

Abstract
Laboratory evolution studies have demonstrated that parallel evolutionary trajectories can lead to genetically distinct enzymes with high activity towards a non-preferred substrate. However, it is unknown whether such enzymes have convergent conformational dynamics and mechanistic features. To address this question, we use as a model the wild-type Homo sapiens kynureninase (HsKYNase), which is of great interest for cancer immunotherapy. Earlier, we isolated HsKYNase_66 through an unusual evolutionary trajectory, having a 410-fold increase in the kcat/KM for kynurenine (KYN) and reverse substrate selectivity relative to HsKYNase. Here, by following a different evolutionary trajectory we generate a genetically distinct variant, HsKYNase_93D9, that exhibits KYN catalytic activity comparable to that of HsKYNase_66, but instead it is a “generalist” that accepts 3’-hydroxykynurenine (OH-KYN) with the same proficiency. Pre-steady-state kinetic analysis reveals that while the evolution of HsKYNase_66 is accompanied by a change in the rate-determining step of the reactions, HsKYNase_93D9 retains the same catalytic mechanism as HsKYNase. HDX-MS shows that the conformational dynamics of the two enzymes are markedly different and distinct from ortholog prokaryotic enzymes with high KYN activity. Our work provides a mechanistic framework for understanding the relationship between evolutionary mechanisms and phenotypic traits of evolved generalist and specialist enzyme species.
Subject terms: Enzyme mechanisms, Molecular evolution, Mass spectrometry
In this work, the authors show that two parallel and distinct evolutionary pathways can lead to genetically diverse enzyme species that harbour similar catalytic activities against the desired substrate, modulated by distinct conformational dynamics during catalysis.
Introduction
More than 40 years ago Jensen proposed his seminal hypothesis1 postulating that the evolution of enzymes with high catalytic activity and selectivity (often called “specialist” enzymes) arises from a progenitor enzyme having lower activity but a high degree of catalytic promiscuity (“generalist” enzymes). There have been numerous reports of natural or laboratory evolution of specialist enzymes to “generalist” enzymes with markedly higher catalytic activity for a non-preferred substrate that is accompanied by relaxed substrate specificity2–6. Elegant recent studies by Tokuriki and others support the notion that the increased promiscuity often observed in enzymes evolved for a new function is the outcome of strong selective pressure for that function in the absence of counterselection7–9. In other words, selected mutations that lead to the largest possible increases in a new function may or may not decrease the primary function of the template enzyme “creating a bias towards initially weak trade-offs and the emergence of generalist enzymes”7,10,11.
In several contexts, it has been shown that parallel, distinct evolutionary trajectories can lead to a comparable threshold of new function and share similar amino acid substitutions12,13. Kaltenbach et al. showed that directed evolution of arylesterase (AE) from phosphotriesterase (PTE) resulted in a highly active and selective specialist AE enzyme with a ∼105-fold increase in arylesterase activity and a comparable (i.e. >104) decrease in phosphotriesterase activity14. By contrast, “reverse evolution” starting with AE led to a generalist “neoPTE” with 104-fold greater phosphotriester hydrolysis activity with minimal reduction in aryltransferase activity. Structural studies revealed that PT and neoPT presented a similar active site environment and the dependence of the kcat/KM on the pKa of the leaving group suggested that the rate-determining step is the same for both enzymes. These findings are of major importance for understanding the reversibility of evolutionary pathways and the nature of epistatic interaction networks in evolved enzymes15–19. However, they do not address the key question of whether genetically distinct enzymes arising from parallel evolutionary trajectories have convergent conformational dynamics and enzymatic mechanisms. How is the chemical step in catalysis optimized in genetically different enzyme species resulting from parallel trajectories, yet showing similar activity against the evolved substrate? How do their respective conformational ensembles during catalysis of the new substrate converge and compare to those from other ortholog enzymes that have naturally evolved to accept this substrate?
As a model system to address these questions we have been focusing on the Homo sapiens kynureninase (HsKYNase), an enzyme with important therapeutic implications in cancer immunotherapy20. Kynureninases (KYNases) are pyridoxal-5’-phosphate (PLP)-dependent enzymes of the aminotransferase superfamily that catalyze the hydrolysis of the Cβ-Cγ bond of L-Kynurenine (KYN) or of 3’-OH-L-Kynurenine (OH-KYN) forming L-alanine (ALA) and either anthranilic acid (AA) or 3’-OH-anthranilic acid (OH-AA), respectively (Fig. 1a). Similar to all animal KYNases, HsKYNase catalyzes the hydrolysis of OH-KYN but has very low activity towards KYN [(kcat/KM)OH-KYN/(kcat/KM)KYN > 400]21,22. Conversely, most bacterial enzymes, strongly prefer KYN over OH-KYN. Recently, we reported a very rugged fitness landscape leading to the evolution of the OH-KYN-selective HsKYNase to a specialist mutant enzyme, [HsKYNase_66; (kcat/KM)KYN/(kcat/KM)OH-KYN ~ 160] which is capable of hydrolyzing KYN with a catalytic efficiency and selectivity towards KYN comparable to that of the parental HsKYNase for its own preferred substrate (OH-KYN)23. In this process, numerous random mutagenesis and structure-guided directed evolution trajectories initially converged to the isolation of enzymes with modest ( ~ 30-fold) increase in KYN catalytic activity and no selectivity for KYN over OH-KYN (HsKYNase_4623) rendering them as OH-KYN-specialists. Along these lines, we note that a parallel evolutionary trajectory, which relied only on the randomization of loops distal from the active site with high crystallographic B-factors led to an enzyme (BF-HsKYNase24) with similar catalytic properties as HsKYNase_46. Crossing the 30-fold threshold in KYN activity was only possible via incorporating two potentiating amino acid substitutions, H102W-N333T identified by their strict phylogenetic conservation among the bacterial KYN-preferring enzymes. While these two amino acid substitutions are detrimental for KYN hydrolysis for the parental HsKYNase enzyme and for the moderately more active variants (HsKYNase_46 and BF-HsKYNase), their introduction enabled a two-step evolutionary trajectory that led to HsKYNase_66 with 410-fold higher KYN activity relative to the parental human enzyme (Fig. 1b). The catalytic proficiency of HsKYNase_66 with KYN and its selectivity for KYN over OH-KYN are comparable to those of the most active, KYN-selective bacterial enzymes (exemplified by the prototypical bacterial enzyme from P. fluorescens)25,26, which shares less than 30% amino acid identity with HsKYNase_66. Surprisingly, the evolution of HsKYNase_66 resulted in a switch in the rate-determining step of the parental HsKYNase to that of KYN-preferring bacterial enzymes, but no change in the identity of the specificity-determining steps. Hydrogen-deuterium exchange coupled to mass-spectrometry (HDX-MS) studies revealed that the active site of Hs-KYNase_66 displayed a heterogeneous dynamic profile during KYN turnover with certain regions showing neutral flexibility whereas others were stabilized or, conversely exhibited higher flexibility. In contrast, the active site of the phylogenetically distant, high-KYN catalytic activity, P. fluorescens enzyme was found to be pre-organized for efficient KYN binding and subsequent productive turnover, underscoring the role of conformational dynamics in acquisition of catalytic proficiency22.
Fig. 1. Evolutionary trajectories that led to the identification of generalist HsKYNase_93D9 and specialist HsKYNase_66.

a Reaction(s) catalyzed by Kynureninases (X denotes either -H or -OH groups). b Schematic showing the directed evolution trajectory leading to the isolation of HsKYNase_6623. Blunted arrows denote dead-end evolutionary intermediates, from which higher-catalytic-activity variants could not be selected from numerous libraries. c Structural representation of the mutations in key intermediates in the evolutionary trajectory to HsKYNase_93D; Intermediate variants comprising HsKYNase_1 (mutated amino acids in magenta), HsKYNase_8 (orange), high B-factor_10 (green) and rationally designed variants (red) were shuffled and then selection led to the isolation of HsKYNase_93 (for more details see main text, Supplementary Fig. 2, Supplementary Tables 1–7). Center: Location of the 15 amino acid substitutions of HsKYNase_93 is shown in spheres color-coded as described above. Right: Mutations in HsKYNase_93D9 showing in blue the two additional substitutions located in a critical active site loop (see Supplementary Fig. 2 and Supplementary Tables 1–7). The PLP co-factor and the 3-hydroxy-hippuric acid inhibitor are shown in yellow and green sticks respectively. The inhibitor is color-coded by atom (N: blue, O: red). d Schematic summarizing the directed evolution trajectory leading to the isolation of HsKYNase_93D9. Combination of mutations found in intermediate variants such as HsKYNase_1 and HsKYNase_8 (both showing KYN improvement of ~17-fold) with those from high B-factor_10 (6-fold) and rationally designed variants (3- and 16-fold) (Table 2 and Supplementary Tables 3, 4) led to the identification of HsKYNase_93 and finally HsKYNase_93D9.
Residues W102 and T333 (numbering based on the amino acid sequence of HsKYNase) in KYNase enzymes confer substrate specificity for KYN with marginal effects on the catalytic turnover. Other aspects of the catalytic mechanism and the key residues associated with catalysis appear to be conserved across all species25,27. Even though H102W and N333T served as potentiating mutations to favor KYN for the evolution of HsKYNase_66, we wondered whether there could be alternate evolutionary trajectories that might also lead to high KYN catalytic activity while preserving the native amino acids H102 and N333, which are nominally the hallmark of OH-KYN-preferring enzymes. Inspired by analysis of lower eukaryotic members of the enzyme family and from our earlier studies on the importance of distal mutations and conformational dynamics in KYN hydrolysis, we identified a bona-fide generalist enzyme, termed HsKYNase_93D9, displaying equally high catalytic activity for both KYN and OH-KYN (substrate specificity ratio [(kcat/KM)KYN/(kcat/KM)OH-KYN] ~1). The generalist HsKYNase_93D9 and the specialist HsKYNase_66 share only 5 amino acid mutations out of 24 and 17, respectively. Pre-steady-state analysis revealed that the rate-determining steps for KYN hydrolysis are different for the two enzymes while HDX-MS analysis of HsKYNase_93D9 generalist during catalysis of KYN or OH-KYN revealed significantly distinct conformational dynamics relative to HsKYNase_66.
