

Abstract
The mammalian nervous system encodes many different forms of pain, from those that arise as a result of short-term low-grade interactions with noxious thermal, chemical, or mechanical sources to more serious forms of pain induced by trauma and disease. In this Review, we highlight recent advances in our understanding of the neural circuits that encode these types of pain. Promising therapeutic strategies based on recent advances are also highlighted.
The ability to sense pain protects us from harm and is thus an essential aspect of our well-being. Patients suffering from channelopathies that eliminate the ability to feel pain have very high rates of early mortality, largely due to self-mutilation and repetitive fractures (1). Pain, of course, is also a source of substantial discomfort, and humans have long sought ways to ameliorate pain, as exemplified by the Ebers Papyrus from ancient Egypt. As a concept, pain has evolved from archaic notions of demonic punishment to more contemporary views of biological circuit–based origins for the sensation (2). The most recent definition of pain proposed by the International Association for the Study of Pain, unchanged since its first publication in 1979, is “an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage.” The full biological complexity of pain and its underlying circuitry is not wholly conveyed by this definition, however. Pain stems from a varied array of peripheral sensors that detect nociceptive stimuli within internal tissues and those produced by the external world. The information is subsequently distributed to a series of complex neural circuits in the spinal cord dorsal horn and then to numerous brain regions, producing a diverse set of emotions, actions, and sensations (Fig. 1). Pain also manifests with different qualities, including stabbing, pricking, burning, or aching, further highlighting the heterogeneity of the underlying neural circuitry. Longer-lasting pain states produced by nerve and tissue damage, which are also heterogeneous in nature, provide ongoing awareness of the injured area. Pain can become chronic and debilitating when the tissue damage persists and, in some conditions, even after the wound has healed. Because of the high prevalence and lack of adequate treatment options, unraveling the biological basis of persistent pain continues to be an area of intense study. The objective of this Review is to provide an overview of pain circuitry, with an emphasis on recent important advances in our understanding of pathological pain. Insights into neural circuits for pain that have therapeutic potential are also discussed.
Fig. 1. Overall organization of somatosensory circuits.
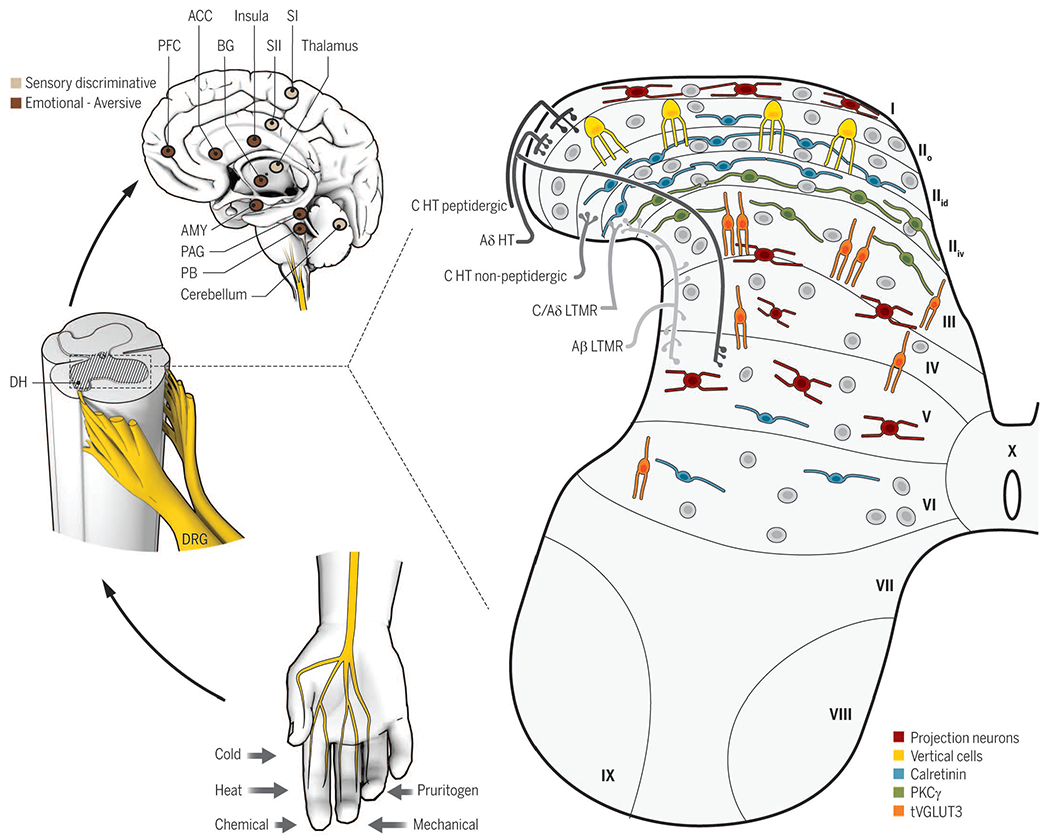
Cutaneous sensory neurons (DRG) are activated by a variety of stimuli (bottom left) and project to the spinal cord dorsal horn (DH, middle left). In the DH (right), the central terminals of high-threshold nociceptors (HT) are located in the most superficial laminae [lamina I to the dorsal part of inner lamina II (IIid)] and lamina V. Low-threshold mechanoreceptors (LTMR) preferentially end in the deep dorsal horn [ventral part of inner lamina II (IIiv) to lamina V]. The spinal cord is divided into 10 laminae (the DH is I to VI) and is composed of numerous neuronal populations. Some identified populations are organized in longitudinal layers (only excitatory neurons are represented): neurons transiently expressing VGLUT3 (tVGLUT3, orange) in laminae III and IV, PKCγ (green) in lamina IIiv, calretinin (blue) in outer lamina II (IIo) and lamina IIid, vertical cells (yellow) in lamina IIo, and projection neurons (red) in laminae I, IV, and V. Projection neurons send information to the brainstem and thalamus and then on to several brain regions implicated in sensory-discriminative (upper left, light brown) and emotional (upper left, dark brown) sensory perception. ACC, anterior cingular cortex; SI (II), primary (secondary) somatosensory cortex; PAG, periaqueductal gray area; PB, parabrachial nucleus; AMY, amygdala; PFC, prefrontal cortex; BG, basal ganglia.
