

Abstract
Most cancer-related deaths are caused by the metastases, which commonly develop at multiple organ sites including the brain, bone, and lungs. Despite longstanding observations that the spread of cancer is not random, our understanding of the mechanisms that underlie metastatic spread to specific organs remains limited. However, metabolism has recently emerged as an important contributor to metastasis. Amino acids are a significant nutrient source to cancer cells and their metabolism which can serve to fuel biosynthetic pathways capable of facilitating cell survival and tumor expansion while also defending against oxidative stress. Compared to the primary tumor, each of the common metastatic sites exhibit vastly different nutrient compositions and environmental stressors, necessitating the need of cancer cells to metabolically thrive in their new environment during colonization and outgrowth. This review seeks to summarize the current literature on amino acid metabolism pathways that support metastasis to common secondary sites, including impacts on immune responses. Understanding the role of amino acids in secondary organ sites may offer opportunities for therapeutic inhibition of cancer metastasis.
INTRODUCTION
The metastatic spread of tumor cells from a primary tumor to distant organs is the main contributor to cancer-related death and is still considered to be largely incurable. Despite the undeniable clinical prevalence and impact of metastases, the metastatic cascade itself is highly inefficient—only 0.02% of circulating tumor cells (CTCs) will go on to produce clinically detectable metastases [1]. In 1889, English surgeon Stephen Paget observed that the secondary sites to which tumors metastasized did not seem to be completely random. He hypothesized a “seed and soil” model of metastasis, postulating that CTCs, the “seeds,” could not successfully take root and establish secondary tumors if the “soil” or the metastatic site, was not a suitable environment for outgrowth. To extend Paget’s metaphor, recent evidence indicates that the prevalence of nutrients in this “soil” could be a key aspect in the fitness of tumor cells.
Since the first observations of the Warburg effect, researchers have been investigating how nutrients influence the behaviors of tumor cells. Today, cancer-associated metabolic adaptation is widely considered to be a hallmark of cancer and an essential component of metastatic capability. Of particular significance, cancer cells utilize amino acids and their various metabolic byproducts in a myriad of critical processes that support the survival and progression of cancer cells (Fig. 1). First, and perhaps most obviously, rapidly dividing cancer cells must maintain a sufficient amino acid pool from which to pull building blocks for protein biosynthesis [2]. These amino acids are capable of entering the tricarboxylic acid (TCA) cycle to contribute to the cell’s supply of ATP [2] and of being converted to lipids and nucleosides [3–5]. Beyond this, amino acids can act as nutrient signals, including as neurotransmitters, to trigger the activation of important signaling pathways, and play key roles in the epigenetic modification and regulation of gene expression [6–8]. Finally, these important biomolecules function in the maintenance of intracellular redox status via the production of the antioxidant glutathione [3, 5, 9].
Fig. 1. Amino acid metabolic pathways critical in metastasis and key Inhibitors.
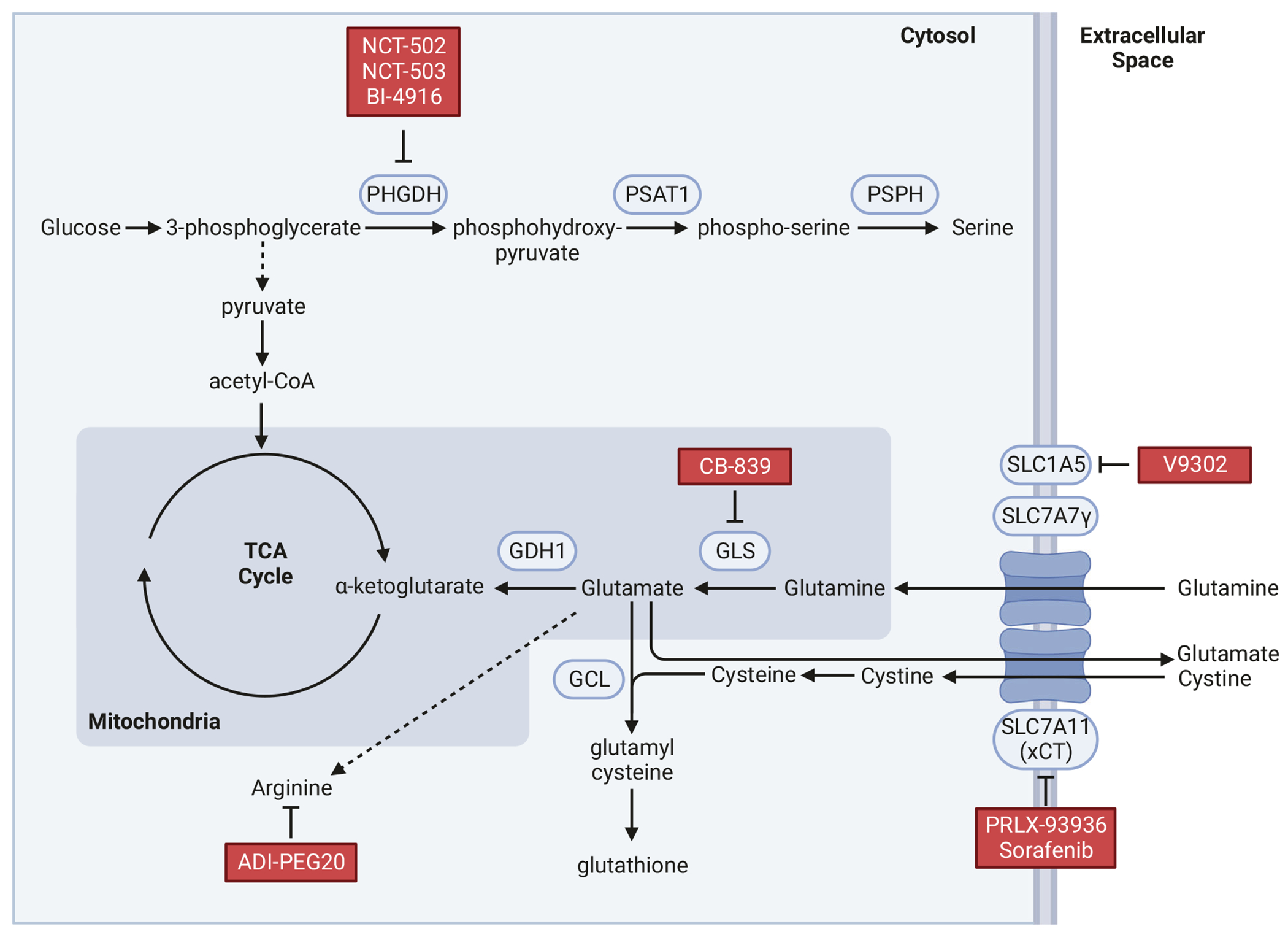
A simplified diagram of major pathways of selected amino acid metabolism relevant to metastasis to the secondary sites. Inhibitors in Table 1 for specific enzymes and nutrient transporters are indicated.
In this review, we will discuss the ways in which tumor cells leverage amino acid metabolism to effectively survive and form macro-metastases in a variety of secondary tissues. While other critical metabolic pathways like glycolysis and fatty acid oxidation contribute to amino acid biosynthesis in cancer cells, their importance in metastasis has been reviewed elsewhere [2–5, 9–11]. We also summarize the impact of amino acid on immune cells in tumor microenvironment and potentials for leveraging this knowledge for therapeutic intervention.
METABOLIC ADAPTATION IN METASTASIS
The metastatic cascade consists of multiple steps, including invasion of tumor cells into surrounding tissues, intravasation and survival in the circulation, and extravasation and outgrowth into distant niches (Fig. 2). In the earliest steps, metastatic cells at the primary tumor acquire migratory and invasive capabilities that are energy demanding, necessitating sufficient ATP supplies [12]. Indeed, enhancing amino acid metabolism can support the necessary elevation in ATP production. Invasive ovarian cancer cell lines are more glutamine dependent than those that are non-invasive, while genetically targeting glutaminase (GLS), an enzyme that converts glutamine to glutamate, reduces the invasive and metastatic capacity of colorectal cancer cells [13–15]. Similarly, overexpression of PSAT1, an important enzyme involved in the serine biosynthetic pathway, promotes invasion and metastatic colonization of non-small cell lung cancer cells [16]. Asparagine utilization supports a migratory and invasive phenotype in breast cancer cells through induction of the epithelial-to-mesenchymal transition (EMT) [17], indicating that metabolic rewiring can directly support metastatic progression. Other evidence suggests that reprogramming of amino acid metabolism may be a consequence of oncogenic signaling or environmental stressors. Increased expression of asparagine synthetase, glutamic oxaloacetic transaminase 2, and GLS are observed upon activation of SOX12, a transcription factor that promotes EMT and is associated with metastatic progression in colorectal and breast cancer patients [18]. Likewise, environmental factors such as hypoxia and purine depletion support elevated glutamine metabolism and serine biosynthesis, respectively [15, 19].
