

Abstract
Background
The thymidine kinase (tk) mutagenesis assay is often utilized to determine the frequency of herpes simplex virus (HSV) replication-mediated mutations. Using this assay, clinical and laboratory HSV-2 isolates were shown to have a 10- to 80-fold higher frequency of spontaneous mutations compared to HSV-1.
Methods
A panel of HSV-1 and HSV-2, along with polymerase-recombinant viruses expressing type 2 polymerase (Pol) within a type 1 genome, were evaluated using the tk and non-HSV DNA mutagenesis assays to measure HSV replication-dependent errors and determine whether the higher mutation frequency of HSV-2 is a distinct property of type 2 polymerases.
Results
Although HSV-2 have mutation frequencies higher than HSV-1 in the tk assay, these errors are assay-specific. In fact, wild type HSV-1 and the antimutator HSV-1 PAAr5 exhibited a 2–4 fold higher frequency than HSV-2 in the non-HSV DNA mutatagenesis assay. Furthermore, regardless of assay, HSV-1 recombinants expressing HSV-2 Pol had error rates similar to HSV-1, whereas the high mutator virus, HSV-2 6757, consistently showed signficant errors. Additionally, plasmid DNA containing the HSV-2 tk gene, but not type 1 tk or LacZ DNA, was shown to form an anisomorphic DNA stucture.
Conclusions
This study suggests that the Pol is not solely responsible for the virus-type specific differences in mutation frequency. Accordingly, it is possible that (a) mutations may be modulated by other viral polypeptides cooperating with Pol, and (b) the localized secondary structure of the viral genome may partially account for the apparently enhanced error frequency of HSV-2.
Background
The replicative polymerase of herpes simplex virus (HSV) is comprised of catalytic (Pol) and processivity subunits (UL42) [9,11]. HSV Pol is responsible, in concert with other viral proteins, for regulating the fidelity of DNA replication by selecting the correct nucleotides for incorporation and by proofreading or editing mispaired nucleotides [20]. The Pol subunit contains intrinsic 3'-5' exonuclease activity [14-16] and the N-terminal region of the polypeptide encoding this function is highly conserved in other polymerases, containing three segments referred to as Exo motifs [3,26].
Thymidine kinase polypeptide (TK) is required for the activation of some antiviral nucleoside analogs e.g. acyclovir (ACV) and penciclovir (PCV), in order to inhibit viral DNA replication. HSV with TK mutations may impair activation of these drugs and confer a drug-resistant phenotype. The natural phenomenon of spontaneous mutation, which occurs in the absence of drug selection, results in the accumulation of approximately six to eight TK-deficient variants per 104 plaque-forming viruses in virus populations that have never been exposed to selective pressure [6,10,12]. Furthermore, HSV-2 clinical isolates were shown to have a higher frequency of spontaneous mutations resistant to ACV and PCV, approximately 30-fold, compared to HSV-1 isolates, and the majority of these mutations are in tk. Consistent with this observation, HSV-2 strains also exhibited approximately a 20- to 80-fold higher spontaneous mutation rate to cidofovir (HPMPC), an inhibitor of HSV Pol, resistance compared with HSV-1 [21]. Therefore, these naturally-occuring mutations are not unique to tk and TK substrates as detected with antiviral agents ACV and PCV, but were extended with the direct Pol inhibitor, HPMPC, to other loci, most likely pol.
Since mutations within the viral pol gene have been previously shown to affect spontaneous viral mutation rates [10], the higher frequency of errors associated with type 2 viruses may be an inherent property of the type 2 Pol. To assess the direct contribution of polymerases from virus types 1 and 2 to mutation frequency, recombinant HSV expressing a type 2 Pol within a type 1 viral genome were generated by marker transfer and examined along with a panel of other HSV isolates in two mutagenesis assays.
