 7 (Ki 0.85 and 0.74 μM, respectively), with >10 times selectivity against FABP3 (Ki 9.75 μM), as compared to L4, while the 1,1′-biphenyl-4-yl isomer considerably lost an affinity to all three FABPs [34]. Accordingly, in the present work, we put a primary focus on the modifications of 1,1′-biphenyl-2-yl and 1,1′-biphenyl-3-yl monoesters of α-truxillic acid (α-TA) and α-(2-MeO)-truxillic acid (α-MeO-TA) for computer-aided drug design, docking analysis and synthesis. Naturally, we also performed systematic in silico modifications of naphthyl and fluorenylmethyl groups in L2 and L3, but the chemical space for those modifications was limited.
Fig. 3.

Structures of L4 and L5, as well as a general structure of biphenylyl-α-TAMEs.
Over 1,900 analogs of L1, L2 and L3 were designed through systematic modifications of the ester moiety and subjected to computational analysis. Although L1, L2 and L3 are all α-TAMEs, the corresponding γ- and ε-TAMEs were also designed and evaluated by molecular docking analysis. Synthetically, γ-TAMEs have an advantage over α-TAMEs since γ-TAMEs can be easily obtained selectively through ring-opening of the intermediary γ-truxillic anhydrides, which are readily prepared from α-TA and α-MeO-TA (see Scheme 1). ε-TAMEs have two planes of symmetry and thus achiral, which means that ε-TAMEs do not have a potential chirality issue in pharmacological properties and their toxicological assessments.
Scheme 1.

Synthesis of α-, γ- and ε-TAMEs.
Reagents and conditions: (i) SOCl2, DMF, reflux, 3 h; (ii) ROH, pyridine, THF, r.t., overnight; (iii) ROH, EDC, DMAP, THF, r.t., overnight; (iv) NaOAc, Ac2O, reflux, 24 h; (v) ROH, DIPEA, THF, r.t., overnight; vi) KOH, 325 °C, 20 min.
Since FABP5 is the primary target for antinociceptive effects, the docking scores for FABP5 were used as a critical factor for funneling down a library of TAMEs designed. Thus, the docking score of −8.0 kcal/mol (AutoDock 4.2) was set as a filter in the first screening step (Fig. 4). [Note: The docking score of (S,S,S,S)-L1 to FABP5 was −8.17 kcal/mol (AutoDock 4.2)] [34]. Another critical factor was the relevant docking pose of designed TAMEs, keeping the canonical interaction of the carboxylic acid moiety with Arg and Tyr residues, identified by X-ray cocrystal structures of FABP5-L1 (PDB: 5UR9) and FABP7-L1 (PDB: 5URA) [33]. Since our original and primary interest was on selective FABP5 inhibitors until potential therapeutic benefits of the inhibitions of FABP3 [24–26] and FABP7 [28–31] emerged recently, we imposed a bias, especially against FABP3 in the funneling process. This was based on a reported negative consequence of FABP3 knockout on heart muscle functions in mice [35]. Thus, the docking scores of −10.5 kcal/mol for FABP3 and −11.0 kcal/mol for FABP7 were set as additional filters. [Note: The docking scores of (S,S,S,S)-L1 to FABP3 and FABP7 were −10.34 and −10.83 kcal/mol, respectively (AutoDock 4.2)]. After the first screening, 617 compounds out of 1,921 designed TAMEs were filtered through (Fig. 5).
Fig. 4.

H-bonding between α-TAME (α-1, α-5, and α-8: see Table 1) and the S3-S4 loop (Ser58, Leu60, and Lys61) of FABP5.
Fig. 5.

A flow chart of computer-aided drug design and selection through multi-factor filters.
Among the α-TAMEs filtered through, a library of biphenyl esters, fused with heterocycles such as dioxolane, dioxane, lactam, etc., emerged with docking scores substantially better than that of L1, i.e., ΔEdock = 1.0 ~ 3.0 kcal/mol, in FABP5 (Table S2, Supporting Information). Computational docking analysis showed that the S3-S4 loop of FABP5 contains a cluster of hydrogen-bond donors, including Ser58, Thr59, and the backbone NH of Leu60 and Lys61 (Fig. 4). This area of the binding site in FABP5 lines up well with the far end of the biphenyl ester moiety of TAME, and thus forms H-bonds to the H-bond acceptors, i.e., oxygen and/or nitrogen-containing functional groups, fused with the far end of the 1,1′-biphenyl ester, as exemplified in Fig. 4. However, a large variation was observed in the case of the corresponding γ- and ε-TAMEs. In general, it was predicted that γ-isomers have a weaker affinity, while ε-isomers showed a stronger affinity than that of the corresponding α-isomer, based on the difference in docking scores (ΔEdock) (Table S2, Supporting Information). Accordingly, a large number of ε-TAMEs were designed at this stage (i.e., 692 of 1,921 compounds, Fig. 5). However, none of ε-TAMEs had been synthesized and their Ki values determined at that time. Thus, we synthesized several ε -TAMEs and evaluated their affinities to FABP5, 3 and 7 to examine the consistency with the predicted affinities based on docking scores, as well as a possible parallel shift in the computational prediction. Then, it was found that the predicted affinities were apparently overestimated for ε-TAMEs as compared to those for α-TAMEs, and thus normalization was necessary for fair comparison based on the docking scores. For example, the docking scores of α-5 and ε-5 were −9.67 kcal/mol and −10.56 kcal/mol, respectively, while the Ki values were 0.36 μM and 3.40 μM, respectively. Similar overestimation in docking scores was observed for two other α- and ε-TAME pairs, arbitrarily selected for this examination. Consequently, we introduced this adjustment of the docking scores for ε-TAMEs to the next step of the screening process.
In the second screening step, ClogP (<7.0, but <6.0 preferred) prediction by ChemDraw [36] and ADMETox predictions by pkCSM [37] (AMES toxicity, hERG inhibition, LD50) were employed to exclude highly hydrophobic compounds, as well as potentially highly toxic compounds. In addition, synthetic feasibility was introduced as a filter. After the second screening, 55 compounds were selected for chemical synthesis and Ki determinations (Fig. 5, Table S2).
2.2. Chemical synthesis
α-Truxillic acid (α-TA) and α-2-MeO-truxillic acid (α-MeO-TA) were synthesized through [2 + 2] photocycloaddition of E-cinnamic acid and E-2-methoxycinnamic acid, respectively, as reported previously [32,34]. A library of hydroxy-1,1′-biphenyls was synthesized from the corresponding aryl halides and arylboronic acids or esters through Suzuki coupling (Schemes S1–S3, Supporting Information), unless the necessary components for designed TAMEs were commercially available.
α-TAMEs were synthesized by reacting the diacid chlorides, in situ generated from α-TA/ α-MeO-TA and thionyl chloride, with hydroxyarenes or alcohols, as previously reported [32,34]. Alternatively, α-TAMEs were also synthesized through condensation of α-TA/ α-MeO-TA with hydroxyarenes or alcohols, using N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide (EDC) as dehydrating agent (Scheme 1) [34].
γ-TAMEs were synthesized from the corresponding γ-truxillic anhydride (γ-TAA) with hydroxyarenes or alcohols in the presence of a base, as reported previously (Scheme 1) [34]. γ-TAA was readily prepared through epimerization-cyclization of α-truxillic acids, using acetic anhydride in the presence of sodium acetate [34]. ε-Truxillic acid (ε-TA) was prepared from α-TA in high yield through alkali fusion at 325 °C, following the literature procedure with modification [38]. ε-TAMEs were synthesized in a manner similar to that used for α-TAMEs (Scheme 1).
2.3. Improved synthesis of α-TAMEs via α-TA/ α -MeO-TA monosilyl esters (α-TAMSEs)
The synthesis of α-TAMEs by conventional methods described above (Scheme 1) suffered from low or at most moderate yields due to the inevitable formation of the corresponding diesters and difficulty in separating these diesters and unreacted α-TA/ α-MeO-TA from α-TAMEs by column chromatography. Thus, an introduction of a more efficient method was deemed necessary. Accordingly, we explored the use of α-TA/ α-MeO-TA monosilyl esters (α-TAMSEs) as the common key intermediates for the synthesis of α-TAMEs and recycle α-TA/ α-MeO-TA disilyl ester side products back to α-TA/ α-MeO-TA (Scheme 2). By taking into account the reasonable stability of silyl esters during column chromatography on silica gel, tert-butyldiphenylsilyl (TBDPS) and triisopropylsilyl (TIPS) groups were examined. Two equivalents of α-TA/ α-MeO-TA to TBDPS-Cl or TIPS-Cl were used in the presence of diisopropylethylamine (DIPEA). The unoptimized yields of TBDPS-esters, i. e., α-TAMSE-1 and α-MeO-TAMSE-1, were 71 % and 73 %, respectively, while those of TIPS-esters, i.e., α-TAMSE-2 and α-MeO-TAMSE-2, were 34 % and 69 %, respectively. In all cases, isolated disilyl esters were desilylated with tetrabutylammonium fluoride (TBAF) to give α-TA/ α-MeO-TA, which were combined into the unreacted α-TA/ α-MeO-TA recovered for use in the next α-TAMSE synthesis.
Scheme 2.

Synthesis of α-TAME through α-TAMSE.
Reagents and conditions: (i) R3SiCl, DIPEA, THF, r.t., 2 h; (ii) TBAF, DCM, r.t., 3 h; (iii) TCFH, NMI, ROH, MeCN, r.t., overnight; (iv) 1 M HCl workup.
α-TAMSE-1/2 and α-MeO-TAMSE-1/2, thus obtained, were subjected to esterification with hydroxyarenes or alcohols in the presence of N,N, N′,N′-tetramethylchloroformamidinium hexafluorophosphate (TCFH) and N-methylimidazole (NMI) in acetonitrile [39], followed by acidic workup (1 M HCl) to give the corresponding α-TAMEs in high to excellent yields (Scheme 2). We found, however, that the yields of TIPS-esters were lower than the corresponding TBDPS-esters and also the solubilities of TIPS-esters were much lower than that of TBDPS-esters. Thus, we selected TBDPS-esters, α-TAMSE-1 and α-MeO-TAMSE-1, as preferred and reliable synthetic key intermediates to prepare and stock for α -TAMEs synthesis.
2.4. Structure-Activity relationship (SAR) analysis
The binding affinities (Ki values) of newly synthesized TAMEs to FABP5, 3 and 7 were determined based on the fluorescence displacement assay, using 11-(dansylamino)undecanoic acid (DAUDA) or 8-anilino-1-naphthalenesulfonic acid (ANS) as a fluorescent probe and arachidonic acid as a positive control, as reported previously [34]. Results are summarized in Table 1 in the order of affinity to FABP5. The Ki values of L1, L2 and L3 are also listed for comparison.
Table 1.
Affinity (Ki, μM) of TAMEs to FABP5, 3 and 7
|
||||||
|---|---|---|---|---|---|---|
|
| ||||||
| Entry | TAME | X | R | FABP5Ki (μM) | FABP3 Ki (μM) | FABP7 Ki (μM) |
|
| ||||||
| 1 | α-1 | H | 3-(2-oxoindolin-6-yl)phenyl | 0.12 ± 0.02 | 4.56 ± 1.12 | 0.54 ± 0.11 |
| 2 | α-2 | H | 2-(2-oxoindolin-6-yl)phenyl | 0.29 ± 0.01 | 3.40 ± 0.41 | 0.12 ± 0.03 |
| 3 | α-3 | H | 4-cyanobiphenyl-3-yl | 0.32 ± 0.08 | 1.06 ± 0.09 | 0.63 ± 0.09 |
| 4 | α-4 | OMe | 4-cyanobiphenyl-3-yl | 0.33 ± 0.06 | 1.60 ± 0.23 | 0.96 ± 0.12 |
| 5 | γ-3 | H | 4-cyanobiphenyl-3-yl | 0.33 ± 0.02 | 0.72 ± 0.03 | 0.46 ± 0.09 |
| 6 | α-5 | H | 3-(benzo[d][1,3]dioxol-5-yl)phenyl | 0.36 ± 0.05 | 6.00 ± 1.07 | 0.57 ± 0.09 |
| 7 | α-6 | OMe | 3-(2,3-dihydrobenzo[b][1,4]dioxin-5-yl)phenyl | 0.41 ± 0.15 | 4.02 ± 1.04 | 0.91 ± 0.03 |
| 8 | α-7 | H | 5-(3,6-dihydro-2H-pyran-4-yl)-2-cyanophenyl | 0.44 ± 0.05 | 5.47 ± 0.59 | 2.00 ± 0.25 |
| 9* | L2 | OMe | 1-naphthyl | 0.55 ± 0.05 | 0.69 ± 0.17 | 0.67 ± 0.04 |
| 10 | α-8 | H | 3-(3,4-dihydroquinolin-2(1H)one-7-yl)phenyl | 0.59 ± 0.08 | 7.67 ± 0.71 | 0.33 ± 0.01 |
| 11 | α-9 | OMe | 3-(benzo[d][1,3]dioxol-5-yl)phenyl | 0.72 ± 0.08 | 3.37 ± 0.26 | 1.04 ± 0.24 |
| 12 | α-10 | H | 3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)phenyl | 0.74 ± 0.06 | 8.11 ± 1.67 | 0.70 ± 0.03 |
| 13 | α-11 | H | 2-(benzo[d][1,3]dioxol-5-yl)phenyl | 0.79 ± 0.18 | 1.13 ± 0.15 | 0.32 ± 0.13 |
| 14 | γ-12 | OMe | biphenyl-2-ylmethyl | 0.79 ± 0.08 | 25.53 ± 3.25 | 5.52 ± 0.75 |
| 15* | L1 | H | 1-naphthyl | 0.81 ± 0.09 | 2.70 ± 0.42 | 0.45 ± 0.07 |
| 16 | α-12 | OMe | biphenyl-2-ylmethyl | 0.87 ± 0.06 | 23.45 ± 1.99 | 3.34 ± 0.62 |
| 17 | γ-9 | OMe | 3-(benzo[d][1,3]dioxol-5-yl)phenyl | 0.89 ± 0.25 | 8.00 ± 0.58 | 1.86 ± 0.39 |
| 18 | α-13 | OMe | 4′-fluorobiphenyl-3-yl | 1.03 ± 0.11 | 2.28 ± 0.36 | 0.99 ± 0.53 |
| 19 | α-14 | OMe | 3-(2-oxoindolin-6-yl)phenyl | 1.06 ± 0.08 | 3.29 ± 0.45 | 1.55 ± 0.14 |
| 20 | γ-6 | OMe | 3-(2,3-dihydrobenzo[b][1,4]dioxin-5-yl)phenyl | 1.26 ± 0.12 | 6.86 ± 0.07 | 1.78 ± 0.45 |
| 21 | α-15 | H | 6-(pyrimidin-5-yl)naphth-2-yl | 1.30 ± 0.09 | 4.78 ± 0.18 | 6.36 ± 1.04 |
| 22 | α-16 | OMe | indan-2-yl | 1.30 ± 0.28 | 53.79 ± 7.28 | 21.79 ± 4.22 |
| 23 | γ-102 | H | 4′-cyanobiphenyl-2-yl | 1.39 ± 0.07 | 5.78 ± 1.30 | 0.95 ± 0.05 |
| 24 | ε-11 | H | 2-(benzo[d][1,3]dioxol-5-yl)phenyl | 1.45 ± 0.26 | 4.55 ± 0.64 | 0.18 ± 0.01 |
| 25 | α-17 | OMe | 3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)phenyl | 1.56 ± 0.19 | 2.79 ± 0.85 | 2.25 ± 0.13 |
| 26 | α-18 | OMe | 2-(benzo[d][1,3]dioxol-5-yl)phenyl | 1.59 ± 0.11 | 1.28 ± 0.28 | 0.26 ± 0.09 |
| 27 | α-19 | OMe | biphenyl-2-yl | 1.59 ± 0.24 | 0.21 ± 0.09 | 1.36 ± 0.23 |
| 28 | ε-201 | H | biphenyl-2-ylmethyl | 1.65 ± 0.22 | 3.40 ± 0.54 | 0.68 ± 0.16 |
| 29* | L3 | OMe | fluoren-9-ylmethyl | 1.72 ± 0.12 | 25.3 ± 4.57 | 14.26 ± 0.41 |
| 30 | α-20 | OMe | 4′-cyano-biphenyl-3-yl | 1.74 ± 0.08 | 4.64 ± 0.04 | 3.16 ± 0.43 |
| 31 | α-21 | H | 3-(3,6-dihydro-2H-pyran-4-yl)phenyl | 1.75 ± 0.07 | 67.91 ± 8.87 | 0.83 ± 0.01 |
| 32 | γ-13 | OMe | 4′-fluorobiphenyl-3-yl | 1.77 ± 0.02 | 6.10 ± 1.34 | 3.59 ± 0.24 |
| 33 | γ-103 | OMe | 1-naphthyl | 1.82 ± 0.66 | 1.66 ± 0.45 | 1.45 ± 0.52 |
| 34 | α-22 | OMe | 9-fluorofluoren-9-ylmethyl | 1.83 ± 0.27 | 25.95 ± 2.40 | 7.72 ± 1.20 |
| 35 | α-23 | OMe | 1-naphthylmethyl | 2.15 ± 0.17 | 52.58 ± 8.62 | 8.04 ± 0.71 |
| 36 | γ-104 | H | 3-(2-oxoindolin-5-yl)phenyl | 2.21 ± 0.43 | 2.74 ± 0.44 | 4.12 ± 0.32 |
| 37 | γ-20 | OMe | 4′-cyanobiphenyl-3-yl | 2.46 ± 0.19 | 27.85 ± 6.37 | 9.11 ± 1.47 |
| 38 | γ-15 | H | 6-(pyrimidin-5-yl)naphth-2-yl | 2.56 ± 0.17 | 1.30 ± 0.18 | 4.19 ± 0.71 |
| 39 | α-24 | H | 1-naphthylmethyl | 2.64 ± 0.01 | 51.23 ± 5.71 | 10.20 ± 1.42 |
| 40 | α-25 | OMe | 2-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)phenyl | 2.81 ± 0.42 | 2.29 ± 0.56 | 0.67 ± 0.15 |
| 41 | γ-105 | OMe | 4′-cyanobiphenyl-2-yl | 2.81 ± 0.57 | 3.39 ± 0.63 | 1.00 ± 0.17 |
| 42 | ε-202 | H | 1-naphthyl | 3.03 ± 0.18 | 0.32 ± 0.03 | 0.38 ± 0.08 |
| 43 | γ-1 | H | 3-(2-oxoindolin-6-yl)phenyl | 3.11 ± 0.62 | 2.63 ± 0.50 | 3.81 ± 0.06 |
| 44 | ε-203 | H | 3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)phenyl | 3.15 ± 0.28 | 7.84 ± 0.70 | 1.49 ± 0.15 |
| 45 | γ-8 | H | 3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)phenyl | 3.39 ± 0.34 | 8.67 ± 1.31 | 7.99 ± 0.83 |
| 46 | ε-204 | H | 4′-fluorobiphenyl-3-yl | 3.40 ± 0.52 | 15.53 ± 1.91 | 2.19 ± 0.04 |
| 47 | ε-5 | H | 3-(benzo[d][1,3]dioxol-5-yl)phenyl | 3.40 ± 0.56 | 11.17 ± 0.75 | 1.75 ± 0.4 |
| 48 | γ-17 | OMe | 3-(2,3-dihydrobenzo[b][1,4]dioxin-6-yl)phenyl | 3.69 ± 0.65 | 8.18 ± 2.67 | 5.12 ± 0.32 |
| 49 | γ-106 | H | 4′-cyanobiphenyl-3-yl | 3.94 ± 0.43 | 5.15 ± 0.65 | 6.73 ± 0.88 |
| 50 | ε-23 | H | 1-naphthylmethyl | 5.08 ± 0.62 | 7.42 ± 1.38 | 0.86 ± 0.04 |
| 51 | ε-205 | H | biphenyl-3-yl | 5.11 ± 0.84 | 18.60 ± 0.73 | 2.15 ± 0.09 |
| 52 | γ-16 | OMe | indan-2-yl | 6.6 ± 0.83 | 135.5 ± 16.5 | 5.14 ± 0.27 |
| 53 | γ-10 | H | 3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)phenyl | 7.59 ± 1.11 | 4.07 ± 0.24 | 3.49 ± 0.49 |
| 54 | γ-107 | OMe | fluoren-9-ylmethyl | 8.20 ± 1.54 | 27.39 ± 6.35 | 1.63 ± 0.10 |
| 55 | ε-206 | H | indan-2-yl | 8.38 ± 0.72 | 4.75 ± 0.57 | 2.07 ± 0.09 |
| 56 | γ-108 | H | 3-(quinolin-2-on-7-yl)phenyl | 10.90 ± 2.81 | 4.02 ± 0.36 | 13.76 ± 1.30 |
| 57 | α-26 | OMe | 9-hydroxyfluoren-9-ylmethyl | 11.80 ± 1.64 | 54.69 ± 3.43 | 30.98 ± 2.47 |
| 58 | γ-109 | OMe | 4-methoxynaphth-1-yl | 26.61 ± 1.88 | 3.50 ± 0.63 | 0.47 ± 0.08 |
Ki values represent an average ± S.E. of at least three independent experiments.
Ki values from Reference [34].
As predicted by the docking analysis, described above (Fig. 4, Table S2), the introduction of H-bond acceptors to the far end of the 1,1′-biphenyl-3-yl and 1,1′-biphenyl-2-yl ester moieties indeed improved the affinity of α -TAMEs to FABP5, as exemplified by α-1 (Ki 0.12 μM) and α-2 (Ki 0.29 μM) (Table 1, entries 1, 2). 4-Cyano-1,1′-biphenyl-3-yl esters, α-3, α-4 and γ-3, also possess high affinities to FABP5 (Table 1, entries 3–5, Ki 0.32–0.33 μM). It is worthy of note that γ-3 is the first γ-TAME, demonstrating a high affinity to FABP5 and competing with α-TAMEs.
As Table 1 shows, there are 13 TAMEs, which possess better affinities to FABP5 than L1, arising from this study, and L2 is ranked 9th. Since L3, bearing a fluoren-9‑ylmethyl ester moiety, which is not a hydroxyarene, exhibited the best FABP5/3 selectivity with a good Ki value for FABP5 in our last SAR study (Table 1, entry 29) [34], TAMEs with aralkyl esters were designed, selected, synthesized and Ki determined (Table 1, entries 14, 16, 22, 28, 35, 39, 50, 52 and 55).