In this work, we collectively show that two parallel and distinct evolutionary pathways can lead to genetically diverse enzyme species that harbor similar catalytic activities against the desired substrate, which, however, are achieved by different conformational dynamics during catalysis. Remarkably, those conformational dynamics profiles are totally distinct from ortholog enzymes that have naturally evolved to catalyze the same reaction. Importantly, active site potentiating mutations (in the case of HsKYNase_66, the H102W-N333T motif) do not only confer substrate specificity in favor to the evolved substrate, but are also imperative for the optimization of the chemical step of the new function, manifested by alterations in the rate-determining step of the reactions.
Results
Evolution of the HsKYNase_93D9 generalist enzyme
We expressed and characterized Saccharomyces cerevisiae KYNase (ScKYNase) which, like all eukaryotic members of the family, contains the signature OH-KYN-preferred active site residues, H102 and N333 (numbering based on HsKYNase). ScKYNase however, also harbors several residues highly conserved among the bacterial KYNases (Supplementary Fig. 1) and has a kcat/KM for KYN which is 20-fold higher than that of the Homo sapiens enzyme (Table 1). Further, the kinetics of ScKYNase with KYN and OH-KYN are quite similar with those of HsKYNase_46 (Fig. 1b, Table 1), (the “dead-end” intermediate identified in the evolution of the high KYN activity HsKYNase_66) even though the two enzymes share only ~45% identity.
Table 1.
Steady-state kinetic parameters for HsKYNase_93, HsKYNase_93D9 and other relevant enzymes as discussed in the Results section
| Enzyme | KYN | OH-KYN | ||||
|---|---|---|---|---|---|---|
| kcat (s−1) | KM (μM) | kcat/KM (M−1s−1) | kcat (s−1) | KM (μM) | kcat/KM (M−1s−1) | |
| HsKYNasea | 0.12 ± 0.02 | 1200 ± 120 | 100 ± 26 | 1.70 ± 0.1 | 29 ± 5 | (6 ± 1.3) × 104 |
| ScKYNase | 1.0 ± 0.08 | 500 ± 63 | (2 ± 0.3) × 103 | 0.75 ± 0.025 | 24 ± 3 | (3 ± 0.39) × 104 |
| MpKYNaseb | 5.5 ± 0.3 | 60 ± 8 | (9 ± 1.3) × 104 | 5.65 ± 0.5 | 1200 ± 175 | (4.7 ± 0.8) × 103 |
| HsKYNase_46c | 0.58 ± 0.01 | 190 ± 20 | (3.1 ± 0.25) × 103 | 1.60 ± 0.17 | 14 ± 0.5 | (11.4 ± 1.0) × 104 |
| HsKYNase_46-H102W/N333Tc | 0.044 ± 0.002 | 210 ± 20 | 210 ± 27 | Non-detectable | ||
| HsKYNase_66 | 1.68 ± 0.1 | 41 ± 7 | (4.1 ± 0.7) × 104 | 0.84 ± 0.04 | 710 ± 60 | (1.2 ± 0.12) × 103 |
| HsKYNase_93 | 1.50 ± 0.1 | 110 ± 15 | (1.36 ± 0.2) × 104 | 1.0 ± 0.075 | 90 ± 12 | (1.11 ± 0.17) × 104 |
| HsKYNase_93D9 | 1.45 ± 0.07 | 85 ± 10 | (1.7 ± 0.22) × 104 | 2.80 ± 0.1 | 180 ± 15 | (1.55 ± 0.14) × 104 |
| HsKYNase_93D9-H102W/N333T | 0.10 ± 0.007 | 670 ± 90 | 150 ± 23 | 0.30 ± 0.01 | 1180 ± 58 | 255 ± 15 |
Based on sequence comparisons of: (i) the ScKYNase, (ii) a high KYN-activity bacterial enzyme (Mucilaginibacter paludis; MpKYNase) and (iii) HsKYNase_66 (Supplementary Fig. 1), we identified positions in the parental HsKYNase sequence that could possibly be altered to afford higher KYN catalytic activity. Sets of 3-4 residues that included amino acid residues upstream and downstream of the identified positions were subjected to combinatorial site saturation mutagenesis. Twenty-one libraries (Supplementary Table 1) were screened using an in-vivo selection for anthranilate complementation of a ΔtrpE E. coli strain that is supplied from the hydrolysis of KYN supplemented to minimal media, followed by 96-well plate assays of clones which could support growth on agar plates, as reported earlier23. Two of these libraries converged to one variant each containing 4 and 3 amino acid substitutions, respectively: HsKYNase_1: F71M/L72N/K81R/M82N and HsKYNase_8: E103F/V104H/G105T (Table 2, Supplementary Fig. 2, Supplementary Table 2). These two variants resulted in a ~ 17-fold increase in (kcat/KM)KYN over the wild-type HsKYNase that served as the original template. The catalytic activity of these enzymes is comparable to ScKYNase. The G105T substitution is particularly notable as G105 is conserved among both bacterial and eukaryotic KYNases (Supplementary Fig. 1). Mutations among these two variants were recombined by DNA shuffling in reactions that also included: (i) genes encoding 2 rationally designed variants showing modest activity improvements (Combo variant_1 and rationally-designed variant_12, Table 2, Supplementary Fig. 2, Supplementary Tables 2, 3) (ii) a set of genes with amino acid substitutions in two high B-factor regions, which while distant from the active site, increase KYN hydrolysis by 20-fold by allosterically affecting the conformational dynamics around the PLP binding pocket24 (Supplementary Table 4). The resulting library was screened first for anthranilate auxotrophy complementation and then by 96-well plate screening, as above. This directed evolution campaign led to the isolation of a variant (termed HsKYNase_93) with 136-fold improved (kcat/KM)KYN and about 5.5-fold lower (kcat/KM)OH-KYN relative to HsKYNase (Fig. 1d, Table 1). HsKYNase_93 harbors mutations that are found in all HsKYNase intermediate variants that were recombined to create a shuffled library as described above. Of note, it contains all the mutations of B-factor_10 highlighting the contribution of distal mutations located at dynamic regions to enzyme catalysis (Table 2, Supplementary Fig. 2, Supplementary Table 4). The location of the amino acid residues of the five key HsKYNase intermediate variants and HsKYNase_93 is shown in Fig. 1c.
Table 2.
HsKYNase variants critical in the evolution of HsKYNase_93D9 and their respective amino acid substitutions
| Enzyme species | Type of mutagenesis | # of mutations | Amino acid substitutions | Fold relative to HsKYNase |
|---|---|---|---|---|
| HsKYNase_1 | NNS combinatorial mutagenesis | 4 | aF71M-L72N-K81R-M82N | 17.2 |
| HsKYNase_8 | NNS combinatorial mutagenesis | 3 | E103F-V104H-G105T | 17 |
| Combo variant_1 | Rational combination of beneficial mutations | 6 | F71M-L72N-K81R-M82N-I331N-F306Y | 16 |
| Rationally-designed variant_12 | Rational combination of beneficial mutations | 6 | E259P-W272G-S274G-A280A-A282P-G287F | 3 |
| B-factor_10 | NNS & rational combination of beneficial mutations | 7 | I183P-E184A-M187L-M189I-K191N-E197T-I201T | 6 |
| HsKYNase_93 | DNA shuffling | 15 | F71M-L72N-E103F-V104H-G105T-I183P-E184A-M187L-M189I-K191N-E197T-I201T-A280S-A282P-F306Y | 131 |
| HsKYNase_93D9 | NNS combinatorial mutagenesis & DNA shuffling | 17 | 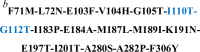 |
170 |
aMutations that are incorporated in HsKYNase_93 through DNA shuffling are shown in underlined and bold font throughout the Table.
bThe two additional amino acid substitutions of HsKYNase_93D9 relative to HsKYNase_93 are shown in blue. All mutations in bold font are present in both HsKYNase_93 and HsKYNase_93D9.
Using HsKYNase_93 as template, further combinatorial site-saturation mutagenesis (Supplementary Tables 1, 5), gene shuffling (Supplementary Table 6) and library screening resulted in the isolation of an even more active variant, HsKYNase_93D9 with a (kcat/KM)KYN ~ 1.7×104 M−1s−1 (170-fold higher than HsKYNase) and slightly lower activity for OH-KYN, (kcat/KM)OH-KYN ~ 1.55 ×104 M−1s−1 (Table 1, Supplementary Fig. 2, Supplementary Table 7). Thus, HsKYNase_93D9 has nearly identical kcat/KM for both of substrates and therefore it represents a bona-fide generalist enzyme. Strikingly, despite carrying the H102-N333 OH-KYN-preferring motif found exclusively in animal enzymes, the apparent (kcat/KM)KYN of HsKYNase_93D9 is comparable (2.4-fold lower) to that of the KYN-specialist HsKYNase_66 ((kcat/KM)KYN ~ 4.1×104 M−1s−1, (kcat/KM)KYN / (kcat/KM)OH-KYN = 34) and also to its bacterial, KYN-preferring counterparts. Only 4/17 amino acid substitutions in HsKYNase_93D9 are identical to residues found in KYN-preferring prokaryotic enzymes (Supplementary Fig. 1), namely E103F-I183P-I201T-A282P. Interestingly, while the substitution F225Y (located close to the active site) was found in all previously KYN-improved HsKYNase variants (HsKYNase_46 and _66), in ScKYNase and in the bacterial MpKYNase, HsKYNase_93D9 maintained the wild-type residue F225. Conversely, HsKYNase_93D9 includes an amino acid substitution in its active site (F306Y) that is not found in other HsKYNase species, ScKYNase or bacterial KYNases (Supplementary Fig. 1). Introduction of the “canonical” KYN substrate specificity residues Trp and Thr at positions H102 and N333 respectively onto HsKYNase_93D9, dramatically reduced its catalytic activity towards both KYN and OH-KYN by 113- and 60-fold respectively (Table 1). These observations illustrate that: (a) the “canonical” KYN-specificity motif W102-T333 which had been deduced by phylogenetic analysis is in fact not biochemically essential for conferring high KYN catalytic activity and (b) the H102W-N333T substitutions can confer KYN over OH-KYN selectivity only in combination with a large network of epistatic mutations that were identified through evolution23. We note that HsKYNase, HsKYNase_93D9 and HsKYNase_66 are all thermostable with their respective Tm values measured by differential scanning fluorometry being ~73, ~76 and 82 °C, respectively.
