

Abstract
Clonal haematopoiesis of indeterminate potential (CHIP) has been associated with many adverse health outcomes. However, further research is required to understand the critical genes and pathways relevant to CHIP subtypes, evaluate how CHIP clones evolve with time, and further advance functional characterisation and therapeutic studies. Large epidemiological studies are well placed to address these questions but often collect saliva rather than blood from participants. Paired saliva- and blood-derived DNA samples from 94 study participants were sequenced using a targeted CHIP-gene panel. The ten genes most frequently identified to carry CHIP-associated variants were analysed. Fourteen unique variants associated with CHIP, ten in DNMT3A, two in TP53 and two in TET2, were identified with a variant allele fraction (VAF) between 0.02 and 0.2 and variant depth ≥ 5 reads. Eleven of these CHIP-associated variants were detected in both the blood- and saliva-derived DNA sample. Three variants were detected in blood with a VAF > 0.02 but fell below this threshold in the paired saliva sample (VAF 0.008—0.013). Saliva-derived DNA is suitable for detecting CHIP-associated variants. Saliva can offer a cost-effective biospecimen that could both advance CHIP research and facilitate clinical translation into settings such as risk prediction, precision prevention, and treatment monitoring.
Keywords: Clonal haematopoiesis, CHIP, Somatic mutations, Next generation sequencing, Saliva, Blood
Subject terms: Haematopoietic stem cells, Cardiology, Molecular medicine, Medical genetics, Genetics, Disease genetics, Genetic testing
Introduction
Age-related clonal haematopoiesis of indeterminate potential (CHIP), usually observed as somatic mosaicism in blood-derived DNA, has been associated with many adverse health outcomes including haematological conditions, cardiovascular disease (CVD), and all-cause mortality1. CHIP is characterised as haematopoietic cells of peripheral blood with at least one driver mutation, and without haematological malignancy or detectable morphological evidence of dysplasia2,3. Haematopoietic stem cells and progenitor cells with mutations that confer a fitness advantage will proliferate in clonal expansion, and the accumulation of these mutations can result in disease1,2.
Research deciphering the molecular and associated clinical features of CHIP has gained considerable momentum via the analysis of large human data sets available from research initiatives such as the UK biobank and All of Us4. These studies have refined both our understanding of CHIP and the bioinformatic approaches required to identify CHIP in a range of genomic datasets including whole genome, whole exome, and targeted gene panel sequencing data. These studies have revealed CHIP to have diverse molecular phenotypes (somatic mutation-driven subtypes), that are associated with a spectrum of germline genetic causes and clinical features5.
Recently, population-scale genomic datasets have enabled further interrogation of the complexities of CHIP and the identification of important differential associations between disease susceptibility and the clone-specific gene mutation. For instance, DNMT3A mutations are not associated with CVD but have been shown to be associated with an increased risk of solid tumours. Kessler et al., further described common genetic variation associated with CHIP5. For example, common germline variants at the CD164 gene regions were associated with decreased risk of DNMT3A CHIP, whereas germline variants in TCL1A were associated with increased risk of DNMT3A CHIP.
More research is required to understand the critical genes and pathways relevant to each CHIP subtype, evaluate how CHIP clones change with time, and further advance functional and therapeutic studies. Population-scale genomic studies rarely involve serial blood sampling of participants and are thus not well placed to address some of these emerging questions in CHIP research. In contrast, large-scale epidemiological studies of human health often take serial biological samples from participants over long periods of time (often decades). These studies can therefore be well positioned to address some of these gaps in CHIP knowledge.
In this context, saliva is often collected as a source of germline DNA from research participants because it can be collected non-invasively at home and shipped at room temperatures at lower cost with no time sensitivities for downstream biobanking (e.g., processing and freezing). Several pieces of evidence suggest that DNA extracted from saliva may be a suitable template for CHIP analysis. First, white blood cells are known to cross the mucosal barrier and have been suggested to make up approximately 75% of the nucleated cells in a saliva specimen6. Second, DNA derived from mouthwashes after allogeneic blood stem cell transplantation have been shown to display chimeric or complete donor genotype supporting a considerable blood-DNA contribution6,7. Third, saliva-derived DNA has been successfully used in targeted gene panel sequencing. Fourth, Soyfer et al., (2024), assessed saliva for haematopoietic cells and were able to successfully quantify somatic variants in families with myeloproliferative neoplasm8. However, there are likely considerable saliva-specific technical and bioinformatic challenges that will need to be overcome to differentiate germline and CHIP-associated genetic variation especially in the context of a potential reduction in CHIP-associated variant allele fraction (VAF) (if the contribution of blood-cell nuclei to the DNA yield is not high in saliva samples). If it can be demonstrated to be a suitable template for CHIP analysis, saliva-derived DNA offers a cost effective, practical alternative biospecimen that could be utilised to both advance research and be a companion to clinical translation into settings such as risk prediction, precision prevention, and treatment monitoring.