Materials and methods
Cell lines, compounds and viruses
Vero (ATCC, passage 15–20), MRC-5 (ATCC, passage 12–15) and PolB3 (kind gift of D. Coen, Harvard University, Boston MA) cell lines were grown in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal calf serum (FCS) and incubated at 37°C, 5%CO2. PolB3 represents a Vero cell line constitutively expressing the HSV-1 Pol [17]. PCV was synthesized at SmithKline Beecham Pharmaceuticals. ACV used in these studies was obtained from Sigma (St. Louis, MO). For cell culture assays, 10 mg/ml stock solutions were prepared in dimethyl sulphoxide (DMSO) and stored at -20°C. Working dilutions were prepared in assay medium immediately before use as described below. HSV-1 strain SC16 (wild type laboratory virus), HSV-2 SB5 (wild type laboratory virus), HSV-2 6652 (wild type clinical virus), HSV-2 83D (wild type clinical virus), HSV-2 6757 (high error mutant clinical virus) were described previously [21] and the HSV-1 PAAr5 (antimutator laboratory virus) utilized in this study was a kind gift of D. Coen (Harvard University, Boston, MA) [10]. HP66 (HSV-1 genome containing a LacZ insertion within the pol coding region) viral DNA was kindly provided by D. Coen (Harvard University, Boston, MA) [17]. HP66 viral stocks were prepared by transfecting HP66 viral DNA into PolB3 cells, which allow for virus propagation by expressing the viral polymerase.
In vitro susceptibility assays
The tk mutagenesis assay was performed in MRC5 and Vero cells as described previously [10,21]. The proportion of resistant virus referred to as mutation frequency or error frequency throughout this study was calculated as follows: (titer in the presence of drug) / (titer in the absence of drug) × 100.
Construction of recombinant viruses
Marker transfer experiments involved co-transfection of HSV-2 pol coding region (HSV-2 SB5, HSV-2 83D, HSV-2 6652 and HSV-2 6757 described in [21]) with HP66 viral DNA into Vero cells. Recombinant virus was plaque purified to homogeneity in the presence of X-Gal. The pol coding region from the recombinant viruses was confirmed to be identical to their respective parental virus pol sequences by terminator cycle sequencing using an automated model 377 DNA sequencer (Perkin Elmer, Applied Biosystems).
Virus growth analysis
To compare the ability of parental viruses and recombinant viruses to replicate in Vero cells with that of the wild type HSV-2 SB5 strain, 3 × 105 Vero cells were infected with each virus at a multiplicity of infection of 5 PFU per cell. Virus progeny were harvested at different time points and titered on Vero cells. All viruses yielded similar PFU/ml (data not shown).
Plasmid construction
A non-HSV gene template (pSV110) was constructed by taking the plasmid pSVβgal (Promega) and inserting the HSV-1 origin of replication (oriS) SmaI fragment [5]. The plasmid pTK1 contains HSV-1 SC16 sequence coordinates 45,055–48,634 subcloned into the HindIII / EcoRI site of pUC19 (Invitrogen). The plasmid pTK2 contains HSV-2 SB5 sequence coordinates 44,469–48,184 subcloned into the HindIII / EcoRI site of pUC19. The plasmid pLacZ contains the LacZ coding region from pSVβgal subcloned into the HindIII / EcoRI site of pUC19.
Transient replication/rescue assay
Vero cells were seeded at 3 × 105 cells per 2 mL well in a six well plate and incubated overnight at 37°C. The following day, cells were transfected with 1.5 μg of pSV110 and 1 μg of salmon sperm DNA using Lipofectamine reagent according to the manufacturer's recommendations (Invitrogen). Following overnight incubation at 37°C, the medium was removed and virus inoculated at an MOI = 5 in 2 mL volume for 1 h. Following overnight incubation at 37°C, the cells demonstrated CPE and the infected-cell monolayer was rinsed twice with 1X phosphate buffered saline. Next, cell monolayers were lysed by scraping into 1.0 mL of lysis buffer (0.6% SDS, 0.01 M EDTA pH 7.5) per well. The infected cell lysate was adjusted to a final concentration of 1 M NaCl, inverted 10X and stored at 4°C overnight. The samples were centrifuged at 14,000 rpm for 10 minutes at 4°C and the supernatant was collected, treated with 100 μg/ml RNAse at 37°C for one hour. Following that incubation, 100 μg/mL Proteinase K was added and samples were incubated at 50°C for 2 hours. After several phenol-chloroform extractions, the DNA was precipitated, centrifuged at 14,000 rpm for 10 minutes at 4°C and the pellets resuspended in 50 μl water and incubated at 37°C for 2 hours. To remove non-replicated templates, 2.5 μg of infected cell DNA was digested with DpnI and precipitated, centrifuged at 14,000 rpm for 10 minutes at 4°C and resuspended in 20 μl dH2O. One-fourth of the DpnI-treated DNA was transformed into DH5α cells (recombination-deficient bacterial cells; Invitrogen). The transformation mix was plated onto several LB-AMP plates containing Xgal and isopropyl β-D thiogalactoside (IPTG). The number of blue colonies (indicating no mutation in β galactosidase gene) and white colonies (indicating mutation within β galactosidase gene or promoter) were determined. An average of 300,000 colonies were recovered from three independent experiments for each virus infection. Two control samples were included in each experiment to normalize for mutations induced by transfection or superinfection processes. For the transfection control, pSV110 was transfected into Vero cells, mock-infected and directly transformed into bacteria for blue/white colony screening. For the superinfection control, pSV110 was transfected into Vero cells, superinfected with HP66 Pol-mutant virus (which is unable to replicate) and transformed into bacteria for blue/white colony screening. For the controls, both DpnI and non-DpnI treated samples were anlayzed. The values in Table 2 are presented as mutation frequency which is (number of white colonies from test sample/number of blue colonies from test sample) - (number of white colonies from HP66 superinfection sample/number of blue colonies from HP66 superinfection sample) × 100.