Pre-steady-state kinetics analysis
We next examined how the distinct catalytic properties of the two high KYN activity enzymes HsKYNase_93D9 and HsKYNase_66, which arose via distinct evolutionary trajectories, may reflect differences in their respective catalytic mechanisms. We determined the rate-determining step for the reaction of HsKYNase_93D9 with KYN or OH-KYN by performing pre-steady-state stopped-flow kinetic analysis to monitoring the fluorescence signal due to the formation and release of the product anthranilate (AA; reaction with KYN) or OH-anthranilate (OH-AA; reaction with OH-KYN) as described previously22. When HsKYNase_93D9 was mixed with KYN, AA generation was linear over time (Fig. 2a) indicating that the rate-determining step is the chemical step (or a step preceding it) of the catalytic cycle. The same kinetic behavior was observed in the reaction of HsKYNase with KYN22,24. Consequently, the 170-fold improvement of (kcat/KM)KYN for HsKYNase_93D9 enzyme does not involve a change in the rate-determining step of the reaction. In contrast, the hydrolysis of KYN by the high catalytic activity specialist HsKYNase_66 resulted in a burst of AA formation after mixing with KYN which indicates that product release is, at least partially, rate-limiting28. In contrast, the absence of a burst (or a lag) implies that the chemical step is rate-limiting for both wild-type HsKYNase22 and HsKYNase_93D9 (Fig. 2a).
Fig. 2. Comparison of pre-steady-state kinetics for HsKYNase_93D9 and HsKYNase_66 with KYN or OH-KYN.

a Substrate concentration-dependence of pre-steady-state formation of AA after mixing of 1 μM HsKYNase_93D9 with different KYN concentrations in the stopped-flow instrument. Smooth curves: simulated traces globally fit to the four-step minimal model shown in panel b. b Four-step minimal kinetic model for the reaction pathway of HsKYNase_93D9 with KYN. Intrinsic rate constants shown in green were calculated by globally fitting the experimental data shown in panel a. Intrinsic rate constants that were locked during global data fitting are shown in Table 3. Species that were experimentally observed are shown in red. E: enzyme, S: substrate, AA: anthranilic acid and P: Alanine. c Pre-steady-state formation of OH-AA upon mixing 5 (red) and 10 (green) μM HsKYNase_93D9 with 500 μM OH-KYN. d Substrate concentration-dependence of pre-steady-state AA burst formation after mixing 5 μM HsKYNase_66 with different concentrations of KYN as indicated in the plot. e Calculated intrinsic rate constants for the HsKYNase_66/KYN reaction pathway after global fitting of the data shown in panel d. Calculated values are colored as in panel b above. f Pre-steady-state formation of OH-AA upon mixing 12.5 μM HsKYNase_66 with 500 μM OH-KYN. All concentrations refer to the final concentration after mixing the solutions in the stopped-flow instrument and each trace represents the average of five measurements. All reactions were performed in PBS, pH 7.4 at 37 °C.
We examined the KYN concentration-dependence of the fluorescence signal to estimate and then compare the intrinsic rate constants of the different reaction steps for HsKYNase_93D9 and HsKYNase_66 by globally fitting the data to a realistic four-step kinetic model using the KinTek Explorer program29 (Fig. 2a, d). Overall, confidence contour analysis30 revealed that the calculated intrinsic rate constants are well-constrained by the data (Supplementary Fig. 3, Supplementary Tables 8, 9). For HsKYNase_93D9 we observed substrate inhibition at high concentrations of substrate, a phenomenon also evident with HsKYNase22 and with BF-HsKYNase24. For this reason, in deriving a kinetic model we used only data with KYN concentrations below the range where substrate inhibition was observed (0-75 μM KYN). This comprehensive analysis supported the conclusion that the observed kcat is determined by the rate of chemistry (formation of AA) for HsKYNase_93D9 (k2 ~ 0.8 s−1; Fig. 2b, Table 3), which is comparable to the observed steady-state turnover rate (1.45 s−1). In contrast, with HsKYNase_66 kcat is limited by the apparent rate of ALA release (k4 ~ 1.5 s−1; Fig. 2e, Table 3). Of note, the calculated second-order rate constant for KYN binding (k1) appeared to be ~25-fold slower for HsKYNase_93D9 (0.24 μM−1s−1) relative to HsKYNase_66 (5.9 μM−1s−1). This finding is consistent with the lag phase observed with HsKYNase_93D9 (Fig. 2a).
Table 3.
Rate constants and kinetic parameters for the reaction of KYN with HsKYNase_93D9 and HsKYNase_66 derived by the global fitting of the experimental data (Fig. 2a, d) to the kinetic models shown in Fig. 2b and e
| Rate constant and kinetic parameter | HsKYNase_93D9 | HsKYNase_66 | ||
|---|---|---|---|---|
| Best fit | 95% Confidence interval | Best fit | 95% Confidence interval | |
| k1 | 0.24 μM−1s−1 | (0.22, 0.27) | 5.9 μM−1s−1 | (5.5, 6.4) |
| k−1 | 4 s−1 | (3.31, 4.83) | 268 s−1 | Locked |
| k2 | 0.79 s−1 | 0.77, 0.81) | 3.1 s−1 | (2.78, 3.28) |
| k−2 | a0.34 s−1 | Locked | 19 s−1 | Locked |
| k3 | 11.5 s−1 | Locked | 21 s−1 | (17.5, 24.8) |
| k−3 | 0.001 μM−1s−1 | Locked | 0.001 μM−1s−1 | Locked |
| k4 | 11 s−1 | Locked | 1.5 s−1 | Locked |
| k−4 | 0.02 μM−1s−1 | Locked | 0.001 μM−1s−1 | Locked |
| kcat (s−1) | 0.68 | - | 0.75 | - |
| KM (μM) | 17.5 | - | 21.1 | - |
| kcat/KM (M−1s−1) | 3.9 × 104 | - | 3.6 × 104 | - |
aRate constants shown in bold font were locked at the displayed value corresponding to either their lower or upper limit during the global fitting process.
Rate constants that were allowed to float during the global data fitting (shown in green font in Fig. 2b and e) are summarized with confidence intervals by contour analysis. Using Eqs. 3, 4 and 5 (Methods) and the rate constants from the pre-steady-state analysis that are listed above, the steady-state kinetic parameters kcat, KM and kcat/KM were estimated respectively.
Using the estimated values of the intrinsic rate constants derived by the global fitting analysis (Table 3) to the four-step kinetic model (Fig. 2b, e), the steady-state parameters kcat, KM and kcat/KM of HsKYNase_93D9 and HsKYNase_66 were calculated using the Eqs. (3), (4) and (5) respectively as described in Methods. The kcat/KM values of HsKYNase_93D9 and HsKYNase_66 were found to be comparable at ~390- and 360-fold higher than that of HsKYNase (steady-state (kcat/KM)KYN ~ 102 M−1s−1) respectively, which is in acceptable agreement with the respective (kcat/KM)KYN values as determined by the steady-state kinetic assay (Table 1). The KM of HsKYNase_93D9 derived from the pre-steady-state kinetic analysis (17.5 μM, Table 3) is ~4.5-fold lower relative to its apparent steady-state KM value (85 μM). This difference is likely due to the different KYN concentration regimes that were employed for the two types of experiments (up to 400 μM for steady-state kinetics whereas up to 75 μM for pre-steady-state analysis due to significant substrate inhibition at KYN concentrations >100 μM). While in the steady-state kinetic assessments kcat/KM is well defined by the data at low substrate concentrations, the estimations of kcat and KM are based on the extrapolation of data to the limit of infinite KYN concentrations. Thus, the values of kcat and KM shown in Table 1 do not account for the substrate inhibition at higher KYN concentrations.
When HsKYNase_93D9 was mixed with 500 μM OH-KYN in the stopped-flow apparatus, a low amplitude burst of OH-AA was evident and was followed by a steady-state turnover (Fig. 2c) indicating that the rate-determining step is ALA release, similar to the parental HsKYNase22. The calculated kinetic parameters yielded burst decay rates (eigenvalues) of 13.7 ± 0.39 s−1 and 14.65 ± 0.75 s−1 for the 10 and 5 μM enzyme concentrations, respectively (Fig. 2c). The respective steady-state rates were found to be 0.95 ± 0.005 s−1 and 1.29 ± 0.01 s−1, in good agreement with the steady-state data (Table 1). In contrast, when 12.5 μM HsKYNase_66 was mixed with 500 μM OH-KYN, OH-AA accumulation was linear over time with a rate of 0.13 ± 0.035 s−1, suggesting that the rate-determining step is OH-AA formation (chemical step or a step preceding it). Finally, we also examined the pre-steady-state kinetics of HsKYNase_93D9-H102W-N333T with both AA and OH-AA. These experiments revealed that the rate-determining step for both reactions is the chemical step, with calculated steady-state rates of 0.015 ± 0.0009 s−1 and 0.03 ± 0.002 s−1 (Supplementary Fig. 4).
Taken together, our mechanistic pre-steady-state kinetic analysis showed that while both HsKYNase_93D9 and HsKYNase_66 catalyze KYN hydrolysis with comparable steady-state (kcat/KM)KYN values that are >170-fold higher than the parental enzyme, their respective rate-determining steps for the KYN reaction differ. Optimization of the chemical step which occurs in bacterial KYN-preferring enzymes as well as in HsKYNase_66, is not the rate-determining step for HsKYNase_93D9, therefore we conclude that it is not a prerequisite for achieving high catalytic activity. However, because the AA release step appears to be largely irreversible (step 3 involving k3 and k−3 in Fig. 2b, e, Table 3) the specificity constant is a function only of the steps preceding and including the AA release step. This is a common feature for both HsKYNase_93D9 and HsKYNase_66.
