

Abstract
This unit includes general protocols for the genetic manipulation of Streptomyces species, including genomic DNA isolation, genomic library preparation, intergeneric conjugation of Streptomyces with E. coli, generation and transformation of Streptomyces protoplasts, electroporation of Streptomyces mycelia, and colony PCR.
Keywords: Streptomyces, protoplast, conjugation, genomic DNA isolation, transformation
INTRODUCTION
Streptomyces are Gram-positive soil bacteria and are the largest representative genus of actinomycetes. They are industrially useful microorganisms that produce a wide variety of antibiotics, and, as such, the processes that are involved in producing these antibiotics are of considerable interest to the pharmaceutical industry. In recent years, much research has been conducted towards cataloging and characterizing the genes that compose the genomes of Streptomyces species, and thus it has become particularly advantageous to develop methods to directly manipulate streptomycete genetics for a wide variety of applications.
The protocols presented in this unit describe genetic manipulation procedures for common Streptomyces species. Basic Protocol 1 describes the isolation procedure for genomic DNA, while Basic Protocol 2 details its use in genomic library preparation. Basic Protocol 3 and Alternate Protocols 1 and 2 provide methods for conjugating different Streptomyces species with E. coli. Basic Protocols 3 and 4 detail transformation methods for transformation of Streptomyces using protoplasts and electroporation, respectively. Lastly, Basic Protocol 5 describes a rapid procedure for amplifying genes from intact Streptomyces.
CAUTION: Streptomyces and all E. coli strains described in this unit are Biosafety Level 1 (BSL-1) organisms. Such organisms are not known to consistently cause disease in healthy adult humans, and are of minimal potential hazard to laboratory personnel and the environment. Standard microbiological practices should be followed when working with these organisms.
BASIC PROTOCOL 1
GENOMIC DNA ISOLATION FROM STREPTOMYCETES
Genetic manipulation of Streptomyces species begins with the isolation of genomic DNA, which is used in sequencing, gene amplification, constructing genomic libraries, and a variety of other protocols. The information obtained from genomic DNA forms the foundation for more advanced genetic manipulation experiments such as gene cluster isolation and metabolic pathway engineering.
Materials
Tryptic Soy Broth (TSB) or appropriate growth medium (see UNIT 10E.1)
Mycelia or spore stock of Streptomyces (see UNIT 10E.1)
Lysis buffer (see recipe)
50 mg/ml lysozyme in 0.01 M Tris·Cl, pH 8.0 (see APPENDIX 2A for Tris buffer)
10% SDS (APPENDIX 2A)
0.5 M EDTA, pH 8.0 (APPENDIX 2A)
20 mg/ml pronase E (Sigma-Aldrich, cat. no. P-6911)
5 M potassium acetate, pH adjusted to 7.5 with 2 M acetic acid (store in aliquots at −20°C)
1:1 (v/v) phenol:chloroform
Chloroform
RNase solution (see recipe)
100% ethanol and 70% ethanol
TE buffer (see recipe)
250-ml baffled Erlenmeyer flasks
28°C shaking incubator
50-ml screw top vials (Fisher, cat. no. 05-539-13)
Centrifuge
Microscope slides
72°C water bath
Blunted Pasteur pipets (see recipe)
Large-bore (with ends cut off) and smaller-bore 1000-μl pipet tips
Grow and lyse cells
-
1
Inoculate 100 ml of TSB medium in a 250-ml baffled Erlenmeyer flask with mycelium or spores and incubate at 28°C with shaking at 250 rpm for 2 to 5 days.
See UNIT 10E.1, Basic Protocol 1, for a detailed protocol on growing Streptomyces species for genetic manipulation.
-
2
Collect the mycelia growth in two 50-ml screw top vials and centrifuge at 10 min at 2000 × g, room temperature.
In our experience a 5 to 7.5 ml wet pellet should be used for each 50-ml screw top vial. Depending on the growth of the Streptomyces, you should divide the pellet into the appropriate number of screw top vials.
-
3
Remove the supernatant and wash each pellet twice more with 25 ml lysis buffer, each time centrifuging as in step 2 and removing the supernatant.
-
4
Add 15 ml of 50 mg/ml lysozyme to each pellet and mix thoroughly.
You can vigorously shake or quickly vortex the vials to mix thoroughly.
-
5
Incubate the vials in a 37°C water bath for 1 hr or until complete lysis has occurred. Test for complete lysis by spotting 20 μl of sample onto a clean microscope slide. Add 20 μl of 10% SDS to sample spot and use pipet tip to mix.
The mixture will become clear when lysis is complete.
-
6
Add 1.2 ml 0.5 M EDTA and 0.13 ml of 20 mg/ml pronase E to each vial and gently invert samples several times to mix. After mixing, incubate for 10 min in a 37°C water bath.
-
7
Add 4 ml of 10% SDS to sample, then mix by inverting vials gently.
As on the microscope slide, after addition of 10% SDS the mixture should become clear.
-
8
Place samples in a 72°C water bath for 10 min.
Avoid vigorous shaking or vortexing past this point, as doing so may shear DNA, resulting in unwanted fragmentation of genomic DNA.
-
9
Place the samples in an ice bath for 5 min.
-
10
Add 4 ml of 5 M potassium acetate. Gently invert to mix thoroughly.
This step precipitates SDS and causes the solution to become cloudy.
Extract DNA
-
11
Add sufficient 1:1 phenol:chloroform for a total volume of 45 ml.
-
12
Gently invert until sample is uniform throughout.
IMPORTANT NOTE: This is the most important step of the protocol and should be executed correctly for desired results. Upon addition of the phenol:chloroform mixture there will be two distinct phases (the clear organic phase and the cloudy water phase). The goal is to mix the samples until the water phase drops to the bottom of the screw-top vial (at least to 5 ml). We have determined that simply inverting the vials manually or mechanically does not mix the solutions properly. Instead, we have found that holding the screw-top vials parallel to your fingers and mixing by following a smooth repetitive oscillating motion allows for complete mixing of the two phases. This may still take up to 30 min but is necessary for complete recovery of all genomic DNA.
-
13
Once the mixture is uniform throughout, centrifuge 15 min at 2000 × g, room temperature.
-
14
Transfer the water phase (top layer) to a new 50-ml screw top vial.
-
15
Add an equal volume of chloroform, mix gently, and centrifuge 5 min at 2000 × g, room temperature.
-
16
Repeat steps 14 and 15.
-
17
Transfer water phase (top layer) to a new 50-ml screw top vial.
Purify DNA
-
18
Add 0.13 ml of RNase solution and incubate in a 37°C water bath for 1 hr.
This will remove unwanted RNA from your sample. If this step is omitted, you will see smearing when the genomic DNA is run on an agarose gel.
-
19
Add ice-cold 100% ethanol to a total volume of 45 ml. Tilt back and forth gently to precipitate the DNA out of solution.
Precipitation of DNA will be complete in 3 to 5 min.
-
20
Dip a blunted Pasteur pipet into the vial and swirl the pipet around the areas where you can see precipitated DNA.
The DNA should attach to the pipet, allowing you to remove the precipitated DNA and transfer it to a 1.5-ml microcentrifuge tube.
-
21
Wash the precipitated DNA by adding 0.5 ml of ice-cold 70% ethanol to each microcentrifuge tube, making sure that the DNA is completely submerged in ethanol. Using a 1000-μl pipet tip, remove excess ethanol without disturbing the DNA. Allow the sample to dry at room temperature overnight in the uncapped 1.5-ml microcentrifuge tube.
-
22
Add 0.5 ml of TE buffer to the dried DNA and allow it to sit at room temperature overnight.
-
23
After the DNA has been allowed to sit overnight, mix the sample by pipetting up and down using large-bore pipet tips (make large-bore by cutting off the end of a 1000-μl pipet tip) to loosen the DNA.
Even though some shearing is unavoidable, you want to try and limit rough pipetting, as this can cause shearing of the DNA.
-
24
Once the sample is mixed well using the large-bore tip, move to a smaller bore size until the sample is mixed thoroughly.
Depending on the amount of DNA recovered, you can add additional TE buffer to help reduce the viscosity of the sample.
BASIC PROTOCOL 2
GENOMIC LIBRARY PREPARATION
Genomic libraries are random subclones of genomic DNA that cover the entire genome of a given species. These libraries are useful for genome sequencing, isolation of large DNA fragments, isolation of entire gene clusters for heterologous expression, gene inactivations, and various other genetic manipulations. There are several ways to produce a genomic library, and the protocols can differ greatly. There are several vectors which can be used for genomic library construction, and some commonly used examples include pOJ446 (Bierman et al., 1992), SuperCos1 (Evans et al., 1989; Invitrogen), pKC505 (Rao et al., 1987), and pWEB (Wahl et al., 1987; Epicentre). We prefer to construct genomic libraries using pOJ446, as it provides additional advantages compared to the previously mentioned vectors, such as an E. coli and Streptomyces origin of replication, oriT, for conjugal transfer into Streptomyces, and an apramycin resistance marker. This protocol describes the preparation of a genomic library using pOJ446.
Preparation of genomic DNA
When preparing genomic DNA, one must be aware of the cosmid vector utilized, as this will dictate which restriction enzyme to use for the preparation. Specifically, BfuCI is used to partially digest genomic DNA because it produces compatible “sticky ends” to BamHI, which is used to digest the cosmid vector pOJ446. We use BfuCI (MboI and Sau3AI are commonly used isoschizomers) because the recognition site is found frequently and homogeneously throughout Streptomyces chromosomal DNA, unlike the recognition site for BamHI, which varies between species. The goal of partial digestion is to reduce the genomic DNA into ~30- to 40-kb fragments that can be readily ligated into pOJ446, producing linear recombinant ~40 to 50-kb DNA fragments which will be preferentially packaged by bacteriophage.
Preparation of vector pOJ446
Cosmid vector pOJ446 carries an apramycin [aac(3)IV] resistance marker and an oriT site (permits conjugal transfer, see Basic Protocol 3) and contains λ cos sites that are utilized during packaging to circularize the vector (see Fig. 10E.3.2). The vector is first linearized using HpaI, and then the 5′ overhang of the “sticky end” is dephosphorylated to ensure the inability to self-ligate during proceeding ligation reactions. A second digestion using BamHI produces two DNA fragments, and is used as the site to insert partially digested genomic DNA. This positions the inserted genomic DNA, apramycin resistance marker, oriT, and the origin of replication (ori) between λ cos sites, which is important for proper packaging by bacteriophage.