Table 2.
Mutation frequency in non-HSV DNA.
| Virusa | Mutation Frequencyb (%) |
| HSV-2 SB5 | 1.7, 2.0, 1.7c |
| HSV-2 SB5PR | 2.0, 1.8, 2.1 |
| HSV-2 6652 | 0.7, 0.9, 1.1 |
| HSV-2 6652PR | 1.3, 1.0, 1.5 |
| HSV-2 83D | 0.8, 1.1, 1.0 |
| HSV-2 83D PR | 1.6, 1.2, 1.8 |
| HSV-2 6757 | 4.1, 3.4, 4.9 |
| HSV-2 6757PR | 1.2, 1.1, 1.1 |
| HSV-1 SC16 | 2.6, 2.2, 2.1 |
| PAAr5 | 3.5, 3.6, 4.1 |
aVirus labeled PR represents pol-recombinant virus. bMutation frequency = (number of white colonies for test sample/ number of blue colonies for test sample) - (number of white colonies in control sample/number of blue colonies in control sample) × 100. cThree independent experiments were performed, with data presented from experiment 1, 2 and 3, respectively.
S1 nuclease digestion assay
Supercoiled plasmids (5 μg) were treated with 3 units S1 nuclease or mock-treated, in S1 nuclease buffer (Invitrogen) containing 100 mM final concentration of NaCl, for 5 min at 37°C. Reactions were stopped by performing two phenol, and two phenol-chloroform extractions. DNA was precipitated with ethanol, centrifuged and resuspended in dH2O for digestion with restriction enzymes as indicated. Restriction digested samples were electrophoresed on a 0.8% agarose gel and stained with ethidium bromide for visualization. Two separate plasmid purifications were utilized for this study, with similar results.
Results
Pol-recombinant viruses
HSV-2 pol (SB5, 83D, 6652 and 6757 described in [21]) or HSV-1 pol (SC16 and PAAr5) coding regions were co-transfected with replication defective HP66 viral DNA containing a LacZ insertion within pol[17] into Vero cells to generate recombinant viruses. Recombinant viruses were successfully obtained for all pols except wild type HSV-1 SC16 and the low error rate or antimutator HSV-1 PAAr5 [10]. The pol-recombinant viruses were plaque-purified and the pol genes completely sequenced along with those of their parental viruses to confirm the recombination event. Three of the HSV-2 viruses, HSV-2 SB5, HSV-2 6652 and HSV-2 83D, had pol sequences consistent with wild type isolates found in GeneBank and were recently characterized [21].
The pol from HSV-2 6757, a virus which showed a 300- to 700-fold higher error rate than HSV-1 SC16 by the tk mutagenesis assay previously [21], was found to contain three mutations compared to the other HSV-2 pols, with one change residing in region VII which is shared among diverse DNA polymerases (Fig. 1). No amino acid changes were identified within the proofreading (Exo) domains. The high frequency of errors generated by HSV-2 6757 did not appear to preclude its ability to replicate efficiently in culture. In fact, no significant differences in the growth curve of parental and recombinant viruses in Vero cells (compared to wild type HSV-2 SB5) were apparent (data not shown).
Figure 1.