This study sought to assess the suitability of saliva-derived DNA in the detection of CHIP associated variants using a custom targeted gene panel (focusing on the 10 genes most frequently detected to carry CHIP-associated variants), a massively parallel sequencing approach, and saliva- and blood-derived DNA samples from 94 cohort study participants.
Results
Library preparation and sequencing
Paired blood and saliva samples were obtained from 94 healthy participants of the Australian Breakthrough Cancer cohort (Table 1) and DNA was extracted from all samples. A total of 192 samples successfully underwent library preparation. This included 188 test samples (94 blood-derived DNA and 94 saliva-derived DNA pairs), two commercial controls, and two in-house high molecular weight (HMW) controls. Quality metrics of all sequenced samples showed a median read duplication rate of 54.2% and, following deduplication, a median off-target base rate of 20.8%. Of the 188 test samples, 33 samples (17.6%) did not reach ≥ 80% target coverage at 500 × depth; 32 of these 33 samples were saliva-derived DNA, with one blood-derived DNA sample (Table 2). Nine of 188 test samples (5%) did not reach > 50% target coverage at 500 × depth; 8 of these 9 samples were saliva-derived DNA and 1 was a blood-derived DNA sample (Table 2). These 9 correspond to samples that, following enzymatic fragmentation, had poor pre-capture DNA library profiles (long fragment sizes, a plateau peak and/or low concentrations).
Table 1.
A demographic representation of the 94 participants selected from the Australian Breakthrough Cancer cohort.
| History | Males (N = 35) | Females (N = 59) |
|---|---|---|
| Age at blood draw, years | ||
| Median | 70 | 69 |
| Range | 66–76 | 65–76 |
| Smoking status | ||
| Never | 19 (54%) | 34 (58%) |
| Former | 16 (46%) | 23 (39%) |
| Current | 0 | 2 (3%) |
| Ethnicity(self-reported) | ||
| Northern European | 30 (86%) | 41 (69%) |
| Southern European | 2 (6%) | 2 (3%) |
| Other European | 2 (6%) | 11 (19%) |
| Other | NA | 2 (3%) |
| Unknown | 1 (3%) | 3 (5%) |
Table 2.
Sequencing alignment metrics of deduplicated reads for 188 samples and 4 controls.
| Criteria | DNA Source | Criteria met | Samples (n) | Median | Mean | Min | Max | |
|---|---|---|---|---|---|---|---|---|
| > 80% of target coverage at 500 × depth | Test DNA samples | Blood | No | 1 | 242.13X | |||
| Yes | 93 | 1341.83X | 1365.04X | 892.58X | 1909.86X | |||
| Saliva | No | 32 | 741.1X | 638.66X | 0.48X | 974.54X | ||
| Yes | 62 | 1167.49X | 1234.51X | 842.97X | 2082.23X | |||
| Horizon control | Yes | 2 | 919.21X | 919.21X | 885.43X | 952.99X | ||
| HMW DNA control | Yes | 2 | 1865.2X | 1865.2X | 1803.96X | 1926.44X | ||
| > 50% of target coverage at 500x | Test DNA samples | Blood | No | 1 | 242.13X | |||
| Yes | 93 | 1341.83X | 1365.04X | 892.58X | 1909.86X | |||
| Saliva | No | 8 | 176.53X | 225.0X | 0.48X | 478.97X | ||
| Yes | 86 | 1044.75X | 1106.7X | 589.74X | 2082.23X | |||
| Horizon control | Yes | 2 | 919.21X | 919.21X | 885.43X | 952.99X | ||
| HMW DNA control | Yes | 2 | 1865.2X | 1865.2X | 1803.96X | 1926.44X | ||
Controls
Variants that were included in the myeloid control, and in the 10 genes assessed, were called down to a VAF of 0.01 (Supplementary Table 1). Sequencing metrics for both our in-house HMW and commercial controls met the > 80% target coverage at 500 × depth criteria (Table 2).
