

Abstract
Adaptive divergence between marine and freshwater (FW) environments is important in generating phyletic diversity within fishes, but the genetic basis of this process remains poorly understood. Genome selection scans can identify adaptive loci, but incomplete knowledge of genotype–phenotype connections makes interpreting their significance difficult. In contrast, association mapping (genome-wide association mapping [GWAS], random forest [RF] analyses) links genotype to phenotype, but offer limited insight into the evolutionary forces shaping variation. Here, we combined GWAS, RF, and selection scans to identify loci important in adaptation to FW environments. We utilized FW-native and brackish water (BW)-native populations of Atlantic killifish (Fundulus heteroclitus) as well as a naturally admixed population between the two. We measured morphology and multiple physiological traits that differ between populations and may contribute to osmotic adaptation (salinity tolerance, hypoxia tolerance, metabolic rate, body shape) and used a reduced representation approach for genome-wide genotyping. Our results show patterns of population divergence in physiological capabilities that are consistent with local adaptation. Population genomic scans between BW-native and FW-native populations identified genomic regions evolving by natural selection, whereas association mapping revealed loci that contribute to variation for each trait. There was substantial overlap in the genomic regions putatively under selection and loci associated with phenotypic traits, particularly for salinity tolerance, suggesting that these regions and genes are important for adaptive divergence between BW and FW environments. Together, these data provide insight into the mechanisms that enable diversification of fishes across osmotic boundaries.
Keywords: adaptation, selection scans, genome wide association mapping, salinity
Introduction
Evolutionary transitions from marine to freshwater (FW) environments have served a vital role in generating the phyletic diversity within ray-finned fishes (Vega and Wiens 2012; Betancur-R et al. 2015). It has been hypothesized that adaptive divergence of estuarine species in different salinities is a major driver of these transitions (Schultz and McCormick 2012). Indeed, adaptive divergence plays a role in the generation of new species across diverse phyla (ecological speciation, Nosil 2012). Therefore, discovering the mechanisms that enable such adaptive divergence between osmotic niches is important for understanding the mechanisms that drive speciation within fishes. To understand the processes involved in this divergence, it is necessary to understand how traits and their underlying molecular pathways and genes facilitate adaptation and isolation in different ecological niches.
Despite its importance, identifying the mechanistic basis of adaptation is often difficult. Many recent successes include morphological traits, for example in stickleback fish, field mice, and butterflies (Colosimo et al. 2005; Manceau et al. 2011; Dasmahapatra et al. 2012), and these studies have implicated a small number of genes of major effect in these processes. However, it has been argued that these classic examples may not be typical of how evolution often proceeds (Rockman 2012). For many adaptive challenges, relevant traits may be highly polygenic, and multiple traits may contribute to a complex adaptive phenotype. This may be especially true along marine to FW gradients where many environmental features vary (e.g., salinity, oxygen, pH, food, predators, pathogens, symbionts, tidal rhythmicity, etc., Lozupone and Knight 2007; Austin et al. 2012; Osborne et al. 2015; Ou et al. 2015) and may affect fitness. For example, while diversification of morphological traits in stickleback is driven by loci of large effect, there is also evidence for complex physiological adaptation to fresh water (Jones et al. 2012; DeFaveri and Merila 2014). Genome wide association mapping (GWAS) is often deployed to identify genetic variation underlying phenotypic traits, but is often inefficient for discovering variation associated with highly polygenic traits (Rockman 2012). In contrast, machine learning algorithms such as random forests (RF) may be better suited to identifying variation associated with complex polygenic phenotypes (Brieuc et al. 2018). However, both GWAS and RF offer limited insight into the fitness effects of that variation. In contrast, genome-wide scans for signatures of selection offer an unbiased approach for discovering the many genomic regions that underlie local adaptation. Yet, selection scans rarely enable inference that links specific genotypic variation with relevant phenotypes. In confronting this conundrum, recent work has suggested integrating association mapping and genome-wide scans for signatures of natural selection (Berg and Coop 2014). By combining these approaches, it is possible to both identify causal variants underlying specific phenotypes and simultaneously address whether or not these variants are the targets of selection (Weigel and Nordborg 2015).
Since adaptation to fresh water should require the modulation of multiple traits, fishes that have colonized environments with different salinities are an ideal system for understanding how complex physiologies evolve to enable divergence across key ecological boundaries. Killifish within the Fundulus genus provide a particularly compelling model system for studying this diversification between marine and FW environments. Within this genus, marine to FW transitions have occurred at least three times independently (Whitehead 2010; Ghedotti and Davis 2013); such lability is rare for closely related species (Vega and Wiens 2012). Fundulus heteroclitus is a particularly good microevolutionary model species in which to identify the genetic and physiological mechanisms important for FW adaptation. This species inhabits environments ranging from marine to fresh water (Hildebrand and Schroeder 1928), and exhibits adaptive divergence along salinity gradients (Whitehead et al. 2011). In the Potomac River and Chesapeake Bay, genetically distinct populations occupy different osmotic niches. A genetic break occurs along a salinity cline where individuals upstream of the FW boundary are distinct from downstream fish in brackish water (BW; Duvernell et al. 2008; Whitehead et al. 2011). FW populations (FW-native) exhibit salinity-dependent physiological differences relative to their BW counterparts (BW-native), potentially representing the incipient stages of speciation across an osmotic boundary (Whitehead et al. 2011; Brennan et al. 2015, 2016), making this an ideal system to identify genomic regions under divergent selection. Simultaneously, GWAS can be used to identify loci that are likely to influence physiological performance and underlie local adaptation to each environment. Integrated together, these approaches can reveal the genes important for adaptation and divergence in FW environments.
In this study, we seek to identify the physiological and genomic basis of adaptation to fresh water. Specifically, we addressed the following questions: 1) Does adaptation account for the divergence of a suite of physiological traits between BW-native and FW-native populations?; 2) What is the genetic basis of FW adaptation in these traits? To address these questions, we sampled F. heteroclitus individuals from BW-native and FW-native populations (“parental” populations) along a salinity gradient in Chesapeake Bay, and individuals that are genetically intermediate to BW- and FW-native fish (“admixed” population; fig. 1). The presence of admixed individuals can be leveraged to identify genomic regions associated with phenotype (admixture mapping, Buerkle and Lexer 2008). Furthermore, we can test for the independent segregation of multiple phenotypic traits in admixed individuals to understand the modularity of traits and how this may influence adaptation. To quantify physiological divergence between populations, all individuals were assessed for salinity tolerance, hypoxia, and metabolic rate (MR)—physiological traits known to differ between populations of this species and that may underlie adaptation to low salinity environments (Fangue et al. 2009; Brennan et al. 2015; McBryan et al. 2016). For example, seawater holds less oxygen than FW (Osborne et al. 2015) and higher hypoxia tolerance should be favored at higher salinity. Conversely, osmoregulation is more costly in fresh water (Kidder et al. 2006) and MR may need to be altered as a consequence. We also compared morphology since it has been shown to adaptively diverge between marine and FW habitats in other fish systems (Colosimo et al. 2004). We genotyped these individuals using RADseq and performed selection scans to identify genomic regions evolving by natural selection. We performed association mapping to identify the genetic variants underlying variation in physiological traits using both genome-wide association for individual genes and a polygenic RF approach. This combination of selection scans and association mapping is especially powerful because regions overlapping between the approaches are those that are the most likely to be involved in adaptive divergence of this suite of physiological traits between marine and FW environments.
Fig. 1.
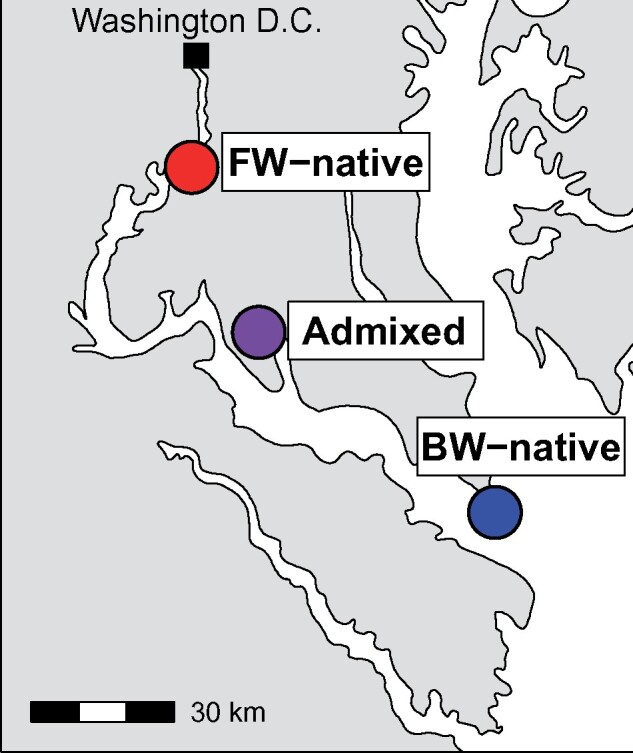
Map of sampling locations in the Potomac River and Chesapeake Bay.
Results
Phenotypic Variation
There was significant variation in each of the physiological phenotypes measured both within and between populations (fig. 2). In low salinity, fish must actively regulate their internal homeostasis to prevent ion loss and limit water influx (Evans et al. 2005). Fish that fail to acclimate will experience a dilution of body fluid and individuals with greater tolerance will maintain higher internal ion concentrations. For our FW challenge, parental populations did not differ significantly in plasma Cl- regulation (all results mean ± standard error; BW-native = 122.0 ± 1.46 mM; FW-native = 126.5 ± 1.64 mM; P = 0.13), but qualitatively the FW-native population maintained higher plasma Cl- when facing FW challenge than BW-native individuals, consistent with previous research that showed similar but significant differences and evidence of adaptive divergence in this trait between these populations (Whitehead et al. 2011; Brennan et al. 2015). The lack of significance here is likely due to the use of only one sampling time point. Admixed individuals lost Cl− homeostasis to a greater degree than either of the parental populations (admixed = 108.1 ± 0.84 mM; P < 0.001; fig. 2A), a pattern that is consistent with negative epistasis. Hypoxia tolerance showed a clinal pattern where FW-native individuals tolerated low oxygen levels for the shortest time (30.3 ± 2.81 min), BW-native for the longest time (89.5 ± 2.71 min), and admixed individuals were intermediate (50.0 ± 1.64 min; fig. 2B). All population comparisons were significant at P < 0.001. MR results differed from hypoxia tolerance; FW-native individuals had high MR (4.7 ± 0.20 μmol O2g−1 h−1), yet BW-native (3.2 ± 0.07 μmol O2g−1 h−1) and admixed populations (3.3 ± 0.03 μmol O2g−1 h−1) were significantly lower (P < 0.001) and not different from each other (P = 0.96; fig. 2C). Finally, regressions between the three phenotypes in admixed individuals revealed no significant correlations, suggesting that phenotypic variation in salinity tolerance, hypoxia tolerance, and MR are inherited independently (P > 0.56 for all comparisons; see supplementary fig. S1, Supplementary Material online).