Docking and MD Simulations of the KYN and OH-KYN Complexes
Multiple attempts to crystallize HsKYNase_93D9 exploring a wide spectrum of crystallization conditions and different construct versions failed to yield crystals that diffract at <6 Å resolution. We suspect that the difficulty in getting high-quality crystals was related to incorporating amino acid substitutions within flexible loops originating from BF-KYNase variants as these proteins also failed to crystallize, unlike HsKYNase_6623 or HsKYNase31. Therefore, a homology model of HsKYNase_93D9 was built using the program YASARA32 using the crystal structure of HsKYNase (PDB entry: 3E9K)31as template. As expected, considering the high degree of sequence identity, between HsKYNase_93D9 and HsKYNase ( ~ 97%) no significant structural changes were observed within the regions that harbor mutated residues (Supplementary Fig. 5). Subsequently, in-silico docking experiments were performed with both KYN and OH-KYN using VINA which is the default search method of YASARA33 (see Methods for more information). We validated the process by docking the 3-hydroxyhippuric acid inhibitor into the active site of HsKYNase_93D9 (the only amino acid differences in the active site of HsKYNase_93D9 that could interact with the ligand are the mutations E103F and F306Y) and compared it against the respective crystal structure complex of the wild-type enzyme31. The inhibitor bound to the active site of HsKYNase_93D9 in a mode very similar to that of the experimentally determined complex, properly forming key H-bonds between the N333 residue and the inhibitor’s aromatic hydroxyl group (Supplementary Fig. 6). For all three enzymes (HsKYNase, HsKYNase_93D9 and HsKYNase_66), docking of KYN and OH-KYN into their active site was followed by fifty-nanosecond molecular dynamics (MD) simulations. The simulation trajectories were analyzed and the protein-ligand complex for each enzyme species at t = 0 was structurally compared against the respective time-averaged frame to gain insights as to how key interactions dynamically alter over the course of the trajectory (both conformations were energy minimized prior to their comparison). All those complexes are collectively shown in Fig. 3 and the RMSD values of different runs for each complex are shown in Supplementary Figs. 7–9.
Fig. 3. Structural models of the active site of HsKYNase, HsKYNase_93D9 and HsKYNase_66 with either KYN or OH-KYN obtained from the 50-ns MD simulations.

a, b Docked KYN and OH-KYN respectively, into the active site of wild-type HsKYNase. The t = 0 and the time-averaged structural frames are shown in salmon and grey color respectively. Similarly docked substrates (KYN and OH-KYN) are shown in salmon and grey sticks representing the t = 0 and the time-averaged frames respectively and PLP is shown as yellow sticks (all sticks are colored by atom; N is blue, O is red, and phosphate is orange). Interactions and distances between residues’ side groups and the ligands are denoted either as dashed salmon or grey lines for the t = 0 and time-averaged structures respectively. This color-coding applies for all panels of this figure. The RMSD plots from the respective trajectories are shown in Supplementary Fig. 7a, d. c, d Docked KYN and OH-KYN in the active site of HsKYNase_93D9 respectively. The RMSD plots from the respective trajectories are shown in Supplementary Fig. 8a, d. e, f Docked KYN and OH-KYN into the active site of HsKYNase_66 respectively. The RMSD plots from the respective trajectories are shown in Supplementary Fig. 9a, d. In all panels residues with asterisk are contributed from the “second” monomer relative to the motif residues H102-N333 or W102-T333 and black arrow indicates conformational changes of either the H102- or W102-active site loop. Distances of the interactions are discussed in the main text.
The analysis of the KYN complexes led to interesting insights for all three enzyme species we examined. KYN bound initially to the active site of HsKYNase and HsKYNase_93D9 in a similar mode with the primary stabilizing interactions being between H102 and the aromatic ring of the substrate (π-stacking) and R434 with KYN’s carboxyl group (4.0 and 4.2 Å for HsKYNase and HsKYNase_93D9 respectively) (Fig. 3a, c, Supplementary Figs. 7a, 8a). Both H102 and R434 have been shown to be critical residues for substrate binding and catalysis respectively21,25. However, a striking difference between the two complexes is the time scale during which the R434-KYN interaction (mediated by a combination of ionic interactions and H-bonding) was maintained during the simulation time. That is, HsKYNase_93D9 interacted for 92% of the total time whereas HsKYNase only for 10% (Table 4). Another difference is related to the hydrogen bond formation between the side chain of S75 and the carbonyl oxygen of KYN in case of HsKYNase_93D9. This interaction formed at 3.5 Å in the HsKYNase_93D9 initial complex and it became stronger in the time-averaged structure (2.8 Å) accounting for 91% of the total simulation time (50 ns) whereas it occurred for 44% of the time in case of HsKYNase (initially forming at 3.8 Å) (Table 4). A third noteworthy distinction between the two complexes is associated with the side chain of E103F substitution that is harbored by HsKYNase_93D9 and forms additional perpendicular stacking interactions with the aromatic ring of KYN during the 26% of the simulation time, thereby further enhancing the hydrophobicity and likely stabilizing the binding of the substrate relative to HsKYNase.
Table 4.
Interactions between key active site residues of different HsKYNase species with either KYN or OH-KYN as a function of total simulation time (50 nanoseconds)
| Residue | HsKYNase | HsKYNase_93D9 | HsKYNase_66 |
|---|---|---|---|
| KYN | |||
| S75 | 44% (H-bonding) | 91% (H-bonding) | 75% (H-bonding) |
| H102 | 33% (Hydrophobic) | 79% (Hydrophobic) | N/A |
| W102 | N/A | N/A | 75% (Hydrophobic) |
| E103 | 0% | N/A | N/A |
| F103 | N/A | 26% (Hydrophobic) | 60% (Hydrophobic) |
| H253 | 0% | 10% (Ionic interactions) | 100% (H-bonding + ionic interactions) |
| W305 | 49% (Hydrophobic) | 92% (Hydrophobic) | 95% (Hydrophobic) |
| F314 | 78% (Hydrophobic) | 84% (Hydrophobic) | 85% (Hydrophobic) |
| R434 | 10% (H-bonding + ionic interactions) | 92% (H-bonding + ionic interactions) | 100% (H-bonding + ionic interactions) |
| F165* | 100% (Hydrophobic) | 100% (Hydrophobic) | 100% (Hydrophobic) |
| OH-KYN | |||
| S75 | 50% (H-bonding) | 3% (H-bonding) | 10% (H-bonding) |
| H102 | 61% (Hydrophobic) | 21% (H-bonding + ionic interactions) | N/A |
| W102 | N/A | N/A | 78% (Hydrophobic) |
| E103 | 64% (H-bonding) | N/A | N/A |
| F103 | N/A | 0% | 20% (Hydrophobic) |
| H253 | 10% (Ionic interactions) | 58% (Ionic interactions) | 0% |
| W305 | 6% (Hydrophobic) | 99% (Hydrophobic) | 33% (Hydrophobic) |
| F314 | 56% (Hydrophobic) | 0% | 0% |
| R434 | 89% (H-bonding + ionic interactions) | 64% (H-bonding) | 5% (H-bonding) |
| F165* | 100% (Hydrophobic) | 31% (Hydrophobic) | 9% (Hydrophobic) |
The type of interactions is also shown. F165* refers to the interactions between F165 and the co-factor PLP and not with the substrate. These data were generated from one representative replicate. The respective structural complexes are shown in main Fig. 3 above and the RMSD plots in the Supplementary Figs. 7–9. N/A: not applicable.
In contrast, with HsKYNase_66, KYN adopted a distinct conformation relative to the other two enzymes. The aromatic ring of the substrate forms a tripartite network of strong hydrophobic interactions involving W102 and F103 that accounted for 75% & 60% of the total simulation time respectively (Table 4). Of note, KYN’s carboxylic group forms ionic contacts with R434 at 3.6 Å and 3.0 Å for the t = 0 and time-averaged conformation respectively (Fig. 3e, Supplementary Fig. 9a) and with H253 (3.2 Å for the time-averaged conformation) during the entire simulation time (Table 4). Interestingly, the conformation and orientation of the side chain of the E103F substitution in HsKYNase_66 is very similar to the E103F in HsKYNase_93D9 (Fig. 3c & e). Key hydrophobic interactions between side chains of important residues (W305 and F314) and KYN as well as F165 with the co-factor PLP showed correct molecular alignment and high stability throughout the entire trajectory of all three enzymes (Table 4).
Importantly, the time-averaged structures of the high KYN-activity species HsKYNase_93D9 and HsKYNase_66 suggest that their active site loop that harbors the H102 and W102 substrate-recognition residues respectively, underwent similar conformational changes upon KYN binding (arrows in Fig. 3c and e). In contrast, the respective loop of HsKYNase appeared to be stable during the simulation. Nearly one third of all the mutations in HsKYNase_93D9 (E103F-V104H-G105T-I110T-G112T out of 17 in total) and in HsKYNase_66 (H102W-E103F-V104E-K106D-R107S-T111H-G112Y out of 24 in total)23 are located in that loop. Taken together, our MD simulations analysis sampled several important interactions between KYN and active site residues in all three enzymes and identified conformational changes of critical loops that collectively may provide a rationale for the observed high-KYN activity of HsKYNase_93D9 and HsKYNase_66.
