

Abstract
Argonaute proteins (AGOs) are loaded with small RNAs as guides to recognize target mRNAs. Since the target specificity heavily depends on the base complementarity between two strands, it is important to identify small guide and long target RNAs bound to AGOs. For this purpose, next-generation sequencing (NGS) technologies have extended our appreciation truly to the nucleotide level. However, identification of RNAs via NGS from scarce RNA samples remains a challenge. Further, most commercial and published methods are compatible with either small RNAs or long RNAs, but are not equally applicable to both. Therefore, a single method that yields quantitative, bias-free NGS libraries to identify small and long RNAs from low levels of input will be of wide interest. Here, we introduce such a procedure that is based several modifications of two published protocols and allows robust, sensitive and reproducible cloning and sequencing of small amounts of RNAs of variable lengths. The method was applied to identification of the small RNAs bound to a purified eukaryotic AGO. Following ligation of a DNA adapter to RNA 3’-end, the key feature of this method is to use the adapter for priming reverse transcription (RT) wherein biotinylated deoxyribonucleotides specifically incorporated into the extended complementary DNA. Such RT products are enriched on streptavidin beads, circularized while immobilized on beads and directly used for PCR amplification. We provide a step-wise guide to generate RNA-Seq libraries, their purification, quantification, validation and preparation for next-generation sequencing. We also provide basic steps in post-NGS data analyses using Galaxy, an open-source, web-based platform.
Keywords: small RNAs, Argonaute, Next-generation sequencing, biotinylated dNTPs, low RNA input
1. Introduction
Small RNAs are non-coding RNAs critical for most eukaryotes across animals, plants, and fungi. They are classified into microRNAs, small interfering RNAs and Piwi-interacting RNAs based on their structure and function[1]. These RNAs range in size from ~19–25 nucleotides (nt) and participate in a wide variety of cellular processes such as heterochromatin formation, messenger RNA destabilization, translational repression and transposon silencing[2,3]. Small RNAs function within ribonucleoprotein complexes called RNA-induced silencing complex (RISC) following their incorporation into Argonaute proteins (AGOs)[4]. The loaded RNA serves as guide to recruit the RISC to target RNAs with full or partial sequence complementarity. In the last decade, next-generation sequencing (NGS) has illuminated the significance of the base complementarity between the guide and target strands at nucleotide level. This technology has enabled researchers to gain a greater understanding of small RNAs found in different biological samples as well as to reveal diversity of small RNA sequences bound to the purified AGOs[5–9]. NGS based approaches have also enabled high-throughput detection of long target RNAs[10]. Concurrently, strides are being made in expression and purification of recombinant AGOs for their biochemical and structural analysis. When purified from heterologous sources, AGOs co-purify small RNAs; identification and analysis of such RNAs holds promise for unveiling the underlying principles of guide RNA selection by AGOs[11].
Here, we present a method for preparation of NGS libraries from small RNAs that co-purify with a yeast AGO. The described workflow is primarily based on a previously published method that extensively optimized each individual enzymatic step in RNA-Seq library preparation[12]. The specificity and sensitivity of the method was further improved by incorporating a feature from another kit-free RNA-Seq library preparation approach wherein complementary DNAs (cDNAs) from adapter-ligated small RNAs are specifically enriched using biotinylated deoxyribonucleotides (dNTPs)[13]. Since this chapter aims for small RNA-seq, the protocol does not include RNA fragmentation step that is required for NGS of long RNAs. Note that, however, this method is applicable to longer RNAs purified from any biological source, via a broad range of approaches such as RNA-protein immunoprecipitation, ribosome footprinting, and is even suitable for extra-cellular RNAs. In cases where long RNAs may need to be fragmented to input into this procedure, chemical methods of RNA fragmentation are much more desirable over enzymatic methods[14,15].
A schematic of the whole procedure is depicted in Figure 1. Isolated RNAs are first enzymatically treated to generate a 3’-hydroxyl group to allow ligation to a pre-adenylated DNA adapter. The adaptor-ligated-RNAs are used to perform reverse transcription (RT) using biotinylated dNTPs and one of the twelve RT primers to produce biotinylated cDNAs containing a specific 5-nucleotide barcode (Table 1). Another feature of the RT primers is the inclusion of five random nucleotides at positions that will be the first five nucleotides sequenced (see Figure 3D). This random sequence ensures sequence complexity during first few sequencing cycles, and also allows for removal of any sequencing reads that arise due to PCR duplication. Following RT, reaction is run on a gel, the desired product is extracted and enriched via streptavidin pulldown. The purified RT product is then circularized where the 3’-end is ligated to the 5’-end by circLigase, and the circular molecule serves as a template for PCRs. We describe the utility of small scale PCRs to determine the appropriate number of cycles for production of the highest concentration of libraries without accumulation of nonspecific PCR products. Large scale PCR under the determined conditions specifically amplifies the cDNA libraries for NGS. The purified PCR product then undergoes series of quantification and quality control steps before submission for NGS. After testing serially diluted synthetic RNAs as input, we have determined that our protocol can successfully generate complex cDNA libraries with as low as 0.01 pmoles of RNA.
Figure 1.
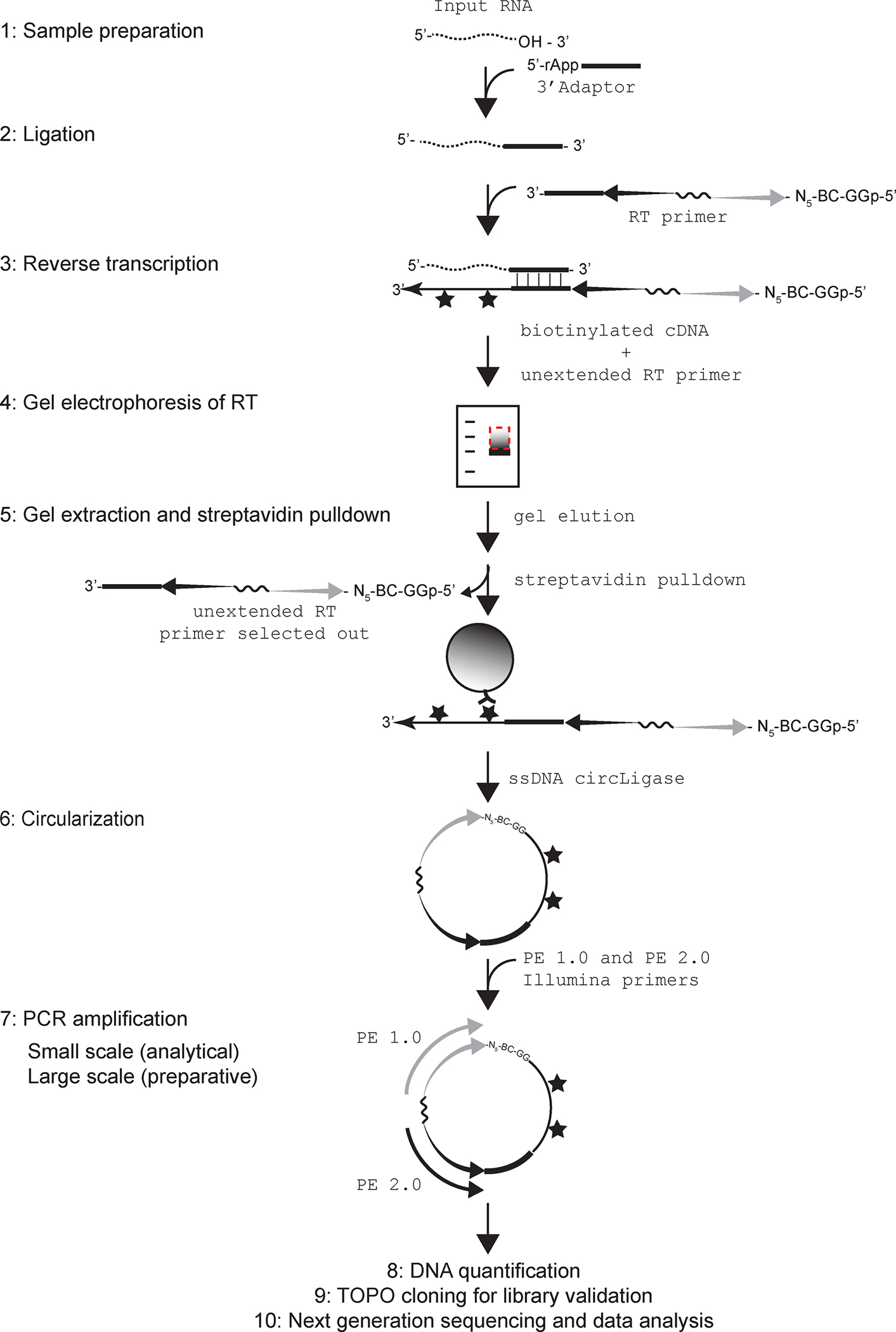
A schematic of the steps in the NGS library prep procedure. 1: Dotted wavy line depicts input RNA with a 3’ hydroxyl. 2: Pre-adenylated 3’ adaptor is shown as a thick black line ligated to RNA 3’-end. 3: 3’ adapter complementary sequences in the RT primer is as thick black line. In the RT primer, sequence corresponding to forward sequencing primer is represented by a grey tapered arrow, and reverse primer is shown as a black tapered arrow. The hexaethylene glycol spacer SP18 that connects the forward and reverse primers is shown as a solid wavy line. At the 5’ end of the forward primer are a random 5-mer, the 5-nt TruSeq barcode sequence (BC) and two Gs. The 5’ phosphate on the terminal G is also shown. Following RT, the incorporated biotinylated dNTPs are represented by stars. 4: Dotted box marks the area of the gel that is excised out for elution of the RT product. 5: The biotinylated RT product is shown captured on the streptavidin conjugated magnetic beads (grey sphere) whereas unextended RT primer is shown to be selected away. 6: The circularized RT product is shown where the G at the 5’ terminus of the forward primer end is shown ligated to the 3’ end of the extended cDNA. 7: Illumina primers PE 1.0 and PE 2.0 are as grey and black arrows, respectively, and are shown bound to their complementary sequences in the circularized RT product. Note that biotinylated nucleotides in the cDNA do not interfere with PCR amplification. 8, 9 and 10 describe main steps post cDNA library preparation.