Fig. 2. Amino acid metabolism in the metastatic cascade.
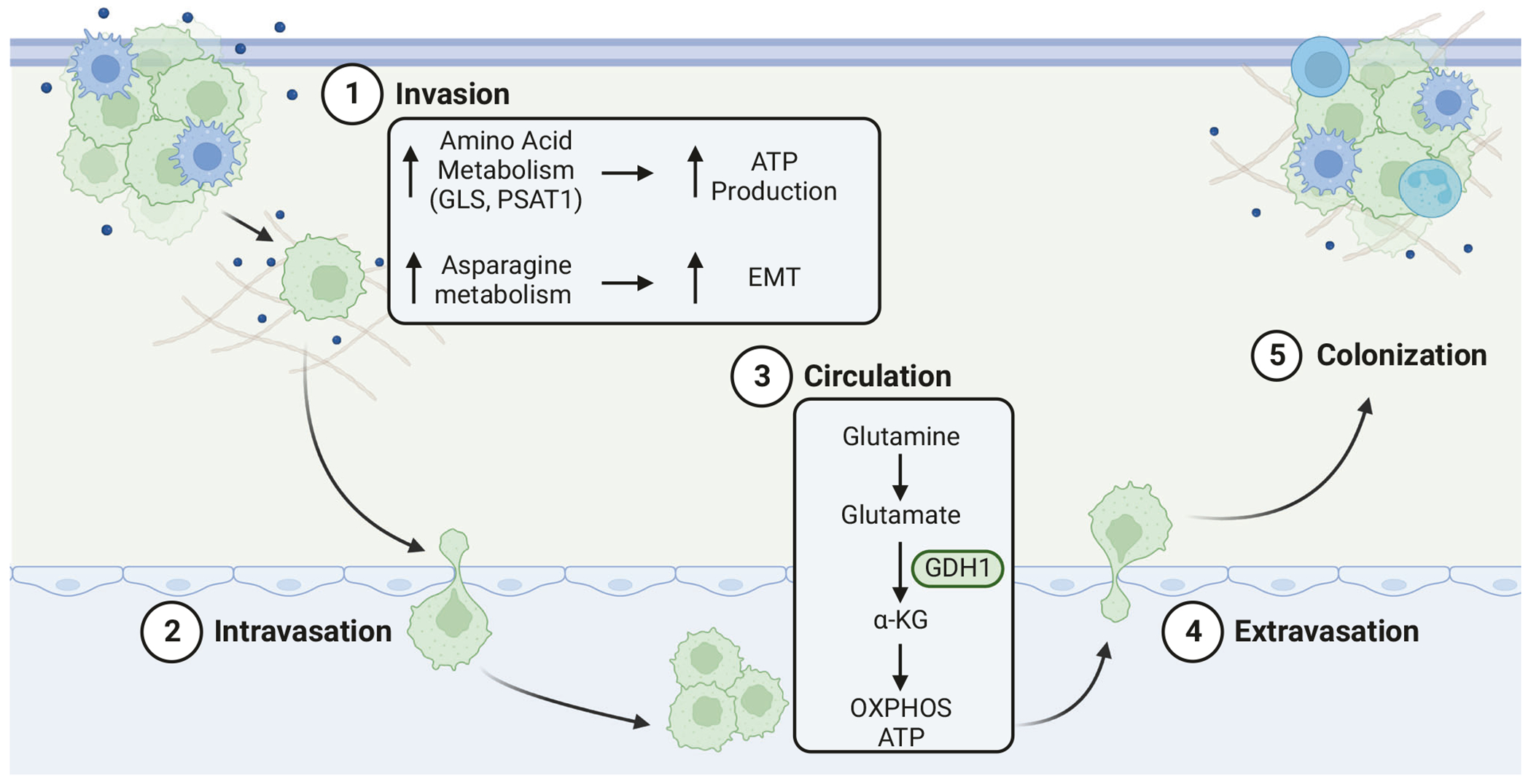
The metastatic cascade consists of multiple steps, including (1) invasion of tumor cells into surrounding tissues, (2) intravasation and (3) survival in the circulation, and (4) extravasation and (5) outgrowth into distant niches.
As disseminating tumor cells enter the vasculature, they encounter a stressful environment where few escaping cells survive. Most circulating tumor cells (CTC) will succumb to anoikis. While evidence related to amino acid metabolism in CTCs is limited, glutamine metabolism may also support survival of CTCs. The conversion of glutamate to α-ketoglutarate by glutamate dehydrogenase promotes anoikis resistance in metastatic lung cancer cells [20]. Consistent with enhanced glutamine metabolism in CTCs, the plasma of metastatic esophageal squamous cell carcinoma patients was found to exhibit reduced glutamine but elevated glutamate levels [21]. Breast cancer CTCs also display a preference for oxidative phosphorylation (OXPHOS) [22–26], possibly reflecting an increased requirement for ATP synthesis. Indeed, glutamine-derived α-ketoglutarate feeds into OXPHOS and ATP production through the TCA cycle. While additional studies are necessary to fully elucidate the role of amino acid metabolism in CTCs, it is intriguing to speculate that many of the same pathways that contribute to survival and migration within the primary tumor may also support metastatic spread through the circulation.
Metastatic tumors that develop at the secondary sites are traditionally thought to behave similarly to the primary tumors. Indeed, in some cases disruption of metabolic pathways leads to similar phenotypes in cancer cells both in primary tumors and secondary metastasis. However, recent studies in single-cell RNA-seq revealed distinct expression profiles in metabolic pathways between primary and secondary lesions [27–29]. Although the mechanism by which different metabolic states arise in primary tumors and metastases is not well understood, at least two possibilities, not mutually exclusive, could be envisioned. Because primary tumors contain heterogenous cancer cell populations, certain metabolic traits previously existing or acquired by subpopulations of cancer cells may have distinct advantages to survive during metastatic spread or during outgrowth in secondary organs. Alternatively, disseminated cancer cells may evolve and adapt to local environment to promote metastatic outgrowth. Regardless of parallel or unique metabolic attributes of primary versus metastatic tumors, organotropism of tumor metastasis could be metabolically affected by many factors, including nutrient availability in organ-specific niches and competition for nutrients between tumor cells and immune cells in the tumor microenvironment.
NUTRIENT AVAILABILITY IN ORGAN-SPECIFIC NICHES
Brain metastasis
Although the brain is an extremely energy-demanding organ, the interstitial environment does not contain a substantial energy reserve, allowing neuronal signaling fidelity [6]. Instead, this critical organ compensates for the lack of reserves through metabolic plasticity, efficiently utilizing a variety of alternative metabolites when blood glucose is low [7, 8]. It is becoming clear that metastatic cancer cells must also demonstrate nutrient flexibility in order to survive in the brain.
While glucose reserves are more limiting, the interstitial space within the brain is highly abundant in both glutamine and branched-chain amino acids (BCAAs) [30–33]. In addition to its role as an energetic and biosynthetic substrate, the brain utilizes glutamine to support a high rate of glutamate synthesis, which can function as a neurotransmitter [34]. It is primarily BCAAs that serve to synthesize glutamate—at least two-thirds of the amino groups incorporated into brain glutamate are taken from BCAA-derived keto acids [32]. There is also evidence that brain metastatic tumor cells utilize these readily available BCAAs. Positron emission tomography detection of brain metastasis revealed improved sensitivity of 11C-BCAA tracers compared to the traditional 18FDG glucose analog, suggesting BCAAs are a more favored fuel source of brain metastatic cells [35]. Indeed, brain-tropic triple-negative breast cancer (TNBC) cells possess more activated branched-chain ketoacid dehydrogenase E1 and oxidize more BCAAs than their parental counterparts [36]. These findings suggest that brain metastatic cells efficiently utilize BCAAs to fulfill their energetic needs.
The importance of glutamine metabolism in brain metastatic cells was recently illustrated by Parida and colleagues in a HER2+ breast cancer brain metastasis model [37]. Cells derived from latent or metachronous metastases relied on glutamine metabolism, taking more time to influence the brain microenvironment. This is in contrast to cells derived from rapidly-forming synchronous tumors, which were highly proficient in glucose metabolism, allowing them to outcompete brain native cells for limited environmental glucose. These findings suggest that the interactions between the brain microenvironment and metabolic programming of brain metastatic tumor cells play a significant role in metastatic outgrowth. Importantly, glutamine-dependent disseminated tumor cells (DTCs) were found to be resistant to HER2-targeted therapies at least partially through improved protection against oxidative stress. Glutamine utilizing DTCs exhibited increased expression of the SLC7A11/xCT cystine/glutamate antiporter to support synthesis of glutathione (GSH), a prominent ROS sink [37]. Sensitivity to HER2 inhibitors was restored through xCT inhibition, suggesting that glutamine metabolism serves to support biosynthetic pathways as well as redox balance during metastatic outgrowth in the brain.
