

Key Clinical Message
This case details a term neonate with Apert syndrome, featuring webbed digits, FGFR2 mutations, skull bone fusion, a rostral nose, and cleft palate. The neonate displayed acrocephaly, a flat skull back, a prominent forehead, and syndactyly, confirming Apert syndrome. It emphasizes the need for early recognition and intervention.
Keywords: Apert syndrome, case report, genetic disorder, neonate
1. INTRODUCTION
Apert syndrome, a rare genetic disorder affecting neonates, has distinctive physical features and potential developmental challenges. Epidemiologically, Apert syndrome occurs in approximately 1 in 65,000–88,000 live births, making it a relatively uncommon condition. 1 The etiology of Apert syndrome lies in mutations in the fibroblast growth factor receptor 2 (FGFR2) gene, leading to the premature fusion of certain skull bones, causing craniosynostosis, and resulting in craniofacial anomalies such as abnormal head shape and distinctive facial features. 2 Additionally, syndactyly, the fusion of fingers and toes, along with other skeletal abnormalities, are common clinical manifestations of this syndrome.
Pathophysiologically, the premature fusion of skull bones in Apert syndrome disrupts normal craniofacial development, impacting facial structure and potentially hindering neurological growth. 3 Fusion of digits results from abnormal tissue development during early embryonic stages. Clinically, neonates with Apert syndrome present with craniosynostosis, characterized by an abnormally shaped head and face, along with fused fingers and toes. 4
Diagnosing Apert syndrome involves a comprehensive approach, including physical examinations, imaging studies to assess skeletal anomalies, and genetic testing to identify specific FGFR2 gene mutations. Early interventions for neonates with Apert syndrome often involve surgical procedures to address craniofacial and limb abnormalities, aiming to improve both function and aesthetics. 4 A multidisciplinary care team, comprising specialists in genetics, craniofacial surgery, orthopedics, and developmental care, plays a crucial role in providing comprehensive management for affected individuals. Supporting families through informational and emotional guidance is essential in navigating the challenges associated with Apert syndrome. 5
The developmental obstacles linked with Apert syndrome highlight the necessity of thorough care and support to enhance outcomes for affected infants. By comprehending the complexities of this condition in its early stages, medical professionals can develop personalized treatment strategies, involving surgical interventions and continuous medical support, to alleviate potential long‐term complexities and improve the overall quality of life for both the affected individuals and their families. The provision of both informational and emotional support is vital in navigating the hurdles presented by Apert syndrome, promoting a comprehensive care approach that caters not solely to the physical aspects but also the emotional well‐being of those affected by this condition. 6
This case contributes to the understanding of Apert syndrome by highlighting its recognizable pattern of physical anomalies and emphasizing the importance of early identification and management. Continued research and documentation of similar cases will further enhance our knowledge of Apert syndrome, potentially revealing variations in its clinical presentation and refining diagnostic and treatment strategies.
2. CASE HISTORY/EXAMINATION
This case study involves a 37‐week and 6‐day‐old female fetus delivered by a 38‐year‐old Kurdish mother at Motahari Hospital, Urmia, Iran, through a normal vaginal delivery. The mother had previously undergone a cesarean delivery at age 32, resulting in a healthy newborn. She hails from a low socioeconomic background, residing in a village distant from Urmia city. Her prenatal care was minimal, with only one visit to a gynecologist during this pregnancy and no prenatal examinations or diagnostic assessments such as ultrasonography. There was no history of smoking, alcohol, or drug abuse, and no reported exposure to teratogens, particularly during the first trimester. Notably, there was no mention of congenital anomalies or consanguineous marriages in the family history. Upon the onset of labor, the mother presented at the hospital and delivered a 37‐week and 6‐day‐old male fetus weighing 1800 g, with a height of 41 cm, and a head circumference of 30 cm.