How is pain detected in the periphery?
Pain is produced through the activation of nociceptive primary sensory neurons categorized into the classes C, Aδ, and to a lesser extent Aβ, depending on their axon caliber, degree of myelination, and conductivity properties. Nociceptors innervate the skin, deep tissues, and internal organs and are tuned to detect a wide variety of noxious mechanical, thermal, and chemical stimuli through the activation of modality-specific sensory transduction molecules. A number of these key molecules have been identified (3). Most if not all modalities are conveyed through the activation of more than one transducer, as exemplified by heat transduction. Genetic deletion of the heat-sensing transient receptor potential channel vanilloid 1 (TRPV1) in mice impaired the response to heat only at high noxious temperatures (4), pointing to the existence of one or more additional heat sensors. Recent efforts have now identified TRPM3 (5) and the calcium-activated chloride channel ANO1 (6) as important contributors to the sensation of heat. As was observed with TRPV1, deletion of either channel alone strongly reduced, but did not completely eliminate, noxious heat sensitivity. In contrast, ablation of TRPV1+ fibers by intrathecal injection of the TRPV1 ligand capsaicin (7) or silencing the cells by selective uptake of the voltage-gated sodium channel blocker QX-314 (8) completely abolished noxious heat sensitivity, demonstrating that the fibers are sufficient to account for heat pain, despitemore limited contribution from each channel Given that TRPV1, TRPM3, and ANO1 show considerable overlap in their distribution, analyses of double and triple genetic deletion of the channels in mice may be required to fully understand at the cellular level how noxious heat is transduced. Cold is also transduced through multiple channels. Mice with a genetic deletion of the menthol-sensitive TRPM8, a critical transducer of innocuous cooling, showed only partial avoidance of noxious cold temperatures whereas selective ablation of TRPM8-expressing cells completely abolished noxious cold sensitivity (9). The molecules that transduce mechanical nociception remain stubbornly elusive. Deletion of the leading candidate molecules in mice unexpectedly produced little to no change in noxious mechanical pain sensitivity (10, 11).
Transduction in the periphery: More than just neurons
Epithelial cells such as keratinocytes and Merkel cells directly interact with peripheral axon terminals and have been implicated in the modulation of sensory transduction. Recently, several elegant studies have demonstrated an active role for epithelial cells in tuning the response of sensory neurons. Merkel cells are the epidermal end organ of slowly adapting type 1 (SA1) mechanoreceptors and are involved in the detection of features such as edges and textures. Depolarization of the Merkel cells by optogenetic stimulation or by touch, which depends on the innocuous mechanotransduction channel Piezo2, directly excites SA1 fibers through still-unidentified neurotransmitter signaling mechanisms (12). Epithelial cells have also been shown to actively contribute to nociceptive transmission. Activation of channelrhodopsin or TRPV1 channels ectopically expressed by keratinocytes in mice induced action potential firing in sensory neurons, neuronal activity in the spinal cord, and nocifensive behaviors (13, 14). Conversely, inhibition of the cells blocked the response of nociceptors and other primary sensory neurons to cutaneous stimulation (14). Studies that identify the salient signaling molecules or establish the extent to which epithelial cells contribute behaviorally to pain will be of interest.
Is pain a labeled line?
Most nociceptors express various combinations of sensory transducers that are specific for different modalities (often heat and mechanical) and therefore are classified as “polymodal.” Large-scale efforts using single-cell RNA transcriptome analyses have sought to better understand the molecular and functional logic of primary sensory neurons by parsing the transcriptome profiles of hundreds of individual cells into groups defined by convergent gene expression patterns (15–17). These cluster analyses offer a richer picture of the diversity of nociceptors than previous molecular marker–based classifications, as well as a deeper understanding of the molecular constituents that give rise to the functional properties of sensory neurons (Fig. 2). Although the precise relationships between the molecular constituents and the response properties of the neurons will require further study, the transcriptome data do show some predictive capacity: For example, the tyrosine hydroxylase (TH+) cluster expresses high levels of Piezo2, but not the heat sensors TRPV1, TRPM3, or ANO1, consistent with the response of the cells to light touch, but not to heat (18–20).
Fig. 2. Organization of primary sensory neurons.