Metabolic adaptation to ROS is not the only way that brain metastatic cells modulate glutamine metabolism to fit into their new niche. In fact, brain metastatic breast cancer cells have been shown to parasitize a glutamate-dependent neuronal signaling pathway to steal nutrients [38]. Glutamate is released by excitatory glutamatergic presynaptic neurons and quickly taken up by postsynaptic neurons [39]. Brain metastatic breast cancer cells appear to co-opt this process, establishing pseudo-tripartite synapses to access glutamate secreted by presynaptic neurons (Fig. 3A). Targeting the glutamate N-methyl-D-aspartate receptor in breast cancer cells significantly decreased brain metastatic burden not affecting the growth of primary orthotopic tumors or lung metastases [38]. Unfortunately, targeting synaptic glutamate theft may be difficult to put into practice due to potential neurotoxicity. Of note, primary glioma cells also appear to “steal” glutamate from presynaptic neurons by upregulating glutamine and glutamate transporters [34, 40, 41]. This similarity in adaptation strategies of brain metastatic and primary brain cancer cells supports the idea that the “soil” governs metabolic adaptations in cancer cells.
Fig. 3. Amino acid metabolism in secondary niches.
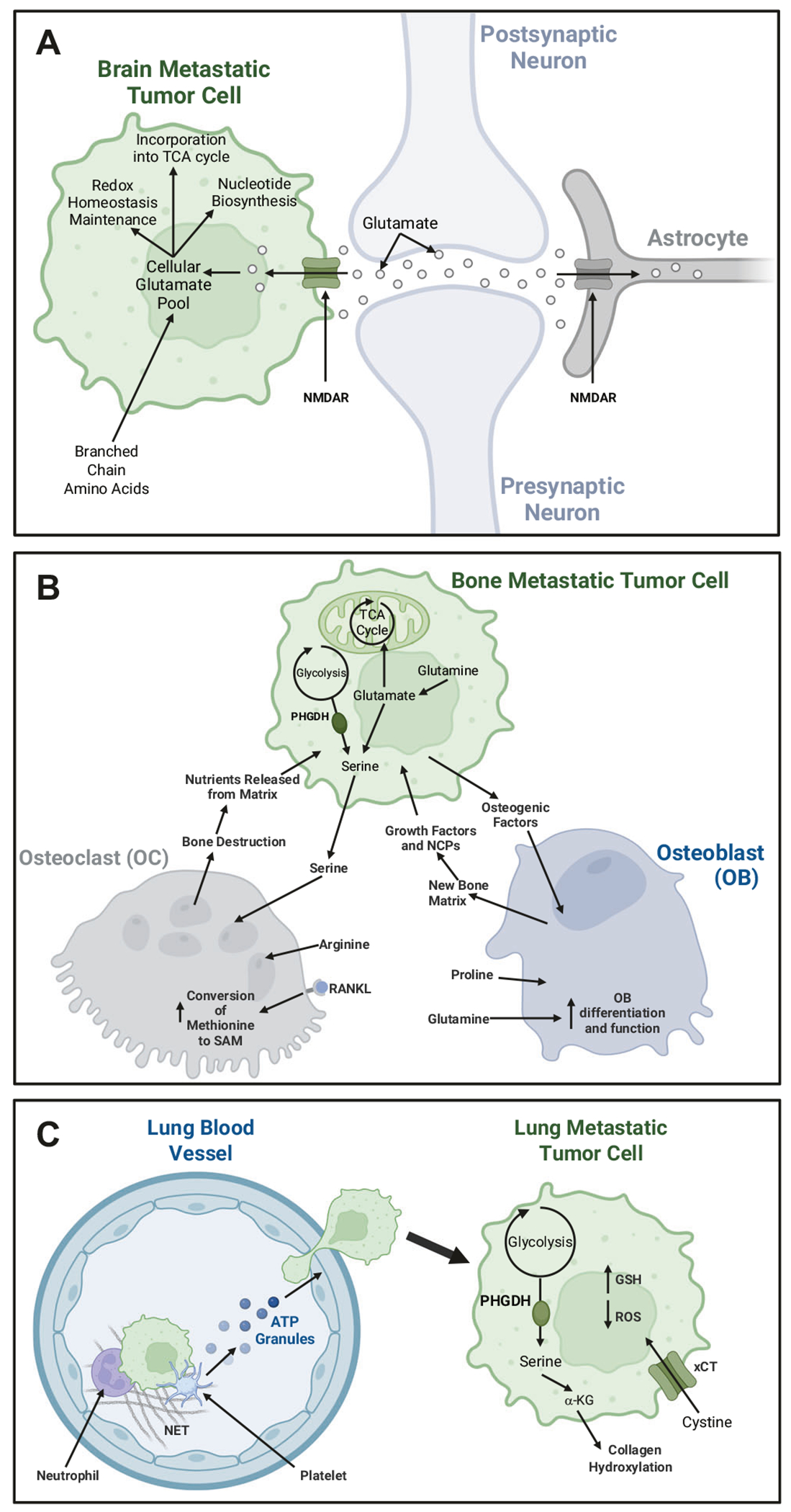
A Amino Acid Metabolism in the Brain Metastatic niche: Brain-metastatic cells take up large amounts of branched-chain amino acids (BCAAs) to meet their energetic needs and upregulate enzymes to oxidize glutamine and BCAAs. The metastatic cells also parasitize a glutamine-dependent neuronal signaling pathway to steal nutrients. B Amino Acid Metabolism in the Bone Metastatic Niche: In the bone niche, glutamine metabolism and proline are essential for osteoblast differentiation and linage commitment. While arginine and methionine appear to be critical in osteoclast differentiation and function. Bone metastatic breast cancer cells have also been shown to promote osteoclast activation through modulation of serine metabolism. C Amino Acid Metabolism in the Lung Metastatic Niche: During metastasis to the lung, modification of arginine residues is essential to the formation of neutrophil extracellular traps (NETs), which allow CTCs to survive in the bloodstream. Lung metastatic breast cancer lesions, but not primary breast tumors, use phosphoglycerate dehydrogenase (PHGDH) and the serine biosynthesis pathway to support mTORC1-mediated growth signaling. Production of α-ketoglutarate (α-KG) activates collagen hydroxylation, allowing DTCs to favorably remodel the extracellular matrix in the lung metastatic niche. Increased xCT expression is also increased in metastatic cells, supporting glutathione (GSH) production to combat oxidative damages.
Despite the prevalence of glutamine and BCAAs in brain tissue, levels of other critical amino acids are greatly restricted, challenging metastatic tumor growth. To generate sufficient nucleotide pools, highly aggressive brain metastatic cells increase de novo biosynthesis of serine by enhancing expression of phosphoglycerate dehydrogenase (PHGDH), which catalyzes the rate-limiting step [42]. Pharmacological inhibition and genetic attenuation of PHGDH suppressed growth of brain metastases but not extracranial metastases or primary tumors [42]. Interestingly, aggressive brain metastatic clonal derivatives of the breast cancer cell line MDA-MB-231 exhibited a higher increase in glucose-derived serine than similar indolent lines. This observation dovetails nicely with Parida and colleagues’ model of aggressive glucose-dependent and more latent glutamine-dependent brain metastatic breast cancer cells [37]. Taken together, these data suggest that brain-tropic DTCs must adopt a similar metabolic cooperativity to cells native to the brain milieu to successfully compete for the limited energy reserves.
Bone metastasis
Bone is among the most common sites of metastasis and bone lesions are a major cause of the morbidity [43]. A greater frequency of bone metastases is osteolytic, which stimulates bone destruction [8]. Breakdown of the hard bone tissue seems to be necessary to provide tumor cells with additional space to expand [44]. Notably, this bone destruction is not directly mediated by bone metastatic tumor cells themselves, but rather through activation of osteoclasts, bone-native cells that mediate bone resorption [45]. This phenomenon is known as the “vicious cycle” of bone metastasis, which is initiated by DTCs that secrete cytokines and other factors such as parathyroid hormone-related protein (PTHrP) to act on osteoblasts and trigger the production of RANKL and other osteolytic factors. These factors drive osteoclast differentiation and increased bone destruction that releases growth factors like TGF-β that further stimulate tumor cell proliferation [46]. While breast-to-bone metastases are often osteolytic, the majority of prostate cancer bone metastases are osteoblastic lesions, which stimulate abnormal bone formation [8]. While the advantages of osteoblastic lesions are less apparent, these cancer cells produce osteogenic factors to activate osteoblasts to deposit a new, but not yet mineralized matrix, that is rich in growth factors [47]. This especially fertile “soil” is quickly co-opted by tumor cells [47].