The analysis of the OH-KYN complexes revealed even more striking differences among the three KYNase enzymes that we examined in detail. Specifically, the complex of HsKYNase/OH-KYN is stabilized by several key interactions which have been previously suggested by the crystal structure of the enzyme in complex with a substrate analogue31. The aromatic ring of OH-KYN faces H102 whereas S75 and Y275/N333 form a hydrogen bond network with the substrate’s carbonyl (3.3 Å) and the aromatic hydroxyl groups at 3.0 and 3.5 Å respectively (Fig. 3b, Supplementary Fig. 7d Table 4). In addition, R434 forms salt bridges (Å) with the OH-KYN’s carboxyl group. The time-averaged structure of HsKYNase/OH-KYN suggests that OH-KYN moves within the enzyme’s active site towards R434 and the PLP in a similar manner as KYN (Fig. 3a). However, in the case of OH-KYN this positioning of the substrate movement is accompanied by a conformational change in the H102 loop (arrow in Fig. 3b) along with significantly more extensive interactions with R434 (89% and 10% for OH-KYN and KYN respectively, Table 4). Such a dynamic movement was also observed in the respective HsKYNase_93D9/KYN and HsKYNase_66/KYN complexes (Fig. 3c, e).
Interestingly, OH-KYN bound to HsKYNase_93D9 very differently relative to HsKYNase with the ligand adopting a totally distinct conformation at t = 0 and lacking important interactions with critical residues such as F103, N333 and R434 except for a hydrogen bond formation with S75 (2.9 Å) (Fig. 3d, Supplementary Fig. 8d). However, the interaction of OH-KYN with S75 appeared to be short-lived (3% of the total simulation time, Table 4) whereas with H102 it was 3-fold shorter relative to the HsKYNase/OH-KYN (61% vs 21%, Table 4). The time-averaged frame suggests that the aromatic ring of the substrate falls in between F165 and the PLP forming a tripartite hydrophobic network. The latter is also evidenced by the fact that the F165-PLP interaction accounted only for 31% of the simulation time in contrast to HsKYNase/OH-KYN and all the KYN complexes (Table 4). R434 interacted with OH-KYN only via hydrogen-bonding throughout the simulation time which was by 25% shorter relative to HsKYNase/OH-KYN complex (Table 4). This specific binding mode of OH-KYN to HsKYNase_93D9 may account for the observed 6-fold weaker KM ( ~ 180 μM) and nearly 2-fold lower burst amplitude (kburst ~ 13 s−1) relative to HsKYNase/OH-KYN reaction (KM ~ 30 μM & kburst ~ 22 s−1 22). Of note, the all-heavy-atom RMSDs analysis of one out of three simulation replicates of HsKYNase_93D9/OH-KYN complex showed some evidence of destabilization during the second half of the simulation (Supplementary Fig. 8f). This observation may suggest that the ligand gradually diffuses away from the active site. However, extensive studies well beyond the scope of the present study would be needed to further investigate such a hypothesis and more broadly their implication, if any, on such fine mechanistic questions to the consequences of divergent evolutionary trajectories, which are the focus of this work.
In case of the HsKYNase_66/OH-KYN complex the t = 0 frame showed hydrophobic interactions between the aromatic ring of OH-KYN and W102 as well as hydrogen bonding of S75 and T333 with the substrate’s carbonyl (3.5 Å) and hydroxyl group (2.7 Å) respectively (Fig. 3f, Supplementary Fig. 9d). However, other key residues including R434 and F165 appeared to be misaligned and disarranged. This molecular misalignment continued throughout the entire trajectory as evidenced by the time-averaged structure and was expanded to additional important residues (F314). More specifically, as shown in Table 4, interactions between OH-KYN with H253 and F314 were not maintained during the simulations and were nearly zero between OH-KYN-R434. In turn, F165’s aromatic ring formed hydrophobic interactions with PLP only during the 9% of the entire simulation time (Table 4). Strikingly, several loops harboring key residues (W102, T333, Y225, P282) displayed major conformational changes. Such an extensive active site conformational instability and lack of key PLP-interacting contacts with F165 was observed only in case of the HsKYNase_66/OH-KYN complex which may explain why this enzyme exhibits the lowest catalytic activity against this substrate relative to HsKYNase and HsKYNase_93D9.
Taken together, our docking and MD simulations analysis afforded some important insights regarding key interactions between active site residues and the two substrates before the transaldimination reaction step with the PLP. Such interactions along with conformational changes of critical active site loops may offer a rationale for either the enhanced or the decreased catalytic activity of HsKYNase_93D9 and HsKYNase_66 against KYN and OH-KYN, respectively.
Conformational dynamics with KYN or OH-KYN by HDX-MS
We used HDX-MS34,35 to monitor the dynamics of HsKYNase_93D9 during steady-state catalysis of either KYN or OH-KYN. We recovered approximately 130 overlapping peptides that were followed throughout the 1-, 10-, and 100-minute exposures to D2O (D) (Supplementary Table 11, Supplementary Data 1). We calculated the D-uptake difference between with and without substrate for each peptide at each timepoint, and defined significance as a difference ≥|0.3| Da with a p-value < 0.01 in a Welch’s t-test (n = 3). Steady-state parameters for HsKYNase_93D9 determined in D-PBS (kcatKYN = 0.16 ± 0.012 s−1, KMKYN = 250 ± 49 μM and kcatOH-KYN = 0.135 ± 0.007 s−1, KMOH-KYN = 50 ± 8 μM, Supplementary Table 10) suggest that the reaction was under steady-state conditions for the first two timepoints (1 and 10 min).
During KYN catalysis, HsKYNase_93D9 exhibited a statistically significant reduction in D-uptake predominantly, but not exclusively, in regions nearby the active site that harbor mutations (Fig. 4a–d). The active site loop that harbors H102 along with the region encompassing the E103F-V104H-G105T-I110T-G112T mutations showed significantly decreased D-uptake during reaction with KYN (Figs. 5a-b, 5e, example peptide 91-114). For comparison, HsKYNase_66 showed no dynamics during catalysis in this region, while HsKYNase and BF-KYNase showed stabilization (Supplementary Fig. 10a, c, e)23. Regions with PLP-interacting residues in HsKYNase_93D9 displayed a short-lived reduction in D-uptake at the 1-minute timepoint only (Fig. 5e, example peptides 135-141, 161-175); and the loop upstream of K276 where PLP is covalently attached had reduced D-uptake at the 10-minute timepoint (Fig. 5e, example peptide 272-289). The most striking difference between HsKYNase_93D9 and HsKYNase_66 is around the PLP-binding pocket and neighboring loops where HsKYNase_66 showed significant destabilization during the entire course of the reaction (Supplementary Fig. 10c). We also observed subtle stabilization of internal loops and distal sites during KYN turnover (Fig. 4a, b). Notably, four of these sites did not overlap with the disordered regions mutated in BF-HsKYNase, while two (391-403 and 419-444) overlapped with disordered region B24.
Fig. 4. Conformational dynamics by HDX-MS for HsKYNase_93D9 during the reaction with either KYN or OH-KYN.

a, b Ribbon diagrams of the HsKYNase_93D9 homology model colored by the difference in fractional D-uptake (%) between no substrate and KYN after 1 or 10 minutes (red to blue, −20% to 20%). Coloring is based on DynamX residue-level scripts without statistical filtering. Residues without coverage are grey and PLP is yellow sticks. Black lines denote regions that exhibited significant differences in exchange. c, d Volcano plots showing the average ΔHDX calculated by subtracting D-uptake in the presence of KYN from no-substrate after 1- or 10-minutes reaction. p-values were calculated using a Welch’s t-test (n = 3); significance cutoffs are p-value < 0.01 with an average ΔHDX > | 0.3| Da. Boundaries of significant peptides and regions that showed statistically significant exchange are labeled. Figure was created using HD-eXplosion44. e, f HsKYNase_93D9 colored by the difference in fractional D-uptake (%) between no substrate and OH-KYN after 1 and 10 minutes similar to panels a and b. g, h Volcano plots showing the average ΔHDX calculated by subtracting D-uptake in the presence of OH-KYN from no-substrate after 1 and 10 minutes reaction respectively similar to panels c and d.
Fig. 5. Conformational dynamics of active site regions monitored by HDX-MS during the reaction of HsKYNase_93D9 with KYN or OH-KYN.

a, b HsKYNase_93D9 active site colored by the difference in fractional D-uptake (%) between no substrate and KYN after 1 and 10 minutes (as described for Fig. 4). Key active site residues are shown including H102, *F165, *D168, *D250, *K276, W305 and N333. The asterisk indicates that residue is contributed from the “second” monomer and the arrow highlights the 272-289 loop region that harbors the *A280S-*A282P mutations. PLP is shown in yellow sticks (N: blue, O: red, P: orange). c, d HsKYNase_93D9 active site colored by the difference in fractional D-uptake (%) between no substrate and OH-KYN after 1 and 10 minutes similar to panels a and b. e Example peptides with significant ΔHDX in the active site and PLP-binding pocket of HsKYNase_93D9. D-uptake plot traces for no substrate, with KYN, or with OH-KYN, are shown with black, orange or grey respectively. Data are presented as mean values ± SD (2σ) from three technical replicates. Y-axis is scaled to 80% of theoretical maximum uptake.
During turnover of OH-KYN, HsKYNase_93D9 showed similar dynamics to those observed during KYN catalysis in the active site, PLP-binding pocket, and loop or distal regions (Fig. 4e–h). The reduced D-uptake seen for OH-KYN, however, was more sustained overtime, often causing less D-uptake at the 10-min timepoint. The H102 active site loop that was stabilized during KYN hydrolysis, showed decreased D-uptake in the hydrolysis of OH-KYN (Fig. 5c–e, example peptide 91-114). Similar dynamics profiles were also observed for peptides surrounding the PLP (Fig. 5e, example peptides 135-141, 161-175, 272-289). The only additional region that showed protection during OH-KYN catalysis is the loop that harbors the second motif residue N333 which forms H-bonds with the aromatic ring of OH-KYN and the PLP (Fig. 5e, example peptide 330-339). For comparison, HsKYNas_93D9 is similar to HsKYNase and BF-KYNase in showing extensive stabilization during OH-KYN turnover, while HsKYNase_66 shows a large degree of flexibility in its entire active site, including the critical PLP-interacting peptides (Supplementary Fig. 10b, d, f).