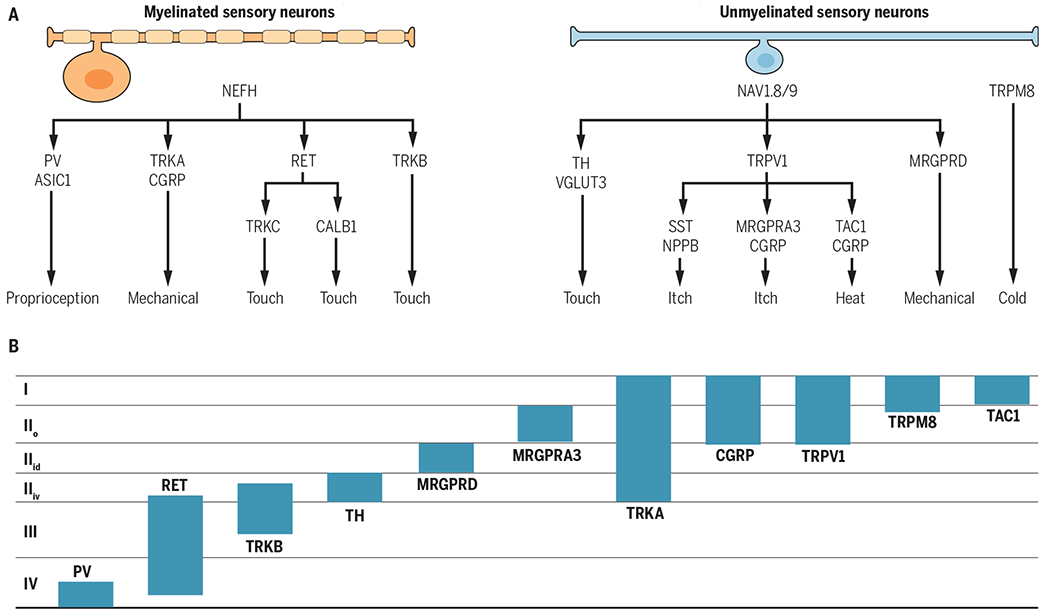
(A) The categories of myelinated and unmyelinated neurons and their respective functional roles are derived from large-scale transcriptional analyses, behavioral analyses, and the literature. (B) The locations of the central terminations of the primary sensory neuron categories are shown. The schematic is based on analyses of gene reporter mouse lines (PV, RET, TRKB, TH, MRGPRD, MRGPRA3, CGRP, and TRPM8) and immunohistological analyses (TRKA, TRPV1, TAC1). Myelinated neurons preferentially express neurofilament heavy chain (NEFH), and unmyelinated neurons preferentially express the sodium channels Nav1.8 and −1.9. Laminae are indicated on the left. RET, ret proto-oncogene; CALB1, calbindin 1; TAC1, tachykinin 1; SST, somatostatin; NPPB; natriuretic peptide type B.
However, limits on the ability to predict the functional output of nociceptors on the basis of their molecular and biophysical properties were also recently demonstrated in studies in which select polymodal populations were ablated in mice. Ablation of nociceptors that express the Mas-related G protein–coupled receptor subtype D (MRGPRD) markedly reduced acute and persistent mechanical pain but had no effect on thermal sensitivity (7). Similarly, ablation of the calcitonin gene–related peptide (CGRP) neurons, a population that partially overlaps with the heat sensors TRPV1, TRPM3, and to a lesser extent ANO1, predictably produced a profound loss of heat but did not alter innocuous or noxious mechanical sensation (21). Loss of CGRP-expressing sensory neurons, which are not responsive to cooling agents (22), greatly increased tonic and evoked activity in spinal neurons associated with cold. This latter finding, mechanistically resembling the activation of dorsal horn itch circuits after silencing pain afferents (pain normally inhibits itch at the level of the spinal cord), further highlights the complexity of primary afferent coding, as well as an important role for central processing in shaping the sensory percept.
Integration in the spinal cord
Primary sensory neurons innervate the six Rexed laminae of the spinal cord dorsal horn, the major locus for the integration of peripheral sensory input and descending supraspinal modulation. The central terminals of sensory neurons are somatotopically organized, forming ventrodorsal-oriented columns similar to the arrangement of the primary somatosensory cortex. Most C- and Aδ-nociceptive afferents form synaptic contacts in the superficial laminae (I and II), whereas low-threshold Aδ- and Aβ-afferents generally project to deeper laminae (III to V) (Fig. 1). Accordingly, in vivo electrophysiological recordings in mammals have shown that laminae I and II respond mainly to noxious peripheral stimulation, whereas neurons in deep layers are more sensitive to touch. The dorsal horn is composed of a large number of excitatory (75%) and inhibitory (25%) interneuron populations, as well as a smaller number of projection populations located in laminae I, III, IV, and V. The projection neurons relay information to numerous supraspinal sites to give rise to both qualitative and affective aspects of the pain sensation. Recent studies of dorsal horn nociceptive circuits have focused largely on persistent pain because of its high prevalence and the need for better treatment options (23, 24).
Mechanical allodynia: Peripheral contributions
Nerve and tissue damage produces dramatic changes in peripheral and central somatosensory circuits. Although acute pain typically follows the activation of nociceptors, light touch–activated neurons can be recruited into the nociceptive network to cause pain after injury. This pathological condition, termed mechanical allodynia, occurs in numerous peripheral neuropathies and central pain disorders, presenting in up to 50% of patients with neuropathic pain (25). Still unknown is the identity of the light touch–sensitive primary sensory population(s) that conveys mechanical allodynia. A large number of primary afferents respond to light mechanical touch and are thus potential candidates (26).
