

Abstract
Mosquitoes, the major vectors of viruses like dengue, are naturally host to diverse microorganisms, which play an important role in their development, fecundity, immunity and vector competence. The composition of their microbiota is strongly influenced by the environment, particularly their aquatic larval habitat. In this study we used 2×300 bp 16s Illumina sequencing to compare the microbial profiles of emerging adult Aedes aegypti mosquitoes and the water collected from common types of aquatic habitat containers in Puerto Rico, which has endemic dengue transmission. We sequenced 141 mosquito and 46 water samples collected from plastic containers, septic tanks, discarded tires, underground trash cans, tree holes or water meters. We identified 9 bacterial genera that were highly prevalent in the mosquito microbiome, and 77 for the microbiome of the aquatic habitat. The most abundant mosquito-associated bacterial OTUs were from the families Burkholderiaceae, Pseudomonadaceae Comamonadaceae and Xanthomonadaceae. Microbial profiles varied greatly between mosquitoes and there were few major differences explained by container type, however the microbiome of mosquitoes from plastic containers was more diverse and contained more unique taxa than the other groups. Container water was significantly more diverse than mosquitoes, and our data suggest that mosquitoes filter out many bacteria, with Alphaproteobacteria in particular being far more abundant in water. These findings provide novel insight into the microbiome of mosquitoes in the region and provide a platform to improving our understanding of fundamental mosquito-microbe interactions.
Keywords: Aedes aegypti, microbiome, dengue, bacteria, 16s sequencing, aquatic container habitat
Introduction
Mosquitoes are major vectors of arthropod-borne viruses (arboviruses), which cause hundreds of millions of infections each year. The yellow fever mosquito, Aedes aegypti, is responsible for the transmission of dengue (DENV), chikungunya, yellow fever and Zika viruses, which have recently caused major outbreaks of human disease [1–3]. Transmission occurs when a mosquito bites someone who is already infected, the virus then replicates within the mosquito for a period of 7–14 days before it invades the salivary glands and is passed to a new host when the mosquito subsequently feeds. Increasing urbanization and changing climate have contributed to an expansion of Ae. aegypti’s geographic distribution [4–6]. While greater global connectivity has led to an increased frequency of imported cases of arboviral disease and subsequent local outbreaks [7,8]. These have been major driving forces in the increased incidence of arboviral disease over the past few decades [9], and collectively, these diseases represent a major threat to human health.
Broadly effective vaccines or treatments against key diseases such as dengue are not currently available to the general public. Accordingly, most intervention efforts focus on controlling mosquito populations, typically through the use of chemical insecticides. However, widespread usage has led to the emergence of genetic resistance against many insecticides amongst mosquito populations in areas with endemic disease transmission [10,11]. Researchers are currently exploring alternative mosquito control strategies, including those that exploit mosquito-associated bacteria. These novel approaches include biopesticides developed from bacteria [12,13], paratransgenesis [14,15], and the suppression or replacement of target mosquito populations by releasing microbiologically-altered mosquitoes [16,17].
Mosquitoes are naturally host to a diverse range of microorganisms, with their microbiota typically including bacteria, fungi, viruses and protists [18]. Hundreds of thousands of bacterial cells, representing many bacterial species, can inhabit a single mosquito. Most of these reside within the gut, but they can also be found in other somatic and germline tissues. Mosquito-bacteria interactions are complex and can influence many aspects of mosquito biology including their susceptibility to different pathogens [19].
Mosquito larvae acquire their microbiota through vertical transmission, or environmentally, predominantly from the water of their immature aquatic habitats [20]. The presence of microorganisms, or of live eukaryotic cells has been demonstrated to be essential for larval development [21–23]. However, axenic development under laboratory conditions is also possible if larvae are provided with sterile liver and yeast extract [24]. The bacteria that mosquitoes are exposed to as larvae can be carried over into the adult stage, and can be re-acquired from the larval environment by recently-emerged adults [25,26]. The adult microbiome is important for blood digestion and egg development [27–29], and also plays a role in immunity. The presence of the microbiota is linked to the activity of the Toll and IMD innate immune pathways, and clearance through antibiotic treatment affects resistance to DENV and SINV [30,31]. Similarly, the presence of the microbiome also stimulates production of reactive oxygen species, which can influence susceptibility to viral infection [32]. The presence of specific bacteria in adult mosquitoes can alter susceptibility to arboviral infection. For instance, transinfection with the bacterial endosymbiont Wolbachia pipientis restricts infection with multiple arboviruses [33,34]. Oral exposure to Serratia marcescens can enhance DENV infection, exposure to Serratia odorifera enhances infection with either CHIKV or DENV [35,36], while exposure to Chromobacterium species Panama (Csp_P) reduces DENV infection [37,38].
Given the potential impact of the microbiota on vector competence and host fitness, understanding which bacteria infect mosquitoes in different regions, how these bacteria affect their mosquito hosts, and how different environmental factors affect community composition could allow us to assess whether certain mosquito populations are more likely to contribute to disease transmission than others. Meta-analysis of mosquito microbiomes indicates that mosquitoes do not become infected with all of the environmental bacteria to which they are exposed [18]. Studies highlight Pseudomonas, Aeromonas, Enterobacter, Serratia, Stenotrophomonas, and Burkholderia as key taxa within the Ae. aegypti microbiota [39–42], but also suggest that there is a high level of inter-individual and inter-container habitat variability in microbiome composition [20].
There is also evidence that mosquitoes may have a ‘core microbiome’ – a cohort of bacterial taxa that are commonly found across different populations of the same species [43–45]. In laboratory-reared Aedes aegypti there is evidence of low microbial diversity, with mosquito colonies from Latin America, Asia, Australia and Africa sharing the same 12 most-abundant bacterial genera [46]. Conversely, studies of the microbiome in field-collected Aedes mosquitoes show higher levels of mosquito-to-mosquito variation in community composition [39,21]. Some studies have demonstrated high levels of microbial community structure linked to the site of larval development. For instance, Anopheles gambiae mosquitoes from neighboring villages in Burkina Faso had distinct microbial profiles [47]. While a study of mosquitoes that emerged from tires or tree holes in Mississippi, USA determined that aquatic habitat type significantly affected community composition [48].
In light of these observations, we sought to assess microbial diversity of Ae. aegypti in eastern Puerto Rico, which had not previously been characterized. This region has endemic dengue transmission, and past mosquito control programs have determined that Ae. aegypti in the area develop in diverse container habitats, including septic tanks, which might contribute to inter-epidemic dengue virus transmission [49,50]. Consequently, we were interested in determining whether adult mosquitoes that emerged from six common types of container habitats had distinct microbial profiles, and whether these profiles could be reliably linked to the microbial composition of their container habitats. Additionally, we compared the microbial profiles of container water and the mosquitoes that emerged from those sites, in order to identify environmental bacteria that were differentially effective at colonizing Ae. aegypti, and thereby improve understanding of fundamental mosquito-microbe interactions.
Methods
Sample collection
Between October 2017 and October 2019, we collected newly-eclosed adult mosquitoes and water samples from 106 containers across eastern Puerto Rico, from San Juan in the north to Salinas in the south (Fig. 1). Samples were collected from six different types of water-filled containers (Fig. 1): Large 19L plastic buckets (plastic containers), septic tanks, discarded tires (tires), inground trash cans (trash cans), tree holes, and water meter pits (water meters), all common aquatic habitats for Ae. aegypti in Puerto Rico. When located, these containers were assessed visually to confirm the presence of mosquitoes. Emergence traps were used to cover apertures, as described below, and container water and adult mosquitoes were collected daily from each container, within a 24–72-hour window, so that all adult mosquitoes were collected within 24 hours of eclosion. This time point was selected to maximize similarities between mosquito and water samples, which would allow us to determine if any container-specific bacterial operational taxonomic units (OTUs) came to be associated with mosquitoes. Collected mosquitoes had no opportunity to leave the vicinity of their aquatic habitat, or become contaminated with microorganisms from other sources. During this time, emerged adult mosquitoes were not prevented from drinking from, or coming into physical contact with the water. We chose not to surface sterilize adult mosquito samples in order to preserve the cuticle microbiome.
Fig. 1: Container collection map and example site type images.
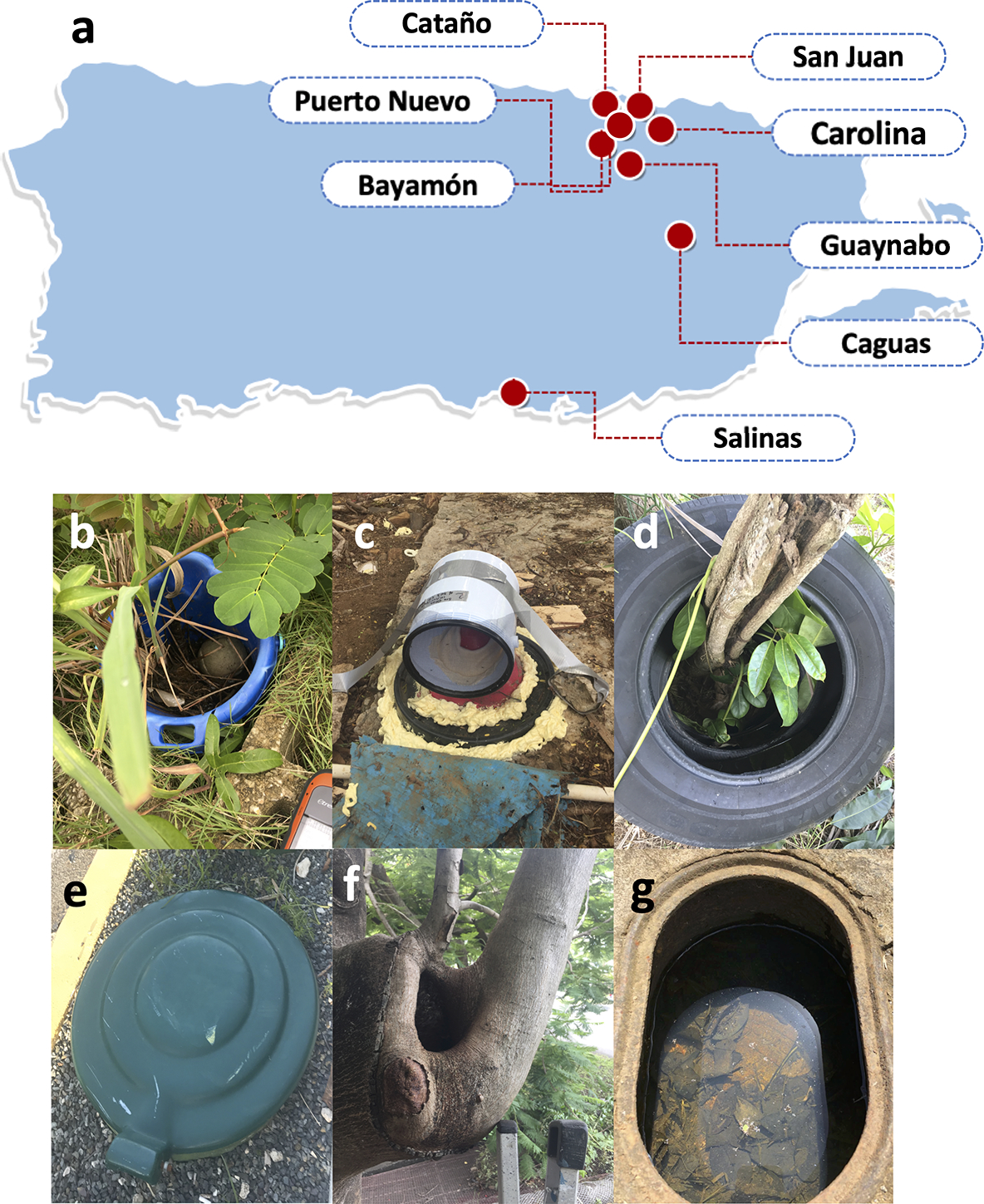
Mosquito and water samples were collected for 16S sequencing from different natural mosquito aquatic habitats in eastern Puerto Rico between October 2017 and October 2019 (a). These samples were collected from six distinct types of containers, represented here by type photographs taken on-site at the time of collection: large plastic buckets (b), septic tanks (c), discarded tires (d), inground trash cans (e), tree holes (f), and water meter pits (g).
The emergence traps used to collect adult mosquito samples were fabricated at the Centers for Disease Control (CDC). Collection protocols varied slightly depending on the size and location of the container habitat. Plastic containers and their contents, including water and mosquito larvae/pupae were physically transported to a greenhouse at the CDC Dengue Branch in San Juan in order to prevent the sites from being tampered with. Within the greenhouse, an emergence trap was placed over the container entrance in order to collect adult mosquitoes as they emerged. For larger unmovable containers, which included all containers from the in-ground trash cans, septic tanks, tires, water meters and tree holes categories, emergence traps were placed over the entrance on site. The emergence traps consisted of one plastic 3.8L cylindrical container with a screen on top, and a hole in the side with a fabric sleeve. This sleeve was used to surround exit points from containers, forcing emerging adults to fly into the traps [49,51].