Variants identified with VAFs between 0.02 and 0.2
In our cohort of healthy participants between the age of 64–75 (Table 1), twenty-one variants (VAF 0.02–0.20) were identified in 18 participants. Thirteen were detected in both blood and saliva-derived DNA pairs. Six variants appeared to be present only in blood-derived DNA, within the VAF thresholds, while two were detected only in saliva-derived DNA (Supplementary Table 2). Upon further investigation, five of these six variants found only in the blood-derived DNA were found below the 0.02 threshold in the saliva DNA pair (ranging between 0.007 – 0.019). The two variants observed in one saliva-derived DNA sample were not detected in the blood-derived DNA pair.
Only one artifact was identified (NM_004972.4:c.1777-7del) in 30/188 samples (15.9%), 14 in blood & 16 in saliva-derived DNAs (VAF ~ 0.03). This artifact was removed. No artifacts were observed in the manual inspection of CHIP associated variants in IGV.
Variants associated with CHIP
Fourteen of the twenty-one variants (VAF 0.02–0.20) were found to be associated with CHIP (Table 3). Ten variants were identified in DNMT3A; two variants in TP53; and two variants in TET2. No putative CHIP-associated variants were identified in the other seven genes assessed. Eleven of fourteen (79%) CHIP associated variants were found in both the blood and saliva-derived DNA pairs when applying the VAF 0.02—0.20 and variant depth (VD) ≥ 5 read thresholds. For a given variant, the VAFs were very similar between the blood and saliva-derived DNA pairs with a largest difference of ~ 3% (Table 3). Three of the fourteen (21%) CHIP associated variants were found in only the blood-derived DNA samples using the thresholds of VD ≥ 5 and VAF 0.02–0.20 (Table3; Fig. 1). However, they were detected in their paired saliva-derived DNA with a VD ≥ 5 and VAFs 0.008 – 0.013 (Table 3).
Table 3.
Fourteen CHIP-associated genetic variants identified in 94 paired saliva and blood-derived DNA samples. Three variants fell below the VAF 0.02 threshold as indicated in bold.
| GRCh genomic coordinates | Ref | Alt | Gene | HGVSc | HGVSp | Blood | Saliva | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| DP | VD | VAF | DP | VD | VAF | ||||||
| 2:25246163 | T | A | DNMT3A | NM_022552.5:c.1426A > T | p.Arg476Ter | 2107 | 137 | 0.07 | 1360 | 90 | 0.07 |
| 2:25241561 | CAT | C | DNMT3A | NM_022552.5:c.2081_2082del | p.Ile695ProfsTer17 | 1073 | 95 | 0.09 | 1403 | 83 | 0.06 |
| 4:105272592 | G | A | TET2 | NM_001127208.3:c.4211_4214delinsAAT* | p.Arg1404GlnfsTer44 | 1388 | 71 | 0.05 | 1152 | 59 | 0.05 |
| 2:25247053 | G | A | DNMT3A | NM_022552.5:c.1120C > T | p.Gln374Ter | 1184 | 25 | 0.02 | 1386 | 48 | 0.04 |
| 2:25246747 | G | GA | DNMT3A | NM_022552.5:c.1151_1152insT | p.Val386GlyfsTer7 | 1665 | 87 | 0.05 | 902 | 50 | 0.06 |
| 2:25241682 | GC | G | DNMT3A | NM_022552.5:c.1961del | p.Gly654AlafsTer51 | 2139 | 45 | 0.02 | 1573 | 40 | 0.03 |
| 2:25247710 | TC | T | DNMT3A | NM_022552.5:c.894del | p.Lys299AsnfsTer17 | 3416 | 105 | 0.03 | 3216 | 79 | 0.03 |
| 2:2548216 | C | CA | DNMT3A | NM_022552.5:c.675_676insT | p.Ala226CysfsTer27 | 710 | 76 | 0.11 | 506 | 40 | 0.08 |
| 4:105276152 | A | G | TET2 | NM_001127208.3:c.5642A > G | p.His1881Arg | 3123 | 212 | 0.07 | 2087 | 120 | 0.06 |
| 2:25240363 | A | C | DNMT3A | NM_022552.5:c.2261 T > G | p.Leu754Arg | 2225 | 64 | 0.03 | 2176 | 62 | 0.03 |
| 17:7673802 | C | T | TP53 | NM_000546.6:c.818G > A | p.Arg273His | 2367 | 115 | 0.05 | 1734 | 75 | 0.04 |
| 2:25235726 | A | G | DNMT3A | NM_022552.5:c.2578 T > C | p.Trp860Arg | 1410 | 41 | 0.03 | 913 | 7 | 0.008 |
| 2:25244257 | G | T | DNMT3A | NM_022552.5:c.1749C > A | p.Cys583Ter | 1952 | 59 | 0.03 | 1273 | 17 | 0.013 |
| 17:7675217 | T | C | TP53 | NM_000546.6:c.395A > G | p.Lys132Arg | 2334 | 48 | 0.02 | 1273 | 11 | 0.009 |
*Upon visual reassessment of the genomic alignment the variant nomenclature was adjusted.