Although the identity of the mechanosensory neurons engaged during mechanical allodynia remains unclear, progress has been made in our understanding of innocuous sensory mechanotransduction (27). As mentioned previously, innocuous tactile sensitivity is primarily dependent on the cation channel Piezo2 (28). In primary sensory neurons, the channel is almost exclusively expressed by low-threshold mechanoreceptors. Conditional deletion of Piezo2 in all sensory neurons in mice resulted in markedly reduced mechanical activation of low-threshold mechanoreceptors, as well as reduced behavioral touch sensation, with no change in acute mechanical pain (29). Because low-threshold mechanoreceptors are thought to convey some forms of mechanical allodynia, Piezo2 could have a role in this type of pain—although after an inflammatory insult, mice with a conditional deletion of Piezo2 in primary sensory neurons did not show evidence of a defect in their mechanical pain threshold under the conditions tested. In contrast, a separate study, which demonstrated sensitization of Piezo2 by the cyclic adenosine monophosphate sensor EPAC1 after neuropathic injury, pointed to a potential role for the mechanotranducer in mechanical allodynia (30).
Mechanical allodynia: The silent dorsal horn circuit
Changes in the dorsal horn circuitry contribute to the expression of mechanical allodynia. One of the principal concepts to explain these changes is based on the gate control theory in which touch inhibits pain through a feed-forward inhibitory circuit within the superficial layers of the dorsal horn (31). In the case of mechanical allodynia, injury impairs the feed-forward inhibitory circuit, allowing light touch to engage the nociceptive network. A seminal study, performed in rodent spinal cord slices, showed that low-threshold mechanosensory A-fiber input is able to activate lamina I pain projection neurons through a polysynaptic network when inhibitory receptor antagonists are present (mimicking the injury-induced decrease in inhibition) (32). Importantly, the study established that the mechanical allodynia circuit, which is normally silent, is already in place under physiological conditions.
Dorsal horn mechanical allodynia circuits: Excitatory neurons
The dorsal horn circuit for mechanical allodynia was originally modeled as a dorsally directed pathway that begins with excitatory interneurons at the border between laminae II and III that express the γ isoform of protein kinase C (PKCγ) and ends with lamina I pain projection neurons (33) (Fig. 3). Two intermediary populations in this pathway were recently identified through paired recordings, the transient central cells in inner lamina II and vertical cells in outer lamina II (34). The dendrites of vertical cells extend deep into lamina III and receive input from most classes of primary afferents, suggesting a potential role as integrators of the network (35). An expansion of the circuit into deeper laminae was shown by studies of the vesicular glutamate transporter 3 (VGLUT3). A population of excitatory interneurons was identified in lamina III that transiently expresses VGLUT3 and receives almost exclusively low-threshold input. Selective activation of the cells by using a designer receptor strategy (DREADD) further demonstrated their important role in the transmission of touch as painful (36). Calretinin-expressing excitatory interneurons in inner lamina II were also identified as an essential element of the circuit. A functional study of the calretinin and PKCγ populations that used c-Fos analysis revealed the existence of distinct excitatory microcircuits for mechanical allodynia, which are differentially engaged depending on whether the injury is inflammatory or neuropathic in nature (36).
Fig. 3. Organization of the dorsal horn circuit for pain.
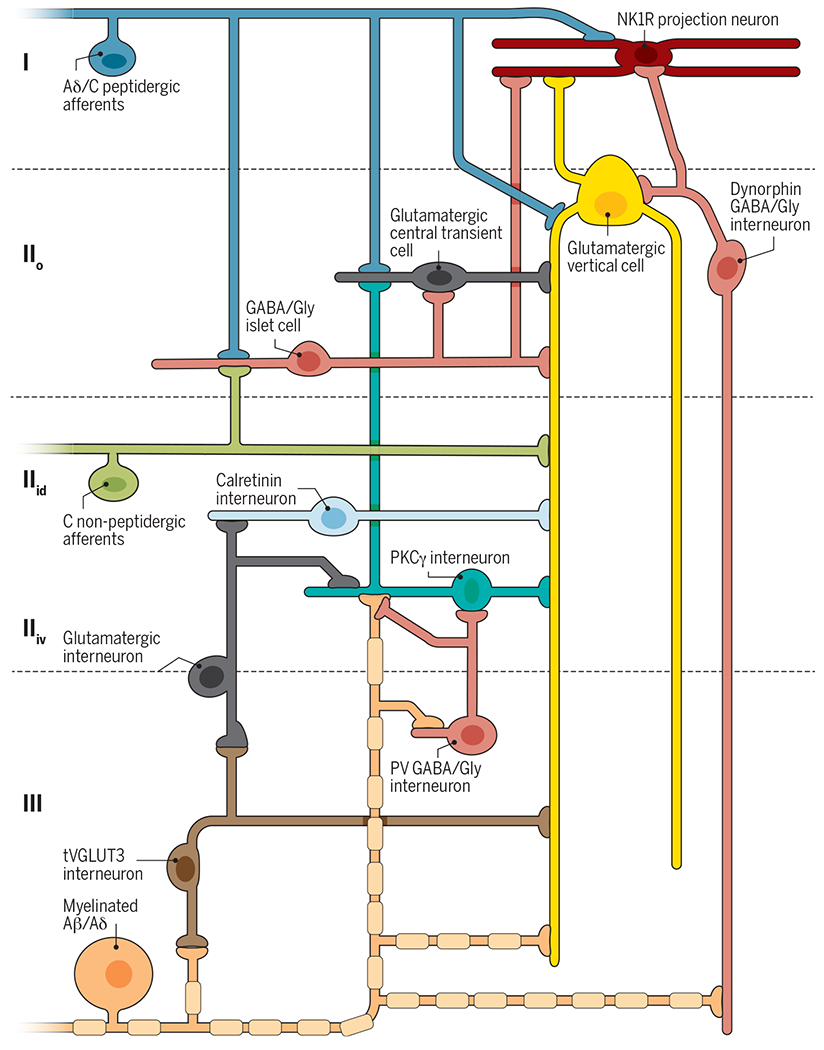
Peripheral nociceptors (blue) project onto excitatory interneurons in lamina IIo (central cell, dark gray; vertical cells, yellow) and onto neurokinin 1 receptor (NK1R) projection neurons (red) in lamina I. Nonpeptidergic afferents expressing MRGPRD (green) project to lamina IIid, including to excitatory vertical cells with ventrally directed elongated dendrites. Both primary afferents also contact inhibitory islet cells (horizontally elongated, pink). Stimulation of nociceptive afferents activates excitatory central cells, vertical cells, and NK1R projection neurons to mediate noxious pain. Inhibitory islet cells modulate this activity. Innocuous afferents (orange) project onto excitatory interneurons expressing tVGLUT3 in lamina III (brown), PKCγ in lamina IIiv (teal), and vertical cells in lamina IIo. Myelinated afferents also contact PV inhibitory interneurons in