Table 1.
Top: Name and full sequence of each of the twelve RT oligos used in step 3.3. The hexaethylene glycol spacer SP18 and 5’−3’ orientation of DNA sequences linked on its each end are indicated. The barcode sequence is underlined. Bottom: Name and full sequence of the PCR oligos used for amplification of cDNA libraries in step 3.7. The position of the phosphorothioate linkage in each primer is shown.
| RT primer sequences | |
|---|---|
| Name | Sequence |
| TruSeq_SE1 | 5’ - pGGCACTANNNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT-SPACER 18-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCTTGGCACCCGAGAATTCCA – 3’ |
| TruSeq_SE2 | 5’ - pGGGTAGCNNNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT-SPACER 18-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCTTGGCACCCGAGAATTCCA – 3’ |
| TruSeq_SE3 | 5’ - pGGTCGATNNNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT-SPACER 18-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCTTGGCACCCGAGAATTCCA – 3’ |
| TruSeq_SE4 | 5’ - pGGCCTCGNNNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT-SPACER 18-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCTTGGCACCCGAGAATTCCA – 3’ |
| TruSeq_SE5 | 5’ - pGGTGACANNNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT-SPACER 18-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCTTGGCACCCGAGAATTCCA – 3’ |
| TruSeq_SE6 | 5’ - pGGTAGACNNNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT-SPACER 18-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCTTGGCACCCGAGAATTCCA – 3’ |
| TruSeq_SE7 | 5’ - pGGGCCCTNNNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT-SPACER 18-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCTTGGCACCCGAGAATTCCA – 3’ |
| TruSeq_SE8 | 5’ - pGGATCGGNNNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT-SPACER 18-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCTTGGCACCCGAGAATTCCA – 3’ |
| TruSeq_SE9 | 5’ - pGGACTGANNNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT-SPACER 18-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCTTGGCACCCGAGAATTCCA – 3’ |
| TruSeq_SE10 | 5’ - pGGTGTTCNNNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT-SPACER 18-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCTTGGCACCCGAGAATTCCA – 3’ |
| TruSeq_SE11 | 5’ - pGGTAAGTNNNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT-SPACER 18-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCTTGGCACCCGAGAATTCCA – 3’ |
| TruSeq_SE12 | 5’ - pGGAGATGNNNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT-SPACER 18-CTCGGCATTCCTGCTGAACCGCTCTTCCGATCTCCTTGGCACCCGAGAATTCCA – 3’ |
| PCR Primers (Illumina PE primers; * indicates location of phosphorothioate bond) | |
| Name | Sequence |
| PE1.0 PCR Primer | 5’–AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC*T–3’ |
| PE2.0 PCR Primer | 5’–CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATC*T–3’ |
Figure 3.
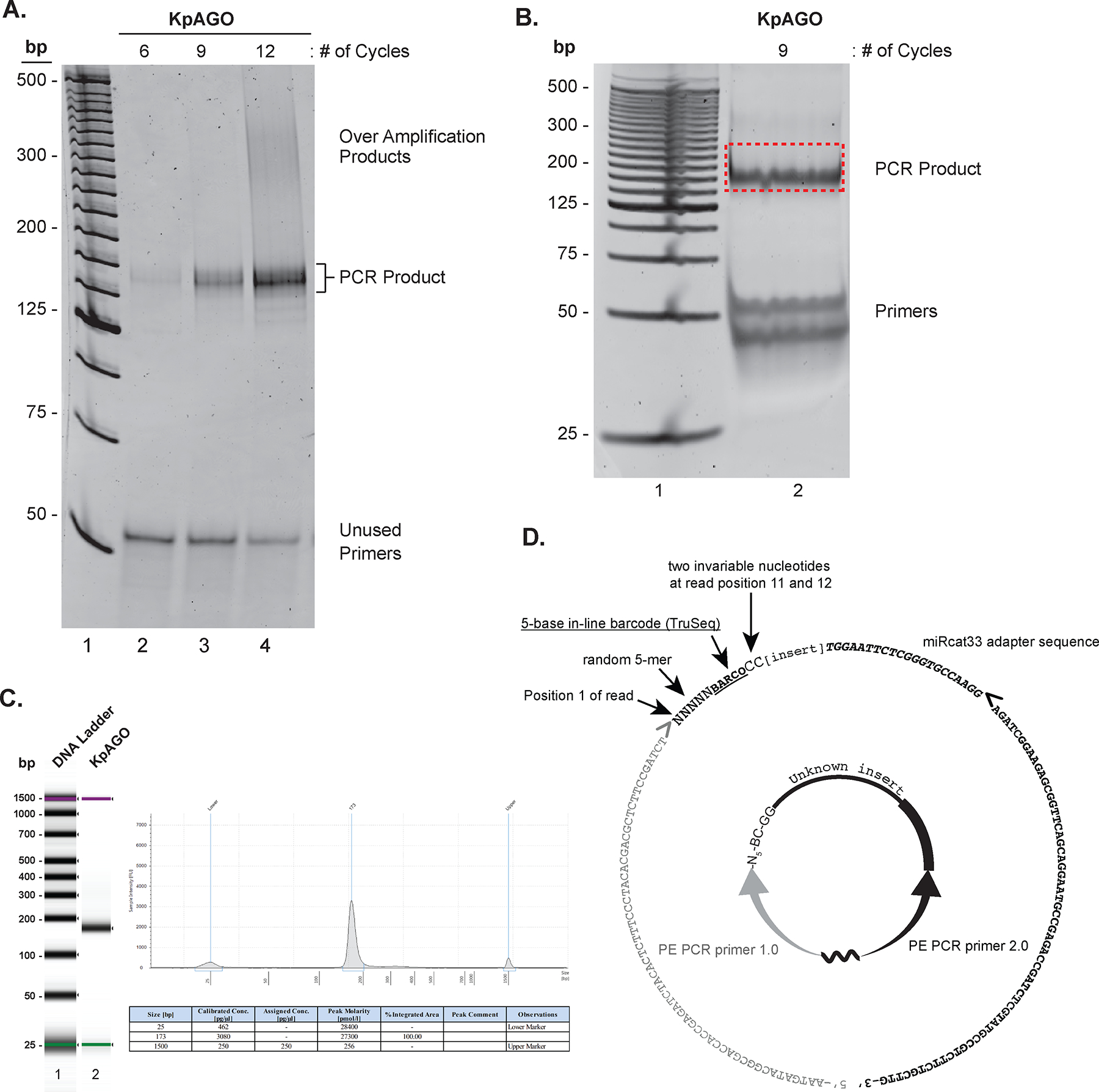
PCR amplification and size estimation of NGS libraries. A. SYBR® gold stained 8% nondenaturing PAGE-gel showing specific PCR products, over-amplification products, and unused primers (lanes 2–4; each species is indicated on the right) in small scale PCRs performed for increasing number of cycles indicated on top of each lane. The 25-bp DNA ladder is in lane 1. B. SYBR® gold stained 8% nondenaturing PAGE showing the product band and unused primers (labeled on the right) from large scale PCR (lane 2). The 25-bp DNA ladder is in lane 1. The gel fragment excised to purify DNA for NGS is indicated vy a red-dotted rectangle. C. The Bioanalyzer report of the purified PCR product from B. On the left is a virtual gel image of the DNA ladder (lane 1) and cDNA library of KpAGO-bound RNAs (lane 2). On the right is the histogram showing the size of the cDNA library along with size markers (top) and a table with summary of size and amount quantification of the library (bottom). Note that while Bioanalyzer analysis also provides sample concentration (see table), fluorescent DNA binding dye based quantification provides more accurate quantification of DNA amount. D. The circularized cDNA product as in Figure 1 is shown in the center. The corresponding sequence of the final PCR product in 5’ to 3’ direction is shown as the outer circle, and its different segments are individually labeled. Right and left pointing angled brackets indicate the start position of DNA synthesis during PCR. The grey right-pointing angled bracket also indicates where the Illumina first-read sequencing primer begins extension.