Fig. 2.

Variation in physiological traits for all populations. Plots depict freshwater tolerance (A), hypoxia tolerance (B), and metabolic rate (C) for the FW-native, admixed, and BW-native populations. Data presented in Tukey boxplots and all values have been mass corrected to a fish size of 4g (for visualization purposes only). Letters above each box indicate statistically significant differences following post hoc tests.
Morphological data suggest that all populations are distinct. Principal component analysis (PCA) separated the admixed individuals from both parental populations along principal component 1 (PC1) (46% of the variation) whereas PC2 distinguished the FW-native and BW-native populations (17% of the variation; see supplementary fig. S2, Supplementary Material online). The shape changes between populations appears to depend on the relative location of the caudal and anal fin relative to the tail for PC1; there was also a subtle shift in eye location. PC2 suggested similar shifts; however, the ratio of the tail fin itself was also elongated and shortened in FW-native versus BW-native individuals. Permutation tests from a canonical variate analysis revealed significant differences for Mahalanobis distances for all populations (P < 0.001) but Procrustes distance permutations showed only differences between admixed and the two parental populations (P < 0.001) but no difference between the parental populations (P = 0.065).
Population Genetics
Population genetic results demonstrate that the admixed population is genetically intermediate to both parental populations (fig. 3A and B). PC1 explains 20% of the variation among individuals and distinguishes populations along the salinity cline. PC2 separates admixed individuals from both parental populations and explains 7% of the interindividual variation (fig. 3A). For admixture analysis, genotypes from parental populations clustered into different groups while individuals sampled from the zone of admixture have an average admixture proportion of 0.62 (range 0.52–0.70; fig. 3B). Genome-wide average FST values between populations were: FW-native versus BW-native 0.082, FW-native versus Admixed 0.035, BW-native versus Admixed 0.022.
Fig. 3.

Population structure of the FW-native, admixed, and BW-native populations as determined by PCA (A) and Admixture (B) analyses. (A) Each point represents an individual genotype where shapes and colors distinguish populations. The amount of variation explained by PC1 and PC2 is 20% and 7%, respectively. (B) Inferred ancestry proportions for each individual where the number of ancestral populations (K) is 2. Each bar represents an individual and x-axis location specifies sampling location.
Selection Scans
Selection scans revealed regions of elevated divergence across the genome. We identified 1387 outlier loci when using an LD value of 27 kb. These loci collapsed into 679 outlier windows with an average size of 3.5 kb. 285 of the windows contained >1 outlier SNP. The largest outlier window contained 18 outlier variants with a length of 128.9 kb, falling on chromosome 8 from 2.61 to 2.74 Mb. See supplementary figure S3, Supplementary Material online for distribution of outlier window lengths.
Genotype–Phenotype Association
GWAS results using typical genome-wide Bonferroni-corrected significance cutoffs yielded significantly associated loci for only the MR phenotype, which had one SNP passing the threshold (see supplementary figs. S4 and S5, Supplementary Material online). Hypoxia tolerance had one variant passing the LD thinned threshold. Proportion of variance explained (PVE ± standard error) by all loci in the GWAS for each trait was as follows: salinity 0.83 ± 0.15; MR 0.52 ± 0.17; hypoxia 0.27 ± 0.22; PC1 0.48 ± 0.17; PC2 0.14 ± 0.39. The 0.1% most significant GWAS hits resulted in 139 candidate loci for each phenotype and these were distributed throughout the genome (see supplementary fig. S6, tables S2–S6, Supplementary Material online). The best fit model from RF analysis detected 26, 23, 36, 37, and 45 SNPs for salinity tolerance, hypoxia tolerance, MR, morphology PC1 and PC2, respectively. These sets explained 0.54, 0.46, 0.58, 0.79, and 0.61 of the variation. To make the RF analysis comparable to the GWAS, we consider the top 139 variants as potentially influencing phenotype (see supplementary fig. S7, tables S7–S11, Supplementary Material online). Comparing these sets of 139 variants, there were 28, 23, 19, 13, and 16 overlaps between the top GWAS and RF SNPs for salinity tolerance, hypoxia, MR, morphology PC1 and PC2, respectively (see supplementary tables S7–S11, Supplementary Material online). For all phenotypes, this overlap is significantly more than would be expected by chance (P < 0.001).
Overlap between Association Mapping and Selection Scans
We identified 18 GWAS variants associated with physiological traits that fell within genomic regions showing signatures of natural selection between marine and FW populations (fig. 4A). Permutation tests show that these top phenotype-associated loci were more likely to show signatures of selection than by chance (fig. 4B). That is, genomic signatures of selection were enriched for top GWAS loci for each of the physiological traits. Salinity and hypoxia showed six overlapping variants between GWAS and selection scan outlier regions (P = 0.005; P = 0.002). The four overlapping loci for MR were also more than expected by chance (P = 0.05). See table 1 for a complete list of variants overlapping with selection scan outlier regions. Of the 16 candidate SNPs, 13 fell in coding regions of genes whereas the rest were in noncoding regions. Morphology analysis showed 0 and 2 overlapping variants between GWAS (fig. 4A) and selection scan outlier regions, which is no more than is expected by chance (P > 0.05). It should be noted that the method for LD estimation slightly influences our results. Using the 27 kb LD cutoff, our variants are informative of ∼73% of the genome. The more conservative 5 kb cutoff provides information for ∼25% of the genome and the outlier windows collapse into 839 windows with an average of 0.2 kb and a max of 4.8 kb. However, this only slightly alters our main findings. Salinity tolerance, hypoxia tolerance, and MR drop to 4, 3, and 2 variants that overlap between phenotype association and selection scan regions (P = 0.04, P = 0.05, P = 0.31). See table 1 for details.
Fig. 4.

Integration of genome-wide scans for signatures of natural selection and genome-wide association mapping of physiological traits. (A) Signatures of selection as represented by Mahalanobis distance across the genome, where alternating black and gray dots represent genetic variants associated with different chromosomes. Horizontal solid red line is the 99% cut-off for the selection scan. Each black or grey point represents a variant and its location in the genome. Unplaced scaffolds are placed on the far right. Vertical dashed lines signify overlap between loci associated with phenotypes and loci showing signatures of selection between BW-native and FW-native populations. The color of the symbol above each line specifies the relevant phenotype. Open shapes are variants identified with GWAS, closed shaped from RF. Where multiple variants are associated with phenotype and fall within a single region showing a signal of natural selection, points are stacked. Variants significant for both GWAS and RF are indicated by *. (B, C, D) Results from 2,500 permutations to test for nonrandom overlap between variants showing signatures of natural selection and phenotype-associated variant from (B) GWAS, (C) RF, and (D) intersection of GWAS and RF. Grey bars represent the distribution of the number of variants expected to overlap from random permutations. Each shape and dashed line are the number of empirically discovered overlaps. Note that some points have been jittered for clarity.
Table 1.
Candidate SNPs Overlapping between Association Mapping and Selection Scans.
| Phenotypea | Gene Name | Gene Abbreviation | SNP Location (BP) | Location Relative to Gene | 5kb Sig.b | Statisticc |
|---|---|---|---|---|---|---|
| Osmoregulation | TBC1 domain family member 9 | TBC1D9 | Chr4: 257,976 | Coding | No | GWAS + RF |
| Osmoregulation | Lysosomal thioesterase PPT2 | PPT2 | Chr6: 9,463,694 | Coding | Yes | GWAS |
| Osmoregulation | Histone-lysine N-methyltransferase | EHMT2 | Chr6: 9,524,839 | Coding | Yes | RF |
| Osmoregulation | MARVEL domain-containing protein 2 | MARVELD2 | Chr11: 34,323,063 | Coding | No | GWAS + RF |
| Osmoregulation | Potassium channel, subfamily K, member 3 | KCNK3/ TASK1 | Chr19: 34,342,868 | 29 kb upstream | Yes | GWAS + RF |
| Osmoregulation | Potassium channel, subfamily K, member 3 | KCNK3/ TASK1 | Chr19: 34,342,975 | 29 kb upstream | Yes | GWAS |
| Osmoregulation | Serine/threonine-protein kinase 26 | STK26 | Chr23: 26,009,786 | 1 kb upstream | Yes | RF |
| Osmoregulation | U4/U6 small nuclear ribonucleoprotein Prp4 | PRPF4 | NW_012225290.1: 53,815 | Coding | No | GWAS |
| Hypoxia | Uncharacterized | na | Chr3: 7,631,466 | Coding | No | GWAS |
| Hypoxia | Uncharacterized | na | Chr6: 766,051 | Coding | Yes | RF |
| Hypoxia | Uncharacterized | na | Chr6: 766,080 | Coding | Yes | RF |
| Hypoxia | Hyaluronan synthase 1 | HAS1 | Chr8: 20,253,186 | 8.5 kb upstream | Yes | GWAS |
| Hypoxia | Amnionless | AMN | Chr9: 18,223,455 | Coding | No | GWAS |
| Hypoxia | wu: fd14a06 (uncharacterized) | na | Chr21: 1,992,631 | Coding | Yes | GWAS |
| Hypoxia | wu: fd14a06 (uncharacterized) | na | Chr21: 1,992,713 | Coding | Yes | GWAS |
| Hypoxia | wu: fd14a06 (uncharacterized) | na | Chr21: 1,992,747 | Coding | Yes | GWAS |
| Metabolic rate | YrdC domain-containing protein, mitochondrial | YRDC | Chr4: 25,567,990 | Coding | Yes | GWAS |
| Metabolic rate | Collagen alpha-6(VI) chain | COL6A6 | Chr8: 2,696,598 | Coding | Yes | GWAS |
| Metabolic rate | Deafness Associated Tumor Suppressor | DFNA5 | Chr11: 1,047,562 | Coding | No | GWAS |
| Metabolic rate | Arrestin domain-containing protein 3 | ARRDC3 | Chr24: 26,089,526 | Coding | No | GWAS |
| Metabolic rate | Ferric-chelate reductase 1 | FRRS1 | NW_012225290.1: 60,216 | Coding | Yes | RF |
| Morphology: PC1 | Myeloperoxidase | MPO | Chr24: 21,343,691 | Coding | Yes | RF |
| Morphology: PC2 | Heterogeneous nuclear ribonucleoprotein A3 | HNRNPA3 | Chr1: 20,964,738 | 56 kb upstream | Yes | RF |
| Morphology: PC2 | Oxysterol-binding protein-related protein 6 | OSBPL6 | Chr1: 21,194,584 | Coding | Yes | RF |
| Morphology: PC2 | Sentan | SNTN | Chr8: 1,969,596 | Coding | Yes | GWAS + RF |
| Morphology: PC2 | Semaphorin-4B | SEMA4B | Chr8: 2,032,200 | Coding | Yes | RF |
| Morphology: PC2 | Thyroglobulin-like protein | TG | Chr8: 2,434,973 | Coding | Yes | RF |
| Morphology: PC2 | Calcitonin | CALCA | NW_012224574.1: 519,435 | 29 kb upstream | Yes | RF |
| Morphology: PC2 | Interleukin-34 | IL34 | NW_012234311.1: 4,689,696 | Coding | Yes | GWAS |
Table based on an estimated LD of 27 kb.