The neonate in this case study displayed a range of clinical manifestations indicative of Apert syndrome, encompassing distinct craniofacial anomalies and limb abnormalities. Craniofacially, the infant exhibited acrocephaly, characterized by a tall, sharply tapered head with a flat back and a prominent or wide forehead, often showing visible furrows. Notable features included a rostral nose and cleft palate. Limb abnormalities were evident through the presence of webbed fingers and toes (syndactyly), with three fused digits on each hand and foot. Furthermore, the infant had short fingers with broad tips, and fusion of fingers and toes was observed, illustrating the classic presentation of Apert syndrome, a condition associated with mutations in the FGFR2 gene leading to premature fusion of skull bones and skeletal anomalies.
3. METHODS
The methodology used for diagnosing Apert syndrome in the neonate typically involves a combination of clinical evaluation and genetic testing.
3.1. Clinical evaluation
Physical examination to assess characteristic features such as webbed fingers and toes, craniofacial abnormalities like acrocephaly, cleft palate, and distinctive facial features. Radiological imaging to examine premature fusion of skull bones, syndactyly, and other skeletal abnormalities.
3.2. Genetic testing process
Genetic testing often involves sequencing the FGFR2 gene to identify specific mutations associated with Apert syndrome. Specific mutations identified in the FGFR2 gene in Apert syndrome often involve the substitution of amino acids at specific positions. The most common mutation is a missense mutation in exon 7 (c.755C > G, p.Ser252Trp) of the FGFR2 gene. 7
We present the case of a neonate, born at term, with a confirmed diagnosis of Apert syndrome (Figure 1). The infant displayed distinctive features associated with the condition, including webbed fingers and toes (Figure 2), a genetic test revealing mutations in the FGFR2 gene, and premature fusion of skull bones. The neonate presented with a rostral nose (Figure 3) and cleft palate. Notably, the infant exhibited acrocephaly, where the head appeared taller than normal and tapered sharply at the top. The back of the skull appeared flat, and the forehead was either prominent or wide, often with visible furrows. Furthermore, the infant had short fingers with broad tips, and fusion of fingers and toes (syndactyly) was observed, with three digits being fused on each hand and foot. This combination of features strongly supported the diagnosis of Apert syndrome in this neonate.
FIGURE 1.
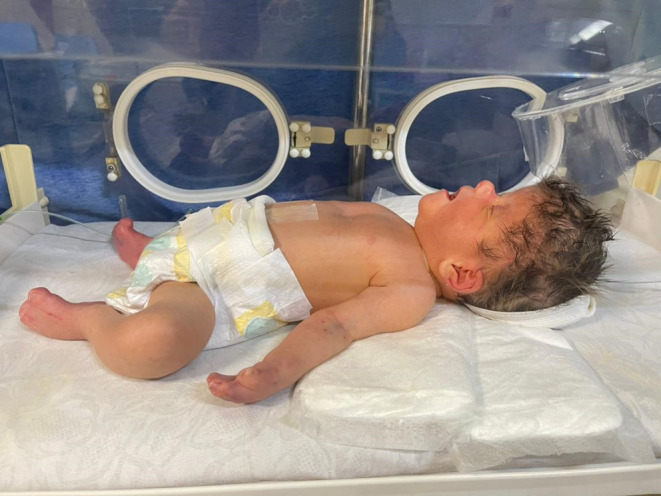
A newborn with Apert syndrome.
FIGURE 2.
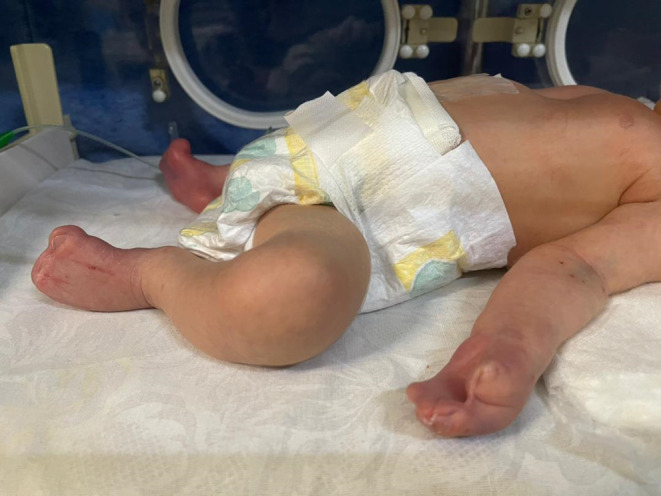
Webbed fingers and toes in a newborn with Apert syndrome.