After collection, mosquito samples from containers in the CDC Dengue Branch greenhouse were immediately frozen at −20°C. For containers that remained in the field, emergence traps were placed on ice and transported back to the laboratory. Once there, mosquito samples were sorted on ice, taxonomically identified by trained taxonomists. Ae. aegypti, Ae. mediovittatus and Culex spp. adults were identified, however only female Ae. aegypti samples were processed and sequenced. Ae. aegypti samples were stored individually in sterile, DNase and RNAse free 1.5mL tubes at −80°C. On each day of adult collection, a 50–100mL sample of water was collected from the containers in a sterile 50mL conical tube or a sterile 100mL cup using a sterile serological pipet. Water samples were stored at −80°C, or on ice and thereafter at −80°C, as detailed above. Subsequently, groups of containers were selected for further analysis based on the collection of sufficient numbers of mosquito samples within a 1–3 day time period.
Datasets
Over the course of the study, three groups of samples were sequenced and analyzed independently. The main dataset utilized in this work included 92 adult female Ae. aegypti mosquito samples, and 23 water samples from 19 containers, representing the six container types of interest (Table 1). These samples were collected between December 2018 through October 2019 and were the focus of most of the analyses in this paper. Two smaller groups of samples, which we have referred to as ‘supplementary’ datasets were sequenced prior to the main dataset to help validate collection and extraction methodologies. These data (File S1) were used to validate key findings from the analysis of the main dataset.
Table 1:
List of sequenced breeding site samples
| Site code | Collection location | Collection time | Mosquito samples | Water samples |
|---|---|---|---|---|
| Site type: Underground trash cans | ||||
| C1 | San Juan | Sep 11th 2019 | 3 | 1 |
| C2 | San Juan | Sep 12th 2019 | 4 | 1 |
| C3 | San Juan | Sep 17th - 19th 2019 | 2 | 1 |
| Site type: Tree holes | ||||
| H1 | Salinas | Sep 4th - 5th 2019 | 5 | 1 |
| H2 | Salinas | Sep 9th - 10th 2019 | 5 | 2 |
| H3 | San Juan | Oct 3rd - 4th 2019 | 4 | 2 |
| Site type: Large plastic buckets | ||||
| P1 | Cataño | Dec 24th - 26th 2018 | 5 | 2 |
| P2 | Puerto Nuevo, San Juan | Jan 30th - Feb 1st 2019 | 4 | 1 |
| P3 | Puerto Nuevo, San Juan | Feb 6 – 8th 2019 | 4 | 1 |
| P4 | Puerto Nuevo, San Juan | Feb 5th 2019 | 4 | 1 |
| Site type: Septic tanks | ||||
| S1 | Carolina | Jun 14th 2019 | 5 | 1 |
| S2 | Salinas | Aug 1st 2019 | 5 | 1 |
| S3 | Salinas | Aug 9th 2019 | 5 | 1 |
| Site type: Discarded tires | ||||
| T1 | Guaynabo | Sep 1st - 3rd 2019 | 6 | 1 |
| T2 | Puerto Nuevo, San Juan | Sep 6th - 9th 2019 | 3 | 2 |
| T3 | Salinas | Sep 10th 2019 | 6 | 1 |
| Site type: Water meters | ||||
| W1 | San Juan | Aug 27th 2019 | 5 | 1 |
| W2 | San Juan | Sep 17th 2019 | 4 | 1 |
| W3 | San Juan | Oct 4th 2019 | 5 | 1 |
Initial collections performed between October 2017 and March 2018, used ethanol. Here, container water was collected using sterile pipets. 20mL of each sample was added to sterile, DNase and RNase free 100mL containers and 80mL of molecular grade ethanol added. Mosquito samples were collected individually or in groups in sterile, DNase and RNase free 1.5mL tubes containing approximately 1mL of 80% ethanol. Samples stored in groups were collected from the same container on the same day. A selection of 37 mosquito samples and 17 water samples, representing 17 septic tanks, tree holes, tires or different plastic containers were sequenced and analyzed independently, these samples are referred to as supplementary dataset A in the manuscript.
Due to issues with DNA quality in some of the early collections with ethanol, this protocol was changed, and all subsequent mosquito samples were collected using emergence traps and water samples were frozen directly upon collection, as described above. To assess the change in methodology, 12 mosquito samples and 6 water samples from 4 plastic buckets or septic tanks were collected between January and February 2019, and then sequenced. These samples make up supplementary dataset B.
DNA extraction
Separate DNA extraction protocols were used for mosquito and water samples. Individual mosquito samples and 100μL squash buffer (10mM Tris pH 8.2, 1mM EDTA, 50mM NaCl) were added to tubes and homogenized using sterile pestles. Samples were placed on ice for 5 mins, heated at 100°C for 5 mins using a dry bath, placed on ice again for 2 mins, and then centrifuged at 14,000 rcf for 5 mins. The supernatant from each sample was cleaned using the DNA Clean & Concentrator kit (Zymo Research Cat. No. D4013), according to the manufacturer’s instructions. Water samples were passed through 47mm diameter nitrocellulose filter membrane with a pore size of 0.22μm (Millipore, Cat. No. N8645–100EA) under vacuum. DNA on these filter papers was extracted using the DNeasy PowerWater kit (Qiagen Cat. No. 14900–50-HF), according to manufacturer’s instructions. At the end of both protocols, the samples were dried using a vacuum centrifuge, and shipped to Johns Hopkins University, Baltimore, MD, USA, resuspended in 100μL sterile DNase- and RNase-free water, and aliquots sent for 16s sequencing. Negative controls in the form of no sample extractions for each kit, and the DNase-free water used to resuspend sample DNA were also sequenced and analyzed.
Sample preparation, 16S sequencing, and taxonomic analysis
PCR amplification, sample processing, sequencing of the 16s rRNA gene and taxonomic analysis were performed commercially for all samples by MR DNA, Shoalwater, TX, USA (www.mrdnalab.com), the protocols described below are summarized from the MR DNA website. Samples were amplified via PCR with the 515F/806R primers for the V4 region of the 16S rRNA gene, and using the HotStarTaq Plus Master Mix Kit (Qiagen). Forward primers were barcoded in order to permit sample pooling. PCR conditions were as follows: 3 mins at 94°C, followed by 30 cycles of 30 sec at 94°C, then 40 sec at 53°C, then 60 sec at 72°C, and finally 5 mins at 72°C. PCR products for all samples were then run on 2% agarose gels in order to gauge band size and intensity. Amplified DNA for each sample was purified using Ampure XP beads. Samples were pooled in equal proportions, according to the concentration of DNA and the molecular weight, and this mixture was used to prepare Illumina DNA libraries. Libraries were then sequenced using Illumina MiSeq in three independent runs to generate 2× 300bp paired end reads. All raw sequence data are available through google drive: https://drive.google.com/drive/folders/17aUjQy8fwFOkepyDv0GRcHbtj5HHkaW5?usp=sharing.
All data generated from sequencing were processed using the MR DNA analysis pipeline (detailed in [52]). Barcode sequences were removed, and then raw sequences were assembled. Short assemblies of less than 150bp were removed from the data set, as were sequences that contained ambiguity in base calls. Sequences were denoised and assigned to an operational taxonomic unit (OTU) by clustering at the 3% divergence (or 97% similarity) level. Chimeric sequences not conclusively assigned to an OTU were removed. OTUs were then classified taxonomically using BLASTn comparison against a custom, curated database that was derived from both NCBI (www.ncbi.nlm.nih.gov) and RDPII (http://rdp.cme.msu.edu). OTUs that could not be classified taxonomically were marked as ‘unclassified’ and were not considered in downstream analyses. FASTAQ mapping files were produced at different taxonomic levels, and these were used to describe the count of the reads associated with each OTU, and the percentage of the total reads associated with each OTU, for individual samples. We received these data from MR DNA.
Bacterial profiles
Read count and abundance data for bacterial OTUs were analyzed at the class (File S2), family (File S3) and genus (File S4) levels, and for both supplementary data sets at the family level (File S5). The maximum percentage abundance for each bacterial OTU was calculated across all samples in the dataset, and low abundance taxa where the value was less than 0.05% were removed. Abundance data were then recalculated based on the revised read count data with those taxa absent. The analyses described below were performed using family level data unless otherwise specified, as this level provided the greatest level of certainty about taxonomic identification given the amplicon length. For negative controls, bacterial OTUs that had an average read count equating to greater than 5% of the average reads of mosquito and water samples were considered to be contaminants. As contaminants could still be associated with mosquitoes and/or their aquatic habitats in nature, we did not remove contaminant OTUs from the total reads or microbial profiles, but instead acknowledged their identity as likely contaminants and did not consider them in subsequent analyses.
Highly abundant bacterial families were identified through assessment of average and maximum abundance of bacterial families in the main mosquito dataset. Twenty-one of these families were part of 7 major classes – Actinobacteria, Alphaproteobacteria, Bacilli, Bacteroidia, Betaproteobacteria, Clostridia and Gammaproteobacteria. Other less-abundant families within these classes were grouped together in a single category, e.g., Other Actinobacteria. Two further Family groups, Unclassified Oscillatoriales family, and Flavobacteriaceae were highly abundant but not from any of these 7 classes. All remaining families, which occurred at lower abundance (average abundance of <1% across all samples), were grouped together into a category termed ‘Other Bacteria’. These 31 groupings were used to construct bacterial profiles of mosquito and water samples for all three datasets, and for QIIME2 pipeline analysis. Abundance levels of these key bacterial families were compared between mosquitoes from the six container types using Kruskal-Wallis ANOVA.
Differentially abundant families between mosquito and water samples were identified using a volcano plot. Briefly, average mosquito and average water abundances were calculated and the LOG2 ratio of mosquito: water abundance was calculated. These were plotted against negative LOG10-transformed P values resulting from one-tailed t tests comparing the abundance of each bacterial family between all mosquito and water samples. Families with significantly higher abundance in water samples had a LOG2 abundance ratio greater than −1, and a LOG10-transformed P value greater than 1.30103. Families with significantly higher abundance in water samples met the same P value criterion, but had a LOG2 abundance ratio greater than 1.
Lists of highly prevalent bacterial genera associated with mosquito and water samples were generated in order to identify bacterial OTUs widely associated with these sample types in Puerto Rico, under the hypothesis that these OTUs might form part of the core microbiome for mosquitoes and aquatic habitats in this region. To be included in these lists, bacteria had to present in all samples of that particular type at abundance of greater than 0%. Bacterial OTUs were not exclusive to either list. These lists were then compared against each other, and against similar lists generated from the two supplementary data sets.
Bacterial diversity
Multiple measures were used to compare the bacterial diversity of mosquitoes and container water from the different types of containers, and between water and mosquito samples. Total families per sample at a minimum abundance of 0.05% were compared for mosquito samples from the different container types using One-way ANOVA, with pairwise comparisons performed using the Holm-Šídák multiple test correction. Family abundance data for water samples were compared using Kruskal-Wallis ANOVA. Similar analyses were performed for the two supplementary data sets. Shannon and Simpson diversity indices for aquatic habitat and mosquito samples were computed for bacterial OTU count data using the diversity() function in the Vegan package (version 2.5–6) in R. These values were compared across container types using Kruskal-Wallis ANOVA. Between and within group variance metrics across container types and across individual sites within each of the six container types were generated from univariate ANOVAs of these diversity indices. Rarefied family counts for water and mosquito samples were generated using the rarefy() function in the Vegan package in R, and compared between container types using Kruskal-Wallis ANOVA.
Non-metric multidimensional scaling & hierarchical clustering
Bray-Curtis distance matrices for mosquito and water data were generated and used to perform NMDS using the metaMDS() function in R. MDS values were exported and visualized using Microsoft Excel v16.32. A subsequent NMDS analysis was performed on a joint Mosquito-Water Bray-Curtis distance matrix. NMDS analysis was used to examine ordination for mosquito and water samples from the larger of the two supplementary data sets. A Euclidean distance matrix from family level data was generated for all mosquito and water samples using the dist() function. This matrix was used to build a dendrogram from using the hclust() hierarchical clustering function using Ward’s method to minimize within cluster variance.
PERMANOVA analysis
Mosquito bacterial OTU count data were square root transformed, and then a Bray-Curtis dissimilarity matrix was created using the vegdist() function in R. PERMANOVA was performed to compare differences in mosquito profiles between containers, and between types of containers using the adonis2() function. Pairwise PERMANOVA was performed using the pairwise.adonis() function in R to identify pairs of container types where mosquito samples had significant differences in their bacterial profiles. Key families underlying these differences were identified using volcano plot analysis.
Venn diagrams
Genus level abundance data for water and mosquito samples were transformed to binary presence/absence data to determine which genera were present in all mosquito and water samples, or mosquito and water samples from each of the six container types. A genus was considered to be present if it was detected in at least one sample from that group. Pairwise Venn diagrams were generated in Microsoft Excel (Fig. S4). Six-way Venn diagrams comparing mosquito or water samples by site type were generated using the Venn package and venn() function in R.
