

Abstract
Based on DNA‐methylation, ependymomas growing in the spinal cord comprise two major molecular types termed spinal (SP‐EPN) and myxopapillary ependymomas (MPE(‐A/B)), which differ with respect to their clinical features and prognosis. Due to the existing discrepancy between histomorphogical diagnoses and classification using methylation data, we asked whether deep neural networks can predict the DNA methylation class of spinal cord ependymomas from hematoxylin and eosin stained whole‐slide images. Using explainable AI, we further aimed to prospectively improve the consistency of histology‐based diagnoses with DNA methylation profiling by identifying and quantifying distinct morphological patterns of these molecular ependymoma types. We assembled a case series of 139 molecularly characterized spinal cord ependymomas (n MPE = 84, n SP‐EPN = 55). Self‐supervised and weakly‐supervised neural networks were used for classification. We employed attention analysis and supervised machine‐learning methods for the discovery and quantification of morphological features and their correlation to the diagnoses of experienced neuropathologists. Our best performing model predicted the DNA methylation class with 98% test accuracy and used self‐supervised learning to outperform pretrained encoder‐networks (86% test accuracy). In contrast, the diagnoses of neuropathologists matched the DNA methylation class in only 83% of cases. Domain‐adaptation techniques improved model generalization to an external validation cohort by up to 22%. Statistically significant morphological features were identified per molecular type and quantitatively correlated to human diagnoses. The approach was extended to recently defined subtypes of myxopapillary ependymomas (MPE‐(A/B), 80% test accuracy). In summary, we demonstrated the accurate prediction of the DNA methylation class of spinal cord ependymomas (SP‐EPN, MPE(‐A/B)) using hematoxylin and eosin stained whole‐slide images. Our approach may prospectively serve as a supplementary resource for integrated diagnostics and may even help to establish a standardized, high‐quality level of histology‐based diagnostics across institutions—in particular in low‐income countries, where expensive DNA‐methylation analyses may not be readily available.
Keywords: DNA‐methylation, ependymoma, hematoxylin and eosin stain, morphology, neural networks, whole‐slide image
We established a deep learning framework that accurately predicts the DNA methylation class of spinal cord ependymomas (SP‐EPN, MPE(‐A/B)) from H&E‐stained whole slide images. Our approach may serve as a supplementary resource for integrated diagnostics and may prospectively help to establish a standardized, high‐quality level of ependymoma diagnostics across institutions—in particular in low‐income countries, where expensive DNA‐methylation analyses may not be readily available. Moreover, the novel, morphological analyses in this study address the discrepancy between histological diagnoses by human experts as well as classification based on DNA‐methylation data and provide neuropathologists with quantitatively obtained characteristics of ependymoma types, which may eventually enable rapid, and accurate decisions on patient‐specific treatment without requiring prior, expensive DNA‐methylation analyses.

1. INTRODUCTION
Ependymal tumors (ependymomas, EPNs) are clinically heterogeneous neoplasms of neuroepithelial origin and can occur in all three compartments of the central nervous system (supratentorial, in the posterior fossa, and in the spinal cord) [1]. The 2021 WHO classification defines 10 types of ependymomas by their molecular characteristics, anatomical compartment, and (for some types) immunohistochemical criteria [2, 3]. Spinal cord ependymomas comprise four different types, labeled spinal ependymoma (SP‐EPN), spinal ependymoma with MYCN amplification (SP‐MYCN), myxopapillary ependymoma (MPE) and subpendymoma (SE) [2, 3]. In particular, authors have reported more than 20% of primary tumors in the spinal cord to be of ependymal origin [1, 4]. Of note, SE and MPE may also occur in other anatomical compartments than the spine [2, 5, 6]. The spinal ependymoma types SE and SP‐MYCN represent very rare ependymoma types—thus, this manuscript exclusively focuses on MPE and SP‐EPN, which together represent the majority of spinal cord ependymomas [1, 2, 7].
Myxopapillary ependymomas are CNS WHO grade 2 tumors that are histologically characterized by myxoid changes in microcysts or around blood vessels and by the presence of papillary‐arranged tumor cells around vascularized fibromyxoid cores [2]. SP‐EPN are mainly identified from the absence of morphological features of MPE or SE [2]. Although the overall prognosis of patients SP‐EPN and MPE ependymomas is good (5‐year overall survival of nearly 100%), more frequent relapses have been reported for myxopapillary ependymomas than for SP‐EPNs (50% vs. 88% 5‐year progression‐free survival (PFS)) [1, 2]. This underlines the necessity of the assignment of the correct tumor type in ependymoma diagnostics. Of note, methylation profiling was recently used to define two molecularly distinct MPE subtypes, MPE‐A and MPE‐B, that showed association to papillary or tanycytic morphology, respectively, and to specific clinical features such as tumor localization and patient outcome [8]. In particular, MPE‐A cases were demonstrated to exhibit lower PFS than MPE‐B.
In combination with histological assessment of hematoxylin and eosin (H&E) stained tissue, DNA‐methylation based classification has become an integral part of diagnostics and is even mandatory for the EPN type PF‐B (posterior fossa B) according to the WHO [2, 3, 9, 10, 11]. Moreover, ependymomas histologically represent a challenging differential diagnosis for other tumor entities as well. In particular, a recent publication on a subset of histologically characterized ependymomas reported frequent match of DNA methylation profiles to other, non‐ependymal tumor types [12]. Due to their vast expressiveness, neural networks have significantly advanced image classification over the last years [13, 14, 15, 16], but no image‐based classifier for ependymomas exists to date. Thus, we aimed to employ neural networks to elicit the limits of molecular EPN classification based on classical H&E‐stained whole‐slide images. In a second step, characteristic morphological patterns of each molecular EPN type need to be identified as well as quantified in order to provide quantitative guidance for future histology‐based molecular EPN diagnostics.
Whole‐slide images (WSIs) are often multiple gigapixels in size and are therefore typically processed as a set of small sub‐images, so‐called patches (cf. [17, 18]). Since many of these patches may show irrelevant contents (e.g., background, artifacts), until recently, tumor classification based on WSIs required human experts to manually label representative tumor areas (regions‐of‐interest) or even individual pixels [19, 20].
This manual annotation is laborious and Ilse et al. proposed a weakly‐supervised classification algorithm which required only slide‐level labels (e.g., the molecular type of the tumor for the WSI) and demonstrated excellent performance on binary classification tasks [21]. Their approach trains a neural network (aggregation function) to combine sets of unlabeled patches into a meaningful representation of the entire slide, which is used together with the slide‐level label to train established classification architectures. The relative contribution of each patch to the slide‐level representation depicts the respective attention and can be used for feature discovery, cf. [22]. Recently, clustering‐constrained attention multiple instance learning (CLAM) was introduced, which extends the algorithm of Ilse et al. to the multi‐class case [23], hence allowing discrimination of not only two but many different diagnoses/molecular subgroups. CLAM computes one or multiple slide‐level representations (single‐branch/CLAM‐SB, multi‐branch/CLAM‐MB) for multi‐class classification and additionally introduces a linear classification objective in a compressed input space. Compared to other methods, this approach was demonstrated to work well with limited amounts of data. Thus, we focused on CLAM throughout this study, since EPNs represent a relatively rare tumor entity.
Prior to classification, these deep‐learning architectures utilize other deep‐learning models, so‐called encoders, to embed each patch in a lower‐dimensional latent space. Multiple architectures for encoders exist, including residual networks (ResNets) and vision transformers [13, 24]. Recent results indicate that domain‐specific encoders that were trained using self‐supervised learning (SSL) on histology datasets are suited better for WSI classification than encoders that were trained on the classical ImageNet dataset [22, 25, 26, 27, 28]. Compared to other methods [29, 30, 31, 32], simple siamese networks are a particularly efficient SSL method (wrt. memory and computation) and can be used to train residual networks of various depths [33]. Therefore, we focus on SimSiam and ResNets in this study.
In summary, we aim to predict the molecular type of major spinal cord ependymomas types and subtypes from hematoxylin and eosin stained whole‐slide images using explainable AI and to strive to extract morphological properties of these DNA methylation types of ependymomas from the classifier. Thus, our study addresses the challenge of improving the consistency between histological diagnoses by neuropathologists and tumor classification by DNA methylation profiling. Prospectively, our work may serve as a supplementary resource for integrated diagnostics of MPE/SP‐EPN and could help to establish a comparable level of diagnostic quality across institutions—which could be especially beneficial in low‐income countries, where expensive DNA‐methylation analyses may not be readily available. Of note, the methodology employed in this study was explicitly chosen to be agnostic of the considered ependymoma entities and may prospectively be extended other ependymoma types as well.
2. MATERIALS AND METHODS
Cases with DNA methylation classes MPE or SP‐EPN were obtained from previous publications [8] and from the archive of the Institute of Neuropathology at the University Medical Center Hamburg‐Eppendorf (Figure 1). The spinal ependymoma types SE and SP‐MYCN had to be excluded, since only a very small number of cases could be collected for them (<10 cases in total). Of note, the vast majority of cases had been histologically diagnosed as ependymomas (mostly grade 2) in our clinical databases. Furthermore, reference cases of glioblastomas (n = 24), medulloblastomas (n = 109), and PF‐A (posterior‐fossa A) ependymomas (n = 100) were gathered from the archive as well. Consent to collect demographic data and samples was obtained from each patient, following the protocols approved by the respective institutional review boards of the participating institutions. H&E‐stained slides and DNA methylation profiling data were obtained using standard protocols (cf. Supporting Information). The Heidelberg brain tumor classifier v11b6 [9] was used to assign the methylation class used for our analyses.