2.4.1. FABP5-Selective inhibitors
As described above, the design and discovery of highly FABP5-selective TAMEs was another aim of this study. Therefore, we reorganized the results in Table 1 based on the FABP5/3 selectivity index (SI), and identified the best 20 TAMEs, as shown in Table 2. L3 is ranked 7th (FABP5/3 SI: 14.7) in this list with a high FABP5/7 SI of 8.3 and a suboptimal ClogP of 6.26 (Table S2, entry 29). The clear front runner is α-16, bearing a 2‑indanyl (i.e., aralkyl) ester moiety (Table 2, entry 1), which has FABP5/3 SI of 41.3, as well as FABP5/7 SI of 16.7 with ClogP of 4.72 and Ki (FABP5) of 1.30 μM. Although α-21 is ranked 2nd, FABP5/7SI is 0.47, and thus α-1, γ-12 and α-12 (Table 2, entries 3–5), exhibiting FABP5/7 SI of 3.8–7.0 with Ki (FABP5) of 0.12–0.87 μM appear more promising. It should also be noted that γ-12 is ranked 4th, competing with α-TAMEs favorably (Table 2, entry 4), while no ε-TAMEs made the top 20 in this list.
Table 2.
Top 20 TAMEs based on FABP5/3 selectivity index (SI).
| Entry | TAME | FABP5/3 SI | FABP5 KI (μM) | FABP5/7 SI |
|---|---|---|---|---|
|
| ||||
| 1 | α-16 | 41.3 | 1.30 ± 0.28 | 16.7 |
| 2 | α-21 | 36.2 | 1.75 ± 0.07 | 0.47 |
| 3 | α-1 | 38.0 | 0.12 ± 0.02 | 4.5 |
| 4 | γ-12 | 32.3 | 0.79 ± 0.08 | 7.0 |
| 5 | α-12 | 26.9 | 0.87 ± 0.06 | 3.8 |
| 6 | α-5 | 16.7 | 0.36 ± 0.05 | 1.6 |
| 7* | L3 | 14.7 | 1.72 ± 0.12* | 8.3 |
| 8 | α-22 | 14.2 | 1.83 ± 0.27 | 4.2 |
| 9 | α-8 | 13.0 | 0.59 ± 0.08 | 0.56 |
| 10 | α-7 | 12.4 | 0.44 ± 0.05 | 4.5 |
| 11 | α-2 | 11.7 | 0.29 ± 0.01 | 0.41 |
| 12 | α-10 | 11.0 | 0.74 ± 0.06 | 0.95 |
| 13 | α-6 | 9.8 | 0.41 ± 0.15 | 2.2 |
| 14 | γ-9 | 9.0 | 0.89 ± 0.25 | 2.1 |
| 15 | γ-6 | 5.4 | 1.26 ± 0.12 | 1.4 |
| 16 | α-4 | 4.8 | 0.33 ± 0.06 | 2.9 |
| 17 | α-9 | 4.7 | 0.72 ± 0.08 | 1.4 |
| 18 | γ-102 | 4.2 | 1.39 ± 0.07 | 0.64 |
| 19 | α-15 | 3.7 | 1.30 ± 0.09 | 0.68 |
| 20 | γ-13 | 3.4 | 1.77 ± 0.02 | 2.0 |
Ki values represent an average ± S.E. of at least three independent experiments.
Ki values from Reference [34].
2.4.2. FABP3-Selective inhibitors
Since our original focus in this study was to design the FABP5-selective TAMEs by imposing a bias against FABP3, it is logical to think that FABP3-selective TAMEs should have been screened out. Nevertheless, it would be still possible that some TAMEs that were predicted to possess a good affinity to FABP5 and selected for synthesis and affinity assay, might show a high affinity to FABP3. Accordingly, we selected 10 TAMEs, which possess a good affinity to FABP3 (i.e., Ki FABP3 <2 μM), from Table 1, as shown in Table 3. As anticipated, there are 4 TAMEs that exhibit a better affinity to FABP3 than that to FABP5 (Table 3, entries 1, 2, 7, 8 and 10). Rather surprisingly, two TAMEs, α-19 (Table 3, entry 1) and ε-202 (Table 3, entry 2), possess an excellent affinity to FABP3 (Ki FABP3: 0.21 and 0.32 μM, respectively) with good selectivity (FABP3/5 SI: 7.6 and 9.5, respectively), and α-19 also has fairly good selectivity against FABP7 (FABP3/7SI: 6.5). It should be noted that ε-202 is the first ε-TAME that exhibited an excellent Ki value to any of FABP3, 5 and 7, which turned out to be FABP3-selective. In addition, another “non–α” TAME, γ-15 (Table 3, entry 8), shows a good affinity (Ki FABP3 1.30 μM) with modest FABP3/5 and FABP3/7 SI. It is worthy of note that α-19 and ε-202 have been identified as potent FABP3-selective inhibitors, which have applications related to their possible use in the protection of cardiac myocytes from apoptosis after myocardial infarction, as well as the reduction of α-synuclein accumulation in Parkinson’s disease [24–26].
Table 3.
TAMEs as FABP3 inhibitors.
| Entry | TAME | X | R | FABP3 Ki (μM) | FABP3/5 SI | FABP3/7 SI |
|---|---|---|---|---|---|---|
|
| ||||||
| 1 | α19 | OMe | biphenyl-2-yl | 0.21 ± 0.09 | 7.6 | 6.5 |
| 2 | ε-202 | H | 1-naphthyl | 0.32 ± 0.03 | 9.5 | 1.2 |
| 3* | L2 | OMe | 1-naphthyl | 0.69 ± 0.17* | 0.80 | 0.97 |
| 4 | γ-3 | H | 4-cyano-biphenyl-3-yl | 0.72 ± 0.03 | 0.46 | 0.64 |
| 5 | α-3 | H | 4-cyano-biphenyl-3-yl | 1.06 ± 0.09 | 0.30 | 0.59 |
| 6 | α-11 | H | 2-(benzo[d][1,3] dioxol-5-yl)phenyl | 1.13 ± 0.15 | 0.70 | 0.28 |
| 7 | α-18 | OMe | 2-(benzo[d][1,3] dioxol-5-yl)phenyl | 1.28 ± 0.28 | 1.2 | 0.20 |
| 8 | γ-15 | H | 6-(pyrimidin-5-yl)-naphth-2-yl | 1.30 ± 0.18 | 2.0 | 3.2 |
| 9 | α-4 | OMe | 4-cyano-biphenyl-3-yl | 1.60 ± 0.23 | 0.21 | 0.60 |
| 10 | γ-103 | OMe | 1-naphthyl | 1.66 ± 0.45 | 1.1 | 0.87 |
Ki values represent an average ± S.E. of at least three independent experiments.
Ki values from Reference [34].
2.4.3. FABP7-Selective inhibitors
In a manner similar to that for FABP3, we imposed a bias against FABP7 in the initial computational design of TAMEs that were predicted to have high affinities to FABP5, as well as in the subsequent filtering process. In the previously reported SAR study [34], there was a general observation that α -TAMEs possessing a high affinity to FABP5 tend to have a comparable affinity to FABP7. Nevertheless, we reorganized the results in Table 1 in the order of affinity to FABP7 and selected top 20 TAMEs, as shown in Table 4. Then, 13 out of 20 TAMEs in Table 4 were found to be FABP7-selective, which was not anticipated. Furthermore, there are 5 out of 13 TAMEs, exhibiting FABP7/3 SI of >10 (Table 4, entries 1, 2, 5, 11, 16 and 17), with α-21 as the most selective (FABP7/3 SI: 81.8), followed by α-2, ε-11 and α-8 (FABP7/3SI: 28.3, 25.3 and 23.2, respectively). It should be noted that γ-109, a L2 analog, exhibits an unexpectedly high FABP7/5 selectivity (SI: 56.6) with good FABP7/3 selectivity (SI: 7.4) (Table 4, entry 9). It is noteworthy that this study identified several TAMEs as FABP7-selective inhibitors, which would have potentially beneficial therapeutic effects in diseases such as Down’s syndrome, schizophrenia, breast cancer, and astrocytoma [28–31].
Table 4.
Top 20 TAMEs as FABP7 inhibitors.
| Entry | TAME | X | R | FABP7Ki (μM) | FABP7/3 SI | FABP7/5 SI |
|---|---|---|---|---|---|---|
|
| ||||||
| 1 | α-2 | H | 2-(2-oxoindolin-6-yl) phenyl | 0.12 ± 0.03 | 28.3 | 2.4 |
| 2 | ε-11 | H | 2-(benzo[d][1,3] dioxol-5-yl)phenyl | 0.18 ± 0.01 | 25.3 | 8.1 |
| 3 | α-18 | OMe | 2-(benzo[d][1,3] dioxol-5-yl)phenyl | 0.26 ± 0.09 | 4.9 | 6.1 |
| 4 | α-11 | H | 2-(benzo[d][1,3] dioxol-5-yl)phenyl | 0.32 ± 0.13 | 3.5 | 2.5 |
| 5 | α-8 | H | 3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)phenyl | 0.33 ± 0.01 | 23.2 | 1.8 |
| 6 | ε-202 | H | 1-naphthyl | 0.38 ± 0.08 | 0.84 | 8.0 |
| 7* | L1 | H | 1-naphthyl | 0.45 ± 0.07* | 6.0 | 1.8 |
| 8 | γ-3 | H | 4-cyano-biphenyl-3-yl | 0.46 ± 0.09 | 1.6 | 0.72 |
| 9 | γ-109 | OMe | 4-methoxynaphth-1-yl | 0.47 ± 0.08 | 7.4 | 56.6 |
| 10 | α-1 | H | 3-(2-oxoindolin-6-yl) phenyl | 0.54 ± 0.11 | 8.4 | 0.22 |
| 11 | α-5 | H | 3-(benzo[d][1,3] dioxol-5-yl)phenyl | 0.57 ± 0.09 | 10.5 | 0.63 |
| 12 | α-3 | H | 4-cyano-biphenyl-3-yl | 0.63 ± 0.09 | 1.7 | 0.51 |
| 13 | α-25 | OMe | 2-(2,3-dihydrobenzo [b][1,4]dioxin-6-yl) phenyl | 0.67 ± 0.15 | 3.4 | 4.2 |
| 14* | L2 | OMe | 1-naphthyl | 0.67 ± 0.04* | 1.03 | 0.82 |
| 15 | ε-201 | H | biphenyl-2-ylmethyl | 0.68 ± 0.16 | 5.0 | 2.4 |
| 16 | α-8 | H | 3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl)phenyl | 0.70 ± 0.03 | 11.6 | 1.1 |
| 17 | α-21 | H | 3-(3,6-dihydro-2H-pyran-4-yl)phenyl | 0.83 ± 0.01 | 81.8 | 2.1 |
| 18 | ε-206 | H | 1-naphthylmethyl | 0.86 ± 0.04 | 8.6 | 5.9 |
| 19 | α-6 | OMe | 3-(2,3-dihydrobenzo [b][1,4]dioxin-5-yl) phenyl | 0.91 ± 0.03 | 4.4 | 0.45 |
| 20 | γ-102 | H | 4′-cyanobiphenyl-2-yl | 0.95 ± 0.05 | 6.1 | 1.5 |
Ki values represent an average ± S.E. of at least three independent experiments.
Ki values from Reference [34].
2.4.4. L3 derivatives
Although L3 was identified as the most FABP5-selective lead compound arising from the previous SAR study, mainly due to the fluoren-9-yl ester moiety in the molecule, there was a concern about its metabolic stability, especially against hydroxylation by cytochrome P450 oxidases. Apparently, the most likely site for hydroxylation is the benzylic methine at C9 of the fluorenyl moiety. Accordingly, we prepared the most probable metabolite, α-26, bearing a 9-hydroxyfluoren-9‑ylmethyl ester moiety to examine its affinity to FABP5, as well as FABP3 and FABP 7 (Table 1, entry 57). As anticipated, α26 exhibited a substantially reduced affinity to all three FABPs. Consequently, it should be beneficial to block the specific hydroxylation at this specific site of the L3 molecule to prolong and thus enhance its efficacy. Therefore, we strategically introduced fluorine to the fluoren-9‑ylmethyl ester moiety at C9 to block the plausible hydroxylation by cytochrome P450. Thus, α-22, a fluorinated derivative of L3, was synthesized and examined its affinity to FABP5, 3 and 7 (Fig. 6). As Table 1 (entry 34) shows, α-22 maintains a good affinity and selectivity to FABP5, comparable to the parent L3, which clearly demonstrates an excellent mimicking of hydrogen with fluorine, and success in design. For the synthesis of α-22 and α-26, see Supporting Information.
Fig. 6.

Comparison of L3, α-22, and α-26.
2.5. Computational analysis of SAR
The molecular docking analysis of α -TAMEs with FABP5 indicates that the 1,1′-biphenyl ester moiety in those TAMEs binds to FABP5 in a pose similar to that of the 1‑naphthyl ester moiety of L1. However, the flexibility in the dihedral angle of two phenyl rings of the 1,1′-biphenyl system obviously has an advantage over rigid fused aromatic systems to accommodate structural variations, especially on the far end phenyl ring. The H-bonding interactions between the backbone NH of Leu60 and Lys61 in the S3-S4 loop and the H-bond acceptors on the far end of 1,1′-biphenyl-3-yl ester moiety should significantly enhance the binding of α-1, α-5, α-7, α-8, α-9, α-10 and TAMEs bearing similar H-bond acceptors (Figure S2, Supporting Information). In the case of α-1, the NH of the lactam moiety forms an additional H-bonding with the hydroxy group of Ser58 as H-bond donor, which further enhances binding (Fig. 7). One of the α-TAMEs bearing 1,1′-biphenyl-2-yl ester group with an oxoindoline moiety, α-2, binds FABP5 with a pose different from that of α-1, wherein the H-bond donor (lactam NH) interacts with Met35 in the α-helix-1 (Fig. 7).
Fig. 7.

Docking pose of α-1 (purple) and α-2 (pink) at the canonical binding site (Tyr131 and Arg129) of FABP5 with additional H-bonding interactions of the lactam moieties with the protein (Ser58, Leu60 and Met35). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Three TAMEs, α-3, α-4, and γ-3, bearing a 4-cyano-1,1′-biphenyl-3-yl ester moiety, possess a high affinity to FABP5 (Ki: 0.32–0.33 μM), as well as a fairly high to high affinity to FABP3 (Ki: 0.72–1.60 μM) and FABP7 (Ki: 0.46–0.96 μM) (Table 1, entries 3–5). The docking pose analysis indicates that the 4-cyano group extends the ligand surface to increase van der Waals contact near α-helix-1 (Fig. S3a, Supporting Information). It is characteristic of these three TAMEs that the 4-cyano-1‑phenyl moiety and 1′-phenyl moiety are almost orthogonal in the binding pose (Figure S3, Supporting Information). A strong H-bonding interaction of the cyano-nitrogen with Tyr22 appears to explain a clear benefit of introducing a cyano group to the biphenyl ester moiety (Fig. S3b, Supporting Information).
TAME α-16 exhibits strong canonical interactions with Tyr131 and Arg129 in both FABP5 and FABP3, while the ester moiety mainly contributes to the hydrophobic interactions with the neighboring α-helixes Table 4 and S3–S4 loop (Fig. 8a and 8b). By considering its structural simplicity, it is natural to assume that the observed FABP5/3 selectivity (SI: 41.3) of α-16 is attributed to van der Waals clashes in FABP3. In fact, the docking analysis of α-16 in FABP3 indicates that the 2‑indanyl ester moiety causes rather serious clashes with amino acid residues, Thr29, Phe57 and Lys58. The open structure of FABP5′s S3-S4 loop allows for the 2‑indanyl ester to lay flat on top of the α-helix-2 (Fig. 8a), whereas in FABP3 this area is occupied by Phe57, which causes one of the serious clashes mentioned above (Fig. 8b).
Fig. 8.

Docking poses of α-16 at the canonical binding site of (a) FABP5 and (b) FABP3 with protein surface, indicating van der Waals clashes (magenta). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
As described above, α-19 exhibited an unexpectedly high affinity (FABP3 Ki: 0.21) and good selectivity to FABP3 (Table 3, entry 1). Since the binding pocket of FABP3 is considerably more compact than that of FABP5, a rather small and flexible 1,1-biphenyl-2-yl ester moiety of α-19 is favorably accommodated into the binding pocket, while keeping the strong canonical interactions of the α-TA’s carboxylic acid moiety (Fig. 9a). Furthermore, the ether oxygens of two 2-MeO-phenyl groups of the α-MeO-TA moiety act as H-bond acceptors with nearby H-bond doners, Arg126, Arg106 and Thr53, which should enhance the binding (Fig. 9a). In contrast, the 1,1-biphenyl-2-yl ester moiety of α-19 is too small and lacks H-bond acceptors to make a tight binding in FABP5 with a more spacy binding pocket (Fig. 9b). Partly because of the size of the binding pocket of FABP7, which is between those of FABP5 and FABP3, as well as to avoid crash with Phe58 in FABP7, the 1,1′-biphenyl-2-yl ester takes a binding pose in which the far end phenyl group is directed to the spacy area without H-bonding interactions. It should be noted that the binding poses of α-19 are similar in FABP3 and 7, but the ester group is almost 180° flipped in FABP5 (Fig. 9b).
Fig. 9.

(a) FABP3-selective α-19 (pink) docked in FABP3. (b) α-19 docked in FABP5 (light blue protein and blue ligand) and FABP7 (light pink protein and magenta ligand). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
As the SAR study described above clearly indicated, ε-TAMEs are poor binders to FABP5 in general, but some of them are strong binders to FABP3 and 7, e.g., ε-202 (Ki: 0.32 and 0.38 μM, respectively). The binding pose analysis of ε-202 has revealed that all four substituents of the cyclobutane core of this TAME are quasi-equatorial to minimize steric strains, and the carbonyl group of the 1‑naphthyl ester moiety is oriented towards the α -helix-1 to enable water-mediated H-bonding with Tyr19, while keeping the strong canonical interaction of the carboxylic acid moiety with Tyr128 and Arg126 (Fig. S4a, Supporting Information). In contrast to the binding pose in FABP3, ε-202 shows substantial deviation, especially the positions of two phenyl groups, from the binding pose of L1 in FABP7 (Fig. S4b, Supporting Information). Nevertheless, the canonical interactions with Tyr129 and Arg127 are retained, and the 1‑naphthyl group has an excellent overlap with that of L1, although the ester moiety is 180° flipped. This flipping enables the ester carbonyl group water-mediated H-bonding with Lys59 in the S3-S4 loop (Fig. S4b, Supporting Information).
The docking pose analysis of α-2, which exhibited the highest binding affinity to FABP7 (Ki: 0.12 μM) in this study (Table 4, entry 1), has revealed a solid H-bonding between the lactam oxygen of the 2-(2-oxoindolin-6-yl)phenyl ester moiety and Gln96 of FABP7, while keeping the canonical interaction of the carboxylic acid moiety with Tyr129 and Arg127 (Fig. 10a). As discussed above, α-2 possesses a high affinity to FABP5 (Ki: 0.29 μM), but shows high selectivity against FABP3 (Ki: 3.40 μM; FABP7/3 SI: 28.3). The docking pose analysis of α-2 with FABP5 is illustrated in Fig. 7 and that with FABP3 in Fig. 10b. As Fig. 10b shows, the bulky 2-oxoindolin-6-yl group in the ester moiety clashes with the protein surface in the absence of any H-bonding partners, although the canonical interaction of the carboxylic acid moiety with Tyr128 is preserved, which explains the observed high FABP7/3 SI.
Fig. 10.

Docking poses of α-2 and L1 (cocrystal structure for comparison) at the canonical binding site (Tyr129, Arg127) of (a) FABP7, wherein the lactam oxygen of α-2 forms a H-bond with Gln96, and (b) FABP3, wherein the bulky 2-oxoindolin-6-yl group of the ester moiety clashes with Thr30 and Leu104.
As Table 4 shows, five α-TAMES, α-2, α-18, α-11, α-8 and α-1 (entries 1, 3–5 and 10, respectively), are ranked highly as FABP7 inhibitors, bearing H-bond acceptors in the far ends of 1,1′-biphenylyl ester moieties. However, there is a difference between α -TAMEs bearing a 1,1′-biphenyl-2-yl ester moiety (α-2, α-18 and α-11) and those bearing a 1,1′-biphenyl-3-yl ester moiety (α-8 and α-1). Thus, the oxygen (H-bond acceptor) of 2-(2-oxoindolin-6-yl)phenyl and 2-(benzo[d][1,3]dioxol-5-yl)phenyl groups in α-2, α-18 and α-11 forms H-bonding with Arg79 and Gln96, while that of 3-(2-oxo-1,2,3,4-tetrahydroquinolin-7-yl) phenyl and 3-(2-oxoindolin-6-yl)phenyl groups interact with Thr54 and Thr60 through H-bonding (Figure S5, Supporting Information).
Among the top five FABP7-selective TAMEs listed in Table 4, ε-11 (entry 2) is the only ε-TAME, exhibiting a superior affinity (Ki: 0.18 vs 0.32 μM) and selectivity (FABP7/3 SI: 25.3 vs 3.5; FABP7/5 SI: 8.1 vs 2.5) to FABP7 than its α-TAME counterpart, α-11 (entry 4). The docking pose analysis of ε-11 and α-11 in FABP7 has shown that both TAMEs fit into the canonical binding site well, and the ether oxygen of the 2-(benzo [d][1,3]dioxole-5-yl)phenyl ester moiety of ε-11 forms H-bondings with Arg79 and Gln96, while that of α-11 forms H-bonding only with Arg79 (Fig. 11a). In FABP3, however, the benzodioxolylphenyl ester moiety of ε-11 (KiFABP3 = 4.55 μM) is pushed to the hydrophobic site, clashing with Tyr19 and Val25 (Fig. 11b). In contrast, the same ester moiety of α-11 (KiFABP3 = 1.13 μM) keeps H-bonding interactions with Gln95 and Ala75 in spite of a minor clash with Tyr19 (Fig. 11b).
Fig. 11.

(a) Docking poses of ε-11 (magenta) and α-11 (purple) in FABP7, showing the H-bonding interactions between the oxygens of the 2-(benzo[d][1,3]dioxol-5-yl)phenyl ester moiety with Gln96 and Arg79; (b) Docking poses of ε-11 (magenta), clashing with Tyr19, Leu23 and Val23, and α-11 (purple), forming H-bonds with Gln96 and Ala75 in FABP3. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
It should be noted that γ-109 exhibited by far the highest FABP7/5 selectivity (SI: 56.6) (Table 4, entry 9). Thus, this γ-TAME warrants molecular docking pose analysis. The docking pose of γ-109 in FABP7 (Ki: 0.47 μM) shows that it fits into the canonical binding site well and has a very good overlay with the L1 (cocrystal structure for comparison; Ki: 0.45 μM). The 4-MeO group of the naphthyl ester moiety forms attractive van der Waals interactions with Thr30, Val33 and Phe58. Also, it is very likely that the hydroxyl group of Thr30 rotates to form a H-bond with the oxygen of the 4-MeO group (Fig. S6a, Supporting Information). In sharp contrast, γ-109in FABP5 (Ki: 26.61 μM) suffers from the clash between the 4-MeO group and Leu32 and Met35 in the α -helix-2. This clash leads to substantial weakening of the canonical interaction of the carboxylic acid moiety with Arg129 and Tyr131 (Fig. S6b, Supporting Information).
