

Abstract
It is currently unclear at which point during viral replication that RNA genomes are first recognized as nonself by the immune system. In this issue of Cell Host & Microbe, Weber et al. show that incoming nucleocapsid-bound genomes are sufficient to bind and activate innate immune sensors.
In the constant struggle between pathogens and their hosts, the evolution of the immune system was a critical turning point. Most vertebrates can mount adaptive immune responses in which specific recognition of the invading microbe often leads to long-term protection from reinfection. In contrast, almost all living organisms exhibit some form of innate immunity. The innate immune system provides a more generic and limited response to infection and, in vertebrates, serves to augment the subsequent adaptive response. The effectors of the innate immune response include many types of receptors, both membrane bound and soluble in the cytoplasm and extracellular milieu, which bind to structural motifs known as pathogen-associated molecular patterns (PAMPs) (Janeway and Medzhitov, 2002). One of the more well studied of these groups of pathogen recognition receptors (PRRs) is the family of RIG-I-like proteins, consisting of RIG-I, MDA5, and LGP2, that recognize viral PAMPs. Although these molecules have closely related sequences and structures, they recognize PAMPs from different viruses. For example, MDA5 can interact with long double-stranded RNA (dsRNA) and is important for resistance to picornavirues (Kato et al., 2006). In contrast, RIG-I can interact with short dsRNA, as well as both single- and double-stranded RNA containing a 5′ triphosphate (5′ PPP) group, and is critical for the response to infection by influenza and other negative sense single-stranded RNA viruses (Kato et al., 2006). Ligand-receptor binding induces a conformational change that facilitates interactions between the PRR and downstream signaling molecules (Figure 1) (Cui et al., 2008). These binding partners act as scaffolds to assemble large complexes at internal organelle membranes. Current evidence suggests that the location of the assembled complexes plays an important role in the cellular response. In the case of RIG-I and the downstream adaptor protein IPS-1 (also referred to as MAVS, VISA, and Cardiff), an interaction has been detected at both peroxisomes and mitochondria (Dixit et al., 2010). Signaling from the mitochondrial location leads to activation of numerous transcription factors necessary for expression of interferon-beta (IFN-β). After secretion, IFN-β binds to its receptor at the cell surface and activates the expression of interferon-stimulated genes (ISGs) in both an autocrine and paracrine manner. Binding of RIG-I and IPS-1 at peroxisomes yields a faster response by directly activating select ISGs independent of interferon production (Dixit et al., 2010). The net result of signaling from these organelle membranes is the production of an antiviral state in both the infected and in the surrounding cells (Figure 1).