FIGURE 1.
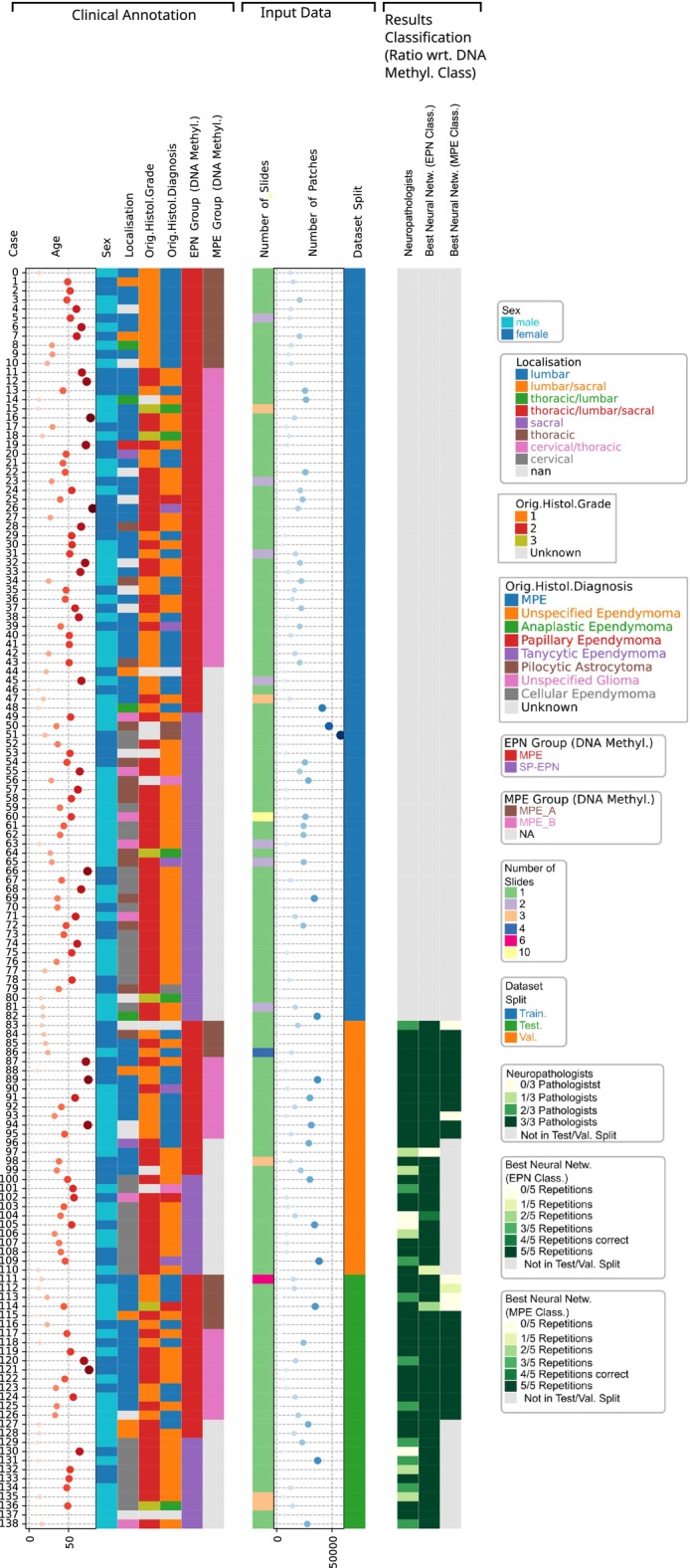
Cohort overview. Left column: Clinical annotations (age, sex, tumor localization, histological grade and histological tumor type as per neuropathology archive, ependymoma and MPE type as per DNA methylation profile). Centre column: Input data characteristics (number of available slides, patches per slide at 2× downscale). Right column: Ratio of neuropathologists or (independently trained) neural networks that predict the DNA methylation class from the H&E image.
Our method is summarized in Figure 2. Slides were digitized on a Hamamatsu C9600‐12 slide scanner at 40× magnification (226 nm per pixel). The median area of the slides was pixels, and the scan quality of each WSI was verified by manual inspection. For tissue segmentation, each WSI was first scaled to 1.25× magnification (32× downscale) and then converted into HSV color space. Following [23], we then computed a binary mask for the tissue regions by thresholding the saturation (S) color channel, followed by morphological opening to smooth the corners of the contours. We then inverted the resulting segmentation mask so that the tissue area was in the foreground and filtered out small artifacts (see Table S5 for details). Finally, tissue was identified as foreground contours with an area of at least 51,200 square pixels. Per contiguous tissue area, up to 15 holes with a minimal area of 3840 square pixels were identified and removed from further consideration. We then sampled overlapping RGB patches of 224 × 224 pixels with variable step size (stride) from the identified tissue area. If not explicitly stated otherwise, experiments were conducted at 4× downscale (10× magnification) with a stride of 112 pixels. For this configuration, we sampled 2,493,112 patches in total (median of 12,378 patches per WSI).
FIGURE 2.
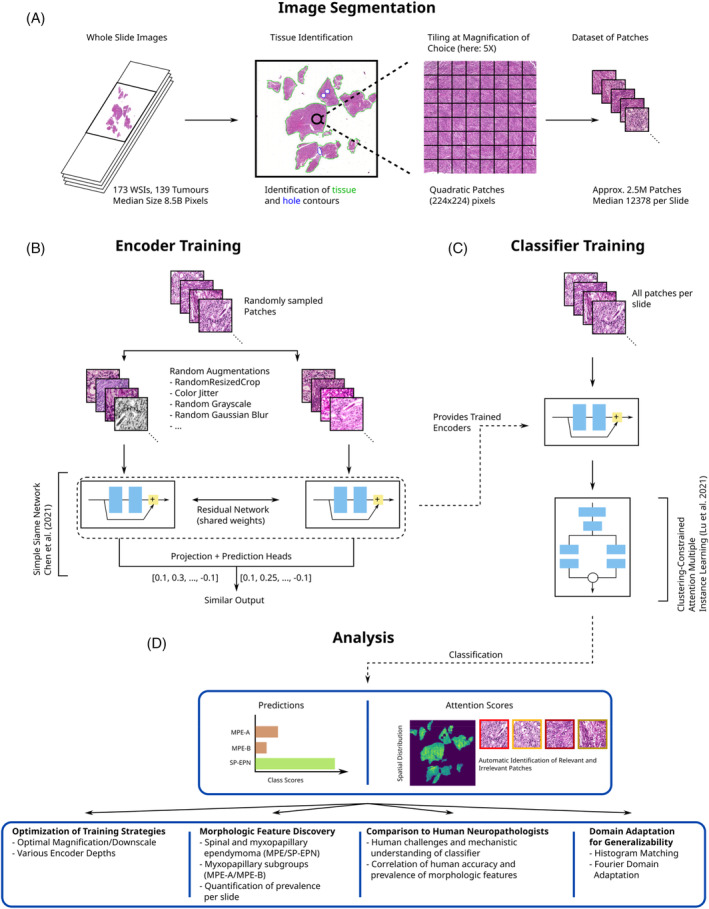
Method for the morphology‐based molecular classification of spinal cord ependymomas using deep neural networks. (A) Slide background and tissue holes are identified for each whole‐slide image (WSI) and patches are sampled from a regular grid over the tissue area. (B) Simple siamese networks are used to train residual networks on patches of WSIs from the training set. (C) The trained encoders are used for slide‐level classification using clustering‐constrained attention multiple instance learning (CLAM). (D) CLAM provides the molecular type prediction per slide, as well as the attention score per patch, for further analyses.
We trained domain‐specific ResNet encoders on patches of the training WSIs using SimSiam, cf. Table S6. Encoder training was distributed over 5 GPU nodes of the supercomputing cluster HSUper, each node featuring 256 GB DDR4 RAM, 2 Nvidia A100 GPUs and 2 Intel Icelake sockets with an Intel Xeon Platinum 8360Y processor (36 cores) each [34]. Features for each patch were extracted from the final average pooling layer (cf. [33]). For comparison, we also considered pretrained ResNets from ImageNet with 50 trainable layers (ResNet50), which represent a common choice in literature (cf. [23, 35]). Following [23], features were extracted using mean‐average pooling after the third residual block (conv4x).
The extracted features were used by CLAM models to predict the molecular EPN type (MPE/SP‐EPN). We used the ADAM optimization algorithm [36] to train the models for 50–200 epochs using early stopping with a patience of 20 epochs. Hyperparameters were manually tuned once for a pretrained ImageNet encoder (ResNet50) on a single‐branch model of CLAM and kept constant in all other experiments for comparability (cf. Table S7).
In some cases, our dataset contained multiple slides of the same tumor (stained for another study or collected from a different block). During training, these WSIs were considered independently by CLAM to increase the size of the training set. During validation/testing however, the patches from all available WSIs were considered jointly, so that CLAM predicts one unique diagnosis per case, just like a neuropathologist would do. Moreover, WSIs of both classes were sampled with balanced probabilities during training in order to encourage unbiased models. In order to estimate model variability, we trained CLAM models in five independent repetitions for each experiment and evaluated them using the metrics (1) accuracy with respect to the DNA methylation class, (2) area under receiver‐operating characteristic curve (ROC AUC) and (3) cross‐entropy [37].