Indicates variants that remain overlapping with association hits when an estimated LD of 5 kb is used.
Statistic column indicates the association mapping approach that identifies each SNP.
RF revealed 15 trait-associated variants overlapping with selection scan outlier regions (fig. 4A and C;table 1). For physiological traits, only salinity tolerance showed significant enrichment with selection scan outlier regions (five overlapping variants, P = 0.02, fig. 4C). Morphology PC2 was also significantly enriched with six variants in selection scan outlier regions (P = 0.016, fig. 4C). When considering trait-associated variants that were detected by both GWAS and RF, hypoxia tolerance, MR, and morphology PC1 showed no overlap with selection scan outlier regions (table 1). Morphology PC2 had one variant detected by both approaches (P = 0.27). For salinity tolerance, there were three trait-associated variants detected in both GWAS and RF that were also identified as putatively under selection, which is significantly more than expected by chance (fig. 4D;P = 0.003).
Discussion
When studying adaptation to a complex environment, phenotypes that are the targets of natural selection may be many and complex. For inferring the genetic basis of such adaptations, phenotype agnostic approaches such as genome scans provide a path forward but interpreting the phenotypic relevance of the implicated genes can be difficult. Conversely, approaches such as GWAS and RF focus specifically on the variants underlying phenotypes of interest, but provide no information concerning the adaptive significance of these variants. We attempted to confront and ameliorate the limitations of these two approaches by integrating them; an idea that has only recently begun to gain traction (Via et al. 2012; Chaves et al. 2016; McGirr and Martin 2016; Brelsford et al. 2017; Pfeifer et al. 2018). Our approach is unique in that we leverage LD generated by natural admixture between populations to identify genotype–phenotype associations (see also Brelsford et al. 2017). We first located potential adaptive regions of the genome using scans for selection then used GWAS and a polygenic RF approach to reveal genomic regions that are associated with multiple potentially adaptive phenotypes. By identifying the regions where GWAS, RF, and selection scans overlap, we isolate the genomic regions under selection while simultaneously inferring physiological function of these regions. At the same time, we provide additional evidence that natural selection is governing the divergence of these physiological phenotypes between marine and FW environments. This study extends our understanding of the genotypes and phenotypes that are important for adaptation to alternate osmotic environments, which has heretofore been primarily focused on morphological traits. We interrogate multiple, complex physiological phenotypes that are known to diverge between FW and marine environments. This focus enables us to identify mechanisms of adaptive divergence that have likely been important in enabling speciation events across osmotic boundaries in fishes.
We find strong evidence that salinity tolerance is diverging through adaptive processes. There is significant enrichment in overlap between top-ranked GWAS loci and top-ranked selection scan regions, and between top-ranked RF loci and selection scan regions, where at least three of these loci showed trait association detected using both the GWAS and RF approaches. This suggests that the regions of the genome influencing the key phenotype of salinity tolerance are disproportionally divergent between the FW-native and BW-native populations (fig. 4). It should be noted that it is possible that selection on correlated traits or pleiotropic effects could be driving the signal of selection rather than direct selection on the measured trait, but the overlap between the association mapping results and the signatures of selection strongly implies a direct association. Further, whereas the salinity tolerance data are not significantly different between the BW- and FW-native populations, the pattern of increased tolerance in FW-native individuals is consistent with previous studies that have identified adaptation (Whitehead et al. 2011; Brennan et al. 2015). We conclude that divergence in salinity tolerance is important for enabling evolutionary transitions from marine to FW environments.
We detected evidence of selection on a subset of the SNPs identified using GWAS for hypoxia tolerance and MR. However, these SNPs did not overlap between the GWAS and RF approaches. In contrast, the RF approach identified morphology PC2 variants that show evidence of selection that was not evident using a GWAS approach. The differences between approaches may be driven by the different strengths of each method; GWAS detects single locus associations while RF is better suited for identifying the more complex genetic basis of quantitative traits such as nonadditive interactions and epistatic interactions (Brieuc et al. 2018). Furthermore, the overlap between the selection scan and association analyses may be an underestimate of the true value due to conditional neutrality. That is, because all individuals were common gardened at 20 ppt, the GWAS and RF will miss or underestimate variants that influence phenotype at low salinity but are neutral at higher salinity; this phenomenon appears to be common in natural populations (Fournier-Level et al. 2011). Therefore, while we provide modest evidence that hypoxia tolerance, MR, and morphology are under selection between BW and FW environments, additional work is needed to verify this finding.
Physiological and genomic results suggest that there is limited overlap in the genetic underpinnings of the three physiological traits. Covariance between traits could be due to LD between underlying genes, epistatic interactions between genes, or by pleiotropy, where one gene influences multiple phenotypes (Wolf et al. 2005; Saltz et al. 2017). We find no correlations between the phenotypes in admixed individuals (P > 0.05, see supplementary fig. S1, Supplementary Material online). Similarly, there is no overlap among adaptive candidate genes between the three phenotypes. As such, the lack of correlations between the phenotypes in admixed individuals suggest that there is minimal shared genetic basis influencing these traits. A shared genetic basis between traits can limit or accelerate the efficiency of natural selection and adaptation, depending on the direction of correlations (positive or negative). The modular nature of these phenotypes implies that each trait can be altered independently, thereby promoting the evolvability of a diversity of complex physiological phenotypes (Hansen 2003; Van Belleghem et al. 2017).
Genetic Basis of Divergence in Osmoregulatory Physiology
The variation between populations in the ability to regulate plasma chloride suggests that, while the parental populations are adapted to their native salinity, admixed individuals have reduced osmoregulatory capabilities relative to the parents (fig. 2). This pattern may represent hybrid breakdown due to negative epistasis (Burke and Arnold 2001), which is indicative of Bateson–Dobzhansky–Muller incompatibilities (Bateson 1909; Dobzhansky 1936; Muller 1942). Sets of parental alleles that typically do not co-occur due to purging through natural selection may be brought together in hybrids. Because evolutionarily derived alleles tend to be those responsible for incompatibilities (Orr 1995), adaptation to FW may result in the reduction of performance seen in admixed individuals. This process would impede gene flow across the hybrid zone, enabling adaptive divergence between the parental populations.
In fish, gills function as the primary site of osmoregulation. Upon transitioning from high to very low salinity, killifish reversibly remodel their gill morphology as part of a strategy to maintain osmotic homeostasis. This process involves regulating cell volume, replacing or remodeling the morphology of the cells responsible for ion transport (ionocytes; fig. 5), expressing appropriate ion transport proteins and inserting them in the appropriate surface (apical or basolateral) of ionocytes, and modulating junctions between epithelial cells to prevent or allow ion loss (Wilson and Laurent 2002; Hwang et al. 2011). Previous work in F. heteroclitus has established that the ability to transition between alternate gill morphologies is essential during acclimation to different osmotic conditions (Scott, Richards, et al. 2004; Whitehead et al. 2012; Brennan et al. 2015). More specifically, previous comparative physiology and transcriptomics research led to a prediction that adaptive divergence between marine and FW killifish is linked to divergence in both cell junction regulation and ion transport (Whitehead et al. 2011; Brennan et al. 2015). Consistent with this a priori prediction, here we identify five genes involved in these processes, three of which overlap between the GWAS and RF analyses. These genes (table 1) are associated with variation in osmoregulatory physiology among individuals and show signatures of selection between BW-native and FW-native populations, and are therefore likely contributing to adaptive osmoregulatory divergence.
Fig. 5.

Hypothesized model of adaptive sodium uptake metabolon in freshwater fish. Genes (bold black) that show signatures of selection in genome scans and that contribute to ionoregulation. The model on the left is a fish ionocyte, where proteins constitute a metabolic unit that regulates sodium uptake in fresh water. NHE and CA facilitate sodium uptake from water, and NBC and NKA facilitate export of sodium into the blood. RHCG may also work with NHE to facilitate both ammonia excretion and sodium uptake. The ionocyte and an adjacent cell (right) are joined with paracellular tight junction proteins that regulate paracellular permeability.
TASK1 is an outwardly rectifying K+ channel and may be involved in cell volume regulation and was identified by both GWAS and RF. This gene plays a role in shark osmoregulation by establishing the driving force for apical Cl− excretion via CFTR (Telles et al. 2016). CFTR is an important ion channel in killifish at high salinity (Marshall et al. 2002; Scott et al. 2008), but our experiment focused on acclimation to low salinity, and it is unlikely that an outwardly rectifying K+ channel is functioning in the same manner here. TASK2 channels have been found to influence cell volume regulation, particularly in response to hypotonic stress and cell swelling (Stutzin and Hoffmann 2006). Regulatory volume decrease (RVD) involves the activation of Cl− and K+ channels (Hoffmann and Dunham 1995). It is possible that in F. heteroclitus orthologs to TASK1 facilitate RVD and variation in the structure or regulation of this gene results in differential osmoregulatory capabilities. This hypothesis merits further investigation.