The binding pose analysis of α-21, exhibiting the highest FABP7/3 selectivity (SI: 81.8) in this study, has disclosed a significant van der Waals clash of the 3-(3,6-dihydro-2H-pyran-4-yl)phenyl ester moiety with the protein surface in the absence of any attractive interactions, including H-bonding. Consequently, the carboxylic acid moiety of α-21 is not accommodated to the canonical binding site, which should be the primary reason for the observed weak affinity (Ki: 67.91 μM) to FABP3 (Fig. 12a). In contrast, α-21 is well accommodated to FABP7 (Ki: 0.83 μM) at the canonical binding site with additional H-bonding between the ether oxygen of the 3-(3,6-dihydro-2H-pyran-4-yl)phenyl ester moiety with Gln96 and Arg79 in the S5-S6 loop (Fig. 12b). The affinity of α-21 to FABP5 is fairly good (Ki: 1.75 μM) with the sustained canonical interaction of the carboxylic acid moiety and additional H-bonding of the ester moiety to Leu60 and Lys61 in the S3-S4 loop of FABP5 (Fig. 12b).
Fig. 12.

(a) Docking pose of α-21 (white) in FABP3, showing substantial dislocation of the whole molecule in the binding site, cutting off canonical interactions with Arg126 and Tyr128. (b) Docking pose of α-21 in FABP5 (protein: light blue; ligand: blue) and FABP7 (protein: pink; ligand: magenta). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.6. Efficacy evaluation of FABP5 inhibitors
Of the three FABP subtypes examined, we previously reported that selective inhibition of FABP5 produces antinociceptive effects [4,11,32,34,40,41]. Consequently, inflammatory hyperalgesia provides an ideal platform to assess the bioactivity of FABP5 inhibitors in vivo following acute administration. To date, the antinociceptive profiles of γ-TAMEs have not been examined and therefore a subset of such compounds was included in the analysis. Administration of inhibitors (10–20 mg/kg, i.p.) reduced thermal hyperalgesia, with α-16 and γ-3 exhibiting full reversal of hyperalgesia at the 20 mg/kg dose (Fig. 13). Administration of the remaining compounds at 40 mg/kg resulted in enhanced efficacy, with α-4 and γ-9 completely reversing hyperalgesia. These results confirm the bioavailability of α- and γ-TAMEs. Consequently, FABP3, FABP5, or FABP7 selective inhibitors based on these scaffolds can serve as in vivo active tools to modulate FABP function in diverse disease models.
Fig. 13.

Antinociceptive effects of FABP5 inhibitors. Thermal withdrawal latencies at baseline and after intraplantar injection of complete Freund’s adjuvant in mice receiving FABP5 inhibitor or vehicle. **, p < 0.01; ***, p < 0.001 vs vehicle. #, p < 0.05; ##, p < 0.01; ###, p < 0.001 vs baseline. ns = not significantly different from vehicle (n = 6).
2.7. Assessment of potential off-target interactions in silico
Interactions of the TAME-based FABP inhibitors with potential off-targets were assessed by using the “Swiss Target Prediction” program [42]. Selected TAMEs, i.e., α-1 (FABP5 selective), α-2 (FABP7 selective), γ-3 (non-selective, pan active), α-16 (FABP5 selective) and α-19 (FABP3 selective) were screened against 100 potential targets other than FABPs. Results are summarized in Table S3 and Table S4 in the Supplementary Material. The screening suggested that the most likely off-target for α-1, α-2 and γ-3 would be GPCR44, while that for α-16 and α-19 would be peroxisome proliferator-activated receptor gamma (PPAR-γ). However, the predicted probability for these targets is 0.103–0.122, i.e., very low probability in all cases, which strongly suggests that the TAME-based FABP inhibitors are selective to FABPs and very likely to have negligible off-target effects.
3. Conclusions
The design and discovery of highly potent and FABP5-selective TAMEs was an initial aim of this study. The introduction of H-bond acceptors to the far end of the 1,1′-biphenyl-3-yl and 1,1′-biphenyl-2-yl ester moieties improved the affinity of α -TAMEs to FABP5, as exemplified by α-1 and α-2 (Table 1). 4-Cyano-1,1′-biphenyl-3-yl esters, α-3, α-4 and γ-3, also possess high affinities to FABP5 (Table 1). It is worthy of note that γ-3 is the first γ-TAME to demonstrate a high affinity to FABP5 comparable to α-TAMEs.
In order to better visualize the FABP5-selectivity, we reorganized the results in Table 1 based on the FABP5/3 selectivity index, and identified the 20 best TAMEs (Table 2). The clear front runner is α-16, bearing a 2‑indanyl ester moiety (FABP5/3 SI = 41.3, FABP5/7SI = 16.7, Clog P = 4.72, KiFABP5 = 1.30 μM). Three other TAMEs, α-1, γ-12 and α-12 (FABP5/7 SI = 3.8–7.0, KiFABP5 = 0.12–0.87 μM) are promising as well. TAME γ-12 is ranked 4th, competing with α -TAMEs favorably, but in sharp contrast, no ε-TAMEs made the top 20 in this list.
Since our original focus in this study was to design the FABP5-selective TAMEs by imposing a bias against FABP3, FABP3-selective TAMEs should have been eliminated by in silico screening. Neverthe-less, some TAMEs predicted to possess a good affinity to FABP5 and selected for synthesis and affinity assay, showed a high affinity to FABP3. Indeed, we found that 4 TAMEs, listed in Table 1, exhibited a better affinity to FABP3 than that to FABP5 (Table 3). Rather surprisingly, two TAMEs, α-19 and ε-202 (Table 3), possess an excellent affinity to FABP3 (KiFABP3 = 0.21 and 0.32 μM, respectively) with good selectivity (FABP3/5 SI = 7.6 and 9.5, respectively), and α-19 also has fairly good selectivity against FABP7 (FABP3/7 SI = 6.5). It should be noted that ε-202 is the first ε-TAME that exhibited an excellent Ki value to any of FABP3, 5 and 7, which turned out to be FABP3-selective. It is worthy of note that α-19 and ε-202 have been identified as potent FABP3-selective inhibitors for research and applications related to their possible use in the protection of cardiac myocytes from apoptosis after myocardial infarction, as well as the reduction of α-synuclein accumulation in Parkinson’s disease [24–26].
In our previous SAR study [34], α-TAMEs bearing high affinity to FABP5 tend to have a comparable affinity to FABP7. To our surprise, however, when we reorganized the results in Table 1 to select the best 20 TAMEs based on the affinity to FABP7, 13 out of 20 TAMEs were found to be FABP7-selective. Moreover, 5 out of these 13 TAMEs exhibited > 10 FABP7/3 SI (Table 4), with α-21 as the most selective (FABP7/3 SI = 81.8), followed by α-2, ε-11 and α-8 (FABP7/3 SI = 28.3, 25.3 and 23.2, respectively). Accordingly, this study identified several TAMEs as FABP7-selective inhibitors, which would have potentially beneficial therapeutic effects in diseases such as Down’s syndrome, schizophrenia, breast cancer, and astrocytoma [28–31].
The synthesis of α-TAMEs by conventional methods (Scheme 1) suffered from low to most moderate yields due to the inevitable formation of diesters and difficulty in separating these diesters and unreacted α-TA/ α-MeO-TA from α -TAMEs by column chromatography. Therefore, we successfully introduced the α-TAMSE-mediated protocol to dramatically improve the overall yields of α-TAMEs. α-TAMSEs with TBDPS as the silyl group is isolated in good yields and unreacted α-TA/ α-MeO-TA, as well as disilyl esters, α-TADSEs, are fully recycled. This was a notable improvement in the chemical synthesis of α-TAMEs.
The molecular docking analysis of α-TAMEs with FABP5 has confirmed that the flexibility in the dihedral angle of two phenyl rings of the 1,1′-biphenyl system obviously has an advantage over rigid fused aromatic systems to accommodate structural variations, especially on the far end phenyl ring. The H-bonding interactions between the backbone NH of Leu60 and Lys61 in the S3-S4 loop and the H-bond acceptors on the far end of 1,1′-biphenyl-3-yl ester moiety should significantly enhance the binding of α -TAMEs. The docking pose analysis has also revealed that the 4-cyano group extends the ligand surface to increase van der Waals contact near α-helix-1. A strong H-bonding interaction of the cyano-nitrogen with Tyr22 appears to explain a clear benefit of introducing a cyano group to the biphenyl ester moiety. The high FABP5/3 selectivity (SI: 41.3) of α-16 is attributed to van der Waals clashes in FABP3. The docking analysis of α-16 in FABP3 has indicated that the 2‑indanyl ester moiety causes rather serious clashes with amino acid residues, Thr29, Phe57 and Lys58, while the open structure of the S3-S4 loop in FABP5 allows for the 2‑indanyl ester to lay flat on top of the α-helix-2.
As the SAR study clearly indicated, ε-TAMEs are poor binders to FABP5 in general. However, ε-202 has a very strong affinity to FABP3 and FABP7 (Ki: 0.32 and 0.38 μM, respectively). The binding pose analysis of ε-202 has revealed that the carbonyl group of the 1‑naphthyl ester moiety is oriented towards α-helix-1 to enable water-mediated H-bonding with Tyr19, while keeping the strong canonical interaction of the carboxylic acid moiety with Tyr128 and Arg126. In FABP3, ε-202 shows substantial deviation from the binding pose of L1. Nevertheless, the canonical interactions with Tyr129 and Arg127 are retained, and the carbonyl group of 1‑naphthyl ester moiety forms water-mediated H-bonding with Lys59 in the S3-S4 loop.
As exemplified by the cases described above, the molecular docking analysis has successfully provided rational explanations for the observed binding affinity and selectivity of the FABP3, 5 and 7 inhibitors in this study, including their α, γ and ε isomers. In silico assessment of potential off-target interactions using the Swiss Target Project program [42] revealed that this class of compounds have a very low probability of having off-target effects with 100 major targets.
4. Experimental section
4.1. General methods
The melting points of all new TAMEs were determined on a Thomas Hoover Capillary melting point apparatus and were uncorrected. 1H and 13C NMR spectra were collected on a Bruker Ascend 700 spectrometer operating at 700 MHz for 1H and 175 MHz for 13C, a Bruker 500 Advance spectrometer operating at 500 MHz and 125 MHz for 1H and 13C, respectively, or a Bruker 400 Nanobay spectrometer operating at 400 MHz, 100 MHz, and 376 MHz for 1H, 13C, and 19F, respectively. Chemical shifts were recorded in ppm on the δ scale and referenced to the residual solvents used for 1H and 13C NMRs respectively (1H: CDCl3, δ 7.26; (CD3)2SO, δ 2.50; CD3OD, δ 3.31; CD3CN, δ 1.94; acetone‑d6; δ 2.05; 13C: CDCl3, δ 77.16; DMSO‑d6, δ 39.52; CD3OD, δ 49.00; acetone‑d6 δ 29.84, 206.26). Multiplicity was designated as s = singlet, br = broad, d = doublet, t = triplet, q = quartet, m = multiplet; J-coupling constants in Hz and integrations. The progress of a reaction was monitored by thin-layer chromatography (TLC) using Agela Technologies TLC plates pre-coated with 250 mm thickness silica gel 60 F254 plates and visualized under UV light. Flash chromatography was performed on SiliaFlash® Silica Gel 40–63 mm 60 Å particle size. All air- and moisture-insensitive reactions were carried out under an ambient atmosphere, magnetically stirred, while all air- and moisture-sensitive reactions were carried out using oven-dried glassware under nitrogen. High-resolution mass spectrometry (HRMS) analysis was carried out on an Agilent LC-UV-TOF mass spectrometer at the Institute of Chemical Biology and Drug Discovery, Stony Brook, NY. The purity of synthesized compounds was determined by a Shimadzu LC-2010A HT series HPLC assembly or Agilent 1100 series HPLC assembly. Purities of all new compounds were > 95 %. Conditions used for HPLC analysis: (1) Kinetex evoc18, 2.6 μm, 100 Å, 100 × 2.1 mm column, solvent A: H2O (5 mM ammonium acetate, pH 7), solvent B: CH3CN/H2O, 9:1 (5 mM ammonium acetate, pH 7), flow rate: 0.50 mL/min, t: 0–30 min, gradient: 5–100 % solvent B, 45 °C; (2) Kinetex F5, 2.7 μm, 100 Å, 100 × 2.1 mm column, solvent A: H2O (10 mM ammonium acetate), solvent B: CH3CN/H2O, 95:5 (10 mM ammonium acetate), flow rate: 0.50 mL/min, t: 0–46 min, gradient: 5–95 % solvent B, 45 °C; (3) Agilent Poroshell 120 EC-C18, 2.7 μm, 30 × 50 mm column, solvent A: H2O, solvent B: CH3CN, flow rate: 0.30 mL/min, t: 0–35 min, gradient: 40–95 % solvent B, 25 °C; (4) Kinetex F5, 2.6 μm, 100 Å, 150 × 2.1 mm column, solvent A: H2O (10 mM ammonium acetate), solvent B: CH3CN/H2O, 95:5 (10 mM ammonium acetate), flow rate: 0.50 mL/min, t: 0–46 min, gradient: 5–95 % solvent B, 45 °C; (5) Kinetex F5, 2.6 μm, 100 Å, 100 × 4.6 mm column, solvent A: H2O, solvent B: CH3CN, flow rate: 1.0 mL/min, t: 0–35 min, gradient: 5–95 % solvent B, 25 °C; (6) Kinetex F5, 2.6 μm, 100 Å, 100 × 4.6 mm column, solvent A: H2O, solvent B: CH3OH, flow rate: 1.0 mL/min, t: 0–35 min, gradient: 5–95 % solvent B, 25 °C; (7) Luna 5u Silica, 5.0 μm, 100 Å, 150 × 3.0 mm column, 1 % isopropanol in hexanes (isocratic), flow rate: 1.0 mL/min, t: 0–15 min, 25 °C.
4.2. Materials
Diethyl ether and tetrahydrofuran were distilled from deep purple sodium benzophenone ketyl. Dichloromethane, chloroform, and acetonitrile were dried over CaH2 and distilled. Dichloromethane was degassed via three freeze–pump-thaw cycles before use. All deuterated solvents were purchased from Cambridge Isotope Laboratories. 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium bromide (MTT) was obtained from VWR. 12-N-methyl-(7-nitrobenz-2-oxa-1,3-diazo)aminostearic acid (NBD-stearate)was purchased from Avanti Polar Lipids (Alabaster, AL, USA). 11-[5-(Dimethylamino)-1-naphthalenesulfonylamino]undecanoic acid (DAUDA) was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). 1-Anilinonaphthalene-8-sulfonic acid (ANS) was purchased from Cayman Chemical Company (Ann Arbor, MI, USA). All other chemicals were used as received.
4.3. Interference compound analysis
All 58 compounds in the present work were analyzed for their potential as pan assay interference compounds (PAINS) using the publicly available ZINC15 web tool [43]. None of the compounds were identified to contain PAINS patterns, nor were there any alerts for predicted high risk of aggregation.
4.4. Chemistry
4.4.1. Chemical synthesis and characterization
Note: For the nomenclatures of α-, γ- and ε-truxillic acid, their derivatives and their monoesters, IUPAC nomenclatures are used, but with the sign “α-“, “γ-“ and “ε-” designations, wherein “α-“ stands for (1S*,2S*,3S*,4S*) or (1R*,2R*,3R*,4R*) stereochemistry, i.e., racemic with all four carbon centers bearing the same S or R configuration, “γ-“ stands for (1s*,2R*,3r*,4S*) or (1r*,2S*,3s*,4R*) stereochemistry, i.e., racemic with only C2 carbon center bearing opposite configuration, and “ε-” stands for (1s*,2R*,3r*,4S*) or (1r*,2S*,3s*,4R*), which achiral due to the existence of a plane of symmetry.
4.4.1.1. Synthesis of α-3-(4-cyano-1,1′-biphenyl-3-yloxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (α-3) via acid dichloride route.
To a round-bottomed flask with a reflux condenser and a magnetic stir bar was added α-truxillic acid (α- TA) (164 mg, 0.55 mmol) under nitrogen. Then, thionyl chloride (4 mL) was added along with 3 drops of DMF and the reaction mixture was heated under reflux for 3 h with stirring. Excess thionyl chloride was removed in vacuo using a protecting column packed with sodium hydroxide pellets to give α-truxillic acid dichloride as a yellow solid, which was directly used in the subsequent reaction. To this solid was added dry THF (15 mL), followed by 3-hydroxy-1,1′-biphenyl-4-carbonitriles (61 mg, 0.44 mmol) and pyridine (0.5 mL) under nitrogen. The mixture was stirred for 3 h at room temperature, and then the reaction was quenched with water (12 mL) and the reaction mixture was stirred for an additional 30 min. The reaction mixture was diluted with ethyl acetate (15 mL) and extracted thrice with ethyl acetate (15 mL × 3). The combined organic layers were washed with brine (15 mL), dried over anhydrous magnesium sulfate, and concentrated in vacuo. The resulting crude product was purified by column chromatography on silica gel, using 50 % EtOAc/hexanes as eluant, to afford the title compound mixed with a trace of α-TA. The resulting crude product was further purified by column chromatography on C18-coated silica gel, using acetonitrile/water (gradient from 30 % to 100 %), to afford α-3 (70 mg, 45 % yield) as a white solid: m.p. 196–198 °C; 1H NMR (400 MHz, acetone‑d6) δ 4.20 (ddd, J = 10.8, 7.1, 1.1 Hz, 1H), 4.44 (ddd, J = 10.8, 6.9, 1.1 Hz, 1H), 4.67 (dd, J = 10.7, 6.9 Hz, 1H), 4.75 (dd, J = 10.8, 7.1 Hz, 1H), 6.23 (d, J = 1.7 Hz, 1H), 7.26 – 7.33 (m, 1H), 7.35 – 7.42 (m, 3H), 7.45 – 7.59 (m, 9H), 7.61 – 7.66 (m, 2H), 7.69 (dd, J = 8.1, 1.7 Hz, 1H), 7.85 (d, J = 8.1 Hz, 1H); 13C NMR (176 MHz, acetone‑d6) δ 41.2, 42.00, 45.87, 46.58, 105.44, 114.80, 120.98, 124.88, 127.05, 127.21, 127.56, 127.68, 128.23, 128.35, 128.70, 129.13, 133.53, 137.78, 138.92, 139.08, 146.91, 152.41, 169.82, 171.96; HRMS (ESI-TOF) calcd. for : 491.1965. Found: 491.1976 ; HPLC purity 96.0 % at 210 nm, Condition (5).
In a manner similar to that of α-3, the following α-TAMEs were synthesized and characterized.
4.4.1.2. α-3-(4-Cyano-1,1′-biphenyl-3-yloxycarbonyl)-2,4-di-(2-methoxyphenyl)cyclobutane-1-carboxylic acid (α-4).
White solid; 45 % yield; m. p. 169 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.87 (s, 3H), 3.88 (s, 3H), 4.20 (dd, J = 10.4, 8.3 Hz, 1H), 4.29 (dd, J = 10.5, 6.4, 1H), 4.81 (dd, J = 10.5, 6.4, 1H), 4.92 (dd, J = 10.4, 8.4, 1H), 6.20 (d, J = 1.5 Hz, 1H), 6.99 (m, 2H), 7.08 (m, 2H), 7.27 (t, J = 7.8 Hz, 1H), 7.36 (t, J = 7.8 Hz, 1H), 7.45–7.60 (m, 7H), 7.68 (dd, J = 8.1, 1.5 Hz, 1H), 7.85 (d, J = 8.1 Hz, 1H), 10.55 (bs, 1H); 13C NMR (125 MHz, acetone‑d6) δ 37.5, 38.0, 44.7, 46.3, 55.7, 56.0, 106.4, 111.3, 111.4, 115.7, 121.1, 121.3, 121.8, 125.6, 128.1, 128.2, 128.5, 129.1, 129.6, 129.96, 129.98, 134.4, 138.7, 147.8, 153.5, 158.6, 158.9, 171.2, 173.2; HRMS (ESI-TOF) calcd. For : 532.1766. Found: 532.1757 ; HPLC purity 96.8 % at 210 nm, Condition (1).
4.4.1.3. α-3-(3-Benzo[d][1,3]dioxol-5-ylphenoxycarbonyl-2,4-diphenylcyclobutane-1-carboxylic acid (α-5).
White solid; 24 % yield; m.p. > 230 °C; 1H NMR (500 MHz, DMSO‑d6) δ 3.98 (dd, J = 10.7, 7.5 Hz, 1H), 4.22 (dd, J = 10.8, 6.7 Hz, 1H), 4.46 (dd, J = 10.7, 6.7 Hz, 1H), 4.55 (dd, J = 10.8, 7.5 Hz, 1H), 6.09 (s, 2H), 6.35 (t, J = 1.8 Hz, 1H), 6.44 (dd, J = 8.0, 1.5 Hz, 1H), 6.97 (dd, J = 8.1, 1.7 Hz, 1H), 7.03 (d, J = 8.1 Hz, 1H), 7.07 (d, J = 1.7 Hz, 1H), 7.26–7.33 (m, 2H), 7.33 – 7.49 (m, 7H), 7.52 (d, J = 7.4, 2H), 12.22 (s, 1H); 13C NMR (175 MHz, acetone‑d6) δ 37.5, 38.0, 41.3.41.9, 46.0, 46.6, 101.4, 107.1, 108.4, 119.8, 120.1, 120.5, 123.7, 126.9, 127.3, 127.8, 128.2, 128.2, 128.5, 129.4, 133.9, 139.2, 139.3, 141.8, 147.6, 148.4, 151.2, 170.5, 172.1; HRMS (ESI-TOF) calcd. for : 493.1573. Found: 493.1644 ; HPLC purity 97.4 % at 215 nm, Condition (1).
4.4.1.4. α-3-(3-Naphthalen-1-ylmethoxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutane-1-carboxylic acid (α-23).
White solid; 25 % yield; m.p. 182–184 °C; 1H NMR (400 MHz, acetone‑d6) δ 3.58 (s, 3H), 3.82 (s, 3H), 3.96 – 4.05 (m, 2H), 4.62 (dd, J = 10.6, 7.1 Hz, 1H), 4.62 (dd, J = 10.4, 7.7 Hz, 1H), 5.10 (d, J = 12.5 Hz, 1H), 5.34 (d, J = 12.5 Hz, 1H), 6.80 (d, J = 8.1 Hz, 1H), 6.87 – 6.97 (m, 3H), 7.16 – 7.23 (m, 2H), 7.25 – 7.34 (m, 3H), 7.38 – 7.44 (m, 1H), 7.49 – 7.57 (m, 2H), 7.75 – 7.80 (m, 1H), 7.85 – 7.93 (m, 2H), 10.38 (bs, 1H); 13C NMR (175 MHz, CD3CN) δ 37.81, 37.82, 45.32, 45.91, 55.70, 55.94, 65.04, 111.28, 111.44, 121.11, 121.21, 124.59, 126.30, 126.91, 127.52, 128.10, 128.15, 128.31, 128.40, 128.44, 129.19, 129.20, 129.45, 129.95, 132.40, 132.52, 134.59, 158.35, 158.55, 173.23, 173.73; HRMS (ESI-TOF) calcd. for : 495.1813. Found: 495.1812 ; HPLC purity 93 % at 254 nm, Condition (2).