In order to enable the identification of characteristic morphological features for each molecular EPN type, we employed MeanShift clustering [38] to the 50 patches with highest attention per WSI and CLAM attention branch (maximum of 500 iterations). In pursuance of further manual analysis, the closest patch per cluster center was extracted. In order to quantify the relative prevalence of the identified morphological patterns per slide, supervised machine learning (RandomForest, [39]) was used. Hyperparameters were optimized by a randomized grid search in 5‐fold stratified cross‐validation (cf. Supporting Information, as well as Table S8).
3. RESULTS
3.1. Classification of major molecular spinal ependymoma types MPE/SP‐EPN
With this study, we aimed to predict the molecular (DNA‐methylation) ependymoma type from H&E‐stained whole‐slide images. For this, we trained domain‐specific encoders using simple siamese networks and classified the respective patch feature vectors using explainable neural networks (CLAM classification algorithm). The resulting predictions and attention scores per patch led to a mechanistic understanding of the classification model and allowed for comprehensive analyses of the morphologic, inter‐class heterogeneity of spinal cord ependymomas.
We collected a case series of 139 patients (173 WSIs), who were molecularly diagnosed with ependymoma of either myxopapillary (MPE) or spinal (SP‐EPN) type, as assigned by the Heidelberg brain tumor classifier using DNA‐methylation data [9] (cf. Figure 1). Histological diagnosis confirmed glial origin of all cases (136/136 cases, three cases censored due to missing histological information in clinical databases). In particular, the vast majority of cases was histologically diagnosed as ependymomas (132/136 cases) of mostly CNS WHO grade 1 or 2 (123/129 cases, 10 cases censored). Between 1 and 10 WSIs were available per patient. Eighty‐four tumors (102 WSIs) were classified as MPE and the remaining 55 cases (71 WSIs) as SP‐EPN. Median age at diagnosis was 43 years for both molecular types. Of the MPE cases, 36/48 patients were female/male respectively, whereas the sex distribution was 19/36 for SP‐EPN. In concordance with previously reported findings [1, 2], relapses occurred more often in MPE cases than for SP‐EPN cases. Significant differences in patient outcome (PFS) could be confirmed for our cohort () using a robust Peto log‐rank test [40], which confirmed the necessity for MPE/SP‐EPN patient stratification, cf. Figure S8. Patients were distributed into stratified training/validation/test sets with a ratio of 3:1:1 (49/34, 17/11 and 18/10 cases per class, respectively). In particular, all WSIs of a tumor were assigned to the same data split.
3.2. Model comparisons
In order to obtain optimal classification results, detailed comparisons of different encoders and training strategies were necessary. We trained domain‐specific ResNets as described in Section 2 with 18, 34, 50, 101, and 152 layers (denoted ResNet18, ResNet34, etc. in the following) using stochastic gradient descent (SGD) with batch sizes of 8192, 4096, 2048, 1024, and 1024, respectively. Deeper encoders (50 trainable layers and more) had smooth loss curves and the training time of the encoders increased by up to 7× from the shallowest to the deepest network (cf. Figure 3A). Smaller batch sizes or layer‐wise adaptive rate scaling (LARS) [41] smoothed the loss curves for the shallow ResNet18 encoder (Figure 3B).
FIGURE 3.
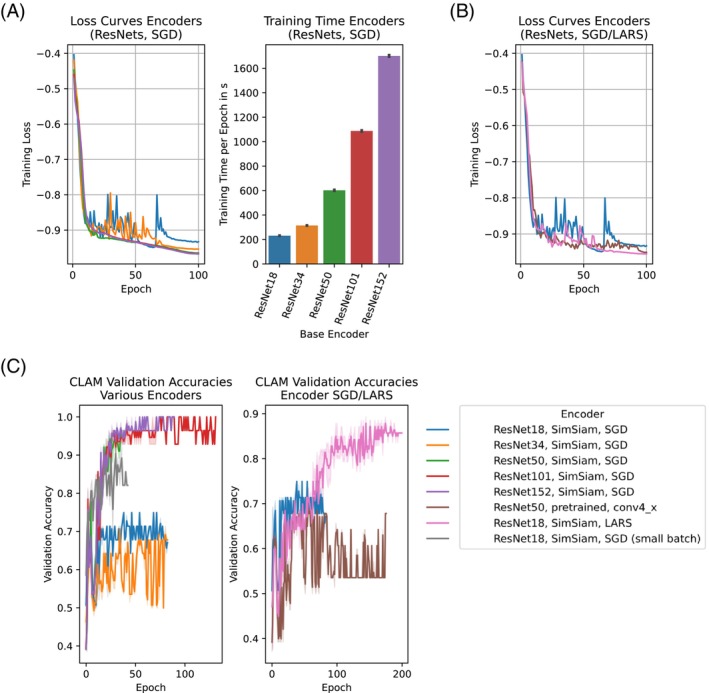
Encoder and classifier training. (A) Loss curves (left panel) and training time per epoch (right panel) for ResNet encoders of various depths. Error bars indicate 95% confidence intervals. (B) Loss curves for ResNet18 encoder trained using stochastic gradient descent (SGD) and layer‐wise adaptive rate scaling (LARS). The large batch optimizer LARS reduces the oscillations of the loss curves observed for SGD. (C) Validation accuracies of CLAM models trained using custom (SimSiam) and pretrained ResNet encoders. Solid lines indicate averages and shaded areas represent 95% confidence intervals. (D) Validation accuracies of CLAM models using domain‐specific ResNet18 encoders trained using LARS and SGD with small batch size. Solid lines indicate averages and shaded areas represent 95% confidence intervals.
Based on preemptive comparisons of single‐branch and multi‐branch CLAM architectures (cf. Table S9), we trained multi‐branch CLAM classifiers using encoded patches from each of the trained encoders (Figure 3C and Table 1).
TABLE 1.
Validation metrics (accuracy, cross‐entropy and ROC AUC) of multi‐branch CLAM models for different encoders and encoder training strategies (stochastic gradient descent—SGD, layer‐wise adaptive rate scaling—LARS).
| Metric | Accuracy | Cross‐entropy | ROC AUC |
|---|---|---|---|
| Encoder | |||
| ResNet18, SimSiam, SGD | 0.67 ± 0.01 | 0.6 ± 0.0 | 0.73 ± 0.0 |
| ResNet34, SimSiam, SGD | 0.68 ± 0.0 | 0.63 ± 0.0 | 0.67 ± 0.01 |
| ResNet50, SimSiam, SGD | 0.96 ± 0.01 | 0.2 ± 0.04 | 0.96 ± 0.04 |
| ResNet101, SimSiam, SGD a | 0.97 ± 0.03 | 0.09 ± 0.05 | 0.99 ± 0.01 |
| ResNet152, SimSiam, SGD | 0.98 ± 0.02 | 0.07 ± 0.04 | 1.0 ± 0.0 |
| ResNet50, pretrained, conv4x | 0.9 ± 0.04 | 0.35 ± 0.03 | 0.9 ± 0.03 |
| ResNet50, pretrained, last avg‐pool b | 0.83 ± 0.01 | 0.38 ± 0.02 | 0.91 ± 0.01 |
| ResNet18, SimSiam, SGD (small batch) | 0.86 ± 0.0 | 0.35 ± 0.02 | 0.92 ± 0.01 |
| ResNet18, SimSiam, LARS | 0.64 ± 0.05 | 0.63 ± 0.01 | 0.7 ± 0.0 |
Note: The domain‐specific ResNet152 performed best, closely followed by the domain‐specific ResNet101. For comparability, we also report the result for a pretrained ResNet50, where features were extracted after the last average‐pooling layer (similar to the domain‐specific encoders) instead after the typical conv4x block. Mean and standard deviations refer to 5 independently trained CLAM models and the two models with best validation results are highlighted.
Selected for all further comparisons due to close to perfect validation metrics and good encoder training time (36% faster than ResNet152).
Similar to the domain specific encoders, features were extracted after the last average‐pooling layer for this model.
Using our default training strategy (cf. Section 2), the shallow ResNet18 and ResNet34 encoders yielded the worst evaluation metrics among the tested models (e.g., a respective average validation accuracy of 67% and 68%, respectively). The deeper, domain‐specific ResNets (50 trainable layers and more) significantly outperformed a pretrained ResNet50 encoder, domain‐specific ResNet18 and ResNet34 encoders, as well as the domain‐specific ResNet18 encoder that was trained using smaller batches or LARS optimization. The best classification performance was achieved by the ResNet152 (average validation accuracy of 98%), closely followed by the ResNet101 (97%). Since the latter achieved close to perfect validation metrics as well as exhibited considerably lower encoder training times than the ResNet152, we focused on the ResNet101 in all further experiments. Of note, automatic mixed‐precision and re‐structuring of the data layout in GPU memory could even accelerate the ResNet101 training time by 1.7× without loss of validation accuracy (cf. Table S10).
In order to analyze the influence of patch magnification on the CLAM models, we sampled patches at 2×, 4× and 8× downscale (20×, 10× and 5× magnification, respectively) from the WSIs. The dataset size was roughly maintained by varying the stride (224, 112 and 56 pixels, respectively). We trained domain‐specific encoders (ResNet101) and respective CLAM models for each magnification and compared the results to classification based on a pretrained ResNet50 encoder (cf. Table 2). We observed the classifiers based on domain‐specific encoders to outperform their pretrained counterparts (wrt. magnification) on both the validation and the test set with respect to all evaluation metrics. Furthermore, the test accuracy, test cross‐entropy and test ROC AUC score of the CLAM models generally improved with increasing downscale factor for the domain‐specific encoders. In particular, the best model at 8× downscale (5× magnification) exhibited an average test accuracy of 98%, a cross‐entropy of 0.14 and a ROC AUC score of 0.99.
TABLE 2.