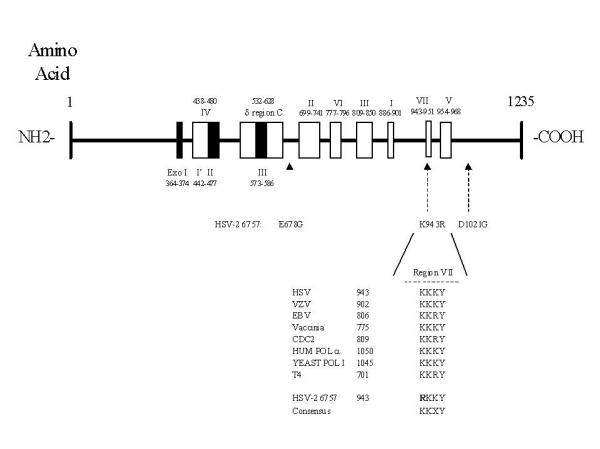
Schematic of HSV pol locus and conserved regions. Regions I through VII, similar among α-like DNA polymerases, are indicated as boxed areas with amino acid residue boundaries. Region VII purported to function in substrate recognition and binding is shown with the amino acid sequences from various DNA polymerases. The following polymerases are represented: HSV [28]; varicella-zoster virus (VZV) [7]; Epstein-Barr virus (EBV) [2]; Vaccinia [8]; Saccharomyces cerevisiae Pol δ (CDC2) [4]; human Pol α [27]; S. cerevisiae Pol α (Yeast Pol 1) [18]; T4 DNA polymerase (T4) [25]. The changes identified in HSV-2 6757 DNA polymerase are indicated. The numbers adjacent to the various polymerase indicate the residue number for the beginning of shared exonuclease domain VII.
tk mutagenesis assay
The proportions of virus resistant to ACV or PCV, which arise during amplification of virus stocks in the absence of drug selection, were measured by the method described by Hall et al. [10]. Plating efficiency assays were performed in MRC-5 cells at 3 μg/ml ACV, which is 10-times higher than the average IC50 for HSV-1 SVC16, and at 8 μg/ml ACV, which is 10-times higher than the average IC50 for the panel of HSV-2 viruses. Notably, whereas the four HSV-2 parental viruses had spontaneous mutation frequencies significantly higher than HSV-1 isolates (Table 1), the frequency of errors from pol-recombinant viruses expressing an HSV-2 Pol in an HSV-1 background were indistinguishable from HSV-1 SC16 when tested in MRC-5 cells. In fact, three of the type 2 pol-recombinant viruses, SB5PR, 6652PR and 83DPR, had error rates equal to or slightly lower than HSV-1 SC16 (0.05%). Results were similar for plating efficiency with PCV (data not shown). In this assay, the HSV-2 Pol does not appear to be solely responsible for the difference in drug-resistant spontaneous tk mutations observed between HSV-1 and HSV-2 strains.
Table 1.
Percent of ACVr mutants from tk mutagenesis assaya.
| %ACVr | ||
| Virusb | 3 μg/ml | 8 μg/ml |
| HSV-2 SB5 | 0.8 | 0.7 |
| HSV-2 SB5PR | 0.06 | 0.04 |
| HSV-2 6652 | 1.0 | 0.8 |
| HSV-2 6652PR | 0.04 | 0.01 |
| HSV-2 83D | 0.7 | 0.5 |
| HSV-2 83D PR | 0.18 | 0.06 |
| HSV-2 6757 | 28 | 20 |
| HSV-2 6757PR | 5.0 | 3.0 |
| HSV-1 SC16 | 0.06 | 0.05 |
| PAAr5 | 0.001 | 0.0006 |
a The proportion of resistant virus was calculated as follows: (titer in the presence of drug) / (titer in the absence of drug) × 100. b Virus labeled PR represents pol-recombinant virus.
Interestingly, unlike the other pol-recombinant viruses, 6757PR, containing the pol coding region from the supermutator clinical isolate HSV-2 6757, retained a high spontaneous mutation frequency. This recombinant virus showed a sixty-fold increase in mutation frequency compared to HSV-1 SC16 (Table 1). This increase in errors is most likely the direct result of the mutations identified within the pol coding region and suggests that this virus may indeed possess a Pol defect in replication fidelity. The fact that the 6757PR mutation frequency was 3–5%, whilst parental HSV-2 6757 was 20–28%, implicates the target/template or other viral proteins in modulating spontaneous mutations.