Figure 10E.3.2.
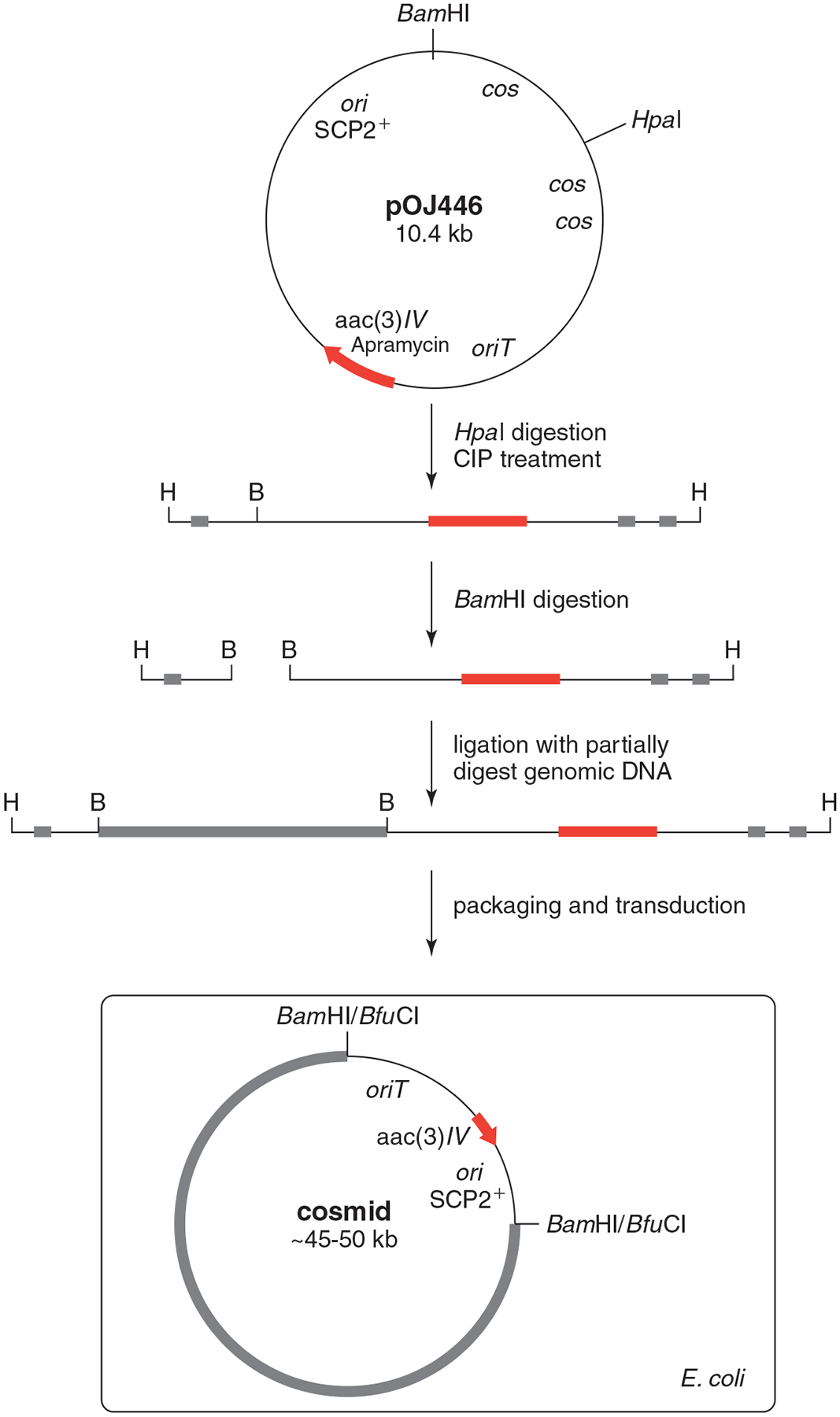
Overview of genomic library construction utilizing pOJ446. The λ cos sites (black box), apramycin-resistance marker (red arrow), and genomic DNA (gray box) are represented for illustration purposes only and are not drawn to scale.
Ligation of pOJ446 and genomic DNA
After completing preparation of genomic DNA and preparation of vector pOJ446 (see steps below), both need to be ligated together. BamHI (from pOJ446) and the BfuCI (from partial digestion of genomic DNA) form compatible “sticky ends” and are used to produce complete linear DNA fragments, upon ligation, which will be packaged and inserted into E. coli by bacteriophage.
Packaging and transduction of ligated sample
Ligation of pOJ446/HpaI/CIP/BamHI with partially digested genomic DNA results in the formation of various linear DNA fragments, as shown in Figure 10E.3.2. This linear DNA is packaged within a bacteriophage and is introduced into E. coli through transduction. There are commercially available kits that provide all materials needed for packaging and transduction. We prefer to use Gigapack III XL from Agilent Technologies, which preferentially packages large recombinant fragments between 47 and 51 kb.
Storing the genomic library
Once the genomic library is complete, collect all the colonies from the LB plates and store them at −80°C until needed.
Materials
BfuCI restriction endonuclease with 10× NEB Buffer 4 (New England Biolabs)
Genomic DNA (Basic Protocol 1)
DNA loading buffer (see recipe)
0.8% agarose gel (Voytas, 2000)
1:1 (v/v) phenol:chloroform
TE buffer (APPENDIX 2A)
Chloroform
100% and 70% ethanol
~0.4 μg/μl purified pOJ446 (The John Innes Centre; http://www.jic.ac.uk/)
HpaI restriction endonuclease (New England Biolabs)
DNA miniprep kit (available from various suppliers; optional)
10 U/μl calf intestinal alkaline phosphatase (CIP; New England Biolabs)
10× NEB Buffer 2 (New England Biolabs)
BamHI restriction endonuclease with 10 NEB Buffer 3 (New England Biolabs)
400 U/μl T4 DNA ligase and 10× buffer (New England Biolabs)
Gigapack III XL (Agilent Technologies), or similar kit
LB agar plates with appropriate antibiotics (APPENDIX 4A)
LB liquid medium (APPENDIX 4A)
50% (v/v) glycerol
50°C drying oven
1.5 ml microcentrifuge tubes
16°C water bath
50-ml screw-cap vials (Fisher, cat. no. 05-539-13)
Sterile spreader or wire loop
Additional reagents and equipment for agarose gel electrophoresis (Voytas, 2000)
Prepare genomic DNA
-
1
Prepare 1:0, 1:3, 1:10, 1:20, 1:50, and 1:100 dilutions of BfuCI with 1× NEB Buffer 4.
-
2Digest isolated genomic DNA with each dilution to determine appropriate enzyme concentration using the following reaction mixture:
- 8 μl genomic DNA
- 1 μl BfuCI dilution
- 1 μl 10× NEB Buffer 4.
Incubate 5 min at 37°C.
-
3
Immediately add 3 μl DNA loading buffer to stop reaction after 5 min, and immediately check digestion on a 0.8% agarose gel (Voytas, 2000).
When running the agarose gel, always run standard genomic DNA at the same concentration as in the digestion as a control.
-
4
Determine the enzyme concentration that gives the best partial digestion in 5 min.
A typical digestion pattern for testing restriction enzyme concentration is shown in Figure 10E.3.1. As the restriction enzyme concentration is decreased, less smearing is visible and the major band resembles the undigested sample (lane 1). The ideal concentration will give digestion patterns similar to that of lane 4. The genomic DNA has reduced in size and there is only slight smearing visible.
-
5Digest genomic DNA using the previously determined BfuCI concentration for 0, 3, 4, 5, 6, 7, and 8 min; prepare the following reaction mixture for each of these time points:
- 16 μl genomic DNA
- 2 μl BfuCI at optimal dilution determined in step 4
- 2 μl 10× NEB Buffer 4.
Incubate the respective reaction mixtures for the abovementioned time periods at 37°C
-
6Immediately stop the reaction at each timepoint using the phenol-chloroform method described below:
- After desired time at 37°C, immediately quench the reaction with 200 μl 1:1 phenol:chloroform. Mix well.
- Add 250 μl of TE buffer and mix well. Microcentrifuge 5 min at 13,000 rpm, room temperature.
- Take aqueous phase (top layer) and transfer to a new 1.5-ml microcentrifuge tube.
- Add 250 μl of chloroform and mix well. Microcentrifuge 5 min at 13,000 rpm, room temperature.
- Repeat steps c and d.
- Take aqueous (top layer) and transfer to a new 1.5-ml microcentrifuge tube.
- Add 1 ml of ice-cold 100% ethanol and mix gently. Place at −80°C for 15 min.
- Microcentrifuge the samples 10 min at 13,000 rpm, room temperature.
- Wash pellet (DNA) with ice-cold 70% ethanol.
- Dry in 50°C incubator for 5 min or until dry.
- Dissolve DNA in 20 μl TE buffer.
-
7
Take 3 μl of each purified digested sample and run on a 0.8% agarose gel (Voytas, 2000).
-
8
Determine the sample that has the desired amount of partial digestion.
Again, the digestion pattern will resemble that of Figure 10E.3.1. This time you may choose a few samples with appropriate partial digestion to continue the cosmid library construction.
-
9
Select the samples that have the appropriate amount of partial digestion (as seen by running the agarose gel). Save the remaining 17 μl of DNA that corresponds to those samples for further ligation experiments.
Figure 10E.3.1.