Validation and test metrics of multi‐branch CLAM models based on features from domain‐specific (SimSiam) and pretrained encoders at multiple resolutions.
| Accuracy | Cross‐entropy | ROC AUC | ||||||
|---|---|---|---|---|---|---|---|---|
| Encoder type | Downscale | Magnification | Test | Valid. | Test | Valid. | Test. | Valid. |
| ResNet50 pretrained | 2× | 20× | 0.86 ± 0.03 | 0.88 ± 0.02 | 0.36 ± 0.07 | 0.35 ± 0.01 | 0.96 ± 0.02 | 0.91 ± 0.02 |
| 4× | 10× | 0.82 ± 0.07 | 0.9 ± 0.04 | 0.38 ± 0.06 | 0.35 ± 0.03 | 0.91 ± 0.02 | 0.9 ± 0.03 | |
| 8× | 5× | 0.75 ± 0.03 | 0.83 ± 0.01 | 0.82 ± 0.3 | 0.42 ± 0.02 | 0.86 ± 0.04 | 0.9 ± 0.01 | |
| ResNet101, SimSiam, SGD | 2× | 20× | 0.85 ± 0.04 | 0.9 ± 0.01 | 0.43 ± 0.11 | 0.28 ± 0.02 | 0.9 ± 0.04 | 0.92 ± 0.03 |
| 4× | 10× | 0.9 ± 0.01 | 0.97 ± 0.03 | 0.26 ± 0.05 | 0.09 ± 0.05 | 0.98 ± 0.0 | 0.99 ± 0.01 | |
| 8× | 5× | 0.98 ± 0.02 | 0.93 ± 0.0 | 0.14 ± 0.08 | 0.13 ± 0.04 | 0.99 ± 0.01 | 0.99 ± 0.0 | |
Note: The best test metrics were achieved at 8× downscale using a domain‐specific ResNet101 (98% accuracy, highlighted in bold font). Mean and standard deviations refer to 5 independently trained CLAM models.
3.3. Interpretable analysis of classification results
For each patch, the classifier assigns an attention score, which represents the relative contribution of the patch to the slide‐level representation of the WSI (cf. Section 1). These attention scores may be interpreted as the relevance of the respective patch to classification and can be used for the discovery of characteristic morphology per EPN type (cf, Figure 4A).
FIGURE 4.
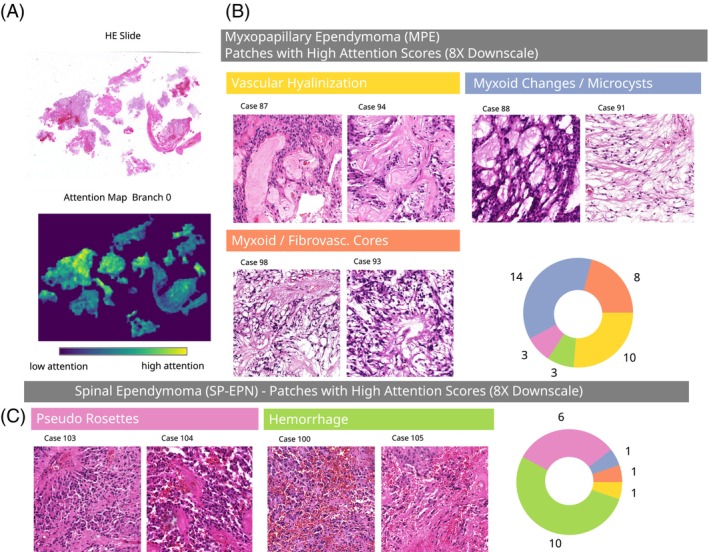
Attention analyses reveal distinct morphology associated with molecular EPN types. (A) Exemplary HE slide (left) and the corresponding spatial distribution of attention scores. (B, C) Patches with high attention scores at 8× downscale for myxopapillary and spinal ependymomas, grouped by the manually assigned morphological pattern. The pie charts indicate the number of validation cases with the respective morphology among the considered 50 highly attended patches. Additional cross‐validation experiments on the full cohort (all 139 cases) confirmed these findings, cf. appendix Figures S10 and S11.
Here, we selected the domain‐specific ResNet101 at 8× downscale for further analysis, since it yielded the best test metrics in prior analysis (cf. Section 3.2). For our analyses, we selected the 50 patches with highest attention scores per WSI from a single CLAM model and retrieved a subset of patches with representative morphology using a clustering method as described in Section 2. Here, we focused on the classifier's attention branch for the SP‐EPN class, since supplementary analyses showed that other attention branches yielded very similar attention scores (cf. Figure S9). By manual inspection of the representative patches obtained by this method, we were able to extract respective morphological characteristics of MPE and SP‐EPN ependymomas (Figure 4B,C). Of note, the selected patches represent tumor locations that were highly attended by our algorithm and may therefore differ from morphological features that are classically considered by neuropathologists (e.g., the type of pseudo rosettes).
The classifier characterized WSIs of MPE ependymomas using patches with dominant hyalinized vessels (HV), myxoid changes/microcysts (MM) and myxoid/fibrovascular cores (MFC). Based on the highly attended patches of the validation WSIs, these features occurred in 59% (n HV = 10), 82% (n MM = 14) and 47% (n MFC = 8) of validation patients with MPE EPNs and in only 9% (n HV = 1), 9% (n MM = 1) and 9% (n MFC = 1) of the validation patients with SP‐EPN, respectively. This indicates that these features may serve as positive indicators for the molecular MPE type. SP‐EPN cases were typified by the classifier using tumor tissue with hemorrhages (H), as well as by a type of pseudo rosettes (PR) morphology. These features were found in 91% (n H = 10) and 55% (n PR = 6) of the validation patients of SP‐EPN type, respectively, and in only 18% (n H = 3) and 18% (n PR = 3) of the validation MPE cases. Therefore, these features may represent positive correlates of the molecular SP‐EPN type. Additional cross‐validation experiments on the entire cohort (all 139 cases) confirmed our findings for the entire cohort (cf. Figures S10 and S11).
We aimed to additionally validate these findings on the correlation of morphological features and EPN types. In order to retrieve automated patch‐level annotations (e.g., hyalinized vessel present/not present on patch) we used supervised machine‐learning (RandomForest) to classify each patch with respect to presence of the identified morphological features (cf. Supporting Information for implementation details). The corresponding RandomForest‐classifiers achieved average validation F1 scores of 0.93, 0.92, and 0.89 on non‐training data, respectively. We applied these classifiers to all patches at 8× downscale and computed the relative prevalence (percentage of positive tissue area) for each feature and WSI (Figure 5A) as well as the Hausdorff distance (HD) between positively/negatively labeled patches (Figure 5B). The HD may be interpreted as a measure of non‐focality (low HD—high focality and strongly interleaved positive/negative patches; high HD—low focality and contiguous areas of positive/negative patches). For WSIs of MPE ependymomas, one‐tailed t‐tests confirmed statistically significant enrichment of myxoid changes/microcysts () and hyalinized vessels (), as well as significantly lower prevalence of patches with hemorrhages (). For each morphological feature, the HD was higher for WSIs of the corresponding molecular EPN type (, and (ns.), respectively). This indicates that the spatial distribution of the respective characteristics corresponded to larger, contiguous areas, whereas they occurred only very focally in WSIs of the other EPN type.
FIGURE 5.
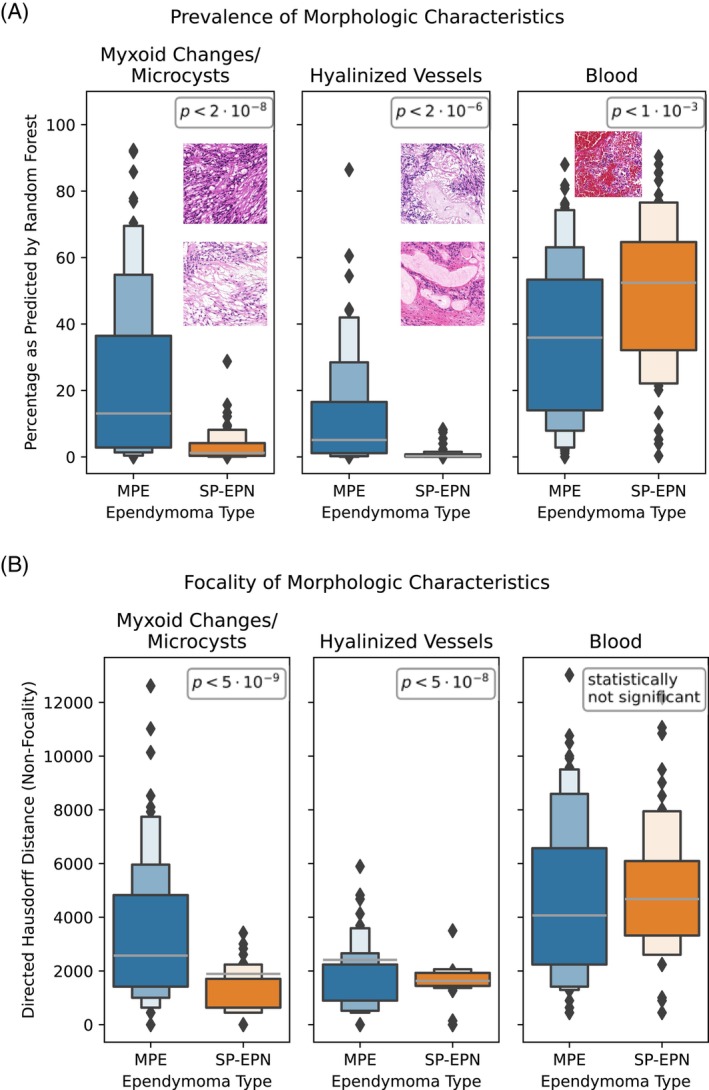
Validation of identified ependymoma morphology. (A) Percentage of tissue area per slide labeled to exhibit predominant myxoid changes/microcysts, hyalinized vessels or bleedings by RandomForest. p‐Values of one‐sided t‐tests between the ependymoma types MPE/SP‐EPN indicate significant differences (p < 0.05). (b) Non‐focality of the morphologies, as measured by directed Hausdorff distance. p‐values of one‐sided t‐tests indicate significant differences for myxoid changes/microcysts and vascular hyalinization.