Figure 1.
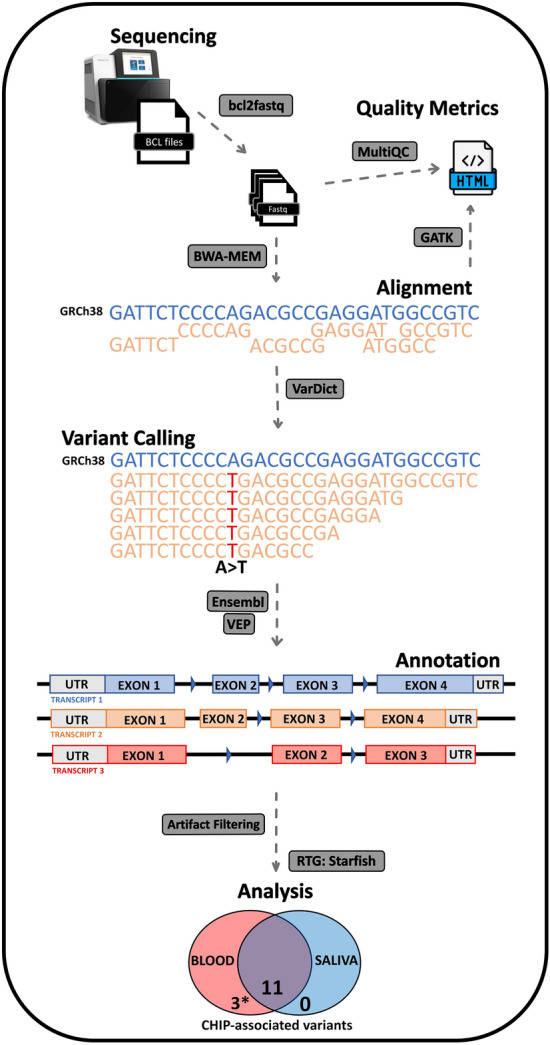
A graphic representation of our bioinformatic workflow used in this study to identify somatic variants (VAFs 0.02–0.2) in blood and saliva-derived DNA pairs. Three CHIP-associated variants detect in blood only, indicated by *, were detected in the saliva-derived DNA pair after exploring below the VAF threshold 0.02 (ranging between 0.008—0.013).
Discussion
Our study demonstrates high concordance between CHIP-associated variants called in pairs of DNAs sourced from blood and saliva, illustrating the suitability of saliva-derived DNA for the detection of CHIP.
This study focused on the analysis of 10 genes that have been reported in large studies to be the most frequently involved in CHIP-associated somatic mutation4. Vlasschaert et al., examined the distribution of genes carrying CHIP variants in 19,921 individuals and found that these ten genes carried the most CHIP-associated variants. Consistent with this, and other literature4,9–11, our small study only identified variants in DNMT3A, TP53, and TET2, with DNMT3A being the most mutated gene.
Prior to this study, there was some evidence to support saliva-derived DNA being a suitable biological resource for detecting somatic mutations in clonal haematopoiesis and other haematologic malignancies. Soyfer et al. recently presented data that examined the feasibility of using DNA prepared from saliva specimens to measure somatic variation at low VAFs (≤ 0.1)8. However, challenges were still anticipated relating to the poorer quality of saliva-derived DNA and the proportion of blood cell nuclei represented in the DNA yield. Indeed, eight of nine DNA samples that did not meet the quality metric threshold of 50% coverage at 500X were from saliva and corresponded to pre-capture libraries with poor TapeStation profiles and/or low concentrations after pre-capture PCR. However, vast majority of saliva-derived DNA samples performed very well and had similar metrics to their paired blood-derived DNA sample.