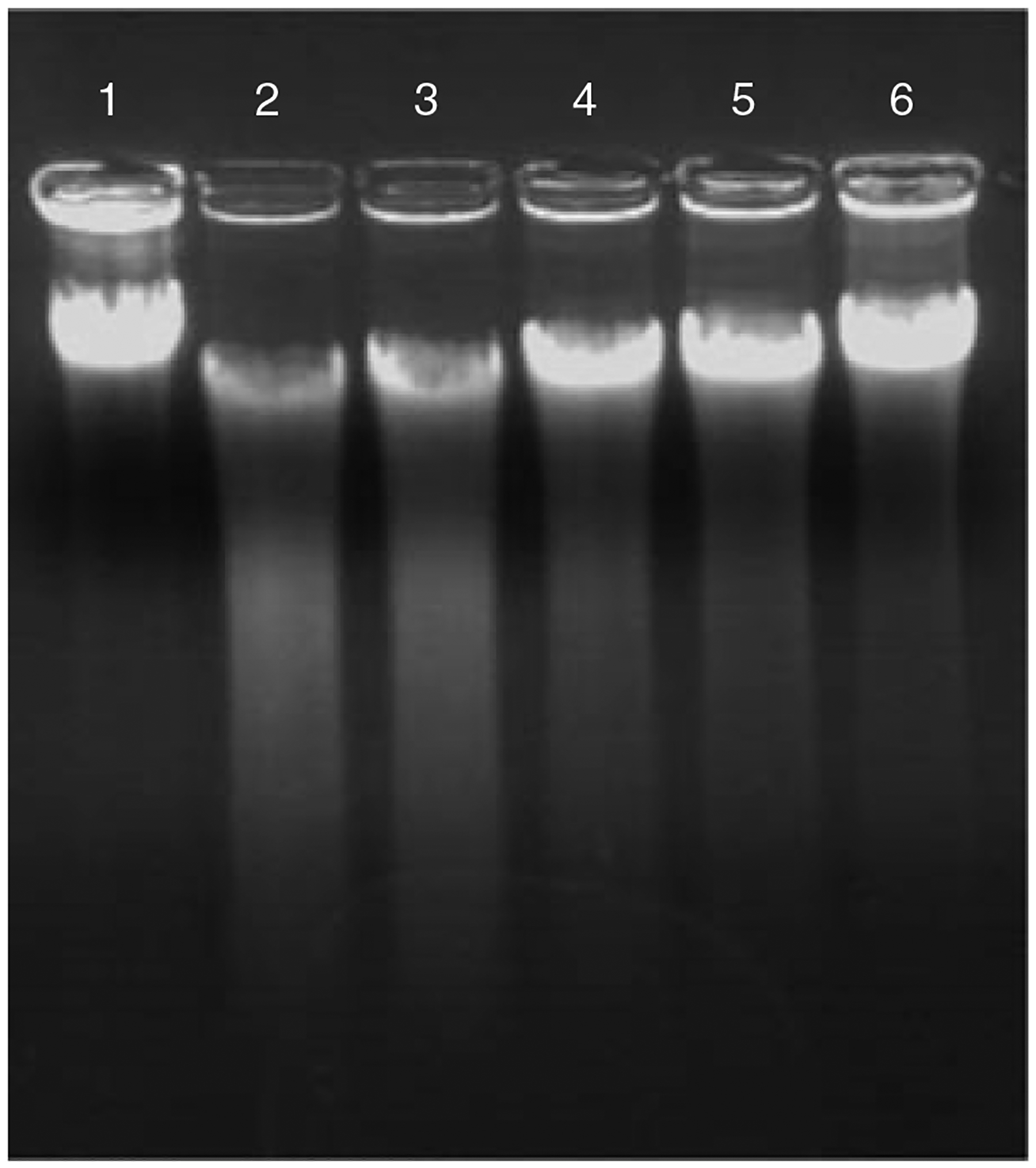
Partial digestion of genomic DNA (lane 1) using diluted BfuCI; 1:0 (lane 2), 1:3 (lane 3), 1:10 (lane 4), 1:20 (lane 5), and 1:50 (lane 6).
Prepare vector pOJ446
-
10Digest pOJ446 with HpaI using the following reaction mixture:.
- 41 μl pOJ446
- 4 μl HpaI
- 5 μl 10× NEB Buffer 4.
Incubate 1 hr at 37°C.
-
11
After digestion, remove the restriction enzyme using the phenol:chloroform method described under step 6, above, or using a DNA miniprep kit. Make the final elution of purified DNA up to 50 μl using TE buffer.
-
12
Run 3 μl of purified pOJ446/HpaI DNA on a 0.8% agarose gel (Voytas, 2000) to check for complete digestion.
A single ~10.4 kb band should be seen on the gel.
-
13Treat the pOJ446/HpaI DNA digest with calf intestinal alkaline phosphatase (CIP) to prevent self ligation of the vector using the following reaction mixture:
- 44 μl pOJ446/HpaI digest from step 11
- 2 μl 10 U/μl CIP
- 4 μl 10× NEB Buffer 2.
Incubate 1 hr at 37°C.
-
14
After dephosphorylation, remove CIP using the phenol:chloroform method (see step 6 of this protocol) or using a DNA miniprep kit.
Alternatively, it is possible to run the reaction on a 0.8% agarose gel (Voytas, 2000) and recover the CIP-treated pOJ446/HpaI band using a gel-extraction kit.
-
15Digest purified pOJ446/HpaI/CIP DNA with BamHI.
- 41 μl pOJ446/HpaI/CIP digest from step 12
- 4 μl BamHI
- 5 μl 10× NEB Buffer 3.
Incubate 1 hr at 37°C.
-
16
After digestion, remove the restriction using the phenol:chloroform method described under step 6, above, or using a DNA miniprep kit. Make the final elution of purified DNA up to 20 μl using TE buffer.
The final purified vector (pOJ446/HpaI/CIP/BamHI) will be used for further ligation reactions.
It may be necessary to use CIP after BamHI digestion, as self ligation at this site is also unwanted; however, our experience has shown that vectors with non-dephosphorylated BamHI 5′ overhangs give a higher final recombinant colony count than if you use vectors with dephosphorylated BamHI 5′ overhangs.
Ligate pOJ446 and genomic DNA
-
17Ligate partially digested genomic DNA (see step 9) with prepared pOJ446 (see step 16) using the following reaction mix:
- 12 μl partially digested genomic DNA from step 9
- 4 μl pOJ446/HpaI/CIP/BamHI digest from step 16
- 2 μl 400 U/μl T4 DNA ligase
- 2 μl 10× T4 DNA ligase buffer.
Incubate at 16°C overnight.
-
18
Take 3 μl of ligation reaction and 3 μl of control pOJ446/HpaI/CIP/BamHI and run a 0.8% agarose gel (Voytas, 2000).
Figure 10E.3.3 shows a typical ligation reaction (lane 2) alongside a control pOJ446/HpaI/CIP/BamHI (lane 1).
-
19
Save the remaining 17 μl of ligated sample at 4°C, as it will be used for packaging and transduction reactions.
Figure 10E.3.3.
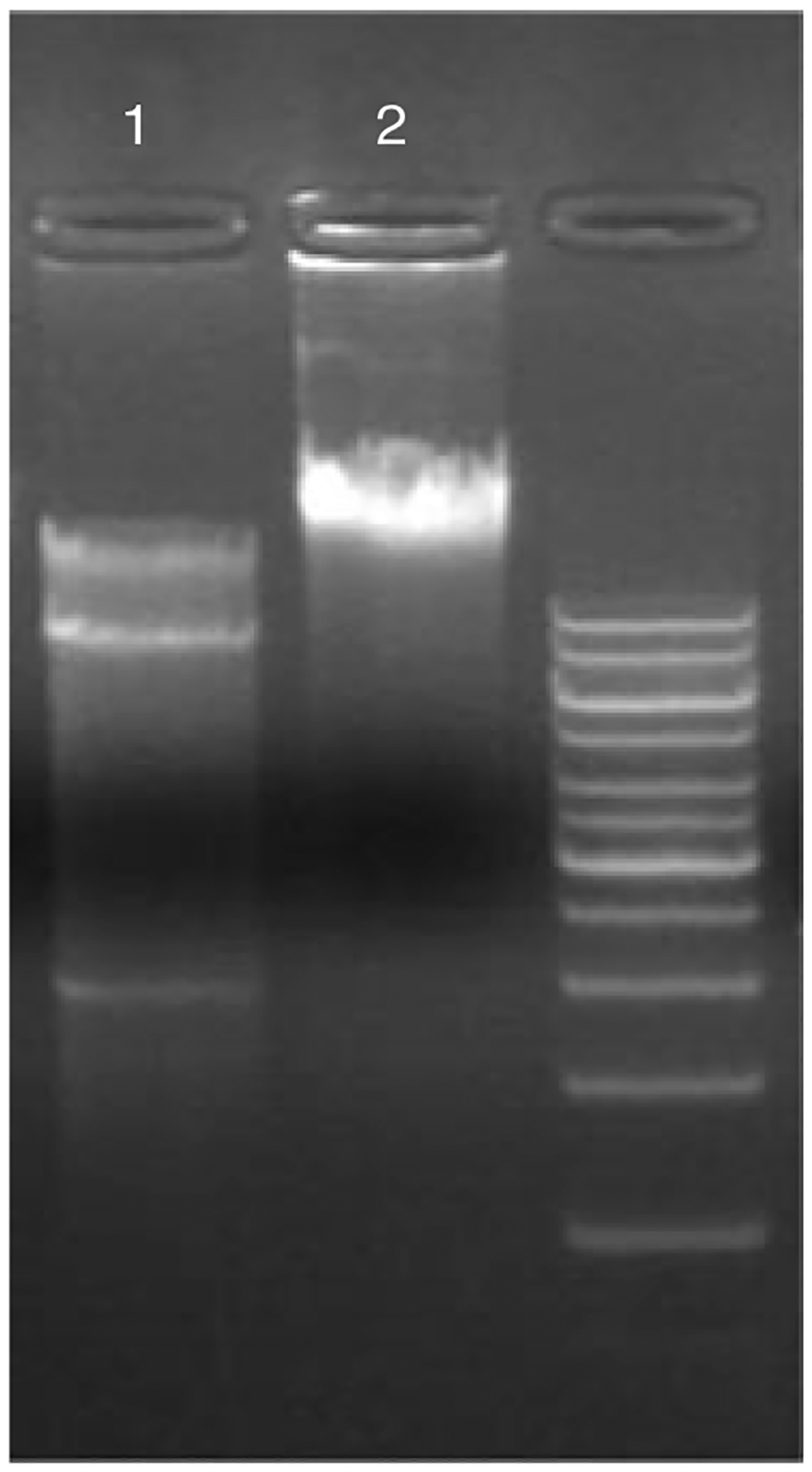
Control pOJ446/HpaI/CIP/BamHI (lane 1) with a ligation reaction of partially digested genomic DNA (lane 2).