Of note, we analyzed the patches with lowest attention scores per slide (both MPE and SP‐EPN cases) as well and predominantly found low‐quality patches with a large proportion of background (e.g., at tissue boundaries), with smudges on the slide, with artifacts and also unfocused patches (cf. Figures S12 and S13). This was consistent with our expectations for unimportant patches and thus supported the validity of the proposed attention mechanism.
For comparison to our classifier, three experienced human neuropathologists were asked to label the validation and test WSIs as either MPE or SP‐EPN in a blinded experiment based on histomorphology (Figure 6C, Venn diagrams). On average, their histological diagnosis matched the molecular, DNA methylation class in 82.7% of all validation and test cases. An average 92% of the molecular MPE cases were assigned to the histological MPE class, whereas only 67% of the molecular SP‐EPN cases were histologically assigned to the histological SP‐EPN class. For MPE, the ratio of neuropathologists that assigned a matching histological diagnosis was significantly positively correlated to the relative abundance of myxoid changes/microcysts () as predicted by the RandomForests, whereas for SP‐EPN it was significantly negatively correlated to the prevalence of hyalinized vessels () and myxoid changes/microcysts () (Figure 6G).
FIGURE 6.
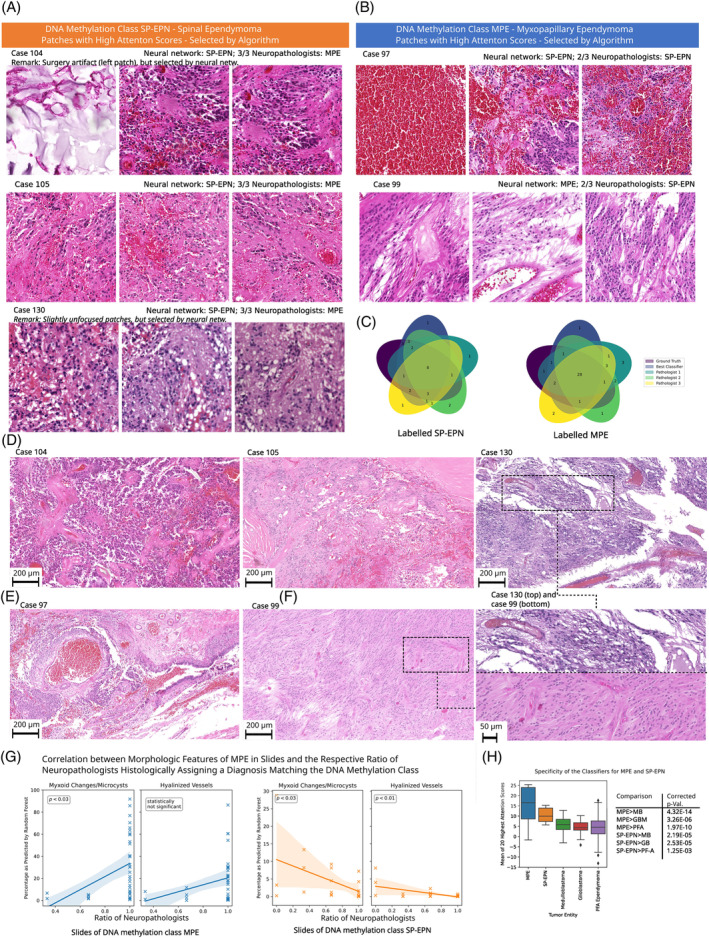
Comparison to neuropathologists. (A) Patches with high attention scores from cases with DNA methylation class MPE, that were histologically assigned differently by the majority of participating neuropathologists. Here, the patches are algorithmically selected—the reader is referred to (D) and (E) for representative overview images as selected by neuropathologists. (B) Corresponding patches for cases with DNA methylation class SP‐EPN. (C) Venn diagrams showing the number of concordantly and dis‐concordantly classified cases of the participating neuropathologists, our classifier and the DNA methylation class for MPE and SP‐EPN. (D) High‐quality images representative for the cases from (A), as selected by human experts. (E) Corresponding images for cases from (B). (F) Representative high‐resolution images of exemplary cases from (D) and (E), as selected by human experts. (G) Linear regression models for the relationship between the average accuracy of human neuropathologists per slide and the relative prevalence of myxoid changes/microcysts and hyalinized vessels (left panel: slides with MPE ground truth, right panel: slides with SP‐EPN ground truth). 95% confidence intervals are indicated and the p‐values demonstrate statistical significance. (H) Average attention over the 20 highest attended patches for different tumor entities (left panel; boxes range from the first to the third quartile of the data and the whiskers extend from the box by 1.5× interquartile range). The specificity of our classifier is visible by significantly higher average attention for MPE/SP‐EPN ependymomas than for medulloblastomas (MB), glioblastomas (GB) and PF‐A ependymomas (right panel, p‐values represent one‐tailed t‐tests).
Of note, only for three molecular SP‐EPN cases all neuropathologists assigned the histological class MPE. For these cases, the neuropathologists reported the WSIs to lack distinctive features of MPE/SP‐EPN (cf. Discussion), but the classifier's attention mechanism was able to identify areas with hemorrhages characteristic to molecular SP‐EPN ependymomas in all of the three cases (Figure 6A). Accordingly, the cases were assigned to the correct molecular class by our classifier. For the molecular MPE cases, two cases were assigned to the histological SP‐EPN class by 2/3 neuropathologists and one of these cases was correctly classified (w.r.t. molecular class) by our classifier using patches with myxoid changes/microcysts and myxoid/fibrovascular cores. The other case, however, exhibited dominant bleedings and was accordingly mis‐classified as molecular SP‐EPN by CLAM (Figure 6B). Recently, HOXB13 has been reported as a novel immunohistochemical marker for MPE ependymomas and we confirmed the molecularly assigned class for these cases as far as tissue availability allowed (cf. Figure S16).
Of note, the patches selected by the classifier for the aforementioned cases with dis‐concordant histological diagnosis and DNA methylation class were of lower quality than usual (case 130: mildly unfocused tumor tissue, case 104: surgery artifact resembling microcysts, case 97: patches consisting purely of blood). This highlights that these cases were challenging for the classifier and demonstrate that human verification of the highly attended patches is still necessary when employing the classifier as supplementary resource in an integrated diagnostics setting. For the considered cases, additional overview images, which were selected by human experts, can be found in Figure 6D–F.
Additional cross‐validation experiments indicated that a score of less than 0.9 represents a suitable rejection criterion for WSIs, which strikes a balance between predicting as many samples as possible (approx. 84% and 88% of the validation and test cases respectively) and obtaining accurate predictions of the DNA methylation class (cf. Figure S14). Such a quantitative quality estimate is highly necessary for a future application in diagnostics and would have (correctly) rejected the mis‐classified molecular MPE case 97 in the considerations above (the respective score was 0.62 for SP‐EPN and 0.38 for MPE).
In order to test discrimination of the trained EPN types to other brain tumor entities unknown to the classifier, we also applied our classifier to WSIs of glioblastomas, medulloblastomas, as well as PF‐A ependymomas. Based on the previously determined rejection threshold of 0.9, 55% of the medulloblastomas, 39% of the PF‐A ependymomas and 17% of the glioblastomas were rejected by our classifier. Additionally, we observed the mean raw attention scores of highly attended patches (n = 20) per WSI to be significantly lower for those previously unseen entities than for MPE/SP‐EPN ependymomas (one‐tailed t‐test, for MPE and for SP‐EPN, cf. Figure 6H). This suggests, that mean attention scores can be used as an additional proxy for the distinction of other brain tumor entities in general. We therefore conducted proof‐of‐concept experiments using a calibrated support‐vector classifier on the mean raw attention scores per attention branch (cf. Supporting Information for implementation details). This method yields an improved rejection rate of 79%, 70% and 73% for glioblastomas, medulloblastomas and PF‐A ependymomas, respectively. Combining this method with the previously defined rejection threshold of 0.9 improves the rejection rate to 86%, 85% and 74%, respectively (Figure S15). Of note, the CLAM classifier was not trained to identify and reject any non‐MPE/SP‐EPN tumor entities—yet, this approach successfully rejects medulloblastomas/glioblastomas/PF‐A ependymomas, which are all brain tumors of neuro‐ectodermal origin.
3.4. Generalization to an external validation cohort
In order to obtain the best results, neural networks may specialize to properties of the dataset at hand, causing sub‐optimal performance on external cases, which is an important hurdle for prospective applications in diagnostics [42]. A particularly prominent challenge is the distortion of the color distribution caused by digitization with other slide scanners. Common domain‐adaptation techniques to reduce this technical bias include histogram matching (cf. [43]) and Fourier domain adaptation (FDA, [44, 45]).