Analysis in QIIME2
To further examine key findings, we also analyzed the sequence data using the QIIME2 pipeline (https://qiime2.org; [53]). All raw reads were imported in QIIME artifact format using a FASTQ processor (available on http://www.mrdnalab.com/mrdnafreesoftware/fastq-processor.html). The DADA2 pipeline was used to denoise and trim the reads [54]. Samples from the three independent runs were denoised independently and then merged for further analysis. Alpha and beta diversity indices were calculated using the QIIME2 diversity plugin. A sampling depth of 4000 was used. For taxonomic analysis, QIIME2 feature classifier trained on GreenGenes 13_8 at 99% similarity was used to call OTUs. This pipeline differed from the MR DNA pipeline in the databases queried, with the MR DNA pipeline analyzing a database that contained information from GreenGenes, but also RDP and NCBI. Similarity for calling OTUs also differed with the MR DNA pipeline using 97% rather than 99%. Level 2 (Class), Level 5 (Family) and Level 6 (Genus) data were assessed. Unassigned sequences and sequences associated with Archaea, chloroplasts, mitochondria, or unclassified bacterial OTUs at the phylum or class levels were removed from data sets prior to analysis. QIIME2 data (File S6) were used to compare bacterial abundance with MR DNA data at the Class level, to generate bacterial profiles, and to perform NMDS ordination at the family level, and to compare highly prevalent bacteria at the genus level.
Statistical analysis
The statistical analyses described above were performed using either Prism v 6.0h (Graphpad) or R version 3.6.1 in RStudio Version 1.2.1335. ANOVA and Kruskal-Wallis ANOVA were performed in Prism. Input data were checked for normality using the D’Agostino & Pearson omnibus normality test, and then either parametric or non-parametric statistical analyses were employed, as appropriate. All other analyses were performed in R, with the exception of the one-tail t tests used in the volcano plot and pipeline comparison analyses, which were generated using the Excel t.test function. R scripts used to analyze data are provided in File S7.
Results
Aedes aegypti bacteria profiles
We collected 92 emerging adult females of Ae. aegypti, and 23 water samples from 6 different container types across eastern Puerto Rico (Table 1) and described their microbiomes using 16s rRNA sequencing (Fig. 1). Bacterial profiles of water and associated mosquitoes were prepared at the family level for mosquito (Fig. 2) and water samples (Fig. 3). We observed a total of 268 bacterial families from 61 distinct bacterial classes represented within the dataset. Two hundred and fifty-three of those families were observed in at least one mosquito sample and 267 were observed in at least one water sample. The five most abundant bacterial families (by average abundance) across the mosquito dataset were: Burkholderiaceae (Class: Betaproteobacteria, Av. abundance: 19.64%), Pseudomonadaceae (Gammaproteobacteria, 14.95%), an unclassified family from Order Oscillatoriales (Cyanophyceae, 14.75%), Comamonadaceae (Betaproteobacteria, 4.17%), and Xanthomonadaceae (Gammaproteobacteria, 3.33%). Of these families, only Comamonadaceae was present in a similar list of the five most abundant families in water samples, which was as follows: Rhodocyclaceae (Betaproteobacteria, 9.46%), Hyphomicrobiaceae (Alphaproteobacteria, 6.57%), Comamonadaceae (Betaproteobacteria, 5.83%), Microbacteriaceae (Actinobacteria, 3.67%), and an unclassified family from order Burkholderiales (Betaproteobacteria, 3.37%). 23 mosquito samples had a single bacterial family representing over 50% of the sequenced reads. For 19 of these samples, that family was Burkholderiaceae, other families represented in this manner were Burkholderiales (2 mosquitoes), the unclassified Oscillatoriales family, and Enterobacteriaceae (1 mosquito each).
Fig. 2: Bacterial profiles of adult Ae. aegypti mosquitoes.
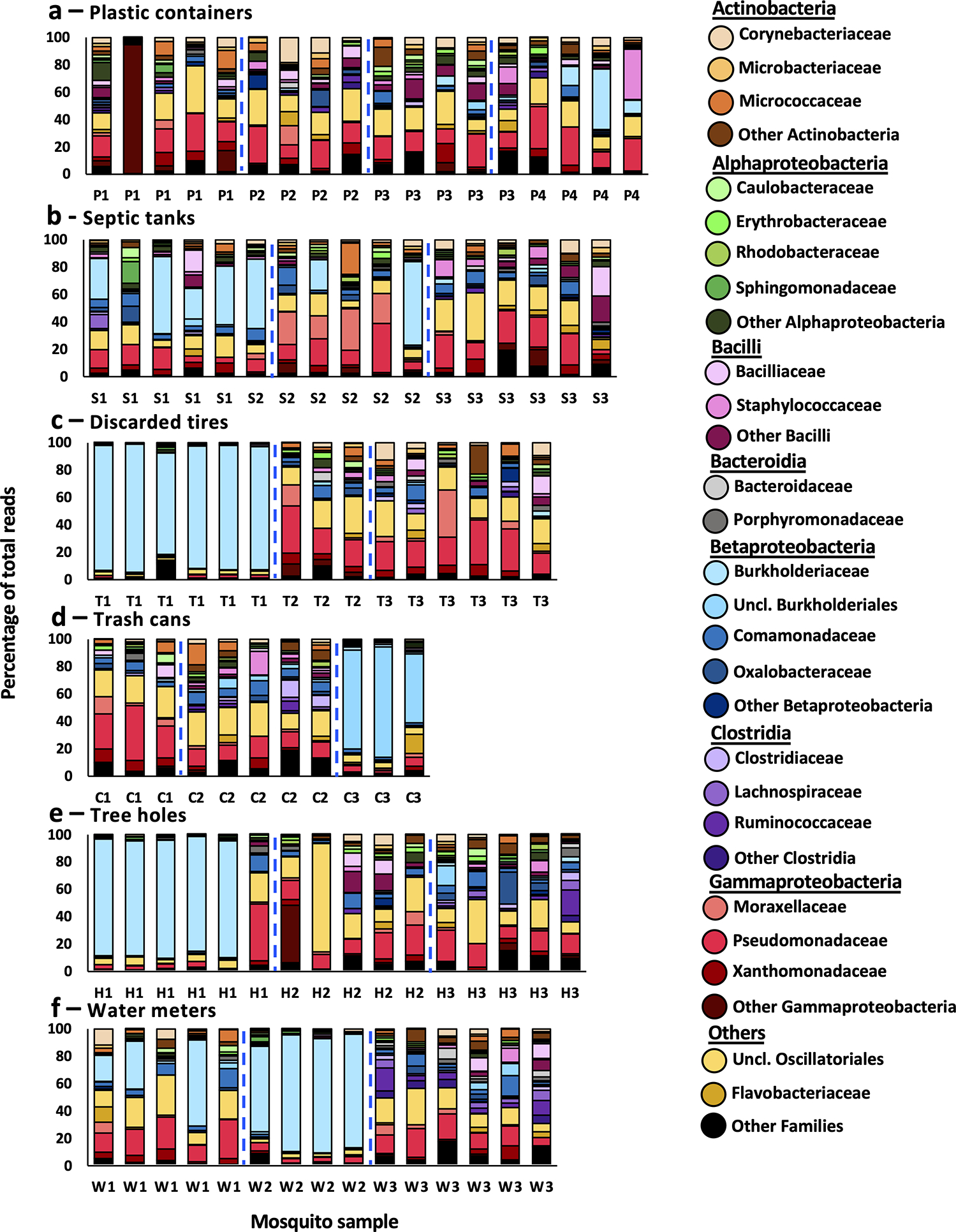
Bar plots represent the bacterial profiles of adult female Ae. aegypti mosquitoes collected within 24 hours post-emergence from six different container types: large plastic buckets (a), septic tanks (b), discarded tires (c), inground trash cans (d), tree holes (e), and water meter pits (f). Each bar represents the profile of a single mosquito sample. Bacterial taxa of high abundance are depicted in different colors at the family level, or as an unclassified family (Uncl.) belonging to a bacterial order. Common colors represent families from the same class. Families of lower abundance have been grouped together as ‘Other’. Dashed blue lines separate mosquitoes collected from different containers. x-axis labels indicate container of origin (Table 1).
Fig. 3: Bacterial profiles of container-habitat water.
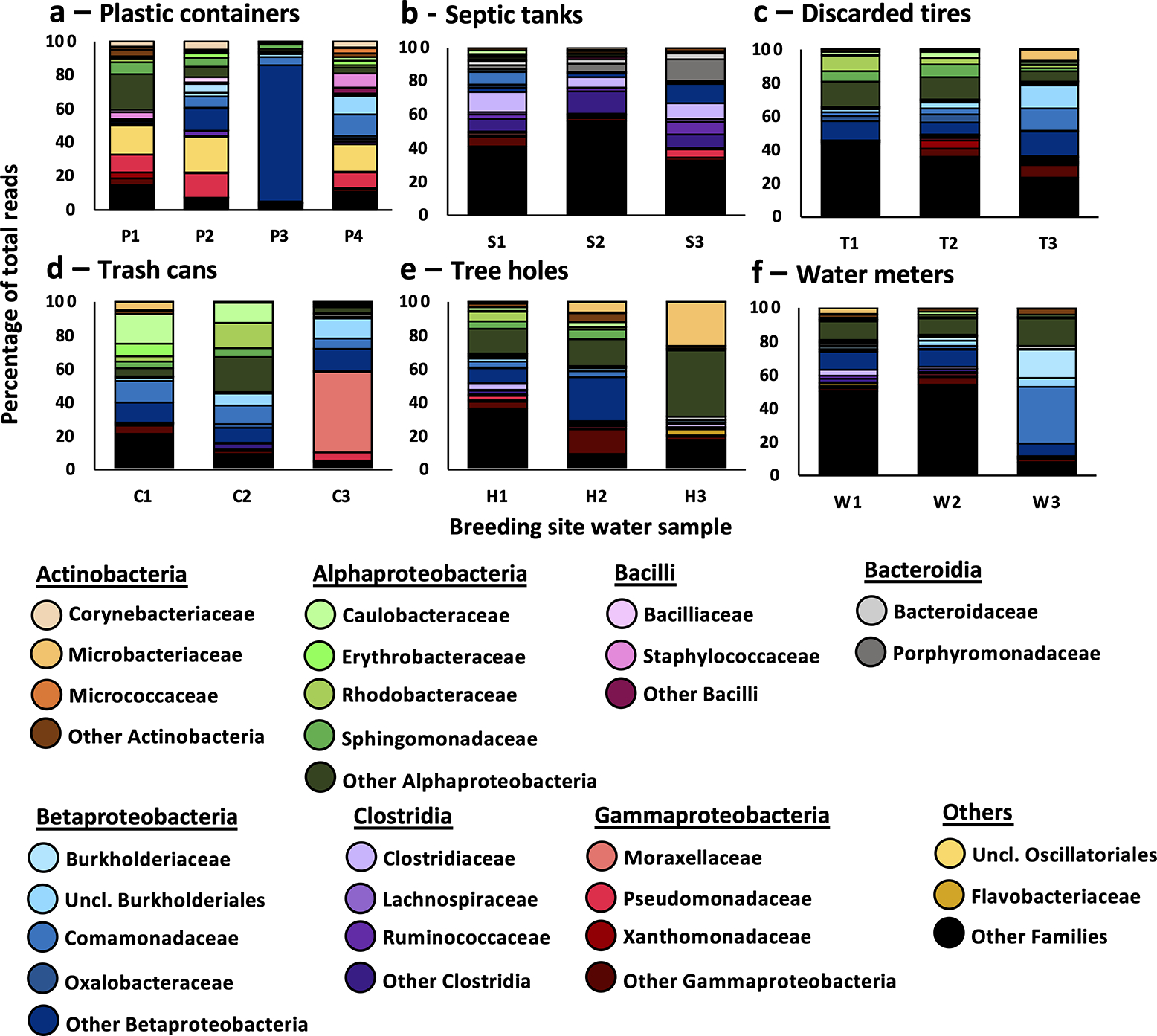
Bar plots represent the bacterial profiles of container water samples collected from large plastic buckets (a), septic tanks (b), discarded tires (c), inground trash cans (d), tree holes (e), and water meter pits (f). Each bar represents the profile of 1–2 water samples. Bacterial taxa of high abundance are depicted in different colors at the family level, or as an unclassified family belonging to an order (Uncl.). Common colors represent families from the same Class. Families of lower abundance have been grouped together as ‘Other’. X-axis labels indicate container of origin (Table 1).
We also assessed genus level data in order to identify bacterial OTUs present in all mosquito and/or water samples (Table 2). We observed a total of 738 genera across all samples, with 689 of these detected in at least one mosquito sample, 693 observed in at least one water sample, and 644 of these shared between both sample types (File S8). We observed that 9 genera were present in all mosquito samples, while 77 genera were present in all water samples (File S9), with that number including all of the 9 mosquito genera. Assessing the two supplementary datasets (Fig. S1 & S2), 7/9 of the highly prevalent mosquito bacterial OTUs, were present across all of the other 48 mosquito samples, with the exclusions being Altererythrobacter and Delftia. However, only 45/77 genera from container water-associated OTUs were present in the 21 water samples from the supplementary data sets.