Like cancer cells, amino acids are integral to the identity and function of bone-remodeling cells. During differentiation, osteoblasts increase expression of the glutamine transporters SLC1A5/ASCT2 and SLC7A7/γ(+)-LAT1 to stimulate glutamine uptake [48]. Once inside the cell, glutamine catabolism by GLS has been shown to regulate lineage allocation of skeletal stem cells. Conditional ablation of GLS expression in mesenchymal progenitors decreased their proliferation and bone formation in mice [49]. Specifically, GLS was identified as a requirement for commitment to the osteoblast lineage since loss of GLS appears to bias toward adipogenesis [49]. While glutamine appears to be critical for osteoblast differentiation, evidence suggests that glutamine serves as an important precursor to proline, which is necessary for translation of proline-rich bone matrix proteins (OCN, COL1A1) and other osteoblast-associated proteins (OSX, RUNX2). Expression was found to be dependent on sufficient exogenous glutamine and proline supplied by SLC38A2/SNAT2 uptake [50], suggesting that environmental supplies of proline or glutamine can significantly impact osteoblast differentiation and bone formation (Fig. 3B). Tumor cells have been demonstrated to be one of the primary consumers of glutamine in tumors, resulting in a limited supply of interstitial glutamine [51, 52]. Therefore, it is reasonable to postulate that amino acids may become limiting during metastatic outgrowth to alter normal osteoblast function [53].
Similar to osteoblasts, osteoclasts upregulate expression of SLC1A5/ASCT2 and GLS to enhance glutamine uptake and utilization required for activation [54]. However, α-ketoglutarate supplementation epigenetically promotes SLC7A11/xCT, reducing osteoclastogenesis through ROS-dependent induction of NFATc1 osteoclast transcriptional program [55]. While counterintuitive to osteolytic progression, the relative levels of glutamate and α-ketoglutarate may be limited through BCAA metabolism. RANKL-induced osteoclast differentiation steadily accumulates intracellular BCAAs, which are catalyzed by branched-chain aminotransferase 1 (BCAT1) to generate glutamate and alpha-keto acids [56]. Osteoclasts also appear to readily utilize arginine and methionine for energy production through anaplerosis or epigenetic repression of anti-osteoclastogenic genes upon RANKL stimulation [57, 58] (Fig. 3B). Lastly, evidence suggests a potential cooperation for serine may exists between osteoclasts and cancer cells. A bone tropic variant of the MDA-MB-231 human breast cancer cell line significantly upregulated expression of key genes involved in the serine synthesis pathway, including PHGDH, phosphoserine aminotransferase 1 (PSAT1), and phosphoserine phosphatase [59]. Excess serine exported into the tumor interstitium by ASCT2 [60] can serve to stimulate the differentiation of osteoclasts and bone resorption in vitro [59, 61]. Although bone resorption is a energetically demanding process, evidence supports the concept that osteoclasts are more metabolically adaptable to limiting nutrients within the bone metastatic microenvironment.
Lung metastasis
As with other organ sites, CTCs must successfully extravasate through the lung endothelium and adapt themselves to the nutrient milieu of their new home. Circulating neutrophils and platelets in the lung microvasculature interact with platelets and CTCs to support successful extravasation of CTCs into the lungs [62, 63]. Neutrophils can release web-like extracellular chromatin networks, called neutrophil extracellular traps (NETs), that can facilitate the clustering of CTCs by “catching” and holding them together, thus improving their chances of survival in circulation [63, 64] (Fig. 3C). Circulating NET levels were significantly increased in advanced esophageal, gastric, and lung cancer patients compared to patients with local disease or healthy controls [63]. Interestingly, the process of NETosis is highly dependent on amino acids, controlled by peptidyl arginine deaminase type IV, which converts arginine to citrulline on histones to support NET formation and nuclear membrane disruption [63, 65]. In platelets, the purine nucleotide adenine is converted to ATP and packaged in dense granules that are released upon interaction with CTCs [66]. Released adenine binds to purinergic receptors (P2Y2) on endothelial cells to facilitate extravasation [66, 67]. Consistently, targeting this cellular interaction using chronic low-dose aspirin therapy or blockade of the adenosine-P2Y2 signaling axis reduced tumor cell metastasis in mouse models [66, 67]. Taken together, these findings indicate how amino acid metabolism may contribute to the high rate of lung metastases despite the relative competence of the lung endothelial barrier.
Recent data has shown that lung metastatic breast cancer lesions upregulate the serine biosynthetic pathway compared to the primary tumor. The lung metastatic niche is enriched with the glycolytic product pyruvate, which lung metastatic cells utilize via MCT2 upregulation to fuel serine synthesis [68]. A key byproduct of PSAT1 in the serine biosynthetic pathway is α-ketoglutarate (α-KG), an important metabolic activator of mTORC1 signaling. Expression of the rate-limiting enzyme PHGDH was required for sensitivity to mTORC1 inhibitors in breast cancer lung metastases, but not primary tumors [68], indicating the importance of this pathway in lung metastasis. Consistent with this concept, the PHGDH inhibitor BI-4916 has no effect on the growth of primary breast tumors, but strongly inhibits pulmonary metastases in mice [69]. Further insight into the mechanism of how this metabolic switch facilitates breast cancer lung metastasis was provided in a study by Elia et al. where α-KG produced in lung-metastatic breast cancer cells activated collagen hydroxylation, allowing DTCs to favorably remodel the extracellular matrix in the lung metastatic niche [70]. In agreement, several studies have also indicated the importance of GLS and glutamine metabolism, an important producer of α-ketoglutarate, in establishment of lung metastasis in mouse models [52, 71]. Taken together, these data demonstrate how increased de novo serine synthesis is advantageous to lung-metastatic cells (Fig. 3C). It is interesting to note that while upregulation of glutamine metabolism and serine biosynthesis appears to be important in all three major metastatic sites discussed in this review, the mechanisms through which it exerts pro-metastatic effects do appear to be somewhat organ-specific.
The lung’s function as the primary organ of respiration is one of the factors that makes this microenvironment unique. In this capacity, the lung is exposed to high levels of oxygen as well as inhaled toxins, leading to elevated levels of tissue-intrinsic oxidative stress [8, 72]. Logically, this implies that lung metastatic cancer cells would require a dependable system for dealing with ROS. Indeed, lung metastatic breast cancer cells upregulate expression of xCT, which supports GSH production in order to combat ROS [73]. Additionally, a study in colorectal cancer revealed that the BRAFV600E mutation induced expression of glutamate-cysteine ligase (GCL), the rate-limiting enzyme in GSH synthesis [74]. Quenching of ROS through GSH production was shown to be critical for lung metastasis formation but did not affect primary tumor growth or peritoneal metastasis [74]. Interestingly, the GSH pathway may also support development of lung metastasis by promoting cell adhesion. Glutathione peroxidase-1, which serves to reduce peroxides using GSH, acts as a redox safeguard of FAK kinase activation and cell attachment of TNBC cells, a pathway that is necessary for lung metastasis in vivo [75]. These data indicate that DTCs must develop the capacity to metabolically adapt to the increased oxidative stress intrinsic to the lung.
TUMOR–IMMUNE CELL INTERACTION IN THE MICROENVIRONMENT
Tumor cells at different metastatic sites encounter and interact with varied populations of tissue-specific cells. Aside from several specific examples of these interactions discussed above, there is an additional population of “neighbors” that DTCs must contend with no matter where in the body they end up – immune cells. This becomes significant as many of the metabolic pathways that are so integral to cancer cell growth and survival are important to all highly proliferative cells, including both pro- and antitumorigenic immune cells [76–78]. This shared demand for increasing amounts of cellular “fuel” can create a potentially competitive microenvironment as tumor cells and immune cells battle to take up greater amounts of dwindling nutrients. Notably, current work directly probing tumor-immune interactions in specific metastatic sites is limited. This is largely due to experimental challenges posed by specific metastatic models and sites. However, the possible crosstalk between tumor cells and immune cells cannot be discounted when conceptualizing the metastatic niche. For example, the availability of oxygen in the lung is likely not only to impact tumor cells as discussed above, but also affect immune cells. Consequently, investigating the interaction between organ-specific niche cells and immune cells will open up new avenues for targeting organ-specific metastasis.