We collected an external validation series of 18 WSIs (5 SP‐EPN, 13 MPE as per DNA methylation profile), which were assembled at the Charité Berlin and digitized on a Hamamatsu C13140 slide scanner at 40× magnification. We then applied the aforementioned domain‐adaptation methods to these external WSIs and classified them using our best model (multi‐branch CLAM classifier, domain‐specific ResNet101 encoder, 5× magnification, cf. Section 3.2) as well as a classifier based on a pretrained ResNet50, cf. Table 3.
TABLE 3.
Evaluation metrics for multi‐branch CLAM models on an external validation cohort using different encoders at 8× downscale.
| Encoder type | Method | Accuracy | Cross‐entropy | ROC AUC |
|---|---|---|---|---|
| ResNet101, SimSiam, SGD | FDA, | 0.79 ± 0.04 | 0.86 ± 0.12 | 0.79 ± 0.04 |
| Histogram matching | 0.79 ± 0.02 | 0.43 ± 0.03 | 0.91 ± 0.02 | |
| No domain adaptation | 0.77 ± 0.04 | 0.83 ± 0.1 | 0.8 ± 0.01 | |
| ResNet50, pretrained | FDA, | 0.73 ± 0.05 | 0.48 ± 0.05 | 0.85 ± 0.03 |
| Histogram matching | 0.63 ± 0.08 | 0.68 ± 0.1 | 0.82 ± 0.05 | |
| No domain adaptation | 0.6 ± 0.06 | 0.64 ± 0.06 | 0.57 ± 0.07 |
Note: The hyperparameter β was set to 1% for the Fourier domain adaptation (FDA) technique. The best results (highlighted in bold print) were achieved using a domain‐specific ResNet101 and histogram matching. Mean and standard deviation refer to 5 independently trained CLAM models.
Compared to naive classification (no domain‐adaptation), the aforementioned techniques improved the generalization of the classifiers to external cases by 22% and 3% accuracy for pretrained and domain‐specific encoders, respectively. For all methods, classifiers based on domain‐specific encoders showed better generalization to the validation cohort than their pretrained counterparts. Among the former, FDA (hyperparameter ) and histogram matching achieved an equivalent generalization accuracy of 79%, but the cross‐entropy and ROC AUC scores were better for histogram matching (50% and 15% improvement over FDA, respectively).
Of note, the best average accuracy obtained using these domain‐adaptation techniques (79%) is very similar to the average consistency of neuropathologists' diagnosis with DNA methylation profiles, as measured on the in‐house validation and test WSIs (83%, cf. Section 3.3). In summary, these results demonstrate that our classifier was able to generalize towards external cases with human‐grade performance and achieved even better results when specialized to the respective in‐house slide‐scanner.
3.5. Interpretable classification of myxopapillary ependymomas type A/B
Recently, two novel and clinically relevant subtypes of myxopapillary ependymomas (MPE‐A/MPE‐B) were defined using DNA‐methylation analyses [8]. Motivated by our previous results, we aimed to extend our method towards MPE‐A/MPE‐B classification and to elicit the limits of their classifiability based on H&E‐stained slides. Moreover, our method allowed for the automated, quantitative analysis of their respective, distinctive morphological features We used the previously trained, domain‐specific ResNet101 encoders (cf. Section 2) to train classifiers for the distinction of MPE‐A/MPE‐B at 2×, 4× and 8× downscale (Table 4 and Figure 7A, left panel). The best validation metrics were obtained at 8× downscale (e.g., 71% average validation accuracy), whereas the results were less clear for the test set (potentially due to limited dataset size). While the best classifier typically assigned the MPE‐B cases to the correct class (89% average validation accuracy), the mis‐classification rate was high for MPE‐A (only 30% accuracy). Again, we used the softmax‐probability for the predicted class as a measure of certainty for our classifier and rejected ‘uncertain’ samples based on a cutoff. The optimal cutoff was found to be 0.71, at which the classifier rejected 23% and 6% of the validation and test cases, respectively, and achieved a validation and test accuracy of 89% and 80%, respectively (Figure 7A, right panel). In particular, this thresholded classifier assigned all MPE‐B cases to the correct class (validation and test set, 100% accuracy), whereas the mis‐classification rate of validation (test) MPE‐As improved by 29% (10%), respectively.
TABLE 4.
Classification metrics of multi‐branch CLAM models for MPE‐A/MPE‐B classification using domain‐specific ResNet101 encoders at 2×, 4× and 8× downscale.
| Metric | Accuracy | Cross‐entropy | ROC AUC | |
|---|---|---|---|---|
| Set | Downscale | |||
| Test | 2× | 0.75 ± 0.0 | 0.47 ± 0.0 | 0.96 ± 0.0 |
| 4× | 0.75 ± 0.0 | 0.49 ± 0.1 | 0.93 ± 0.0 | |
| 8× | 0.75 ± 0.0 | 0.53 ± 0.02 | 0.93 ± 0.01 | |
| Validation | 2× | 0.68 ± 0.03 | 0.59 ± 0.0 | 0.69 ± 0.0 |
| 4× | 0.69 ± 0.0 | 0.58 ± 0.0 | 0.72 ± 0.0 | |
| 8× | 0.71 ± 0.03 | 0.47 ± 0.01 | 0.86 ± 0.0 |
Note: The best validation results were achieved at 8× downscale (highlighted in bold), whereas the results were less consistent for the test set (probably due to dataset size). Mean and standard deviation refer to 5 independently trained CLAM models.
FIGURE 7.
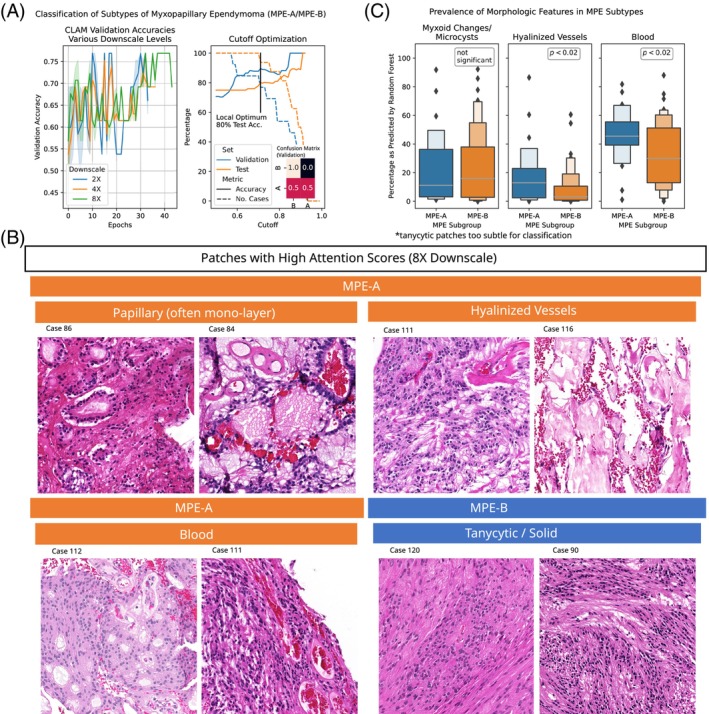
Histology‐based classification of MPE subtypes MPE‐A and MPE‐B. (A) Validation accuracies of CLAM models during training for various downscale levels (left panel). The accuracy can be further optimized by rejecting samples with high uncertainty (right panel). (B) Representative patches for the two MPE subtypes with high attention scores from the test and validation set at 8× downscale. (C) Percentage of tissue area per slide labeled to exhibit predominant myxoid changes/microcysts, hyalinized vessels or blood by RandomForest. Hyalinized vessels and blood show statistically significant differences (one‐sided t‐tests, p < 0.05), whereas myxoid changes/microcysts are not specific to a MPE subgroup.
Moreover, we used the previously described methodology (Sections 2 and 3.3) to explore morphological features of the two MPE subtypes on the validation and test set (Figure 7B). In particular, we found vascular hyalinization, bleedings, and papillary (often mono‐layered) structures to be distinctive for the MPE‐A subtype. For the MPE‐B cases, we found tumor tissue with tanycytic morphology to be particularly prominent. We used the RandomForests from Section 3.3 to quantify the prevalence of hyalinized vessels and bleedings in the respective WSIs and confirmed significant enrichment of these features in MPE‐A WSIs (both ). Of note, no significant differences were found for the relative abundance of myxoid changes/microcysts, which was in concordance with [8] and supported the validity of our findings.
4. DISCUSSION
We demonstrated the accurate prediction of the DNA methylation class of spinal cord ependymomas (MPE/SP‐EPN) from H&E‐stained whole‐slide images using neural networks. We trained, validated and tested our models on a case series of 139 patients and extracted quantitative and human‐interpretable morphological information about the respective ependymoma subgroups. Our approach achieved up to 98% test accuracy, whereas the histological diagnosis by neuropathologists matched the DNA‐methylation class in 83% of cases. Moreover, our method even generalized to an external validation cohort with up to human‐grade performance.
Seizing the particular interpretability of our weakly‐supervised approach, we were able to correlate morphological features with molecular attributes of spinal cord ependymomas. The morphological features arising from the employed attention mechanism were consistent with histo‐morphological criteria used by neuropathologists to date. To our knowledge, this study thus comprises the first algorithmic confirmation of established morphological criteria of MPE/SP‐EPN ependymomas. Moreover, the validity of the proposed methodology was confirmed by the analysis of patches with low attention scores, which revealed low‐quality features. Besides, quantitative attention analyses and additional supervised machine‐learning revealed insights on significant differences in prevalence and spatial distributions of histo‐morphological features in H&E‐slides that go beyond classical attributes that are considered by neuropathologists to date.