Tight junctions form at intercellular junctions and work to regulate the flow of solutes through the paracellular pathway (Schneeberger and Lynch 2004). Tight junction proteins are well-known components of the osmoregulatory apparatus in fish (Hwang et al. 2011), and have been identified as regulators of variation in osmotic tolerance in stickleback (Gibbons et al. 2017), killifish (Whitehead et al. 2013), alewives (Velotta et al. 2017), silverside (Hughes et al. 2017), and sea bass (Tine et al. 2014), among others. At low salinity, tight junctions prevent the loss of Cl− whereas leaky junctions at high salinity enable the excretion of Na+. Tri-cellular junction proteins, which occur where any three cells join, influence paracellular permeability in fish gills (Kolosov and Kelly 2013; Kolosov et al. 2014). MARVELD2 (also known as tricellulin—a tri-cellular tight junction protein) was identified as a target of selection and was associated with trait variation by both GWAS and RF, suggesting that tri-cellular tight junctions are likely important in adaptive divergence in osmoregulatory physiology between FW-native and BW-native populations. This adds mechanistic insight into previous research identifying an evolutionary trade-off between BW-native and FW-native populations in the physiological ability to regulate tight versus leaky junctions during osmotic acclimation (Brennan et al. 2015).
We measured the ability to maintain plasma chloride homeostasis as a representation of the phenotypic variation in osmoregulatory abilities among individuals. However, other components of ionoregulatory physiology may be subject to adaptive divergence between marine and FW populations. Indeed, many genes show signatures of selection between parental populations that are consistent with this expectation. For example, outlier loci from selection scans are enriched for GO terms tight junction, regulation of intracellular pH, and bicarbonate transport. These include a number of genes that are core components of fish gill ionocytes, including multiple sodium bicarbonate cotransporters (NBC), sodium potassium ATPase (NKA), sodium hydrogen exchanger (NHE), carbonic anhydrase (CA), anion exchange protein (SLC4A3), rhesus glycoprotein (RHCG), and aquaporin (see supplementary table S12, Supplementary Material online). This is consistent with an evolved sodium uptake metabolon in the FW-native population, where apical sodium uptake is through NHE and CA, and basolateral sodium export is through NBC and NKA (fig. 5), as reported in rainbow trout (Parks et al. 2007). Rhesus glycoproteins interact with the same molecular machinery to pair ammonia excretion with sodium uptake in larval FW fish (Zimmer et al. 2017). Selection scan outliers also include multiple tight junction proteins (claudins and occludin) that may be involved in adaptive changes in paracellular ion transport pathways. Furthermore, the hormone prolactin is important for regulating ion retention and water excretion in FW, and prolactin receptor (PRLR) is within a selection scan outlier region (Breves et al. 2014). We hypothesize that derived mechanisms of sodium uptake, perhaps paired with ammonia excretion, are an important component of adaptive divergence between marine and FW populations of killifish.
Genetic Basis of Divergence in Hypoxia
Hypoxic environments occur regularly in marsh systems, primarily due to isolation of pools (i.e., during tidal cycles) and nocturnal respiration (Congleton 1980; Innes and Wells 1985; Timmerman and Chapman 2004). However, the oxygen availability in fresh versus salt tidal wetland differs where low salinity habitats typically have higher oxygen concentrations than those of higher salinity (Osborne et al. 2015). Therefore, one might predict that BW-native and FW-native populations regularly experience environments with different oxygen availability and have diverged in their relative ability to acclimate to hypoxia challenge. Population differences are consistent this expectation where FW-native individuals harbor lower hypoxia tolerance than the BW-native fish (fig. 2).
We identify eight SNPs that contribute to adaptive divergence in hypoxia tolerance between populations (table 1). Of these, none overlap between GWAS and RF analyses and six are uncharacterized, making biological interpretation difficult. However, GWAS identified a variant 8.5 kb upstream of HAS1 as a promising candidate. HAS1 synthesizes cellular hyaluronan, which is a polysaccharide that is one of the main components of the extracellular matrix (Siiskonen et al. 2015). Hyaluronan has been directly implicated as a negative regulator of hypoxia-inducible factor-1alpha (HIF-1alpha) under hypoxic conditions (Chou et al. 2011). HIF genes are transcriptionally responsive to hypoxia in F. heteroclitus (Flight et al. 2011; Townley et al. 2017). If variation in HAS1 influences the production of hyaluronan, this could lead to a direct effect on hypoxia tolerance.
Genetic Basis of Divergence in MR
We identify variation in five genes that contributes to phenotypic variation in MR and that fall in selection scan outlier regions (table 1). However, none of these putatively adaptive variants overlap between GWAS and RF. Additionally, the adaptive significance of MR divergence between osmotic environments is unclear. For example, this phenotype does not seem to correspond to population variation in swimming performance in FW and BW conditions (Brennan et al. 2016). However, there is evidence that at least four of these five identified genes may be mechanistically linked to MR, including for ARRDC3 (Patwari et al. 2011; Carroll et al. 2017), COL6A6 (Khan et al. 2009), and DFNA5 (de Beeck et al. 2011). Though there has been little work linking candidate YRDC to metabolism, since it controls mitochondrial tRNA synthesis (Harris et al. 2011) it is plausible that it may influence energy production. Healy et al. (2017) showed that YRDC expression is increased in cold temperatures in coastal populations of F. heteroclitus. Increased metabolic activity at low temperatures is necessary to ensure sufficient energy available for cellular functions (Guderley 2004) and the up-regulation of YRDC at low temperature may indicate a link with MR. These findings suggest that further study of the adaptive relevance of alternate MR physiologies between marine and FW environments is merited.
Genetic Basis of Divergence in Morphology
In stickleback and alewife, body shape diverges between marine and FW populations (Colosimo et al. 2004; Jones et al. 2013) and Baltic Sea herring morphology shifts in parallel with salinity adaptation (Jørgensen et al. 2008). Similarly, we identify subtle shifts in body shape between osmotic environments, though we observe no enrichment for overlap between the selection scan and phenotypic associations identified through GWAS. In contrast, we find that phenotype associations identified through RF (PC2) appear to be evolving by natural selection across osmotic environments. Of the six variants that overlap between RF and selection scans, four are associated with genes that have been directly implicated in aspects of bone structure and function including chondrocyte and osteoclast differentiation (SNTN; IL34, Saito et al. 2007; Hwang et al. 2012), and bone formation (CALCA; SEMA4B; Dacquin et al. 2004; Alto and Terman 2017). While these populations may be diverging in body shape, the functional significance of this divergence is currently unknown.
Conclusion
Selection scans are a powerful method to identify putatively adaptive loci between populations diverging across different environments. However, determining the phenotypic effects of these loci is dependent on inferring function from gene annotations. While this approach is a useful first step, it is limited by the fact that many genes have species-specific, ambiguous, or unknown functions thereby reducing our ability to link these genes to phenotypes. Making these links becomes even more difficult as multiple, polygenic, phenotypes are involved in the process of adaptation, a characteristic that is likely representative of adaptation to many environments (Rockman 2012). To overcome these issues, it is necessary to understand the identity and nature of specific phenotypes that are important in adaptation. We addressed this by integrating association mapping using GWAS and RF with selection scans. This approach was useful for two reasons. First, we were able to identify genetic variation that is both under selection and that explains phenotypic variation. Second, we can begin to disentangle traits that are diverging though adaptive processes in contrast to neutral drift. If phenotypic variation is underpinned with genetic variation that is not under selection, it is likely that neutral rather than adaptive processes are the main drivers underlying population differentiation of phenotypes.
We have demonstrated the strengths of this integrative approach by identifying the genetic variation that is important for adaptation to a FW environment. Indeed, nearly all of our identified genes are promising candidates. In particular, the identification of tricellulin as a gene that contributes to adaptive divergence in osmoregulatory physiology is consistent with a priori hypotheses generated by our previous research findings. Focusing on multiple traits allowed us to gain a more integrative understanding of how fish evolve upon radiating into fresh water. Together, these data identify the genetic basis of adaptation to a FW environment and, more generally, demonstrate a promising approach with which to identify the mechanisms important in adaptive divergence between niches.
Materials and Methods
Fish Collection and Lab Acclimation
Adult killifish were collected from the FW-native (n = 40), BW-native (n = 40), and admixed (n = 257) populations in June 2014 (fig. 1). FW-native individuals were sampled from Piscataway Park, near Accokeek, MD (38°41′42.18′′N, 77°3′10.38′′W) and BW-native fish were from Point Lookout State Park, near Scotland, MD (38°3′10.90′′N, 76°19′34.38′′W). The admixed population was collected at Allen’s Fresh Run, MD (38°21′54.54′′N, 76°58′52.02′′W). Water parameters have been measured by the Chesapeake Bay Program since 1984 and are freely available at http://data.chesapeakebay.net/ (last accessed August 14, 2017). The sampling stations closest to our collection sites were used to calculate dissolved oxygen and salinity for each population. The corresponding sampling locations names for BW-native, admixed, and FW-native populations are LE2.3, RET2.4, and TF2.1, respectively. Mean dissolved oxygen levels over the past 30+ years for each site are as follows (in mg/l O2 ± standard deviation): BW-native 7.14 ± 3.75, Admixed 7.19 ± 2.97, FW-native 8.69 ± 2.53. Salinity was calculated from the same locations (in parts per thousand[ppt] ± standard deviation): BW-native 14.86 ± 3.15, Admixed 7.80 ± 3.42, FW-native 0.00 ± 0.01. Additionally, the maximum salinity at the FW-native site over the past 30+ years of monitoring is 0.12 ppt demonstrating that this site is exclusively fresh water.
Fish were shipped to the University of British Columbia, Canada and held in a recirculating aquarium system in 200 L tanks at 15 °C and 12L:12D. Animal care and experimental procedures were in accordance with approved University of British Columbia animal care protocol A11-0372. Acclimation salinity was 20 ppt, made using dechlorinated City of Vancouver tap water adjusted with Instant Ocean Sea Salt (Instant Ocean, Spectrum Brands, Blacksburg, VA). Each individual was marked with a unique fluorescent elastomer tag (Northwest Marine Technology, Shaw Island, WA) and held separated by population. Nutrafin Max Tropical Fish Flakes (Hagen, Mansfield, MA) were used for feeding, though a 24 h fast was implemented prior to any experimental measurements. Following each phenotypic measure, acclimation conditions were re-established for at least one month before the next measure.