When considering all variants identified with VAFs between 0.02 and 0.20, six variants were identified in blood-derived DNA, but not in the corresponding saliva-derived DNA pair, for six individuals. Five of these variants were found below the 0.02 threshold in saliva-derived DNA while one variant was not detected in saliva. Three of these five variants were identified as CHIP-associated variants (Table 3). There were two variants detected in saliva that were not detected in the paired blood samples (Supplementary Table 2). Interestingly, these were from the same individual, a woman with a prior history of smoking but who had ceased smoking 40 years before providing these samples. It is possible given their absence in blood, that these two variants could be derived from mucosal epithelia8. Further development of methodologies aimed at reducing the epithelial content of saliva, such as that described by Soyfer et al. (2024), could help to refine a saliva derived based assay for CHIP.
When considering all CHIP-associated variants with VAFs between 0.02 and 0.20, eleven of the fourteen variants were detected in both the blood and saliva-derived DNA pairs with these thresholds. The VAFs of these variants in blood and saliva were similar between pairs and there was no suggestion that the VAF measured in the saliva-derived DNA was consistently reduced compared to blood—consistent with the DNA being predominantly from blood cell nuclei. There was no identifiable technical reason why three CHIP associated variants identified in different saliva-derived DNA samples had lower VAFs (between 0.008—0.013). TapeStation profiles were consistent with other well performing saliva-derived DNA samples, and all three of these saliva-derived DNA samples had at least 50% coverage at 500x (one had as high as 88% target coverage at 500x). The time between sampling of the three saliva and blood sample pairs ranged between 2 months and 34 months. However, given that CHIP progression seems to be ~ 0.5–1.0% per year2, it is unlikely CHIP clones evolved enough during this time between biological sampling to reflect observed changes in CHIP clone frequency in these VAF.
The small number of artifacts found in this study is likely a result of a combination of the small sample size; assessing only ten specific genes, none of which present technical sequencing challenges; and deep sequencing (average 1196x).
This study has a number of strengths: The Horizon’s myeloid control was diluted with a wildtype reference to provide confidence that variants would be called if present in the samples. All variants that were in this control, and in the assessed 10 genes, were successfully called after applying our pipeline and filtering methods. The participants included in this work were 64–75 years old, given the age relatedness of CHIP, the number of CHIP-associated variants in this group was anticipated to be ~ 10–15%11,12, which was consistent with our results. Variants were detected below the VAF threshold of 0.02 in saliva samples, indicating this method could be applied to variants present below this frequency. There is some evidence that supports clinical relevance for detecting CHIP-associated variants below the standard 2% threshold13–15. A limitation of this study due to the technical design, is that the study does not capture large chromosomal alterations and thus cannot detect mosaic chromosomal alterations.
Conclusion
This study has demonstrated that saliva-derived DNA is a suitable template for CHIP analysis. Saliva-derived DNA offers a cost effective, practical alternative biospecimen that could be utilised to both advance research and be a companion to clinical translation into settings such as risk prediction, precision prevention and treatment monitoring.
Methods
Ethical statement
The Australian Breakthrough Cancer Study is approved by the Cancer Council Victoria Human Ethics Review Committee (#1403). The conduct of our study is consistent with The National Health and Medical Research Council of Australia’s National Statement on ethical conduct in human research and performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants.
Source material
Paired saliva and blood samples were collected from 94 participants aged 64–75 years at enrolment into the Australian Breakthrough Cancer Study, a prospective cohort of over 56,000 Australians aged 40–74 and unaffected by cancer when recruited in 2014–18. Study participants were provided an at-home saliva collection kit, Oragene OG-500 (DNAGenotek), and returned the sample to Biobanking Victoria via a postal service. Blood samples were collected in EDTA tubes at local pathology services and processed centrally within 72 h of blood draw. Duration between collection of paired saliva and blood samples ranged from 2 to 34 months.
Reference standards were utilised including 100% wildtype (Catalogue ID: HD752) and a myeloid DNA reference standard (Catalogue ID: HD829) (Horizon Discovery, UK) to identify if this platform could detect variants at a VAF of at least 0.01. This control mix was included in each of the two 96 well plates.