In particular, MPE cases were characterized by vascular hyalinization, myxoid changes/microcysts and myxoid/fibrovascular cores, whereas distinctive features for SP‐EPN cases were pseudo‐rosettes and hemorrhages. Although the latter could be confirmed as distinct feature for SP‐EPN within our cohort, further validation studies are necessary. Of note, a recent report indicated that hemorrhages occur more often in a certain type of primary brain tumors, termed WNT medulloblastomas, than in other medulloblastomas which may hint towards bleedings being a potential marker for some molecular tumor entities [46]. To date, neuropathologists should include further morphological features (or immunohistochemical methods, such as HOXB13 stain [47]) into their considerations for the discrimination of the two ependymoma types or any differential diagnoses.
Finally, our analyses shed light on the morphological intra‐class heterogeneity of myxopapillary ependymomas and elucidated their correlation to recently defined, molecular MPE subgroups, for which our classifier achieved up to 80% test accuracy. This finding confirmed that these DNA methylation subgroups may also be reflected on a morphological level, although further validation of the subgroups may be necessary.
Prospectively, our findings might improve the diagnostic understanding of the considered groups of spinal cord ependymomas and might stimulate further research in the field of weakly‐supervised, neural‐network based discovery of CNS tumor morphology. As a future perspective for our method, we envision the integrated consideration of highly attended patches, their mean attention scores and the WSI‐based classification score to support neuropathologists in their diagnostic workflows in addition to classical methods. In particular, a combination of the WSI‐based score and a classifier that uses the mean attention scores was able to reject three other brain tumor entities with a true positive rate of up to 86%. Of note, the histology‐based classifier had not been trained to exclude non‐MPE/SP‐EPN tumor entities and still achieved these high true‐positive rate. Training the classifier with a heterogeneous set of differential diagnoses (brain and non‐brain tumors) may prospectively yield further improvements of the classifier's specificity.
Of remark, our cohort was initially selected by DNA methylation profiling instead of the histomorphological diagnoses. Although the latter matched the DNA methylation type in almost all cases (>97%), many factors (incl. tumor microenvironment and tumor location) have been reported to affect the tumor's DNA methylation profile [9, 48, 49]. Thus, the accurate predictions of our classifier are primarily intended as a prospective surrogate for DNA methylation analysis—in particular in low‐income countries or as a supplementary resource in integrated diagnostics. Especially for myxopapillary ependymomas, this mismatch between DNA methylation profiles and histomorphology has not been fully resolved to date. In particular, the molecular subgroup of MPEs includes cases of histologically classical ependymomas and for histologically unresolved lesions, the alignment of DNA methylation with the molecularly defined EPN type is mandatory for a diagnosis of MPE, according to the 2021 WHO classification of CNS tumors [2, 3]. In this context, a peculiar advantage of our attention‐based approach is that it is able to identify even very small tissue sections of representative morphology (e.g., a single patch), whereas it may be challenging for a neuropathologist to identify very focal alterations.
In our experiments, only three cases (all SP‐EPN by DNA methylation profiling) were histologically assigned to the other considered EPN type (MPE) by the three participating neuropathologists. Closer investigation of these three cases revealed, that these tumors mostly lacked decisive, morphological features indicative of SP‐EPN and MPE and both options were conceivable to the neuropathologists. In all cases, the tumor tissue fit well to SP‐EPN but alterations of the vessels (cases 104 and 105) and minor myxoid/microcystic changes (case 130) had determined the neuropathologists' split decisions. All three neuropathologists recommended additional immunohistochemical or molecular analyses for the questionable cases and would have been willing to reconsider their diagnoses given the results of these additional methods. Their exact statements are reported in Supporting Information. Among those cases with sufficient amounts of available tissue, supplementary immunohistochemical methods (HOXB13) confirmed the molecularly assigned ependymoma type. From our experience, such cases can often be resolved using an integrated diagnostic approach that combines, for example, clinical information, histomorphological features, immunohistochemical markers and DNA methylation profiling.
Of note, our dataset is relatively small (even smaller for the MPE‐A/MPE‐B classification) and slightly imbalanced towards MPE ependymomas. This class imbalance could also have influenced the predictions of the human neuropathologists who were tasked with classifying the H&E‐stained WSIs from the validation/test set as either MPE or SP‐EPN. Future validation studies should contain more cases (in particular SP‐EPN) and could also be extended towards the other spinal cord EPN types (SE, SP‐MYCN), since collecting a sufficient number of cases to facilitate reliable prediction was not possible in the current study. Since the methods employed in this study were explicitly chosen to be agnostic of the considered types of ependymomas (MPE/SP‐EPN), the prospective extension to other EPN types should be straight‐forward. We conducted initial proof‐of‐concept experiments and were able to confirm the conceptual extendibility of our approach to ZFTA and PF‐A ependymomas and we aim to report detailed experiments in follow‐up research.
In summary, we established a deep learning framework that accurately predicts the DNA methylation class of spinal cord ependymomas (SP‐EPN, MPE(‐A/B)) from H&E‐stained whole slide images. Thus, our approach may serve as a supplementary resource for integrated diagnostics and may prospectively help to establish a standardized, high‐quality level of ependymoma diagnostics across institutions—in particular in low‐income countries, where expensive DNA‐methylation analyses may not be readily available. Moreover, the novel, morphological analyses in this study address the discrepancy between histological diagnoses by human experts as well as classification based on DNA‐methylation data and provide neuropathologists with quantitatively obtained characteristics of ependymoma types, which may eventually enable rapid, and accurate decisions on patient‐specific treatment without requiring prior, expensive DNA‐methylation analyses. As an important step towards the translation into clinical practice, our model generalized to an external validation cohort with human‐grade performance. We suspect that increasing the variability of the training set (e.g., by additional slide‐level augmentation, or by adding WSIs from different scanners, laboratories and time points) or the dataset size will further increase the model generalization.
Current and future research include the extension of our cohort to more EPN types in order to establish a broader tool for molecular EPN classification. Moreover, we aim to improve the generalization of our classifier to other image capturing modalities in order to further improve the potential, future integration of our classifier into routine diagnostic workflows.
AUTHOR CONTRIBUTIONS
Y.S. conducted the experiments, analyzed the data and wrote the manuscript. L.S., M.K., T.L., U.S. and J.E.N. collected and digitized the data. M.D., U.S. and J.E.N. reviewed the cases and assigned histological diagnoses. P.N. and J.E.N. supervised the study and share the last authorship. All authors have read and approved the final manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests.
ETHICS STATEMENT
Consent to collect clinical data and samples was obtained from each patient, following the protocols approved by the respective institutional review boards of the participating institutions.
Supporting information
Appendix S1. Supporting Information
ACKNOWLEDGMENTS
Computational resources (HPC‐cluster HSUper) have been provided by the project hpc.bw, funded by dtec.bw—Digitalization and Technology Research Center of the Bundeswehr. dtec.bw is funded by the European Union—NextGenerationEU. J.E.N. is funded by the DFG Emmy Noether Program. U.S. is supported by the DFG and the Fördergemeinschaft Kinderkrebszentrum Hamburg. We highly appreciate the tremendous efforts of Michael Bockmayr and Swenja Goedicke who were so kind as to collect and share their whole‐slide images of glioblastomas, medulloblastomas and PF‐A ependymomas. Open Access funding enabled and organized by Projekt DEAL.
Schumann Y, Dottermusch M, Schweizer L, Krech M, Lempertz T, Schüller U, et al. Morphology‐based molecular classification of spinal cord ependymomas using deep neural networks. Brain Pathology. 2024;34(5):e13239. 10.1111/bpa.13239
Philipp Neumann and Julia E. Neumann shared last authorship.
Contributor Information
Yannis Schumann, Email: schumany@hsu-hh.de.
Julia E. Neumann, Email: ju.neumann@uke.de.
DATA AVAILABILITY STATEMENT
All data and source code are available on reasonable request from the corresponding authors.
REFERENCES
- 1. Pajtler K, Witt H, Sill M, Jones DTW, Hovestadt V, Kratochwil F, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27(5):728–743. 10.1016/j.ccell.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kresbach C, Neyazi S, Schüller U. Updates in the classification of ependymal neoplasms: the 2021 WHO classification and beyond. Brain Pathol. 2022;32(4):e13068. 10.1111/bpa.13068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. WHO Classification of Tumours Editorial Board . Central nervous system tumours – WHO classification of tumours editorial board. Lyon, France: World Health Organization; 2022. [Google Scholar]
- 4. Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J, et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro Oncol. 2014;16(suppl 4):iv1–iv63. 10.1093/neuonc/nou223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tseng YC, Hsu HL, Jung SM, Chen CJ. Primary intracranial myxopapillary ependymomas: report of two cases and review of the literature. Acta Radiol. 2004;45(3):344–347. 10.1080/02841850410004931 [DOI] [PubMed] [Google Scholar]
- 6. Katz SY, Cachia D, Kamiya‐Matsuoka C, Olar A, Theeler B, Prado MP, et al. Ependymomas arising outside of the central nervous system: a case series and literature review. J Clin Neurosci. 2018;47:202–207. 10.1016/j.jocn.2017.10.026 [DOI] [PubMed] [Google Scholar]
- 7. Ghasemi DR, Sill M, Okonechnikov K, Korshunov A, Yip S, Schutz PW, et al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol. 2019;138(6):1075–1089. 10.1007/s00401-019-02056-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bockmayr M, Harnisch K, Pohl LC, Schweizer L, Mohme T, Körner M, et al. Comprehensive profiling of myxopapillary ependymomas identifies a distinct molecular subtype with relapsing disease. Neuro Oncol. 2022;24(10):1689–1699. 10.1093/neuonc/noac088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Capper D, Jones DTW, Sill M, Hovestadt V, Schrimpf D, Sturm D, et al. DNA methylation‐based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. 10.1038/nature26000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mack SC, Witt H, Wang X, Milde T, Yao Y, Bertrand KC, et al. Emerging insights into the ependymoma epigenome. Brain Pathol. 2013;23(2):206–209. 10.1111/bpa.12020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ellison DW, Aldape KD, Capper D, Fouladi M, Gilbert MR, Gilbertson RJ, et al. cIMPACT‐NOW update 7: advancing the molecular classification of ependymal tumors. Brain Pathol. 2020;30(5):863–866. 10.1111/bpa.12866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Neumann JE, Spohn M, Obrecht D, Mynarek M, Thomas C, Hasselblatt M, et al. Molecular characterization of histopathological ependymoma variants. Acta Neuropathol. 2019;139(2):305–318. 10.1007/s00401-019-02090-0 [DOI] [PubMed] [Google Scholar]
- 13. Dosovitskiy A, Beyer L, Kolesnikov A, Weissenborn D, Zhai X, Unterthiner T, et al. An image is worth 16 × 16 words: transformers for image recognition at scale. ArXiv. 2020;abs/2010.11929.