Table 2:
Bacteria present in all mosquito samples
| Class | Family | Genus |
|---|---|---|
|
| ||
| Actinobacteria | Corynebacteriaceae | Corynebacterium |
| Micrococcaceae | Micrococcus | |
|
| ||
| Alphaproteobacteria | Caulobacteraceae | Brevundimonas |
| Erythrobacteraceae | Altererythrobacter a | |
|
| ||
| Bacilli | Bacilliaceae | Bacillus |
|
| ||
| Betaproteobacteria | Comamonadaceae | Delftia a |
| Oxalobacteraceae | Massilia | |
|
| ||
| Gammaproteobacteria | Moraxellaceae | Acinetobacter |
| Xanthomonadaceae | Stenotrophomonas | |
Not present in all mosquito samples from secondary data sets
Analysis of negative control samples (Supplementary File 10) revealed five bacterial genera that were likely contaminants, linked to different stages of the collection and extraction processes [55]. OTUs linked to genus Halospirulina, corresponding to the unclassified Oscillatoriales family, and genus Pseudomonas were likely contaminants of extraction reagents. Oscillatoriales does not have a well described relationship with mosquitoes, and we consider this taxa to be a contaminant, rather than a real biological association with the mosquitoes we sampled. OTUs linked to genus Bradyrhizobium and genus Sediminibacterium were associated with the kit used for aquatic habitat sample DNA extraction. Neither of these bacteria were highly abundant in mosquito or water samples. An OTU associated with genus Staphylococcus was highly abundant in one blank extraction from the kit used to extract mosquito DNA, but was not seen in the other samples. This OTU was present at low abundance in the main dataset. Genus-level NMDS analysis indicated that control samples clustered independently of mosquito and water samples, indicating they had distinct microbial profiles Supplementary File 10.
Differences between container types
We observed a significant difference in the number of families present in mosquitoes from the six different container types (Fig. 4a, One-Way ANOVA: f = 2.947, df = 91, P = 0.0167), and this effect was mostly due to a higher diversity amongst mosquitoes from plastic containers (Mean Families ± s.e.m.: 35.61 ± 2.79) compared to those from tires (25.53 ± 3.08) (Holm-Sidak test: P = 0.0164). However, we observed no significant difference in family numbers between the water of the different container types (Fig. 4b, Kruskal-Wallis: W = 10.01, df = 18, P = 0.075). We then assessed metrics of alpha diversity. Comparison of Shannon (Fig. 4c) and Simpson (Fig. 4d) diversity indices across site types revealed no significant difference for mosquito samples (Kruskal-Wallis: P > 0.05). ANOVA revealed higher levels of variance between samples from different container types than within container types (Table S1). Likewise, for each individual container type, expect septic tanks, we observed higher variance between mosquitoes collected from different sites than within sites. Analysis of mosquito data rarefied to a read count of 12606 reads revealed a significant overall difference in diversity by container type (Fig. 4e, Kruskal-Wallis: W = 37.00, df = 5, P < 0.0001), with higher taxa richness associated with plastic container-derived mosquitoes than those from every other site type (Dunn’s test: vs. septic tanks/trash cans – P < 0.05; vs. water meters – P < 0.001; vs. tires/tree holes – P < 0.0001). For water data rarefied to a read count of 26797 reads, we observed an overall difference in species richness between site types (Fig. 4f, Kruskal-Wallis: W = 11.95, df = 5, P = 0.0354). This effect was associated with a trend towards lower species richness for plastic container and septic water-emerged mosquitoes compared to the other four site types, however pairwise comparisons were not statistically significant.
Fig. 4: Bacterial diversity at the family level in mosquito and water samples.
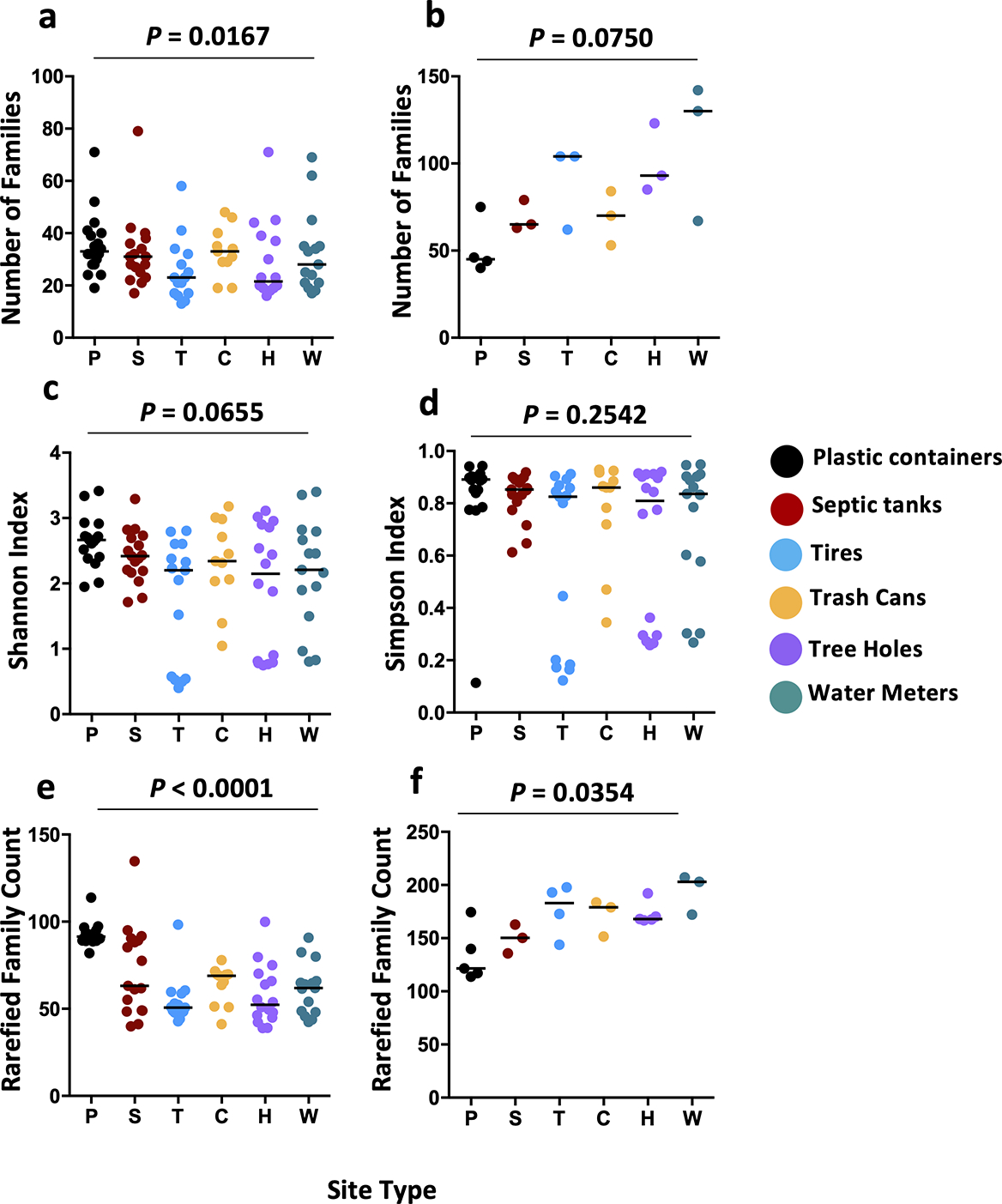
Each dot plot depicts a different measure of bacterial diversity for mosquito or water samples, separated by container type. Panel (a) depicts the number of families in each mosquito sample with a significant difference overall difference due to container type (One-Way ANOVA: f = 2.947, df = 91, P = 0.0167) largely explained by greater diversity in mosquitoes from plastic buckets (P). Panel (b) depicts the number of families in each water sample, with no significant difference observed between groups (Kruskal-Wallis: W = 10.01, df = 18, P = 0.075). Panels (c) and (d) depict the Shannon and Simpson diversity indices for mosquito samples, respectively. Data were generated using the Vegan function diversity(). Higher values indicate greater diversity. No significant differences between groups were observed. Panels (e) and (f) depict family counts for mosquito and water samples, respectively, rarefied to the minimum read count for either mosquito or water samples, with the rarefy() function. A significant difference between mosquito groups (Kruskal-Wallis: W = 37.00, df = 5, P < 0.0001) was driven by higher diversity amongst plastics mosquitoes, while a similar difference amongst water samples (Kruskal-Wallis: W = 11.95, df = 5, P = 0.0354), was driven by lower diversity amongst plastic containers. Each dot represents a single mosquito or water sample. Black lines represent group medians. Treatment codes: P - large plastic buckets, S - septic tanks, T - discarded tires, C - inground trash cans, H - tree holes, and W - water meter pits.
Venn diagram analysis revealed that 197/689 genera (28.59%) were present in at least one mosquito sample from each of the six container types (Fig. 5a). The greatest number of unique genera were present in plastic container-emerged mosquitoes (70), while plastic container and septic-emerged mosquito mosquitoes shared 110 genera that were not present in the other four groups of mosquitoes. Tire, trash can-, tree hole- and water meter-emerged mosquitoes all had fewer than five unique genera. A similar analysis of water samples across site types indicated that 311/693 genera (44.88%) were present in at least one water sample of each site type (Fig. 5b). There were few unique genera associated with water from any site type, with the maximum being 14 for water meters and septic tanks. A total of 62 genera were present in all container types except septic tanks.
Fig. 5: Differences between mosquito and water samples from different types of containers.
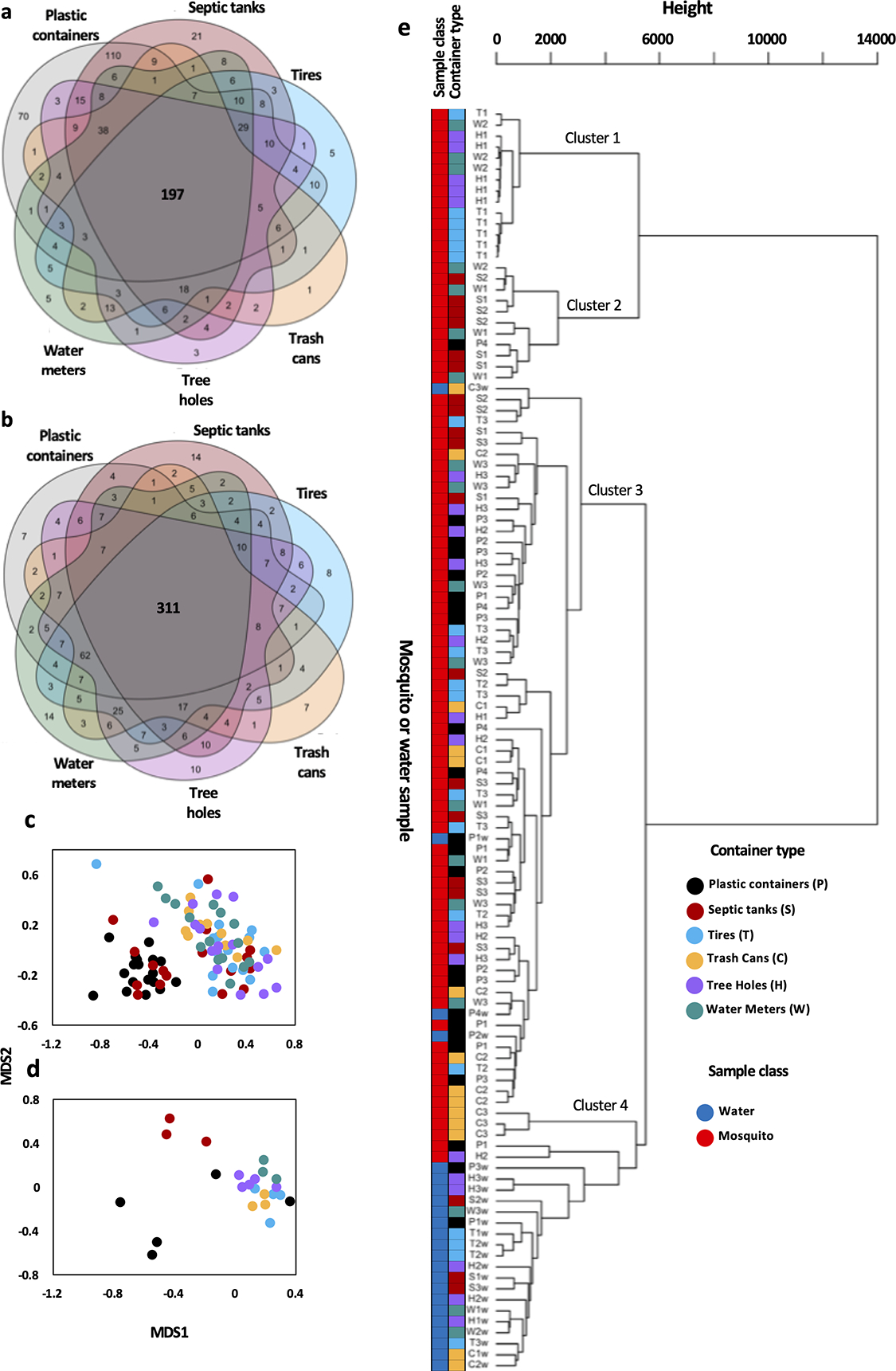
Six-way venn diagrams for mosquito (a) and water samples (b) genus level presence/absence data were produced using the venn() function from the R package Venn. Numbers represent total genera unique to samples from each site type or held in common between different groups of site types: plastics (grey), septics (dark red), tires (light blue), trash cans (orange), tree holes (purple), and water meters (green). NMDS ordination plots for mosquito (c) and water (d) samples generated through non-metric multidimensional scaling (NMDS) using the metaMDS() function of family level data. Each dot depicts one sample, with samples from the same container type sharing colours. Samples that are closer together in space have more similar bacterial profiles. Dendrogram (e) produced using the hclust() function depicting hierarchical clustering between mosquito and water samples across the six site types. Sample codes followed by a ‘w’ represent water samples (blue), all others are mosquitoes (red).