2. Materials
Equipment and general supplies
-
1
Thermocycler
-
2
Blue light transilluminator (VWR)
-
3
Typhoon scanner (or a similar imager compatible with phosphorescent screens and fluorescent DNA binding stains)
-
4
Phoshor-imager screens and cassettes
-
5
Gel dryer (for vacuum and heat-assisted gel drying)
-
6
Access to Qubit fluorometer and Bioanalyzer or TapeStation
-
7
0.2 ml PCR tubes
-
8
Vertical gel electrophoresis:
Apparatus (C.B.S. scientific)
Metal heat sink (C.B.S. scientific)
Large glass plates, 20 cm W x 28 cm H (C.B.S. scientific)
Short glass plates, 20 cm W x 10 cm H (Moliterno)
Spacers, 0.15 cm (C.B.S. scientific)
Spacers, 0.1cm (C.B.S. scientific)
Combs
8-well, 0.15 cm thick (C.B.S. scientific)
16-well, 0.1 cm thick (C.B.S. scientific)
8-well, 0.1 cm thick (C.B.S. scientific)
-
9
0.5× Tris/Borate/EDTA buffer (5× TBE: 54 g of Tris base, 27.5 g of boric acid, 20 ml of 0.5 M EDTA (pH 8.0))
-
10
40% (w/v) acrylamide:bisacrylamide (29:1 Accugel, National Diagnostics)
-
11
TEMED
-
12
10% Ammonium persulphate (APS)
-
13
SYBR® gold nucleic acid gel stain (ThermoFisher Scientific)
-
14
25 bp DNA ladder (Invitrogen)
-
15
Glass baking tray for gel staining
-
16
Standard clear sheet protector (e.g. Staples)
-
17
Razor blades (VWR)
-
18
Luer-lok™ Tip syringes (3 ml and 10 ml) and needles (22 guage)
-
19
Magnetic rack (e.g. DynaMag™−2; ThermoFisher Scientific)
-
20
Corning® Costar® Spin-X columns (Sigma Aldrich)
-
21
RNase-free water (ThermoFisher Scientific)
-
22
10 μg/μl Glycogen (ThermoFisher Scientific)
-
23
100% Ethanol (200 proof)
Materials to be prepared ahead of time in lab:
-
24
3 M sodium acetate (pH 5.2)
-
25
2 M magnesium chloride (MgCl2)
-
26
2× Denaturing Load Buffer: 3 ml 5× TBE, 1.8 g Ficoll Type 400, 6.3 g Urea, 3 mg bromophenol blue, 3 mg xylene cyanol, up to 15 ml ddH20. To get into solution, place tube in water in a beaker and boil on hot plate for 10–15 minutes. Add dyes after adjusting the volume to 15 ml. Store at 4 °C. Before use, heat to 50–60 °C to fully solubilize contents if necessary.
2.1. RNA end curing and quantification
-
1
10× CutSmart® buffer (NEB)
-
2
Calf intestine phosphatase (NEB)
-
3
Acid Phenol mix (Phenol:chloroform:iso-amyl alcohol, pH 4.5; ThermoFisher Scientific)
-
4
Low molecular weight single-stranded (ss) DNA ladder (Alfa Aesar)
-
5
Synthetic RNA oligo (any 20–40 nucleotide long sequence)
-
6
1 mM ATP
-
7
γ32P-ATP 3000Ci/mmol (Perkin Elmer)
-
8
10× T4 Polynucleotide Kinase (PNK) buffer (NEB)
-
9
T4 PNK enzyme (NEB)
-
10
Saran wrap
-
11
3M Whatman paper
-
12
Items 7–10 from the equipment and general supplies section.
Materials to be prepared ahead of time in lab:
-
13
20% Urea-PAGE gel mix (20% acrylamide:bisacrylamide (stock: 40% (w/v)), 6M Urea, 0.5× TBE)
2.2. Ligation
50% PEG8000 (NEB; PEG8000 is currently supplied with T4RNL2 Tr. K227Q)
10× Ligation Buffer (10× T4RNL2 Tr. K227Q Buffer; NEB)
T4RNL2 Tr. K227Q enzyme (NEB)
20 mM DTT
-
7 μM Pre-adenylated adaptor (miRCat-33® Conversion Kit from IDT) for single-end Sequencing:
5’ – rAppTGGAATTCTCGGGTGCCAAGGddC – 3’ (see Note 6)
2.3. Reverse Transcription
-
1
100 mM dATP, dCTP, dTTP, dGTP (NEB)
-
2
100 mM DTT
-
3
1 mM Biotin-dATP (Metkinen Biotin-11-dATP or PerkinElmer Biotin-11-dATP,)
-
4
5 mM Biotin-dCTP (Trilink Biotin-16-AA-2’dCTP)
-
5
10 μM TruSeq RT primers (see Table 1 for sequences)
-
6
Superscript III (SSIII) reverse transcriptase (Invitrogen)
Materials to be prepared ahead of time in lab:
-
7
5× First-Strand Buffer w/o MgCl2: 250 mM Tris-HCl (pH 8.3 at room temp), 375 mM KCl
-
8
4× dNTP mix: dGTP- 0.25mM; dTTP- 0.25 mM; dATP- 0.175 mM; biotin-dATP- 0.075mM; dCTP- 0.1625 mM; biotin-dCTP- 0.0875 mM
2.4. Gel electrophoresis of RT product
-
1
Items 6–14 from the equipment and general supplies section.
Materials to be prepared ahead of time in lab:
-
2
10% Urea-PAGE gel mix (10% acrylamide:bisacrylamide (stock: 40% (w/v)), 6M Urea, 0.5× TBE)
-
3
SYBR® gold staining solution: 2 μl of 10000× SYBR® gold in 200 ml of 0.5× TBE
2.5. Gel elution and streptavidin pulldown
-
1
Items 16–19 from the equipment and general supplies section.