- 14. Chen X, Liang C, Huang D, Real E, Wang K, Liu Y, et al. Symbolic discovery of optimization algorithms. ArXiv. February 2023.
- 15. Öztürk S, Akdemir B. HIC‐net: a deep convolutional neural network model for classification of histopathological breast images. Comput Electr Eng. 2019;76:299–310. 10.1016/j.compeleceng.2019.04.012 [DOI] [Google Scholar]
- 16. Yu J, Wang Z, Vasudevan V, Yeung L, Seyedhosseini M, Wu Y. CoCa: contrastive Captioners are image‐text foundation models. ArXiv. May 2022.
- 17. Hou L, Samaras D, Kurc TM, Gao Y, Davis JE, Saltz JH. Patch‐based convolutional neural network for whole slide tissue image classification. 2016 IEEE conference on computer vision and pattern recognition (CVPR). New York: IEEE; 2016. 10.1109/cvpr.2016.266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Coudray N, Ocampo PS, Sakellaropoulos T, Narula N, Snuderl M, Fenyö D, et al. Classification and mutation prediction from nonsmall cell lung cancer histopathology images using deep learning. Nat Med. 2018;24(10):1559–1567. 10.1038/s41591-018-0177-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen PHC, Gadepalli K, MacDonald R, Liu Y, Kadowaki S, Nagpal K, et al. An augmented reality microscope with real‐time artificial intelligence integration for cancer diagnosis. Nat Med. 2019;25(9):1453–1457. 10.1038/s41591-019-0539-7 [DOI] [PubMed] [Google Scholar]
- 20. Bengs M, Bockmayr M, Schüller U, Schlaefer A. Medulloblastoma tumor classification using deep transfer learning with multi‐scale EfficientNets. ArXiv. September 2021.
- 21. Ilse M, Tomczak JM, Welling M. Attention‐based deep multiple instance learning. ArXiv. February 2018.
- 22. Fremond S, Andani S, Wolf JB, Dijkstra J, Melsbach S, Jobsen JJ, et al. Interpretable deep learning model to predict the molecular classification of endometrial cancer from haematoxylin and eosin‐stained whole‐slide images: a combined analysis of the PORTEC randomised trials and clinical cohorts. Lancet Digit Health. 2023;5(2):e71–e82. 10.1016/s2589-7500(22)00210-2 [DOI] [PubMed] [Google Scholar]
- 23. Lu MY, Williamson DFK, Chen TY, Chen RJ, Barbieri M, Mahmood F. Data‐efficient and weakly supervised computational pathology on whole‐slide images. Nat Biomed Eng. 2021;5(6):555–570. 10.1038/s41551-020-00682-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He K, Zhang X, Ren S, Sun J. Deep residual learning for image recognition. 2016 IEEE conference on computer vision and pattern recognition (CVPR). New York: IEEE; 2016. p. 770–778. 10.1109/CVPR.2016.90 [DOI] [Google Scholar]
- 25. Chen RJ, Krishnan RG. Self‐supervised vision transformers learn visual concepts in histopathology. ArXiv. March 2022.
- 26. Dehaene O, Camara A, Moindrot O, de Lavergne A, Courtiol P. Self‐supervision closes the gap between weak and strong supervision in histology. ArXiv. December 2020.
- 27. Chitnis SR, Liu S, Dash T, Verlekar TT, Di Ieva A, Berkovsky S, et al. Domain‐specific pre‐training improves confidence in whole slide image classification. ArXiv. February 2023. [DOI] [PubMed]
- 28. Russakovsky O, Deng J, Su H, Krause J, Satheesh S, Ma S, et al. ImageNet large scale visual recognition challenge. Int J Comput Vis. 2015;115(3):211–252. 10.1007/s11263-015-0816-y [DOI] [Google Scholar]
- 29. Caron M, Touvron H, Misra I, Jégou H, Mairal J, Bojanowski P, et al. Emerging properties in self‐supervised vision transformers. 2021 IEEE/CVF International Conference on Computer Vision (ICCV). 2021:9630–9640.
- 30. Chen T, Kornblith S, Norouzi M, Hinton GE. A simple framework for contrastive learning of visual representations. ArXiv. 2020;abs/2002.05709.
- 31. Chen X, Fan H, Girshick RB, He K. Improved baselines with momentum contrastive learning. ArXiv. 2020;abs/2003.04297.
- 32. He K, Fan H, Wu Y, Xie S, Girshick RB. Momentum contrast for unsupervised visual representation learning. 2020 IEEE/CVF conference on computer vision and pattern recognition (CVPR). 2019:9726–9735.
- 33. Chen X, He K. Exploring simple Siamese representation learning. 2021 IEEE/CVF conference on computer vision and pattern recognition (CVPR). 2020:15745–15753.
- 34. HSUper C . HSUper Documentation. Available from: https://www.hsu-hh.de/hpc/en/hsuper/.
- 35. Shao Z, Bian H, Chen Y, Wang Y, Zhang J, Ji X, et al. TransMIL: transformer based correlated multiple instance learning for whole slide image classification. ArXiv. June 2021.
- 36. Kingma DP, Ba J. Adam: a method for stochastic optimization. CoRR 2014;abs/1412.6980.
- 37. Grandini M, Bagli E, Visani G. Metrics for multi‐class classification: an overview. ArXiv 2020;abs/2008.05756.
- 38. Comaniciu D, Meer P. Mean shift: a robust approach toward feature space analysis. IEEE Trans Pattern Anal Mach Intell. 2002;24(5):603–619. 10.1109/34.1000236 [DOI] [Google Scholar]
- 39. Breiman L. Random forests. Mach Learn. 2001;45(1):5–32. 10.1023/a:1010933404324 [DOI] [Google Scholar]
- 40. Peto R, Peto J. Asymptotically efficient rank invariant test procedures. J R Stat Soc Ser A. 1972;135(2):185. 10.2307/2344317 [DOI] [Google Scholar]
- 41. You Y, Gitman I, Ginsburg B. Large batch training of convolutional networks. ArXiv. 2017.
- 42. Aubreville M, Stathonikos N, Bertram CA, Klopfleisch R, ter Hoeve N, Ciompi F, et al. Mitosis domain generalization in histopathology images the MIDOG challenge. Med Image Anal. 2023;84:102699. 10.1016/j.media.2022.102699 [DOI] [PubMed] [Google Scholar]
- 43. Nikolova M, Steidl G. Fast hue and range preserving histogram specification: theory and new algorithms for color image enhancement. IEEE Trans Image Process. 2014;23(9):4087–4100. 10.1109/tip.2014.2337755 [DOI] [PubMed] [Google Scholar]
- 44. Yang Y, Soatto S. FDA: Fourier domain adaptation for semantic segmentation. 2020 IEEE/CVF conference on computer vision and pattern recognition (CVPR). New York: IEEE; 2020. 10.1109/cvpr42600.2020.00414 [DOI] [Google Scholar]
- 45. Yang S, Luo F, Zhang J, Wang X. Sk‐Unet model with Fourier domain for mitosis detection. In: Aubreville M, Zimmerer D, Heinrich M, editors. Biomedical image registration, domain generalisation and out‐of‐distribution analysis. Basel: Springer International Publishing; 2022. p. 86–90. [Google Scholar]
- 46. Reisinger D, Gojo J, Kasprian G, Haberler C, Peyrl A, Azizi AA, et al. Predisposition of wingless subgroup Medulloblastoma for primary tumor hemorrhage. Neurosurgery. 2020;86(4):478–484. 10.1093/neuros/nyz148 [DOI] [PubMed] [Google Scholar]
- 47. Barton VN, Donson AM, Kleinschmidt‐DeMasters BK, Birks DK, Handler MH, Foreman NK. Unique molecular characteristics of pediatric myxopapillary ependymoma. Brain Pathol. 2010;20(3):560–570. 10.1111/j.1750-3639.2009.00333.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Galbraith K, Snuderl M. DNA methylation as a diagnostic tool. Acta Neuropathol Commun. 2022;10(1):71. 10.1186/s40478-022-01371-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chakravarthy A, Furness A, Joshi K, Ghorani E, Ford K, Ward MJ, et al. Pan‐cancer deconvolution of tumour composition using DNA methylation. Nat Commun. 2018;9(1):3220. 10.1038/s41467-018-05570-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information
Data Availability Statement
All data and source code are available on reasonable request from the corresponding authors.