To assess Beta diversity, we compared differences between container types using Non-metric multidimensional scaling ordination (NMDS), which allowed for visualization of complex data sets in two dimensions. In interpreting these plots, dots that are closer together represent samples with more similar bacterial profiles. For mosquito samples (Fig. 5c) (NMDS: k = 2, stress = 0.19965), we observed two main clusters, one containing all plastic container-derived mosquito samples and 8/17 septic tank-derived mosquito samples, and the other containing all of the remaining samples. For water samples (Fig. 5d), we observed three main clusters (NMDS: k = 2, stress = 0.11321). A cluster containing 3/5 plastic container samples, one containing all three septic tank samples, and the final larger cluster containing all tire, trash can, tree hole and water meter samples. NMDS analysis of the larger supplementary dataset (A) revealed no clustering by site type amongst mosquito or water samples, but did reveal near-complete separation of mosquito and water samples (Fig. S3).
Similar to the NMDS analyses, hierarchical clustering of all water and mosquito samples (Fig. 5e) did not show major groupings of samples by container type of origin. The major difference in clustering amongst mosquito samples was explained by levels of family Burkholderiaceae, with cluster 1 consisting of samples where Burkholderiaceae represented the dominant family, such as those from sites H1, T1 and W2. Cluster 2 consisted of the samples with moderate levels of Burkholderiaceae. Cluster 3 contained the majority of mosquito samples, which had high levels of Pseodomnadaceae and Oscillatoriales. Cluster 4 largely consisted of water samples.
Permutational multivariate analysis of variance indicated there were significant differences in the microbial profiles of mosquitoes collected from the different individual containers (PERMANOVA: F = 3.3287, df = 18, R2 = 0.45078, P = 0.001), and those collected from the six different container types (PERMANOVA: F = 2.2278, df = 5, R2 = 0.11467, P = 0.001). These analyses revealed that a much higher percentage of the variation in the data set was explained by individual container (45.08%) than by container type (11.47%). Pairwise PERMANOVA analysis (File S10) indicated that plastic container-derived mosquitoes had distinct profiles from mosquitoes of all other container types, while septic tank-derived mosquitoes and trash can-derived mosquitoes also had significantly different profiles from each other. We used volcano plot analysis to identify families that were differentially abundant for each of those six comparisons. A total of 101 families were significantly more abundant for plastic container-derived mosquitoes than for mosquitoes from at least one other type of container (File S11). Of those, ten families were significantly more abundant for plastics mosquitoes than for mosquitoes from all of the other container types. Conversely, there were 20 families that were significantly less abundant for mosquitoes emerging from plastic containers than at least one of the other five container types, and only two of these were shared across all five comparisons. For the comparison between septic tank- and trash can-derived mosquitoes, 30 families were significantly more abundant for septic tank-derived mosquitoes and 14 were significantly less abundant (File S11).
Differences between container water and mosquitoes
Comparing all mosquito samples with all water samples, we observed that the median number of bacterial families represented at greater than 0.05% of total reads was 29.5 for mosquito samples, and 75 for water samples (Fig. 6a). There was significantly higher diversity observed amongst water samples than in the mosquitoes that emerged from those waters (Mann Whitney U test: U = 65, P < 0.0001). We observed a total of 693 genera present across all water samples, and 689 in all mosquito samples. A total of 644 of these were observed in both water and mosquito samples. Comparison of mosquito and water samples for each of the six container types (Fig. S4) revealed a high degree of overlap between the two classes of sample with an average of 53.98% of the identified genera shared, 15.02% being observed only in mosquitoes and 31.00% only in water. However, these proportions varied depending on container type. Plastic container-derived mosquitoes (20.03%) and septic tank-derived mosquitoes (23.93%) had a greater percentage of exclusive taxa compared to the other groups and had a higher proportion of taxa shared between adult mosquitoes and waters (71.96% and 64.75%, respectively). Conversely, for tires, trash cans, tree holes and water meters, at least 40% of all bacteria genera were only observed in the water.
Fig. 6: Comparison of water and mosquito samples.
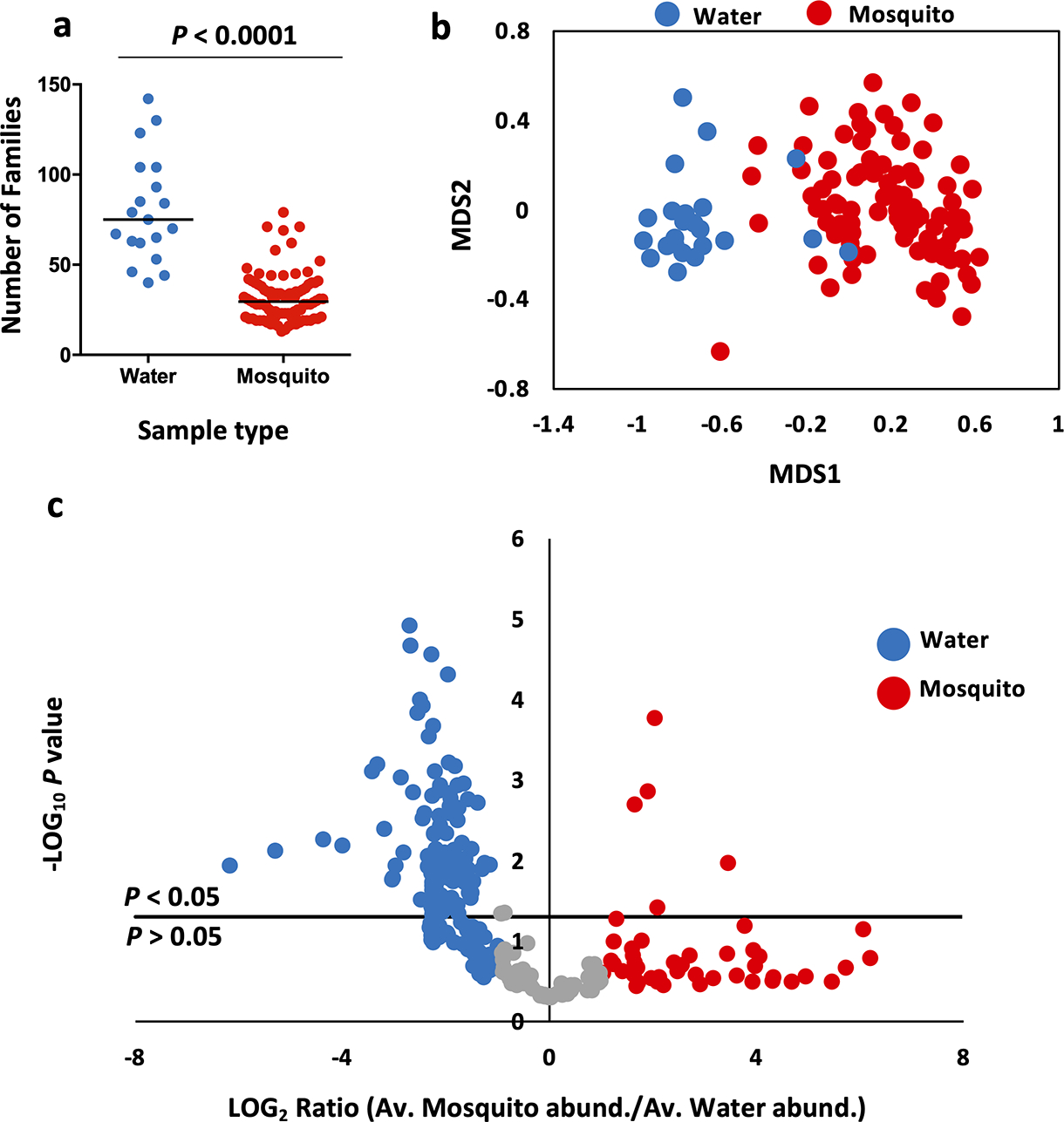
(a) Bacterial diversity, as measured by the total number of bacterial families represented in water (blue) samples is higher than that in mosquito (red) samples (Unpaired t test: t = 9.903, df = 109, P < 0.0001). Black lines represent group medians. (b) NMDS ordination plot of water (blue) and mosquito (red) samples. Each point represents one water or mosquito sample. Samples that are closer together in space have more similar bacterial profiles. Samples clustered into groups dominated either by water (blue) or mosquito (red) samples, with minimal overlap between them suggesting that the two groups had distinct microbial profiles. (c) Volcano plot depicting bacterial families with significantly greater abundance in either water or mosquito samples. Each point represents one bacterial family. Families above the P < 0.05 line on the plot’s y-axis differ significantly in their abundance between the two treatments. Those colored in blue have a LOG2 abundance ratio of >−1 and are more abundant in water samples, while those in red have a LOG2 abundance ratio of >1 and are more abundant in mosquito samples. A total of 105 families were significantly more abundant in water samples, while 5 were more abundant in mosquito samples.
The profiles of mosquito and water samples were compared using NMDS, which revealed near complete separation of the two groups (Fig. 6b, NMDS: k = 2, stress = 0.17585), similar to what was demonstrated in the hierarchical clustering analysis. Follow-up analysis using a volcano plot (Fig. 6c) revealed 110 families significantly associated with either mosquito (five families) or water (105 families) samples. For mosquito samples, these five families were: Micrococcaceae (Actinobacteria), Burkholderiaceae (Betaproteobacteria), the unclassified Oscillatoriales family (Cyanophyceae), Pseudomonadaceae and Xanthomonadaceae (both Gammaproteobacteria). The water-associated families represented 39 classes of bacteria, with Deltaproteobacteria (18 families), Alphaproteobacteria (16) and Gammaproteobacteria (8) the most highly represented classes. A similar volcano plot analysis of class-level data (File S12) indicated that Gammaproteobacteria and Cyanophyceae were the only two classes significantly more prevalent within mosquitoes. In contrast, 18 classes were significantly more abundant in water samples, with Opitutae, Alphaproteobacteria and Sphingobacteriia displaying the strongest association to container water.
Comparison between pipelines
To further evaluate our findings, we analyzed the sequence data using the QIIME2 pipeline. Comparison of mosquito sequences between the two pipelines at the class level revealed that Alphaproteobacteria, Anaerolineae, Cytophagia, Deltaproteobacteria, and Sphingobacteriia sequences were comparatively enriched in the MR DNA pipeline, while Bacilli, Clostridia, and Gammaproteobacteria sequences were enriched in the QIIME2 pipeline. Comparison of the pipelines at the family level revealed many similarities amongst the most abundant taxa. For mosquito samples (Fig. S5), four of the five most abundant families were shared between the two pipelines. The 5th, the unclassified Oscillatoriales family was not present in the QIIME2 pipeline analysis, and in its place was Moraxellaceae, which was ranked 7th in the MR DNA analysis. For the container water data (Fig. S6), 4/5 of the most abundant families identified with the MR DNA pipeline were in the top six most abundant with the QIIME2 pipeline. Sequences associated with an unclassified Burkholderiales family in the former pipeline were called as Comamonadaceae in the latter.
Comparison of the list of highly prevalent bacterial OTUs in mosquitoes between the pipelines revealed that only 2/9 genera were found in all samples through QIIME2 analysis, with these being Stenotrophomonas and an unclassified Comamonadaceae family that likely corresponds to Delftia in the MR DNA pipeline. Six of the other seven genera were present in greater than 80% of samples, with the exceptions being Bacillus, which was detected in 60/92 samples. For the water microbiome, QIIME2 analysis indicated that only 14 genera were present in all 23 samples, as opposed to 77 with the MR DNA pipeline. However, an additional nine genera were detected in 22 samples. Three of these families were not part of the MR DNA container water microbiome. NMDS analysis of water and adult mosquito samples with the QIIME2 pipeline data revealed the same clustering pattern identified with the MR DNA pipeline data (Fig. S7). The mosquito data displayed two major clusters, with no site-specific clustering. The water data displayed three major grouping, with septic tank water isolated from the other types of containers.
The differences we observed here could have arisen due to the relatively short length of the sequencing product that results from 2×300bp 16s sequencing, or differences in OTU calling tools between pipelines. Database content may also have been an issue. The GreenGenes database, which was the sole database queried by the QIIME2 pipeline, and part of the custom database queried by the MR DNA pipeline is no longer being updated, which could also have affected OTU calls. For this reason, we primarily utilized data generated through the Mr DNA pipeline, and used this as our major determinant of OTU calls for highly-prevalent bacterial OTUs associated with this project. However, in spite of these limitations, and because QIIME2 is a tool commonly utilized in mosquito microbial ecology, we felt that this provides a useful point of comparison, particularly as we observed high concordance between the results of the two pipelines for more abundant bacterial OTUs.
Discussion
The microbiota of Puerto Rican Ae. aegypti
In this study, we sought to characterize the microbiome of Aedes aegypti mosquitoes emerging from container habitats from eastern Puerto Rico, a highly urbanized region with endemic transmission of dengue virus. Through 16S sequencing, we identified 253 of bacterial OTUs at the family level associated with the mosquitoes we collected, with the average mosquito microbiome containing 31.42 families, and the most diverse mosquito microbiome containing 79 families. These numbers reflect a high level of microbial diversity, in line with what has been observed in other studies of the microbiomes of field mosquitoes [39,20,21,45].