lamina III (radial, pink) and dynorphin inhibitory vertical cells in lamina IIo (vertical, pink). tVGLUT3 interneurons project onto excitatory vertical cell dendrites and intermediate excitatory interneurons in lamina III. Intermediate interneurons project onto PKCγ and calretinin excitatory interneurons in lamina IIiv. Inhibitory interneurons prevent A-fiber–mediated activation of the nociceptive network through feed-forward circuits that act on PKCγ interneurons, vertical cells, and NK1R projection neurons. After nerve injury, inhibition by PV and dynorphin interneurons is reduced, allowing A-fiber–mediated activation of a dorsally directed circuit that includes tVGLUT3 and PKCγ neurons. After inflammatory injury, reduction in a still-unknown mechanism of disinhibition allows A-fiber–mediated activation of a dorsally directed circuit that includes tVGLUT3 and lamina IIiv calretinin neurons.
Mechanical allodynia circuits: The “gates”
Inhibitory neurons are a fundamental element of the mechanical allodynia network in the spinal cord, forming “gates” that prevent the recruitment of low-threshold fibers into the nociceptive network under physiological conditions (Fig. 3). Using paired recordings in spinal cord slices, a recent study showed that the PKCγ interneurons, which receive Aβ-fiber input, are normally under feed-forward glycinergic disynaptic inhibition (34). After neuropathic injury, which is known to impair inhibitory transmission in the dorsal horn, Aβ-fibers were able to drive action potentials in PKCγ neurons and presumably the rest of the nociceptive network. Consistent with this finding, selective ablation of lamina III parvalbumin (PV)–expressing interneurons, which form contacts with the PKCγ neurons, induced mechanical allodynia, indicating a role for PV interneurons in gating the allodynic pathway (37). Conversely, selective activation of the PV interneurons by the excitatory DREADD strategy reversed mechanical allodynia induced by nerve injury. In addition to PKCγ neurons, PV interneurons were also proposed to act directly on the Aβ-afferents innervating these neurons, suggesting alternative mechanisms for inhibitory control of the circuit (38). The involvement of a second population of inhibitory interneurons in the feed-forward gating of mechanical allodynia was also recently demonstrated. Ablation of dynorphin-expressing interneurons in outer lamina II and at the border of laminae II and III induced mechanical allodynia. The dynorphin gate was further demonstrated to control somatostatin-expressing interneurons, a major population of excitatory neurons that partially overlaps with PKCγ neurons and potentially also vertical and transient central cells (39). Studies of the mechanical allodynia network are beginning to not only reveal individual elements but also produce conceptual advances, including the understanding that there are distinct excitatory microcircuits and multiple distinct gates related to specific types of injuries.
Mechanisms of disinhibition underlying mechanical allodynia
The central sensitization responsible for mechanical allodynia produces various forms of disinhibition in the dorsal horn (40). One mechanism involves the injury-induced release of brain-derived neurotrophic factor (BDNF), which triggers down-regulation of the potassium chloride exchanger 2 (KCC2) in tropomyosin receptor kinase B (TRKB)–expressing pain-transmitting neurons, reducing the driving force for chloride and thus the strength of inhibitory transmission (41, 42). The mechanism of injury-induced BDNF release was recently shown to differ between males and females. Males require the activation of microglia, whereas females require adaptive immune cells, possibly T lymphocytes (43). Sex differences are frequently observed in animal models of chronic pain and may provide insight into the many clinically observed sex differences in pain in humans (44).
Investigations into the mechanisms underlying BDNF release in the dorsal horn of male rodents after injury have focused on adenosine triphosphate signaling through purinergic P2X4 receptors, which requires microglia activation, and on the involvement of chemokine signaling cascades that promote and maintain microglia activation (45). De novo production and release of colony-stimulating factor 1 (CSF1) by injured primary sensory neurons is required for the initial activation of the microglia, as well as for cell proliferation and self-renewal. Deletion of CSF1 specifically from primary sensory neurons or deletion of its downstream effector, the microglial membrane adaptor protein DAP12, was sufficient to markedly reduce microglial activation, as well as the behavioral expression of mechanical allodynia induced by nerve injury (46).