Figure 1. Nature and Availability of RIG-I Ligands during the Virus Replication Cycle.
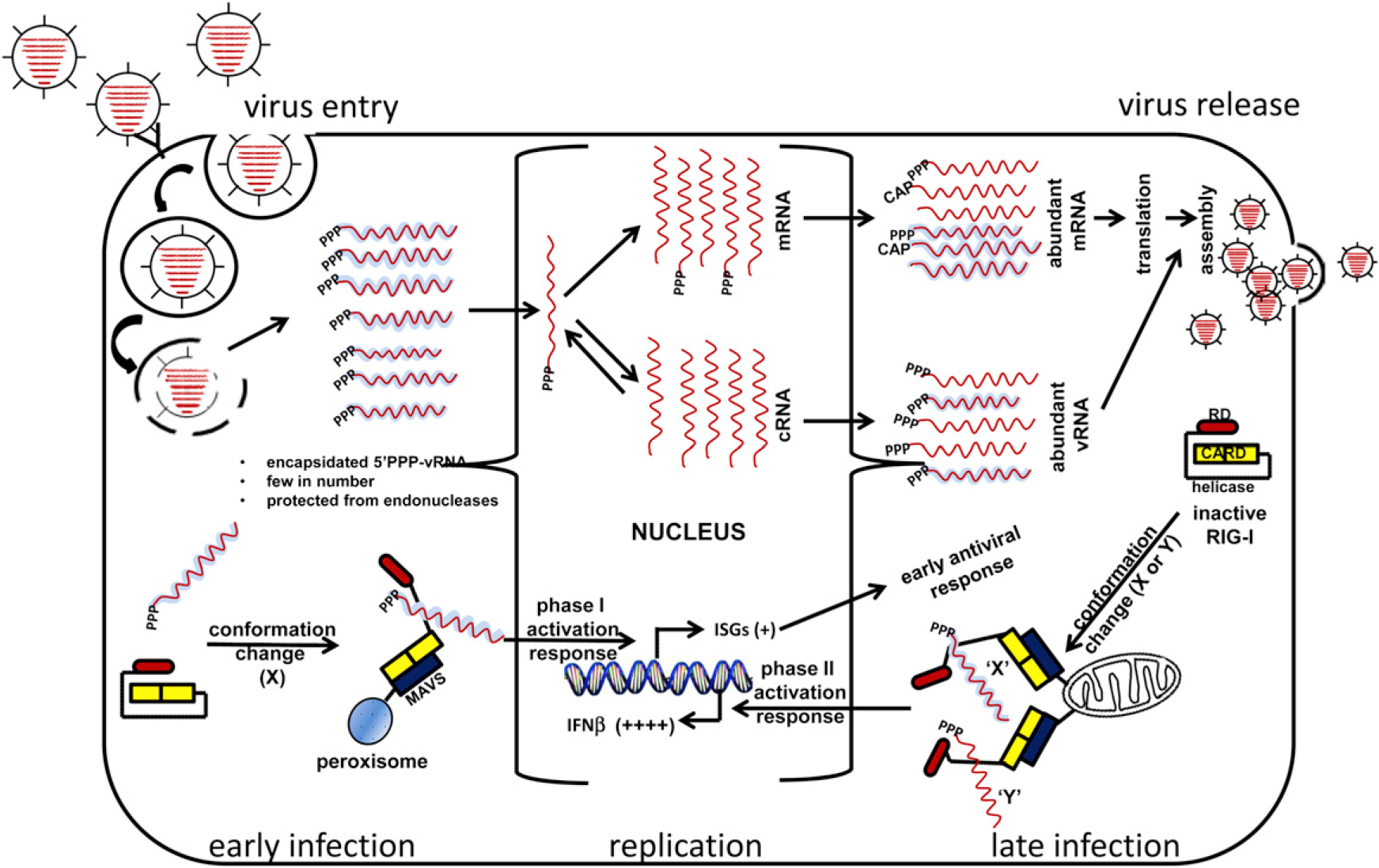
Encapsidated viral RNA with 5′ PPP groups and proper secondary structures for RIG-I recognition are relatively few in number shortly after virus entry. The coated viral RNA (vRNA) interacts with the C-terminal domain of RIG-I to allow activation of RIG-I and association with IPS-1 present on peroxisomes to initiate ISGs mediated early antiviral response. vRNA is replicated and transcribed in the nucleus (in the case of influenza A virus, illustrated here) or in the cytoplasm (many other RNA viruses) to produce abundant messenger RNA (mRNA) (coated or capped?) and vRNA with 5′ PPP (coated?) available for optimal RIG-I activation and IFN production via IPS interaction present on mitochondria. Although RIG-I can be activated by either naked or encapsidated viral RNA, whether the different ligands induce similar or different structural rearrangements (“X” or “Y” conformation) is not clear.
To initiate these complex signaling pathways, RIG-I must sense the presence of an invading virus and bind to the recognized PAMP. The RNA viruses that RIG-I responds to synthesize their genomes with RNA-dependent RNA polymerases (RdRps) (Makeyev and Bamford, 2001). Unlike DNA polymerases, most RdRps do not need a primer to initiate synthesis, resulting in transcripts that are initiated by a 5′ PPP group. The requirements for RIG-I binding to synthetic ligands containing 5′ PPP have been dissected extensively under carefully controlled conditions. Mutagenesis of synthetic ligands followed by transfection into cells and observation of RIG-I activation has provided great insight into the minimal requirements necessary for the induction of innate immunity (Hornung et al., 2006; Pichlmair et al., 2006; Davis et al., 2012). In addition, structural analyses of RIG-I bound to cognate substrates have yielded information about critical contacts between ligand and receptor and led to models of possible mechanisms of RIG-I activation. However, much less is known about the nature of the authentic substrate for RIG-I during an actual infection. For instance, influenza A virus packages eight genomic segments containing 5′ PPP. Although these RNA molecules should be ideal substrates for RIG-I, many studies indicate that viral replication is a requirement for a robust RIG-I-mediated immune response (Rehwinkel et al., 2010). An explanation for these findings has not yet been determined, but possibilities include that early during infection available targets are much less abundant and that nucleocapsid proteins bound to the viral genome interfere with RIG-I access. Now, in this issue of Cell Host & Microbe, Weber et al. (2013) provide evidence that suggests that not only are viral genomes from incoming virions sufficient for induction of the RIG-I-mediated immune response, but also that these RIG-I ligands are capable of initiating the innate response while still bound by nucleocapsids.