Based on evidence from the first oncogenic drivers that were found to dysregulate cell metabolism, it was originally hypothesized that cancer cells were the predominant metabolic consumers within the tumor, serving to deplete nutrients in the tumor microenvironment. Early studies suggested that excessive glucose consumption by cancer cells could suppress antitumor functions of T cells [79, 80]. However, later experiments utilizing the labeled glucose analog 18F-2DG showed that cancer cells were not, in fact, the primary consumers of glucose in the tumor microenvironment (TME);rather, experiments showed that they lagged behind both myeloid cells and, to a lesser degree, T cells in glucose uptake. Cancer cells were found to competitively out-consume immune cells for glutamine [51]. Several studies have indicated that targeting glutamine metabolism can increase glutamine availability and improve antitumor immune responses in solid tumors [52, 81, 82]. Inhibition of glutamine metabolism has been shown to further increase glucose uptake by immune cells, suggesting this metabolic partitioning may be a consequence of glutamine restriction [51].
Similarly, cancer cells exhibit elevated consumption of other environmental amino acids, potentially depriving immune cells of these nutrients. It has recently been shown that cancer cells increase their uptake of extracellular methionine by upregulating the SLC43A2/LAT4 methionine transporter, leading to a decrease in available methionine and reduced intracellular SAM in CD8+ T cells. Mechanistically, this results in loss of dimethylation at lysine 79 of histone H3 and a consequent decrease in expression of STAT5 and impaired antitumor CD8+ T cell function [83]. Cancer cell consumption of extracellular arginine reduces arginine availability to T cells and consequently downregulates T cell mTORC1 activity, reducing T cell effector functions [84–87]. Finally, cancer cells have also been shown to take up the majority of extracellular serine and cysteine, the former of which is integral to T cell expansion and effector function and the latter of which is required for T cell activation [88–90].
Altered amino acid metabolism of cancer cells can not only impact immune cell responses through depleting nutrient supply but also by generating immunosuppressive metabolites. The most notable example comes from tryptophan, an essential amino acid that cannot be synthesized by the body and must instead be obtained entirely from the diet. The amount of extracellular tryptophan is an important factor in determining the magnitude and quality of a T cell response, and T cell proliferation and activation are strongly suppressed when cells are cultured in tryptophan-free media [91–93]. Cancer cells do not only take up the limited tryptophan in the microenvironment, but they also metabolize it to produce kynurenine. Kynurenine is the ligand for the aryl hydrocarbon receptor (AHR) which exerts a pro-tumor effect on T cells, including stimulating differentiation of CD4+ T cells to immunosuppressive Tregs [94–96]. In CD8+ T cells, kynurenine and AhR activation has been shown to induce expression of the immune checkpoint receptor PD-1 [94–98]. This example showcases a phenomenon wherein cancer cells “steal” essential nutrients from immune cells and release a processed form to further encourage immune suppression. Another example of this can be seen in the increased expression of Arginase-1 in pro-tumorigenic tumor-associated macrophages (TAMs). This leads to enhanced consumption of extracellular L-arginine, which, as previously stated, is essential for effective induction of T cell effector functions [84–87]. Additionally, the arginine that is consumed is used in the synthesis of highly immunosuppressive polyamines [99–102].
These effects and those more completely reviewed elsewhere [4, 103, 104] are important to consider in the context of metastatic tumors, but are likely significant to different degrees in distinct metastatic sites because of their other properties, including oxygen saturation and extracellular pH, which can also influence metabolic programming. Further research into tumor-immune metabolic communication in specific metastatic sites is necessary to more completely characterize these interactions and to identify metastasis-specific therapeutic targets.
TARGETING AMINO ACID METABOLISM IN METASTASIS
The critical nature of amino acid metabolism in the process of metastasis, including roles in cancer cell outgrowth and interactions with niche cells at various metastatic sites present attractive potential therapeutic targets. Such therapies have great potential to be used in concert with existing therapies, adding to the antitumor effect without substantially contributing to the side effect profile of the combination. Furthermore, influencing microenvironmental metabolism could theoretically lead to tumor cell starvation without indiscriminately inducing cytotoxicity. As this potential has become more apparent preclinically, an increasing number of new targeted drugs and drug combinations are being tested (Table 1, Fig. 1). Even though these novel therapies have not yet been widely tested in metastatic disease, the mechanisms through which they act are not constrained to primary tumors and thus may provide some benefits in the treatment of metastatic cancer.
Table 1.
Selected agents targeting amino acid metabolism for cancer treatment.
| Target | Treatment | Stage | Cancer Type | Reference(s) | |
|---|---|---|---|---|---|
| Glutamine | GLS1 | CB-839+cabozantinib | Phase II | Renal cell carcinoma | NCT03428217 |
| CB-839 + talazoparib | Phase Ib/II | Solid tumors | NCT03875313 | ||
| CB-839 + paclitaxel | Phase II | TNBC | NCT03057600 | ||
| CB-839 hydrochloride + sapanisertib | Phase I/Ib | NSCLC | NCT04250545 | ||
| CB-839 + Panitumumab and Irinotecan | Phase I/II | Metastatic and refractory RAS wildtype colorectal | NCT03263429 | ||
| CB-839 + Azacitidine | Phase I/II | Advanced myelodysplastic syndrome | NCT03047993 | ||
| CB-839 + capecitabine | Phase I/II | Advanced solid tumors, colorectal cancer | NCT02861300 | ||
| ACB-839 + palbociclib | Phase Ib/II | Solid tumors | NCT03965845 | ||
| ASCT2 | V-9302 | Preclinical | In vivo mouse models and in vitro | [52, 105, 113, 114] | |
| Cysteine | xCT | Sorafenib | In clinic | Kidney, liver, and thyroid cancer | 207012 |
| PRLX 93936 (erastin analog) | Phase I | Solid tumors | NCT00528047 | ||
| Serine | PHGDH | NCT-502 | Preclinical | In vitro | [107, 115] |
| NCT-503 | Preclinical | In vitro and mouse | [107] | ||
| Arginine | Arginine auxotrophic cancer cells | ADI-PEG-20 | Phase II | Metastatic Melanoma | NCT00450372 |
| ADI-PEG 20 | Phase II | Non-Hodgkin’s Lymphoma | NCT01910025 | ||
| ADI-PEG 20 | Phase I | Cutaneous Melanoma, Uveal Melanoma, Ovarian Carcinoma or Other Advanced Solid Tumors | NCT01665183 | ||
| ADI-PEG 20 | Phase II | Acute Myeloid Leukemia | NCT01910012 | ||
| ADI-PEG 20 | Phase I | HER2 Negative Metastatic Breast Cancer | NCT01948843 |
Most therapies targeting amino acid metabolism are aimed at stymying glutamine metabolism in cancer cells, including reducing glutamine uptake or blocking its utilization. It has recently been shown in preclinical models that co-treatment with the S6K inhibitor PF-4708671 and the glutamine uptake inhibitor V-9302 reduces glutamine uptake in paclitaxel-resistant ovarian cancer cells and re-sensitizes them to chemotherapy [105]. Furthermore, a separate study showed that treatment with V-9302 was able to selectively block glutamine uptake in triple-negative breast cancer cells but not in tumor-associated CD8+ T cells [52]. This shows a particularly promising angle for targeting glutamine metabolism—not only can it work to starve tumors, but it appears to actively benefit nearby immune cells, thereby driving cancer cell death in multiple ways. Notably, another preclinical drug targeting glutamine metabolism has demonstrated similar effects on both cancer cells and immune cells by targeting glutamine utilization. DRP-104 is a novel pro-drug of the broad-acting glutamine antagonist 6-diazo-5-oxo-L-norleucine (DON), which had shown high antitumor activity but was hampered by a particularly strong complement of side effects. DRP-104 is injected in an inactive form and preferentially converted to active DON at tumor sites, thereby mitigating side effects [106]. More targeted approaches include telaglenastat (CB-839), an orally bioavailable inhibitor of the GLS. Although it has not shown much promise as a monotherapy, it is currently part of several clinical trials in combination with various existing chemo- and immunotherapy drugs (Table 1, Fig. 1).
In addition to these approaches to target glutamine metabolism, some novel treatments targeting the uptake and metabolism of other amino acids are also being tested, including serine biosynthesis. NCT-502 and NCT-503, novel small molecule inhibitors of PHGDH, have each shown preclinical efficacy in suppressing the growth of PHGDH-dependent tumors in both cell culture and xenograft models [107]. Other approaches have sought to induce oxidative-induced ferroptotic cell death by blocking cysteine uptake, but with disappointing results. The erastin analog PRLX-93936 and the kinase inhibitor Sorafenib inhibit xCT to block cysteine uptake. Unfortunately, PRLX-93936 showed poor tolerability in Phase I clinical trials, while sorafenib, which is FDA approved for the treatment of unresectable HCC and advanced renal cell carcinoma, showed poor induction of ferroptosis in most cancer cell lines [108, 109]. Better clinical success has been observed through pharmacological arginine depletion using a PEGylated form of arginine deiminase (ADI-PEG-20). Preclinical studies demonstrated that ADI-PEG-20 enhanced the antitumor efficacy of doxorubicin in melanoma and breast cancer cell lines lacking arginosuccinate synthase 1 [110, 111]. A recent Phase I clinical trial (Table 1) indicated that this combination had some clinical benefit in metastatic HER2- breast cancer patients [112].