Drug-independent mutation frequency using non-HSV DNA
To eliminate any contribution of drug selection or tk gene template on the spontaneous mutation frequency in the tk mutagenesis assay, non drug-selected HSV replication-dependent mutations within an unrelated prokaryotic gene were measured using a LacZ shuttle plasmid assay. This assay employed transfection of a plasmid containing the 230 bp SmaI fragment of oris, the HSV origin of replication, and a LacZ reporter gene, into Vero cells. This plasmid was confirmed to generate DpnI-resistant progeny in Vero cells upon superinfection with the wild type HSV-1 and HSV-2 (data not shown). After superinfection, DpnI-resistant (non-methylated; replicated) plasmid DNA was rescued in E. coli. In the presence of X-Gal, bacterial colonies which turn blue do not contain mutations in the LacZ promoter or LacZ coding region. However, colonies remaining white contain mutations to impair LacZ expression.
Mutations can be introduced during replication by the viral proteins or also by the mutagenic processes of transfection and viral infection. Since the process of transfection is often mutagenic, pSV110 was transfected into Vero cells, mock-infected and rescued into bacteria to determine background frequency of mutation from the experimental process. Transfection was found to confer a mutation frequency less than 0.002% (6 white colonies in over 300,000 total colonies) after analyzing non-DpnI treated samples. No colonies were recovered when treating the rescued DNA with DpnI. Moreover, to ensure that viral replication, and not simply the process of viral infection, is responsible for the mutations in a rescued plasmid, a control transfection-superinfection was performed using the pol mutant, HP66, which is unable to replicate. The mutation frequency contributed directly from the process of viral infection was 0.12% (335 in over 280,000 colonies) after analyzing non-DpnI treated samples. Less than 25 colonies were recovered when treating the rescued DNA with DpnI. The data presented in Table 2 was normalized by subtracting the mutation frequency for HP66 infection (0.12 %).
The relative mutation frequency for the panel of viruses tested in the non-HSV DNA mutagenesis assay are presented in Table 2. Consistent with recent data from Hwang et al. [13], PAAr5 showed a minor increase in mutation frequency, compared with wild type HSV-1 and HSV-2 (Table 2). The presence of an antimutator phenotype with this viral polymerase in the tk mutagenesis assay was not apparent when using the non-HSV DNA mutagenesis assay, and therefore phenotype-labelling may be dependent upon both the assay method and/or the target gene. Moreover, the panel of wild type HSV-2 viruses and the PR viruses in this LacZ phenotypic assay had mutation frequencies similar or below that of wild type HSV-1 SC16. Thus, the high proportion of errors associated with HSV-2 viruses appears specific to the tk mutagenesis assay.
HSV-2 6757, although not 6757PR, conferred a slightly higher proportion of mutations in non-HSV DNA in the plasmid assay, than wild type HSV-1 and HSV-2, consistent with a mutator phenotype. Sequencing of the Pol encoded from HSV-2 6757 confirmed several residue changes which may contribute to the higher error rate, although the direct relationship of these individual mutations within the pol coding region to decreased Pol fidelity has not been confirmed (Figure 1). It remains unclear whether a conserved residue change (Lys to Arg at 943) in region VII, a motif purported to participate in the recognition of the PPi and nucleotide substrates [27], or whether one or more of the other three residue changes identified in HSV-2 6757 pol cooperate to affect mutation frequency.
Anisomorphic DNA conformations
Reiterated sequence elements are often observed to form an unwound S1 nuclease-sensitive DNA conformation, termed anisomorphic or left-handed DNA, when present in negatively supercoiled plasmids [22,24]. Nuclease-sensitive secondary structures within the HSV origin of replication are believed to be recognized by the viral origin binding protein, and mutations that reduce this complex formation in vitro also reduce viral replication, thereby indicating that localized torsional strain may occur during viral DNA replication [1]. The tk mutagenesis assay measures replication-dependent mutation frequency within the tk gene, whereas the non-HSV DNA mutagenesis assay measures errors within a bacterial LacZ gene. Three pUC-based plasmids were examined for the potential to form S1 nuclease sensitive structures: pTK1 which contains a 3.6 Kb HSV-1 DNA fragment including the tk coding region, pTK2 containing a 3.7 Kb HSV-2 DNA fragment including the tk coding region, and pLacZ which contains the 3.4 Kb LacZ coding region. These negatively supercoiled plasmids were treated with S1 nuclease or mock-treated for 5 min. After removal of the nuclease, the plasmids were linearized with HindIII. Figure 2 shows that treatment of pTK1 and pLacZ with S1 nuclease did not alter the restriction enzyme profile, suggesting that in this assay, the formation of S1 nuclease-sensitive structures was below the level of detection (Fig. 2, lanes 3–6). However, pTK2 was readily digested with S1 nuclease, which resulted in the appearance of four novel bands suggesting two potential nuclease-hypersensitive sites (Fig. 2, lane 2). Additional restriction mapping studies suggest that the hypersensitive regions are within the viral DNA fragment, and not in pUC DNA (data not shown).