4.4.1.5. Synthesis of α-3-(3-(2,3-dihydrobenzo[b][1,4]dioxin-5-yl)phenoxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutane-1-carboxylic acid (α-6) via EDC coupling route.
1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDC⋅HCl) (242 mg, 1.26 mmol) and 4-N,N-dimethylaminopyridine (DMAP) (153 mg, 1.26 mmol) were added to a solution of 2,2′-dimethoxy-α-truxillic acid (α-MeO-TA) (410 mg, 1.15 mmol) and 6-(3-hydroxyphenyl)indolin-2-one (210 mg, 0.92 mmol) in anhydrous THF (20 mL) under nitrogen. The reaction mixture was stirred at room temperature for 20 h. To the reaction mixture was added ethyl acetate and water (20 mL each), followed by 5 % NaH2PO4 solution to bring the pH from 9 to 5, and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous magnesium sulfate, and concentrated in vacuo. The resulting crude product was purified by column chromatography on silica gel, using 25 % EtOAc/hexanes as eluant, to afford α-6 (152 mg, 29 % yield) as a white solid: m.p. 199–200 °C; 1H NMR (500 MHz, DMSO‑d6) δ 3.80 (s, 3H), 3.81 (s, 3H), 3.92 (dd, J = 10.5, 8.0 Hz, 1H), 4.13 (dd, J = 10.5, 6.8 Hz, 1H), 4.20–4.28 (m, 4H), 4.58 (dd, J = 10.5, 6.8 Hz, 1H), 4.66 (dd, J = 10.5, 8.0 Hz, 1H), 6.36–6.46 (m, 2H), 6.71 (dd, J = 7.2, 1.9 Hz, 1H), 6.82–6.94 (m, 2H), 6.98 (t, J = 8.4 Hz, 3H), 7.04 (d, J = 8.1 Hz, 1H), 7.27 (dt, J = 9.9, 4.9 Hz, 4H), 7.33 (d, J = 7.1 Hz, 1H), 7.39 (d, J = 7.0 Hz, 1H), 12.00 (s, 1H); 13C NMR (125 MHz, DMSO‑d6) δ 36.2, 36.76, 44.58, 45.18, 55.87, 56.00, 64.27, 64.59, 111.06, 111.14, 117.26, 120.53, 120.64, 120.80, 121.34, 122.46, 122.66, 127.03, 127.37, 127.54, 127.73, 127.84, 128.57, 128.84, 129.19, 129.26, 138.94, 140.88, 144.23, 150.40, 157.70, 157.85, 171.39, 173.52; HRMS (ESI-TOF) calcd. for : 565.1868. Found: 565.1873 ; HPLC purity 98.0 % at 210 nm, Condition (1).
In a manner similar to that of α-6, the following α-TAMEs were synthesized and characterized.
4.4.1.6. α-3-(2-(2-Oxoindolin-6-yl)phenoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (α-2).
White solid; 24 % yield; m.p. > 230 °C; 1H NMR (700 MHz, DMSO‑d6) δ 3.48 (s, 2H), 3.86 (dd, J = 10.6, 6.2 Hz, 1H), 4.01 (dd, J = 10.3, 8.2 Hz, 1H), 4.30–4.24 (m, 1H), 4.37 (dd, J = 10.6, 6.2 Hz, 1H), 5.92 (d, J = 8.0 Hz, 1H), 6.77 (s, 1H), 6.84 (d, J = 7.4 Hz, 1H), 7.31–7.14 (m, 8H), 7.35 (dd, J = 10.3, 7.6 Hz, 2H), 7.48–7.39 (m, 4H), 10.44 (s, 1H), 12.25 (s, 1H); 13C NMR (176 MHz, DMSO‑d6) δ 36.07, 40.78, 41.73, 46.08, 46.27, 109.53, 121.92, 122.59, 124.80, 125.64, 126.94, 127.16, 127.56, 127.62, 128.31, 128.53, 128.88, 130.76, 134.75, 136.63, 139.29, 139.32, 144.32, 147.47, 170.33, 173.42, 176.80; HRMS (ESI-TOF) calcd. for : 502.1660. Found: 502.1682 ; HPLC purity 99.7 % at 210 nm, Condition (5).
4.4.1.7. α-3-(2-Cyano-5-(3,6-dihydro-2H-pyran-4-yl)phenoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (α-7).
White solid; 39 % yield; m.p. 167–168 °C (decomp.); 1H NMR (700 MHz, acetone‑d6) δ 2.32–2.37 (m, 2H), 3.86–3.91 (t, J = 5.2 Hz, 2H), 4.19 (dd, J = 10.7, 7.2 Hz, 1H), 4.31 (dd, J = 5.4, 2.6 Hz, 2H), 4.38–4.43 (dd, J = 10.7, 7.2 Hz, 1H), 4.65 (dd, J = 10.7, 7.2 Hz, 1H), 4.72 (dd, J = 10.7, 7.2 Hz, 1H), 6.03 (d, J = 1.5 Hz, 1H), 6.29 (d, J = 1.5 Hz, 1H), 7.29 (t, J = 7.4 Hz, 1H), 7.39 (dd, J = 10.7, 4.7 Hz, 2H), 7.43 (t, J = 7.4 Hz, 1H), 7.48 (dd, J = 8.2, 1.7 Hz, 1H), 7.51 (dd, J = 7.5, 5.5 Hz, 4H), 7.62 (d, J = 7.5 Hz, 2H), 7.73 (d, J = 8.2 Hz, 1H), 10.80 (s, 1H); 13C NMR (176 MHz, acetone‑d6) δ 26.57, 41.22, 41.95, 45.90, 46.49, 63.59, 65.22, 105.00, 114.86, 118.68, 122.58, 127.03, 127.50, 127.65, 128.16, 128.34, 128.70, 131.89, 133.01, 138.93, 139.06, 146.20, 152.21, 169.77, 171.97; HRMS (ESI-TOF) calcd. for : 478.1660. Found: 478.1667 ; HPLC purity 99.2 % at 210 nm, Condition (5).
4.4.1.8. α-2,4-Bis(2-methoxyphenyl)-3-(2-oxoindolin-6-yl)phenoxycarbonylcyclobutane-1-carboxylic acid (α-9).
White solid; 20 % yield; m.p. 187 °C (decomp.); 1H NMR (500 MHz, acetone‑d6) δ 3.88 (s, 3H), 3.89 (s, 3H), 4.13 (dd, J = 10.4, 8, 1H), 4.25 (dd, J = 10.6, 6.8, 1H), 4.78 (dd, J = 10.5, 6.7, 1H), 4.87 (dd, J = 10.6, 7.9, 1H), 6.08 (s, 2H), 6.44 (dd, J = 1.9, 1.9, 1H), 6.54 (dd, J = 8.0, 1.4, 1H), 6.95–7.10 (m, 7H), 7.25–7.35 (m, 2H), 7.37 (m, 2H), 7.47 (m, 2H), 10.50 (s, 1H); 13C NMR (125 MHz, acetone‑d6) δ 36.3, 36.9, 44.2, 45.2, 54.9, 55.1, 101.4, 107.2, 108.4, 110.4, 110.5, 119.7, 120.1, 120.2, 120.4, 120.5, 123.6, 127.42, 127.44, 127.5, 127.6, 128.1, 128.5, 129.4, 134.0, 141.8, 147.6, 148.4, 151.4, 157.7, 158.0, 170.9, 172.5; HRMS (ESI-TOF) calcd. for : 553.1857. Found: 553.1864 ; HPLC purity 95.7 % at 254 nm, Condition (1).
4.4.1.9. α-3-(3-(2-Oxo-1,2,3,4-tetrahydroquinolin-6-yl)phenoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (α-10).
White solid; 19 % yield; m.p. > 230 °C; 1H NMR (500 MHz, acetone‑d6) δ 2.98 (t, J = 7.5 Hz, 2H).), 3.32 (t, J = 7.5 Hz, 2H), 3.97 (dd, J = 10.8, 7.2 Hz, 1H), 4.22 (dd, J = 10.9, 7.0 Hz, 1H), 4.45 (dd, J = 10.8, 7.0 Hz, 1H), 4.54 (dd, J = 10.9, 7.2 Hz, 1H), 6.35 (t, J = 2.0 Hz, 1H), 6.43 (dd, J = 8.0, 2.3 Hz, 1H), 7.04 (d, J = 8.8 Hz, 1H), 7.29 (dt, J = 19.2, 5.6 Hz, 4H), 7.35 (t, J = 7.4 Hz, 1H), 7.38 (t, J = 7.6 Hz, 2H), 7.41 (d, J = 7.8 Hz, 1H), 7.46 (dd, J = 16.5, 7.9 Hz, 4H), 7.52 (d, J = 7.5 Hz, 2H), 10.2 (s, 1H), 12.23 (s, 1H); 13C NMR (175 MHz, DMSO‑d6) δ 25.3, 30.8, 40.5, 41.1, 46.6, 115.9, 119.4, 120.3, 123.8, 124.6, 125.8, 126.4, 127.2, 127.6, 128.3, 128.6, 128.7, 128.9, 130.1, 132.9, 138.6, 139.58, 139.60, 141.5, 151.1, 170.7, 171.1, 173.1; HRMS (ESI-TOF) calcd. for : 518.1962. Found: 518.1948, ; HPLC purity 99.9 % at 210 nm, Condition (1).
4.4.1.10. α-3-(2-Benzo[d][1,3]dioxol-5-ylphenoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (α-11).
White solid; 28 % yield; m.p. 175–177 °C; 1H NMR (500 MHz, DMSO‑d6) δ 3.90 (dd, J = 10.6, 6.4 Hz, 1H), 4.00 (dd, J = 10.6, 7.7 Hz, 1H), 4.29 (dd, J = 10.6, 7.7 Hz, 1H), 4.39 (dd, J = 10.6, 6.4 Hz, 1H), 5.93 (d, J = 8.0 Hz, 1H), 6.03 (d, J = 5.9 Hz, 2H), 6.78 (dd, J = 8.0, 1.7 Hz, 1H), 6.86 (d, J = 1.7 Hz, 1H), 6.91 (d, J = 8.0 Hz, 1H), 7.14–7.28 (m, 5H), 7.30–7.37 (m, 4H), 7.38–7.46 (m, 4H), 12.29 (bs, 1H); 13C NMR (175 MHz, acetone‑d6) δ 41.1, 41.8, 46.0, 46.3, 101.3, 108.1, 109.0, 122.4, 122.6, 126.1, 126.7, 127.1, 127.3, 127.9, 128.0, 128.2, 128.5, 130.3, 131.3, 134.5, 139.2, 139.3, 147.2, 147.7, 169.9, 172.4; HRMS (ESI-TOF) calcd. for : 493.1646. Found: 493.1661 ; HPLC purity 98.4 % at 210 nm, Condition (1).
4.4.1.11. α-3-(2,3-Dihydro-1H-inden-2-yloxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutane-1-carboxylic acid (α-16).
White solid; 40 % yield; m.p. 152–154 °C; 1H NMR (500 MHz, CD3CN) δ 1.88 (d, J = 17.0 Hz, 1H), 2.77 (d, J = 17.0 Hz, 1H), 2.89 (dd, J = 17.1, 6.1 Hz, 1H), 3.12 (dd, J = 17.1, 6.3 Hz, 1H), 3.52 (s, 3H), 3.78–3.82 (m, 1H), 3.83 (s, 3H), 3.97 (dd, J = 10.4, 8.0 Hz, 1H), 4.51 (dd, J = 10.4, 8.0 Hz, 1H), 4.55 (dd, J = 10.7, 6.7 Hz, 1H), 5.18 (tt, J = 6.1, 2.0 Hz, 1H), 6.82 (d, J = 8.1 Hz, 1H), 6.94–7.02 (m, 3H), 7.12 (d, J = 6.3 Hz, 1H), 7.15–7.23 (m, 3H), 7.24–7.35 (m, 4H), 8.70 (s, 1H); 13C NMR (125 MHz, CD3CN) δ 37.4, 37.5, 40.0, 40.1, 45.0, 45.9, 55.6, 56.0, 76.0, 111.5, 111.6, 121.1, 121.3, 125.4, 126.0, 127.4, 127.5, 128.0, 128.1, 128.4, 128.5, 129.2, 129.3, 141.8, 158.4, 158.6, 173.2, 173.7; HRMS (ESI-TOF) calcd. for : 473.1959. Found: 473.1954 ; HPLC purity at 210 nm 99.1 %, Condition (2).
4.4.1.12. α-3-(3-(3,6-Dihydro-2H-pyran-4-yl)phenoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (α-21).
White solid; 20 % yield; m.p. 195–197 °C; 1H NMR (700 MHz, acetone‑d6) δ 2.36 (s, 4H), 3.85 (t, J = 5.4 Hz, 4H), 4.12 (dd, J = 10.7, 7.3 Hz, 2H), 4.25 (d, J = 2.5 Hz, 4H), 4.29 (dd, J = 10.8, 7.0 Hz, 2H), 4.59 (dd, J = 10.6, 7.1 Hz, 2H), 4.65 (dd, J = 10.7, 7.3 Hz, 2H), 6.12 (s, 2H), 6.33 (s, 2H), 6.46 (d, J = 7.7 Hz, 2H), 7.23 (dt, J = 15.5, 7.8 Hz, 4H), 7.28 (t, J = 7.3 Hz, 2H), 7.35–7.42 (m, 6H), 7.47 (t, J = 7.5 Hz, 4H), 7.50 (d, J = 7.6 Hz, 4H), 7.57 (d, J = 7.6 Hz, 4H), 10.70 (s, 1H); 13C NMR (500 MHz, acetone‑d6) δ 26.8, 41.27, 41.95, 46.12, 46.61, 63.79, 65.22, 117.85, 120.33, 121.69, 123.62, 126.85, 127.23, 127.81, 128.16, 128.22, 128.52, 128.94, 132.90, 139.32, 139.36, 141.55, 150.95, 170.47, 172.25; HRMS (ESI-TOF) calcd. for : 455.1853. Found: 455.1865 ; HPLC purity 96.9 % at 210 nm, Condition (1).
4.4.1.13. α-3-(9-Fluoro-9H-fluoren-9-yl)methoxycarbonyl-2,4-bis(2-methoxyphenyl)-cyclobutane-1-carboxylic acid (α-22).
White solid; 31 % yield; m.p. 93–95 °C; 1H NMR (500 MHz, CDCl3): δ 3.73 (s, 3H), 3.76 (s, 3H), 4.01 (dd, J = 10.4, 7.4 Hz, 1H), 4.15 – 4.07 (m, 2H), 4.24 (dd, J = 21.5, 12.3 Hz, 1H), 4.26 (dd, J = 17.9, 10.4 Hz, 2H), 6.80 (dd, J = 13.0, 8.2 Hz, 2H), 6.89–7.00 (m, 2H), 7.19–7.28 (m, 6H), 7.36–7.45 (m, 3H), 7.53 (d, J = 7.5 Hz, 1H), 7.59 (d, J = 6.8 Hz, 2H); 13C NMR (125 MHz, CDCl3): δ 177.75, 172.14, 157.46, 157.36, 141.31 (d, 2JCF = 16.6 Hz), 141.16 (d, 2JCF = 16.2 Hz), 140.27, 130.61, 128.24, 128.13, 127.68, 127.43, 126.96, 126.83, 125.45, 125.11, 120.48, 120.34, 120.24, 110.14, (d, 2JCF = 16.8 Hz), 97.79 (d, 1JCF = 186.0 Hz), 55.15, 55.12, 44.78, 44.51, 37.64, 37.28; 19F NMR (376 MHz, CDCl3): δ −166.6; HRMS (ESI-TOF) calcd. for : 570.2304. Found: 570.2286. ; HPLC purity 98.0 % at 210 nm, Condition (5).
4.4.1.14. α-3-(Naphthalen-1-ylmethoxycarbonyl-2,4-diphenylcyclobutane-1-carboxylic acid (α −24).
White solid; 78 % yield; m.p. 174–176 °C; 1H NMR (500 MHz, acetone‑d6) δ 4.01–4.07 (m, 2H), 4.43–4.53 (m, 2H), 5.10 (d, J = 12.4 Hz, 1H), 5.36 (d, J = 12.4 Hz, 1H), 7.21–7.35 (m, 7H), 7.37 (d, J = 7.1 Hz, 2H), 7.42 (t, J = 8.0 Hz, 3H), 7.51–7.58 (m, 2H), 7.67–7.74 (m, 1H), 7.90 (d, J = 8.3 Hz, 1H), 7.92–7.98 (m, 1H), 10.61 (s, 1H); 13C NMR (500 MHz, acetone‑d6) δ 41.53, 41.73, 46.29, 46.75, 64.26, 123.66, 125.23, 125.83, 126.48, 126.82, 126.95, 127.56, 127.59, 127.80, 128.20, 128.25, 128.48, 129.06, 131.43, 131.69, 133.77, 139.28, 139.35, 171.6, 172.03; HRMS (ESI-TOF) calcd. for 454.2013. Found = 454.2009 ; HPLC purity: 99.8 % at 210 nm, Condition (1).
4.4.1.15. Synthesis of α-3-tert-butyldiphenylsiloxycarbonyl-2,4-diphenylcyclobutane-1-carboxylic acid (α-TAMSE-1).
N,N-Diisopropylethylamine (1.94 mL, 11.1 mmol) was added to a solution of α-truxillic acid (3.00 g, 10.1 mmol) in anhydrous THF (67 mL), followed by drop-wise addition of tert-butylchlorodiphenylsilane (1.33 mL, 5.06 mmol) under nitrogen. The mixture was stirred at room temperature for 2 h. The reaction mixture was concentrated in vacuo to afford a beige crude product. The crude product was purified by flash chromatography on silica gel, using methanol/dichloromethane (gradient from 0 % to 2 %), to give α-TAMSE-1 (1.911 g, 71 % yield) as a white solid: m.p. 65–67 °C (decomp.); 1H NMR (500 MHz, acetone‑d6) δ 0.94 (s, 9H), 4.00 (dd, J = 10.6, 6.1 Hz, 1H), 4.32 (dd, J = 10.6, 8.1 Hz, 1H), 4.51 (dd, J = 10.7, 6.1 Hz, 1H), 4.58 (dd, J = 10.4, 8.3 Hz, 1H), 7.25 (t, J = 7.3 Hz, 1H), 7.27 – 7.47 (m, 17H), 7.53 (d, J = 7.0 Hz, 2H), 10.66 (s, 1H); 13C NMR (176 MHz, acetone‑d6) δ 18.43, 26.13, 42.01, 46.55, 47.90, 126.75, 127.15, 127.48, 127.56, 127.60, 128.03, 128.17, 128.62, 129.72, 129.82, 131.57, 131.72, 135.01, 135.02, 139.38, 139.58, 170.77, 172.43; HRMS (ESI-TOF) calcd. for : 552.2565. Found: 552.2560 ; HPLC purity: 96.0 % at 210 nm, Condition (5).
Bis(tert-butyldiphenylsilyl) α-2,4-diphenylcyclobutane-1,3-dicarboxylate (α-TADSE-1) (468 mg, 0.64 mmol, 25 % yield) was recovered at the beginning of the chromatography using dichloromethane as eluent, while α-TA (1.649 g, 5.57 mmol, accounting for 55 % of the α-TA used) was recovered at the end of the chromatography using 10 % methanol/dichloromethane as eluent.
4.4.1.16. Bis(tert-butyldiphenylsilyl) α-2,4-diphenylcyclobutane-1,3-dicarboxylate (α-TADSE-1).
White solid; m.p. 145–147 °C; 1H NMR (700 MHz, acetone‑d6) δ 0.96 (s, 18H), 4.32 (dd, J = 10.2, 7.6 Hz, 2H), 4.63 (dd, J = 10.2, 7.6 Hz, 2H), 7.45–7.31 (m, 26H), 7.52 (d, J = 7.3 Hz, 4H); 13C NMR (176 MHz, acetone‑d6) δ 18.52, 26.21, 42.39, 48.11, 127.23, 127.55, 127.63, 127.91, 128.66, 129.80, 129.90, 131.53, 131.71, 135.04, 135.08, 139.24, 170.80; HRMS (ESI-TOF) calcd. for : 790.3742. Found: 790.3775 ; HPLC purity: 99.6 % at 210 nm, Condition (7).
In a manner similar to that of α-TAMSE-1, the following α-TAMSEs were synthesized and characterized.
4.4.1.17. α-3-tert-Butyldiphenylsiloxycarbonyl-2,4-bis(2-methoxyphenyl) cyclobutane-1-carboxylic acid (α-2-MeO-TAMSE-1).
White solid; 73 % yield; m.p. 57–60 °C; 1H NMR (500 MHz, acetone‑d6) δ 0.93 (s, 9H), 3.77 (s, 3H), 3.85 (s, 3H), 4.06 (t, J = 8.7 Hz, 1H), 4.14 (t, J = 8.9 Hz, 1H), 4.69–4.81 (m, 2H), 6.94–7.03 (m, 4H), 7.25 (t, J = 7.4 Hz, 1H), 7.33 (q, J = 7.3 Hz, 5H), 7.37 (d, J = 6.8 Hz, 2H), 7.38–7.51 (m, 6H), 10.50 (s, 1H); 13C NMR (176 MHz, acetone‑d6) δ 18.44, 26.15, 36.77, 37.08, 44.72, 47.13, 54.82, 54.92, 110.36, 110.47, 120.15, 120.44, 127.09, 127.51, 127.52, 127.70, 127.83, 127.95, 128.25, 129.69, 129.74, 131.83, 131.86, 134.99, 135.00, 157.64, 171.29, 172.87; HRMS (ESI-TOF) calcd. for : 595.2510. Found: 595.2523 ; HPLC purity 99.2 % at 210 nm, Condition (5).
Bis(tert-butyldiphenylsilyl) α-2,4-bis(2-methoxyphenyl)cyclo-butane-1,3-dicarboxylate (α-2-MeO-TADSE-1) (211 mg, 0.25 mmol, 18 % yield) was recovered at the beginning of the chromatography using dichloromethane as eluent, while α-TA (894 mg, 3.02 mmol, accounting for 53 % of the α-TA used) was recovered at the end of the chromatography using 10 % methanol/dichloromethane as eluent.