Targeting amino acid metabolism is already showing tangible clinical potential in variety of tumor types. As more is learned about cancer metabolism and the different ways that the metabolism of the tumor microenvironment can be manipulated, these therapies will only improve. It remains to be determined, however, if these agents have efficacy in metastatic cancer. Due to the importance of amino acid metabolism in metastatic dissemination and outgrowth, it is intriguing to speculate that these same therapies may have a clinical benefit in the metastatic setting. Therapies that inhibit glutamine metabolism are particularly interesting, especially with availability of pre-clinical and clinical candidates. Bone metastatic tumors may be responsive to glutamine metabolism inhibition, particularly since these metastatic tumors are also characteristically reliant on glutamine metabolism [49, 50, 54]. One possible caveat to using these inhibitors in bone is the essential nature of glutamine for osteoblast lineage commitment [49]. However, it could be possible that impeding osteoblast activation could hinder further osteoclast activation to suppress the “vicious cycle” of bone metastasis. While targeting glutamine metabolism may be beneficial in treating bone metastases, evidence suggests that these inhibitors may induce unfavorable neurotoxicity in brain metastatic cancers due to dysregulation of glutamate homeostasis [38]. However, it is also easy to see where inhibitors of PHGDH and de novo serine synthesis could be applied to metastatic disease at brain, bone, and lung. Elevated serine biosynthesis has been observed in cancer cells associated with all three organ sites [37, 42, 59–61, 68, 69], suggesting that PHGDH inhibition may be universally effective in metastatic disease. These inhibitors could potentially serve a dual purpose in the bone, not only preventing unchecked proliferation of tumor cells, but by reducing levels of osteoclast-activating serine in the extracellular space. However, it remains to be determined how these inhibitors will impact antitumor T cell activities. Collectively, despite the lack of specific testing of amino acid-targeted drugs in clinical setting, targeted therapies of amino acid metabolism have potential for specific metastatic targeting in combination with established regimens of care.
CONCLUSIONS AND FUTURE DIRECTIONS
A growing body of emerging evidence indicates the presence of organ-specific metabolic vulnerabilities in metastatic tumors. Targeting these vulnerabilities could lead to more successful treatments for existing metastatic lesions and, potentially, to amelioration or prevention of further metastatic spread. In this review, we have summarized evidence indicating that DTCs must adapt to the metabolic demands of the metastatic organ to survive and expand. Current treatment modalities targeting metastatic tumors do not take into consideration the different environments and vulnerabilities of distinct host tissues. Fortunately, as the potential of therapies targeting amino acid metabolism have been increasingly noted preclinically, an increasing number of promising new targeted drugs and drug combinations are being tested (Table 1). In addition, metabolic remodeling at metastatic sites affects the immune microenvironment and impacts antitumor immune response [4]. Understanding of how amino acids are utilized at secondary organs may also provide opportunities to improve immunotherapies. Although further work is inarguably necessary to understand the determinants of organotropism more completely in metastasis, our current knowledge has already resulted in the preliminary development of novel targeted therapies for largely incurable diseases. Metastasis is still the primary cause of cancer-associated death, but further insight into this process could lead to a world in which the successful treatment of metastatic tumors will become commonplace.
ACKNOWLEDGEMENTS
This work was supported by NIH grants R01 CA250506 and R01 CA266767 (to JC), T32 CA009592 (to BK); a VA Merit Award 5101BX000134 and a VA Senior Research Career Scientist Award IK6BX005391 (to JC); and a Department of Defense CDMRP Award W81XWH220109 (to DNE).
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, et al. Multistep nature of metastatic inefficiency. Am J Pathol. 1998;153:865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mosier JA, Schwager SC, Boyajian DA, Reinhart-King CA. Cancer cell metabolic plasticity in migration and metastasis. Clin Exp Metastasis. 2021;38:343–59. [DOI] [PubMed] [Google Scholar]
- 3.Zanotelli MR, Zhang J, Reinhart-King CA. Mechanoresponsive metabolism in cancer cell migration and metastasis. Cell Metab. 2021;33:1307–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arner EN, Rathmell JC. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell. 2023;41:421–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bian X, Liu R, Meng Y, Xing D, Xu D, Lu Z. Lipid metabolism and cancer. J Exp Med. 2021;218:e20201606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang C, Luo D. The metabolic adaptation mechanism of metastatic organotropism. Exp Hematol Oncol. 2021;10:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ebert D, Haller RG, Walton ME. Energy contribution of octanoate to intact rat brain metabolism measured by 13 C nuclear magnetic resonance spectroscopy. J Neurosci. 2003;23:5928–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schild T, Low V, Blenis J, Gomes AP. Unique metabolic adaptations dictate distal organ-specific metastatic colonization. Cancer Cell. 2018;33:347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang T, Suo C, Zheng C, Zhang H. Hypoxia and metabolism in metastasis. In: Gilkes DM (ed). Hypoxia and cancer metastasis. Springer International Publishing: Cham, 2019, pp 87–95. [Google Scholar]
- 10.Vaghari-Tabari M, Ferns GA, Qujeq D, Andevari AN, Sabahi Z, Moein S. Signaling, metabolism, and cancer: an important relationship for therapeutic intervention. J Cell Physiol. 2021;236:5512–32. [DOI] [PubMed] [Google Scholar]
- 11.Ghanavat M, Shahrouzian M, Deris Zayeri Z, Banihashemi S, Kazemi SM, Saki N. Digging deeper through glucose metabolism and its regulators in cancer and metastasis. Life Sci. 2021;264:118603. [DOI] [PubMed] [Google Scholar]
- 12.Zanotelli MR, Goldblatt ZE, Miller JP, Bordeleau F, Li J, VanderBurgh JA, et al. Regulation of ATP utilization during metastatic cell migration by collagen architecture. Mol Biol Cell. 2018;29:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang L, Moss T, Mangala LS, Marini J, Zhao H, Wahlig S, et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol Syst Biol. 2014;10:728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodrigues MF, Obre E, De Melo FHM, Santos GC, Galina A, Jasiulionis MG, et al. Enhanced OXPHOS, glutaminolysis and β-oxidation constitute the metastatic phenotype of melanoma cells. Biochem J. 2016;473:703–15. [DOI] [PubMed] [Google Scholar]
- 15.Xiang L, Mou J, Shao B, Wei Y, Liang H, Takano N, et al. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019;10:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan Y-C, Chang Y-C, Chuang H-H, Yang Y-C, Lin Y-F, Huang M-S, et al. Over-expression of PSAT1 promotes metastasis of lung adenocarcinoma by suppressing the IRF1-IFNγ axis. Oncogene. 2020;39:2509–22. [DOI] [PubMed] [Google Scholar]
- 17.Knott SRV, Wagenblast E, Khan S, Kim SY, Soto M, Wagner M, et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature. 2018;554:378–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du F, Chen J, Liu H, Cai Y, Cao T, Han W, et al. SOX12 promotes colorectal cancer cell proliferation and metastasis by regulating asparagine synthesis. Cell Death Dis. 2019;10:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Soflaee MH, Kesavan R, Sahu U, Tasdogan A, Villa E, Djabari Z, et al. Purine nucleotide depletion prompts cell migration by stimulating the serine synthesis pathway. Nat Commun. 2022;13:2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin L, Chun J, Pan C, Kumar A, Zhang G, Ha Y, et al. The PLAG1-GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2-AMPK signaling in LKB1-deficient lung cancer. Mol Cell. 2018;69:87–99.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin H, Qiao F, Chen L, Lu C, Xu L, Gao X. Serum metabolomic signatures of lymph node metastasis of esophageal squamous cell carcinoma. J Proteome Res. 2014;13:4091–103. [DOI] [PubMed] [Google Scholar]