Package and transduce ligated sample
-
20
Follow the Gigapack III XL protocol to obtain 50 μl of cells.
The Gigapack III XL takes the double-stranded linear DNA prepared in step 17, above, and packages it inside a lambda phage particle, which then infects E. coli cells, producing a library of cosmids containing ~30- to 40-kb fragments of your genomic DNA sequence.
-
21
Plate the cells on LB agar with the appropriate antibiotics at several diluted samples (from the original 50 μl of transformed cells) on individual LB plates in order to count individual colonies.
For example plate 0.01 μl, 0.1 μl, 1 μl, and the remainder of the cells in 10-μl portions.
It is important to know the amount of recombinant colonies recovered from the packaging protocol so that total genomic coverage can be determined.
Antibiotics used will depend on the vector used in library preparation. For example, pOJ446 and pKC505 would require apramycin, while SuperCos1 would require ampicillin.
-
22
Calculate the total number of recombinant colonies obtained from packaging and transduction.
-
23Determine if the cosmid library covers the entire genome, using the following formula to calculate the number of colonies needed to cover an entire genome within a desired probability (see Kieser et al., 2000):
where N is the number of recombinant colonies needed to cover an entire genome within a desired probability, P, and F is the fraction of genome (in base pairs) in one insert.If you want your cosmid library to cover 99% of your genomic DNA sequence, you should set P = 0.99. The value of F will be dependent on the size of the genomic DNA fragment found in each cosmid. Using this protocol, we expect genomic DNA fragments of ~30 to 40 kb and, therefore, we would set F = 35,000, where 35,000 is the average size of the expected genomic DNA fragments found in each cosmid.
Store the genomic library
-
24
Add 1 ml of LB liquid medium to each LB plate used to count and collect the recombinant colonies (step 21).
-
25
Using a sterile spreader or wire loop, lightly scrape the surface of the LB plate to dislodge the colonies.
-
26
Tilt the LB plate so the LB liquid accumulates at one side.
-
27
Using a sterile pipet, remove the LB liquid (with dislodged colonies) and transfer to a sterile 50-ml screw tip vial.
-
28
Aliquot 250 μl of the dislodged colonies into sterile 1.5-ml microcentrifuge tubes.
-
29
To each tube, add 250 μl of sterile 50% glycerol.
-
30
Mix by inverting the tubes and store at −80°C until needed.
BASIC PROTOCOL 3
INTERGENERIC CONJUGATION BETWEEN STREPTOMYCES COELICOLOR CH999 OR STREPTOMYCES LIVIDANS TK 64 AND E. COLI
Intergeneric conjugation remains a robust means to introduce DNA into Streptomyces species from an E. coli host. The horizontal gene transfer of plasmids between Streptomyces species and E. coli was made possible with some modifications to prior conjugation protocols between E. coli and other Gram-positive strains (Mazodier et al., 1989). This method takes advantage of an E. coli host with a transfer function (tra gene), which allows for mobilization of plasmids (e.g., E. coli ET12567/pUZ7002 or E. coli S17-1) and a plasmid with an origin of transfer (oriT). Conjugation is advantageous over other Streptomyces transformation protocols for two reasons: (1) it can readily transform large plasmids with an oriT into a Streptomyces recipient (e.g., cosmid DNA >10 kb) and (2) many oriT-bearing plasmids permit direct integration into a chromosomal locus (e.g., via int-attP on pKC1138).
A general protocol suitable for the transformation of Streptomyces lividans TK 64 or Streptomyces coelicolor CH999 is included here. It utilizes the nonmethylating E. coli ET12567/pUZ8002 strain as the host to bypass the restriction system of Streptomyces coelicolor CH999. This protocol is modified from a previously reported procedure (Kieser et al., 2000).
Materials
E. coli ET12567/pUZ8002 competent cells (ATCC BAA-525; plasmid pUX8002 is obtained from The John Innes Centre; http://www.jic.ac.uk/)
LB liquid medium (APPENDIX 4A) containing 25 μg/ml chloramphenicol (add from 25 mg/ml stock in ethanol) and 25 μg/ml kanamycin (add from 25 mg/ml stock in sterile double-distilled H2O)
LB agar plates (APPENDIX 4A) containing 25 μg/ml chloramphenicol (add from 25 mg/ml stock in ethanol) and 25 μg/ml kanamycin (add from 25 mg/ml stock in sterile double-distilled H2O)
Plasmid containing oriT (for this example, pOJ446; The John Innes Centre, http://www.jic.ac.uk/; see Basic Protocol 2)
Antibiotic to select for plasmid with oriT (antibiotic no. 3; for this example, apramycin; prepare 50 mg/ml stock in sterile H2O)
LB liquid medium (APPENDIX 4A) without antibiotics
Streptomyces recipient strain (Streptomyces lividans TK 64 or Streptomyces coelicolor CH999; The John Innes Centre, http://www.jic.ac.uk/), well-sporulated, growing on oatmeal agar (see recipe) plates
2× YT broth (see recipe)
Sterile mannitol soya flour agar plates (MS agar; see recipe)
Nalidixic acid: dissolve 50 mg nalidixic acid (Sigma-Aldrich) in 1 ml 0.15 M NaOH
R2YE medium (see recipe) supplemented with antibiotic no. 3
37°C incubator with orbital shaker
15-ml culture tubes
Centrifuge
Sterile toothpicks
50°C water bath
28°C incubator
Sterile spreaders
Additional reagents and equipment for preparing competent E. coli (Sambrook and Russell, 2001)
-
Prepare chemically competent cells of E. coli ET12567/pUZ8002 grown in LB liquid medium supplemented with kanamycin (25 μg/ml) and chloramphenicol (25 μg/ml), according to the protocol of Sambrook and Russell (2001).
These antibiotics maintain selection for the nonmethylating phenotype and the nontransmissible pUZ8002 “driver plasmid.”
-
Transform E. coli ET12567/pUZ8002 competent cells with the plasmid containing oriT (in this example, POJ446) and select for positive clones on LB agar supplemented with kanamycin (25 μg/ml), chloramphenicol (25 μg/ml), and antibiotic no. 3 for oriT-containing plasmid selection. Grow these transformations at 37°C for 12 to 16 hr.
In this example, pOJ446 requires apramycin, 50 μg/ml final concentration added from 50 mg/ml stock in water, as antibiotic no. 3.
-
Inoculate 15-ml culture tubes of 7 ml LB liquid medium supplemented with kanamycin (25 μg/ml) and chloramphenicol (25 μg/ml) plus antibiotic no. 3 by picking single colonies of E. coli ET12567/pUZ8002 transformants containing the desired plasmid. Grow cultures overnight.
Be sure to select for pUZ8002 and the dam− mutation by using both chloramphenicol and kanamycin. It is a good idea to make these cultures in triplicate, because ET12567/pUZ8002 sometimes loses resistance spontaneously, and one or more cultures may not achieve growth.
Centrifuge overnight cultures for 5 min at 1157 × g, room temperature, to collect cells at the bottom. Wash the cells with 7 ml of sterile LB liquid medium without antibiotics, centrifuge again as before, and remove the supernatant to eliminate residual antibiotics. Repeat this step one more time.
-
With a sterile toothpick, gently scrape spores from the top of a well-sporulated plate of the Streptomyces recipient strain, and add them to a sterile 1.5-ml centrifuge tube with 500 μl of 2× YT broth. Place the spores in a water bath at 50°C for 10 min.
A well-sporulated plate will be covered with dark gray spores that are easily scraped off with the side of a toothpick.
This heat-shock treatment causes the spores to germinate.
Place the Streptomyces spores on ice and resuspend the E. coli cells in 500 μl of 2× YT broth. Mix the E. coli in with the Streptomyces in the 1.5-ml microcentrifuge tube thoroughly by pipetting up and down.
-
Plate 200 μl of the Streptomyces–E. coli mixture on a sterile MS agar plate and spread the mixture evenly with a sterile spreader. Let it dry in the hood for 10 to 15 min.
It is good practice to spread this mixture until the plate has a smooth appearance. It can be difficult to initially distinguish potential exconjugants if aggregated spores remain on the plate.
Incubate the MS agar plates in a 28°C incubator for 16 to 20 hr.
-
Add 50 μl of 50 mg/ml nalidixic acid stock and 50 μl of 50 mg/ml apramycin stock to 1 ml of water, and mix by pipetting thoroughly. Add this mixture to the MS agar plate (step 8) to flood it, and spread the mixture evenly using a sterile spreader. Let the plate dry in the biological safety cabinet.
Nalidixic acid selectively kills the E. coli donor strain to select for Streptomyces exconjugants; streptomycetes are naturally resistant to nalidixic acid.
-
Incubate the plate in a 28°C incubator for 3 to 5 days until exconjugant colonies appear. Pick these individual colonies with the head of a sterile toothpick and then replicate to R2YE medium supplemented with antibiotic no. 3 (for this example, apramycin at 50 μg/ml final concentration) to select for transformants with the desired vector.
The original protocol by Kieser et al. (2000) recommends inoculating a fresh 100-ml culture of LB using 1% (v/v) seed culture and monitoring growth until absorbance at 600 nm reaches 0.4 to 0.6. In our experience, this protocol is very forgiving with regard to the growth phase of the E. coli ET12567/pUZ8002 donor cells, because even performing the conjugation with a 7-ml culture grown overnight for 10 to 12 hr can yield a suitable number of transformants. Also, this E. coli strain grows at a slower rate than other commonly used E. coli strains (e.g., BL21, XL1 Blue).
Optional
Successful transformants on replicative media can be further analyzed through genomic DNA extraction and PCR analysis and/or Southern Blot analysis to confirm successful integration.
ALTERNATE PROTOCOL 1
INTERGENERIC CONJUGATION BETWEEN S. GLOBISPORUS 1912, S. KANAMYCETICUS, AND S. CYANOGENUS S136, AND E. COLI
The Streptomyces used in Basic Protocol 3, S. lividans TK64 and S. coelicolor CH999, are hosts that are amenable to several forms of genetic manipulation: protoplast transformation, electroporation, and conjugation. Some strains, such as S. globisporus 1912, S. kanamyceticus, or S. cyanogenus S136 can only be transformed through conjugation. The following method is modified from the protocol developed by Fedorenko, which creates a genetic system to manipulate these industrially useful Streptomyces species (Luzhetskii et al., 2001, 2006).