FIGURE 3.
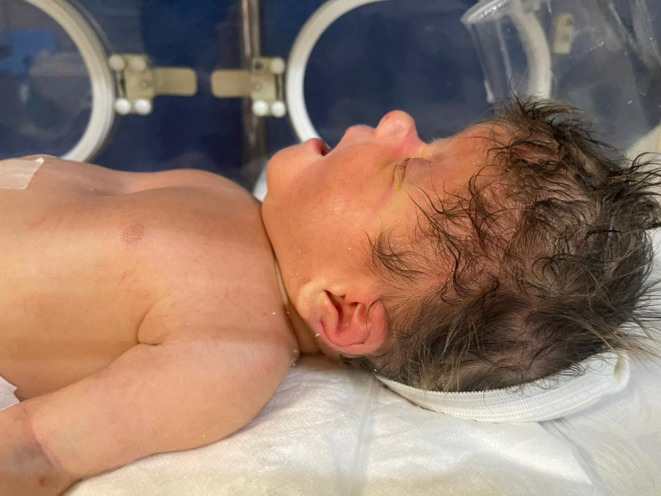
Rostral nose in a newborn with Apert syndrome.
4. CONCLUSION AND RESULTS
In summary, Apert syndrome affects multiple aspects of health beyond physical traits. Timely diagnosis is vital for effective management, with personalized surgical procedures enhancing quality of life. A holistic approach encompassing medical, surgical, and supportive care is crucial for meeting the varied needs of individuals with Apert syndrome. Further research and treatment progress are necessary to enhance the well‐being of those affected. Future investigations should focus on improving diagnostics and developing innovative treatment approaches to enhance outcomes for individuals with this condition.
5. DISCUSSION
The presented case of a neonate with Apert syndrome showcases a typical array of physical manifestations associated with the condition. The distinctive features observed, such as webbed fingers and toes, premature fusion of skull bones, rostral nose, cleft palate, acrocephaly, and syndactyly, align with previously documented cases of Apert syndrome. These observations are consistent with the established literature on the syndrome and contribute to the collective understanding of its clinical presentation.
Comparing these features to previous cases, we find a recurring pattern of characteristic traits, reinforcing the consistency of the syndrome's phenotype across different individuals. The presence of mutations in the FGFR2 gene, as evidenced by genetic testing, further corroborates the diagnosis and aligns with the known genetic underpinnings of Apert syndrome. Genetic counseling in families with a history of Apert syndrome or related conditions plays a pivotal role in assessing genetic risks, providing information on inheritance patterns and testing options, offering emotional support, aiding in family planning decisions, coordinating care with healthcare providers, and facilitating referrals to specialists for comprehensive management, ensuring families are well‐informed and supported in navigating the complexities of these genetic conditions. 1 , 2 , 3 , 4 , 5
A genetic mutation in the FGFR2 gene, responsible for skeletal growth, leads to Apert syndrome. 2 When gene mutation occurs, the receptors fail to interact with fibroblast growth factors, resulting in the premature closure of joints (sutures) between bones during fetal growth. 3 As these joints close prematurely, the child's brain continues to grow, causing changes in the shape of the skull, particularly in the forehead and the sides of the skull. Abnormal signaling leads to rapid maturation of cells, resulting in premature fusion of the skull, hands, and feet bones. The irregular formation of bones leads to physical abnormalities. 4
The diagnosis of Apert syndrome typically occurs after birth, but early detection during pregnancy is possible through two‐dimensional or three‐dimensional ultrasound or MRI to track fetal skeletal growth. 8 , 9 When considering differential diagnoses for a neonate presenting with features suggestive of Apert syndrome, healthcare providers may evaluate other conditions with overlapping symptoms such as craniosynostosis syndromes like Crouzon syndrome or Pfeiffer syndrome, both characterized by craniofacial abnormalities and limb anomalies. Conditions like Carpenter syndrome or Saethre–Chotzen syndrome could also be considered due to shared features. The rationale for confirming Apert syndrome involves a comprehensive assessment combining clinical findings, radiological imaging, and genetic testing. Clinical examination would reveal characteristic features like acrocephaly and syndactyly, while radiological imaging would support the diagnosis by demonstrating premature fusion of skull bones. Genetic testing, identifying mutations in the FGFR2 gene, would definitively confirm Apert syndrome. Through this integrated diagnostic approach, healthcare providers can confidently establish the presence of Apert syndrome in the neonate. 10
Apert syndrome affects the child's physical characteristics as they grow, but it can also impact other parts of the child's body, including:
Brain: Apert syndrome can exert pressure on the brain, potentially affecting the child's cognitive abilities, which may range from normal cognitive function to mild to moderate intellectual disability. Craniosynostosis also affects brain growth, potentially impacting cognitive development. 11 , 12
Ears: Typically, the sides of the child's head are among the first areas to fuse during growth, potentially altering the formation of the child's ears. Some individuals with Apert syndrome may experience hearing impairment or frequent ear infections due to incomplete ear structures. 11 , 12
Eyes: Due to protrusion, misalignment, or wideness of the eyes, the child may experience visual impairments. Ocular irregularities may lead to visual problems, including eye protrusion (exophthalmos), wide‐set eyes (hypertelorism), downward slanting of the outer corners of the eyes (downward palpebral fissures), lack of inward eye movement, eye deviation (strabismus), and shallow eye sockets (ocular proptosis). 11 , 12
Lungs: The abnormal growth of facial and cranial structures can partially obstruct airways, leading to respiratory issues in individuals with Apert syndrome. Depending on the severity of facial abnormalities, nasal formation may lead to breathing difficulties or sleep apnea due to airway obstruction. 11 , 12
Skin: The child may produce excessive oil, leading to severe acne. They may also experience excessive sweating (hyperhidrosis) and have patches of hairless skin (e.g., missing eyebrows). 11 , 12
Teeth: The child's teeth may appear overcrowded in the mouth and may lead to dental issues during formation. Additionally, tooth loss and irregular enamel formation may occur. 11 , 12
Apert syndrome's impact on affected individuals and families can be substantial due to the physical and developmental hurdles it poses. Challenges may arise concerning self‐esteem, body image, social interactions, and educational achievements. Effective coping strategies like therapy, engagement with support communities, and cultivating inner strength through positivity and self‐acceptance can be valuable. Support systems encompassing healthcare providers, family, peers, and advocacy organizations play a pivotal role in offering emotional backing, practical aid, and necessary resources. Improving the quality of life for those with Apert syndrome involves timely interventions, access to specialized care, education on adaptive techniques, and creating a nurturing environment that fosters inclusivity and acceptance. 6
Treatment for Apert syndrome varies based on the severity of the child's condition and typically involves surgical interventions to alleviate symptoms. If the child exhibits symptoms that impact their skull or brain (craniosynostosis or hydrocephalus), surgical intervention is planned between 2 to 4 months after birth to alleviate pressure and normalize brain conditions. Reconstructive or corrective surgeries can address any part of the child's body affected by abnormal formation, including:
eye correction surgery
jaw reconstruction surgery (osteotomy)
chin plastic surgery (genioplasty)
nose plastic surgery (rhinoplasty)
separation of fused fingers and toes
There is no known cure for Apert syndrome, but surgery significantly reduces the child's symptoms, aiding them in leading a normal life. Moreover, The comprehensive management of Apert syndrome underscores the collaborative efforts of various specialties, including genetics, craniofacial surgery, pediatric neurology, and other critical disciplines. 4
6. LIMITATIONS AND BIASES
Lack of longitudinal data: Since it is a case report, there might be a lack of follow‐up information over time, limiting insights into the progression and long‐term outcomes of the condition.
Limited context: The case study might not provide a comprehensive overview of all factors influencing the diagnosis and management of Apert syndrome in newborns, potentially overlooking crucial details that could affect decision‐making.
Expertise bias: The diagnosis and management could be influenced by the specific expertise or experience of the healthcare professionals involved, potentially leading to variations in interpretation and treatment decisions.