DNA extraction
DNA was extracted from paired whole blood and saliva samples using either a Qiagen Symphony or Chemagic™ platform following manufacturers protocols (Qiagen, Valencia, CA; PerkinElmer, Waltham, MA, United States).
Sequencing panel design
The panel design consisted of 39 genes and covered 57.111 kbp. This study considered ten specific genes and gene regions (~ 28,805 kbp of the design) that where most likely to contain somatic variants associated with CHIP: DNMT3A, TET2, ASXL1, JAK2, GNB1, PPM1D, TP53, NF1, SRSF2, SF3B11,4,9,16.
Library preparation and sequencing
Agilent’s SureSelect XT HS2 DNA System was utilised using the automated Agilent NGS Workstation Option B (SureSelect; Agilent Technologies, Santa Clara, CA, USA). Input genomic DNA was 200 ng for both blood and saliva-derived DNA samples and 100 ng for the prepared horizon control. DNA enzymatic fragmentation and library preparation followed the SureSelect protocol with minor modification including extension of the fragmentation incubation time from 25 to 30 min to accommodate the target size of 2 × 75 bp. Pre-capture PCR conditions involved 8 cycles with unique dual-indexed primers, and sample libraries were assessed on Agilent’s 4200 TapeStation system using a D1000 ScreenTape. Libraries with poor profiles or low concentrations were noted but not excluded from sequencing to understand the impact that poor libraries had on variant calling between the source materials. Multiplex hybridisation (16x) and capture method for enrichment of targeted genes was applied before sequencing on NextSeq 550 using Illumina’s high output kit v2.5 (150 CYS) with the aim of reaching 80% coverage of target region at 500X. Sequencing methods followed Illumina’s NextSeq System: Denature and Dilute Libraries Guide17.
Bioinformatic pipeline for variant calling
Bioinformatic pipelines (Fig. 1) were written in Nextflow (v23.10.1)18 (https://github.com/Prec-Med/bldsal-analysis/tree/main) and executed on the ‘The Multi-modal Australian ScienceS Imaging and Visualisation Environment (MASSIVE) high performance computing infrastructure’ established by Monash University and partners19.
Raw sequence data conversion from bcl files to fastq used illumina’s bcl2fastq (v2.20) to achieve this. SureSelect adapters were trimmed with Agilent’s AGeNT tools v3.0.6 trimmer (Agilent Technologies, Santa Clara, CA, USA), before alignment to human genome reference build GRCh38 using BWA-MEM v0.7.1720. Unique Molecular Index (UMI) deduplication was performed with Agilent’s AGeNT CReaK in hybrid mode (Agilent Technologies, Santa Clara, CA, USA). Metrics for Fastqs and BAMs were generated with FastQC (v 012.1)21 and Genome Analysis Toolkit (GATK v4.4.0.0)22 before aggregating using MultQC (v1.18)23.
VarDict-java (v1.8.3)24 was used to call somatic variants as the caller can be used to call single nucleotide variants, multi-nucleotide variants, insertions/deletions, complex, and even structural variants13,24,25. However, this study focused specifically on insertions/deletions and single nucleotide variants. Variant calling thresholds were set at a VAF ≥ 0.005 before applying secondary thresholds later in the pipeline. Indel normalisation and multiallelic site decomposition, along with general VCF file manipulation, was conducted using bcftools (v1.18)26 before annotating with Ensembl-VEP v11127. Variants were then filtered with slivar (v0.3.0)28 using a threshold requiring a minimum of 5 reads per variant, and VAF between 0.02—0.20 (2—20%).
Agreement between variants called in the paired blood-saliva samples was evaluated using Starfish (https://github.com/dancooke/starfish) which uses Real Time Genomics (RTG)29 engine for VCF intersections. Blood/saliva VCF pairing, parallel execution of intersections, and aggregation of variant statistics from intersected VCFs (Supplementary Material) were performed in Python using pysam (https://github.com/pysam-developers/pysam).26 Sequence artifacts were identified and removed by applying a threshold of variant detected in greater than 10% of samples, other studies have used similar cut-offs (6%)13.
Variant filtering and identifying putative CHIP variants
Only variants identified in the genetic regions reported by Vlasschaert, et al. were assessed excluding premature truncating variants 3’ to the last 50 bases of the penultimate exon—to distinguish bona fide CHIP variants from somatic variants that have not been previously associated with clonal expansion of haematopoietic stem cells4.