-
2
Hydrophilic Streptavidin magnetic beads (NEB)
Materials to be prepared ahead of time in lab:
-
3
DNA Elution Buffer: 300 mM NaCl, 1 mM EDTA
-
4
Streptavidin bead wash buffer: 0.5 M NaOH, 20mM Tris-HCl pH 7.5, 1mM EDTA
-
5
Streptavidin bead resuspension buffer: 10 mM Tris HCl pH7.5, 0.1 mM EDTA, 0.3 M NaCl
2.6. Circularization
CircLigase™ reaction kit (CircLigase reaction buffer, CircLigase enzyme and 50 mM MnCl2) (Epicentre Biotechnologies)
1 mM ATP
5 M Betaine solution (SigmaAldrich)
2.7. PCR amplification
Phosphorothioate Illumina PE1.0 and PE2.0 primers (see Table 1 for sequences)
5× Q5 reaction buffer (NEB)
Q5 DNA polymerase (NEB)
10 mM dNTPs
6× DNA gel loading dye (NEB)
SYBR® gold staining solution: 2 μl of 10,000× SYBR® gold in 200 ml of 0.5× TBE
Items 8–13 from the equipment and general supplies section
2.8. DNA quantification and sample preparation
Items 8–13 from the equipment and general supplies section
100 bp DNA ladder (NEB)
2.9. TOPO cloning to validate deep seq libraries
-
1
TOPO® TA Cloning® Kit for Subcloning, without competent cells (Invitrogen)
-
2
10× Taq buffer (NEB)
-
3
10mM dATP
-
4
GoTaq® Hot Start Green Master Mix (Promega)
-
5
Taq polymerase (5000 units/ml, NEB)
-
6
Bacterial Competent cells (TOP10 or DH5α)
-
7
PCR purification columns (e.g. Qiagen)
-
8
M13 forward and M13 reverse primers
Materials to prepared ahead of time in lab:
-
9
Luria-Bertani + Ampicillin (LB+Amp) plates: 10 g/l Tryptone, 10 g/l NaCl, 5 g/l Yeast Extract, 15 g/l Agar; pH 7.4; add 100 μg/ml Ampicillin after autoclaving
2.10. Next-generation sequencing and data analysis using Galaxy
User account on Galaxy (https://usegalaxy.org), an open source, web-based environment for analysis and manipulation of NGS data.
3. Methods
3.1. RNA end curing and quantification
-
1
There are two important considerations for the RNAs that will be input into the method described here. First, this method requires a hydroxyl group at the RNA 3’-ends. Most small RNAs have a 3’-hydroxyl group and hence can be directly input into ligation reaction in step 1 of section 3.2. RNAs used for the NGS library preparation in our laboratories were co-purified with a fragment of Kluyveromyces polysporus Argonaute1 (KpAGO). The recombinant protein was expressed and purified from E. coli cells as reported previously[11]. The size of these RNAs fall in two distinct size classes that peak around 13 and 19 nucleotides (Figure 2A). Consistent with previous crystal structures of eukaryotic AGOs, these RNAs possess a 5’-monophosphate[16,11,17–19]. However, the exact nature of these RNA 3’-ends (3’-hydroxyl or 3’-phosphate) is not known. Therefore, we first describe a Calf Intestine phosphatase (CIP)-mediated dephosphorylation step to ensure that all RNAs end in a 3’-hydroxyl. This treatment will be necessary in other cases when a small RNA (or any RNA for that matter) does not end in a 3’-hydroxyl group (e.g. chemical or enzymatic nucleolysis yields RNAs with 3’-phosphate). Second, this method can be executed most reliably if input RNA concentration is precisely known. To quantify input RNA, we prefer to use a radioactive method that compares the number of 5’-ends in the RNA sample to a small synthetic RNA oligo of known size and concentration. This is a very sensitive approach that is particularly beneficial when input RNA amounts are low (nanomolar range) and cannot be reliably quantified using spectroscopic methods. As small RNAs often have a 5’-monophosphate, the CIP treatment described here also converts the 5’-phosphate to a 5’-hydroxyl group, which can be subsequently labeled with a radioactive phosphate using T4 polynucleotide kinase (PNK) (see Note 7).
Figure 2.
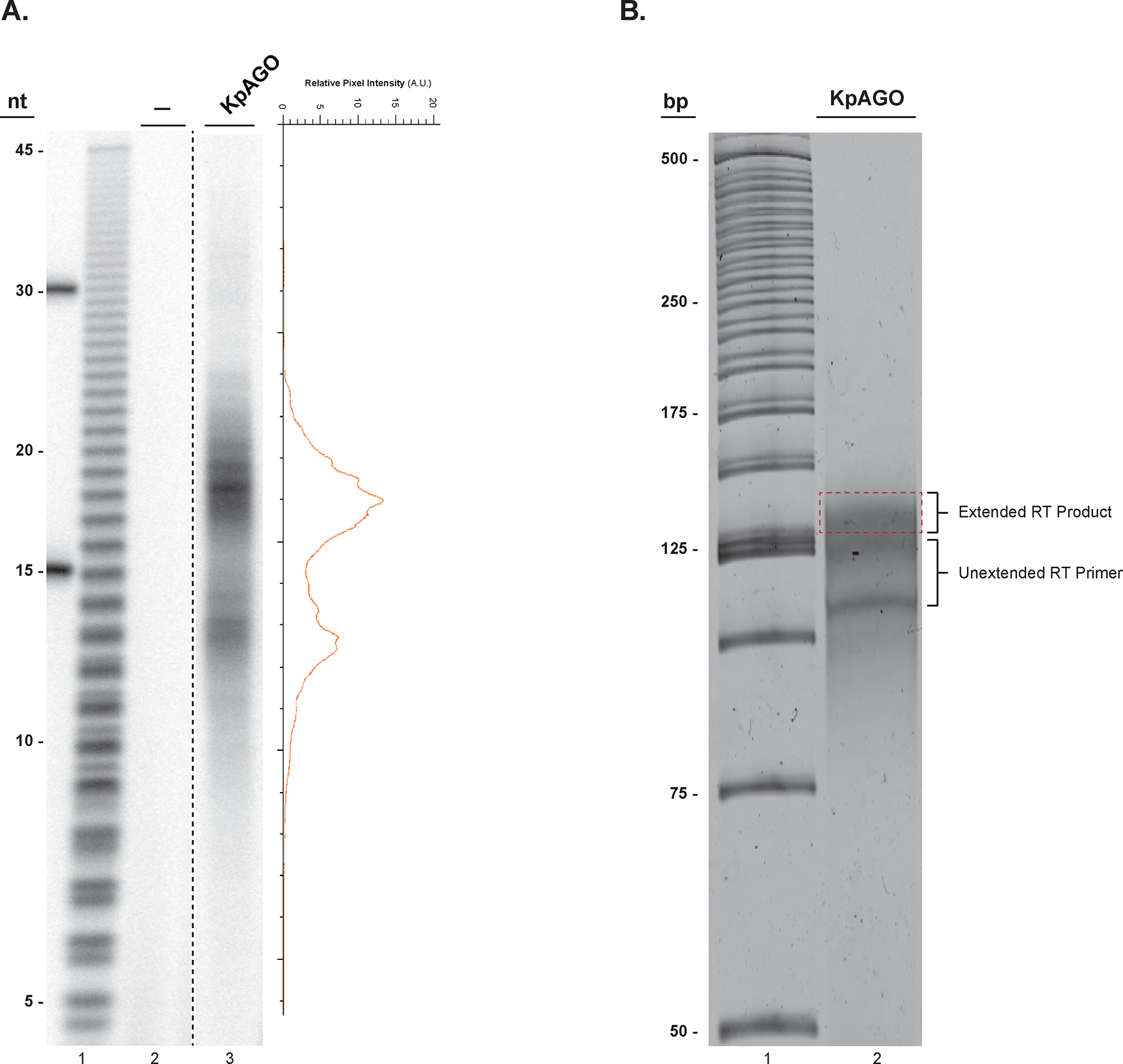
Input RNA size estimation and reverse transcription product size selection. A. A 16% denaturing urea PAGE of 32P-labeled endogenous E. coli RNAs co-purified from KpAGO (lane 3) and a mock sample (lane 2). On the right the pixel intensity profile of lane 3 shows the presence of two distinct RNA populations centered at 13 and 19 nts in length from the purified KpAGO. A hydrolyzed poly uridine 45-mer was used to estimate relative size of the input RNA (lane 1). B. A SYBR® gold stained 10% denaturing urea-PAGE gel showing the extended RT product smear located above the unextended RT primer (lane 2). The excised gel region for elution of the DNA product is indicated by a dashed red rectangle. The 25-bp DNA ladder is in lane 1.