To address these issues, the authors infected cells with a variety of viruses in the presence of the protein synthesis inhibitor cycloheximide (CHX). Some of the viruses used, such as influenza A virus, are able to transcribe their genomes to generate primary transcripts but are not able to accomplish full-genome replication in the absence of protein synthesis. Others, such as Rift Valley fever virus, cannot even complete primary transcription. In each case, however, innate immune activation, as indicated by the induction of ISGs, was observed. That the innate receptor involved was RIG-I was determined through several lines of experimentation. First, the ability to induce interferon in the presence of CHX was not common to all viruses tested. Only those exhibiting 5′ PPP groups were able to induce IFN expression in the presence of CHX. Two viruses without free 5′ PPP groups were not able stimulate interferon production when protein synthesis was inhibited, suggesting that the innate response to these viruses is not signaled through the RIG-I pathway. Second, abrogation of RIG-I expression, either through small-interfering-RNA-mediated knockdown or by use of knockout cell lines, eliminated the IFN response. Finally, RIG-I was directly shown to be activated, based on conformational changes and oligomerization, in response to the infecting viruses.
To further examine the interaction between RIG-I and incoming viral genomes, Weber et al. used immunofluorescence to colocalize nucleocapsids and RIG-I. Under conditions of arrested protein synthesis, as well as during full viral replication, nucleocapsids and RIG-I colocalized to peroxisomes. As peroxisomes have been shown to be the site of early immune signaling through IPS-1(Dixit et al., 2010), these results support previous evidence of a biphasic innate immune response and raise the possibility that signaling at peroxisomes is stimulated by incoming viral genomes and later mitochondrial signaling facilitated by a higher quantity products of viral replication. The differences in the components of the two signaling platforms at the distinct cellular locations that allow this biphasic response remain to be determined. This colocalization is supported by coimmunoprecipitation experiments in which RIG-I and nucleocapsids formed complexes in both mammalian and insect cell systems.
This study provides provocative new evidence that full viral replication is not necessary for activation of the RIG-I-mediated arm of the innate immune system and raises the possibility that viral nucleocapsid proteins participate directly in early peroxisome-mediated signaling events. Although it was shown with RNase and phosphatase treatments of the nucleocapsids that the 5′ PPP RNA was essential for RIG-I activation by nucleocapsids, the need for viral RNA for the RIG-I/nucleocapsid colocalization was unclear. The genetic tractability of the Drosophila cell system has the potential to provide further insight into host factors required for the RIG-I/nucleocapsid interaction. It will also be of interest to determine whether or not the nucleocapsid proteins are required for the early initiation of peroxisome signaling. Even if direct interactions between RIG-I and nucleocapsids do not take place, nucleocapsids could function to protect RIG-I ligands long enough for RIG-I binding and peroxisomal localization to take place. Answers to these remaining questions should further illuminate the mechanism of essential immune responses critical for mitigation of viral infection.
ACKNOWLEDGMENTS
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of Centers for Disease Control and Prevention.
REFERENCES
- Cui S, Eisenächer K, Kirchhofer A, Brzózka K, Lammens A, Lammens K, Fujita T, Conzelmann KK, Krug A, and Hopfner KP (2008). Mol. Cell 29, 169–179. [DOI] [PubMed] [Google Scholar]
- Davis WG, Bowzard JB, Sharma SD, Wiens ME, Ranjan P, Gangappa S, Stuchlik O, Pohl J, Donis RO, Katz JM, et al. (2012). PLoS ONE 7, e32661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, et al. (2010). Cell 141, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzózka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, et al. (2006). Science 314, 994–997. [DOI] [PubMed] [Google Scholar]
- Janeway CA Jr., and Medzhitov R (2002). Annu. Rev. Immunol. 20, 197–216. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. (2006). Nature 441, 101–105. [DOI] [PubMed] [Google Scholar]
- Makeyev EV, and Bamford DH (2001). RNA 7, 774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, and Reis e Sousa C (2006). Science 314, 997–1001. [DOI] [PubMed] [Google Scholar]
- Rehwinkel J, Tan CP, Goubau D, Schulz O, Pichlmair A, Bier K, Robb N, Vreede F, Barclay W, Fodor E, and Reis e Sousa C (2010). Cell 140, 397–408. [DOI] [PubMed] [Google Scholar]
- Weber M, Gawanbacht A, Habjan M, Rang A, Borner C, Schmidt AM, Veitinger S, Jacob R, Devignot S, Kochs G, et al. (2013). Cell Host Microbe 13, this issue, 336–346. [DOI] [PMC free article] [PubMed] [Google Scholar]