Figure 2.
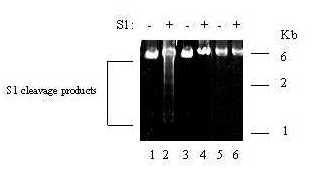
S1 nuclease sensitivity assay. Supercoiled plasmids pTK2 (lanes 1–2), pTK1 (lanes 3–4) and pLacZ (lanes 5–6) were treated with or without S1 nuclease prior to linearization with HindIII. DNA fragments were electrophoresed on a 0.8% agarose gel and visualized by staining with ethidium bromide. Treatment with or without S1 nuclease is indicated at the top of the gel. S1 nuclease cleavage products and a DNA ladder (in Kb) are indicated.
Discussion
The tk mutagenesis assay has been utilized by ourselves and others [10,13] to determine both frequency and spectrum of mutations in the HSV tk gene following selection for drug resistance. In this study, we have extended the findings of previous work [13] by using pol-recombinant viruses to examine mutation frequency in HSV. Specifically, this study evaluated the contribution of type 2 polymerase to mutation frequency when measured by the tk mutagenesis and non-HSV DNA mutagenesis assays.
The control viruses, HSV-2 6757 and HSV-1 PAAr5, demonstrated mutator or antimutator phenotypes, respectively, in thetk mutagenesis assay, but not in the non-HSV DNA mutagenesis assay. Thus, classification of a pol gene as mutator (HSV-2 6757) or antimutator (PAAr5) is not a general characteristic, but is likely dependent upon the assay method and target gene. This observation is consistent with the recent report of high mutation frequency within the SupF gene upon replication by PAAr5 [13].
Since the control viruses described above functioned as expected in the tk mutagenesis assay, recombinant viruses expressing the HSV-2 polymerase within an HSV-1 genome were also examined. Interestingly, three wild type HSV-2 polymerases (6652, 83D and SB5) showed mutation frequencies similar to HSV-1 SC16, rather than their parental virus, when placed within an HSV-1 genome. Only HSV-2 6757 maintained a high error rate (60-fold) over that of HSV-1 SC16, when placed within an HSV-1 genome. However, the mutation frequency of this recombinant virus (HSV-2 6757PR) was not completely penetrant when compared to the parental HSV-2 6757 virus, which showed a 400-fold higher percent of resistant mutants compared to HSV-1 SC16.
An important distinguishing factor between the tk mutagenesis and LacZ test systems is the use of antiviral selectors to measure mutation frequency. Silent mutations within the tk gene that do not impair susceptibility to antiviral agents, or mutations resulting in lethality of the virus, would escape detection as a mutation in the tk mutagenesis assay. Futhermore, the DNA sequence being replicated is unique between these systems and this could contribute to the differences in mutation frequency between assay systems. The demonstration of S1 nuclease-sensitive sites within the type 2 viral genome overlapping the tk coding region, and the absence of such sites in the type 1 viral genome, suggest that local topology may be different between these HSV-1 and HSV-2. Although the potential contribution of this supercoiling-induced phenomenon to modulate mutation frequency during viral replication is unclear, it could partially explain why the type 2 polymerases confer a high mutation frequency to HSV-2 genomes and a low mutation frequency when placed within an HSV-1 genome. Changes in local topology could contribute to polymerase slippage and the incorporation of errors.
The inability of type 2 viruses to confer a higher mutation rate than type 1 viruses in the non-HSV DNA mutagenesis assay is consistent with the absence of S1 nuclease hypersensitive sites in the LacZ gene fragment. Furthermore, the HSV-2 tk sequence used herein contained almost 69% G-C content, compared to the HSV-1 tk which has 65% of bases as G or C. Melting temperature alone may not contribute to the difference in S1 nuclease sensitivity. However, alignment of the tk sequences showed that the type 2 gene had at least eight distinct stretches containing between 7 and 13 G-C bases, whereas the overlapping region in the type 1 tk had four or less G-C bases. This differential in more extensive G-C rich regions supports the observations reported herein.
The data presented herein clearly demonstrate that polymerase alone may not account for the high mutation frequency associated with type 2 viruses in the tk mutagenesis assay. In addition to the potential impact of target sequence and secondary structure topology on mutation frequency, other viral replicative proteins including Pol accessory protein, TK, dUTPase, and uracil-DNA glycosylase may cooperate with Pol to modulate polymerase repair and nucleotide selection [12,13,19,23].