Phenotype Data
Three physiological traits were characterized for each fish, including salinity tolerance, hypoxia tolerance, and MR. Routine oxygen consumption was used to as a proxy for MR and measured using closed respirometry (Fangue et al. 2009; Healy and Schulte 2012). Repeatability of individual MR was within 10%, as assessed from preliminary tests. For the FW-native, BW-native, and admixed population, data were collected for 36, 40, and 218 individuals. Hypoxia tolerance was calculated as the amount of time required for an individual to lose equilibrium in 0.2 kPa O2 water (McBryan et al. 2016) where sample size was 40, 40, and 248 for FW-native, BW-native, and admixed populations. Salinity tolerance was measured using plasma chloride homeostasis following an acute fresh water exposure. Fish were transferred from 20 ppt to 0 ppt and allowed to acclimate for 24 h. At 24 h, individuals were sacrificed by pithing and blood was collected by caudal puncture using heparinized hematocrit capillary tubes. Blood was immediately spun at 1,500 × g for 1 min to isolate plasma. Blood plasma was snap frozen in liquid nitrogen for later Cl− quantification. Cl− was used to assess the salinity tolerance as it is the ion that most consistently reveals divergence in regulation between F. heteroclitus populations from different osmotic environments; we sampled fish at 24 h posttransfer because it is at this time that population differences are greatest (Scott, Rogers, et al. 2004; Whitehead et al. 2012). Cl− concentrations were quantified using a colorimetric mercuric thiocyanate method (Zall et al. 1956; Gibbons et al. 2017). Finally, we characterized morphology for 12 landmarks (see supplementary fig. S8, Supplementary Material online) of 24 FW-native, 29 BW-native, and 152 admixed individuals using TpsDig (Rohlf 2006). For individual fish, data were collected for all three physiological parameters from 38, 34, and 210 FW-native, BW-native, and admixed individuals, respectively.
Phenotype Statistics
Physiological data were analyzed in an ANOVA framework in R with population as a main effect. Mass had an effect on all phenotypes and was included as a covariate in the model. However, residuals from these models failed the assumption of normality, therefore we reran all statistics using the nonparametric Kruskal–Wallis rank sum test. Both models showed the same significant results, therefore we present the results from only the parametric analyses. For clarity of figures, we present mass-corrected values from linear models where salinity and hypoxia phenotypes are scaled to a fish size of 4 g. Correlations between phenotypes were assessed in admixed individuals only. Regressions between hypoxia, MR, and salinity tolerance were performed in all possible combinations. Because mass was slightly different for each phenotype (because of growth during the time between physiological measures) we mass corrected phenotypes to a standardized 4 g fish before investigating correlations.
Morphological data were analyzed using MorphoJ (Klingenberg 2011). Landmark positions were imported to MorphoJ and a Procrustes fit was performed. The effects of allometry were controlled for by a regression of Procrustes coordinates and centroid size and residuals were obtained for further analyses. Shape variation was summarized with a PCA on residuals and differences between groups were calculated using canonical variates analysis (CVA). Positions from PC1 and PC2 were used for subsequent association mapping.
Genotyping
A custom DNA extraction method utilizing Agencourt Ampure XP beads (Beckman Coulter) was used to obtain genomic DNA for each individual. Details of the method are provided in Ali et al. (2016). Briefly, fin clips were digested overnight at 56 °C in 4.2 mg/ml Proteinase K followed by an Ampure XP bead cleanup and elution in low TE (10 mM Tris–HCl, pH 7.5, 0.1 mM EDTA). Genotyping was performed using restriction site-associated DNA sequencing (RADseq) following the protocol developed by Ali et al. (2016). For each sample, 250 ng DNA was digested with SbfI. Individual barcodes were annealed to each sample by adding 2 μl indexed SbfI biotinylated RAD adapters (50 nM). These adapters feature 96 unique 8-bp barcodes that distinguish individuals within a library. Libraries of 96 individuals were then pooled and fragmented to 200–500 bp using a Bioruptor NGS sonicator with nine cycles 30 s on/90 s off. We assessed fragment size using a 2% sodium borate gel and further fragmentation was performed as needed. Dynabeads M-280 streptavidin magnetic beads (Life Technologies, 11205D) were used to physically isolate the RAD-tagged DNA fragments from off-target DNA and the final DNA was eluted in 55.5 μl low TE. These “RAD libraries” were then prepared for Illumina sequencing using a NEBNext Ultra DNA Library Prep Kit for Illumina. During this process, each RAD library of 96 individuals receives a unique barcode, which allows multiplexing of RAD libraries during sequencing.
Libraries were sequenced using 150-bp paired end reads on an Illumina HiSeq 4000 at the University of California Davis Genome Center. A single lane was first sequenced to aid with normalization prior to definitive library sequencing. From this preliminary single lane of data, libraries were demultiplexed by Illumina barcode then by RAD barcode. RAD barcode demultiplexing was performed using custom Perl scripts that require a perfect match of the barcode and partial restriction site. Reads that contained barcodes on both the forward and reverse reads were discarded. The number of reads per individual was counted and used to renormalize libraries and resequence. We went back to the original fragmented and barcoded DNA from the RAD preparation (prior to physical isolation of the RAD libraries) and adjusted the amount of DNA taken from each individual. This step ensures equal depth of sequencing across all individuals. This DNA was then repooled as above and sequenced on an additional two lanes of an Illumina HiSeq 4000. These data were demultiplexed as described above.
Reads were aligned to the F. heteroclitus reference genome 3.0.2 (Reid et al. 2017) using BWA-MEM version 0.7.12 (Li and Durbin 2009) and PCR duplicates were marked using SAMBLASTER version 0.1.22 (Faust and Hall 2014). Variants were called using Freebayes (v0.9.21-19-gc003c1) where reads with a mapping quality <30 and discordantly and duplicate reads were discarded (Garrison and Marth 2012). We used GATK (McKenna et al. 2010) to filter SNPs based on coverage, where each site required 80% of the samples to have at least 8× coverage. VCFtools version 0.1.13 (Danecek et al. 2011) was used to filter variants to retain sites with bi-allelic single nucleotide polymorphisms (SNPs), base quality >20, minor allele frequency (MAF) >0.01, and average coverage per individual <100×. Nine individuals had <50% of the average read depth and were removed. Variants were required to be in Hardy–Weinberg equilibrium in at least 2 of the 3 populations. This resulted in 139,721 high quality SNPs that were used in subsequent analyses. Variants were converted to their appropriate position on the F. heteroclitus genetic map (unpublished data). All variants were retained regardless of their inclusion in the genetic map and 126,651 (90.6%) were located on assembled chromosomes.
Population Genetics Data Analysis
To understand the genetic relationships between populations, we took two approaches: PCA and admixture analysis. Variants were thinned to remove linkage disequilibrium (LD) based on the variance inflation factor in PLINK v1.90 (Purcell et al. 2007) with the options –indep 50 5 2. PCA was run using PLINK v1.90. Admixture analysis was run with two ancestral populations in Admixture v1.23 (Alexander et al. 2009), which calculates the maximum likelihood of the ancestry of each individual.
To identify loci putatively under selection, we used a combined FST and π outlier approach, which are common approaches to identify regions under selection (Hoban et al. 2016) and indicate elevated allelic differentiation and reduced genetic diversity, respectively. Weir and Cockerham’s FST was calculated per site using VCFtools v0.1.13. Values for π were calculated using Stacks v1.46 and are similar to expected heterozygosity (Hohenlohe et al. 2010; Catchen et al. 2013). We sought to identify genomic regions with signatures of selection specifically in the derived FW-native population. Therefore, we calculated π within each population then took the difference between the FW-native and the BW-native population. In this case, more negative values (i.e., lower π in the FW-native population) are evidence of a selective sweep after entering fresh water. FST and π were combined using a multivariate outlier approach as implemented in the R package MINOTAUR (Verity et al. 2017). Raw statistics were converted to rank based P-values based on a uniform distribution, reflecting quantile values from the empirical distribution (Lotterhos et al. 2017). To identify selection in fresh water, we based these P-value conversions on right-tailed and left-tailed expectations for FST and π, respectively. This identifies variants that have high FST between the populations and lower π in the FW-native individuals. These P-values were then negative log10 transformed and the Mahalanobis distances for all SNPs were calculated. Mahalanobis distance represents the number of standard deviations a point lies from the center of a multivariate distribution. Importantly, this measure does not assume independence between statistics, though it does assume smooth dispersal from a centroid, making it necessary to perform the aforementioned P-value transformations. This approach follows the Md-rank-P method described in Lotterhos et al. (2017). From this distribution, we considered the top 1% of SNPs to be outliers, and therefore the most likely candidates as targets of natural selection.
Genotype–Phenotype Association for Physiological Traits
We utilized both a GWAS and a RF approach for association mapping as each is likely to identify different types of genetic variation. For example, RF can identify loci with complex genetic interactions (i.e., epistasis) whereas GWAS is suited for single loci of larger effect. For GWAS, association mapping was conducted for each phenotype using genome-wide efficient mixed-model association (GEMMA; Zhou and Stephens 2012). We implemented univariate linear mixed models (LMM) while incorporating a n × n relatedness matrix as a random effect. GEMMA was used to generate the centered relatedness matrix using all markers and all individuals. Because different numbers of individuals were phenotyped for each trait (see “Phenotype Data”, above), a small number of SNPs had MAFs that fell below our 0.01 cutoff and were removed from the analysis. As such, for hypoxia tolerance, MR, and salinity tolerance 139,451, 139,242, and 138,951 SNPs, respectively, were included in the LMM. Admixture proportions and log transformed individual mass were included as covariates for single SNP associations and Wald test P-values for log transformed phenotypes were obtained. QQ plots and genomic inflation factors suggest that the covariates are sufficiently controlling spurious associations (see supplementary fig. S9, Supplementary Material online). Admixture proportions were from the admixture analysis described above. Genome-wide significance was calculated based on the number of SNPs that are not in LD (76,655) and Bonferroni correction with α = 0.05.