We observed broad similarities between our data and previously published mosquito microbiomes. When considering our mosquito data, we observed that several of the more abundant bacterial families; Burkholderiaceae, Pseudomonadaceae, Comamonadaceae, and Xanthomonadaceae were all well described members of the Ae. aegypti microbiota [39,21,41], and the mosquito microbiota, in general [56,20,57]. While a bacterial OTU linked to genus Pseudomonas was a likely contaminant, it had much higher abundance in mosquito samples than in control, and given the known links between mosquitoes and this bacterial family, we consider it likely that this is an association with biological relevance.
At the genus level, we identified 689 bacterial OTUs associated with mosquitoes, and determined that 9 of these were present in every mosquito sample. It remains to be seen if these bacteria are widely found within mosquitoes from this area. Eight of these genera; Acinetobacter, Bacillus, Brevundimonas, Corynebacterium, Delftia, Massilia, Micrococcus, and Stenotrophomonas have previously been described in Aedes and/or Anopheles mosquitoes from the field or laboratory [47,21,22,58,59,44]. In the case of Altererythrobacter, we found no clear evidence linking the genus to infection in mosquitoes, however its family, Erythrobacteraceae, has been observed in Anopheles gambiae [45]. Differential analysis of the these bacteria across the two analytical pipelines revealed the importance of the choice of reference 16S database to OTU calls, particularly for sequencing with shorter amplicons, as in 2× 300bp 16S sequencing.
Comparison of our data with other published mosquito microbiomes and metanalyses [57] indicated that several other common mosquito-associated bacterial OTUs were present in a fairly high proportion of our samples. These include: Aeromonas (39/92 mosquito samples), Comamonas (53) Enterobacter (71), Pantoea (42) and Propionibacterium (61). However, the key taxon of Serratia was only detected in 21 mosquito samples, which is interesting given its widespread occurrence in mosquito microbiomes [60,39,61,62,28]. As some Serratia species and isolates have been linked to mosquito vector competence [35,36,63], it is possible that its absence in Ae. aegypti could be impacting disease transmission in the field. Other key mosquito-associated bacterial OTUs including Asaia, Elizabethkingia and Klebsiella were entirely absent. It is currently unclear why these taxa were not represented in our data set. It is possible that the differences between our data and other published studies reflect the nature of Puerto Rico as an isolated tropical island, which could potentially facilitate distinct mosquito-microorganism associations. A larger-scale, longitudinal study encompassing additional geographic locations might reveal whether taxa like Serratia and Elizabethkingia are underrepresented in mosquitoes from Puerto Rico as a whole. The mosquitoes we sampled were all recently emerged mosquitoes that had not yet taken a carbohydrate-rich nectar meal. Age, diet, and diet by age interactions could all be expected to contribute to differences between our results and the results of other studies. It is also possible that the microbiomes of our mosquitoes contained environmental bacteria that were reacquired from the container water and may not go on to form long-term associations with adult mosquitoes.
Our laboratory is particular interested in bacteria from the genus Chromobacterium, as one species, Chromobacterium Csp_P can inhibit DENV [37,38], the Plasmodium parasites that cause malaria [37,64], and is the basis of a promising new biopesticide [12]. We saw little evidence of Chromobacterium in the main data set, with only 15/92 samples infected. However, that genus was present in all 48 samples from the supplementary datasets, including one sample where Chromobacterium represented 81.29% of all reads, suggesting it may just have been underrepresented in the main dataset, or that there may be temporal or seasonal variations in its abundance. Finally, we saw no evidence of a native Wolbachia infection in our mosquitoes, with Wolbachia sequences detected in 13/92 samples at the low average abundance of 0.19%, likely suggestive of environmental contamination rather than a persistent infection, in line with previous studies of Wolbachia infection in Ae. aegypti [65].
Differences in mosquito microbiome composition due to aquatic habitat of origin
We were interested in determining whether adult female mosquitoes that emerged from six common Ae. aegypti container types found around Puerto Rico could be distinguished from each other based on differences in their microbiomes. We did observe high similarity between mosquito and water samples from the same container, with an average of 53.98% of all bacterial genera identified in container water also detected in mosquitoes. Neither NMDS analysis nor hierarchical clustering of mosquito microbial profiles revealed any stand-alone clusters where mosquitoes from a single container type clustered together in the absence of mosquitoes from another site type, suggesting that there were few major differences between the groups.
Similarly, PERMANOVA analysis indicated that the amount of variation in the dataset explained by differences between mosquitoes from different containers was approximately four times higher than variation between container types, suggesting that container type-specific factors only made minor contributions to microbial community composition. Nevertheless, the analysis suggested that the effect of these factors was still significant, with pairwise PERMANOVA comparisons revealing that this effect was primarily due to differences between plastic container-derived mosquitoes and those from all other container types. This observation was supported by Venn diagram analysis, which revealed plastic container mosquitoes were associated with the highest number of unique bacterial genera. Comparative analysis of the six container types identified a key group of 12 bacterial families that were differentially abundant between plastics mosquitoes and all others. Ten of these families were significantly more abundant in plastic container-derived mosquitoes. These had diverse characteristics including freshwater and marine species, three methanotrophs, a photosynthetic bacterium, and a thermophilic bacterium [66–70]. While of the two families associated with mosquitoes from the other five container types, one was aerobic and the other anaerobic [71,72].
Two of four of the analyses of microbial diversity that we performed suggested that the profiles of plastic container-derived mosquitoes were more diverse than those of other site types. This may reflect a real biological difference, as these containers tended to contain plants and soil, which could potentially be a source of increased bacterial diversity. It could also have been influenced by the fact that plastic containers were relocated before collection while others were not, which could have altered the microbial profile of the containers and mosquitoes within. Interestingly, our hierarchical clustering analysis placed mosquito samples from plastic containers in three distinct clusters, potentially suggesting that relocation did not have a significant impact on the mosquito microbiome. Tire-derived mosquitoes had the lowest diversity, reflecting a previous study that observed few unique bacterial taxa in Aedes and Culex mosquitoes that developed in tires [73]. Additionally, we were unable to control for the number of larvae in the different aquatic habitats, which could influence food availability and microbiome composition. Another potential explanation for the difference relates to the date of collection, as plastic container-derived mosquito and water samples were collected earlier in the collection window due to an inability to locate acceptable sites during the same time period as the other site types. However, it should be noted that the septic tank-derived mosquitoes that clustered with the plastic container-derived mosquitoes in the NMDS analysis were not collected in the same time period, nor were septic tanks relocated prior to collection, suggesting that the microbial differences that defined the plastics/septics NMDS cluster were unlikely to be due to either of these factors in isolation.
We had originally hypothesized that, given the presence of human-derived or associated bacteria, mosquitoes that emerged from septic tank water might be most likely to have a distinct microbiome. However, our analyses indicated that there were few bacterial OTUs exclusively associated with these mosquitoes. Both NMDS and Venn diagram analysis suggested that plastics container- and septic tank-derived mosquitoes were more similar to each other than mosquitoes from other site types. While pairwise PERMANOVA analysis indicated that the septic tank mosquito microbiome was only significantly different from mosquitoes collected from underground trash cans. We identified 44 families with significantly different abundance between septic tank- and trash can-derived mosquitoes, with many of these being anaerobic in nature, and these taxa could underlie biological differences between mosquitoes reared in septic tanks and those from other container types. The lack of distinction between septic tank-derived mosquitoes and other groups was particularly noteworthy given that septic tank water samples formed an isolated cluster in the NMDS analysis. Suggesting that while the septic tank water has a distinct microbiome to the other site types, these differences do not carry over to the mosquitoes that develop within it. Potentially, given the high degree of inter-sample variation we observed, differences could have been revealed if we had been able to sequence a greater number of samples from each type of aquatic habitat. Interestingly, septic tank water samples in the supplementary datasets did not cluster independently, which might indicate that not all septic tanks have unique microbial profiles, or potentially that there is temporal or seasonal variation in the septic tank microbiome. It is also possible that this difference was observed due to differences in sample collection protocols between the main and supplementary datasets.
The literature is split on the influence of geographic or collection location on the composition and diversity of the mosquito microbiome. Several studies have demonstrated the importance of these parameters [74,20,47,75,76], while others show no effect [39,77,78]. Other studies show high inter-individual and inter-site variation in the microbiome, in line with our results [39,21]. Our results provided little evidence to suggest that there were major differences in the Ae. aegypti microbiome due to container of origin. A recently published study of the microbiomes of container water and Ae. aegypti larvae in Guadeloupe and French Guiana observed minimal impact of container type on microbiome composition, but did see an effect of geographic origin on the water microbiome profile [79]. We did not attempt to assess the role of geography in our data as sampling of containers in different locations was not well controlled over time (see Table 1), and because most sites were collected from the San Juan region. The cause of this disparity between studies is currently unclear. It may reflect real biological differences between mosquito populations from different regions, but could also occur due to studies employing different methods of analyzing their data. There are likely numerous other factors that influence mosquito microbial community structure, including mosquito developmental stage, time post-eclosion in adult mosquitoes, genetic background, and behavioral differences, and in some circumstances, this could include the site of origin. Previous studies highlight the importance of environmental factors such as seasonality, which can affect community diversity and the abundance of some community members [74,80,81]. These factors may represent the cause of the 45% of the variation in community composition that was unexplained by the variables we measured in our study.
Mosquitoes as a biological filter
From a biological perspective, understanding the differences between host and environment can tell us what types of bacteria are more likely to infect mosquitoes, and help us to understand functional bacterial niches within a mosquito. Our NMDS analysis indicated that container water and mosquitoes have distinct microbial profiles [56,82]. We also observed greater diversity of container water in comparison with mosquito samples, with the average water sample containing approximately 2.5 times more bacterial families than the average mosquito sample. These findings are in line with previous studies that suggest mosquitoes filter out certain bacterial OTUs [56,21,83,45].
We observed significant differences in the abundance of certain bacterial OTUs between water and adult mosquito samples, with the vast majority of these (18/20 classes and 105/110 families) being more abundant in container water. Two classes, Gammaproteobacteria and Cyanophyceae were more highly represented in mosquitoes, while many others, most notably Sphingobacteriia, Opitutae, Alphaproteobacteria and Deltaproteobacteria were underrepresented compared to container water. Interestingly, in Aedes koreicus adults, Alphaproteobacteria were more abundant than in aquatic habitats [56], suggesting that there are species-specific differences in bacteria that associate with mosquitoes. Our results could suggest a predisposition for colonization of mosquito guts by Gammaproteobacteria and Cyanophyceae, and a tendency to filter out some taxa belonging to those other classes. Gammaproteobacteria, particularly the genera Enterobacter, Serratia and Pseudomonas, are well known members of the mosquito microbiome. Given the predominance of phylum Proteobacteria in mosquitoes [20,84,45,85] it would be interesting to understand the reason why Gammaproteobacteria are more abundant in adult mosquitoes, why Alpha-, Delta and Epsilonproteobacteria remain more abundant in the water, and why there was no difference in abundance in Betaproteobacteria between mosquitoes and water.
Volcano plot analysis indicated that only five bacterial families were more abundant (by relative proportion) in adult mosquito samples than in water samples: Micrococcaceae (Actinobacteria), Burkholderiaceae (Betaproteobacteria), Pseudomonadaceae and Xanthomonadaceae (both Gammaproteobacteria), and the unclassified Oscillatoriales Family (Cyanophyceae), with the first four of these being well described mosquito-associated bacterial families that are likely to be highly adept at colonizing Ae. aegypti [57]. At the genus level, we observed that a larger proportion of the water microbiome was held in common across container types than for adult mosquitoes. This likely reflects the lower diversity of mosquito microbiomes, but could also be seen as further evidence that many of the bacteria in the aquatic habitat are not capable of successfully colonizing a mosquito. When considering the different types of containers, we saw that microbiomes of plastic container- and septic tank-derived mosquitoes were more similar to their water than mosquitoes from other site types. These two groups also had a greater proportion of bacteria that were exclusively associated with mosquito samples, although these typically had low prevalence and abundance. These observations might indicate that the relationship between the aquatic habitats and mosquito microbiomes may differ depending on the container type or environmental factors, or that certain container habitats are more likely to have bacteria in the water that can also infect mosquitoes.
Summary
We observed that the microbiome of Ae. aegypti from Puerto Rico was rich in bacteria from the families Burkholderiaceae and Pseudomonadaceae, as well as an unclassified family from Order Oscillatoriales. However, key members of the microbiota found in other locations, including the genera Asaia, Elizabethkingia, and Serratia were either absent or present only at low abundance. We also observed that there were few major differences between mosquitoes from different container habitat types but did see some evidence of distinct and more diverse profiles for mosquitoes that developed in the water of large plastic containers. These observations should be validated over time, across changes in seasons and at a greater range of locations in Puerto Rico, but the data as they stand should serve as a platform for future investigations of mosquito-microbe interactions and vector competence with Ae. aegypti populations from this region. From a broader perspective, we have provided insight into the nature of mosquito-microbe interactions within aquatic habitats from prominent types of containers in this region. The next step is to determine why those particular taxa are present, and how they are interacting with their mosquito hosts on a functional level, as this will help us to better understand the biology of an organism that is critical to public health across the world.