Read alignment and quality for all variants were manually inspected using Interactive Genomics Viewer (IGV, Broad Institute, MA) to confirm sufficient read depth and allele balance. Variants were also inspected to make sure they were not i) in regions of low genomic complexity (i.e. homopolymer regions), ii) in regions with multiple misaligned reads, iii) in regions with multiple nearby non-reference or poor-quality base calls, or iv) in regions with exon–intron boundary soft clipping. Any variants suspected to be sequencing or mapping artifacts were flagged. Variants that were not identified in both samples were investigated to identify if this was because the VAF fell outside of the 0.02—0.2 cut-off or if the VD was less than 5.
Supplementary Information
Acknowledgements
This study was made possible by the contribution of many people. In particular, we thank the thousands of participants from across Australia who continue to participate in the study. The ABC Study was funded by Cancer Council Victoria, State Trustees, and a generous gift from the Geary Estate. Funding to collect blood samples [LP1] was provided by Gandel Philanthropy, the Ian Potter Foundation and the Harry Secomb Foundation and the Percy Baxter Charitable Trust, managed by Perpetual Trustees. Funding to collect faecal samples was provided by Gandel Philanthropy and Perpetual Trustees, (Winifred & John Webster Charitable Trust Fund, Pf – Alan (Agl), Shaw Endowment and Broomhead Family Foundation. [LP2] Cases and their vital status were ascertained through the Victorian Cancer Registry and the Australian Institute of Health and Welfare, including the Australian Cancer Database. This work was also funded by the Australian Medical Research Future Fund (PI Nicholls) and the National Health Medical Research Council (Investigator Grant GNT2017325; Southey).
Author contributions
R.L.O., J.B., P.H., M.C.S., P.Y., K.H., conceptualised and drafted the manuscript; developed the study design’s logistics; generated, analysed, and interpreted the data. R.L.O., A.R. conducted the lab work. H.T. managed the biological material and collection. M.C.S., S.J.N., K.J.B., G.G.G., R.L.M. provided grant funding and conceptualised the study design. All authors contributed substantially to manuscript preparation. All authors approved the final version.
Data availability
Data presented in this report can be requested via PEDIGREE. https://www.cancervic.org.au/research/epidemiology/pedigree.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-69398-0.
References
- 1.Asada, S. & Kitamura, T. Clonal hematopoiesis and associated diseases: a review of recent findings. Cancer Sci.112, 3962–3971. 10.1111/cas.15094 (2021). 10.1111/cas.15094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Steensma, D. P. et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood.126, 9–16. 10.1182/blood-2015-03-631747 (2015). 10.1182/blood-2015-03-631747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heuser, M., Thol, F. & Ganser, A. Clonal hematopoiesis of indeterminate potential. Dtsch Arztebl Int.113, 317–322. 10.3238/arztebl.2016.0317 (2016). 10.3238/arztebl.2016.0317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vlasschaert, C. et al. A practical approach to curate clonal hematopoiesis of indeterminate potential in human genetic data sets. Blood.141, 2214–2223. 10.1182/blood.2022018825 (2023). 10.1182/blood.2022018825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kessler, M. D. et al. Common and rare variant associations with clonal haematopoiesis phenotypes. Nature.612, 301–309. 10.1038/s41586-022-05448-9 (2022). 10.1038/s41586-022-05448-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiede, C., Prange-Krex, G., Freiberg-Richter, J., Bornhäuser, M. & Ehninger, G. Buccal swabs but not mouthwash samples can be used to obtain pretransplant DNA fingerprints from recipients of allogeneic bone marrow transplants. Bone Marrow Transplant.25, 575–577. 10.1038/sj.bmt.1702170 (2000). 10.1038/sj.bmt.1702170 [DOI] [PubMed] [Google Scholar]
- 7.Endler, G., Greinix, H., Winkler, K., Mitterbauer, G. & Mannhalter, C. Genetic fingerprinting in mouthwashes of patients after allogeneic bone marrow transplantation. Bone Marrow Transplant.24, 95–98. 10.1038/sj.bmt.1701815 (1999). 10.1038/sj.bmt.1701815 [DOI] [PubMed] [Google Scholar]
- 8.Soyfer, E. M. et al. Saliva as a feasible alternative to blood for interrogation of somatic hematopoietic variants. Blood Neoplasia.10.1016/j.bneo.2024.100012 (2024). 10.1016/j.bneo.2024.100012 [DOI] [Google Scholar]