This protocol differs from Basic Protocol 3 in that it uses E. coli ET12567/pUB307 as the donor strain. This E. coli strain is transformed with a derivative of pSET152 (Amr, ori pUC18, int-attP, φC31, oriT) harboring the desired genes for manipulation. pSET152 contains apramycin resistance and is an integrative plasmid with the int-attP genotype, which allows for stable chromosome integration at the attB site(s). Successful integration of the pSET152 derivative into the chromosome can be confirmed by Southern blotting.
Materials
E. coli ET12567/pUB307 competent cells (ATCC BAA-525; plasmid pUB307 is obtained from The John Innes Centre; http://www.jic.ac.uk/)
LB liquid medium (APPENDIX 4A) containing 50 μg/ml chloramphenicol (add from 25 mg/ml stock in ethanol) and 50 μg/ml kanamycin (add from 25 mg/ml stock in sterile double-distilled H2O)
LB agar plates (APPENDIX 4A) containing 50 μg/ml each of kanamycin (add from 25 mg/ml stock in sterile double-distilled H2O), chloramphenicol (add from 25 mg/ml stock in ethanol), and apramycin (add from 50 mg/ml stock in sterile double-distilled H2O)
pSET152 (Amr, ori pUC18, int-attP, φC31, oriT) harboring the desired genes for manipulation (Bierman et al., 1992; obtained from The John Innes Centre; pSET152 has restriction sites to create XbaI, BamHI, EcoRV, and EcoRI restriction fragments)
LB liquid medium (APPENDIX 4A) containing 50 μg/ml each of kanamycin (add from 25 mg/ml stock in sterile double-distilled H2O), chloramphenicol (add from 25 mg/ml stock in ethanol), and apramycin (add from 50 mg/ml stock in sterile double-distilled H2O)
LB liquid medium (APPENDIX 4A) without antibiotics
Oatmeal agar plates (see recipe) of S. globisporus 1912 (Luzhetskii et al., 2001; can be obtained from authors), S. kanamyceticus (ATCC 12853), or S. cyanogenus S136 (DSM 5087; http://www.dsm.com) grown to confluence over 3 to 5 days (well sporulated)
Tryptic Soy Broth (TSB; UNIT 10E.1)
Oatmeal agar plates (see recipe)
50 mg/ml nalidixic acid in 0.15 M NaOH
50 mg/ml apramycin in sterile H2O
- Southern blot kit (optional; Roche) for analysis of transformants including:
- Roche nylon membranes (cat. no. 11-699-075-001 (85 mm) or cat. no. 11-699-083-001 (132 mm)
- Anti-digoxigenin-AP conjugate, Fab fragments (cat. no. 11-093-274-910)
- NBT/BCIP (cat. no. 11-681-451-001 (8 ml)
- DIG Easy Hyb granules [cat. no. 11-796-895-001 (6× 100 ml)]
- Blocking reagent [cat. no. 11-096-176-001 (50 g)]
- Hybridization bags [cat. no. 11-666-649-001 (50 bags)]
- DIG DNA labeling kit [cat. no. 11-175-033-910 (40 labeling reactions)]
37°, 29°, and 28°C incubators with orbital shakers
15-ml culture tubes
Centrifuge
Sterile toothpicks
1.5-ml microcentrifuge tubes
50°C water bath
250-ml baffled Erlenmeyer flask with ventilated screw cap
Sterile spreaders
Additional reagents and equipment for preparing competent E. coli (Sambrook and Russell, 2001)
Transform E. coli
-
1.
Prepare competent cells of E. coli ET12567/pUB307 grown in LB liquid medium containing 50 μg/ml kanamycin and 50 μg/ml chloramphenicol, according to the protocol of Sambrook and Russell (2001).
-
2.
Transform E. coli ET12567/pUB307 competent cells with the modified pSET152 (described above) and select for positive clones on LB agar plates supplemented with 50 μg/ml each of kanamycin, chloramphenicol, and apramycin. Grow overnight (12 to 16 hr) at 37°C.
pSET152 has apramycin resistance.
-
3.
Inoculate 7 ml of LB liquid medium, supplemented with 50 μg/ml each of kanamycin, chloramphenicol, and apramycin, by transferring single colonies of E. coli ET12567/pUB307 transformants containing the pSET152 plasmid from the plate to the liquid medium. Grow overnight at 37°C with shaking (250 rpm) for 14 to 16 hr.
Again, preparing these inoculations in triplicate is highly recommended.
-
4.
Centrifuge overnight cultures 5 min at 1157 × g, room temperature, to collect cells at the bottom.
-
5.
Wash the cells with 7 ml of sterile LB liquid medium (without antibiotics), centrifuge again for 5 min at 1157 × g, room temperature, and remove the supernatant to eliminate any residual antibiotics. Repeat wash and centrifugation step.
Prepare Streptomyces
S. cyanogenus S136 does not readily sporulate.
-
6a.
For S. globisporus 1912 or S. kanamyceticus only: With a sterile toothpick, gently collect spores from well-sporulated oatmeal agar plates of S. globisporus 1912 or S. kanamyceticus recipient strain, and add them to a sterile 1.5-ml microcentrifuge tube with 300 μl of TSB at a concentration of 108 to 109 spores per ml. Place the spores in a water bath at 50°C for 10 min. Afterwards, incubate spores at 29°C for a further 3 to 4 hr in an orbital shaker.
-
6b.
For S. cyanogenus S136 only) S. cyanogenus: Gently scrape vegetative mycelia substrate from a 3- to 5-day-old plate of oatmeal agar and inoculate into a 250-ml baffled Erlenmeyer flask containing 50 ml of sterile TSB. Incubate for 24 to 28 hr at 29°C with shaking on an orbital shaker (250 rpm) to achieve the desired germination.
Do not heat shock the S136 culture.
-
7.
Centrifuge cells for 5 min at 1157 × g, room temperature.
Conjugate E. coli and Streptomyces and select for exconjugants
-
8.
Resuspend the Streptomyces seed pellet (from step 7) and E.coli cells (from step 5) in 500 μl of sterile TSB. Mix the cells at a 1:1 (v/v) ratio and spread 200 μl evenly on a sterile oatmeal agar plate with a sterile spreader. Incubate at 29°C for 16 to 20 hr.
-
9.
Add 50 μl of 50 mg/ml nalidixic acid and 50 μl of 50 mg/ml apramycin to 1 ml of water, and mix by pipetting up and down thoroughly. Add this mixture to the oatmeal agar plate and spread the mixture evenly using a sterile spreader. Let the plate dry in the biological safety cabinet.
-
10.
Incubate the plates for another 5 to 10 hr at 28°C until exconjugants start to appear.
-
11.
Pick and replicate exconjugants with a sterile toothpick on oatmeal agar supplemented with 50 μg/ml (final concentration) nalidixic acid, without apramycin. Grow exconjugants for 2 to 3 days until vegetative mycelia achieves appreciable growth.
-
12.
Inoculate 7 ml of sterile TSB with a small amount of vegetative mycelia from the Streptomyces strain in step 11. Grow for 24 to 28 hr and then inoculate a second 7 ml of TSB with 70 μl of the previous 7-ml culture from this step without apramycin. Repeat this nonselective culturing for 7 to 10 generations for shedding of nonintegrated pSET152.
-
13.
Plate 100 μl of the 10th-generation culture to oatmeal agar supplemented with apramycin (50 μg/ml; add from 50 mg/ml stock in ethanol). Allow 3 to 4 days for transformants to grow.
Optional
Successful transformants on replicative media can be further analyzed through genomic DNA extraction and PCR analysis and/or Southern Blot analysis to confirm successful integration.
ALTERNATE PROTOCOL 2
INTERGENERIC CONJUGATION BETWEEN STREPTOMYCES VERTICILLUS AND ESCHERICHIA COLI
S. verticillus (ATCC, cat. no. 15003), is an industrially useful Streptomyces species that produces the antitumor compound bleomycin. However, genetic manipulation of this strain was not successful until Shen and colleagues transformed this strain via intergeneric conjugation with E. coli S17-1 (Galm et al., 2008). This protocol uses a methylation-proficient E. coli donor (E. coli S17-1) and illustrates the troubles one can encounter in developing genetic systems for novel Streptomyces species or streptomycetes that are particularly difficult to transform via conventional protoplasting and electroporation protocols. This protocol uses derivatives of the integrating plasmids pSET152 and pRT801, and is adapted from previously reported protocols (Kieser et al., 2000; Galm et al., 2008).
NOTE: E. coli S17-1 has an RP4-mediated trans-acting mobilization function integrated directly into its chromosome. Therefore, this strain is a stable mutant that requires no antibiotics to select for its requisite trans function, and it grows much faster than the comparable nonmethylating strain E. coli ET12567/pUZ8002, which is kanamycin- and chloramphenicol-resistant. E. coli S17-1 is also sensitive to kanamycin, which allows it to serve as a host for oriT vectors with a kanamycin-resistance gene. (Simon et al., 1983).