Supplementary Material
Acknowledgements:
This work was funded by grants from the CDC (BAA 2017-N-18041) and USAID (AID-OAA-F-16-00096). This work has also been supported by the Bloomberg Philanthropies. The authors wish to thank María Roubert, Jesús Estudillo, Luis Rivera, Luis Perez, and Orlando Gonzales for their assistance with field work, Betzabel Flores, Glenda Gonzales, and Gilberto Santiago for technical support, Scot O’Dowd from Mr DNA (https://www.mrdnalab.com) for sequencing and data preparation services, and a special thanks to Luis Eduardo Martínez Villegas for helpful discussion of the manuscript, data analysis tools and microbial ecology. We thank the six anonymous reviewers for their helpful comments.
Funding
This work was funded by grants from the CDC (BAA 2017-N-18041) and USAID (AID-OAA-F-16-00096). This work has also been supported by the Bloomberg Philanthropies. CVT was supported by the Johns Hopkins Malaria Research Institutes’ postdoctoral fellowship
Footnotes
Conflicts of interest/Competing interests
The authors declare they have no competing interests.
Consent to participate
Verbal consent was solicited and obtained from home/property owners before entering and collecting mosquito and container water samples when mosquito aquatic habitats were identified on private property.
Code availability
R scripts are available in the supplementary materials.
Availability of data and material
All raw sequence data are available through google drive:
https://drive.google.com/drive/folders/17aUjQy8fwFOkepyDv0GRcHbtj5HHkaW5?usp=sharing
References
- 1.WHO (2017) Chikungunya. Fact Sheets [Google Scholar]
- 2.WHO (2018) Zika virus. Fact Sheets [Google Scholar]
- 3.WHO (2019) Dengue and severe dengue. Fact Sheets [Google Scholar]
- 4.Gould EA, Higgs S (2009) Impact of climate change and other factors on emerging arbovirus diseases. Trans R Soc Trop Med Hyg 103 (2):109–121. doi: 10.1016/j.trstmh.2008.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gubler DJ (2011) Dengue, Urbanization and Globalization: The Unholy Trinity of the 21(st) Century. Trop Med Health 39 (4 Suppl):3–11. doi: 10.2149/tmh.2011-S05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kraemer MU, Sinka ME, Duda KA, Mylne AQ, Shearer FM, Barker CM, Moore CG, Carvalho RG, Coelho GE, Van Bortel W, Hendrickx G, Schaffner F, Elyazar IR, Teng HJ, Brady OJ, Messina JP, Pigott DM, Scott TW, Smith DL, Wint GR, Golding N, Hay SI (2015) The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. Elife 4:e08347. doi: 10.7554/eLife.08347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilder-Smith A (2012) Dengue infections in travellers. Paediatr Int Child Health 32 Suppl 1:28–32. doi: 10.1179/2046904712Z.00000000050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilder-Smith A, Gubler DJ (2008) Geographic expansion of dengue: the impact of international travel. Med Clin North Am 92 (6):1377–1390, x. doi: 10.1016/j.mcna.2008.07.002 [DOI] [PubMed] [Google Scholar]
- 9.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI (2013) The global distribution and burden of dengue. Nature 496 (7446):504–507. doi: 10.1038/nature12060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ranson H, Lissenden N (2016) Insecticide Resistance in African Anopheles Mosquitoes: A Worsening Situation that Needs Urgent Action to Maintain Malaria Control. Trends Parasitol 32 (3):187–196. doi: 10.1016/j.pt.2015.11.010 [DOI] [PubMed] [Google Scholar]
- 11.Vontas J, Kioulos E, Pavlidi N, Morou E, della Torre A, Ranson H (2012) Insecticide resistance in the major dengue vectors Aedes albopictus and Aedes aegypti. Pesticide Biochemistry and Physiology 104 (2):126–131. doi: 10.1016/j.pestbp.2012.05.008 [DOI] [Google Scholar]
- 12.Caragata EP, Otero LM, Carlson JS, Borhani Dizaji N, Dimopoulos G (2020) A Nonlive Preparation of Chromobacterium sp. Panama (Csp_P) Is a Highly Effective Larval Mosquito Biopesticide. Appl Environ Microbiol 86 (11). doi: 10.1128/AEM.00240-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lacey LA (2007) Bacillus thuringiensis serovariety israelensis and Bacillus sphaericus for mosquito control. J Am Mosq Control Assoc 23 (2 Suppl):133–163. doi: 10.2987/8756-971X(2007)23[133:BTSIAB]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- 14.Shane JL, Grogan CL, Cwalina C, Lampe DJ (2018) Blood meal-induced inhibition of vector-borne disease by transgenic microbiota. Nat Commun 9 (1):4127. doi: 10.1038/s41467-018-06580-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S, Dos-Santos ALA, Huang W, Liu KC, Oshaghi MA, Wei G, Agre P, Jacobs-Lorena M (2017) Driving mosquito refractoriness to Plasmodium falciparum with engineered symbiotic bacteria. Science 357 (6358):1399–1402. doi: 10.1126/science.aan5478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryan PA, Turley AP, Wilson G, Hurst TP, Retzki K, Brown-Kenyon J, Hodgson L, Kenny N, Cook H, Montgomery BL, Paton CJ, Ritchie SA, Hoffmann AA, Jewell NP, Tanamas SK, Anders KL, Simmons CP, O’Neill SL (2019) Establishment of wMel Wolbachia in Aedes aegypti mosquitoes and reduction of local dengue transmission in Cairns and surrounding locations in northern Queensland, Australia. Gates Open Res 3:1547. doi: 10.12688/gatesopenres.13061.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zheng XY, Zhang DJ, Li YJ, Yang C, Wu Y, Liang X, Liang YK, Pan XL, Hu LC, Sun Q, Wang XH, Wei YY, Zhu J, Qian W, Yan ZQ, Parker AG, Gilles JRL, Bourtzis K, Bouyer J, Tang MX, Zheng B, Yu JS, Liu JL, Zhuang JJ, Hu ZG, Zhang MC, Gong JT, Hong XY, Zhang ZB, Lin LF, Liu QY, Hu ZY, Wu ZD, Baton LA, Hoffmann AA, Xi ZY (2019) Incompatible and sterile insect techniques combined eliminate mosquitoes. Nature 572 (7767):56–61. doi: 10.1038/s41586-019-1407-9 [DOI] [PubMed] [Google Scholar]
- 18.Guégan M, Zouache K, Demichel C, Minard G, Tran Van V, Potier P, Mavingui P, Valiente Moro C (2018) The mosquito holobiont: fresh insight into mosquito-microbiota interactions. Microbiome 6 (1):49. doi: 10.1186/s40168-018-0435-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caragata EP, Tikhe CV, Dimopoulos G (2019) Curious entanglements: interactions between mosquitoes, their microbiota, and arboviruses. Curr Opin Virol 37:26–36. doi: 10.1016/j.coviro.2019.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boissière A, Tchioffo MT, Bachar D, Abate L, Marie A, Nsango SE, Shahbazkia HR, Awono-Ambene PH, Levashina EA, Christen R, Morlais I (2012) Midgut microbiota of the malaria mosquito vector Anopheles gambiae and interactions with Plasmodium falciparum infection. PLoS Pathog 8 (5):e1002742. doi: 10.1371/journal.ppat.1002742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coon KL, Brown MR, Strand MR (2016) Mosquitoes host communities of bacteria that are essential for development but vary greatly between local habitats. Molecular Ecology 25 (22):5806–5826. doi: 10.1111/mec.13877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coon KL, Vogel KJ, Brown MR, Strand MR (2014) Mosquitoes rely on their gut microbiota for development. Molecular Ecology 23 (11):2727–2739. doi: 10.1111/mec.12771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valzania L, Martinson VG, Harrison RE, Boyd BM, Cooncurrency KL, Brown MR, Strand MR (2018) Both living bacteria and eukaryotes in the mosquito gut promote growth of larvae. Plos Neglected Tropical Diseases 12 (7). doi:ARTN e0006638 10.1371/journal.pntd.0006638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Correa MA, Matusovsky B, Brackney DE, Steven B (2018) Generation of axenic Aedes aegypti demonstrate live bacteria are not required for mosquito development. Nature Communications 9. doi:ARTN 4464 10.1038/s41467-018-07014-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duguma D, Hall MW, Rugman-Jones P, Stouthamer R, Terenius O, Neufeld JD, Walton WE (2015) Developmental succession of the microbiome of Culex mosquitoes. BMC Microbiol 15:140. doi: 10.1186/s12866-015-0475-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galeano-Castañeda Y, Bascunan P, Serre D, Correa MM (2020) Trans-stadial fate of the gut bacterial microbiota in Anopheles albimanus. Acta Trop 201:105204. doi: 10.1016/j.actatropica.2019.105204 [DOI] [PubMed] [Google Scholar]
- 27.Coon KL, Brown MR, Strand MR (2016) Gut bacteria differentially affect egg production in the anautogenous mosquito Aedes aegypti and facultatively autogenous mosquito Aedes atropalpus (Diptera: Culicidae). Parasites & Vectors 9. doi:ARTN 375 10.1186/s13071-016-1660-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaio AD, Gusmao DS, Santos AV, Berbert-Molina MA, Pimenta PFP, Lemos FJA (2011) Contribution of Midgut Bacteria to Blood Digestion and Egg Production in Aedes aegypti (Diptera: Culicidae) (L.). Parasites & Vectors 4. doi:Artn 105 10.1186/1756-3305-4-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma A, Dhayal D, Singh OP, Adak T, Bhatnagar RK (2013) Gut microbes influence fitness and malaria transmission potential of Asian malaria vector Anopheles stephensi. Acta Tropica 128 (1):41–47. doi: 10.1016/j.actatropica.2013.06.008 [DOI] [PubMed] [Google Scholar]
- 30.Barletta ABF, Nascimento-Silva MCL, Talyuli OAC, Oliveira JHM, Pereira LOR, Oliveira PL, Sorgine MHF (2017) Microbiota activates IMD pathway and limits Sindbis infection in Aedes aegypti. Parasites & Vectors 10. doi:ARTN 103 10.1186/s13071-017-2040-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xi Z, Ramirez JL, Dimopoulos G (2008) The Aedes aegypti toll pathway controls dengue virus infection. PLoS Pathog 4 (7):e1000098. doi: 10.1371/journal.ppat.1000098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan X, Zhou G, Wu J, Bian G, Lu P, Raikhel AS, Xi Z (2012) Wolbachia induces reactive oxygen species (ROS)-dependent activation of the Toll pathway to control dengue virus in the mosquito Aedes aegypti. Proc Natl Acad Sci U S A 109 (1):E23–31. doi: 10.1073/pnas.1116932108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dutra HL, Rocha MN, Dias FB, Mansur SB, Caragata EP, Moreira LA (2016) Wolbachia blocks currently circulating Zika virus isolates in Brazilian Aedes aegypti mosquitoes. Cell Host Microbe 19 (6):771–774. doi: 10.1016/j.chom.2016.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, Lu GJ, Pyke AT, Hedges LM, Rocha BC, Hall-Mendelin S, Day A, Riegler M, Hugo LE, Johnson KN, Kay BH, McGraw EA, van den Hurk AF, Ryan PA, O’Neill SL (2009) A Wolbachia symbiont in Aedes aegypti limits infection with Dengue, Chikungunya, and Plasmodium. Cell 139 (7):1268–1278. doi:Doi 10.1016/J.Cell.2009.11.042 [DOI] [PubMed] [Google Scholar]
- 35.Apte-Deshpande A, Paingankar M, Gokhale MD, Deobagkar DN (2012) Serratia odorifera a Midgut Inhabitant of Aedes aegypti Mosquito Enhances Its Susceptibility to Dengue-2 Virus. Plos One 7 (7). doi:ARTN e40401 10.1371/journal.pone.0040401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Apte-Deshpande AD, Paingankar MS, Gokhale MD, Deobagkar DN (2014) Serratia odorifera mediated enhancement in susceptibility of Aedes aegypti for Chikungunya virus. Indian Journal of Medical Research 139:762–768 [PMC free article] [PubMed] [Google Scholar]