RNA end curing
-
2
Add the following to an eppendorf tube:
10× CutSmart® buffer 2 μl
RNA x μl
CIP enzyme 1 μl
RNase-free water to 20 μl
-
3
Incubate at 37 °C for 30 mins (see Note 8).
-
4
To the same tube, add the following:
500 mM EDTA 1 μl
Water 380 μl
Acid Phenol mix 400 μl
-
5
Vortex for at least 1 minute at room temperature.
-
6
Centrifuge at 12,000 ×g at room temperature for 5 mins.
-
7
Carefully pipet 350 μl of the upper aqueous phase into a new eppendorf tube (see Note 9).
-
8
Add the following to the tube:
3M sodium acetate (pH 5.2) 40 μl
2M Magnesium chloride 10 μl
Glycogen 2 μl
100% Ethanol 1000 μl (2.5× of aqueous volume)
-
9
Precipitate RNA overnight at −20 °C.
-
10
Pellet RNA at 12,000 ×g in a refrigerated (4 °C) centrifuge for 30 mins. Carefully discard supernatant and wash with 1 ml of 70% ethanol by spinning at 12,000 ×g at 4 °C for 5 mins.
-
11
Discard supernatant and quick spin the tube. With a 20 μl pipet, carefully remove any residual liquid. Air dry the pellet for 1 min.
-
12
Resuspend RNA pellet in a small volume (≤10 μl ).
RNA quantification
-
13
Add the following to a new eppendorf tube on ice:
10× PNK buffer 1 μl
RNA (from step 12) 1 μl
1mM ATP 0.5 μl
γ32P-ATP 20 μCi
T4 PNK 0.5 μl
RNase-free water to 10 μl
(see Note 10)
-
14
Set-up parallel T4 PNK reactions to label 0.1 pmole of a synthetic RNA oligo, and 1 μl low molecular weight ssDNA ladder.
-
15
Incubate at 37 °C for 30 mins.
-
16
Add 10 μl of 2× Denaturing Load Buffer to each reaction.
-
17
Heat at 65 °C for 2–5 mins.
-
18
Prepare a 20% Urea-PAGE gel (20 × 27 × 0.1 cm) with sixteen wells (see Note 11).
-
19
Pre-run gel at 35 Watts (constant power setting) for at least 30 mins (see Note 12).
-
20
Load 10 μl of the radiolabeled input RNA, 10 μl of the radiolabeled RNA oligonucleotide, and 2 μl of the radiolabeled low molecular weight DNA ladder from step 16 above (see Note 13).
-
21
Run the gel at 35 W until bromophenol blue migrates 3/4th the length of the gel.
-
22
Carefully lift-off gel onto 3 M Whatman filter paper and cover with saran wrap (see Note 14).
-
23
Dry gel for 1 hour at 80 °C in a gel dryer (see Note 15).
-
24
Expose gel to phosphorimager screen overnight to visualize 32P-labeled RNA.
-
25
Quantify the volume of signal of the input RNA and the RNA oligo used as standard.
-
26
Determine molar concentration of the input RNA by comparing its signal to that of the known RNA oligonucleotide.
-
27
If needed, adjust the concentration of the remaining input RNA (step 12) such that 3’-ends are between 0.015–0.5 pmoles/μl (μM). This will yield ~0.05–2 pmol of 3’-ends in 3.8 μl, which is optimal for efficient ligation reaction in the next step, and eventually yields a complex library.
3.2. Ligation
Thaw the following and keep on ice: 50% PEG8000, 10× T4RNL2 Tr. K227Q Buffer, 20 mM DTT, and 7 μM pre-adenylated adaptor.
-
Add the following to a PCR tube on ice:
7 μM pre-adenylated adaptor 1.0 μl
RNA 3.8 μl
RNase-free water to 4.8 μl
Incubate in a thermocycler at 65 °C for 10 minutes. Rapidly cool to 16 °C and hold for 5 minutes. Samples can be on hold at 4°C.
-
Transfer tubes from thermocycler to ice and add the following:
10× Ligation Buffer 1.5 μl
50% PEG8000 7.5 μl
20 mM DTT 0.75 μl
T4RNL2 Tr. K227Q 0.45 μl
Final Volume 15.0 μl
Incubate in a thermocycler at 30 °C for 6 hours.
Heat inactivate the enzyme at 65 °C for 20 minutes. Samples can be on hold at 4 °C.
3.3. Reverse Transcription
Thaw the following and keep on ice: RT Primer, 4× dNTP mix, 5× First-Strand Buffer without MgCl2, 100 mM DTT.
-
To the 15 μl ligation mix from step 6 in section 3.2, add the following:
10 μM TruSeq RT primer 1.0 μl
10 mM dNTP mix 11.25 μl
RNase-free water 6.8 μl
Incubate at 65 °C for 5 minutes. Hold at 4 °C for at least a minute.
-
Transfer tubes to ice and add the following:
5× FS w/o MgCl2 Buffer 9.0 μl
100 mM DTT 2.25 μl
SSIII (200U/μl) 1.2 μl
Final Volume 45.0 μl
Incubate at 55 °C for 30–45 minutes.
Heat inactivate RT reaction at 70 °C for 15 minutes. Sample can be on hold at 4 °C.
3.4. Gel electrophoresis of RT product
To RT reaction (from step 4 in 3.3), add 45 μl of 2× denaturing urea load buffer.
Prepare 10% denaturing Urea-PAGE gel (20 × 28 × 0.15 cm) with eight 1.7 cm-wide wells (see Note 16).
Pre-run the gel at 35 Watts for at least 30 minutes (see Note 17).
Load samples using P20 tips. For size markers, mix 6 μl of 25 bp ladder (Invitrogen) with 34 μl of 2× urea denaturing buffer and load 20 μl on either side of sample (see Note 18).
Run gel at 35 Watts until bromophenol blue is 3/4th from the bottom of the gel.
Stain the gel with SYBR® gold for 5 mins using SYBR® gold staining solution.
Transfer gel to a plastic sheet protector open on three sides (see Note 19).
Scan on a Typhoon scanner using SYBR® gold compatible excitation (520nm) and emission (580 nm) filters.
Print an image of the gel for documentation and to reliably identify the desired bands/regions to be excised. Figure 2B shows a gel where the to-be-excised fragment with the extended RT product is indicated (see Note 20).
To excise the desired size gel fragments, place gel on a blue light transilluminator and cut out bands using razor blades (see Note 21).
3.5. Gel elution and streptavidin pulldown
To crush a gel fragment, place it in the barrel of a 3 ml syringe (piston removed), and extrude the gel fragment using the piston into a 2 ml eppendorf tube.
Add 800 μl of DNA elution buffer to the eppendorf tube with gel pieces. Nutate overnight at room temperature (see Note 22).
For each sample, aliquot 10 μl of streptavidin beads into a 1.5 ml eppendorf tube. Add 200 μl streptavidin bead wash buffer. Flick tube with a finger to uniformly suspend the beads. Place tube in a magnetic rack to capture the beads. Carefully pipet-off the supernatant and discard. Repeat wash steps at least three times (see Note 23).
After final wash, resuspend the magnetic beads in 10 μl of streptavidin bead resuspension buffer.
Transfer gel slurry from step 2 above to Spin-X column placed in its collection tube. Spin at 10,000 ×g for 3 min, at room temperature (see Note 24).
If needed, pool elution into a single 2 ml eppendorf tube. Add streptavidin magnetic beads, and nutate for 2–3 hours at room temperature (see Note 25).
Place tube on a magnetic rack to capture magnetic beads and discard supernatant.