Pain processing in the brain
Functional magnetic resonance imaging (fMRI) studies in humans have demonstrated the coordinated activation of several brain areas in response to noxious somatic and visceral stimuli, including the thalamus, anterior cingulate cortex (ACC), insular cortex, primary and secondary sensory cortices, prefrontal cortex, basal ganglia, cerebellum, and amygdala. This network of brain regions involved in both sensory-discriminative and emotional-affective aspects of pain is termed the “pain matrix” (47).
Several recent studies have questioned whether activation of the pain matrix specifically represents pain or is a more generalized system to detect salient events and the state of the body. The posterior insular cortex encodes nociceptive intensity, and lesions in this brain area disrupt pain, whereas direct stimulation of the area induces pain. However, the insular cortex also has a role in affective, cognitive, and homeostatic functions for nonnociceptive sensory modalities. Intracerebral recordings of the anterior and posterior insular cortex showed activity levels that responded similarly to different sensory modalities (visual, auditory, and somatosensory), as well as to noxious and innocuous somatosensory stimuli, consistent with the idea that the insula is not specific for pain-related information (2). Additionally, patients with loss-of-function mutations in the sodium channel Nav1.7 (SCN9) show congenital insensitivity to pain due to a loss of noxious peripheral sensory drive (1). In a recent study, the response of patients and controls to normally noxious peripheral stimuli was assessed by fMRI. Both groups reported experiencing similar levels of sensation; although controls reported the stimuli as painful and patients reported no pain, the two groups nevertheless showed similar patterns and levels of activity within the pain matrix (48). Gross activity measured in supraspinal structures thus may not reliably predict the pain experience. Rather, pain-related information is likely encoded by specific subregional patterns of activity throughout the pain matrix, which would require analysis by other methods and by experimental designs that allow for causal inference. Machine-learning algorithms to assess fMRI patterns of activity across the pain matrix are showing a high predictive capacity for distinct forms of physical pain, as well as for social pain such as vicarious pain (49).
Complementing these recent findings from brain-imaging studies in humans, work in rodents has focused on synaptic-level mechanisms of pain processing and plasticity in particular regions of the pain matrix. These studies provide further support for the idea that individual brain areas have multiple functional roles. Emotional aspects of pain such as anxiety and fear have been linked to the ACC. A study in mice identified a form of presynaptic longterm potentiation (pre-LTP) at thalamocortical synapses in the ACC that was induced by either anxiety-evoking experiences or chronic pain models (50). It was concluded that the pre-LTP represents anxiety, which can be triggered by chronic pain. The coexistence of this form of pre-LTP with a previously identified form of postsynaptic LTP required for the behavioral expression of chronic pain (51) suggests an important role for these ACC synapses in the mutually enhancing interactions of anxiety and chronic pain.
Mechanisms of plasticity affecting motivation in models of persistent pain were also investigated recently. The nucleus accumbens has been implicated in subjective pain processing in humans and in motivation in rodents. The induction of persistent pain by either nerve injury or an inflammatory mediator impaired motivational behavior in mice, as measured using a progressive ratio operant task (52). The decrease in motivation was linked to enhanced release of the neuropeptide galanin in the nucleus accumbens core, which induced a form of long-term depression (LTD) in dopamine receptor 2–expressing medium spiny neurons (D2-MSNs), thereby reducing their activity. The injury-induced effects on D2-MSNs and motivated behavior were reversed by knockdown of the galanin receptor GALR1 and by interfering with intracellular signaling pathways required for the LTD. Synaptic-level investigations of pain-related mechanisms are thus providing important insights into the role of individual brain areas in the pain experience, including interactions with other brain functions. Such studies are also revealing potential new therapeutic avenues.
Blocking pain circuits at the periphery
Nerve growth factor (NGF) and its major receptor TRKA have essential roles in nociceptor function both during development and in adults. Specific mutations in the genes encoding NGF or TRKA in humans produce phenotypes that include insensitivity to pain, largely resulting from defects in the development of nociceptors (53). In adults, NGF signaling through TRKA receptors at the periphery contributes considerably to the pain associated with many types of injuries and diseases’ including bone cancer, lower back injury, diabetic neuropathy, and osteoarthritis. As a new class of analgesic treatment, humanized antibodies raised against NGF, such as tanezumab (Pfizer), now in phase III clinical trials for osteoarthritis pain, are showing promise. Inhibitors of the TRKA receptor are also of therapeutic interest.
Recent studies have focused also on the therapeutic potential of targeting voltage-gated sodium channels in sensory neurons to relieve pain. In addition to the Nav1.7 loss-of-function mutations that cause congenital insensitivity to pain in humans, studies of Nav1.7 and Nav1.8 in mice indicate a required role for the channel in many forms of acute and pathological pain (54, 55). A highly specific Nav1.7 inhibitor isolated from centipede venom, μ-SLPTX-Ssm6a, when injected intraperitoneally in mice, showed potent analgesic effects on chemical-induced pain with an efficacy equivalent to or exceeding that of morphine (56). Similarly, monoclonal antibodies against Nav1.7, when injected into mice, effectively reduced mechanical allodynia and thermal hyperalgesia induced by inflammation and neuropathic injury (57). Given the promising therapeutic benefit of targeting Nav1.7 channels to relieve pain, several selective inhibitors are now in phase II clinical trials.