4.4.1.18. Bis(tert-butyldiphenylsilyl) α-2,4-bis(2-methoxyphenyl)cyclobutane-1,3-dicarboxylate (α-2-MeO-TADSE-1).
White solid; m.p. 197–198 °C; 1H NMR (500 MHz, CDCl3) δ 0.95 (s, 18H), 3.68 (s, 6H), 4.18 (dd, J = 7.6, 10.3 Hz, 2H), 4.82 (dd, J = 7.6, 10.3 Hz, 2H), 6.80 (d, J = 8.2 Hz, 2H), 6.92–6.97 (m, 2H), 7.24 (d, J = 1.5 Hz, 1H), 7.27–7.29 (m, 10H), 7.33–7.395 (m, 13H); 13C NMR (126 MHz, CDCl3) δ 18.94, 26.73, 37.32, 46.95, 55.14, 110.20, 120.52, 127.47, 127.50, 127.58, 127.69, 128.08, 129.68, 129.71, 131.77, 131.82, 135.28, 135.30, 157.50, 171.78; HRMS (ESI-TOF) calcd. for : 833.3688. Found: 833.3683 ; HPLC purity: 95.0 % at 210 nm, Condition (7).
4.4.1.19. α-2,4-Diphenyl-3-triisopropylsiloxycarbonylcyclobutane-1-carboxylic acid (α-TAMSE-2).
White solid; 34 % yield; m.p. 98–100 °C; 1H NMR (500 MHz, acetone‑d6) δ 0.86–0.93 (m, 18H), 1.03–1.12 (m, 3H), 3.96 (dd, J = 10.6, 6.4 Hz, 1H), 4.12 (dd, J = 10.6, 7.9 Hz, 1H), 4.41 (dd, J = 10.5, 6.5 Hz, 1H), 4.49–4.56 (m, 1H). 7.22–7.29 (m, 2H), 7.35 (d, J = 7.5 Hz, 4H), 7.41 (d, J = 7.6 Hz, 2H), 7.46 (d, J = 7.6 Hz, 2H), 10.64 (s, 1H); 13C NMR (175 MHz, acetone‑d6) δ 11.6, 17.10, 41.84, 42.13, 46.49, 47.92, 126.73, 126.84, 127.55, 127.80, 128.17, 128.43, 139.54, 139.72, 171.43, 172.41; HRMS (ESI-TOF) calcd. for : 453.2456. Found: 453.2459 ; HPLC purity: 98.8 % at 210 nm, Condition (5).
4.4.1.20. Bis(triisopropylsilyl) α-2,4-diphenyl-yclobutane)-1,3-dicarboxylate (α-TADSE-2).
White solid; m.p. 68–69 °C; 1H NMR (700 MHz, CDCl3) δ 0.85–0.92 (m, 36H), 1.08 (sept, J = 7.5 Hz, 6H), 4.04 (dd, J = 7.5, 10.2 Hz, 2H), 4.44 (dd, J = 7.5, 10.2 Hz, 2H), 7.24 (t, J = 7.0 Hz, 2H), 7.31–7.38 (m, 8H); 13C NMR (176 MHz, CDCl3) δ 11.79, 17.57, 17.64, 42.49, 48.23, 126.99, 127.55, 128.59, 139.28, 172.05; HRMS (ESI-TOF) calcd. for : 609.3790. Found: 609.3801 ; HPLC purity: 99.9 % at 210 nm, Condition (7).
4.4.1.21. α-2,4-Bis(2-methoxyphenyl)-3-triisopropylsiloxycarbonyl)cyclobutane-1-carboxylic acid (α-2-MeO-TAMSE-2).
White solid; 198 mg, 69 % yield; m.p. 128–130 °C; 1H NMR (500 MHz, acetone‑d6) δ 0.92 (d, J = 5.7 Hz, 18H), 1.05–1.11 (m, 3H), 3.85 (s, 1H), 3.85 (s, 1H), 4.02–3.94 (m, 2H), 4.60–4.74 (m, 2H), 6.96 (t, J = 7.8 Hz, 4H), 7.23 (t, J = 7.8 Hz, 2H), 7.37 (t, J = 7.6 Hz, 2H); 13C NMR (126 MHz, acetone‑d6) δ 11.69, 17.16, 36.53, 37.14, 44.85, 47.05, 54.79, 54.88, 110.26, 110.33, 120.10, 120.27, 127.00, 127.09, 127.22, 127.32, 127.79, 127.84, 127.90, 127.94, 128.07, 157.56, 157.65, 172.08, 173.03; HRMS (ESI-TOF) calcd. for : 513.2667. Found: 513.2662 ; HPLC purity 99.8 % at 210 nm, Condition (5).
4.4.1.22. Bis(triisopropylsilyl) α-2,4-bis(2-methoxyphenyl)cyclobutane-1,3-dicarboxylate (α-2-MeO-TADSE-2).
White solid; m.p. 104–105; 1H NMR (700 MHz, CDCl3) δ 0.94–0.86 (m, 36H), 1.08 (sept, J = 7.5 Hz, 6H), 3.81 (s, 6H), 4.02 (dd, J = 7.7, 10.2 Hz, 2H), 4.71 (dd, J = 7.7, 10.2 Hz, 2H), 6.82 (d, J = 8.1 Hz, 2H), 6.95 (t, J = 7.3 Hz, 2H), 7.17–7.22 (m, 2H), 7.36 (d, J = 7.1 Hz, 2H); 13C NMR (176 MHz, CDCl3) δ 11.83, 17.55, 17.63, 37.10, 46.98, 55.16, 110.04, 120.42, 127.31, 127.80, 128.08, 157.42, 172.76; HRMS (ESI-TOF) calcd. for : 669.4001. Found: 669.4023 ; HPLC purity: 98.9 % at 210 nm, Condition (7).
4.4.1.23. Synthesis of α-SB-FI-103 (L3) from α-2-MeO-TAMSE-1.
To a round-bottomed flask with a magnetic stir bar was added α-2-MeO-TAMSE-1 (2.081 g, 3.5 mmol), N,N,N′,N′-tetramethyl-chloroformamidinium hexafluorophosphate (1.051 g, 3.85 mmol) under nitrogen. Then, N-methylimidazole (837 uL, 10.5 mmol) was added, followed by 9-fluorenemethanol (1.051 g, 5.25 mmol) along with anhydrous acetonitrile (10.5 mL). The reaction mixture was stirred for 16 h at room temperature, and the reaction mixture was diluted with water and ethyl acetate (20 mL each). The organic layer was separated and washed with water (20 mL × 2), 1 M HCl (20 mL × 2), and brine (20 mL). The resulting solution was dried over anhydrous magnesium sulfate, filtered and concentrated in vacuo to give a crude product. The crude product was purified by column chromatography on silica gel first, using EtOAc/hexanes (gradient from 10 % to 50 %), followed by column chromatography on C18-coated silica gel, using acetonitrile/water (gradient from 30 % to 100 %) to afford α-SB-FI-103 (L3) (1.727 g, 92 % yield) as a white solid: m.p. 160–162 °C (lit. 160–162 °C [34]); 1H NMR (400 MHz, acetone‑d6) δ 3.83 (s, 3H), 3.84 (m, 1H), 3.87 (s, 3H), 4.01 (m, 4H), 4.64–4.74 (m, 2H), 6.96 (dd, J = 14.0, 8.5 Hz, 4H), 7.21–7.37 (m, 6H), 7.41 (t, J = 7.4 Hz, 2H), 7.48 (d, J = 7.2 Hz, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.84 (d, J = 7.4 Hz, 2H), 10.38 (s, 1H); HRMS (ESI-TOF) calcd. for : 535.2115. Found: 535.2115 ; These data were consistent with the literature values.[34] HPLC purity: 98.9 % at 254 nm, Condition (5).
In a manner similar to that of L3, the following α-TAMEs were synthesized and characterized.
4.4.1.24. α-3-(3-(2-Oxoindolin-6-yl)phenoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (α-1).
Off-white solid; 64 % yield; m.p. > 230 °C; 1H NMR (500 MHz, DMSO‑d6) δ 3.53 (s, 2H), 3.98 (dd, J = 10.9, 7.5 Hz, 1H), 4.23 (dd, J = 10.9, 6.8 Hz, 1H), 4.46 (dd, J = 10.7, 6.8 Hz, 1H), 4.55 (dd, J = 10.7, 7.5 Hz, 1H), 6.22 (t, J = 2.04 Hz, 1H), 6.54 (t, J = 4.75 Hz, 1H), 6.83 (t, J = 1.57 Hz, 1H), 7.04 (dd, J = 7.6, 1.7 Hz, 1H), 7.27–7.52 (m, 13H), 10.60 (s, 1H), 12.20 (bs, 1H); 13C NMR (125 MHz, DMSO‑d6) δ 35.7, 40.6, 41.4, 45.6, 46.1, 107.2, 119.5, 119.8, 123.9, 124.7, 125.6, 126.8, 127.3, 127.8, 128.0, 128.2, 128.5, 129.7, 130.2, 138.4, 138.99, 139.0, 141.6, 144.4, 150.5, 170.6, 172.6, 176.6; HRMS (ESI-TOF) calcd. for : 504.1805. Found: 504.1816 ; HPLC purity 97.6 % at 210 nm, Condition (5).
4.4.1.25. α-3-(3-(2-Oxo-1,2,3,4-tetrahydroquinolin-6-yl)phenox-ycarbonyl)-2,4-diphenyl-cyclobutane-1-carboxylic acid (α-8).
White solid; 72 % yield; m.p. 230 °C (decomp.); 1H NMR (500 MHz, DMSO‑d6): δ 2.48 (t, J = 7.5 Hz, 2H), 2.92 (t, J = 7.5 Hz, 2H), 3.98 (dd, J = 7.6, 10.6 Hz, 1H), 4.23 (dd, J = 6.8, 10.8 Hz, 1H), 4.46 (dd, J = 7.0, 10.6 Hz, 1H), 4.54 (dd, J = 7.7, 10.5 Hz, 1H), 6.36 (m, 1H), 6.45–6.47 (m, 1H), 6.98 (s, 1H), 7.00 (d, J = 7.9 Hz, 1H), 7.28 (t, J = 7.0 Hz, 2H), 7.32–7.34 (m, 3H), 7.38 (t, J = 7.5 Hz, 2H), 7.44–7.48 (m, 4H), 7.51 (d, J = 7.5 Hz, 2H), 10.18 (s, 1H), 12.22 (s, 1H); 13C NMR (500 MHz, DMSO‑d6): δ 25.0, 30.4, 40.7, 41.4, 45.7, 46.2, 113.1, 119.9, 120.8, 120.9, 123.8, 124.2, 127.3, 127.7, 128.3, 128.5, 128.7, 128.8, 129.0, 130.2, 138.5, 139.4, 139.5, 139.5, 141.8, 150.5, 170.3, 170.6, 172.6; HRMS (ESI-TOF) calcd. for : 518.1962. Found: 518.1958 ; HPLC purity 95.4 % at 210 nm, Condition (1).
4.4.1.26. α-3-(1,1′-Biphenyl-2-ylmethoxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutene-1-carboxylic acid (α −12).
White solid; 98 % yield; m.p. 73–74 °C; 1H NMR (700 MHz, acetone‑d6) δ 3.70 (s, 3H), 3.84 (s, 3H), 3.99 (dd, J = 10.4, 7.4 Hz, 1H), 4.02 (dd, J = 10.4, 7.4 Hz, 1H), 4.61–4.66 (m, 3H), 4.75 (d, J = 12.5 Hz, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.91–6.99 (m, 3H), 7.10 (d, J = 7.6 Hz, 1H), 7.21–7.29 (m, 5H), 7.29–7.41 (m, 7H); 13C NMR (176 MHz, acetone‑d6) δ 36.70, 36.80, 44.59, 44.95, 54.69, 54.84, 63.57, 110.22, 110.30, 120.12, 120.16, 127.13, 127.22, 127.29, 127.47, 127.65, 127.93, 128.00, 128.02, 128.19, 129.05, 129.66, 129.68, 133.27, 140.37, 141.94, 157.51, 157.68, 171.73, 172.72; HRMS (ESI-TOF) calcd. for : 523.2115. Found: 523.2110 ; HPLC purity 97.0 % at 210 nm, Condition (5).
4.4.1.27. α-3-(4′-Fluoro-1,1′-biphenyl-3-yloxycarbonyl-2,4-bis(2-methoxyphenyl)-cyclobutane-1-carboxylic acid (α-13).
White solid; 92 % yield; m.p. 191–193 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.88 (s, 3H), 3.89 (s, 3H), 4.13 (dd, J = 10.4, 8.0 Hz, 1H), 4.26 (dd, J = 10.7, 6.7 Hz, 1H), 4.78 (dd, J = 10.6, 6.8 Hz, 1H), 4.88 (dd, J = 10.4, 8.0 Hz, 1H), 6.48 (s, 1H), 6.59 (d, J = 8.0 Hz, 1H), 6.99 (dd, J = 12.3, 7.8 Hz, 2H), 7.04–7.12 (m, 2H), 7.27 (dd, J = 10.22, 7.2 Hz, 3H), 7.35 (t, J = 7.9 Hz, 1H), 7.38 (t, J = 7.7 Hz, 1H), 7.43 (d, J = 7.6 Hz, 1H), 7.44–7.50 (m, 2H), 7.58 (dd, J = 8.5, 5.4 Hz, 2H), 10.50 (s, 1H); 13C NMR (125 MHz, acetone‑d6) δ 36.0, 36.73, 44.48, 45.27, 55.87, 56.06, 111.12, 111.16, 116.26 (d, 2JCF = 21.5 Hz), 119.93, 120.66, 120.86, 120.91, 124.34, 127.41, 127.53, 127.85, 128.59, 129.01, 129.15 (d, 3JCF = 8.3 Hz), 130.31, 135.74, 135.76, 140.83, 151.22, 157.72, 157.97, 162.56 (d, 1JCF = 244.9 Hz), 171.43, 173.48; HRMS (ESI-TOF) calcd. for : 527.1873. Found: 527.1864 ; HPLC purity 98.1 % at 210 nm, Condition (2).
4.4.1.28. α-2,4-Bis(2-methoxyphenyl)-3-(3-(2-oxoindolin-6-yl)phenoxycarbonyl)cyclobutane-1-carboxylic acid (α-14).
White solid; 57 % yield; m.p. 211–212 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.52 (s, 2H), 3.88 (s, 3H), 3.89 (s, 3H), 4.12 (dd, J = 10.7, 7.8 Hz, 1H), 4.26 (dd, J = 10.9, 6.7 Hz, 1H), 4.79 (dd, J = 10.4, 6.7 Hz, 1H), 4.87 (dd, J = 10.6, 7.5 Hz 1H), 6.47 (t, J = 1.9 Hz, 1H), 6.60 (d, J = 9.22 Hz, 1H), 7.00 (dd, J = 18.3, 8.5 Hz, 3H), 7.10 (td, J = 14.3, 7.4 Hz, 3H), 7.27 (t, J = 6.97 Hz, 1H), 7.31–7.42 (m, 4H), 7.45 (d, J = 7.5 Hz, 2H), 9.50 (s, 1H); 13C NMR (176 MHz, acetone‑d6): δ 35.2, 36.35, 36.94, 44.33, 45.23, 54.89, 55.09, 107.66, 110.42, 110.48, 120.01, 120.20, 120.28, 120.46, 120.54, 123.87, 124.81, 124.83, 125.45, 125.49, 127.44, 127.55, 128.04, 128.52, 129.48, 139.61, 142.30, 144.45, 151.38, 157.75, 158.01, 170.95, 172.79, 175.98; HRMS (ESI-TOF) calcd. for : 564.2021. Found: 564.2017 ; HPLC purity 96.7 % at 215 nm, Condition (1).
4.4.1.29. α-2,4-Diphenyl-3-(6-(pyrimidin-5-yl)naphthalen-2-yloxycarbonyl)cyclobutane-1-carboxylic acid (α-15).
White solid; 79 % yield; m.p. 215 °C (decomp.); 1H NMR (500 MHz, acetone‑d6) δ 4.17 (dd, J = 10.6, 7.3 Hz, 1H), 4.38 (dd, J = 10.7, 7.1, 1H), 4.64 (dd, J = 10.6, 7.1, 1H), 6.69 (dd, J = 8.8, 2.0 Hz, 1H), 4.70 (dd, J = 10.6, 7.3, 1H), 6.96 (s, 1H), 7.29 (t, J = 7.3 Hz, 1H), 7.39 (t, J = 7.6 Hz, 2H), 7.45–7.55 (m, 5H), 7.62 (d, J = 7.3 Hz, 2H), 7.91 (m, 3H), 8.31 (s, 1H), 9.18 (s, 1H), 9.19 (s, 2H), 10.86 (bs, 1H); 13C NMR (125 MHz, DMSO‑d6) δ 40.4, 41.17, 41.98, 46.22, 46.72, 118.64, 122.39, 125.68, 126.54, 127.33, 127.68, 128.28, 128.56, 128.68, 128.83, 129.02, 130.06, 131.46, 131.47, 133.32, 133.37, 139.55, 148.88, 155.40, 157.81, 171.19, 173.20; HRMS (ESI-TOF) calcd. for : 501.1809. Found: 501.1804 ; HPLC purity 97.5 % at 210 nm; Condition (5).
4.4.1.30. α-3-(3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)phenox-ycarbonyl)-2,4-Di-(2-methoxyphenyl)cyclobutane-1-carboxylic acid (α-17).
White solid; 73 % yield; m.p. 169–171 °C; 1H NMR (700 MHz, acetone‑d6) δ 3.88 (s, 3H), 3.89 (s, 3H), 4.13 (dd, J = 10.5, 8.0 Hz, 1H), 4.25 (dd, J = 10.5, 6.8 Hz, 1H), 4.34 (q, J = 5.0 Hz, 4H), 4.78 (dd, J = 10.5, 6.8 Hz, 1H), 4.87 (dd, J = 10.6, 8.0 Hz, 1H), 6.42 (t, J = 1.8 Hz, 1H), 6.54 (dd, J = 8.0, 1.4 Hz, 1H), 6.91–6.95 (m, 1H), 6.97–7.03 (m, 4H), 7.08 (m, 2H), 7.27 (m, 1H), 7.29 (dd, J = 8.0 Hz, 1H), 7.36–7.42 (m, 2H), 7.47 (d, J = 7.4 Hz, 2H), 10.54 (bs, 1H); 13C NMR (175 MHz, acetone‑d6) δ 37.7, 38.3, 45.6, 46.6, 56.3, 56.5, 65.8, 65.8, 111.8, 111.9, 116.9, 118.9, 121.0, 121.1, 121.4, 121.6, 121.8, 124.7, 128.84, 128.88, 128.89, 128.97, 129.5, 130.0, 130.8, 134.3, 143.0, 145.2, 145.4, 152.8, 159.2, 159.4, 172.4, 173.9; HRMS (ESI-TOF) calcd. for : 567.2013. Found: 567.2023 ; HPLC purity 97.2 % at 210 nm, Condition (2).
4.4.1.31. α-3-(2-(Benzo[d][1,3]dioxol-5-yl)phenoxycarbonyl)-2,4-bis(2-methoxyphenyl)-cyclobutane-1-carboxylic acid (α-18).
White solid; 78 % yield; m.p. 139–141 °C; 1H NMR (700 MHz, acetone‑d6) δ 3.83 (s, 3H), 3.88 (s, 3H), 4.01 (dd, J = 10.4, 6.8 Hz, 1H), 4.09 (dd, J = 10.4, 7.9 Hz, 1H), 4.56 (dd, J = 10.2, 8.0 Hz, 1H), 4.72 (dd, J = 10.5, 6.8 Hz, 1H), 5.98–6.03 (m, 2H), 6.03–6.07 (m, 1H), 6.82 (dt, J = 11.5, 4.9 Hz, 3H), 6.90–6.99 (m, 2H), 7.04 (t, J = 7.4 Hz, 1H), 7.08 (d, J = 8.2 Hz, 1H), 7.15 (ddd, J = 16.7, 11.7, 4.4 Hz, 2H), 7.19–7.26 (m, 2H), 7.32 (dd, J = 7.5, 1.3 Hz, 1H), 7.34–7.38 (m, 1H), 7.43 (d, J = 7.2 Hz, 1H); 13C NMR (175 MHz, acetone‑d6) δ 36.7, 36.80, 44.62, 44.78, 54.80, 55.00, 101.26, 108.04, 109.00, 110.22, 110.45, 120.07, 120.38, 122.29, 122.69, 125.97, 127.11, 127.33, 127.37, 127.47, 127.93, 127.94, 128.26, 130.32, 131.31, 134.48, 147.11, 147.68, 147.86, 157.63, 157.79, 170.39, 172.80; HRMS (ESI-TOF) calcd. for : 553.1857. Found: 553.1868 ; HPLC purity = 91.2 % 210 nm, Condition (1).
4.4.1.32. α-3-(1,1′-Biphenyl-2-yloxycarbonyl)-2,4-bis(2-methoxyphenyl) cyclobutane-1-carboxylic acid (α-19).
White solid; 83 % yield; m. p. 170–171 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.83 (s, 3H), 3.87 (s, 3H), 4.00 (dd, J = 10.5, 6.8 Hz, 1H), 4.04 (dd, J = 10.6, 7.8 Hz, 1H), 4.56 (dd, J = 10.5, 7.8 Hz, 1H), 4.71 (dd, J = 10.6, 6.8 Hz, 1H), 6.07 (dd, J = 8.0, 1.3 Hz, 1H), 6.97 – 6.88 (m, 2H), 7.01–7.10 (m, 3H), 7.16–7.28 (m, 3H), 7.36 (s, 7H), 7.43 (d, J = 7.3 Hz, 1H), 10.43 (bs, 1H); 13C NMR (125 MHz, acetone‑d6) δ 37.5, 37.7, 45.5, 45.7, 55.7, 55.9, 111.1, 111.4, 121.0, 121.3, 123.6, 126.9, 128.0, 128.20, 128.24, 128.28, 128.35, 128.8, 129.1, 129.2, 129.6, 131.2, 135.6, 138.4, 148.7, 158.5, 158.7, 171.3, 173.6; HRMS (ESI-TOF) calcd. for : 509.1959. Found: 509.1966 ; HPLC purity 95.0 % 210 nm, Condition (3).
4.4.1.33. α-3-(4′-Cyano-1,1′-biphenyl-3-yloxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutane-1-carboxylic acid (α-20).