As the phenotypes of interest are likely polygenic and complex, we also used a RF approach in the randomForest package (Liaw and Wiener 2002) to identify potential SNPs associated with phenotypic variation. Missing data are not tolerated in this model so variants were filtered accordingly (111,609 remaining). All phenotypes were log transformed and corrected for the effects of mass and population structure (admixture proportions; Zhao et al. 2012). Genotypes were similarly corrected for population structure. RF were run in an iterative process following (Holliday et al. 2012; Brieuc et al. 2015). First, we ran sets of 10,000 trees using all markers and removed any markers that did not improve the fit of the model (importance score ≤ 0), and repeated this process until <2,000 markers remained. We then ran three RF of 10,000 trees on the remaining variants and removed those where average importance ≤0. This was repeated until the removal of markers did not improve model fit. Finally, we ran further iterations as the previous step, but removed variants with the lowest 10% importance. This was repeated until three consecutive iterations did not improve model fit.
Identification of Candidate Loci
We sought to identify loci that were diverging by natural selection between marine and FW environments and also associated with physiological phenotypes that differed between BW-native and FW-native populations. However, the phenotypes we focused on are complex and likely polygenic, making it difficult to identify genetic markers that are statistically significantly associated with phenotype—a well-known challenge of GWAS (Rockman 2012). Therefore, we chose a liberal approach to identify candidates by identifying the SNPs falling in the lowest 0.1% of the distribution as GWAS outliers, the top 139 loci. The goal in this approach is to provide a subset of loci that are most likely involved in the phenotypes investigated. This 0.1% cutoff corresponds to the following P-values: salinity tolerance: 0.001, hypoxia tolerance: 0.0009, MR: 0.0007. For the RF analysis, we were interested in identifying variants that may be both common and unique from the GWAS. Therefore, we again adopted a liberal cutoff and considered the top 139 loci as candidates for each phenotype.
We determined the distance at which LD decayed to background levels by comparing interchromosome values (R2 = 0.0039) to intrachromosome values. Decay in LD with physical distance was calculated using nonlinear regression (Yang et al. 2014; Miller et al. 2015) implemented in the nls package in R (Baty et al. 2015). Using this approach, R2 values decayed to background within 27 kb. It should be noted that this is likely an upper bound. Using a more conservative measure, the point at which R2 drops below 0.1, LD decays after 5 kb (Chaves et al. 2016). Outlier loci (FST and π signals integrated by Mahalanobis distance) were merged into outlier windows when any two SNPs within 27 kb were top outliers; this value was chosen based on the calculated decay in LD. The overlap between windows showing signatures of selection and GWAS or RF outliers was then identified using Bedtools v2.17.0 (Quinlan and Hall 2010). We calculated the probability of obtaining the observed overlap by chance using a permutation test. MAF may influence the probability that an allele is an outlier in the selection scan and may also influence which phenotype associated variants overlap with outlier regions. Allele frequencies between the GWAS and RF variants and all variants in the selection scan were similar (see supplementary fig. S10, Supplementary Material online). Nevertheless, to control for potential bias, MAFs of variants were split into bins of size 0.025 and the density of the GWAS and RF variants in each bin was calculated (i.e., what proportion of SNPs had a MAF between 0 and 0.025, 0.026 and 0.050, etc.). These densities were used to weight random sampling of 139 selection scan variants (the number of loci identified as outliers) to simulate the MAF distribution in the phenotype associated variants. We then calculated how many of these selected variants overlapped with outlier regions. This was repeated 2,500 times and our observed values were compared with this null distribution and P-values were calculated (fig. 4B). We used Bedtools to identify the closest gene to each outlier SNP using the gene models from Reid et al. (2017).
Variants identified in both the GWAS and RF exhibit the strongest support for trait association. We therefore assessed the overlap between the RF and GWAS variants. From these overlapping variants, we again looked for intersection with selected regions and ran permutations as above, but the random sampling of variants was based on the number of overlapping markers for each phenotype (see Results). However, we still considered trait associations that were not overlapping between GWAS and RF as potential candidates as each makes different assumptions of the genetic architecture underlying phenotypic variation. We therefore tested for overlap between those trait associated markers and selection scan regions.
Supplementary Material
Acknowledgments
We thank Michelle Tse and Ruth Hwang for assistance with DNA extractions. Michael Miller and Sean O’Rourke provided training and assistance for RAD library preparation and analysis. David Metzger, Dillon Chung, and Kyle Crowther provided assistance with fish collection. This work was supported by funding from the National Science Foundation (DEB-1265282 to A.W., and DEB-1601076 to R.S.B. and A.W.), a Henry A. Jastro Research Fellowship (RSB), a Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery grant to P.M.S. and a NSERC Canada Graduate Scholarship to T.M.H. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethics Statement
Animal care and experimental procedures were in accordance with approved University of British Columbia animal care protocol A11-0372.
Data Availability
Sequence data are archived at the National Center for Biotechnology Information (NCBI SRA SRP150513; BioProject PRJNA428529). Phenotype and variant data are available at the European Bioinformatics Institute (EMBL-EBI) BioStudies accession S-BSST168.
Author Contributions
Conceived and designed the experiments: R.S.B., T.M.H., P.M.S., A.W. Performed the experiments: R.S.B., T.M.H., H.J.B., V.L. Analyzed the data: R.S.B. and T.M.H. Wrote the manuscript: R.S.B., T.M.H., P.M.S., A.W.
Contributor Information
Reid S Brennan, Department of Biology, University of Vermont, Burlington, VT; Department of Environmental Toxicology, University of California-Davis, Davis, CA.
Timothy M Healy, Marine Biology Research Division, Scripps Institution of Oceanography, University of California, San Diego, CA; Department of Zoology, The University of British Columbia, Vancouver, BC, Canada.
Heather J Bryant, Department of Zoology, The University of British Columbia, Vancouver, BC, Canada.
Van La, Department of Environmental Toxicology, University of California-Davis, Davis, CA.
Patricia M Schulte, Department of Zoology, The University of British Columbia, Vancouver, BC, Canada.
Andrew Whitehead, Department of Environmental Toxicology, University of California-Davis, Davis, CA.
References
- Alexander DH, Novembre J, Lange K. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19(9):1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali OA, O’Rourke SM, Amish SJ, Meek MH, Luikart G, Jeffres C, Miller MR. 2016. RAD capture (Rapture): flexible and efficient sequence-based genotyping. Genetics 202(2):389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alto LT, Terman JR. 2017. Semaphorins and their signaling mechanisms. In: Walker John M. editor, Semaphorin signaling. Vol. 1493. 3rd ed. Methods in molecular biology. New York: Springer. p. 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin B, Austin DA, Austin B, Austin DA. 2012. Bacterial fish pathogens. Dordrecht: Springer. [Google Scholar]
- Bateson W. 1909. Heredity and variation in modern lights. In: Seward AC, editor. Darwin and Modern Science, pp. 85–101. Cambridge: Cambridge University Press. [Google Scholar]
- Baty F, Ritz C, Charles S, Brutsche M, Flandrois J-P, Delignette-Muller M-L. 2015. A toolbox for nonlinear regression in R: the package nlstools. J Stat Softw. 66(5): 1–21. [Google Scholar]
- Berg JJ, Coop G. 2014. A population genetic signal of polygenic adaptation. PLoS Genet. 10(8): e1004412–e1004425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur-R R, Ortí G, Pyron RA. 2015. Fossil‐based comparative analyses reveal ancient marine ancestry erased by extinction in ray‐finned fishes. Ecol Lett. 18(5): 441–450. [DOI] [PubMed] [Google Scholar]
- Brelsford A, Toews DPL, Irwin DE. 2017. Admixture mapping in a hybrid zone reveals loci associated with avian feather coloration. Proc R Soc B. 284(1866): 20171106–20171109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan RS, Galvez F, Whitehead A. 2015. Reciprocal osmotic challenges reveal mechanisms of divergence in phenotypic plasticity in the killifish Fundulus heteroclitus. J Exp Biol. 218(Pt 8):1212–1222. [DOI] [PubMed] [Google Scholar]
- Brennan RS, Hwang R, Tse M, Fangue NA, Whitehead A. 2016. Local adaptation to osmotic environment in killifish, Fundulus heteroclitus, is supported by divergence in swimming performance but not by differences in excess post-exercise oxygen consumption or aerobic scope. Comp Biochem Physiol A Mol Integr Physiol. 196:11–19. [DOI] [PubMed] [Google Scholar]
- Breves JP, McCormick SD, Karlstrom RO. 2014. Prolactin and teleost ionocytes: new insights into cellular and molecular targets of prolactin in vertebrate epithelia. Gen Comp Endocrinol. 203:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brieuc MSO, Ono K, Drinan DP, Naish KA. 2015. Integration of Random Forest with population-based outlier analyses provides insight on the genomic basis and evolution of run timing in Chinook salmon (Oncorhynchus tshawytscha). Mol Ecol. 24(11):2729–2746. [DOI] [PubMed] [Google Scholar]
- Brieuc MSO, Waters CD, Drinan DP, Naish KA. 2018. A practical introduction to Random Forest for genetic association studies in ecology and evolution. Mol Ecol Resour. 18(4): 755–712. [DOI] [PubMed] [Google Scholar]
- Buerkle CA, Lexer C. 2008. Admixture as the basis for genetic mapping. Trends Ecol Evol. 23(12):686–694. [DOI] [PubMed] [Google Scholar]
- Burke JM, Arnold ML. 2001. Genetics and the fitness of hybrids. Annu Rev Genet. 35:31–52. [DOI] [PubMed] [Google Scholar]
- Carroll SH, Zhang E, Wang BF, LeClair KB, Rahman A, Cohen DE, Plutzky J, Patwari P, Lee RT. 2017. Adipocyte arrestin domain-containing 3 protein (Arrdc3) regulates uncoupling protein 1 (Ucp1) expression in white adipose independently of canonical changes in β-adrenergic receptor signaling. PLoS ONE. 12(3):e0173823–e0173814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. 2013. Stacks: an analysis tool set for population genomics. Mol Ecol. 22(11):3124–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaves JA, Cooper EA, Hendry AP, Podos J, De León LF, Raeymaekers JAM, MacMillan WO, Uy JAC. 2016. Genomic variation at the tips of the adaptive radiation of Darwin's finches. Mol Ecol. 25(21):5282–5295. [DOI] [PubMed] [Google Scholar]