Resuspend beads in 10 μl of RNase-free water.
3.6. Circularization
Thaw the following reagents and keep on ice: CircLigase Reaction Buffer, 1 mM ATP, 50 mM MnCl2.
-
Transfer the bead slurry (10 μl; step 8 in 3.5) to a PCR tube and add the thawed reagents as described below:
RT Product 10.0 μl (Bead slurry)
CircLigase Reaction Buffer 2.0 μl
1 mM ATP 1.0 μl
50 mM MnCl2 1.0 μl
5M Betaine 4.0 μl
CircLigase 1.0 μl
RNase-free water to 20 μl
Mix the contents of the PCR tube well by gently flicking the tube. Quick spin, and incubate the reaction at 60 °C for 4 hours.
Heat inactivate CircLigase by heating to 80 °C for 10 mins in a thermocycler.
3.7. PCR Amplification
-
1
The number of PCR cycles required to generate enough cDNA for deep sequencing will depend on the amount of the RT product, and in turn, on the amount of input RNA. The exact number of cycles needed for each sample will have to be empirically determined. If RT product was abundant enough to be readily visible on the urea-PAGE gel in steps 8–10 of the section 3.4, 6–10 PCR cycles will yields sufficient amount of DNA (Figure 3A). However, if input RNA amounts were lower and RT product was faint or undetectable in step 8 in section 3.4, one will have to test higher number of PCR cycles (between 15–18 cycles). Perform small scale test PCRs first to test the appropriate number of PCR cycles for a sample. Another goal of small scale test PCRs is to identify minimum number of PCR cycles for library amplification. This is very important to preserve the biological complexity of the input RNA sample, and also to prevent appearance of artifactual PCR products (see Figure 3A, lane 4). In our experience, these two conditions can be easily met by limiting amplification to the lowest PCR cycle number that yields distinct PCR product and where free (unused) primer pool is minimally depleted.
Small-scale test PCRs
-
2
Thaw the following and keep on ice once thawed: phosphorothioate PE1.0 and PE2.0 primers and 5× Q5 polymerase buffer, dNTPs.
-
3
Add the following to a PCR tube:
5× Q5 buffer 9.0 μl
10 mM dNTPs 0.9 μl
10 μM PE 1.0 2.25 μl
10 μM PE 2.0 2.25 μl
Circularization reaction 4 to 6 μl
Q5 polymerase 0.45 μl
RNase-free water to 45 μl
(see Notes 26 and 27)
-
4
Split the reaction into 3 equal parts (see Note 28).
-
5
Subject each reaction to a different number of PCR cycles (e.g. 5, 8 and 11 cycles) using the following amplification conditions:
98°C – 30 sec
98°C – 5 sec
65°C – 10 sec
72°C – 15 sec
72°C – 2 min
12°C – hold
(repeat steps in bold for desired number of cycles)
-
6
Mix PCR reactions with 6× DNA loading dye and load on 20 × 28 × 0.1 cm 8% nondenaturing PAGE (see Notes 29 and 30).
-
7
Run gel at 35 Watts until bromophenol blue is 3/4th from the bottom of the gel.
-
8
Stain the gel with SYBR® gold for 5 mins in SYBR® gold staining solution.
-
9
Transfer the gel to a clear plastic sheet protector as in step 7 in 3.4 to scan on a Typhoon scanner. The optimal number of cycles needed for sufficient amplification of the library will correspond to minimum number of cycles that provide readily detectable levels of expected size PCR products such that there is <10% depletion of free primers and complete absence of aberrant slow migrating DNA products (see Notes 31 and 32).
Large-scale PCRs
-
10
Redo 2 × 45 μl PCRs for a fixed number of PCR cycles determined above.
-
11
Separate large scale PCR products on a short (20 × 12 × 0.1cm) 8% nondenaturing PAGE at constant volts (150 V max) (see Note 33).
-
12
Stain the gel in SYBR® gold and image on a Typhoon scanner as described in step 8 in 3.4 (see Note 34).
-
13
Excise gel fragment containing PCR products of expected size. Figure 3B shows a gel with the excised fragment containing the large scale PCR product.
-
14
Crush gel fragment and perform DNA elution as described in step 1 of 3.5 (see Notes 35 and 36).
-
15
Precipitate DNA by adding 2.5 volumes of 100% ethanol and 10 ug glycogen. Store at −20 °C for at least 2 hours.
-
16
Pellet DNA at 12,000 ×g at 4 °C for 30 min. Wash the DNA pellet two times with 70% ethanol. Remove residual ethanol via quick spin and fine pipetting.
-
17
Immediately resuspend pellet in 20 μl water (see Notes 37 and 38).
3.8. DNA quantification and sample prep for next generation sequencing
To prepare DNA sample for next generation sequencing, both the size and amount of PCR product has to be carefully quantified. NGS libraries usually result in low sub-nanogram DNA yields, and therefore require highly sensitive quantification methods. DNA size in a library is best quantified via automated chip-based electrophoresis systems such as Bioanalyzer (high sensitivity DNA chip) or TapeStation (DNA and RNA ScreenTape). The Bioanalyzer trace and stats for the library prepared from the example input sample is shown in Figure 3C. DNA concentration of a library can be accurately quantified using a Qubit fluorometer. These instruments are now part of workflows at most NGS facilities, which often provide access to these services for a small per sample charge. If access to these instruments is not available, precise size and amount quantification of NGS library can be carried out using standard lab equipment as in the following steps.
Prepare a 8% native PAGE short gel as in step 11 of the section 3.7.
Load 2 μl of NGS library DNA along with 1.5, 0.5 and 0.15 μl of 100 bp NEB ladder on the gel. Run gel at 150V until bromophenol blue dye front runs 3/4th length of the gel.
Stain the gel with SYBR® gold as described above (step 8 of the section 3.7).
Scan the gel on a Typhoon scanner.
Quantify DNA using software such as ImageQuant. Use the amount of DNA in a specific band of the ladder (the one most comparable to your PCR bands) as a reference for quantification.
After quantification, a small amount of PCR product can be used for cloning into T-tailed vectors (see section 3.9).
Convert DNA concentration from ng/μl to nM (femtomoles/μl) (see Note 39).
Assuming 40 femtomoles (f moles) of DNA will be loaded in one lane of an Illumina flow cell, different barcoded libraries can be pooled depending on the number of reads desired for each library to obtain 40 f moles of DNA in a particular volume. For example, if three samples are to be mixed to get reads at a ratio of 1:1:2, mix them at 10 f moles: 10 f moles: 20 f moles (see Note 40).
-
If the pooled library is to be quantified again prior to sequencing, obtain average size of the DNA in the pooled mix as follows:
((f moles of library 1 x Average BP Size of Library 1)+ (f moles of library 2 x Average BP Size of Library 2)+ ….(f moles of library n x Average BP Size of Library n))/40 total f moles
Determine the concentration of the pooled library via Qubit, and using equation in Note 39 and average bp size from step 10, verify if nM concentration of your pool matches to that expected.
3.9. TOPO Cloning to validate NGS library
A small amount of PCR product can be cloned into T-tailed TOPO TA-cloning vector and transformed into E. coli. Using universal primers, inserts from a handful of bacterial colonies can be PCR amplified and sequenced via Sanger sequencing to validate the library before deep-sequencing.
-
The Q5 polymerase used to PCR amplify NGS libraries is a proofreading enzyme. So, unlike Taq, this polymerase does not leave a 3’-A overhang. To add an A-overhang at the ends of the PCR product to enable cloning into T-tailed TOPO vector, mix the following in a PCR tube:
PCR product 4.0 μl (see Note 41)
10× Taq Buffer 0.5 μl
10mM dATP 0.125 μl
Taq polymerase 0.25 μl
Water 5 μl (0.125 μl)
Incubate the PCR tube in a thermocycler at 72 °C for 20 mins.
-
Mix the following in a PCR tube:
A-tailed PCR product 4.0 μl
Vector (pCR2.1-TOPO) 1.0 μl
Salt solution 1.0 μl
Incubate the PCR tube at room temperature for 5 min then move it to ice.