- 22.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamb R, Ozsvari B, Bonuccelli G, Smith DL, Pestell RG, Martinez-Outschoorn UE, et al. Dissecting tumor metabolic heterogeneity: telomerase and large cell size metabolically define a sub-population of stem-like, mitochondrial-rich, cancer cells. Oncotarget. 2015;6:21892–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lamb R, Harrison H, Smith DL, Townsend PA, Jackson T, Ozsvari B, et al. Targeting tumor-initiating cells: eliminating anabolic cancer stem cells with inhibitors of protein synthesis or by mimicking caloric restriction. Oncotarget. 2015;6:4585–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan AS, Baty JW, Berridge MV. The role of mitochondrial electron transport in tumorigenesis and metastasis. Biochim Biophys Acta BBA—Gen Subj. 2014;1840:1454–63. [DOI] [PubMed] [Google Scholar]
- 26.Spees JL, Olson SD, Whitney MJ, Prockop DJ. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci. 2006;103:1283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis RT, Blake K, Ma D, Gabra MBI, Hernandez GA, Phung AT, et al. Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat Cell Biol. 2020;22:310–20. [DOI] [PubMed] [Google Scholar]
- 28.Karaayvaz M, Cristea S, Gillespie SM, Patel AP, Mylvaganam R, Luo CC, et al. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat Commun. 2018;9:3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basnet H, Tian L, Ganesh K, Huang Y-H, Macalinao DG, Brogi E, et al. Flura-seq identifies organ-specific metabolic adaptations during early metastatic colonization. eLife. 2019;8:e43627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shestov AA, Emir UE, Kumar A, Henry P-G, Seaquist ER, Öz G. Simultaneous measurement of glucose transport and utilization in the human brain. Am J Physiol-Endocrinol Metab. 2011;301:E1040–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gruetter R, Novotny EJ, Boulware SD, Rothman DL, Mason GF, Shulman GI, et al. Direct measurement of brain glucose concentrations in humans by 13C NMR spectroscopy. Proc Natl Acad Sci. 1992;89:1109–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yudkoff M, Nissim I, Daikhin Y, Lin Z-P, Nelson D, Pleasure D, et al. Brain glutamate metabolism: neuronal-astroglial relationships. Dev Neurosci. 1993;15:343–50. [DOI] [PubMed] [Google Scholar]
- 33.Daikhin Y, Yudkoff M. Compartmentation of brain glutamate metabolism in neurons and glia. J Nutr. 2000;130:1026S–31S. [DOI] [PubMed] [Google Scholar]
- 34.Natarajan SK, Venneti S. Glutamine metabolism in brain tumors. Cancers. 2019;11:1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen W. Clinical applications of PET in brain tumors. J Nucl Med. 2007;48:1468–81. [DOI] [PubMed] [Google Scholar]
- 36.Chen J, Lee H-J, Wu X, Huo L, Kim S-J, Xu L, et al. Gain of glucose-independent growth upon metastasis of breast cancer cells to the brain. Cancer Res. 2015;75:554–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parida PK, Marquez-Palencia M, Nair V, Kaushik AK, Kim K, Sudderth J, et al. Metabolic diversity within breast cancer brain-tropic cells determines metastatic fitness. Cell Metab. 2022;34:90–105.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeng Q, Michael IP, Zhang P, Saghafinia S, Knott G, Jiao W, et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature. 2019;573:526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. [DOI] [PubMed] [Google Scholar]
- 40.Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H, et al. Blockage of Ca2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med. 2002;8:971–8. [DOI] [PubMed] [Google Scholar]
- 41.D’Onofrio M, Arcella A, Bruno V, Ngomba RT, Battaglia G, Lombari V, et al. Pharmacological blockade of mGlu2/3 metabotropic glutamate receptors reduces cell proliferation in cultured human glioma cells. J Neurochem. 2003;84:1288–95. [DOI] [PubMed] [Google Scholar]
- 42.Ngo B, Kim E, Osorio-Vasquez V, Doll S, Bustraan S, Liang RJ, et al. Limited environmental serine and glycine confer brain metastasis sensitivity to PHGDH Inhibition. Cancer Discov. 2020;10:1352–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coleman RE. Clinical features of metastatic bone disease and risk of skeletal morbidity. Clin Cancer Res. 2006;12:6243s–9s. [DOI] [PubMed] [Google Scholar]
- 44.Maurizi A, Rucci N. The osteoclast in bone metastasis: player and target. Cancers. 2018;10:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–64. [DOI] [PubMed] [Google Scholar]
- 46.Esposito M, Guise T, Kang Y. The biology of bone metastasis. Cold Spring Harb Perspect Med. 2018;8:a031252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye L, Kynaston H, Jiang W. Bone metastasis in prostate cancer: molecular and cellular mechanisms (Review). Int J Mol Med. 2007. 10.3892/ijmm.20.1.103. [DOI] [PubMed] [Google Scholar]
- 48.Shen L, Sharma D, Yu Y, Long F, Karner C. Biphasic regulation of glutamine consumption by WNT during osteoblast differentiation. J Cell Sci. 2020;134:jcs.251645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu Y, Newman H, Shen L, Sharma D, Hu G, Mirando AJ, et al. Glutamine metabolism regulates proliferation and lineage allocation in skeletal stem cells. Cell Metab. 2019;29:966–978.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shen L, Yu Y, Zhou Y, Pruett-Miller SM, Zhang G-F, Karner CM. SLC38A2 provides proline to fulfill unique synthetic demands arising during osteoblast differentiation and bone formation. eLife. 2022;11:e76963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature. 2021;593:282–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Investig. 2021;131:e140100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edwards DN. Amino acid metabolism in bone metastatic disease. Curr Osteoporos Rep. 2023;21:344–53. [DOI] [PubMed] [Google Scholar]
- 54.Indo Y, Takeshita S, Ishii K-A, Hoshii T, Aburatani H, Hirao A, et al. Metabolic regulation of osteoclast differentiation and function: metabolic signature of osteoclasts. J Bone Min Res. 2013;28:2392–9. [DOI] [PubMed] [Google Scholar]
- 55.Lee S, Kim HS, Kim MJ, Min KY, Choi WS, You JS. Glutamine metabolite α-ketoglutarate acts as an epigenetic co-factor to interfere with osteoclast differentiation. Bone. 2021;145:115836. [DOI] [PubMed] [Google Scholar]
- 56.Go M, Shin E, Jang SY, Nam M, Hwang G-S, Lee SY. BCAT1 promotes osteoclast maturation by regulating branched-chain amino acid metabolism. Exp Mol Med. 2022;54:825–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brunner JS, Vulliard L, Hofmann M, Kieler M, Lercher A, Vogel A, et al. Environmental arginine controls multinuclear giant cell metabolism and formation. Nat Commun. 2020;11:431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishikawa K, Iwamoto Y, Kobayashi Y, Katsuoka F, Kawaguchi S, Tsujita T, et al. DNA methyltransferase 3a regulates osteoclast differentiation by coupling to an S-adenosylmethionine–producing metabolic pathway. Nat Med. 2015;21:281–7. [DOI] [PubMed] [Google Scholar]
- 59.Pollari S, Käkönen S-M, Edgren H, Wolf M, Kohonen P, Sara H, et al. Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res Treat. 2011;125:421–30. [DOI] [PubMed] [Google Scholar]
- 60.Scalise M, Pochini L, Console L, Losso MA, Indiveri C. The human SLC1A5 (ASCT2) amino acid transporter: from function to structure and role in cell biology. Front Cell Dev Biol. 2018;6:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ogawa T, Ishida-Kitagawa N, Tanaka A, Matsumoto T, Hirouchi T, Akimaru M, et al. A novel role of l-serine (l-Ser) for the expression of nuclear factor of activated T cells (NFAT)2 in receptor activator of nuclear factor κB ligand (RANKL)-induced osteoclastogenesis in vitro. J Bone Min Metab. 2006;24:373–9. [DOI] [PubMed] [Google Scholar]
- 62.Suvilesh KN, Manjunath Y, Pantel K, Kaifi JT. Preclinical models to study patient-derived circulating tumor cells and metastasis. Trends Cancer. 2023;9:355–71. [DOI] [PubMed] [Google Scholar]
- 63.Rayes RF, Mouhanna JG, Nicolau I, Bourdeau F, Giannias B, Rousseau S, et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis-promoting effects. JCI Insight. 2019;4:e128008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583:133–8. [DOI] [PubMed] [Google Scholar]
- 65.Naffah De Souza C, Breda LCD, Khan MA, Almeida SRD, Câmara NOS, Sweezey N, et al. Alkaline pH promotes NADPH oxidase-independent neutrophil extracellular trap formation: a matter of mitochondrial reactive oxygen species generation and citrullination and cleavage of histone. Front Immunol. 2018;8:1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell. 2013;24:130–7. [DOI] [PubMed] [Google Scholar]