Collectively, our HDX-MS data showed that HsKYNase_93D9 exhibited stabilization in a large number of active site regions during KYN catalysis, a property that had not been previously observed with wild-type HsKYNase, which exhibits significant stabilization only with its preferred OH-KYN. In contrast to HsKYNase_93D9, the KYN-specialist HsKYNase_66 acquired a large degree of flexibility in key active site and PLP regions during catalysis of both KYN and OH-KYN. Our data highlight how the conformational dynamics during catalysis co-evolve along with the conversion of a specialist (HsKYNase) to a generalist (HsKYNase_93D9) or to another specialist (HsKYNase_66).
Discussion
Earlier studies have reported that parallel evolutionary trajectories lead to the isolation of enzymes that reach a certain activity level towards a new substrate by introducing similar amino acid substitutions13,36,37. However, as we show here, different evolutionary trajectories can also lead to genetically distinct enzymes that rely on very different mutations to reach a similar threshold of new activity by affecting protein dynamics and the catalytic mechanism in different ways5,14,38–40
Inspired by the necessity to engineer a human kynureninase with high KYN catalytic activity for immunotherapy applications, we previously showed that there is no directly accessible evolutionary trajectory leading from the parental enzyme to a variant with high catalytic activity for KYN. Instead, very extensive exploration of the sequence space encompassing the screening of >109 mutants led only to variants with a modest improvement in KYN hydrolysis kinetics. The introduction of two amino acid substitutions found only in KYN-preferring bacterial enzymes was required to potentiate a mutational path to HsKYNase_6623 a KYN-specialist with (kcat/KM)KYN that is >400-fold higher than the parental enzyme (Fig. 6). The complex evolutionary trajectory that was required in order to generate HsKYNase_66 was unexpected. After all, even though KYN-preferring and OH-KYN-preferring enzymes show <50% amino acid homology, all the key catalytic residues, including those that interact with the PLP are identical in prokaryotic and eukaryotic enzymes. Therefore, motivated by our observation that the Saccharomyces cerevisiae enzyme has 20-fold higher KYN catalytic activity even though it contains the critical OH-KYN selectivity-conferring residues H102 and N333 found in all eukaryotic species, we sought to explore alternative evolutionary trajectories that might lead to high KYN activity. We succeeded by combining beneficial mutations within the active site and mutations that are located at remote flexible sites within high B-factor regions as well as rationally designed variants with favorable KYN kinetics. The resulting HsKYNase_93D9 has catalytic activity toward KYN that is comparable to the specialist HsKYNase_66 although it shares only 5 out of 24 amino acid substitutions and, importantly, it is a catalytically proficient generalist that accepts KYN and OH-KYN equally well [(kcat/KM)KYN/(kcat/KM)OH-KYN ~ 1] (Fig. 6).
Fig. 6. The two parallel evolutionary pathways of HsKYNase that led to the identification of the generalist HsKYNase_93D9 and specialist HsKYNase_66.

The height of the peaks indicates the KYN fitness whereas the planar rectangular represents the sequence space. Solid arrows indicate a single evolutionary or mutational step whereas the dashed arrows describe multiple evolutionary steps. The evolutionary pathways that yielded the generalist HsKYNase_93D9 and specialist HsKYNase_66 are shown in red and green color respectively.
Our docking and MD simulations analysis of all the six complexes provided important insights into key interactions between the two substrates on the one hand and active site residues and the PLP of the three enzyme variants on the other. The enhanced hydrophobicity that is conferred by the H102W and E103F substitutions (H102W present only in HsKYNase_66) which interact with the aromatic ring of KYN, the more stable ionic interactions with R434 which is a crucial residue for substrate coordination and the conformational change of the active site loop that harbors several mutations of HsKYNase_93D9 and HsKYNase_66 appear to be the most important features that distinguish these enzymes from the parental HsKYNase. Of note our analysis additionally showed that OH-KYN bound to HsKYNase by forming several interactions with residues which are evident in the crystal structure of the enzyme complex21. In contrast, HsKYNase_93D9 displayed a significantly different binding mode relative to HsKYNase likely responsible for the higher KM and slower pre-steady-state burst rate as compared to HsKYNase22. The analysis of HsKYNase_66/OH-KYN complex showed that interactions between the co-factor PLP and key residues (e.g. F165) were misaligned while other residues appeared to point outwards from the active site pocket (e.g. F314). Such a broad instability of the complex may explain the 23-fold higher KM relative to HsKYNase/OH-KYN and provides a rationale as to why the chemical step is the rate-determining step for this reaction.
HDX-MS studies indicated that the kinetic properties of the generalist HsKYNase_93D9 are largely reflected by the conformational dynamic profiles of the enzyme in the reaction with KYN or OH-KYN. We show that the enzyme displayed a very similar conformational dynamics profile with both KYN and OH-KYN characterized by stabilization in the active site as well as at remote regions during catalysis. This evidence suggests that the acquired mutations of HsKYNase_93D9 shift the ensemble of conformations toward states that enable it to accommodate equally well KYN and OH-KYN. Conversely, the specialist HsKYNase_66 exhibited a large degree of flexibility in key active site and PLP regions during the turnover of KYN. This conformational flexibility may have been acquired as a way to compensate for the structural strain imposed by the H102W and N333T substitutions which confer KYN selectivity. Collectively, our HDX-MS analyses of all the KYN-evolved HsKYNase variants discussed in our recent22–24 and the present studies have revealed differential conformational dynamics depending upon the location and the nature of the mutations, which in turn, define the respective kcat and kcat/KM against KYN and OH-KYN.
Mechanistic pre-steady-state kinetic analyses further revealed that the rate-determining step of the HsKYNase_93D9/KYN reaction was the chemical step similar to wild-type enzyme and other enzymes with more modest increases in catalytic activity (e.g. HsKYNase_46 and BF-HsKYNase) all of which contain the H102-N333 motif. This differs from HsKYNase_66 and all prokaryotic KYN-preferring enzymes that harbor a Trp and Thr at the structurally equivalent positions of H102-N333. Collectively, these findings reveal that, optimization of the chemical step in the reaction pathway through the incorporation of H102W/N333T motif is not a prerequisite for achieving high KYN catalytic activity. In fact, the predominant role of the H102W/N333T motif mutations in the HsKYNase_66 evolutionary trajectory was to impose a negative selection against the native activity of HsKYNase towards OH-KYN, thus enabling strong trade-offs in favor to the promiscuous KYN activity. The combination of those two motif mutations with a fine-tuned network of additional remote mutations led to rate-determining step reversal and chemistry optimization accompanied with extensive flexibility in the PLP binding pocket.
In summary, our findings demonstrate that the evolution of high catalytic activity can be accomplished through genetically different evolutionary trajectories that, in turn, modulate the conformational dynamics of the enzyme to enable bypassing or accommodating other sequence constraints; in this case the amino acid identity in the critical 102 and 333 positions. Overall, our studies provide insights on the relationship between conformational dynamics, substrate specificity and catalytic activity of evolved enzyme species arising from mutating the same template enzyme.
Methods
Cloning and recombinant enzyme expression and purification
Cloning of HsKYNase genes (codon-optimized for expression in E. coli) was performed as described previously in detail23. Briefly, the modified pMAL-c2x E. coli expression vector (whose maltose-binding fusion protein (MBP) was deleted) harboring a chloramphenicol resistance cassette and the Tac promoter was employed for routine cloning and recombinant enzyme expression using the MC1061 and C41(DE3) E. coli strains respectively. A typical cloning protocol included gene amplification by Polymerase Chain Reaction (PCR) using KAPA HiFi polymerase (Roche) and agarose gel-purification (Zymo Research) of the amplicons followed by overnight restriction digestion with KpnI-HF/XmaI/DpnI enzymes at 37 °C. Subsequent ligations included ~100 ng of digested plasmid and ~100 ng digested gene insert ( ~ 1:3, vector:insert molar ratio) and were performed overnight at 16 °C (T4 DNA ligase from NEB). The following day, heat-deactivated ligations were dialyzed against ultrapure water for 30 minutes before electroporation into freshly prepared E. coli electrocompetent cells. The transformants were allowed to recover in 2 mL SOC medium at 37 °C for 1 hour under vigorous shaking conditions. Upon recovery, the cells were plated onto 2xYT medium supplemented with 33 μg/mL chloramphenicol and single clones were screened for positive inserts by either restriction digestion or PCR. Positive clones were sequence-verified by Sanger sequencing.
Expression of HsKYNase molecules in the present study was performed as described elsewhere22,24 unless otherwise stated. For all the recombinant expression experiments the E. coli strain C41(DE3) was used followed by Immobilized Metal Affinity Chromatography (IMAC) purification (the produced proteins carried a N-terminal 6-His tag). Briefly, a single colony was used to inoculate 20 mL of 2xYT medium supplemented with 33 μg/mL chloramphenicol and was grown overnight at 37 °C under vigorous shaking. The following day, fraction of the overnight 2xYT culture was used to inoculate either 0.5 or 1 L of TB medium (final dilution 1:100) and the culture was incubated at 37 °C until it reached an OD600 ~ 1. Subsequently, the flask was equilibrated at RT for 20 minutes and IPTG was added to a final concentration of 0.5 mM. Finally, after the induction the shake flask was incubated overnight at 25 °C for protein expression.