Materials
E. coli S17-1 competent cells (ATCC 47055)
LB liquid medium (APPENDIX 4A)
pSET152 or pRT801 (Bierman et al., 1992; available from The John Innes Centre)
LB agar plates (APPENDIX 4A) supplemented with 50 μg/ml apramycin (add from 50 mg/ml stock in sterile water)
LB liquid medium supplemented with 50 μg/ml apramycin (add from 50 mg/ml stock in sterile water)
Streptomyces verticillus ATCC15003 spores from a well-sporulated ISP4 plate (see recipe) grown for 10 days
Tryptic Soy Broth (TSB; UNIT 10E.1) supplemented with 10.0% (w/v) sucrose and 0.4% (w/v) glycine (add 10 g sucrose and 0.4 g glycine to 100 ml TSB, then autoclave)
50 mg/ml nalidixic acid in 0.15 M NaOH
50 mg/ml apramycin in sterile double-distilled H2O
ISP4 agar plates (see recipe) supplemented with 20 ml/liter of 1 M 1MgCl2·6 H2O (20 mM final), 1 g/liter of tryptone (0.1% w/v final), and 500 mg/liter yeast extract (0.5% w/v final)
- Southern blot kit (optional; Roche) for analysis of transformants including:
- Roche nylon membranes (cat. no. 11-699-075-001 (85 mm) or cat. no. 11-699-083-001 (132 mm)
- Anti-digoxigenin-AP conjugate, Fab fragments (cat. no. 11-093-274-910)
- NBT/BCIP (cat. no. 11-681-451-001 (8 ml)
- DIG Easy Hyb granules [cat. no. 11-796-895-001 (6× 100 ml)]
- Blocking reagent [cat. no. 11-096-176-001 (50 g)]
- Hybridization bags [cat. no. 11-666-649-001 (50 bags)]
- DIG DNA labeling kit [cat. no. 11-175-033-910 (40 labeling reactions)]
37° and 30° incubator with orbital shaker
Spectrophotometer
15-ml culture tubes
Sterile 100-ml baffled Erlenmeyer flask
Sterile toothpicks
1.5-ml microcentrifuge tubes
50°C water bath
Prepare E. coli
-
1
Prepare competent cells of E. coli S17-1 by culturing strain in sterile LB liquid medium at 37°C, according to the protocol of Sambrook and Russell (2001). Monitor growth until absorbance at 600 nm (OD600) reaches 0.4 to 0.6.
-
2
Transform E. coli S17-1 competent cells with the derivative of pSET152/pRT801 and select for positive clones on LB agar supplemented with 50 μg/ml apramycin.
-
3
Inoculate culture tubes of 7 ml LB liquid medium (supplemented with 50 μg/ml apramycin) by picking single colonies of E. coli S17-1 transformants containing the pSET152/pRT801 derivative plasmid. Grow overnight for 8 to 10 hr.
As the doubling time for E. coli S17-1 is faster than that for E. coli ET12567/pUZ8002, E. coli S17-1 will reach OD600 0.4 to 0.6 more rapidly.
-
4
Take 70 μl of the overnight E. coli culture and inoculate a sterile 100-ml baffled Erlenmeyer flask of LB liquid medium (supplemented with 50 μg/ml apramycin) and grow the culture until OD600 reaches 0.4 to 0.6.
Since the genetic system for S. verticillus ATCC15003 requires a certain degree of optimization, it is better in this protocol to ensure that the E. coli donor strain is at the optimal stage of growth for intergeneric conjugation to achieve the highest number of exconjugants.
-
5
Centrifuge overnight cultures for 5 min at 1157 × g, room temperature, to collect cells at the bottom.
-
6
Wash the cells with 7 ml of sterile LB medium (without antibiotics) and centrifuge again for 5 min at 1157 × g, room temperature, to remove any residual antibiotics.
-
7
Repeat the wash and centrifugation step.
Prepare S. verticillus
-
8
With a sterile toothpick, gently collect spores from well-sporulated ISP4 plates of S. verticillus ATCC15003 recipient strain, and add the spores to a sterile 1.5-ml centrifuge tube with 500 μl of sterile TSB medium supplemented with 10.0% sucrose and 0.4% glycine. Place the spores in a water bath at 50°C for 10 min.
-
9
Afterwards, incubate spores at 30°C for a further 3 to 4 hr in an orbital shaker.
Conjugate E. coli and Streptomyces and select for exconjugants
-
10
Resuspend Streptomyces seed pellet in 500 μl of sterile TSB supplemented with 10.0% sucrose and 0.4% glycine and resuspend E. coli cells separately in 500 μl of TSB supplemented with 10.0% sucrose and 0.4% glycine. Mix cells in a 1:1 ratio (v/v) and spread 200 μl evenly on a sterile ISP4 plate (supplemented with MgCl2, tryptone, and yeast extract) with a sterile spreader. Incubate in a 30°C incubator for 16 to 20 hr at 30°C.
-
11
Add 50 μl of 50 mg/ml nalidixic acid and 100 μl of 50 mg/ml apramycin to 1 ml of water, and mix by pipetting thoroughly.
-
12
Add the mixture (from step 11) to the ISP4 plate to flood it, and spread the mixture evenly using a sterile spreader. Let the plate dry in the biological safety cabinet.
-
13
Incubate the plates at 30°C until exconjugants start to appear.
-
14
Pick exconjugants and plate on ISP4 supplemented with 100 μg/ml apramycin and 50 μg/ml nalidixic acid.
Optional
Genomic DNA from S. verticillus ATC15003 transformants may be analyzed directly to isolate positive clones carrying the desired integration via PCR analysis and Southern blot analysis.
BASIC PROTOCOL 4
GENERATION AND TRANSFORMATION OF STREPTOMYCES PROTOPLASTS
Protoplast transformation quite simply offers the highest frequency of Streptomyces species transformants when a strain is able to suitably generate protoplasts and regenerate them. Conditions for protoplast formation and regeneration were first developed by Okanishi and colleagues (Okanishi et al., 1974). The discovery that polyethylene glycol greatly enhances the transformation efficiency of Streptomyces with circular DNA plasmids firmly established this protocol for manipulating the various genes of streptomycetes (Bibb et al., 1980). This discovery led to a rapid accumulation of plasmid vectors for the cloning of various genes to manipulate Streptomyces species.
The following is a general protocol for the generation of protoplasts of Streptomyces lividans TK 64 and Streptomyces coelicolor CH999 and conditions for transformation. This protocol is modified from a previously described procedure (Kieser et al., 2000).
Materials
Yeast extract malt extract (YEME) medium (see recipe)
2.5 M MgCl2·6H2O
20% (w/v) glycine
R2YE plate of S. lividans TK 64 or S. coelicolor CH999 grown for 3 to 4 days
YEME medium (see recipe)
10.3% (w/v) sucrose: dissolve 103 g sucrose per liter H2O
Lysozyme (powder)
P Buffer (see recipe)
DNA sample
E. coli ET12567/pUZ8002 for passaging of DNA through non-methylating host for S. coelicolor CH999, and/or generic E. coli strain for routine cloning
T Buffer (see recipe)
Sterile R2YE plates (see recipe)
Appropriate antibiotic for selection
Baffled 250-ml Erlenmeyer flasks with ventilated screw caps
28°C incubator with orbital shaker
Sterile 50-ml centrifuge tubes with cotton filter plugs
Centrifuge
15-ml centrifuge tubes
Sterile spreaders
Prepare Streptomyces cultures
-
1
Prepare 1 liter YEME medium, aliquot 100 ml into each of ten 250-ml baffled Erlenmeyer culture flasks, and sterilize via autoclaving.
-
2
Add 200 μl of sterile 2.5 M MgCl2·6H2O and 2.5 ml of 20% glycine solution to each 100-ml flask.
-
3
Inoculate each flask with a small cube (2 cm2) of agar using a sterile toothpick from a freshly prepared R2YE plate of S. lividans TK 64 or S. coelicolor CH999 grown for 3 to 4 days. Place this flask in a 28°C incubator with orbital shaker for 3 to 4 days.
20% glycine has been found to weaken the cell wall of Streptomyces species, and growth should occur until the culture flask takes on a red coloration and the mycelia can be visualized as neat globules that virtually fill the flask. Care should be taken not to over-grow, as too-viscous mycelia may be difficult to lyse.
-
4
Take the well-grown Streptomyces species culture, pour into two 50-ml centrifuge tubes, and centrifuge for 8 min at 1157 × g, room temperature.
-
5
Pour off the supernatant and wash each cell pellet with 20 ml of 10.3% sucrose by dispersing the pellet in solution. Centrifuge again as in step 4.
-
6
Repeat the wash step and centrifugation as in step 5. Pour off supernatant.
Prepare protoplasts
If desired, a small sample of the culture before protoplast lysis may be observed on a slide via light microscopy. The formation of protoplasts may be viewed over the course of the lysis reaction microscopically until the desired level of lysis has been achieved. A sample before lysis should be kept and compared microscopically to samples every 15 min after lysis has been started. Complete lysis is indicated by a total removal of the cell wall (thinner outline of cells as compared to control). Lysis should be stopped before many cells have been observed to burst.
-
7
Prepare 2 mg/ml lysozyme in sterile 15-ml centrifuge tubes, and dissolve in P Buffer.
Generally, one can measure 40 mg lysozyme, dissolve in 15 ml of P Buffer, and dissolve thoroughly.
-
8
Add 5 ml of the lysozyme solution to the cell pellet to achieve a 20-ml volume.
This is generally sufficient for dispersing the cell pellet in the lysozyme mixture.
-
9
Resuspend cell pellet in the lysozyme solution by pipetting the solution up and down with a sterile pipettor.
-
10
Immerse the tube of Streptomyces solution in a water bath set to 37°C. Allow the cells to lyse until debris starts to settle in the bottom of the tube and the solution becomes cloudy. Shake the tube every 5 min to agitate the Streptomyces and prevent clumping.
In general, 30 to 40 min is a sufficient amount of time to lyse the cell wall, depending on concentration of Streptomycetes.
-
11
Remove the centrifuge tubes from the water bath and dilute the lysed Streptomyces mixture with 5 ml P Buffer to free any protoplasts sticking to debris.
-
12
Wet the cotton filter plug of a 50-ml centrifuge tube with 5 to 10 ml P Buffer. Pour the lysed Streptomyces solution over the cotton filter and allow the protoplasts to flow through and collect in the sterile tube. Rinse with 5 ml P Buffer to free any remaining Streptomyces protoplasts that may have become stuck in the filter.
-
13
Centrifuge the collected protoplasts for 8 min at 1157 × g, room temperature.
-
14
Pour off the supernatant and wash the protoplast pellet with 20 ml of P Buffer. Do not pipet the cells vigorously, as this may cause cellular lysis. However, resuspend the cells by tapping the bottom of the centrifuge tube with one finger. Repeat centrifugation as described in step 14.
This wash step is very important, as it serves to remove any residual lysozyme that may be left in solution.
-
15
Repeat the wash step (step 15) and pour off supernatant.