- Chou L-W, Wang J, Chang P-L, Hsieh Y-L. 2011. Hyaluronan modulates accumulation of hypoxia- inducible factor-1 alpha, inducible nitric oxide synthase, and matrix metalloproteinase-3 in the synovium of rat adjuvant-induced arthritis model. Arthritis Res Ther. 13(3):R90.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G, Dickson M, Grimwood J, Schmutz J, Myers RM, Schluter D, Kingsley DM. 2005. Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science 307(5717):1928–1933. [DOI] [PubMed] [Google Scholar]
- Colosimo PF, Peichel CL, Nereng K, Blackman BK, Shapiro MD, Schluter D, Kingsley DM. 2004. The genetic architecture of parallel armor plate reduction in threespine sticklebacks. PLoS Biol. 2(5):e109–e117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congleton JL. 1980. Observations on the responses of some southern California tidepool fishes to nocturnal hypoxic stress. Comp Biochem Physiol A Mol Integr Physiol. 66(4):719–722. [Google Scholar]
- Dacquin R, Davey RA, Laplace C, Levasseur R, Morris HA, Goldring SR, Gebre-Medhin S, Galson DL, Zajac JD, Karsenty G. 2004. Amylin inhibits bone resorption while the calcitonin receptor controls bone formation in vivo. J Cell Biol. 164(4):509–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27(15):2156–2158., [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasmahapatra KK, Walters JR, Briscoe AD, Davey JW, Whibley A, Nadeau NJ, Zimin AV, Hughes DST, Ferguson LC, Martin SH, et al. 2012. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487(7405):94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beeck KO, Van Camp G, Thys S, Cools N, Callebaut I, Vrijens K, Van Nassauw L, Van Tendeloo VF, Timmermans JP, Van Laer L. 2011. The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis-inducing protein. Eur J Hum Genet. 19(9):965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFaveri J, Merila J. 2014. Local adaptation to salinity in the three-spined stickleback?. J Evol Biol. 27(2): 290–302. [DOI] [PubMed] [Google Scholar]
- Dobzhansky T. 1936. Studies on hybrid sterility. II. Localization of sterility factors in Drosophila pseudoobscura hybrids. Genetics 21(2):113–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvernell DD, Lindmeier JB, Faust KE, Whitehead A. 2008. Relative influences of historical and contemporary forces shaping the distribution of genetic variation in the Atlantic killifish, Fundulus heteroclitus. Mol Ecol. 17(5):1344–1360. [DOI] [PubMed] [Google Scholar]
- Evans DH, Piermarini PM, Choe KP. 2005. The multifunctional fish gill: dominant site of gas exchange, osmoregulation, acid-base regulation, and excretion of nitrogenous waste. Physiol Rev. 85(1):97–177. [DOI] [PubMed] [Google Scholar]
- Fangue NA, Richards JG, Schulte PM. 2009. Do mitochondrial properties explain intraspecific variation in thermal tolerance?. J Exp Biol. 212(Pt 4):514–522. [DOI] [PubMed] [Google Scholar]
- Faust GG, Hall IM. 2014. SAMBLASTER: fast duplicate marking and structural variant read extraction. Bioinformatics 30(17):2503–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flight PA, Nacci D, Champlin D, Whitehead A, Rand DM. 2011. The effects of mitochondrial genotype on hypoxic survival and gene expression in a hybrid population of the killifish, Fundulus heteroclitus. Mol Ecol. 20(21): 4503–4520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier-Level A, Korte A, Cooper MD, Nordborg M, Schmitt J, Wilczek AM. 2011. A map of local adaptation in Arabidopsis thaliana. Science 334(6052): 86–89. [DOI] [PubMed] [Google Scholar]
- Garrison E, Marth G. 2012. Haplotype-based variant detection from short-read sequencing. arXiv Preprint arXiv 1207:3907. [Google Scholar]
- Ghedotti MJ, Davis MP. 2013. Phylogeny, classification, and evolution of salinity tolerance of the North American Topminnows and Killifishes, family Fundulidae (Teleostei: cyprinodontiformes). Fieldiana Life Earth Sci. 7:1–65. [Google Scholar]
- Gibbons TC, Metzger DCH, Healy TM, Schulte PM. 2017. Gene expression plasticity in response to salinity acclimation in threespine stickleback ecotypes from different salinity habitats. Mol Ecol. 26(10): 2711–2725. [DOI] [PubMed] [Google Scholar]
- Guderley H. 2004. Metabolic responses to low temperature in fish muscle. Biol Rev. 79(2): 409–427. [DOI] [PubMed] [Google Scholar]
- Hansen TF. 2003. Is modularity necessary for evolvability?: remarks on the relationship between pleiotropy and evolvability. Biosystems 69(2–3): 83–94. [DOI] [PubMed] [Google Scholar]
- Harris KA, Jones V, Bilbille Y, Swairjo MA, Agris PF. 2011. YrdC exhibits properties expected of a subunit for a tRNA threonylcarbamoyl transferase. RNA 17(9): 1678–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy TM, Bryant HJ, Schulte PM. 2017. Mitochondrial genotype and phenotypic plasticity of gene expression in response to cold acclimation in killifish. Mol Ecol. 26(3): 814–830. [DOI] [PubMed] [Google Scholar]
- Healy TM, Schulte PM. 2012. Thermal acclimation is not necessary to maintain a wide thermal breadth of aerobic scope in the common killifish (Fundulus heteroclitus). Physiol Biochem Zool. 85(2): 107–119. [DOI] [PubMed] [Google Scholar]
- Hildebrand SF, Schroeder WC. 1928. Fishes of the Chesapeake Bay. Bull. US Bur. Fish. [Google Scholar]
- Hoban S, Kelley JL, Lotterhos KE, Antolin MF, Bradburd G, Lowry DB, Poss ML, Reed LK, Storfer A, Whitlock MC. 2016. Finding the genomic basis of local adaptation: pitfalls, practical solutions, and future directions. Am Nat. 188(4): 379–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann EK, Dunham PB. 1995. Membrane mechanisms and intracellular signalling in cell volume regulation. Int. Rev. Cytol. 161:173–262. [DOI] [PubMed] [Google Scholar]
- Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA. 2010. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genet. 6(2): e1000862.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holliday JA, Wang T, Aitken S. 2012. Predicting adaptive phenotypes from multilocus genotypes in Sitka spruce (Picea sitchensis) using random forest. G3: Genes|Genomes|Genetics. 2:1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes LC, Somoza GM, Nguyen BN, Bernot JP, González Castro M, Díaz de Astarloa JM, Ortí G. 2017. Transcriptomic differentiation underlying marine‐to‐freshwater transitions in the South American silversides Odontesthes argentinensis and O. bonariensis (Atheriniformes). Ecol Evol. 7(14):5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang P-P, Lee T-H, Lin L-Y. 2011. Ion regulation in fish gills: recent progress in the cellular and molecular mechanisms. Am J Physiol Regul Integr Comp Physiol. 301(1): R28–R47. [DOI] [PubMed] [Google Scholar]
- Hwang S-J, Choi B, Kang S-S, Chang J-H, Kim Y-G, Chung Y-H, Sohn DH, So MW, Lee C-K, Robinson WH, et al. 2012. Interleukin-34 produced by human fibroblast-like synovial cells in rheumatoid arthritis supports osteoclastogenesis. Arthritis Res Ther. 14(1): R14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innes AJ, Wells RMG. 1985. Respiration and oxygen transport functions of the blood from an intertidal fish, Helcogramma medium (Tripterygiidae). Environ Biol Fish. 14(2–3): 213–226. [Google Scholar]
- Jones AW, Palkovacs EP, Post DM. 2013. Recent parallel divergence in body shape and diet source of alewife life history forms. Evol Ecol. 27(6): 1175–1187. [Google Scholar]
- Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, Swofford R, Pirun M, Zody MC, White S, et al. 2012. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484(7392): 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen HB, Pertoldi C, Hansen MM, Ruzzante DE, Loeschcke V. 2008. Genetic and environmental correlates of morphological variation in a marine fish: the case of Baltic Sea herring (Clupea harengus). Can J Fish Aquat Sci. 65(3): 389–400. [Google Scholar]
- Khan T, Muise ES, Iyengar P, Wang ZV, Chandalia M, Abate N, Zhang BB, Bonaldo P, Chua S, Scherer PE. 2009. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol. 29(6): 1575–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidder GW, Petersen CW, Preston RL. 2006. Energetics of osmoregulation: II. water flux and osmoregulatory work in the euryhaline fish, Fundulus heteroclitus. J Exp Zool. 305A(4): 318–327. [DOI] [PubMed] [Google Scholar]
- Klingenberg CP. 2011. MorphoJ: an integrated software package for geometric morphometrics. Mol Ecol Resour. 11(2):353–357. [DOI] [PubMed] [Google Scholar]
- Kolosov D, Chasiotis H, Kelly SP. 2014. Tight junction protein gene expression patterns and changes in transcript abundance during development of model fish gill epithelia. J Exp Biol. 217(Pt 10):1667–1681. [DOI] [PubMed] [Google Scholar]
- Kolosov D, Kelly SP. 2013. A role for tricellulin in the regulation of gill epithelium permeability. Am J Physiol Regul Integr Comp Physiol. 304(12): R1139–R1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14): 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A, Wiener M. 2002. Classification and regression by randomForest. R News. 2:18–22. [Google Scholar]
- Lotterhos KE, Card DC, Schaal SM, Wang L, Collins C, Verity B. 2017. Composite measures of selection can improve the signal‐to‐noise ratio in genome scans. Methods Ecol Evol. 8(6): 717–727. [Google Scholar]
- Lozupone CA, Knight R. 2007. Global patterns in bacterial diversity. Proc Natl Acad Sci USA. 104(27): 11436–11440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manceau M, Domingues VS, Mallarino R, Hoekstra HE. 2011. The developmental role of Agouti in color pattern evolution. Science 331(6020): 1062–1065. [DOI] [PubMed] [Google Scholar]
- Marshall WS, Lynch EM, Cozzi RRF. 2002. Redistribution of immunofluorescence of CFTR anion channel and NKCC cotransporter in chloride cells during adaptation of the killifish Fundulus heteroclitus to sea water. J Exp Biol. 205:1265–1273. [DOI] [PubMed] [Google Scholar]
- McBryan TL, Healy TM, Haakons KL, Schulte PM. 2016. Warm acclimation improves hypoxia tolerance in Fundulus heteroclitus. J Exp Biol. 219(Pt 4): 474–484. [DOI] [PubMed] [Google Scholar]
- McGirr JA, Martin CH. 2016. Novel candidate genes underlying extreme trophic specialization in Caribbean pupfishes. Mol Biol Evol. 34:873–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9): 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JM, Poissant J, Malenfant RM, Hogg JT, Coltman DW. 2015. Temporal dynamics of linkage disequilibrium in two populations of bighorn sheep. Ecol Evol. 5(16): 3401–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. 1942. Isolating mechanisms, evolution and temperature. In: Vol. 6. Biol. Symp. p. 71–125.