Transform 4.0 μl of above mix into 50 μl of chemical competent Top10 or DH5α cells.
Plate the transformed bacteria on pre-warmed LB+Amp plate.
-
To identify insert-containing clones, first prepare the following mix in a PCR tube:
GoTaq Master Mix 10 μl
10 μM M13 forward primer 0.8 μl
10 μM M13 reverse primer 0.8 μl
Water to 20 μl
(see Note 42)
Pick a single colony with a pipet tip or tooth pick and mix into the PCR mix.
-
Perform PCR in a thermocycler using following conditions:
95 °C for 5 min
95 °C for 1 min
55 °C for 30 sec
72 °C for 1 min
Repeat steps in bold, 29 times
72 °C for 10 min
4 °C forever.
Run 5 μl of the product on a 1.5% agarose gel.
For each PCR reaction that yields a single, desired-size PCR product, purify the remaining PCR reaction using PCR purification columns and perform Sanger sequencing with M13 reverse primer.
3.10. Next-generation sequencing and data analysis using Galaxy
Sequence the NGS libraries prepared by this procedure on the HiSeq or MiSeq Illumina platforms in the single-end 50 bp format. These libraries use the standard Illumina first-read sequencing primer for both the barcode and insert sequencing. The final sequence of the DNA product from the procedure is shown in Figure 3D. The data from the sequencing pipeline is delivered in the fastq format.
Log into a user account on the Galaxy server (https://usegalaxy.org/)[20].
Upload the fastq file to Galaxy server with “Get data > Upload file”.
Obtain a basic quality control report for fastq data file using “NGS: QC and manipulation > FastQC”.
Groom the fastq file into fastqsanger format using “NGS: QC and manipulation > Fastq Groomer”.
Select the high quality reads using “NGS: QC and manipulation > Filter by quality” (see Note 43).
Convert fastq file into fasta format using “NGS:Convert Formats > FASTQ to FASTA” (see Note 44).
Remove first five nucleotides (random 5-mer) from each read using “NGS: QC and manipulation > Trim sequences”.
Upload a text file with exact sequence of the barcode sequence (positions 6–12 on the RT primers) using “Get data > Upload file” (see Note 45).
Using the barcode text file and the output from step 5 above, split reads based on barcode using “NGS: QC and manipulation > Barcode splitter”. Specify that the barcodes are at the start of the sequence (see Note 46).
Sequences with or without barcode sequences are output separately in an html file. Convert the output file of barcode-containing sequences into a fasta file for subsequent analysis as follows. In the “Results” pane, click on the “eye” icon to view the html barcode splitter output table. For each individual barcode-containing sequence file, right click on the hyperlink to capture the “Copy Link Location” URL of the file. Go to the tool “Get Data > Upload File” and paste the URL into the “ URL/Text:” box. Submit and the data will load as a fasta format dataset.
Remove first seven nucleotides (barcode + two invariable Cs) from each read using “NGS: QC and manipulation > Trim sequences” by keeping bases 8 through 50.
Remove the 3’-adapter using “NGS: QC and manipulation > Clip” tool. Indicate minimum sequence length after clipping as “1” and supply miRCat-33® sequence as custom sequence to be clipped.
Compute lengths of reads using the tool “FASTA manipulation > Compute sequence length”.
Plot histogram of read lengths using “Graph/Display Data > Histogram”. Figure 4A shows the read length profile for the input sample shown in Figure 2A.
To determine sequence composition of first 10 positions of the inserts, first use “NGS: QC and manipulation > Trim sequences” tool to keep bases 1 through 10. Then use the tool “Motif Tools > Sequence Logo” to generate a web-logo. Figure 4B shows the web-logo for the first 10 positions of the KpAGO-bound RNAs (see Note 47).
The trimmed sequences obtained in step 10 above can be used for alignment to reference genome using tools available in “NGS: Mapping” tool-shed.
Figure 4.

Size and nucleotide composition of KpAGO-bound RNAs from NGS of the cDNA library. A. A histogram showing the read counts (y-axis) corresponding to different lengths (x-axis) in the KpAGO cDNA library. ~1.39 million reads are represented in the plot). B. A web-logo showing the probability distribution (y-axis) of each of the four nucleotide bases at positions 1–10 (x-axis) of the KpAGO-bound RNAs. ~1.29 million reads ≥10 nt were used for this analysis.
Notes:
To keep samples as free from contaminants as possible, all pipetting should be done with filter tips.
Pipet very slowly to accurately pipet viscous solutions containing PEG8000. PEG8000 is susceptible to oxidation in the presence of air. Every time a new vial is opened, make small aliquots and store at −20 °C. Dispose of an aliquot after it has been used a couple times.
All homemade buffers should be sterile filtered before use to remove contaminants.
All primers should be gel purified or HPLC purified.
All reactions should preferably be performed in a thermocycler. For all reactions above 25 °C, use heated lid at 110 °C.
This linker is no longer available from IDT in pre-synthesized form. It can be ordered as a custom oligo from any oligo synthesis service.
If input RNA requires only 3’-end dephosphorylation, it can be achieved using the 3’-phosphatase activity of T4 PNK in a reaction where RNA is incubated with the enzyme in the absence of ATP.
Do not use heat to inactivate CIP as RNA is susceptible to hydrolysis at elevated temperature, particularly in the presence of metal ions present in CIP buffer (Mg2+ and Zn2+). Instead, quench reaction with EDTA and remove CIP by Phenol:Chloroform extraction as described.
Recovering only 350 μl of the aqueous phase prevents contamination by interphase/organic phase constituents.
The phosphorylation steps using T4 PNK (steps 13–27 in 3.1) should be performed only with a fraction of the input RNA and most of the RNA should be saved for input into step 1 of 3.2.
Allow the gel to polymerize for at least 1 hour at room temperature.
Before and after the pre-run, flush wells with running buffer using a syringe fitted with a 22 gauge needle. Clamp metal heat sinks onto the plates during the pre-run and the final run to prevent cracking of plates by overheating.
It is best to skip at least one lane between each sample and RNA/DNA standards.
The bottom chamber buffer will be significantly radioactive as unincorporated radioactive ATP will run-off into it from the gel.
If signal is strong and only few hours of exposure is needed, this step can be skipped and a saran-wrapped gel can be exposed to phosphorimager screen.
Broad wells help reduce the height of loaded sample in the well and reduce sample streaking. Those made using the described 8-well comb on 20 cm wide gel makes 1.7 cm wide well.
Before and after pre-run, flush wells before loading and running sample. Clamp metal heat sinks onto the plates during the pre-run and the final run to prevent cracking of plates by overheating.
The unextended RT primer migrates ~120 nt. Depending on the size of starting RNA sample, the extended RT product will appear above the unextended primer as a specific size band (in case of small RNAs) or as a streak (in case of RNAs of variable length).
A standard sheet protector open from three sides allows easy handling of stained PAGE gels without contributing any background fluorescence.
If input is on the lower end of the recommended amount, the RT product may be hard to visualize. In such a case, adjustment of brightness/contrast on images from fluorescent scanner may help to localize the product. In any case, a print copy of the gel image can serve as a guide for excision of the gel region where the RT product is expected to migrate. Numerous times we have successfully made NGS libraries even when RT product is not readily visible.
Use a new razor for each sample.
The volume of DNA elution buffer used may have to be adjusted to ensure that the gel fragment slurry is mixing well on the nutator. We typically start with 800 μl and add more elution buffer if needed. If gel fragment is too large, split into multiple tubes and add sufficient elution buffer to each tube to allow thorough mixing.
For multiple samples, enough beads for n+1 samples can be washed in the same tube using larger volumes of wash buffer. Washed beads can then be split into aliquots.
To easily pipet gel slurry, cut a 1000 μl pipet tip about 0.5 cm from the tip to widen its bore. Also, more than one Spin-X column may be needed if slurry volume is large.
Pre-washed streptavidin magnetic beads can also be incubated with gel pieces in elution buffer overnight during step 2 above. In this case, DO NOT crush gel pieces in step 1 in 3.5 as beads tend to stick to the jagged edges of gel fragments.