-
16
Resuspend protoplasts in 1 to 2 ml P Buffer depending on the quantity of Streptomyces protoplast samples desired. Resuspend by tapping on the bottom of the centrifuge tube, being careful not to introduce bubbles.
-
17
Aliquot 100 μl of this suspension into sterile 1.5-ml microcentrifuge tubes and place them on ice for 3 min.
These may either then be stored or used for introduction of DNA.
Store protoplasts
-
18
To store protoplasts, place the ice bucket of protoplast samples in a −80°C freezer.
These can be stored and remain viable for up to 1 to 2 months.
The ice bucket serves to gradually cool the protoplast sample to −80°C.
-
19
To revive a protoplast sample, remove the sample from the freezer, and run under warm (~30°C) water to thaw quickly.
Freeze slowly and thaw quickly when working with stored protoplast samples.
Introduce DNA into protoplasts
-
20
Remove a protoplast sample as prepared above from the ice bath and add 5 to 10 μl of DNA sample to the protoplasts by pipetting up and down.
In the case of S. coelicolor CH999 protoplasts, it is important to passage the DNA first through E. coli ET12567/pUZ8002 or some other nonmethylating strain, to bypass S. coelicolor CH999’s DNA restriction system.
-
21
Add 50 μl of T Buffer to the above sample, mix thoroughly by pipetting up and down, and spread this mixture evenly on a fresh R2YE plate with a sterile spreader. Let dry in a laminar flow biological safety cabinet and incubate at 28°C for 14 to 20 hr.
This allows the streptomycetes to recover and regrow their cell walls.
-
22
After incubation, flood the R2YE plate with 1 ml of sterile water and an appropriate concentration of antibiotic. Spread this mixture with a sterile spreader. Continue incubation at 28°C.
Colonies will be visible after ~2 to 4 days and may be plated on nonselective medium.
Optional
Successful transformants on replicative media can be further analyzed through genomic DNA extraction and PCR analysis and/or Southern Blot analysis to confirm successful integration.
BASIC PROTOCOL 5
ELECTROPORATION OF STREPTOMYCES MYCELIA
Electroporation has been a commonly used technique to transform E. coli, and recently has also been used to successfully transform Streptomyces species. This protocol involves the application of a low-voltage shock to streptomycete mycelium, which causes pores to open in cells for a brief moment, allowing the cells to become competent and take up DNA. This is advantageous because it bypasses preparation of an E. coli donor for conjugation and establishing and optimizing conditions to generate protoplasts. For strains that do not protoplast well, electroporation can serve as a useful means by which to introduce recombinant DNA. This protocol is a modification of previously described procedures (Pigac and Schrempf, 1995; Kieser et al., 2000).
Materials
Two plates of Streptomyces lividans TK 64 spores from fresh R2YE plate (suspended in 15% glycerol)
CRM liquid medium (see recipe)
10.3% (w/v) sucrose: dissolve 103 g sucrose per liter H2O
15% (v/v) glycerol, ice cold
Lysozyme
Electroporation solution (see recipe)
Miniprep of plasmid DNA to be electroporated
Selective medium for plasmid
Sterile toothpicks
50-ml centrifuge tubes
50°C water bath
Centrifuge
Electroporator (capable of administering a 2-kV pulse)
Sterilized 2-mm-gapped electroporation cuvettes
1.5-ml microcentrifuge tubes
28°C incubator with orbital shaker
-
Scrape spores of Streptomyces lividans TK 64 with a sterile toothpick and suspend in a 50-ml centrifuge tube with 20 ml sterile CRM liquid medium. Place the spores in a water bath set to 50°C for 10 min.
This is to germinate the spores.
-
Centrifuge the cells 6 to 8 min at 1157 × g, room temperature. Pour off supernatant and resuspend cells in 20 ml 10.3% ice cold sucrose. Centrifuge again as before and pour off the supernatant.
This is a wash step.
Resuspend the cells in 20 ml ice cold 15% glycerol. Centrifuge 6 to 8 min at 1157 × g, 4°C.
-
Resuspend the cells in 10 ml ice-cold 15% glycerol containing 1 mg lysozyme (lysozyme concentration, 100 μg/ml) and incubate at 37°C for 30 min.
This serves to weaken the cell wall to enhance the formation of transient pores during the electroporation.
Wash twice, each time using 10 ml ice-cold 15% glycerol, centrifuging down the cell pellet at 6 to 8 min at 1157 × g, 4°C, and removing the supernatant.
Resuspend pellet in 2 ml of electroporation solution. Take a 100-μl sample and put on ice, adding 5 to 10 μl of plasmid DNA miniprep, avoiding the addition of air bubbles to the Streptomyces cells.
Place the Streptomyces-DNA mixture from step 6 in a sterile ice-cold gapped 2-mm electroporation cuvette. Gently agitate the cuvette to force all of the mixture to settle at the bottom of the cuvette, again being careful to avoid introducing air bubbles. Place the cuvette into the electroporator and introduce a 2-kV electric pulse (10 kV cm−1 overall).
Wash the sample out of the cuvette with 500 μl ice-cold CRM medium into a sterile 1.5-ml microcentrifuge tube. Incubate the sample in a 28°C incubator, with shaking on orbital shaker at 250 rpm for 3 hr.
Plate the cells on selective medium (e.g., R2YE with appropriate concentration of antibiotic to select introduced plasmid).
Optional
Successful transformants on replicative media can be further analyzed through genomic DNA extraction and PCR analysis and/or Southern Blot analysis to confirm successful integration.
BASIC PROTOCOL 6
PREPARATION OF SAMPLES FOR COLONY PCR
Colony PCR is useful for quickly confirming the transformation of recombinant DNA into Streptomyces. It can also be used to amplify genes directly from a Streptomyces host without isolating genomic DNA.
Materials
Agar plate containing Streptomyces mycelia (see UNIT 10E.1, Basic Protocol 2)
PCR-grade H2O
Sterile toothpicks
1.5-ml microcentrifuge tubes
Additional reagents and equipment for PCR (Kramer and Coen, 2001)
Using a sterile toothpick, scrape mycelia from agar plate and place into a 1.5-ml microcentrifuge tube containing 200 μl of PCR-grade water.
Place the tube in boiling water for 10 min.
After 10 min, allow the tube to cool down to room temperature, then microcentrifuge 10 min at 13,000 rpm, room temperature.
Remove the supernatant and use as DNA template for PCR (Kramer and Coen, 2001).
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see APPENDIX 2A; for suppliers, see SUPPLIERS APPENDIX.
Blunted Pasteur pipets
-
Hold the tip of a Pasteur pipet above the flame of a Bunsen burner, spinning the pipet in your fingers to heat the tip evenly. Once heated, the tip should close forming a small ball at the end.
Blunting the tip avoids capillary action and provides a good tool to grab the precipitated DNA.
CRM liquid medium
10 g glucose
103 g sucrose
0.12 g MgCl2·6 H2O
15 g trypticase soy broth (as powder)
5 g yeast extract
-
Water to 1 liter
All liquid media should be placed into 250-ml baffled flasks with appropriate vented tops and then autoclaved (121°C for 45 min) and cooled (25°C) or stored (4°C) before use.
Baffled 250-ml Pyrex Erlenmeyer flasks with cotton or foam stoppers are preferred.
DNA loading buffer
40 g sucrose
0.25 g bromphenol blue
0.25 g xylene cyanol FF
Water to 100 ml
Store up to 3 months at 4°C
Electroporation buffer
100 ml glycerol (10% v/v final)
30 g PEG 1000
65 g sucrose
Water to 1 liter
Store up to 3 months at 4°C
ISP4 agar
10 g soluble starch
1.0 g dipotassium phosphate
1.0 g magnesium sulfate
2.0 g ammonium sulfate
1.0 g calcium carbonate
1 mg ferrous sulfate
1 mg magnesium chloride
1 mg zinc sulfate
Adjust to pH. 7.2 using 1 M NaOH
Add 18 g Bacto agar (BD Difco)
For conjugation plates (Alternate Protocols 1 & 2) add:
Add 20 ml of sterile 1 M MgCl2·6 H2O
1 g of tryptone (0.1% w/v final)
-
500 mg yeast extract (0.5% w/v final)
After autoclaving, the medium should be poured into petri dishes and allowed to cool and solidify. Plates should be poured in a sterile environment, preferably a biological safety cabinet. After solidifying, the plates can be stored at 4°C for several weeks.
ISP4 agar can be stored in bottles until needed for plating. Reheat and pour appropriate portion for plating in a 50-ml tube and add appropriate concentration of antibiotic, if necessary.
Lysis buffer (1 L)
150 g sucrose
12.5 ml 2 M Tris·Cl, pH 7.5 (APPENDIX 2A)
50 ml 0.5 M EDTA (APPENDIX 2A)
Water to 1 liter
Store up to 3 months at 4°C
Mannitol-Soya (MS) flour agar (MS agar)
Add to one autoclavable 1-liter bottle with a magnetic stirring bar:
20 g mannitol
20 g soya flour
20 g agar
1 liter H2O
-
After autoclaving in the 1-liter bottle, add 10 ml of sterile 1 M MgCl2·6H2O solution and stir
The medium should be poured into petri dishes and allowed to cool and solidify. Plates should be poured in a sterile environment, preferably a biological safety cabinet. After solidifying, the plates can be stored at 4°C for several weeks.
This recipe for MS agar is modified from Kieser et al. (2000).
Oatmeal agar (1 L)
60 g oatmeal
12.5 g Bacto agar (BD Difco)
Adjust pH to 6.0
For conjugation plates (Alternate Protocol 2):
-
Add 10 ml of sterile 1 M MgCl2·6H2O solution and stir
After autoclaving, the medium should be poured into petri dishes and allowed to cool and solidify. Plates should be poured in a sterile environment, preferably a biological safety cabinet. After solidifying, the plates can be stored at 4°C for several weeks.
Oatmeal agar can be stored in bottles until needed for plating. Reheat and pour appropriate portion for plating in a 50-ml tube and add appropriate concentration of antibiotic, if necessary. This recipe for oatmeal agar is modified from Kieser et al. (2000).