- Nosil P. 2012. Ecological speciation. New York: Oxford University Press. [Google Scholar]
- Orr HA. 1995. The population genetics of speciation: the evolution of hybrid incompatibilities. Genetics 139(4): 1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne RI, Bernot MJ, Findlay SEG. 2015. Changes in nitrogen cycling processes along a salinity gradient in tidal wetlands of the Hudson River, New York, USA. Wetlands 35(2): 323–334. [Google Scholar]
- Ou M, Hamilton TJ, Eom J, Lyall EM, Gallup J, Jiang A, Lee J, Close DA, Yun S-S, Brauner CJ. 2015. Responses of pink salmon to CO2-induced aquatic acidification. Nat Clim Change. 5(10): 950–955. [Google Scholar]
- Parks SK, Tresguerres M, Goss GG. 2007. Interactions between Na+ channels and Na+-HCO3− cotransporters in the freshwater fish gill MR cell: a model for transepithelial Na+ uptake. Am J Physiol Cell Physiol. 292(2): C935–C944. [DOI] [PubMed] [Google Scholar]
- Patwari P, Emilsson V, Schadt EE, Chutkow WA, Lee S, Marsili A, Zhang Y, Dobrin R, Cohen DE, Larsen PR, et al. 2011. The arrestin domain-containing 3 protein regulates body mass and energy expenditure. Cell Metab. 14(5): 671–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer SP, Laurent S, Sousa VC, Linnen CR, Foll M, Excoffier L, Hoekstra HE, Jensen JD. 2018. The evolutionary history of Nebraska deer mice: local adaptation in the face of strong gene flow. Mol Biol Evol. 35(4): 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81(3): 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6): 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid NM, Jackson CE, Gilbert D, Minx P, Montague MJ, Hampton TH, Helfrich LW, King BL, Nacci DE, Aluru N, et al. 2017. The landscape of extreme genomic variation in the highly adaptable Atlantic killifish. Genome Biol Evol. 9(3): 659–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockman MV. 2012. The QTN program and the alleles that matter for evolution: all that's gold does not glitter. Evolution 66(1): 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohlf FJ. 2006. tpsDig2, version 2.1. http://life.bio.sunysb.edu/morph 8:e63248.
- Saito T, Ikeda T, Nakamura K, Chung U-I, Kawaguchi H. 2007. S100A1 and S100B, transcriptional targets of SOX trio, inhibit terminal differentiation of chondrocytes. EMBO Rep. 8(5): 504–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltz JB, Hessel FC, Kelly MW. 2017. Trait correlations in the genomics era. Trends Ecol Evol. 32(4): 279–290. [DOI] [PubMed] [Google Scholar]
- Schneeberger EE, Lynch RD. 2004. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol. 286(6):C1213–C1228. [DOI] [PubMed] [Google Scholar]
- Schultz ET, McCormick SD. 2012. Euryhaline Fishes. In: Farrell AP, McCormick SD, Brauner CJ, editors. Euryhalinity in an evolutionary context. Vol. 32. Boston: Academic Press. p. 477–533. [Google Scholar]
- Scott GR, Baker DW, Schulte PM, Wood CM. 2008. Physiological and molecular mechanisms of osmoregulatory plasticity in killifish after seawater transfer. J Exp Biol. 211(Pt 15): 2450–2459. [DOI] [PubMed] [Google Scholar]
- Scott GR, Richards JG, Forbush B, Isenring P, Schulte PM. 2004. Changes in gene expression in gills of the euryhaline killifish Fundulus heteroclitus after abrupt salinity transfer. Am J Physiol Cell Physiol. 287(2): C300–C309. [DOI] [PubMed] [Google Scholar]
- Scott GR, Rogers JT, Richards JG, Wood CM, Schulte PM. 2004. Intraspecific divergence of ionoregulatory physiology in the euryhaline teleost Fundulus heteroclitus: possible mechanisms of freshwater adaptation. J Exp Biol. 207(Pt 19): 3399–3410. [DOI] [PubMed] [Google Scholar]
- Siiskonen H, Oikari S, Pasonen-Seppänen S, Rilla K. 2015. Hyaluronan synthase 1: a mysterious enzyme with unexpected functions. Front Immunol. 6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzin A, Hoffmann EK. 2006. Swelling‐activated ion channels: functional regulation in cell‐swelling, proliferation and apoptosis. Acta Physiol. 187(1–2): 27–42. [DOI] [PubMed] [Google Scholar]
- Telles CJ, Decker SE, Motley WW, Peters AW, Mehr AP, Frizzell RA, Forrest JN. 2016. Functional and molecular identification of a TASK-1 potassium channel regulating chloride secretion through CFTR channels in the shark rectal gland: implications for cystic fibrosis. Am J Physiol Cell Physiol. 311(6): C884–C894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman CM, Chapman LJ. 2004. Patterns of hypoxia in a coastal salt marsh: implications for ecophysiology of resident fishes. Florida Sci 67(1): 80–91. [Google Scholar]
- Tine M, Kuhl H, Gagnaire P-A, Louro B, Desmarais E, Martins RST, Hecht J, Knaust F, Belkhir K, Klages S, et al. 2014. European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nat Commun. 5(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townley IK, Karchner SI, Skripnikova E, Wiese TE, Hahn ME, Rees BB. 2017. Sequence and functional characterization of hypoxia-inducible factors, HIF1α, HIF2αa, and HIF3α, from the estuarine fish, Fundulus heteroclitus. Am J Physiol Regul Integr Comp Physiol. 312(3): R412–R425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Belleghem SM, Rastas P, Papanicolaou A, Martin SH, Arias CF, Supple MA, Hanly JJ, Mallet J, Lewis JJ, Hines HM, et al. 2017. Complex modular architecture around a simple toolkit of wing pattern genes. Nat Ecol Evol. 1(3): 0052–0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega GC, Wiens JJ. 2012. Why are there so few fish in the sea?. Proc R Soc B. 279(1737): rspb20120075–rspb20122329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velotta JP, Wegrzyn JL, Ginzburg S, Kang L, Czesny S, O'Neill RJ, McCormick SD, Michalak P, Schultz ET. 2017. Transcriptomic imprints of adaptation to fresh water: parallel evolution of osmoregulatory gene expression in the Alewife. Mol Ecol. 26(3): 831–848. [DOI] [PubMed] [Google Scholar]
- Verity R, Collins C, Card DC, Schaal SM, Wang L, Lotterhos KE. 2017. minotaur: a platform for the analysis and visualization of multivariate results from genome scans with R Shiny. Mol Ecol Resour. 17(1): 33–43. [DOI] [PubMed] [Google Scholar]
- Via S, Conte G, Mason-Foley C, Mills K. 2012. Localizing FST outliers on a QTL map reveals evidence for large genomic regions of reduced gene exchange during speciation‐with‐gene‐flow. Mol Ecol. 21(22): 5546–5560. [DOI] [PubMed] [Google Scholar]
- Weigel D, Nordborg M. 2015. Population genomics for understanding adaptation in wild plant species. Annu Rev Genet. 49:315–338. [DOI] [PubMed] [Google Scholar]
- Whitehead A, Roach JL, Zhang S, Galvez F. 2011. Genomic mechanisms of evolved physiological plasticity in killifish distributed along an environmental salinity gradient. Proc Natl Acad Sci USA. 108(15): 6193–6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead A, Roach JL, Zhang S, Galvez F. 2012. Salinity- and population-dependent genome regulatory response during osmotic acclimation in the killifish (Fundulus heteroclitus) gill. J Exp Biol. 215(8): 1293–1305. [DOI] [PubMed] [Google Scholar]
- Whitehead A, Zhang S, Roach JL, Galvez F. 2013. Common functional targets of adaptive micro- and macro-evolutionary divergence in killifish. Mol Ecol. 22(14): 3780–3796. [DOI] [PubMed] [Google Scholar]
- Whitehead A. 2010. The evolutionary radiation of diverse osmotolerant physiologies in killifish (Fundulus SP.). Evolution 64(7): 2070–2085. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Laurent P. 2002. Fish gill morphology: inside out. J Exp Zool. 293(3): 192–213. [DOI] [PubMed] [Google Scholar]
- Wolf JB, Leamy LJ, Routman EJ, Cheverud JM. 2005. Epistatic pleiotropy and the genetic architecture of covariation within early and late-developing skull trait complexes in mice. Genetics 171(2): 683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Shikano T, Li M-H, Merilä J. 2014. Genome-wide linkage disequilibrium in nine-spined stickleback populations. G3: Genes|Genomes|Genetics. 4:1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zall DM, Fisher D, Garner MQ. 1956. Photometric determination of chlorides in water. Anal Chem. 28(11):1665–1668. [Google Scholar]
- Zhao Y, Chen F, Zhai R, Lin X, Wang Z, Su L, Christiani DC. 2012. Correction for population stratification in random forest analysis. Int J Epidemiol. 41(6): 1798–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Stephens M. 2012. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 44(7): 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer AM, Wright PA, Wood CM. 2017. Ammonia and urea handling by early life stages of fishes. J Exp Biol. 220(Pt 21):3843–3855. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data are archived at the National Center for Biotechnology Information (NCBI SRA SRP150513; BioProject PRJNA428529). Phenotype and variant data are available at the European Bioinformatics Institute (EMBL-EBI) BioStudies accession S-BSST168.