Animal venoms that elicit pain have been particularly useful for identifying and characterizing therapeutically relevant nociceptive sensory molecules. MitTx, from the venom of the Texas coral snake Micrurus tener tener, is a highly specific and strong potentiator of the acid-sensing channel ASIC1. Injection of this peptide into the hind paw of mice induced pronounced nocifensive behaviors (58). Conversely, central and peripheral delivery of the isopeptides mambalgin 1 and 2, from the black mamba snake, reversed mechanical allodynia and heat hyperalgesia induced by inflammatory agents by inhibiting ASIC channels, highlighting the therapeutic potential of this family of ion channels (59).
Recent advances in the in vivo application of optogenetic methods also provide attractive therapeutic opportunities for pain. One of the first studies to report the use of optogenetics to evaluate pain circuits in vivo targeted channelrhodopsin to Nav1.8-expressing primary sensory neurons in mice. Cutaneous light-mediated activation of the neurons elicited robust nocifensive behaviors and conditioned place aversion (60). In testing of opsins for therapeutic purposes, the inhibitory opsin NpHR was targeted to small-diameter primary sensory neurons by injection of AAV6-NpHR into the sciatic nerve. Cutaneous light-mediated inhibition of the infected primary sensory neurons alleviated both inflammatory and neuropathic pain (61). The low penetration of light through the skin may be technically limiting; however, implantable miniaturized optoelectronic systems for wireless control of primary sensory and spinal cord neurons are now being developed (62).
Another innovative approach to inhibit nociceptors targets the voltage-gated sodium channel blocker QX-314 to select populations, thereby blocking nerve conduction. Coapplication of capsaicin, which permits entry of QX-314 through the cation channel TRPV1, to the mouse plantar hind paw markedly reduced the response to heat and mechanical pressure (8). QX-314 has also been used to ameliorate persistent pain. The Toll-like receptor TLR5 was identified as a receptor specific to A-fibers (63). Coapplication of the TLR5 ligand flagellin and QX-314 to the mouse plantar hind paw blocked A-fiber activity, as measured electrophysiologically in vitro and in vivo. Although the precise mechanism by which QX-314 enters the cells is still unclear, the approach successfully reversed mechanical allodynia and ongoing pain in a number of chronic neuropathic pain models in mice without producing obvious deficits in touch sensation.
Lastly, the use of gene therapy to restore the normal expression of dysregulated proteins in sensory neurons has become another attractive strategy to treat pain. The expression of the microRNA miR-7a is dramatically reduced after neuropathic injuries. Increasing its expression by intraganglionic injection of AAV6-miR-7a abolished mechanical allodynia and thermal hyperalgesia induced by nerve injury, although not by inflammation (64). In contrast, nerve injury substantially up-regulates the sodium channel Nav1.3. Intraganglionic injection of AAV5 expressing a small hairpin RNA against Nav1.3 reduced its expression and partially reversed nerve injury–induced mechanical allodynia (65).
Blocking pain circuits centrally
Because mechanisms of disinhibition in the dorsal horn have a critical role in the expression of mechanical allodynia and spontaneous pain, therapeutic efforts have been directed toward the restoration of inhibitory tone. Administration of a small-molecule enhancer of KCC2 activity (CLP290) to rodents restored inhibitory drive in dorsal horn neurons, as measured electrophysiologically in spinal cord slices, and reversed behavioral evidence of mechanical allodynia induced by nerve injury. Similarly, overexpression of the cotransporter KCC2 in the spinal dorsal horn and primary sensory neurons of rodents reversed behavioral evidence of mechanical allodynia induced by peripheral nerve injury (66). Humans with a mutation in the HSN2-containing splice variant of the kinase WNK1 suffer from congenital insensitivity to pain. WNK1/HSN2 plays a role in nerve injury–induced mechanical allodynia by reducing KCC2 activity in lamina II neurons of the dorsal horn (67). Antagonizing WNK activity in rodents restored GABA (γ-aminobutyric acid)–mediated inhibition of spinal cord lamina II neurons and reversed the behavioral evidence of mechanical allodynia induced by peripheral nerve injury, but not by inflammatory injury.
Using an entirely different strategy, based on the observation that GABAergic precursors from the medial ganglion eminence grafted into the forebrain correct neurological disorders of hyperexcitability, such as epilepsy, Bráz et al. tested whether injecting the telencephalic GABAergic precursors directly into the dorsal horn of nerve-injured mice could restore inhibitory tone and reverse mechanical allodynia (68). Although the precursor cells maintained their cortical identity, they integrated successfully into the dorsal horn, forming local synaptic connections with primary sensory and spinal cord neurons. By 3 weeks after transplantation, mechanical thresholds were elevated to pre–nerve injury levels. In contrast, inflammation-induced hypersensitivity was not altered by cell transplantation. This work demonstrates that strategies to reverse or compensate for injury-induced disinhibition in the dorsal horn show promise for the treatment of pain.
Conclusions and the road ahead
Recent advances at the molecular, cellular, and systems level are transforming our understanding of how the complex sensation of pain is encoded in the nervous system and providing exciting new avenues for therapeutic intervention. Several key aspects of the nociceptive network and the sensation of pain, however, remain to be addressed. With respect to physiological sensory processing, major outstanding questions include: What is the identity of the noxious mechanotransducer(s)? What are the specific contributions of molecularly distinct primary sensory neurons to modality coding? What is the logic of nociceptor coding of pain? How do spinal cord circuits contribute to modality coding? What is the logic of the projection neuron populations? Which neuronal populations in the brain process the different aspects of pain? In terms of pathological pain, major outstanding questions include: What are the neural networks for spontaneous pain? What are the peripheral and central components of the neural networks that underlie mechanical allodynia induced by different types of injuries, such as inflammatory and neuropathic? How do the neural circuits for pain differ as a function of chronicity? How does chronic pain manifest in the brain? Understanding the precise contributions of the neurons and molecules that underlie each form of pain will greatly facilitate therapeutic drug development.