- 37.Ramirez JL, Short SM, Bahia AC, Saraiva RG, Dong Y, Kang S, Tripathi A, Mlambo G, Dimopoulos G (2014) Chromobacterium Csp_P reduces malaria and dengue infection in vector mosquitoes and has entomopathogenic and in vitro anti-pathogen activities. PLoS Pathog 10 (10):e1004398. doi: 10.1371/journal.ppat.1004398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saraiva RG, Fang J, Kang S, Anglero-Rodriguez YI, Dong Y, Dimopoulos G (2018) Aminopeptidase secreted by Chromobacterium sp. Panama inhibits dengue virus infection by degrading the E protein. PLoS Negl Trop Dis 12 (4):e0006443. doi: 10.1371/journal.pntd.0006443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bennett KL, Gomez-Martinez C, Chin Y, Saltonstall K, McMillan WO, Rovira JR, Loaiza JR (2019) Dynamics and diversity of bacteria associated with the disease vectors Aedes aegypti and Aedes albopictus. Sci Rep 9 (1):12160. doi: 10.1038/s41598-019-48414-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.David MR, Santos LM, Vicente AC, Maciel-de-Freitas R (2016) Effects of environment, dietary regime and ageing on the dengue vector microbiota: evidence of a core microbiota throughout Aedes aegypti lifespan. Mem Inst Oswaldo Cruz 111 (9):577–587. doi: 10.1590/0074-02760160238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hegde S, Khanipov K, Albayrak L, Golovko G, Pimenova M, Saldana MA, Rojas MM, Hornett EA, Motl GC, Fredregill CL, Dennett JA, Debboun M, Fofanov Y, Hughes GL (2018) Microbiome Interaction Networks and Community Structure From Laboratory-Reared and Field-Collected Aedes aegypti, Aedes albopictus, and Culex quinquefasciatus Mosquito Vectors. Front Microbiol 9:2160. doi: 10.3389/fmicb.2018.02160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yadav KK, Bora A, Datta S, Chandel K, Gogoi HK, Prasad GB, Veer V (2015) Molecular characterization of midgut microbiota of Aedes albopictus and Aedes aegypti from Arunachal Pradesh, India. Parasit Vectors 8:641. doi: 10.1186/s13071-015-1252-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Segata N, Baldini F, Pompon J, Garrett WS, Truong DT, Dabiré RK, Diabaté A, Levashina EA, Catteruccia F (2016) The reproductive tracts of two malaria vectors are populated by a core microbiome and by gender-and swarm-enriched microbial biomarkers. Scientific reports 6 (1):1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villegas LM, Pimenta PFP (2014) Metagenomics, paratransgenesis and the Anopheles microbiome: a portrait of the geographical distribution of the anopheline microbiota based on a meta-analysis of reported taxa. Memórias do Instituto Oswaldo Cruz 109 (5):672–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Gilbreath TM 3rd, Kukutla P, Yan G, Xu J (2011) Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya. PLoS One 6 (9):e24767. doi: 10.1371/journal.pone.0024767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dickson LB, Ghozlane A, Volant S, Bouchier C, Ma L, Vega-Rua A, Dusfour I, Jiolle D, Paupy C, Mayanja MN, Kohl A, Lutwama JJ, Duong V, Lambrechts L (2018) Diverse laboratory colonies of Aedes aegypti harbor the same adult midgut bacterial microbiome. Parasit Vectors 11 (1):207. doi: 10.1186/s13071-018-2780-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Buck M, Nilsson LKJ, Brunius C, Dabire RK, Hopkins R, Terenius O (2016) Bacterial associations reveal spatial population dynamics in Anopheles gambiae mosquitoes. Scientific Reports 6. doi:ARTN 22806 10.1038/srep22806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yee DA, Allgood D, Kneitel J, Kuehn K (2012) Constitutive differences between natural and artificial container mosquito habitats: vector communities, resources, microorganisms, and habitat parameters. Journal of medical entomology 49 (3):482–491 [DOI] [PubMed] [Google Scholar]
- 49.Barrera R, Amador M, Diaz A, Smith J, Munoz-Jordan J, Rosario Y (2008) Unusual productivity of Aedes aegypti in septic tanks and its implications for dengue control. Medical and veterinary entomology 22 (1):62–69 [DOI] [PubMed] [Google Scholar]
- 50.Burke R, Barrera R, Lewis M, Kluchinsky T, Claborn D (2010) Septic tanks as larval habitats for the mosquitoes Aedes aegypti and Culex quinquefasciatus in Playa-Playita, Puerto Rico. Medical and veterinary entomology 24 (2):117–123 [DOI] [PubMed] [Google Scholar]
- 51.Irving-Bell R, Okoli E, Diyelong D, Lyimo E, Onyia O (1987) Septic tank mosquitoes: competition between species in central Nigeria. Medical and veterinary entomology 1 (3):243–250 [DOI] [PubMed] [Google Scholar]
- 52.Glassing A, Dowd SE, Galandiuk S, Davis B, Jorden JR, Chiodini RJ (2015) Changes in 16s RNA Gene Microbial Community Profiling by Concentration of Prokaryotic DNA. J Microbiol Methods 119:239–242. doi: 10.1016/j.mimet.2015.11.001 [DOI] [PubMed] [Google Scholar]
- 53.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodriguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Kang KB, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS 2nd, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vazquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG (2019) Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37 (8):852–857. doi: 10.1038/s41587-019-0209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP (2016) DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13 (7):581–583. doi: 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hornung BVH, Zwittink RD, Kuijper EJ (2019) Issues and current standards of controls in microbiome research. FEMS Microbiol Ecol 95 (5). doi: 10.1093/femsec/fiz045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alfano N, Tagliapietra V, Rosso F, Manica M, Arnoldi D, Pindo M, Rizzoli A (2019) Changes in Microbiota Across Developmental Stages of Aedes koreicus, an Invasive Mosquito Vector in Europe: Indications for Microbiota-Based Control Strategies. Front Microbiol 10:2832. doi: 10.3389/fmicb.2019.02832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gendrin M, Christophides GK (2013) The Anopheles mosquito microbiota and their impact on pathogen transmission. In: Anopheles mosquitoes-New insights into malaria vectors. IntechOpen, [Google Scholar]
- 58.Hughes GL, Garay JAR, Koundal V, Rasgon JL, Mwangi MM (2016) Genome sequence of Stenotrophomonas maltophilia strain SmAs1, isolated from the Asian malaria mosquito Anopheles stephensi. Genome Announc 4 (2):e00086–00016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scolari F, Casiraghi M, Bonizzoni M (2019) Aedes spp. and their microbiota: a review. Frontiers in microbiology 10:2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bando H, Okado K, Guelbeogo WM, Badolo A, Aonuma H, Nelson B, Fukumoto S, Xuan X, Sagnon NF, Kanuka H (2013) Intra-specific diversity of Serratia marcescens in Anopheles mosquito midgut defines Plasmodium transmission capacity. Scientific reports 3:1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen S, Blom J, Walker ED (2017) Genomic, physiologic, and symbiotic characterization of Serratia marcescens strains isolated from the mosquito Anopheles stephensi. Frontiers in microbiology 8:1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cirimotich CM, Dong Y, Clayton AM, Sandiford SL, Souza-Neto JA, Mulenga M, Dimopoulos G (2011) Natural microbe-mediated refractoriness to Plasmodium infection in Anopheles gambiae. Science 332 (6031):855–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu P, Sun P, Nie K, Zhu Y, Shi M, Xiao C, Liu H, Liu Q, Zhao T, Chen X (2019) A gut commensal bacterium promotes mosquito permissiveness to arboviruses. Cell host & microbe 25 (1):101–112. e105 [DOI] [PubMed] [Google Scholar]
- 64.Saraiva RG, Huitt-Roehl CR, Tripathi A, Cheng YQ, Bosch J, Townsend CA, Dimopoulos G (2018) Chromobacterium spp. mediate their anti-Plasmodium activity through secretion of the histone deacetylase inhibitor romidepsin. Sci Rep 8 (1):6176. doi: 10.1038/s41598-018-24296-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ross PA, Callahan AG, Yang Q, Jasper M, Arif MAK, Afizah AN, Nazni WA, Hoffmann AA (2020) An elusive endosymbiont: Does Wolbachia occur naturally in Aedes aegypti? Ecol Evol 10 (3):1581–1591. doi: 10.1002/ece3.6012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kalyuhznaya MG, Martens-Habbena W, Wang T, Hackett M, Stolyar SM, Stahl DA, Lidstrom ME, Chistoserdova L (2009) Methylophilaceae link methanol oxidation to denitrification in freshwater lake sediment as suggested by stable isotope probing and pure culture analysis. Environmental microbiology reports 1 (5):385–392 [DOI] [PubMed] [Google Scholar]
- 67.Lee K-B, Liu C-T, Anzai Y, Kim H, Aono T, Oyaizu H (2005) The hierarchical system of the ‘Alphaproteobacteria’: description of Hyphomonadaceae fam. nov., Xanthobacteraceae fam. nov. and Erythrobacteraceae fam. nov. International Journal of Systematic and Evolutionary Microbiology 55 (5):1907–1919 [DOI] [PubMed] [Google Scholar]
- 68.Levett PN (2015) Systematics of Leptospiraceae. In: Leptospira and Leptospirosis. Springer, pp 11–20 [DOI] [PubMed] [Google Scholar]
- 69.Neuenschwander SM, Ghai R, Pernthaler J, Salcher MM (2018) Microdiversification in genome-streamlined ubiquitous freshwater Actinobacteria. The ISME journal 12 (1):185–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stein LY, Roy R, Dunfield PF (2001) Aerobic methanotrophy and nitrification: processes and connections. e LS [Google Scholar]
- 71.Ravcheev DA, Gerasimova AV, Mironov AA, Gelfand MS (2007) Comparative genomic analysis of regulation of anaerobic respiration in ten genomes from three families of gamma-proteobacteria (Enterobacteriaceae, Pasteurellaceae, Vibrionaceae). BMC genomics 8 (1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Takahashi Y, Matsumoto A, Morisaki K, Ōmura S (2006) Patulibacter minatonensis gen. nov., sp. nov., a novel actinobacterium isolated using an agar medium supplemented with superoxide dismutase, and proposal of Patulibacteraceae fam. nov. International journal of systematic and evolutionary microbiology 56 (2):401–406 [DOI] [PubMed] [Google Scholar]
- 73.Kim C-H, Lampman RL, Muturi EJ (2015) Bacterial communities and midgut microbiota associated with mosquito populations from waste tires in East-Central Illinois. Journal of medical entomology 52 (1):63–75 [DOI] [PubMed] [Google Scholar]
- 74.Akorli J, Gendrin M, Pels NAP, Yeboah-Manu D, Christophides GK, Wilson MD (2016) Seasonality and locality affect the diversity of Anopheles gambiae and Anopheles coluzzii midgut microbiota from Ghana. PloS one 11 (6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gimonneau G, Tchioffo MT, Abate L, Boissiere A, Awono-Ambene PH, Nsango SE, Christen R, Morlais I (2014) Composition of Anopheles coluzzii and Anopheles gambiae microbiota from larval to adult stages. Infection Genetics and Evolution 28:715–724. doi: 10.1016/j.meegid.2014.09.029 [DOI] [PubMed] [Google Scholar]
- 76.Muturi EJ, Lagos-Kutz D, Dunlap C, Ramirez JL, Rooney AP, Hartman GL, Fields CJ, Rendon G, Kim C-H (2018) Mosquito microbiota cluster by host sampling location. Parasites & vectors 11 (1):468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Osei-Poku J, Mbogo C, Palmer W, Jiggins F (2012) Deep sequencing reveals extensive variation in the gut microbiota of wild mosquitoes from Kenya. Molecular ecology 21 (20):5138–5150 [DOI] [PubMed] [Google Scholar]
- 78.Zoure AA, Sare AR, Yameogo F, Somda Z, Massart S, Badolo A, Francis F (2020) Bacterial communities associated with the midgut microbiota of wild Anopheles gambiae complex in Burkina Faso. Molecular biology reports 47 (1):211–224 [DOI] [PubMed] [Google Scholar]
- 79.Hery L, Guidez A, Durand AA, Delannay C, Normandeau-Guimond J, Reynaud Y, Issaly J, Goindin D, Legrave G, Gustave J, Raffestin S, Breurec S, Constant P, Dusfour I, Guertin C, Vega-Rua A (2020) Natural Variation in Physicochemical Profiles and Bacterial Communities Associated with Aedes aegypti Breeding Sites and Larvae on Guadeloupe and French Guiana. Microb Ecol. doi: 10.1007/s00248-020-01544-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Duguma D, Hall MW, Smartt CT, Neufeld JD (2017) Temporal variations of microbiota associated with the immature stages of two Florida Culex mosquito vectors. Microbial ecology 74 (4):979–989 [DOI] [PubMed] [Google Scholar]
- 81.Novakova E, Woodhams DC, Rodríguez-Ruano SM, Brucker RM, Leff JW, Maharaj A, Amir A, Knight R, Scott J (2017) Mosquito microbiome dynamics, a background for prevalence and seasonality of West Nile virus. Frontiers in microbiology 8:526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Duguma D, Rugman-Jones P, Kaufman MG, Hall MW, Neufeld JD, Stouthamer R, Walton WE (2013) Bacterial communities associated with Culex mosquito larvae and two emergent aquatic plants of bioremediation importance. PLoS One 8 (8) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dada N, Jumas-Bilak E, Manguin S, Seidu R, Stenström T-A, Overgaard HJ (2014) Comparative assessment of the bacterial communities associated with Aedes aegypti larvae and water from domestic water storage containers. Parasites & vectors 7 (1):391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rani A, Sharma A, Rajagopal R, Adak T, Bhatnagar RK (2009) Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi - an Asian malarial vector. BMC microbiology 9 (1):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zouache K, Raharimalala FN, Raquin V, Tran-Van V, Raveloson LHR, Ravelonandro P, Mavingui P (2011) Bacterial diversity of field-caught mosquitoes, Aedes albopictus and Aedes aegypti, from different geographic regions of Madagascar. FEMS Microbiology Ecology 75 (3):377–389 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw sequence data are available through google drive:
https://drive.google.com/drive/folders/17aUjQy8fwFOkepyDv0GRcHbtj5HHkaW5?usp=sharing