White solid; 40 % yield; m.p. = 203–204 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.88 (s, 3H), 3.89 (s, 3H), 4.14 (dd, J = 10.5, 7.9 Hz, 1H), 4.27 (dd, J = 10.7, 6.7 Hz, 1H), 4.79 (dd, J = 10.6, 6.8 Hz, 1H), 4.89 (dd, J = 10.6, 7.9 Hz, 1H), 6.54 (t, J = 1.9 Hz, 1H), 6.69 (dd, J = 7.6, 1.7 Hz, 1H), 6.96–7.04 (m, 2H), 7.09 (dd, J = 13.5, 7.6 Hz, 2H), 7.28 (t, J = 7.8 Hz, 1H), 7.41 (dd, J = 15.9, 7.9 Hz, 2H), 7.47 (t, J = 6.9 Hz, 2H), 7.54 (d, J = 7.8 Hz, 1H), 7.75 (d, J = 8.4 Hz, 2H), 7.91 (d, J = 8.4 Hz, 2H), 10.45 (s, 1H); 13C NMR (125 MHz, acetone‑d6) δ 36.32, 36.95, 44.20, 45.20, 54.89, 55.11, 110.44, 110.51, 111.27, 118.41, 120.23, 120.36, 120.46, 121.91, 124.22, 127.41, 127.45, 127.48, 127.62, 127.79, 128.11, 128.56, 129.91, 132.69, 140.14, 144.05, 151.55, 157.76, 158.06, 170.95, 172.49; HRMS (ESI-TOF) calcd. for : 534.1911. Found: 534.1914 ; HPLC purity 99.9 % at 215 nm, Condition (1).
4.4.1.34. α-3-(2-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)phenoxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutane-1-carboxylic acid (α-25).
White solid; 78 % yield; m.p. 139–141 °C; 1H NMR (700 MHz, acetone‑d6) δ 3.79 (s, 3H), 3.84 (s, 3H), 3.96 (dd, J = 10.5, 6.7 Hz, 1H), 4.05 (dd, J = 10.6, 7.7 Hz, 1H), 4.28–4.19 (m, 4H), 4.53 (dd, J = 10.5, 7.7 Hz, 1H), 4.69 (dd, J = 10.6, 6.7 Hz, 1H), 5.98 (dd, J = 8.1, 1.3 Hz, 1H), 6.74–6.82 (m, 3H), 6.88–6.93 (m, 2H), 7.00 (td, J = 7.5, 1.1 Hz, 1H), 7.04 (d, J = 8.2 Hz, 1H), 7.08–7.13 (m, 2H), 7.16–7.22 (m, 2H), 7.28 (dd, J = 7.6, 1.4 Hz, 1H), 7.32 (td, J = 7.8, 1.4 Hz, 1H), 7.40 (dd, J = 7.6, 1.6 Hz, 1H); 13C NMR (176 MHz, acetone‑d6) δ 37.6, 37.7, 45.6, 45.7, 55.7, 55.9, 65.2, 65.2, 111.1, 111.3, 117.8, 118.3, 121.0, 121.3, 122.5, 123.6, 126.8, 128.1, 128.3, 128.5, 128.7, 128.8, 129.1, 131.1, 131.5, 135.1, 144.2, 144.4, 148.7, 158.5, 158.7, 171.4; HRMS (ESI-TOF) m/z calcd. for : 567.2013. Found: 567.2011 ; HPLC purity 97.4 % at 254 nm, Condition (1).
4.4.1.35. α-3-(9-Hydroxy-9H-fluoren-9-ylmethoxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutane-1-carboxylic acid (α-26).
White solid; 86 % yield; m.p. 74–76 °C; 1H NMR (700 MHz, DMSO‑d6) δ 3.60 (s, 3H), 3.64 (dd, J = 10.4, 7.2 Hz, 1H), 3.71–3.75 (m, 2H), 3.77 (s, 3H), 4.11 (d, J = 10.9 Hz, 1H), 4.18–4.23 (m, 1H), 4.27–4.34 (m, 1H), 5.89 (s, 1H), 6.84 (t, J = 7.1 Hz, 2H), 6.92 (t, J = 7.3 Hz, 1H), 6.96 (d, J = 8.3 Hz, 1H), 7.02 (d, J = 7.3 Hz, 1H), 7.08 (d, J = 7.2 Hz, 1H), 7.17 (t, J = 7.7 Hz, 1H), 7.25 (dt, J = 13.0, 6.7 Hz, 3H), 7.38 (q, J = 7.2 Hz, 2H), 7.45 (d, J = 7.3 Hz, 2H), 7.75 (d, J = 7.5 Hz, 2H), 11.90 (s, 1H); 13C NMR (176 MHz, DMSO‑d6) δ 36.72, 36.81, 44.61, 44.90, 55.62, 55.82, 68.76, 79.63, 110.72, 110.97, 120.44, 120.46, 120.53, 124.71, 124.91, 127.20, 127.25, 127.51, 127.60, 128.05, 128.32, 128.39, 129.31, 129.37, 139.69, 139.76, 147.29, 147.37, 157.35, 157.59, 171.76, 173.77; HRMS (ESI-TOF) calcd. for : 573.1884. Found: 573.1878 ; HPLC purity 96.8 % at 210 nm, Condition (6).
4.4.1.36. Synthesis of γ-3-(4-cyano-1,1′-biphenyl-3-yloxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (γ-3).
N,N-Diisopropylethylamine (200 μL, 1.2 mmol) was added to a solution of γ-truxillic acid anhydride (280 mg, 1.0 mmol) and 2-hydroxy-4-phenylbenzonitrile (220 mg, 1.13 mmol) in anhydrous THF (5.0 mL) under nitrogen. The mixture was stirred at room temperature for 24 h. To the reaction mixture was added ethyl acetate and water (10 mL each), followed by 5 % NaH2PO4 solution to bring the pH from 9 to 5, and extracted with ethyl acetate (3 × 30 mL). The combined organic layer was washed with brine, dried over anhydrous magnesium sulfate, filtered, and concentrated in vacuo to give a crude product. The crude product was purified by flash chromatography on silica gel, using hexanes:EtOAC:AcOH (75:24:1) as eluant, to give γ-3 (220 mg, 47 % yield) as a white solid: m.p. 203–204 °C; 1H NMR (300 MHz, acetone‑d6) δ 4.03 (t, J = 10.4 Hz, 1H), 4.30 (t, J = 10.4, Hz, 1H), 4.60 (t, J = 10.1, Hz, 1H), 4.82 (t, J = 10.7, Hz, 1H), 6.09 (s, 1H), 7.24–7.62 (m, 15H), 7.69 (d, J = 8.1 Hz, 1H), 7.81 (d, J = 8.1 Hz, 1H); 13C NMR (126 MHz, acetone‑d6) δ 42.59, 43.38, 47.27, 47.95, 106.80, 116.17, 122.33, 126.22, 128.39, 128.56, 128.90, 129.03, 129.58, 129.70, 130.06, 130.48, 134.87, 134.93, 139.13, 140.29, 140.45, 148.25, 153.76, 171.17, 173.42; HRMS (ESI-TOF) calcd. for : 474.1700. Found: 474.1701 ; HPLC purity: 99.2 % at 210 nm, Condition (5).
In a manner similar to that of γ-3, the following γ-TAMEs were synthesized and characterized. In the case of slow reaction, elevated temperature and extended reaction were applied.
4.4.1.37. γ-3-(3-(2-Oxoindolin-6-yl)phenoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (γ-1).
White solid; 89 % yield (recrystallized from methanol/water); m.p. > 230 °C; 1H NMR (700 MHz, DMSO‑d6) δ 3.53 (s, 2H), 3.85 (t, J = 10.5 Hz, 1H), 4.14 (t, J = 10.4 Hz, 1H), 4.39 (t, J = 10.1 Hz, 1H), 4.54 (t, J = 10.6 Hz, 1H), 6.16 (s, 1H), 6.53 (d, J = 8.3 Hz, 1H), 6.82 (s, 1H), 7.04 (d, J = 7.8 Hz, 1H), 7.25–7.47 (m, 13H), 10.59 (s, 1H), 12.29 (bs, 1H); 13C NMR (175 MHz, DMSO‑d6) δ 36.1, 42.3, 44.5, 46.1, 46.2, 107.7, 119.9, 120.3, 120.9, 124.4, 125.3, 126.1, 127.16, 127.21, 127.9, 128.91, 128.94, 129.1, 130.3, 138.87, 138.91, 142.1, 142.4, 144.9, 150.9, 170.8, 173.0, 177.1; HRMS (ESI-TOF) calcd. for : 504.1806. Found 504.1804 ; HPLC purity 99.9 % at 210 nm, Condition (1).
4.4.1.38. γ-3-(3-(2,3-Dihydrobenzo[b][1,4]dioxin-5-yl)phenoxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutane-1-carboxylic acid (γ-6).
White solid; 47 % yield; m.p. 194–195 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.82 (s, 3H), 3.93 (s, 3H), 4.13 (t, J = 10.6 Hz, 1H), 4.20 (t, J = 10.4 Hz, 1H), 4.26 (m, 2H), 4.31 (m, 2H), 4.79 (t, J = 10.4 Hz, 1H), 5.02 (t, J = 10.6 Hz, 1H), 6.47 (dd, J = 8.0, 1.2 Hz, 1H), 6.56–6.64 (m, 1H), 6.78 (dd, J = 7.5, 1.6 Hz, 1H), 6.85 (dd, J = 8.0, 1.6 Hz, 1H), 6.88–6.99 (m, 3H), 7.03 (t, J = 8.6 Hz, 2H), 7.20–7.34 (m, 4H), 7.37 (d, J = 7.4 Hz, 1H), 7.53 (dd, J = 7.5, 0.8 Hz, 1H), 10.53 (s, 1H); 13C NMR (126 MHz, acetone‑d6) δ 39.9, 44.16, 45.30, 54.85, 55.01, 64.05, 64.24, 110.79, 111.03, 116.71, 120.04, 120.17, 120.41, 120.80, 122.41, 122.50, 126.39, 126.82, 128.07, 128.14, 128.36, 129.17, 129.56, 129.75, 138.98, 140.78, 144.17, 150.62, 158.04, 158.18, 170.02; HRMS (ESI-TOF) calcd. for : 567.2013. Found: 567.1997 ; HPLC purity 98.3 % at 215 nm, Condition (1).
4.4.1.39. γ-3-(3-(2-Oxo-1,2,3,4-tetrahydroquinolin-6-yl)phenoxycarbonyl)-2,4-diphenyl-cyclobutane-1-carboxylic acid (γ-8).
Gray solid; 42 % yield (recrystallized from methanol/water); m.p. 230 °C (decomp.); 1H NMR (500 MHz, DMSO‑d6): δ 2.48 (t, J = 7.5 Hz, 2H), 2.92 (t, J = 7.5 Hz, 2H), 3.84 (t, J = 10.4 Hz, 1H), 4.14 (t, J = 10.3 Hz, 1H), 4.39 (t, J = 10.1 Hz, 1H), 4.55 (t, J = 10.7 Hz, 1H), 6.30 (s, 1H), 6.39–6.47 (m, 1H), 6.93–7.02 (m, 2H), 7.23–7.34 (m, 5H), 7.35–7.47 (m, 8H), 10.16 (s, 1H), 12.28 (bs, 1H); 13C NMR (125 MHz, DMSO‑d6) δ 25.0, 30.8, 42.3, 44.5, 46.1, 46.3, 113.6, 119.8, 120.8, 123.8, 124.3, 127.17, 127.21, 127.7, 128.8, 128.9, 129.0, 130.3, 138.4, 138.9, 139.3, 141.8, 142.3, 151.0, 170.7, 173.0; HRMS (ESI-TOF) calcd. for : 518.1962. Found: 518.1966 ; HPLC purity 97.0 % at 210 nm, Condition (1).
4.4.1.40. γ-3-(3-(Benzo[d][1,3]dioxol-5-yl)phenoxycarbonyl)-2,4-bis(2-methoxyphenyl)-cyclobutane-1-carboxylic acid (γ-9).
White solid; 83 % yield; m.p. 167–168 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.82 (s, 3H), 3.93 (s, 3H), 4.15 (t, J = 10.6 Hz, 1H), 4.23 (t, J = 10.4 Hz, 1H), 4.79 (t, J = 10.3 Hz, 1H), 5.05 (t, J = 10.7 Hz, 1H), 6.08 (s, 2H), 6.43 (t, J = 1.9 Hz, 1H), 6.53 (dd, J = 8.0, 1.2 Hz, 1H), 6.91–6.99 (m, 3H), 7.03 (ddd, J = 7.2, 6.8, 3.2 Hz, 4H), 7.24–7.34 (m, 3H), 7.34–7.41 (m, 2H), 7.51–7.59 (m, 1H), 10.52 (s, 1H); 13C NMR (176 MHz, DMSO-d6) δ 39.0, 44.48, 45.46, 48.50, 55.89, 55.96, 101.75, 107.42, 109.15, 111.45, 111.72, 119.66, 120.40, 120.43, 120.80, 120.87, 124.01, 126.86, 128.55, 128.68, 129.22, 129.86, 130.16, 133.52, 141.58, 147.64, 148.46, 151.15, 158.01, 158.07, 170.48, 173.40; HRMS (ESI-TOF) calcd. for : 553.1867. Found: 553.1867 ; HPLC purity 96.5 % at 210 nm, Condition (5).
4.4.1.41. γ-3-(3-(2-Oxo-1,2,3,4-tetrahydroquinolin-6-yl)phenoxycarbonyl)-2,4-diphenyl-cyclobutane-1-carboxylic acid (γ-10).
White solid; 72 % yield; m.p. > 230 °C; 1H NMR (700 MHz, acetone‑d6) δ 2.58 (t, J = 7.4 Hz, 4H), 3.08 (t, J = 7.3 Hz, 4H), 3.98 (t, J = 10.5 Hz, 2H), 4.17 (t, J = 10.5 Hz, 2H), 4.53 (t, J = 10.2 Hz, 2H), 4.78 (t, J = 11.0 Hz, 2H), 6.41 (s, 2H), 6.49 (d, J = 7.8 Hz, 2H), 7.04 (d, J = 8.5 Hz, 2H), 7.30 (t, J = 7.2 Hz, 4H), 7.34 (d, J = 6.2 Hz, 4H), 7.41 (ddd, J = 23.0, 15.3, 7.6 Hz, 12H), 7.52 (dd, J = 24.0, 7.6 Hz, 8H), 9.17 (s, 2H), 10.77 (s, 1H); 13C NMR (176 MHz, DMSO‑d6) δ 25.3, 30.8, 42.3, 44.6, 46.1, 46.3, 115.9, 119.3, 120.3, 123.9, 124.6, 125.8, 126.4, 127.2, 127.2, 127.7, 128.9, 128.9, 129.1, 130.2, 132.8, 138.7, 138.9, 141.6, 142.3, 151.0, 170.7, 170.7, 173.0; HRMS (ESI-TOF) calcd. for : 518.1962. Found: 518.1966 ; HPLC purity 99.3 % at 210 nm: Condition (1).
4.4.1.42. γ-3-(1,1′-Biphenyl-2-ylmethoxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutene-1-carboxylic acid (γ-12).
White solid; 90 % yield; m. p. 69–70 °C; 1H NMR (700 MHz, acetone‑d6) δ 3.70 (s, 3H), 3.84 (s, 3H), 3.99 (dd, J = 10.5, 7.2 Hz, 1H), 4.03 (dd, J = 10.3, 7.4 Hz, 1H), 4.64 (dd, J = 15.4, 8.2 Hz, 3H), 4.75 (d, J = 12.5 Hz, 1H), 6.88 (d, J = 8.1 Hz, 1H), 6.92–6.99 (m, 3H), 7.10 (d, J = 7.6 Hz, 1H), 7.21–7.29 (m, 5H), 7.31–7.41 (m, 7H), 10.40 (s, 1H); 13C NMR (176 MHz, acetone‑d6) δ 36.69, 36.79, 44.58, 44.94, 54.69, 54.84, 63.57, 110.22, 110.29, 120.12, 120.16, 127.13, 127.21, 127.29, 127.46, 127.64, 127.93, 127.99, 128.02, 128.19, 129.05, 129.66, 129.68, 133.27, 140.37, 141.94, 157.51, 157.68, 171.72, 172.68; HRMS (ESI-TOF) calcd. for : 523.2115. Found: 523.2112 ; HPLC purity 95.5 % at 210 nm, Condition (5).
4.4.1.43. γ-3-(4′-Fluoro-1,1′-biphenyl-3-yloxycarbonyl)-2,4-bis(2-methoxyphenyl)-cyclobutane-1-carboxylic acid (γ-13).
White solid; 93 % yield; m.p. 187–188 °C; 1H NMR (500 MHz, CDCl3): δ 3.70 (s, 3H), 3.88 (s, 3H), 4.12 (t, J = 10.4 Hz, 1H), 4.24 (t, J = 10.4 Hz, 1H), 4.76 (t, J = 10.0 Hz, 1H), 4.85 (t, J = 10.8 Hz, 1H), 6.42 (s, 1H), 6.44 (d, J = 7.6 Hz, 1H), 6.84 (d, J = 8.2 Hz, 1H), 6.91–6.98 (m, 3H), 7.10 (t, J = 8.6 Hz, 2H), 7.23–7.43 (m, 8H), 11.11 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 36.7, 40.5, 44.2, 44.9, 55.3, 110.7, 110.8, 128.5, 128.4, 126.1, 124.0, 120.8, 120.6, 120.2, 120.0, 115.5 (d, 2JC,F = 21.4 Hz), 130.0, 129.6, 129.5, 129.1, 128.71 (d, 3JC,F = 8.0 Hz), 158.0, 157.8, 150.9, 141.4, 136.2 (d, 3JC,F = 3.3 Hz), 177.8, 170.5, 162.6 (d, 1JC,F = 246.8 Hz); HRMS (ESI-TOF) calcd. for : 527.1864. Found: 527.1871 ; HPLC purity 99.6 % at 210 nm, Condition (4).
4.4.1.44. γ-2,4-Diphenyl-3-(6-(pyrimidin-5-yl)naphthalen-2-yloxycarbonyl)cyclobutane-1-carboxylic acid (γ-15).
Off-white solid; 75 % yield; m.p. 230 °C (decomp.); 1H NMR (500 MHz, DMSO‑d6) δ 3.87 (t, J = 10.5 Hz, 1H), 4.20 (t, J = 10.4 Hz, 1H), 4.43 (t, J = 10.1 Hz, 1H), 4.58 (t, J = 10.7 Hz, 1H), 6.53 (dd, J = 8.9, 2.3 Hz, 1H), 6.90 (d, J = 2.3 Hz, 1H), 7.29 (t, J = 7.2 Hz, 1H), 7.36–7.48 (m, 9H), 7.87 (t, J = 7.9 Hz, 2H), 7.96 (dd, J = 8.5, 1.7 Hz, 1H), 8.29–8.47 (m, 1H), 9.22 (s, 1H), 9.26 (s, 2H), 12.30 (bs, 1H); 13C NMR (125 MHz, DMSO‑d6) δ 42.3, 44.6, 46.2, 46.3, 118.6, 122.3, 125.2, 125.6, 126.5, 127.2, 127.7, 128.8, 128.9, 129.0, 129.1, 130.1, 131.5, 133.3, 133.4, 138.9, 142.3, 148.8, 155.4, 157.8, 170.8, 172.9; HRMS (ESI-TOF) calcd. for : 501.18088. Found: 501.18181 ; HPLC purity 95.9 % at 210 nm, Condition (6).
4.4.1.45. γ-3-(2,3-Dihydro-1H-inden-2-yloxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutane-1-carboxylic acid (γ-16).
White solid; 40 % yield; m.p. 185–186 °C; 1H NMR (700 MHz, DMSO‑d6) δ 1.74 (d, J = 16.8 Hz, 1H), 2.68 (d, J = 16.8 Hz, 1H), 2.81 (dd, J = 17.0, 6.0 Hz, 1H), 3.06 (dd, J = 17.0, 6.2 Hz, 1H), 3.47 (s, 3H), 3.70 (t, J = 10.4 Hz, 1H), 3.76 (s, 3H), 3.82 (t, J = 10.7 Hz, 1H), 4.50 (t, J = 10.5 Hz, 1H), 4.63 (t, J = 10.5 Hz, 1H), 5.07 (tt, J = 6.2, 2.1 Hz, 1H), 6.86 (d, J = 8.2 Hz, 1H), 6.87–6.93 (m, 2H), 6.97 (d, J = 8.0 Hz, 1H), 7.04–7.07 (m, 1H), 7.10–7.31 (m, 7H), 11.98 (bs, 1H); 13C NMR (175 MHz, DMSO‑d6) δ 38.5, 38.70, 38.75, 40.0, 43.9, 44.9, 54.8, 55.3, 74.2, 110.9, 111.2, 119.6, 120.3, 124.3, 124.6, 126.1, 126.2, 126.3, 127.8, 127.9, 128.1, 128.7, 129.5, 140.3, 140.4, 157.1, 157.5, 170.7, 172.9; HRMS (ESI-TOF) calcd. for : 473.1959. Found: 473.1963 ; HPLC purity 99.3 % at 210 nm, Condition (1).
4.4.1.46. γ-3-(3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)phenoxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutane-1-carboxylic acid (γ-17).
White solid; 75 % yield; m.p. 197–198 °C; 1H NMR (700 MHz, acetone‑d6) δ 3.82 (s, 3H), 3.93 (s, 3H), 4.13 (t, J = 10.6 Hz, 1H), 4.21 (t, J = 10.4 Hz, 1H), 4.26 (dd, J = 5.1, 2.5 Hz, 2H), 4.28–4.33 (m, 2H), 4.79 (t, J = 10.3 Hz, 1H), 5.02 (t, J = 10.6 Hz, 1H), 6.47 (dd, J = 8.0, 1.2 Hz, 1H), 6.57–6.64 (m, 1H), 6.78 (dd, J = 7.5, 1.6 Hz, 1H), 6.85 (dd, J = 8.1, 1.6 Hz, 1H), 6.87–6.99 (m, 3H), 7.03 (t, J = 8.6 Hz, 2H), 7.20–7.34 (m, 4H), 7.37 (d, J = 7.4 Hz, 1H), 7.53 (d, J = 7.5 Hz, 1H), 10.53 (s, 1H); 13C NMR (176 MHz, acetone‑d6) δ 39.91, 44.13, 45.28, 54.84, 55.00, 64.04, 64.24, 110.78, 111.02, 116.71, 120.04, 120.17, 120.41, 120.80, 122.41, 122.49, 126.40, 126.80, 128.08, 128.14, 128.37, 129.10, 129.17, 129.55, 129.72, 138.97, 140.77, 144.16, 150.61, 158.03, 158.17, 170.03, 172.36; HRMS (ESI-TOF) calcd. for : 567.2013. Found: 567.2023 ; HPLC purity 99.3 % at 210 nm, Condition (4).
4.4.1.47. γ-3-(4′-Cyano-1,1′-biphenyl-3-yloxycarbonyl)-2,4-bis(2-methoxyphenyl)-cyclobutane-1-carboxylic acid (γ-20).