- 67.Woods LT, Forti KM, Shanbhag VC, Camden JM, Weisman GA. P2Y receptors for extracellular nucleotides: contributions to cancer progression and therapeutic implications. Biochem Pharm. 2021;187:114406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rinaldi G, Pranzini E, Van Elsen J, Broekaert D, Funk CM, Planque M, et al. In vivo evidence for serine biosynthesis-defined sensitivity of lung metastasis, but not of primary breast tumors, to mTORC1 inhibition. Mol Cell. 2021;81:386–97.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kiweler N, Delbrouck C, Pozdeev V, Neises L, Soriano-Baguet L, Eiden K, et al. Mitochondria preserve an autarkic one-carbon cycle to confer growth-independent cancer cell migration and metastasis. Nat Commun. 2022;13. 10.1038/s41467-022-30363-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Elia I, Rossi M, Stegen S, Broekaert D, Doglioni G, van Gorsel M, et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature. 2019;568:117–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Oh M-H, Sun I-H, Zhao L, Leone RD, Sun I-M, Xu W, et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J Clin Investig. 2020;130:3865–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alvarado A, Arce I. Metabolic functions of the lung, disorders and associated pathologies. J Clin Med Res. 2016;8:689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lanzardo S, Conti L, Rooke R, Ruiu R, Accart N, Bolli E, et al. Immunotargeting of antigen xCT attenuates stem-like cell behavior and metastatic progression in breast cancer. Cancer Res. 2016;76:62–72. [DOI] [PubMed] [Google Scholar]
- 74.Laoukili J, Van Schelven S, Küçükköse E, Verheem A, Goey K, Koopman M, et al. BRAFV600E in colorectal cancer reduces sensitivity to oxidative stress and promotes site-specific metastasis by stimulating glutathione synthesis. Cell Rep. 2022;41:111728. [DOI] [PubMed] [Google Scholar]
- 75.Lee E, Choi A, Jun Y, Kim N, Yook JI, Kim SY, et al. Glutathione peroxidase-1 regulates adhesion and metastasis of triple-negative breast cancer cells via FAK signaling. Redox Biol. 2020;29:101391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li X, Sun X, Carmeliet P. Hallmarks of endothelial cell metabolism in health and disease. Cell Metab. 2019;30:414–33. [DOI] [PubMed] [Google Scholar]
- 77.Ryan DG, O’Neill LAJ. Krebs cycle reborn in macrophage immunometabolism. Annu Rev Immunol. 2020;38:289–313. [DOI] [PubMed] [Google Scholar]
- 78.Makowski L, Chaib M, Rathmell JC. Immunometabolism: from basic mechanisms to translation. Immunol Rev. 2020;295:5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ho P-C, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162:1217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer. 2020;20:516–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leone RD, Zhao L, Englert JM, Sun I-M, Oh M-H, Sun I-H, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366:1013–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature. 2020;585:277–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Patil MD, Bhaumik J, Babykutty S, Banerjee UC, Fukumura D. Arginine dependence of tumor cells: targeting a chink in cancer’s armor. Oncogene. 2016;35:4957–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li F, Simon MC. Cancer Cells Don’t Live Alone: Metabolic Communication Within Tumor Microenvironments. Dev Cell. 2020;54:183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chantranupong L, Scaria SM, Saxton RA, Gygi MP, Shen K, Wyant GA, et al. The CASTOR proteins are arginine sensors for the mTORC1 pathway. Cell. 2016;165:153–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167:829–42.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab. 2017;25:345–57. [DOI] [PubMed] [Google Scholar]
- 89.Martínez-Reyes I, Chandel NS. Cancer metabolism: looking forward. Nat Rev Cancer. 2021;21:669–80. [DOI] [PubMed] [Google Scholar]
- 90.Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8+ T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281:1191–3. [DOI] [PubMed] [Google Scholar]
- 92.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22:633–42. [DOI] [PubMed] [Google Scholar]
- 94.Rothhammer V, Quintana FJ. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol. 2019;19:184–97. [DOI] [PubMed] [Google Scholar]
- 95.Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185:3190–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mondanelli G, Ugel S, Grohmann U, Bronte V. The immune regulation in cancer by the amino acid metabolizing enzymes ARG and IDO. Curr Opin Pharm. 2017;35:30–39. [DOI] [PubMed] [Google Scholar]
- 97.Amobi-McCloud A, Muthuswamy R, Battaglia S, Yu H, Liu T, Wang J, et al. IDO1 expression in ovarian cancer induces PD-1 in T cells via aryl hydrocarbon receptor activation. Front Immunol. 2021;12:678999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-repopulating cells induce PD-1 expression in CD8+ T cells by transferring kynurenine and AhR activation. Cancer Cell. 2018;33:480–94.e7. [DOI] [PubMed] [Google Scholar]
- 99.Shearer JD, Richards JR, Mills CD, Caldwell MD. Differential regulation of macrophage arginine metabolism: a proposed role in wound healing. Am J Physiol-Endocrinol Metab. 1997;272:E181–90. [DOI] [PubMed] [Google Scholar]
- 100.Ye C, Geng Z, Dominguez D, Chen S, Fan J, Qin L, et al. Targeting ornithine decarboxylase by α-difluoromethylornithine inhibits tumor growth by impairing myeloid-derived suppressor cells. J Immunol. 2016;196:915–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mills CD, Shearer J, Evans R, Caldwell MD. Macrophage arginine metabolism and the inhibition or stimulation of cancer. J Immunol. 1992;149:2709–14. [PubMed] [Google Scholar]
- 102.Hayes CS, Shicora AC, Keough MP, Snook AE, Burns MR, Gilmour SK. Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol Res. 2014;2:274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yang L, Chu Z, Liu M, Zou Q, Li J, Liu Q, et al. Amino acid metabolism in immune cells: essential regulators of the effector functions, and promising opportunities to enhance cancer immunotherapy. J Hematol Oncol. 2023;16:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zou W, Green DR. Beggars banquet: Metabolism in the tumor immune microenvironment and cancer therapy. Cell Metab. 2023;35:1101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kim G, Jang S-K, Kim YJ, Jin H-O, Bae S, Hong J, et al. Inhibition of glutamine uptake resensitizes paclitaxel resistance in SKOV3-TR ovarian cancer cell via mTORC1/S6K signaling pathway. Int J Mol Sci. 2022;23:8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yokoyama Y, Estok TM, Wild R. Sirpiglenastat (DRP-104) induces antitumor efficacy through direct, broad antagonism of glutamine metabolism and stimulation of the innate and adaptive immune systems. Mol Cancer Ther. 2022;21:1561–72. [DOI] [PubMed] [Google Scholar]
- 107.Rohde JM, Brimacombe KR, Liu L, Pacold ME, Yasgar A, Cheff DM, et al. Discovery and optimization of piperazine-1-thiourea-based human phosphoglycerate dehydrogenase inhibitors. Bioorg Med Chem. 2018;26:1727–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zheng J, Sato M, Mishima E, Sato H, Proneth B, Conrad M. Sorafenib fails to trigger ferroptosis across a wide range of cancer cell lines. Cell Death Dis. 2021;12:698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reed JC, Pellecchia M. Ironing out cell death mechanisms. Cell. 2012;149:963–5. [DOI] [PubMed] [Google Scholar]
- 110.Qiu F, Chen Y-R, Liu X, Chu C-Y, Shen L-J, Xu J et al. Arginine starvation impairs mitochondrial respiratory function in ASS1-deficient breast cancer Cells. Sci Signal. 2014; 7. 10.1126/scisignal.2004761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stelter L, Fuchs S, Jungbluth AA, Ritter G, Longo VA, Zanzonico P, et al. Evaluation of arginine deiminase treatment in melanoma xenografts using 18F-FLT PET. Mol Imaging Biol. 2013;15:768–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Yao S, Janku F, Koenig K, Tsimberidou AM, Piha-Paul SA, Shi N, et al. Phase 1 trial of ADI-PEG 20 and liposomal doxorubicin in patients with metastatic solid tumors. Cancer Med. 2022;11:340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Park HY, Kim M-J, Kim YJ, Lee S, Jin J, Lee S, et al. V-9302 inhibits proliferation and migration of VSMCs, and reduces neointima formation in mice after carotid artery ligation. Biochem Biophys Res Commun. 2021;560:45–51. [DOI] [PubMed] [Google Scholar]
- 114.Adhikary G, Shrestha S, Naselsky W, Newland JJ, Chen X, Xu W, et al. Mesothelioma cancer cells are glutamine addicted and glutamine restriction reduces YAP1 signaling to attenuate tumor formation. Mol Carcinog. 2023;62:438–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Shen L, Zhang J, Zheng Z, Yang F, Liu S, Wu Y, et al. PHGDH inhibits ferroptosis and promotes malignant progression by upregulating SLC7A11 in bladder cancer. Int J Biol Sci. 2022;18:5459–74. [DOI] [PMC free article] [PubMed] [Google Scholar]