7. RECOMMENDATION FOR FUTURE STUDIES
Future studies on Apert syndrome should focus on long‐term research to better understand its progression and lasting impacts. Implementing genetic screening programs can help identify and intervene early, leading to better outcomes for patients. Additionally, developing new treatment strategies such as precision therapies and surgical advancements could enhance the quality of care for those with Apert syndrome. Collaboration among researchers, healthcare professionals, and genetic counselors is vital for advancing research and improving the management of this condition.
AUTHOR CONTRIBUTIONS
Navid Faraji: Supervision; validation; writing – original draft. Rasoul Goli: Data curation; writing – review and editing. Reza Atharifar: Data curation; writing – review and editing. Noushin Shahmirza: Data curation.
FUNDING INFORMATION
No specific funding was received from any bodies in the public, commercial, or not‐for‐profit sectors to carry out the work described in this article.
CONSENT
Written informed consent was obtained from the patient to publish this report in accordance with the journal's patient consent policy.
ACKNOWLEDGMENTS
We would like to acknowledge all the study investigators for conducting this study, and Rasoul Goli for manuscript review and edit support.
Faraji N, Goli R, Atharifar R, Shahmirza N. Apert syndrome in a newborn with premature fusion of skull bones, a rostral nose, and cleft palate: A case report. Clin Case Rep. 2024;12:e9298. doi: 10.1002/ccr3.9298
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Kumari K, Saleh I, Taslim S, et al. Unraveling the complexity of Apert syndrome: genetics, clinical insights, and future Frontiers. Cureus. 2023;15(10):e47281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Das S, Munshi A. Research advances in Apert syndrome. J Oral Biol Craniofac Res. 2018;8(3):194‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shin HR, Kim BS, Kim HJ, et al. Excessive osteoclast activation by osteoblast paracrine factor RANKL is a major cause of the abnormal long bone phenotype in Apert syndrome model mice. J Cell Physiol. 2022;237(4):2155‐2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Raposo‐Amaral CE, Denadai R, de Oliveira YM, Ghizoni E, Raposo‐Amaral CA. Apert syndrome management: changing treatment algorithm. J Craniofac Surg. 2020;31(3):648‐652. [DOI] [PubMed] [Google Scholar]
- 5. Pius S, Ibrahim HA, Bello M, Mbaya K, Ambe JP. Apert syndrome: a case report and review of literature. Open J Pediatr. 2016;6(2):175‐184. [Google Scholar]
- 6. Netherton J, Horton J, Stock NM, Shaw R, Noons P, Evans MJ. Psychological adjustment in apert syndrome: parent and young person perspectives. Cleft Palate Craniofac J. 2023;60(4):461‐473. [DOI] [PubMed] [Google Scholar]
- 7. Kilcoyne S, Luscombe C, Scully P, et al. Hearing, speech, language, and communicative participation in patients with Apert syndrome: analysis of correlation with fibroblast growth factor receptor 2 mutation. J Craniofac Surg. 2022;33(1):243‐250. [DOI] [PubMed] [Google Scholar]
- 8. Hilton C. An exploration of the cognitive, physical and psychosocial development of children with Apert syndrome. Int J Disabil Dev Educ. 2017;64(2):198‐210. [Google Scholar]
- 9. Tan AP, Mankad K. Apert syndrome: magnetic resonance imaging (MRI) of associated intracranial anomalies. Childs Nerv Syst. 2018;34:205‐216. [DOI] [PubMed] [Google Scholar]
- 10. Bailey CT, Zelaya R, Kayder OO, Cecava ND. Acro‐osteolysis: imaging, differential diagnosis, and disposition review. Skeletal Radiol. 2023;52(1):9‐22. [DOI] [PubMed] [Google Scholar]
- 11. López‐Estudillo AS, Rosales‐Bérber MÁ, Ruiz‐Rodríguez S, Pozos‐Guillén A, Noyola‐Frías Á, Garrocho‐Rangel A. Dental approach for Apert syndrome in children: a systematic review. Med Oral Patol Oral Cir Bucal. 2017;22(6):e660‐e668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fearon JA, Podner C. Apert syndrome: evaluation of a treatment algorithm. Plast Reconstr Surg. 2013;131(1):132‐142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.