White solid; 62 % yield; m.p. 203–204 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.80 (s, 3H), 3.91 (s, 3H), 4.12 (t, J = 10.3 Hz, 1H), 4.21 (t, J = 10.1 Hz, 1H), 4.82 (t, J = 10.1 Hz, 1H), 5.03 (t, J = 10.3 Hz, 1H), 6.58 (s, 1H), 6.68 (d, J = 8.0 Hz, 1H), 6.96 (t, J = 7.4 Hz, 2H), 6.98–7.06 (m, 2H), 7.28 (dt, J = 23.8, 7.7 Hz, 2H), 7.38 (t, J = 7.7 Hz, 2H), 7.49 (d, J = 7.6 Hz, 1H), 7.56 (d, J = 7.6 Hz, 1H), 7.72 (d, J = 8.0 Hz, 2H), 7.85 (t, J = 7.5 Hz, 2H); 13C NMR (126 MHz, acetone‑d6) δ 36.2, 39.89, 44.21, 45.32, 54.88, 55.06, 110.86, 111.05, 111.24, 118.42, 120.13, 120.41, 120.43, 121.93, 124.10, 126.97, 127.78, 128.10, 128.31, 129.22, 129.27, 129.71, 129.88, 132.68, 140.11, 144.10, 151.57, 158.18, 170.10; HRMS (ESI-TOF) calcd. for : 534.1911. Found: 534.1914 ; HPLC purity 99.6 % at 215 nm, Condition (1).
4.4.1.48. γ-3-(4′-Cyano-1,1′-biphenyl-2-yloxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (γ-102).
White solid; 77 % yield; m.p. 224–225 °C; 1H NMR (700 MHz, acetone‑d6) δ 3.84 (t, J = 10.4 Hz, 1H), 3.88 (t, J = 10.4 Hz, 1H), 4.43 (t, J = 10.1 Hz, 1H), 4.65 (t, J = 10.8 Hz, 1H), 6.09 (d, J = 7.7 Hz, 1H), 7.15 (d, J = 7.0 Hz, 2H), 7.25 – 7.30 (m, 2H), 7.30 – 7.40 (m, 6H), 7.43 (d, J = 7.6 Hz, 3H), 7.56 (d, J = 7.5 Hz, 2H), 7.77 (d, J = 8.1 Hz, 2H), 10.77 (s,1); 13C NMR (176 MHz, acetone‑d6) δ 41.5, 44.42, 45.86, 46.06, 118.39, 122.58, 126.23, 126.50, 126.60, 127.23, 128.31, 128.37, 128.81, 129.48, 129.75, 130.15, 132.15, 133.18, 138.16, 142.07, 142.26, 147.57, 169.47, 171.78; HRMS (ESI-TOF) calcd. for : 474.1700. Found: 474.1703 ; HPLC purity 97.1 % at 210 nm, Condition (1).
4.4.1.49. γ-2,4-Bis(2-methoxyphenyl)-3-(naphthalen-1-yloxycarbonyl) cyclobutane-1-carboxylic acid (γ-103).
White solid; 60 % yield; m.p. 179–180 °C; 1H NMR (500 MHz, CDCl3) δ 3.74 (s, 3H), 3.89 (s, 3H), 4.15 (t, J = 10.5 Hz, 1H), 4.38 (t, J = 10.4 Hz, 1H), 4.88 (t, J = 10.0 Hz, 1H), 4.95 (t, J = 10.8 Hz, 1H), 6.47 (d, J = 7.5 Hz, 1H), 6.91 (d, J = 8.2 Hz, 1H), 6.94–7.03 (m, 3H), 7.23 (d, J = 8.4 Hz, 1H), 7.25–7.34 (m, 4H), 7.37 (d, J = 7.2 Hz, 1H), 7.41 (t, J = 7.4 Hz, 1H), 7.50 (d, J = 7.3 Hz, 1H), 7.62 (d, J = 8.3 Hz, 1H), 7.77 (d, J = 8.3 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 38.3, 40.4, 44.3, 45.4, 55.0, 55.2, 110.8, 117.6, 120.8, 121.4, 125.3, 125.5, 125.9, 126.0, 126.1, 126.8, 127.6, 128.3, 128.7, 129.2, 129.4, 130.1, 134.4, 146.6, 157.8, 158.0, 170.3, 178.0; HRMS (ESI-TOF) calcd. for : 483.1802. Found: 483.1794 ; HPLC purity 99.8 % at 210 nm, Condition (4).
4.4.1.50. γ-3-(3-(2-Oxoindolin-5-yl)phenoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (γ-104).
Off-white solid; 81 % yield (refluxed at 65 °C for 2 days); m.p. 230 °C (decomp.); 1H NMR (700 MHz, acetone‑d6) δ 3.57 (s, 2H), 3.97 (t, J = 10.5 Hz, 1H), 4.17 (t, J = 10.4 Hz, 1H), 4.53 (t, J = 10.1 Hz, 1H), 4.78 (t, J = 10.7 Hz, 1H), 6.38 (s, 1H), 6.49 (d, J = 8.0 Hz, 1H), 6.99 (d, J = 7.9 Hz, 1H), 7.23–7.33 (m, 2H), 7.35 (d, J = 8.0 Hz, 1H), 7.36–7.41 (m, 5H), 7.43 (t, J = 7.2 Hz, 2H), 7.50 (d, J = 7.5 Hz, 2H), 7.53 (d, J = 7.2 Hz, 2H), 9.46 (s, 1H), 10.78 (s, 1H); 13C NMR (500 MHz, acetone‑d6) δ 35.4, 42.08, 44.54, 46.03, 46.28, 109.29, 109.34, 119.51, 119.78, 123.16, 123.47, 126.33, 126.68, 127.43, 128.43, 128.45, 128.99, 129.43, 133.15, 138.57, 142.24, 142.28, 143.52, 143.63, 151.19, 170.14, 171.77, 175.75; HRMS (ESI-TOF) calcd. for : 504.1805. Found: 504.1809 ; HPLC purity 96 % at 215 nm, Condition (5).
4.4.1.51. γ-3-(4′-Cyano-1,1′-biphenyl-2-yloxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutane-1-carboxylic acid (γ-105).
White solid; 42 % yield; m.p. 168–169 °C; 1H NMR (700 MHz, acetone‑d6) δ 3.74 (s, 3H), 3.82 (s, 3H), 3.96–4.02 (m, 2H), 4.71 (t, J = 10.4 Hz, 1H), 4.93 (t, J = 10.7 Hz, 1H), 6.23 (d, J = 7.9 Hz, 1H), 6.90–6.96 (m, 2H), 7.01 (dd, J = 16.4, 8.5 Hz, 2H), 7.12 (d, J = 7.4 Hz, 1H), 7.25–7.33 (m, 4H), 7.43 (dd, J = 7.3, 2.0 Hz, 1H), 7.47 (d, J = 5.4 Hz, 1H), 7.53 (d, J = 8.3 Hz, 2H), 7.66 (d, J = 8.3 Hz, 2H), 10.48 (s, 1H); 13C NMR (126 MHz, acetone‑d6) δ 36.24, 39.56, 44.50, 44.95, 54.68, 55.02, 110.87, 111.01, 111.07, 118.39, 120.02, 120.39, 123.00, 126.23, 126.55, 128.12, 128.17, 128.79, 129.11, 129.36, 129.50, 129.76, 130.18, 131.99, 132.83, 142.14, 147.81, 158.02, 158.05, 169.61, 171.88; HRMS (ESI-TOF) calcd. for :534.1911. Found: 534.1918, ; HPLC purity 100.0 % at 210 nm, Condition (1).
4.4.1.52. γ-3-(4′-Cyano-1,1′-biphenyl-3-yloxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (γ-106).
White solid; 75 % yield; m.p.: 203–204 °C; 1H NMR (700 MHz, acetone‑d6) δ 3.96 (t, J = 10.5 Hz, 1H), 4.16 (t, J = 10.4 Hz, 1H), 4.52 (t, J = 10.1 Hz, 1H), 4.77 (t, J = 10.7 Hz, 1H), 6.44 (s, 1H), 6.62 (d, J = 7.95 Hz, 1H), 7.38–7.52 (m, 12H), 7.70 (d, J = 8.2 Hz, 2H), 7.88 (d, J = 8.2 Hz, 2H), 10.81 (s, 1H); 13C NMR (126 MHz, acetone‑d6) δ 43.5, 45.94, 47.46, 47.68, 112.68, 119.79, 121.73, 123.29, 125.74, 128.08, 128.09, 128.85, 129.15, 129.86, 130.41, 131.31, 134.07, 140.00, 141.56, 143.58, 145.36, 152.70, 171.53, 173.22; HRMS (ESI-TOF) calcd. for : 474.1700. Found: 474.1696 ; HPLC purity 99.0 % at 215 nm, Condition (3).
4.4.1.53. γ-3-(9H-Fluoren-9-ylmethoxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutane-1-carboxylic acid (γ-107).
White solid; 58 % yield; m.p. 186–187 °C; 1H NMR (500 MHz, CDCl3) δ 3.65–3.69 (m, 1H), 3.70 (s, 3H), 3.82 (s, 3H), 3.88 (t, J = 9.3 Hz, 1H), 3.99 (t, J = 10.4 Hz, 1H), 4.08 (t, J = 10.4 Hz, 1H), 4.19 (dd, J = 10.6, 6.7 Hz, 1H), 4.75 (t, J = 10.0 Hz, 1H), 4.82 (t, J = 10.6 Hz, 1H), 6.76 (d, J = 8.2 Hz, 1H), 6.85 (t, J = 7.4 Hz, 1H), 6.90 (d, J = 8.1 Hz, 1H), 6.97 (t, J = 7.4 Hz, 1H), 7.13 (t, J = 7.6 Hz, 1H), 7.17–7.32 (m, 4H), 7.35 (t, J = 8.8 Hz, 5H), 7.71 (d, J = 7.5 Hz, 2H); 13C NMR (500 MHz, CDCl3) δ 26.9, 37.81, 40.37, 44.68, 46.76, 55.00, 55.18, 66.45, 110.40, 110.70, 119.82, 119.85, 120.42, 120.78, 125.02, 125.29, 125.95, 126.97, 127.05, 127.57, 127.63, 128.26, 128.39, 129.18, 129.27, 129.48, 141.03, 141.15, 143.53, 144.33, 157.52, 157.85, 172.13; HRMS (ESI-TOF) calcd. for : 535.2115. Found: 535.2112 ; HPLC purity: 99.9 % at 210 nm, Condition (4).
4.4.1.54. γ-3-(3-(2-Oxo-1,2-dihydroquinolin-7-yl)phenoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (γ-108).
White solid; 12 % yield; m.p. > 230 °C; 1H NMR (700 MHz, DMSO‑d6) δ 3.80–3.86 (m, 1H), 4.14 (t, J = 10.0 Hz, 1H), 4.35–4.41 (m, 1H), 4.55 (t, J = 10.5 Hz, 2H), 6.38 (s, 1H), 6.50 (d, J = 7.5 Hz, 1H), 6.53 (d, J = 9.43 Hz, 1H), 7.24–7.31 (m, 3H), 7.33–7.51 (m, 11H), 7.77 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 9.5 Hz, 1H), 11.83 (s, 1H), 12.32 (s, 1H); 13C NMR (176 MHz, DMSO‑d6) δ 40.48, 42.32, 44.58, 46.08, 113.36, 119.16, 120.33, 121.13, 121.58, 122.61, 124.76, 127.13, 127.21, 127.67, 128.90, 128.99, 129.06, 130.49, 138.99, 139.76, 140.33, 141.25, 141.27, 142.43, 151.03, 162.53, 170.78, 173.05; HRMS (ESI-TOF) calcd. for : 516.1806. Found: 516.1810 ; HPLC purity 95 % at 210 nm, Condition (3).
4.4.1.55. γ-3-(4-Methoxynaphthalen-1-yloxycarbonyl)-2,4-bis(2-methoxyphenyl)cyclobutane-1-carboxylic acid (γ-109).
Light brown solid; 75 % yield (refluxed at 65 °C for 3 days); m.p. 210 °C (decomp.); 1H NMR (500 MHz, acetone‑d6) δ 3.85 (s, 3H), 3.92 (s, 3H), 3.97 (s, 3H), 4.17 (t, J = 10.6 Hz, 1H), 4.40 (t, J = 10.5 Hz, 1H), 4.86 (t, J = 10.3 Hz, 1H), 5.14 (dd, J = 10.5 Hz, 1H), 6.39 (d, J = 8.3 Hz, 1H), 6.74 (d, J = 8.3 Hz, 1H), 6.94 (dd, J = 7.5, 7.5 Hz, 1H), 6.98 (dd, J = 7.3, 7.3 Hz, 1H), 7.06 (d, J = 8.3 Hz, 1H), 7.10 (d, J = 8.0 Hz, 1H), 7.20 (d, J = 8.4 Hz, 1H), 7.29 (d, J = 8.3 Hz, 1H), 7.31–7.33 (m, 2H), 7.39 (d, J = 7.5 Hz, 1H), 7.44 (dd, J = 7.0, 7.0 Hz, 1H), 7.58 (dd, J = 8.0, 1.8 Hz, 1H), 8.14 (d, J = 8.6 Hz, 1H), 10.50 (bs, 1H); 13C NMR (125 MHz, acetone‑d6) δ 39.8, 44.4, 45.5, 54.9, 55.1, 55.2, 103.0, 111.0, 111.1, 117.6, 120.2, 120.4, 121.2, 121.7, 125.3, 125.7, 126.5, 126.9, 127.6, 128.1, 128.3, 129.1, 129.3, 129.8, 140.1, 152.9, 158.1, 158.2, 170.3, 172.2; HRMS (ESI-TOF) calcd. for : 513.1908. Found: 513.18955 ; HPLC Purity 96.4 % at 210 nm, Condition (5).
4.4.1.56. Synthesis of ε-3-(2,3-dihydro-1H-inden-2-yloxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (ε-206).
EDC⋅HCl (76 mg, 0.40 mmol) and DMAP (49 mg, 0.34 mmol) were added to a solution of ε-truxillic acid (100 mg, 0.359 mmol) and 2-indanol (54.0 mg, 0.40 mmol) in anhydrous dichloromethane (5 mL) under nitrogen. The reaction mixture was stirred at room temperature for 15 h. To the reaction mixture was added dichloromethane and water (20 mL each), followed by 5 % NaH2PO4 solution to bring the pH from 9 to 5, and extracted with dichloromethane (3 × 20 mL). The combined organic layer was washed with brine (20 mL), dried over anhydrous magnesium sulfate, and concentrated in vacuo. The resulting crude product was purified by column chromatography on silica gel, using EtOAc/hexanes (gradient from 25 % to 50 %), to afford ε-206 (40 mg, 27 % yield) as a white solid: m.p. 147–148 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.01 (dd, J = 16.9, 2.7 Hz, 2H), 3.20 (t, J = 9.7 Hz, 1H), 3.36 (dd, J = 16.9, 6.1 Hz, 2H), 3.40 (t, J = 9.7 Hz, 1H), 3.90 (t, J = 9.7 Hz, 2H), 5.62 (tt, J = 6.1, 2.7 Hz, 1H), 7.13–7.33 (m, 9H), 7.38 (dd, J = 7.5, 1.7 Hz, 4H), 11.07 (bs, 1H); 13C NMR (125 MHz, acetone‑d6) δ 39.3., 43.1, 47.1, 49.0, 75.9, 124.5, 126.6, 127.7, 126.8, 128.4, 140.5, 141.1, 172.2, 173.2; HRMS (ESI-TOF) calcd. for : 413.1747. Found: 413.1751 ; HPLC purity 99.6 % at 210 nm, Condition (1).
In a manner similar to that of ε-206, the following ε-TAMEs were synthesized and characterized.
4.4.1.57. ε-3-(3-(Benzo[d][1,3]dioxol-5-yl)phenoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (ε-5).
White solid; 28 % yield; m.p. 134–135 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.49 (t, J = 9.7 Hz, 1H), 3.65 (t, J = 9.7 Hz, 1H), 4.13 (t, J = 9.7 Hz, 2H), 6.06 (s, 2H), 6.94 (d, J = 8.0 Hz, 1H), 7.11–7.20 (m, 3H), 7.31 (t, J = 7.4 Hz, 2H), 7.37–7.55 (m, 7H), 7.58 (d, J = 7.7 Hz, 4H); 13C NMR (126 MHz, acetone‑d6) δ 43.3, 48.66, 100.00, 101.41, 107.21, 108.49, 119.98, 120.20, 120.60, 124.05, 127.01, 128.57, 129.72, 134.13, 141.12, 142.25, 147.63, 148.45, 151.42, 171.18; HRMS (ESI-TOF) calcd. for : 493.1646. Found: 493.1657 ; HPLC purity 98.5 % at 215 nm, Condition (1).
4.4.1.58. ε-3-(1,1′-Biphenyl-2-ylmethoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (ε-11).
White solid; 19 % yield; m.p. 61–62 °C; 1H NMR (500 MHz, CD3CN) δ ppm 3.28 (t, J = 9.8 Hz, 1H), 3.42 (t, J = 9.8 Hz, 1H), 3.82 (t, J = 9.8 Hz, 2H), 5.95 (s, 2H), 6.76 (d, J = 8.0 Hz, 1H), 6.80 (t, J = 8.0, 1.6 Hz, 2H), 6.85 (d, J = 1.3 Hz, 1H), 7.13–7.19 (m, 1H), 7.27–7.43 (m, 13H); 13C NMR (500 MHz, CD3CN) δ 42.9, 47.90, 48.02, 101.51, 108.17, 109.12, 117.34, 122.39, 122.85, 126.61, 126.87, 127.17, 128.59, 130.82, 130.96, 134.84, 140.57, 147.24, 147.68, 147.76, 171.14, 173.20; HRMS (ESI-TOF) calcd. for : 493.1646. Found: 493.1637 ; HPLC purity 98.3 % at 210 nm, Condition (6).
4.4.1.59. ε-3-(Naphthalen-1-ylmethoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (ε-23).
White solid; 31 % yield; m.p. 156–158 °C; 1H NMR (500 MHz, CD3CN) δ ppm 3.27–3.34 (m, 2H), 3.85 (t, J = 9.77 Hz, 2H), 5.64 (s, 2H), 7.22–7.37 (m, 10H), 7.44–7.50 (m, 1H), 7.51–7.60 (m, 3H), 7.92 (d, J = 8.2 Hz, 1H), 7.93–7.98 (m, 1H), 7.99–8.05 (m, 1H); 13C NMR (500 MHz, CD3CN) δ 43.20, 47.59, 48.71, 64.86, 117.33, 123.83, 125.33, 126.07, 126.62, 126.98, 127.09, 127.64, 128.50, 128.56, 129.22, 131.46, 131.65, 133.71, 140.71, 172.29, 173.12; HRMS (ESI-TOF) calculated for 454.2018. Found = 454.2013 ; HPLC purity 98.5 % at 215 nm: condition (1).
4.4.1.60. ε-3-(1,1′-Biphenyl-2-ylmethoxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (ε-201).
White solid; 22 % yield; m.p. 121–122 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.28 – 3.38 (m, 1H), 3.41 (t, J = 9.8 Hz, 1H), 3.90 (t, J = 9.8 Hz, 2H), 5.12 (s, 2H), 7.25–7.33 (m, 5H), 7.48–7.34 (m, 13H), 7.51 (d, J = 7.1 Hz, 1H), 11.07 (bs, 1H); 13C NMR (176 MHz, acetone‑d6) δ 43.2, 48.17, 48.40, 64.47, 126.91, 126.98, 127.31, 127.56, 128.23, 128.38, 128.51, 129.02, 129.73, 129.96, 133.30, 140.41, 141.08, 142.39, 171.98, 173.27; HRMS (ESI-TOF) calcd. for : 463.1904. Found: 463.1921 ; HPLC purity 99.9 % at 215 nm, Condition (1).
4.4.1.61. ε-3-(Naphthalen-1-yloxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (ε-202).
White solid; 43 % yield; m.p. 191–192 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.53 (t, J = 9.7 Hz, 1H), 3.93 (t, J = 9.8 Hz, 1H), 4.20 (t, J = 9.7 Hz, 2H), 7.34 (t, J = 7.4 Hz, 2H), 7.41 (d, J = 7.5 Hz, 1H), 7.42–7.58 (m, 7H), 7.65 (d, J = 7.5 Hz, 4H), 7.77 (d, J = 8.4 Hz, 1H), 7.85 (d, J = 8.3 Hz, 1H), 7.97 (d, J = 8.2 Hz, 1H); 13C NMR (126 MHz, acetone‑d6) δ 45.09, 49.91, 119.80, 122.51, 126.95, 127.40, 127.93, 127.96, 128.58, 128.64, 129.44, 130.11, 136.14, 142.37, 148.11, 172.69; HRMS (ESI-TOF) calcd. for : 423.1591. Found: 423.1601 ; HPLC purity 99.6 % at 215 nm, Condition (5).
4.4.1.62. ε-3-(3-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)phenoxycarbonyl)-2,4-diphenyl-cyclobutane-1-carboxylic acid (ε-203).
White solid; 30 % yield; m.p. 153–154 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.49 (t, J = 9.7 Hz, 1H), 3.66 (t, J = 9.7 Hz, 1H), 4.13 (t, J = 9.7 Hz, 2H), 4.31 (s, 4H), 6.90–6.94 (m, 1H), 7.10–7.17 (m, 3H), 7.31 (t, J = 7.4 Hz, 2H), 7.41 (t, J = 7.8 Hz, 5H), 7.46 (t, J = 7.8 Hz, 1H), 7.50 (t, J = 7.9 Hz, 1H), 7.58 (d, J = 7.6 Hz, 4H); 13C NMR (126 MHz, acetone‑d6) δ 43.29, 48.10, 48.66, 64.34, 64.39, 115.46, 117.53, 119.74, 119.77, 120.07, 123.83, 127.00, 127.02, 128.57, 129.70, 133.12, 141.12, 141.98, 143.84, 144.07, 151.44, 171.18, 173.27; HRMS (ESI-TOF) calcd. for : 507.1802. Found: 507.1814 ; HPLC purity 98.6 % at 254 nm, Condition (1).
4.4.1.63. ε-3-(4′-Fluoro-1,1′-biphenyl-3-ylcarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (ε-204).
White solid; 13 % yield; m.p. 143–144 °C; 1H NMR (700 MHz, acetone‑d6) δ 3.5 (t, J = 9.7 Hz, 1H), 3.67 (t, J = 9.7 Hz, 1H), 4.13 (t, J = 9.7 Hz, 2H), 7.17–7.22 (m, 1H), 7.22–7.27 (m, 2H), 7.31 (t, J = 7.4 Hz, 2H), 7.42 (t, J = 7.7 Hz, 4H), 7.47–7.60 (m, 7H), 7.69–7.75 (m, 2H); 13C NMR (176 MHz, acetone‑d6) δ 173.24, 171.18, 162.67 (d, 1JC,F = 245.3 Hz) 151.46, 141.43, 141.05, 136.24, 136.23, 129.90, 128.9 (d, 3JC,F = 8.2 Hz), 128.60, 127.07, 127.01, 124.27, 120.67, 120.21, 115.6 (d, 2JC,F = 21.6 Hz), 115.55, 48.61, 48.09, 43.28; 19F NMR (176 MHz, acetone‑d6) δ −116.62; HRMS (ESI-TOF) calcd. for : 467.1653. Found: 467.1653 ; HPLC purity 95.3 % at 210 nm, Condition (5).