ACKNOWLEDGMENTS
Funding was provided by the Rita Allen Foundation and the American Pain Society to R.P.S.
REFERENCES AND NOTES
- 1.Bennett DL, Woods CG, Lancet Neurol. 13, 587–599 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Sabatowski R, Schäfer D, Kasper SM, Brunsch H, Radbruch L, Curr. Pharm. Des 10, 701–716 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Julius D, Carlson JR, Curr. Opin. Neurobiol 34, v–vi (2015). [DOI] [PubMed] [Google Scholar]
- 4.Caterina MJ et al. , Science 288, 306–313 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Vriens J et al. , Neuron 70, 482–494 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Cho H et al. , Nat. Neurosci 15, 1015–1021 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Cavanaugh DJ et al. , Proc. Natl. Acad. Sci. U.S.A 106, 9075–9080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brenneis C et al. , J. Neurosci 33, 315–326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knowlton WM et al. , J. Neurosci 33, 2837–2848 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roudaut Y et al. , Channels 6, 234–245 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharif-Naeini R, Prog. Mol. Biol. Transl. Sci 131, 53–71 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Maksimovic S et al. , Nature 509, 617–621 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pang Z et al. , Pain 156, 656–665 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Baumbauer KM et al. , eLife 4, e09674 (2015).26329459 [Google Scholar]
- 15.Usoskin D et al. , Nat. Neurosci 18, 145–153 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Chiu IM et al. , eLife 3, e04660 (2014).25525749 [Google Scholar]
- 17.Li CL et al. , Cell Res. 26, 83–102 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delfini MC et al. , Cell Rep. 5, 378–388 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Li L et al. , Cell 147, 1615–1627 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seal RP et al. , Nature 462, 651–655 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCoy ES et al. , Neuron 78, 138–151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McCoy ES, Taylor-Blake B, Zylka MJ, PLOS ONE 7, e36355 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Todd AJ, Nat. Rev. Neurosci 11, 823–836 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Braz J, Solorzano C, Wang X, Basbaum AI, Neuron 82, 522–536 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jensen TS, Finnerup NB, Lancet Neurol. 13, 924–935 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Abraira VE, Ginty DD, Neuron 79, 618–639 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zimmerman A, Bai L, Ginty DD, Science 346, 950–954 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coste B et al. , Science 330, 55–60 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ranade SS et al. , Nature 516, 121–125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eijkelkamp N et al. , Nat. Commun 4, 1682 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Melzack R, Wall PD, Science 150, 971–979 (1965). [DOI] [PubMed] [Google Scholar]
- 32.Torsney C, MacDermott AB, J. Neurosci 26, 1833–1843 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miraucourt LS, Dallel R, Voisin DL, PLOS ONE 2, e1116 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu Y et al. , J. Clin. Invest 123, 4050–4062 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yasaka T et al. , Mol. Pain 10, 3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peirs C et al. , Neuron 87, 797–812 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petitjean H et al. , Cell Rep. 13, 1246–1257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hughes DI et al. , J. Physiol 590, 3927–3951 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duan B et al. , Cell 159, 1417–1432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zeilhofer HU, Wildner H, Yévenes GE, Physiol. Rev 92, 193–235 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coull JA et al. , Nature 438, 1017–1021 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Coull JAM et al. , Nature 424, 938–942 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Sorge RE et al. , Nat. Neurosci 18, 1081–1083 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mogil JS, Nat. Rev. Neurosci 13, 859–866 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Beggs S, Trang T, Salter MW, Nat. Neurosci 15,1068–1073 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guan Z et al. , Nat. Neurosci 19, 94–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schweinhardt P, Bushnell MC, J. Clin. Invest 120, 3788–3797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salomons TV, lannetti GD, Liang M, Wood JN, JAMA Neurol. 73, 755–756 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Krishnan A et al. , eLife 5, e15166 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koga K et al. , Neuron 85, 377–389 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu H et al. , J. Neurosci 28, 7445–7453 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schwartz N et al. , Science 345, 535–542 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carvalho OP et al. , J. Med. Genet 48, 131–135 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Minett MS et al. , Cell Rep. 6, 301–312 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nassar MA et al. , Proc. Natl. Acad. Sci. U.S.A 101, 12706–12711 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang S et al. , Proc. Natl. Acad. Sci. U.S.A 110,17534–17539 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee JH et al. , Cell 157, 1393–1404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 58.Bohlen CJ et al. , Nature 479, 410–414 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diochot S et al. , Nature 490, 552–555 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Daou I et al. , eNeuro 10.1523/ENEUR0.0140-15.2016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iyer SM et al. , Nat. Biotechnol 32, 274–278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park SI et al. , Nat. Biotechnol 33, 1280–1286 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu ZZ et al. , Nat. Med 21, 1326–1331 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sakai A et al. , Brain 136, 2738–2750 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Samad OA et al. , Mol. Ther 21, 49–56 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li L et al. , Cell Rep. 15, 1376–1383 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kahle KT et al. , Sci. Signal 9, ra32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bráz JM et al. , Neuron 74, 663–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]