Following the overnight expression, the cells were spun down, resuspended in Lysis/Washing buffer A (100 mM Na2HPO4/NaH2PO4, 300 mM NaCl, 25 mM imidazole, 0.1 % Tween, 1 mM PMSF inhibitors, 1 mM PLP, pH 8.0) and were directly lysed by one pass through a French pressure cell followed by centrifugation at 40,000 g for 1 h. The resulting supernatant (soluble fraction) was filtered using a 0.45 μm filter and loaded onto a 0.5 mL (volume of the slurry mixture) of pre-equilibrated nickel-nitrilotriacetic acid (Ni+2-NTA) resin (Qiagen) which was previously transferred to a 5 mL polypropylene column (Pierce). The supernatant was allowed to pass through the column by gravity and the resin was washed with 25 bed volumes of buffer A before eluting the bound enzymes with 5 mL elution buffer B (100 mM Na2HPO4/NaH2PO4, 300 mM NaCl, 300 mM Imidazole, 0.1 % Tween, 1 mM PLP, pH 8) in a dropwise fashion. Next, the eluted enzyme fractions were incubated with buffer C (100 mM Na2HPO4/NaH2PO4, 300 mM NaCl, 300 mM Imidazole, 0.1 % Tween, 5 mM PLP, pH 8) at 37 °C for 4 h for achieving full loading of PLP and finally were applied to an Amico Ultra 10 K MWCO filter for 4 cycles of buffer exchange against PBS, pH 7.4. The purity of the buffer exchanged eluted fractions was evaluated by SDS-PAGE (consistently > 90% pure) and the enzymes were mixed with 15% final concentration of glycerol and flash-frozen with liquid nitrogen for storage at −80 °C. All the variants were prepared according to the protocol described above unless otherwise stated.
Fed-batch fermentation of HsKYNase_93D9
Due to the relatively low expression levels of HsKYNase_93D9 ( ~ 10 mg/L) and the high amounts required for the pre-steady-state kinetic analyses, we developed a fed-batch fermentation protocol that yielded >1.5 g/L of soluble and fully active enzyme. The detailed protocol has been described previously for the production of HsKYNase22 and no modifications were made.
Library design and construction
The design of the different libraries in the present study was aided by a combination of phylogenetic (Geneious program) and structural analyses (PyMol and YASARA Structure) and by utilizing mutational information from our previous studies23,24. The phylogenetic analysis allowed the determination of highly-conserved amino acid residues among prokaryotic and eukaryotic KYNases including ScKYNase (OH-KYN-specific eukaryotic KYNase) and MpKYNase (KYN-specific prokaryotic KYNase) and upon comparison with HsKYNase, mutant libraries were designed focusing primarily on residues located within the 1st and 2nd shell from the active site as described in the text. Four different methodologies for library design and construction were employed in the present study: i) combinatorial saturation mutagenesis where selected residues were randomized using degenerate oligonucleotides following the NNS scheme (N = A, T, G, C and S = G, C) in order to allow all 20 possible amino acids to be encoded but simultaneously minimizing the stop-codons (only one stop-codon (UAG) is present in the NNS combinations); ii) fixed mutations were combined with partial and pairwise mutagenesis where beneficial mutations identified from previous studies23,24 and mutations that were identified from phylogenetic analysis were fixed and combined with either full (NNS) or partial randomization of neighboring residues. These first two types of libraries were constructed following the overlap extension PCR methodology41; iii) error-prone PCR (using Taq polymerase) targeting an error rate of ~ 2-3 mutations/kb. Mutational rate was adjusted primarily by number of PCR cycles (20-25) and amount of parental DNA (20-100 ng plasmid DNA) and was experimentally determined by Sanger sequencing of several variants; and iv) DNA shuffling. Several HsKYNase variants with improved KYN activity which were both isolated from the present and previous studies23,24 were subjected to DNA shuffling following standard protocols42. Proper cross-overs and recombined mutations were confirmed by Sanger sequencing. Detailed information about the types of mutagenesis, the parental templates that were used and the sequence of isolated variants are shown in the Supplementary Tables.
Large-scale library cloning and preparation was done as described above in the first cloning section with the main difference being that the E. coli transformants (in MC1061 strain) were grown overnight at 37 °C in 50 mL 2xYT supplemented with 33 μg/mL chloramphenicol instead of plating them. The following day the plasmids harboring HsKYNase mutants were purified by midi-prep and were used to transform the E. coli auxotrophic strain (ΔtrpE) for subsequent genetic selection as described in the following section. Library size was determined by plating 0.01, 0.1 and 1 μL cells recovered in SOC and counting individual colonies.
Genetic selection of HsKYNase mutant libraries
The detailed development and validation of the genetic selection screen was described recently23. The E. coli trpE deletion mutant (strain genotype = F-,Δ(araD-araB)567,ΔlacZ4787(::rrnB-3), λ-, ΔtrpE772::kan, rph-1, Δ(rhaD-rhaB)568, hsdR514) was obtained from the E. coli Genetic Stock Center at Yale (thereafter referred simply as ΔtrpE). This strain is tryptophan auxotrophic due to the deletion of the gene that encodes the trpE subunit of anthranilate synthase (anthranilate is a precursor molecule for tryptophan biosynthesis). The cell growth was rescued by the produced anthranilate from the KYN hydrolysis reaction. A typical genetic selection process proceeded as follows: Transformed ΔtrpE cells with a library were grown overnight in 2xYT medium at 37 °C supplemented with 33 μg/mL chloramphenicol and 35 μg/mL kanamycin. The following day an aliquot of cells from the saturated culture was spun down for 5 min at 12,000 x g, washed three times with 0.9% ice-cold sterile NaCl and used to inoculate 25 mL of M9 minimal medium consisting of: 1X M9 salts, 0.4% glucose, 1 mM MgSO4, 0.1 mM CaCl2, 33 μg/mL chloramphenicol, 35 μg/mL kanamycin, 0.5 mM IPTG and the substrate L-KYN at a final concentration of 0.5 mM at the beginning of the selection. The number of cells that were used for the inoculation of the first selection culture (selection round #1) was 5-fold larger than the theoretical diversity of each library, while for the following rounds, cells accounting for ~ 20% of the total library size were used for passaging. Depending on the library size, the cells were subjected to genetic selection for 5-10 rounds. After the first 3 selection rounds the KYN concentration was decreased to 0.25 mM for all the subsequent passages until the completion of the experiment. Several clones were sequenced from the final and intermediate rounds in order to determine the existence of possible converged consensus mutant sequences.
96-well plate enzyme screening
Following the final genetic selection round, the ΔtrpE cells were cultured overnight in 2xYT medium supplemented with 33 μg/mL chloramphenicol and 35 μg/mL kanamycin and plasmid DNA was extracted using mini-prep kit (Zymo Research). Next, C41(DE3) E. coli cells were transformed using the isolated plasmid DNA and were plated onto chloramphenicol plates. Single clones were picked randomly from the plates and used to inoculate wells of sterile 96-well plates containing 150 μL of 2xYT medium per well, with 33 μg/mL chloramphenicol. Typically, 2 plates per library were screened after 5-10 rounds of genetic selection. The plates were incubated at 37 °C under vigorous shaking overnight and the next day 5 μL were transferred to a second 96-well plate containing 130 μL fresh 2xYT medium containing 33 μg/mL chloramphenicol and 0.5 mM IPTG per well. The second plate was incubated at 37 °C for ~6 h, followed by centrifugation for 20 min at 3000 x g. The supernatant was discarded, and the pelleted cells were resuspended in B-PER protein extraction solution (Pierce) which was previously diluted 1:1 with PBS, pH 7.4. The cell pellets were resuspended and incubated at RT for 10 min to achieve maximum lysis, followed by centrifugation at 3,000xg for 20 min. Fraction of the supernatants (50 μL) was mixed with 50 μL of PBS, pH 7.4 and ultimately 100 μL of 1 mM KYN dissolved in PBS (pH 7.4, 1% v/v DMSO) were added in each well resulting in final volume of 200 μL and final concentration of 0.5 mM KYN for activity determination. The activity was monitored continuously by recording the decrease of the absorbance at 365 nm (maximum absorbance of KYN has a λmax = 365 nm with ε = 4.50 mM−1cm−1).
Steady-state kinetics analyses
For the steady-state kinetic characterization of enzymes, the catalytic activity assays were performed in PBS, pH 7.4 at 37 °C by monitoring spectrophotometrically the disappearance of the substrate (KYN and OH-KYN at 365 and 370 nm respectively) using a BioTek Synergy H1 96-well microplate reader. For experiments that were performed in D2O, purified enzymes were buffer exchanged for 3 rounds against D2O-PBS, pD 7.4 using buffer exchange Amicon spin columns (10,000 MWCO) from Millipore while the substrates were dissolved in D2O-PBS, pD 7.4. KYN and OH-KYN concentrations were varied in the range of 0 − 2 mM in a final volume of 200 μL. The final enzyme concentration in the reaction mixture ranged from 0.01-0.5 μM depending on the catalytic activity of the variant that was measured. The obtained v/[E] (initial velocity/total enzyme concentration) values from the linear region of the reaction progress curves with <10% of substrate conversion, were plotted against the respective substrate concentrations and the steady-state kinetic parameters kcat and kcat/KM were calculated by nonlinear regression using the Michaelis−Menten model (Eq. 1 below) analyzed by the SoftZymics software (Igor Pro, Wavemetrics). The molar absorption coefficients of KYN and OH-KYN were used for the conversion of absorbance to substrate concentration (4.5 mM−1cm−1 for KYN and 4.7 mM−1cm−1 for OH-KYN).
| 1 |
Where v is the initial velocity of the reaction, S is the substrate concentration, kcat is the steady-state maximal rate under saturating substrate concentration and KM is the substrate concentration at which the enzyme operates at its half velocity.
Pre-steady-state kinetics and simulations of the rate equations
Pre-steady-state kinetics traces for the reactions of HsKYNase_93D9 and HsKYNase_66 with KYN and OH-KYN were followed by stopped-flow fluorescence spectroscopy (SF-300X instrument from KinTek Corporation) as described previously22. Briefly, for the reaction of each enzyme with KYN, the rate of formation of the first product anthranilic acid (AA) was measured by exciting at 314 nm and recording the emitted light at 393 nm using single band bandpass filters (Semrock, Rochester, NY). Similarly, the produced 3-hydroxy-anthranilic acid (OH-AA) during the reaction with OH-KYN was measured by exciting at 318 nm and detecting fluorescence at 407 nm. Reactions were monitored in the range of 0.4-2 seconds and different substrate and enzyme concentrations were used as follows: for KYN concentration-dependence titrations HsKYNase_93D9 and HsKYNase_66 were mixed with KYN at 1 and 5 μM final enzyme concentration respectively. KYN was titrated at concentrations that are shown in the main figure panels. For the reaction with OH-KYN the following concentrations were mixed: 5 and 10 μM HsKYNase_93D9 with 500 μM OH-KYN and 12.5 μM HsKYNase_66 with 500 μM OH-KYN. The reaction traces for the HsKYNase_93D9-H102W/N333T species were obtained after mixing 9 μM enzyme with either 500 μM KYN or 1000 μM OH-KYN. All the reactions were performed in PBS, pH 7.4 at 37 °C and each trace represents the average of at least five different measurements collecting 2,000 data points for each trace. Single burst traces were fit to single-exponential burst Eq. (2) shown below by non-linear regression analysis using the KinTek Explorer program29 and the burst parameters were calculated.