4.4.1.64. ε-3-(1,1′-Biphenyl-3-yloxycarbonyl)-2,4-diphenylcyclobutane-1-carboxylic acid (ε-205).
White solid; 25 % yield; m.p. 149–150 °C; 1H NMR (500 MHz, acetone‑d6) δ 3.50 (t, J = 9.7 Hz, 1H), 3.68 (t, J = 9.7 Hz, 1H), 4.14 (t, J = 9.8 Hz, 2H), 7.17–7.21 (m, 1H), 7.31 (t, J = 7.4 Hz, 2H), 7.37–7.44 (m, 5H), 7.45–7.53 (m, 4H), 7.55–7.61 (m, 5H), 7.67 (d, J = 7.1 Hz, 2H), 11.14 (s, 1H); 13C NMR (126 MHz, acetone‑d6) δ 43.29, 48.05, 48.63, 120.23, 120.63, 124.32, 126.92, 127.01, 127.05, 127.74, 128.58, 128.90, 129.83, 139.87, 141.07, 142.53, 151.47, 171.17, 173.16; HRMS (ESI-TOF) calcd. for : 449.1747. Found: 449.1757 ; HPLC purity 97.5 % at 254 nm, Condition (1).
4.5. Protein grid preparation
Co-crystal structures of FABP5/L1 (PDB ID: 5UR9) and FABP7/L1 (PDB ID: 5URA) in addition to a stearic acid-bound structure of FABP3 (PDB ID: 3WVM) were used as the basis for modeling the protein grid for all docking calculations. For each protein structure, all water molecules and ligands were removed from the binding site model and the remaining protein was processed through AutoDockTools in order to assign gasteiger charges, determine the grid-box center and merge non-polar hydrogens to their respective bonded carbon atoms. The protein grid was calculated with AutoGrid4 using default input parameters (Pg. S2) specified grid-box center coordinates and a box size of 40x40x40 Å to generate the protein grid for all subsequent docking calculations. Due to the lack of a FABP3 structure co-crystalized with L1, the chosen 3WVM protein structure was validated by docking L1 to produce a well behaved structure consistent with binding conformations of L1 in FABP5 and FABP7.
4.6. Molecular structure design and docking
Docking followed a four step protocol to design, parameterize, dock and evaluate the binding affinity of various small molecule inhibitors in the three protein targets aforementioned. (i) 2D molecular structures for inhibitor design were created via PerkinElmer’s ChemDraw and saved in the MOL format. These were then parsed through the Avogadro molecular editor [44] where rough 3D structures were generated in their biologically relevant protonation state using parameters of the integrated Open Babel toolkit. The internal energy was minimized by the Merck Molecular ForceField [45–49] (MMFF94) until an energy gradient of<0.01 kJ/mol was reached and coordinates were written in the MOL2 format. (ii) MOL2 structures were processed through MGLTools [50] to parameterize them for use with AutoDock4.2, merging non-polar hydrogens, assigning gasteiger charges, defining rotatable bonds and writing output in the PDBQT format. (iii) Docking calculations were carried out via the Scripps Research Institute AutoDock4.2 program. The default settings (Pg. S3) of the Lamarckian genetic algorithm were used for the sampling method where ten solutions were written for evaluation. (iv) Finally, the selection of the representative docking solution was automated by a python script developed in-house which identified the common carboxylate moiety necessary to exhibit interactions at the canonical binding site and subsequently calculated its RMSD using co-crystal carboxylate coordinates as the reference. The most favorable docking score under a 2 Å RMSD cutoff was chosen and solutions which did not meet this cutoff were selected by the lowest carboxylate RMSD. The visualization and depiction of selected docking results were accomplished through UCSF Chimera. Designed ligands retaining the canonical interaction with improved scores were selected for synthesis.
4.7. Protein purification
Recombinant human FABP3, FABP5, and FABP7 were expressed as N-terminal His-tagged proteins using a pET-28a vector (Novagen, Madison, WI, USA). The protein purification and delipidation were performed as previously described [3].
4.8. Fluorescence displacement binding assay
Purified FABPs (3 μM) were incubated with 11-(dansylamino) undecanoic acid (DAUDA) or 8-anilino-1-naphthalenesulfonic acid (ANS) (both at 500 nM) in the presence or absence of FABP inhibitors (0.01–200 μM) in the binding assay buffer (30 mM Tris, 100 mM NaCl, pH 7.5). Each assay included arachidonic acid (10 μM) to account for maximal probe displacement and L1 (5 μM with FABP3 and 1 μM with FABP5 and FABP7, corresponding to their approximate Ki values for each protein). Loss of fluorescence intensity was monitored with a F5 Filtermax Multi-Mode Microplate Reader (Molecular Devices, Sunnyvale, CA, USA) using the following excitation and emission wavelengths (DAUDA: ex./em. = 360/535 nm; ANS: ex./em. = 360/465 nm). Following background subtraction, the fluorescence intensity values were normalized and analyzed via a one-site binding model using the GraphPad Prism (version 9.2). Ki values of each compound were determined using the following equation: Ki = IC50/(1 + ([Probe]/Kd).
4.9. CFA-induced thermal hyperalgesia
Thermal paw withdrawal was measured as previously described [11]. Briefly, mice were habituated to the experimental room and Hargreaves plantar apparatus (Ugo Basile) for at least two days before baseline measurements. Testing began 30 min after mice were placed in the testing chambers. To induce chronic inflammation, mice received unilateral intraplantar injections of 20 μL complete Freund’s adjuvant (CFA) (#F5881, Sigma) in the hind paw. The opposite hind paw was injected with saline. Three to seven days later withdrawal latencies to a focused beam (intensity 3.0) directed at the plantar surface of the hind paw were assessed 90 min after intraperitoneal administration of FABP5 inhibitor or vehicle (DMSO:cremophor-EL:saline at a ratio of 1:1:8).
The experiments performed in this study were approved by the Stony Brook University Animal Care and Use Committees in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (#277150). Ten to twelve week old male C57BL/6J mice (Jackson Laboratory) were housed in a temperature- and humidity-controlled AAALAC-certified facility on a 12 h light/dark cycle with ad libitum access to food and water.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health, R01 DA035949, DA042545 and R01 CA237154, a Fusion Award from the Renaissance School of Medicine, Stony Brook University, and a grant from Artelo Biosciences. The authors are grateful to Professor Robert Rizzo, Department of Applied Mathematics and Statistics, for his helpful discussions. The authors also thank Dr. Bela Ruzsicska, Institute of Chemical Biology and Drug Discovery, and Robert Rieger, Proteomics Center, of the Stony Brook University for mass spectrometry analyses.
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Iwao Ojima reports financial support was provided by National Institutes of Health. Martin Kaczocha reports financial support was provided by National Institutes of Health. Iwao Ojima reports financial support was provided by Stony Brook University Renaissance School of Medicine. Martin Kaczocha reports financial support was provided by Stony Brook University Renaissance School of Medicine. Iwao Ojima reports financial support was provided by Artero Biosciences. Martin Kaczocha reports financial support was provided by Artero Biosciences. Iwao Ojima has patent issued to Research Foundation of State University of New York. Martin Kaczocha has patent issued to Research Foundation of State University of New York. Nothing in particular.
Abbreviations:
- 2-AG
2-arachidonoylglycerol
- AEA
arachidonoyl ethanolamide
- ANS
1-anilinonaphthalene-8-sulfonic Acid
- CB1R
cannabinoid receptor type 1
- CB2R
cannabinoid receptor type 2
- CFA
complete Freund’s adjuvant
- DAUDA
11-[5-(dimethylamino)-1-naphthalenesulfonylamino]undecanoic acid
- DIPEA
N,N-diisopropylethylamine
- EDC
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
- FABP
fatty acid binding protein
- FABP3
heart fatty acid binding protein
- FABP5
epidermal fatty acid binding protein
- FABP7
brain fatty acid binding protein
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium bromide
- NAE
N-acylethanolamine
- NBD-stearate
12-N-methyl-(7-nitrobenz-2-oxa-1,3-diazo)aminostearic acid
- NMI
N-methylimidazole
- TA
truxillic acid
- TAA
truxillic acid anhydride
- TADE
truxillic acid diester
- TADSE
truxillic acid disilyl ester
- TAME
truxillic acid monoester
- TAMSE
truxillic acid monosilyl ester
- TBDPS
tert-butyldiphenylsilyl
- TCFH
N,N,N’,N’-tetramethylchloroformamidinium hexafluorophosphate
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioorg.2022.106184.
Data availability
Data will be made available on request.
References
- [1].Kaczocha M, Lin Q, Nelson LD, McKinney MK, Cravatt BF, London E, Deutsch DG, Anandamide externally added to lipid vesicles containing trapped fatty acid amide hydrolase (FAAH) is readily hydrolyzed in a sterol-modulated fashion, ACS Chem. Neurosci 3 (2012) 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kaczocha M, Glaser ST, Deutsch DG, Identification of intracellular carriers for the endocannabinoid anandamide, PNAS 106 (2009) 6375–6380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kaczocha M, Vivieca S, Sun J, Glaser ST, Deutsch DG, Fatty acid-binding proteins transport N-acylethanolamines to nuclear receptors and are targets of endocannabinoid transport inhibitors, J. Biol. Chem 287 (2012) 3415e3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kaczocha M, Rebecchi MJ, Ralph BP, Teng YH, Berger WT, Galbavy W, Elmes MW, Glaser ST, Wang L, Rizzo RC, Deutsch DG, Ojima I, Inhibition of fatty acid binding proteins elevates brain anandamide levels and produces analgesia, PLoS ONE 9 (2014) e94200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sanson B, Wang T, Sun J, Wang L, Kaczocha M, Ojima I, Deutsch D, Li H, Crystallographic study of FABP5 as an intracellular endocannabinoid transporter, Acta Crystallogr. D Biol. Crystallogr 70 (2014) 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Haunerland NH, Spener F, Fatty acid-binding proteins - insights from genetic manipulations, Prog. Lipid Res. 43 (2004) 328–349. [DOI] [PubMed] [Google Scholar]
- [7].Chmurzyńska A, The multigene family of fatty acid-binding proteins (FABPs) Function, structure and polymorphism, J. Appl. Genetics 47 (2006) 39–48. [DOI] [PubMed] [Google Scholar]
- [8].Dennis EA, Norris PC, Eicosanoid Storm in Infection and Inflammation, Nat. Rev. Immunol 15 (2015) 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Furuhashi M, Tuncman G, Gorgun CZ, Makowski L, Atsumi G, Vaillancourt E, Kono K, Babaev VR, Fazio S, Linton MF, Sulsky R, Robl JA, Parker RA, Hotamisligil GS, Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2, Nature 447 (2007) 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Maeda K, Cao H, Kono K, Gorgun CZ, Furuhashi M, Uysal KT, Cao Q, Atsumi G, Malone H, Krishnan B, Minokoshi Y, Kahn BB, Parker RA, Hotamisligil GS, Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes, Cell Metab. 1 (2005) 107–119. [DOI] [PubMed] [Google Scholar]
- [11].Kaczocha M, Glaser ST, Maher T, Clavin B, Hamilton J, O’Rourke J, Rebecchi M, Puopolo M, Owada Y, Thanos PK, Fatty acid binding protein deletion suppresses inflammatory pain through endocannabinoid/N-acylethanolamine-dependent mechanisms, Mol Pain 11 (2015) 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Owada Y, Yoshimoto T, Kondo H, Spatio-temporally differential expression of genes for three members of fatty acid binding proteins in developing and mature rat brains, J. Chem. Neuroanat 12 (1996) 113–122. [DOI] [PubMed] [Google Scholar]
- [13].Pertwee RG, Ligands that target cannabinoid receptors in the brain: from THC to anandamide and beyond, Addict. Biol 13 (2008) 147–159. [DOI] [PubMed] [Google Scholar]
- [14].Pertwee RG, The therapeutic potential of drugs that target cannabinoid receptors or modulate the tissue levels or actions of endocannabinoids, AAPS J. 7 (2005) E625–E654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Cravatt BF, Lichtman AH, The endogenous cannabinoid system and its role in nociceptive behavior, J. Neurobiol 61 (2004) 149–160. [DOI] [PubMed] [Google Scholar]
- [16].Ahn K, Johnson DS, Cravatt BF, Fatty acid amide hydrolase as a potential therapeutic target for the treatment of pain and CNS disorders, Expert Opin. Drug Discov. 4 (2009) 763–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].van der Stelt M, van Kuik JA, Bari M, van Zadelhoff G, Leeflang BR, Veldink GA, Oxygenated Metabolites of Anandamide and 2-Arachidonoylglycerol: Conformational Analysis and Interaction with Cannabinoid Receptors, Membrane Transporter, and Fatty Acid Amide Hydrolase, J. Med. Chem 45 (2002) 3709–3720. [DOI] [PubMed] [Google Scholar]
- [18].Godlewski G, Alapafuja SO, Batkai S, Nikas SP, Cinar R, Offertaler L, Osei-Hyiaman D, Liu J, Mukhopadhyay B, Harvey-White J, Tam J, Pacak K, Blankman JL, Cravatt BF, Makriyannis A, Kunos G, Inhibitor of fatty acid amide hydrolase normalizes cardiovascular function in hypertension without adverse metabolic effects, Chem. Biol 17 (2010) 1256–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Galal AM, Slade D, Gul W, El-Alfy AT, Ferreira D, Elsohly MA, Naturally occurring and related synthetic cannabinoids and their potential therapeutic applications, Recent Pat CNS Drug Discov 4 (2009) 112–136. [DOI] [PubMed] [Google Scholar]
- [20].Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulamt R, Isolation and Structure of a Brain Constituent That Binds to the Cannabinoid Receptor, Science 258 (1992) 1946–1949. [DOI] [PubMed] [Google Scholar]
- [21].Clapper JR, Moreno-Sanz G, Russo R, Guijarro A, Vacondio F, Duranti A, Tontini A, Sanchini S, Sciolino NR, Spradley JM, Hohmann AG, Calignano A, Mor M, Tarzia G, Piomelli D, Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism, Nat. Neurosci 13 (2010) 1265–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Aceto MD, Scates SM, Razdan RK, Martin BR, Anandamide, an endogenous cannabinoid, has a very low physical dependence potential, J. Pharmacol. Exp. Ther 287 (1998) 598–605. [PubMed] [Google Scholar]
- [23].Thanos PK, Clavin BH, Hamilton J, O’Rourke JR, Maher T, Koumas C, Miao E, Lankop J, Elhage A, Haj-Dahmane S, Deutsch D, Kaczocha M, Examination of the Addictive and Behavioral Properties of Fatty Acid-Binding Protein Inhibitor SBFI26, Front. Psychiatry 7 (2016) 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhuang L, Li C, Chen Q, Jin Q, Wu L, Lu L, Yan X, Chen K, Fatty acid-binding protein 3 contributes to ischemic heart injury by regulating cardiac myocyte apoptosis and MAPK pathways, Am. J. Physiol. Heart. Circ. Physiol 316 (2019) H971–H984. [DOI] [PubMed] [Google Scholar]
- [25].Matsuo K, Cheng A, Yabuki Y, Takahata I, Miyachi H, Fukunaga K, Inhibition of MPTP-induced alpha-synuclein oligomerization by fatty acid-binding protein 3 ligand in MPTP-treated mice, Neuropharmacology 150 (2019) 164–174. [DOI] [PubMed] [Google Scholar]
- [26].Haga H, Yamada R, Izumi H, Shinoda Y, Kawahata I, Miyachi H, Fukunaga K, Novel fatty acid-binding protein 3 ligand inhibits dopaminergic neuronal death and improves motor and cognitive impairments in Parkinson’s disease model mice, Pharmacol. Biochem. Behav 191 (2020), 172891. [DOI] [PubMed] [Google Scholar]
- [27].Shimizu F, Watanabe TK, Shinomiya H, Nakamura Y, Fujiwara T, Isolation and expression of a cDNA for human brain fatty acid–binding protein (B-FABP), BBA 4 (1997) 24–28. [DOI] [PubMed] [Google Scholar]
- [28].Sanchez-Font MF, Bosch-Comas A, Gonzalez-Duarte R, Marfany G, Overexpression of FABP7 in Down syndrome fetal brains is associated with PKNOX1 gene-dosage imbalance, Nucleic Acids Res. 31 (2003) 2769–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Watanabe A, Toyota T, Owada Y, Hayashi T, Iwayama Y, Matsumata M, Ishitsuka Y, Nakaya A, Maekawa M, Ohnishi T, Arai R, Sakurai K, Yamada K, Kondo H, Hashimoto K, Osumi N, Yoshikawa T, Fabp7 maps to a quantitative trait locus for a schizophrenia endophenotype, PLoS Biol. 5 (2007) e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Watanabe R, Fujii H, Yamamoto A, Hashimoto T, Kameda K, Immunohistochemical distribution of cutaneous fatty acid–binding protein in human skin, J. Dermatol. Sci 16 (1997) 17–22. [DOI] [PubMed] [Google Scholar]
- [31].Mita R, Coles JE, Glubrecht DD, Sung R, Sun X, B-FABP-expressing radial glial cells: the malignant glioma cell of origin? Neoplasia 9 (2007) 734–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Berger WT, Ralph BP, Kaczocha M, Sun J, Balius TE, Rizzo RC, Haj-Dahmane S, Ojima I, Deutsch DG, Targeting fatty acid binding protein (FABP) anandamide transporters - a novel strategy for development of anti-inflammatory and anti-nociceptive drugs, PLoS ONE 7 (2012) e50968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hsu HC, Tong S, Zhou Y, Elmes MW, Yan S, Kaczocha M, Deutsch DG, Rizzo RC, Ojima I, Li H, The Antinociceptive Agent SBFI-26 Binds to Anandamide Transporters FABP5 and FABP7 at Two Different Sites, Biochemistry 56 (2017) 3454–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yan S, Elmes MW, Tong S, Hu K, Awwa M, Teng GYH, Jing Y, Freitag M, Gan Q, Clement T, Wei L, Sweeney JM, Joseph OM, Che J, Carbonetti GS, Wang L, Bogdan DM, Falcone J, Smietalo N, Zhou Y, Ralph B, Hsu HC, Li H, Rizzo RC, Deutsch DG, Kaczocha M, Ojima I, SAR studies on truxillic acid mono esters as a new class of antinociceptive agents targeting fatty acid binding proteins, Eur. J. Med. Chem 154 (2018) 233–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Binas B, Danneberg H, McWhir J, Mullins L, Clark AJ, Requirement for the heart-type fatty acid binding protein in cardiac fatty acid utilization, FASEB J. 13 (1999) 805–812. [DOI] [PubMed] [Google Scholar]
- [36].Owada Y, Abdelwahab SA, Kitanaka N, Sakagami H, Takano H, Sugitani Y, Sugawara L, Kawashima H, Kiso Y, Mobarakeh JI, Yanai K, Kaneko K, Sasaki H, Kato H, Saino-Saito S, Matsumoto N, Akaike N, Noda T, Kondo H, Altered emotional behavioral responses in mice lacking brain-type fatty acid-binding protein gene, Eur. J. Neurosci 24 (2006) 175–187. [DOI] [PubMed] [Google Scholar]
- [37].Shimamoto C, Ohnishi T, Maekawa M, Watanabe A, Ohba H, Arai R, Iwayama Y, Hisano Y, Toyota T, Toyoshima M, Suzuki K, Shirayama Y, Nakamura K, Mori N, Owada Y, Kobayashi T, Yoshikawa T, Functional characterization of FABP3, 5 and 7 gene variants identified in schizophrenia and autism spectrum disorder and mouse behavioral studies, Hum. Mol. Genet 23 (2014) 6495–6511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vellturo FA, Griffin GW, Electrolytic Oxidation of Cyclobutane-1,3-dicarboxylic Acids. An Electrochemical Synthesis of 2,4-Dicarbomethoxybicyclobutane, J. Org. Chem 31 (1966) 2241–2244. [Google Scholar]
- [39].Beutner GL, Young IS, Davies ML, Hickey MR, Park H, Stevens JM, Ye Q, TCFH–NMI: Direct Access to N-Acyl Imidazoliums for Challenging Amide Bond Formations, Org. Lett 20 (2018) 4218–4222. [DOI] [PubMed] [Google Scholar]
- [40].Peng X, Studholme K, Kanjiya MP, Luk J, Bogdan D, Elmes MW, Carbonetti G, Tong S, Gary Teng YH, Rizzo RC, Li H, Deutsch DG, Ojima I, Rebecchi MJ, Puopolo M, Kaczocha M, Fatty-acid-binding protein inhibition produces analgesic effects through peripheral and central mechanisms, Mol. Pain 13 (2017), 1744806917697007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bogdan D, Falcone J, Kanjiya MP, Park SH, Carbonetti G, Studholme K, Gomez M, Lu Y, Elmes MW, Smietalo N, Yan S, Ojima I, Puopolo M, Kaczocha M, Fatty acid-binding protein 5 controls microsomal prostaglandin E synthase 1 (mPGES-1) induction during inflammation, J. Biol. Chem 293 (2018) 5295–5306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Gfeller D, Grosdidier A, Wirth M, Daina A, Michielin O, Zoete V, SwissTargetPrediction: a web server for target prediction of bioactive small molecules, Nucleic Acids Res. 42 (2014) W32–W38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sterling T, Irwin JJ, ZINC 15–Ligand Discovery for Everyone, J. Chem. Inf. Model 55 (2015) 2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Hanwell MD, Curtis DE, Lonie DC, Vandermeersch T, Zurek E, Hutchison GR, Avogadro: an advanced semantic chemical editor, visualization, and analysis platform, J Cheminform 4 (2012) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Halgren TA, Merck molecular force field.1. Basis, form, scope, parameterization, and performance of MMFF94, J. Comput. Chem 17 (1996) 490–519. [Google Scholar]
- [46].Halgren TA, Merck molecular force field.2. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions, J. Comput. Chem 17 (1996) 520–552. [Google Scholar]
- [47].Halgren TA, Merck molecular force field.3. Molecular geometries and vibrational frequencies for MMFF94, J. Comput. Chem 17 (1996) 553–586. [Google Scholar]
- [48].Halgren TA, Nachbar RB, Merck molecular force field.4. Conformational energies and geometries for MMFF94, J. Comput. Chem 17 (1996) 587–615. [Google Scholar]
- [49].Halgren TA, Merck molecular force field.5. Extension of MMFF94 using experimental data, additional computational data, and empirical rules, J. Comput. Chem 17 (1996) 616–641. [Google Scholar]
- [50].Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ, AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility, J. Comput. Chem 30 (2009) 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.