Competing interests
None declared.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
We thank D. Coen for generous gift of reagents; R. Boon for support from GlaxoSmithKline Consumer Healthcare; and H. Cowley and K. Esser for support.
Contributor Information
Karen E Duffy, Email: karen_e_duffy@gsk.com.
Matthew R Quail, Email: mquail@tektagen.com.
Tammy T Nguyen, Email: tammy_t_nguyen@gsk.com.
Robert J Wittrock, Email: robert_j_wittrock@gsk.com.
Joan O Bartus, Email: joan_o_bartus@gsk.com.
Wendy M Halsey, Email: wendy_s_halsey@gsk.com.
Jeffry J Leary, Email: jjl27773@gsk.com.
Teresa H Bacon, Email: teresa_h_bacon@gsk.com.
Robert T Sarisky, Email: robert_t_sarisky@gsk.com.
References
- Aslani A, Simonsson S, Elias P. A novel conformation of the herpes simplex virus origin of DNA replication recognized by the origin binding protein. J Biol Chem. 2000;275:5880–5887. doi: 10.1074/jbc.275.8.5880. [DOI] [PubMed] [Google Scholar]
- Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Seguin C, Tuffnell PS, Barrell BG. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature. 1984;310:207–211. doi: 10.1038/310207a0. [DOI] [PubMed] [Google Scholar]
- Bernad A, Blanco L, Lazaro JM, Martin G, Salas M. A conserved 3'-5' exonuclease active site in prokaryotic and eukaryotic DNA polymerases. Cell. 1989;59:219–228. doi: 10.1016/0092-8674(89)90883-0. [DOI] [PubMed] [Google Scholar]
- Boulet A, Simon M, Faye G, Bauer A, Burgers PMJ. Structure and function of the Saccharomyces cerevisiae CDC2 gene encoding the large subunit of DNA polymerase III. EMBO J. 1989;8:1849–1854. doi: 10.1002/j.1460-2075.1989.tb03580.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challberg MD. A method for identifying the viral genes required for herpesvirus DNA replication. Proc Natl Acad Sci U S A. 1986;83:9094–9098. doi: 10.1073/pnas.83.23.9094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta UB, Summers WC. Ultraviolet reactivation of herpes simplex virus is mutagenic and inducible in mammalian cells. Proc Natl Acad Sci U S A. 1978;75:2378–2381. doi: 10.1073/pnas.75.5.2378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison AJ, Scott JE. The complete DNA sequence of varicella-zoster virus. J Gen Virol. 1986;67:1759–1816. doi: 10.1099/0022-1317-67-9-1759. [DOI] [PubMed] [Google Scholar]
- Earl PL, Jones EV, Moss B. Homology between DNA polymerases of poxviruses, herpesviruses, and adenoviruses: nucleotide sequence of the vaccinia virus DNA polymerase gene. Proc Natl Acad Sci USA. 1986;83:3659–3663. doi: 10.1073/pnas.83.11.3659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb J, Marcy AI, Coen DM, Challberg MD. The herpes simplex virus type 1 UL42 gene product: a subunit of DNA polymerase that functions to increase processivity. J Virol. 1990;64:5976–5987. doi: 10.1128/jvi.64.12.5976-5987.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JD, Coen DM, Fisher BL, Weisslitz M, Randall S, Almy RE, Gelep PT, Schaffer PA. Generation of genetic diversity in herpes simplex virus: an antimutator phenotype maps to the DNA polymerase locus. Virology. 1984;132:26–37. doi: 10.1016/0042-6822(84)90088-6. [DOI] [PubMed] [Google Scholar]
- Hernandez TR, Lehman IR. Functional interaction between the herpes simplex-1 DNA polymerase and UL42 protein. J Biol Chem. 1990;265:11227–11232. [PubMed] [Google Scholar]
- Hwang CC, Chen HH. An altered spectrum of herpes simplex virus mutations mediated by an antimutator DNA polymerase. Gene. 1995;152:191–193. doi: 10.1016/0378-1119(94)00712-2. [DOI] [PubMed] [Google Scholar]
- Hwang YT, Liu BY, Hong CY, Shillitoe EJ, Hwang CC. Effects of exonuclease activity and nucleotide selectivity of the herpes simplex virus DNA polymerase on the fidelity of DNA replication in vivo. J Virol. 1999;73:5326–5332. doi: 10.1128/jvi.73.7.5326-5332.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopf CW. Properties of herpes simplex virus DNA polymerase and characterization of its associated exonuclease activity. Eur J Biochem. 1979;98:231–244. doi: 10.1111/j.1432-1033.1979.tb13181.x. [DOI] [PubMed] [Google Scholar]