- 9.Bolton, K. L. et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet.52, 1219–1226 (2020). 10.1038/s41588-020-00710-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coombs, C. C. et al. Therapy-related clonal hematopoiesis in patients with non-hematologic cancers is common and associated with adverse clinical outcomes. Cell Stem Cell.21, 374–382. 10.1016/j.stem.2017.07.010 (2017). 10.1016/j.stem.2017.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaiswal, S. et al. Age-related clonal hematopoiesis associated with adverse outcomes. New Engl. J. Med.371, 2488–2498. 10.1056/NEJMoa1408617 (2014). 10.1056/NEJMoa1408617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park, S. J. & Bejar, R. Clonal hematopoiesis in aging. Curr. Stem Cell Rep.4, 209–219. 10.1007/s40778-018-0133-9 (2018). 10.1007/s40778-018-0133-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan, I. C. C. et al. ArCH: Improving the performance of clonal hematopoiesis variant calling and interpretation. Bioinformatics.10.1093/bioinformatics/btae121 (2024). 10.1093/bioinformatics/btae121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman, D. N. et al. Clonal hematopoiesis in survivors of childhood cancer. Blood Adv.7, 4102–4106. 10.1182/bloodadvances.2023009817 (2023). 10.1182/bloodadvances.2023009817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Young, A. L., Tong, R. S., Brenda, M. B. & Todd, E. D. Clonal hematopoiesis and risk of acute myeloid leukemia. Haematologica.104, 2410–2417. 10.3324/haematol.2018.215269 (2019). 10.3324/haematol.2018.215269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marnell, C. S., Bick, A. & Natarajan, P. Clonal hematopoiesis of indeterminate potential (CHIP): Linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J. Mol. Cell Cardiol.161, 98–105. 10.1016/j.yjmcc.2021.07.004 (2021). 10.1016/j.yjmcc.2021.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Illumina. NextSeq system: denature and dilute libraries guide. https://support.illumina.com/content/dam/illumina-support/documents/documentation/system_documentation/nextseq/nextseq-denature-dilute-libraries-guide-15048776-09.pdf (2018).
- 18.Di Tommaso, P. et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol.35, 316–319. 10.1038/nbt.3820 (2017). 10.1038/nbt.3820 [DOI] [PubMed] [Google Scholar]
- 19.Goscinski, W. J. et al. The multi-modal Australian sciences imaging and visualization environment (MASSIVE) high performance computing infrastructure: Applications in neuroscience and neuroinformatics research. Front. Neuroinformatics.10.3389/fninf.2014.00030 (2014). 10.3389/fninf.2014.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv1303, (2013).
- 21.Andrews, S. FastQC a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
- 22.O'Connor, B. D. & van der Auwera, G. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. (O'Reilly Media, Incorporated, 2020).
- 23.Ewels, P., Magnusson, M., Lundin, S. & Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics.32, 3047–3048. 10.1093/bioinformatics/btw354 (2016). 10.1093/bioinformatics/btw354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai, Z. et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res.44, e108. 10.1093/nar/gkw227 (2016). 10.1093/nar/gkw227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soerensen, M. et al. Clonal hematopoiesis and epigenetic age acceleration in elderly danish twins. HemaSphere.6, e768. 10.1097/hs9.0000000000000768 (2022). 10.1097/hs9.0000000000000768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Danecek, P. et al. Twelve years of SAMtools and BCFtools. Gigascience.10.1093/gigascience/giab008 (2021). 10.1093/gigascience/giab008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McLaren, W. et al. The ensembl variant effect predictor. Genome Biol.17, 122. 10.1186/s13059-016-0974-4 (2016). 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pedersen, B. S. et al. Effective variant filtering and expected candidate variant yield in studies of rare human disease. NPJ Genom. Med.6, 60. 10.1038/s41525-021-00227-3 (2021). 10.1038/s41525-021-00227-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cleary, J. G. et al. Comparing variant call files for performance benchmarking of next-generation sequencing variant calling pipelines. bioRxiv. 023754, 10.1101/023754 (2015).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data presented in this report can be requested via PEDIGREE. https://www.cancervic.org.au/research/epidemiology/pedigree.