P buffer (protoplast buffer)
103 g sucrose
10.12 g MgCl2·6H2O
0.25 g K2SO4
2 ml trace element solution (see recipe)
Combine the above constituents in an appropriately sized flask, and add water to a total volume of 800 ml. Next, autoclave the buffer and the additives listed below. These additives are to be added to the above solution, after autoclaving, and in order listed:
10 ml 0.5% (w/v) KH2PO4
100 ml 3.68% (w/v) CaCl2
100 ml 5.73% (w/v) TES buffer, pH 7.2 (see recipe)
R2YE liquid and solid media
Medium A (add to one autoclavable 1-liter bottle with a magnetic stirring bar):
103 g sucrose
0.25 g K2SO4
10.12 g MgCl2·6H2O
10 g glucose
0.1 g casamino acids (BD Difco)
Distilled H2O to 800 ml
5 g yeast extract (BD Difco)
15 g Bacto-agar (BD Difco; for solid agar medium)
Add the following autoclaved solutions to solution A, in the order indicated:
10 ml 0.5% (w/v) KH2PO4
80 ml 3.68% (w/v) CaCl2 2H2O
15 ml 20% (w/v) l-proline·(20%, w/v)
100 ml 5.73% (w/v) TES buffer (see recipe)
2 ml trace element solution (see recipe)
-
5 ml 1 M NaOH
This will give 1 liter of R2YE liquid medium.
After sterilization, the solid medium should be poured into petri dishes and allowed to cool and solidify. Plates should be poured in a sterile environment, preferably a biological safety cabinet. After solidifying, the plates can be stored at 4°C for several weeks.
R2YE agar can be stored in bottles until needed for plating. Reheat and pour appropriate portion for plating in a 50-ml tube and add appropriate concentration of antibiotic, if necessary. The recipe for R2YE agar is modified from Kieser et al. (2000).
RNase solution
0.1 g RNase A (10 mg/ml final)
50 μl 2 M Tris·Cl, pH 7.5 (APPENDIX 2A)
30 μl 5 M NaCl
H2O to 10 ml
Heat to 100°C for 15 min
Cool slowly to room temperature
Store at −20°C until needed
T Buffer (transformation buffer)
Prepare these stocks:
100 ml of 5 M CaCl2
100 ml of Tris-maleic acid buffer, pH 8.0 (1 M Tris base adjusted to pH 8.0 with maleic acid)
2.5% (w/v) K2SO4
Trace element solution (see recipe)
10.3% (w/v) sucrose
Next, make T Buffer preparation solution no. 1
25 ml 10.3% (w/v) sucrose
0.2 ml trace element solution
1 ml 2.5% (w/v) K2SO4
75 ml H2O
Sterilize by autoclaving
Finally, prepare the T Buffer:
0.5 ml 5 M CaCl2,
1.5 ml Tris-maleic acid buffer, pH 8.0
27.9 ml of T Buffer preparation solution no. 1
Combine the above in a sterile tube to give 30 ml of solution. To this, add 10 g PEG 1000 and autoclave in a 50-ml centrifuge tube, covered in aluminum foil. After autoclaving, combine the 30 ml solution with the 10 g PEG 1000, mix thoroughly, and aliquot 1 ml of the resulting T Buffer solution per sterile 1.5-ml microcentrifuge tubes. Store at 4°C.
TES buffer
Dissolve 5.73 g N-tris(hydroxymethyl)methyl-2-aminoethanesulfonic acid (TES) powder in 100 ml water. Adjust to pH 7.2 by dropwise addition of 2 M NaOH. Sterilize by autoclaving. Store up to 3 months at 4°C.
Trace element solution
40 mg/liter ZnCl2
200 mg/liter FeCl3 6H2O
10 mg/liter CuCl2·2H2O
10 mg/liter MnCl2·4H2O
10 mg/liter Na2B4O7·10H2O
10 mg/liter (NH4)6Mo7O24·4H2O
Store up to 3 months at 4°C·
YEME (yeast extract malt extract)
3 g yeast extract
5 g Bacto-peptone
3 g malt extract
10 g/ml glucose
103 g sucrose
H2O to 1 liter
Aliquot 100 ml of the above solution into 250-ml baffled flasks and autoclave. After autoclaving, add the following autoclaved solutions to each flask before inoculating cultures for protoplasting:
0.2 ml 2.5 M MgCl2·6H2O
-
2.5 ml 20% (w/v) glycine
All liquid media should be placed into 250-ml baffled flasks with appropriate vented tops and then autoclaved (121°C for 45 min) and cooled (25°C) or stored (4°C) before use.
Baffled 250-ml Pyrex Erlenmeyer flasks with cotton or foam stoppers are preferred.
2× YT broth
16 g tryptone
10 g yeast extract
0.5 g sodium chloride
H2O to 1 liter
Transfer 100 ml solution into each of 10 (200-ml) glass bottles
-
Autoclave
All liquid media should be placed into 250-ml baffled flasks with appropriate vented tops and then autoclaved (121°C for 45 min) and cooled (25°C) or stored (4°C) before use.
Baffled 250-ml Pyrex Erlenmeyer flasks with cotton or foam stoppers are preferred.
COMMENTARY
Background Information
There are three primary means indicated in this unit for transformation of streptomycetes: protoplastation, electroporation, and intergeneric conjugation. Attention should be paid to employ sterile technique and to make note of the limitations of the particular strain and vector that are to be used. In general, Streptomyces strains require a fair amount of experimentation when determining which method is suitable for establishing a genetic system. The procedures outlined within merely serve as a starting point for genetic manipulation of Streptomyces species, and, likely, individualized protocols will need to be optimized for each strain.
Critical Parameters and Troubleshooting
Genomic library preparation requires many steps and is a difficult protocol to perfect. It is our experience that the preparation of genomic DNA causes the greatest difficulty during genomic library construction. Take extra care to appropriately select the genomic DNA with the desired amount of partial digestion as described in Basic Protocol 2.
The greatest difficulty in working with Streptomyces is contamination. Sterile technique should be employed at all times when performing any of these protocols. Failure to achieve transformants is often due to employing harsh conditions during preparation, using dilute DNA, or failure to achieve good protoplast formation. Increasing lysis time and increasing DNA concentration (or ratio of E. coli host used to recipient spores) can improve transformation efficiency.
Anticipated Results
These general protocols should allow for the basic manipulation of many Streptomyces species including the isolation of genomic DNA, constructing a genomic library, and various forms of transformation procedures.
Time Considerations
Isolation of genomic DNA (Basic Protocol 1) contains two overnight steps that should be taken into consideration when starting the protocol. When following any of the transformation protocols (Basic Protocols 3, 4, and 5), special time considerations should be given to the growth of the Streptomyces species involved as well as the preparation and sterilization of their growth media.
Acknowledgment
This work was supported by the NIH (CA91901 and CA102102).
Literature Cited
- Bibb M, Schottel JL, and Cohen SN 1980. A DNA cloning system for interspecies gene-transfer in antibiotic-producing Streptomyces. Nature 284:526–531. [DOI] [PubMed] [Google Scholar]
- Bierman M, Logan R, O’Brien K, Seno ET, Rao RN, and Schoner BE 1992. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116:43–49. [DOI] [PubMed] [Google Scholar]
- Evans GA, Lewis K, and Rothenberg BE 1989. High efficiency vectors for cosmid microcloning and genomic analysis. Gene 79:9–20. [DOI] [PubMed] [Google Scholar]
- Galm U, Wang L, Wendt-Pienkowski E, Yang R, Liu W, Tao M, Coughlin JM, and Shen B 2008. In vivo manipulation of the bleomycin biosynthetic gene cluster in Streptomyces verticillus ATCC15003 revealing new insights into its biosynthetic pathway. J. Biol. Chem 283:28236–28245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieser T, Bibb MJ, Buttner MJ, Chater KF, and Hopwood DA 2000. Practical Streptomyces Genetics. John Innes Foundation, Norwich, U.K. [Google Scholar]
- Kramer MF and Coen DM 2001. Enzymatic amplification of DNA by PCR: Standard procedures and optimization. Curr. Protoc. Mol. Biol 56:15.1.1–15.1.14. [DOI] [PubMed] [Google Scholar]
- Luzhetskii AN, Ostash BE, and Fedorenko VA 2001. Interspecies conjugation of Escherichia coli–Streptomyces globisporus 1912 using integrative plasmid pSET152 and its derivatives. Genetika 37:1340–1347. [PubMed] [Google Scholar]
- Luzhetskii A, Fedoryshyn M, Gromyko O, Ostash B, Rebets Y, Bechthold A, and Fedorenko V 2006. IncP plasmids are most effective in mediating conjugation between Escherichia coli and streptomycetes. Genetika 42:595–601. [PubMed] [Google Scholar]
- Mazodier P, Petter R, and Thompson C 1989. Intergeneric conjugation between Escherichia coli and Streptomyces species. J. Bacteriol 171:3583–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okanishi M, Suzuki K, and Umezawa H 1974. Formation and reversion of Streptomycete protoplasts—Cultural condition and morphological study. J. Gen. Microbiol 80:389–400. [DOI] [PubMed] [Google Scholar]
- Pigac J and Schrempf H 1995. A simple and rapid method of transformation of Streptomyces rimosus R6 and other Streptomycetes by electroporation. Appl. Environ. Microb 61:352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RN, Richardson MA, and Kuhstoss S 1987. Cosmid shuttle vectors for cloning and analysis of Streptomyces DNA. Methods Enzymol. 153:166–198. [DOI] [PubMed] [Google Scholar]
- Sambrook J and Russell DW 2001. Molecular Cloning: A Laboratory Manual, 3rd ed. Vol. 1, pp. 1.117–1.118. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. [Google Scholar]
- Simon R, Priefer U, and Puhler A 1983. A broad host range mobilization system for in vivo genetic engineering—Transposon mutagenesis in Gram-negative bacteria. Bio-Technol. 1:784–791. [Google Scholar]
- Voytas D 2000. Agarose gel electrophoresis. Curr. Protoc. Mol. Biol 51:2.5A.1–2.5A.9. [DOI] [PubMed] [Google Scholar]
- Wahl GM, Lewis KA, Ruiz JC, Rothenberg B, Zhao J, and Evans GA 1987. Cosmid vectors for rapid genomic walking, restriction mapping, and gene transfer. Proc. Natl. Acad. Sci. U.S.A 84:2160–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]