- Knopf CW, Weisshart K. Comparison of exonucleolytic activities of herpes simplex virus type-1 DNA polymerase and DNase. Eur J Biochem. 1990;191:263–273. doi: 10.1111/j.1432-1033.1990.tb19119.x. [DOI] [PubMed] [Google Scholar]
- Marcy AI, Olivo PD, Challberg MD, Coen DM. Enzymatic activities of overexpressed herpes simplex virus DNA polymerase purified from recombinant baculovirus-infected insect cells. Nucleic Acids Res. 1990;18:1207–1215. doi: 10.1093/nar/18.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcy AI, Yager DR, Coen DM. Isolation and characterization of herpes simplex virus mutants containing engineered mutations at the DNA polymerase locus. J Virol. 1990;64:2208–2216. doi: 10.1128/jvi.64.5.2208-2216.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzagalli A, Valsasnini P, Plevani P, Lucchini G. DNA polymerase I gene of Saccharomyces cerevisiae: nucleotide sequence, mapping of a temperature-sensitive mutation and protein homology with other DNA polymerases. Proc Natl Acad Sci USA. 1988;85:3772–3776. doi: 10.1073/pnas.85.11.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyles RB, Thompson RL. Mutations in accessory DNA replicating functions alter the relative mutation frequency of herpes simplex virus type 1 strains in cultured murine cells. J Virol. 1994;68:4514–4524. doi: 10.1128/jvi.68.7.4514-4524.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JD, Kunkel TA. Eukaryotic DNA replication fidelity,. In: ML DePamphilis, editor. In DNA replication in eukaryotic cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York.; 1996. pp. 217–247. [Google Scholar]
- Sarisky RT, Nguyen TT, Duffy KE, Wittrock RJ, Leary JJ. Difference in incidence of spontaneous mutations distinct between herpes simplex virus types 1 and 2. Antimicrob Agents Chemother. 2000;44:1524–1529. doi: 10.1128/AAC.44.6.1524-1529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarisky RT, Weber PC. Role of anisomorphic DNA conformations in the negative regulation of a herpes simplex virus type 1 promoter. Virology. 1994;204:569–579. doi: 10.1006/viro.1994.1571. [DOI] [PubMed] [Google Scholar]
- Shillitoe EJ, Zhang S, Wang G, Hwang CBC. Functions and proteins of herpes simplex virus type-1 that are involved in raising the mutation frequency of inefcted cells. Virus Res. 1993;27:239–251. doi: 10.1016/0168-1702(93)90036-M. [DOI] [PubMed] [Google Scholar]
- Singleton CK, Klysik J, Stirdivant SM, Wells RD. Left-handed Z-DNA is induced by supercoiling in physiological ionic conditions. Nature. 1982;299:312–316. doi: 10.1038/299312a0. [DOI] [PubMed] [Google Scholar]
- Spicer EK, Rush J, Fung C, Reha-Krantz LJ, Karam JD, Konigsberg WH. Primary structure of T4 DNA polymerase. Evolutionary relatedness to eucaryotic and other procaryotic DNA polymerases. J Biol Chem. 1988;263:7478–7486. [PubMed] [Google Scholar]
- Weisshart K, Kuo AA, Hwang CC, Kumura C, Coen DM. Structural and functional organization of herpes simplex virus DNA polymerase investigated by limited proteolysis. J Biol Chem. 1994;269:22788–22796. [PubMed] [Google Scholar]
- Wong SW, Wahl AF, Yuan PM, Arai N, Pearson BE, Arai KI, Korn D, Hunkapiller MW, Wang TSF. Human DNA polymerase alpha gene expression is cell proliferation dependent and its primary structure is similar to both prokaryotic and eukaryotic replicative DNA polymerases. EMBO J. 1988;7:37–47. doi: 10.1002/j.1460-2075.1988.tb02781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X, Yang H, Westland CE, Zou R, Gibbs CS. In vitro evaluation of epatitis B virus polymerase mutations associated with famciclovir resistance. Hepatology. 2000;31:219–224. doi: 10.1002/hep.510310132. [DOI] [PubMed] [Google Scholar]