Mix the circularization reaction well by pipetting before adding to PCR reaction.
Do NOT exceed the suggested amount of circularization reaction as template in PCR reaction as it may affect PCR conditions.
To test a wider range of PCR cycles, reaction can be split into 4 equal parts.
Use 18-well comb or one that provides several narrow wells per gel.
Run a parallel negative control PCR without circularized template to use as a comparison for size and starting amount of PCR primers.
The PCR product from unextended but circularized RT is expected to be 151 bp. Hence, insert containing PCR product will be equivalent to the sum of 151 and expected insert size in bp.
Under PCR over-amplification conditions, both depletion of primers and appearance of aberrant slow migrating PCR products can be seen readily; compare lanes 2 and 3 with lane 4 in Figure 3A for the primer depletion and over-amplification artifacts.
Use a wide well comb so as to fit all the 90 μl of the PCRs in a single well sized about 1.7 cm. It is extremely important to perform electrophoresis under conditions that minimize heat generation. Heat produced during electrophoresis can lead to denaturation of double stranded DNA library, and confound DNA quantifications.
Scanning the gel provides documentation for records and enables careful analysis of PCR product sizes before proceeding ahead with next-generation sequencing.
Use of blue-light transilluminator minimizes DNA damage that may be caused by short-wave UV.
As the volume of gel pieces is similar to those in steps in 3.5, steps 1 to 2 of 3.5 can be followed exactly.
Air drying of DNA pellet for even a short duration can lead to DNA denaturation and confound precise quantification in subsequent steps.
Glycogen added during DNA precipitation may interfere with sequencing reaction. To remove glycogen completely, purify the DNA using Zymo Research DNA clean & concentrator™ columns as described by the manufacturer.
- A rough conversion factor for 200bp DNA product is . Use the following conversion formula for a more precise calculation:
Directly consult with the NGS sequencing facility regarding volume and concentration of DNA needed for sequencing. Aim for 1.5–2 times the required volume at the given concentration so that additional pooled sample is available for quantification via Bioanalyzer and Qubit, if necessary.
DNA from multiple barcoded libraries can be pooled and cloned together.
To screen colonies, prepare a master-mix for samples.
As seen in the FastQC output, quality score >28 will keep only the highest quality reads.
There are two tools with the same name in the galaxy tool-shed. We recommend to use the one developed by the Galaxy team.
The required format of the text file is described in the galaxy tool “NGS: QC and manipulation > Barcode splitter”.
Number of mismatches allowed and number of deletions allowed in barcodes can be changed to optimize the output.
Sequence Logo tool requires all input sequences to be of the same length.
Acknowledgements
Our research is supported by start-up funds from The Ohio State University (to G.S. and K.N.), a seed-grant from the Center for RNA Biology, OSU (to G.S. and K.N.), a Center for RNA Biology Fellowship, OSU (to D.M.D.) and a Graduate Student Pelotonia Fellowship (to D.M.D). We acknowledge Erin Heyer and Melissa Moore from University of Massachusetts Medical School, Worcester for their critical insights to streamline this procedure in our laboratory.
References
- 1.Hirose T, Mishima Y, Tomari Y (2014) Elements and machinery of non-coding RNAs: toward their taxonomy. EMBO Rep 15 (5):489–507. doi: 10.1002/embr.201338390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chu CY, Rana TM (2007) Small RNAs: regulators and guardians of the genome. J Cell Physiol 213 (2):412–419. doi: 10.1002/jcp.21230 [DOI] [PubMed] [Google Scholar]
- 3.Kim VN, Han J, Siomi MC (2009) Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol 10 (2):126–139. doi: 10.1038/nrm2632 [DOI] [PubMed] [Google Scholar]
- 4.Nakanishi K (2016) Anatomy of RISC: how do small RNAs and chaperones activate Argonaute proteins? Wiley Interdiscip Rev RNA 7 (5):637–660. doi: 10.1002/wrna.1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu XS, Fan BY, Pan WL, Li C, Levin AM, Wang X, Zhang RL, Zervos TM, Hu J, Zhang XM, Chopp M, Zhang ZG (2016) Identification of miRNomes associated with adult neurogenesis after stroke using Argonaute 2-based RNA sequencing. RNA Biol:1–12. doi: 10.1080/15476286.2016.1196320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spornraft M, Kirchner B, Haase B, Benes V, Pfaffl MW, Riedmaier I (2014) Optimization of extraction of circulating RNAs from plasma--enabling small RNA sequencing. PLoS One 9 (9):e107259. doi: 10.1371/journal.pone.0107259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vongrad V, Imig J, Mohammadi P, Kishore S, Jaskiewicz L, Hall J, Gunthard HF, Beerenwinkel N, Metzner KJ (2015) HIV-1 RNAs are Not Part of the Argonaute 2 Associated RNA Interference Pathway in Macrophages. PLoS One 10 (7):e0132127. doi: 10.1371/journal.pone.0132127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang T, Li R, Wen L, Fu D, Zhu B, Luo Y, Zhu H (2015) Functional Analysis and RNA Sequencing Indicate the Regulatory Role of Argonaute1 in Tomato Compound Leaf Development. PLoS One 10 (10):e0140756. doi: 10.1371/journal.pone.0140756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao J, Luo R, Xu X, Zou Y, Zhang Q, Pan W (2015) High-throughput sequencing of RNAs isolated by cross-linking immunoprecipitation (HITS-CLIP) reveals Argonaute-associated microRNAs and targets in Schistosoma japonicum. Parasit Vectors 8:589. doi: 10.1186/s13071-015-1203-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chi SW, Zang JB, Mele A, Darnell RB (2009) Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460 (7254):479–486. doi: 10.1038/nature08170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakanishi K, Weinberg DE, Bartel DP, Patel DJ (2012) Structure of yeast Argonaute with guide RNA. Nature 486 (7403):368–374. doi: 10.1038/nature11211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heyer EE, Ozadam H, Ricci EP, Cenik C, Moore MJ (2015) An optimized kit-free method for making strand-specific deep sequencing libraries from RNA fragments. Nucleic Acids Res 43 (1):e2. doi: 10.1093/nar/gku1235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sterling CH, Veksler-Lublinsky I, Ambros V (2015) An efficient and sensitive method for preparing cDNA libraries from scarce biological samples. Nucleic Acids Res 43 (1):e1. doi: 10.1093/nar/gku637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ (2012) The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell 151 (4):750–764. doi: 10.1016/j.cell.2012.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wery M, Descrimes M, Thermes C, Gautheret D, Morillon A (2013) Zinc-mediated RNA fragmentation allows robust transcript reassembly upon whole transcriptome RNA-Seq. Methods 63 (1):25–31. doi: 10.1016/j.ymeth.2013.03.009 [DOI] [PubMed] [Google Scholar]
- 16.Nakanishi K, Ascano M, Gogakos T, Ishibe-Murakami S, Serganov AA, Briskin D, Morozov P, Tuschl T, Patel DJ (2013) Eukaryote-specific insertion elements control human ARGONAUTE slicer activity. Cell Rep 3 (6):1893–1900. doi: 10.1016/j.celrep.2013.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schirle NT, MacRae IJ (2012) The crystal structure of human Argonaute2. Science 336 (6084):1037–1040. doi: 10.1126/science.1221551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Elkayam E, Kuhn CD, Tocilj A, Haase AD, Greene EM, Hannon GJ, Joshua-Tor L (2012) The structure of human argonaute-2 in complex with miR-20a. Cell 150 (1):100–110. doi: 10.1016/j.cell.2012.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faehnle CR, Elkayam E, Haase AD, Hannon GJ, Joshua-Tor L (2013) The making of a slicer: activation of human Argonaute-1. Cell Rep 3 (6):1901–1909. doi: 10.1016/j.celrep.2013.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, Grüning B, Guerler A, Hillman-Jackson J, Von Kuster G, Rasche E, Soranzo N, Turaga N, Taylor J, Nekrutenko A, Goecks J, (2016). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res 44, W3–W10. doi: 10.1093/nar/gkw343 [DOI] [PMC free article] [PubMed] [Google Scholar]