The global fitting analysis and the 4-step minimal model development has been described in detail recently22. Briefly, the KYN concentration-dependence data for HsKYNase_93D9 and HsKYNase_66 were globally fit to a 4-step minimal kinetic model where: i) the substrate (S) binds to the enzyme forming the complex E ~ S, ii) E ~ S is followed by the formation of E ~ P ~ AA complex (P represents the second product alanine and AA is anthranilate), iii) AA is released forming E ~ P + AA and finally iv) P is released liberating the enzyme E for the next catalytic cycle. Rate constants in green and black in the reaction pathway in Fig. 2 were allowed to float and were fixed constant respectively during the global fitting of the data. The calculated values were subjected to confidence condour analysis to determine whether they are constrained by the data30. The derived intrinsic rate constants were used to calculate the steady-state parameters using Eqs. (3), (4), and (5) for a 4-step model as shown below.
| 2 |
| 3 |
| 4 |
| 5 |
Homology modeling of HsKYNase_93D9
Homology modeling of HsKYNase_93D9 was done with the YASARA Structure program (19.12.14). The model was built using as template the structure of the wild-type HsKYNase (PDB entry: 3E9K) and using the macro “hm_build” with the following modification “FixModelRes”. The modification was implemented so that the program will not re-predict all the different side chain conformations of the residues in HsKYNase_93D9 considering the 95% identity between the variant and the template HsKYNase. The side chain rotamers of the mutated residues were fine-tuned considering electrostatic and packing interactions along with solvation effects. The model’s hydrogen bonding network was optimized while a full refinement process was followed for additional optimization of the final HsKYNase_93D9 model that included a high-resolution energy minimization in the presence of explicit solvent using the latest knowledge-based YASARA force field.
Docking of substrates into the active site of HsKYNase species
The substrates L-Kynurenine (KYN) and 3’-OH-L-Kynurenine (OH-KYN) were generated using the “Small Molecule Building Mode” of the YASARA Structure program (19.12.14). Docking of the ligands into the active site of HsKYNase (PDB entry: 3E9K), HsKYNase_66 (PDB entry: 7S3V) and HsKYNase_93D9 was performed using the default search method of YASARA Structure called VINA33 according to the program’s guidelines. The macro “dock_runensemble” was used for the initiation of the docking simulation process. The complex with the highest score was further validated by analyzing the interactions between the ligand and the enzyme’s active site and was subjected to molecular dynamics simulations as described in the following section.
Molecular dynamics simulations & analysis of trajectories
Molecular dynamics simulations were performed for all the highest-score complexes that were generated from the docking analysis (all three enzyme species with KYN and OH-KYN) to sample more conformations of the ligand and key active site residues. All-atom molecular dynamics simulations were chosen in order to obtain a more accurate estimation of the interaction strengths, flexibility and entropic effects, given the presence of a large number of water molecules. The simulations were run with YASARA Structure (19.12.14)43. The setup included an optimization of the hydrogen bonding network to increase the solute stability, and a pKa prediction to fine-tune the protonation states of protein residues at the chosen pH of 7.4. NaCl ions were added with a physiological concentration of 0.9%, with an excess of either Na or Cl to neutralize the cell. The system was parametrized and the enzyme molecules were solvated in 10 Å octahedral box. After steepest descent and simulated annealing minimizations to remove clashes, the simulation was run for 50 nanoseconds using the AMBER14 force field for the solute, GAFF2 and AM1BCC for ligands and TIP3P for water. The cutoff was 8 Å for Van der Waals forces (the default used by AMBER) whereas no cutoff was applied to electrostatic forces (using the Particle Mesh Ewald algorithm). The equations of motions were integrated with a multiple timestep of 1.25 fs (used macro md_run) for bonded interactions and 2.5 fs for non-bonded interactions at a temperature of 298 K and a pressure of 1 atm (NPT ensemble) using algorithms described in detail previously43. The total number of water molecules in the different simulations was in the range of 60,000-70,000 whereas the total number of atoms ~180,000-210,000 respectively. Three different simulations were performed for each species and each simulation was initiated using a different random seed number (initial velocity). Finally, the simulation trajectories were analyzed by executing the “md_analyze” macro while information about individual residues was obtained from running the “md_analyze_res” macro. The types of contacts made between the substrates and active site residues as well as the co-factor PLP with the residue F165 as a function of simulation time were analyzed with the “md_analyze” macro by defining the ligand of interest in the macro. The time-averaged structures were calculated by superposing each snapshot on the initial energy-minimized structure at time 0, and then averaging the resulting atom positions. The t = 0 and the time-average structures were energy-minimized and visualized by PyMol for further structural analysis.
Hydrogen-deuterium exchange coupled to mass-spectrometry
Experiments were performed as per our previously published work22–24. Briefly, >200 μM HsKYNase_93D9 was incubated with 5 mM PLP at 37 °C for 3 h. Labeling buffer (D2O-PBS, pD 7.4) was prepared with/out 3.0 mM KYN or OH-KYN. HsKYNase_93D9 was diluted 1:25 (v/v) with labeling buffer to give a final concentration of 1.6 μM enzyme and 3 mM substrate. Labeling reactions were quenched at 1, 10, and 100 minutes by a 1:1 (v:v) dilution with 2 M GuHCl, 0.8% (v/v) FA at 1 °C, pH 2.6. Each time course was performed in triplicate with freshly prepared labeling buffer. Samples were injected into a Waters HDX Manager connected to a Synapt G2-Si Q-TOF. Buffers A and B consisted of 0.1% (v/v) formic acid and 0.1% (v/v) formic acid in 100% acetonitrile, respectively. An Enzymate Pepsin Column (Waters, 300 Å, 5 μm, 2.1 ×30 mm) was used for on-column digestion at 15 °C. Digestion and trapping occurred for 3 minutes at 100 μL/min. All peptides were desalted via reverse-phase trap (Waters Protein BEH C4 VanGuard Pre-column, 300 Å, 1.7 μm, 2.1 ×5 mm). Peptides were then separated on a C18 column (Waters BEH C18 Column, 130 Å, 1.7 μm, 1 ×100 mm) at 1 °C using an isocratic gradient from 3-40% buffer B at 40 μL/min for 7 minutes. Following peptide digestion, a pepsin wash solution consisting of 2 M GuHCl, 4% acetonitrile, 0.8% (v/v) formic acid, was injected to minimize carryover. Blank injections were performed after each sample injection to ensure low carryover. We use Protein Lynx Global Server 3.0.2 (Waters) for peptide identification. Low energy, high energy, and intensity thresholds were set to 250, 50, and 750 counts, respectively, and minimum fragment ion matches per peptide were set to 3. PLGS peptide lists were exported to DynamX 3.0 (Waters). DynamX 3.0 thresholds were set to 0.3 for minimum products per amino acid and 1 minimum consecutive product. All DynamX 3.0 results were manually verified and filtered by applying a Welch’s t-test at 99% confidence to identify significant peptides using HD-eXplosion44.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Description of Additional Supplementary Files
Source data
Acknowledgements
This work was supported by the Cancer Prevention, Research Institute of Texas grant DP150061, and Ikena Oncology (to G.G.), funding from the University of Texas System Proteomics network (to S.D.). We would like to thank Elizabeth Miller for expert administrative assistance and Associate Professor Everett Stone for useful discussions on the project.
Author contributions
C.S.K. designed and performed key experiments; C.S.K. designed and performed directed evolution experiments and KYNase enzyme characterizations; C.S.K. designed and performed pre-steady-state kinetic experiments and analyzed pre-steady-state kinetic data; C.S.K. designed and performed molecular modeling, substrate docking & MD-simulations in-silico experiments; C.S.K., K.M., and S.D. designed and performed HDX experiments; C.S.K. and Y. K. expressed and prepared enzymes for stopped-flow kinetic studies; C.S.K., G.G., K.A.J. and S.D. interpreted the data; and C.S.K. and G.G. wrote the manuscript.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
The data that support the findings of this study are available in the Supplementary Information. All in-silico datasets have been deposited in the Zenodo.org database and can be accessed by using the following 10.5281/zenodo.12600974. PDB accession entries for the structures that were used in this work can be found here 3E9K, and here 7S3V. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD053893. Source data are provided as a Source Data file.
Competing interests
C.S.K. & G.G. are inventors on intellectual property related to this work including the active patent US20190350975A1. The remaining authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-51133-y.
References
- 1.Jensen, R. A. Enzyme recruitment in evolution of new function. Annu Rev. Microbiol30, 409–425 (1976). 10.1146/annurev.mi.30.100176.002205 [DOI] [PubMed] [Google Scholar]
- 2.Matsumura, I. & Patrick, W. M. Dan Tawfik’s Lessons for Protein Engineers about Enzymes Adapting to New Substrates. Biochemistry62, 158–162 (2023). 10.1021/acs.biochem.2c00230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tracewell, C. A. & Arnold, F. H. Directed enzyme evolution: climbing fitness peaks one amino acid at a time. Curr. Opin. Chem. Biol.13, 3–9 (2009). 10.1016/j.cbpa.2009.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amar, D. et al. The transition of human estrogen sulfotransferase from generalist to specialist using directed enzyme evolution. J. Mol. Biol.416, 21–32 (2012). 10.1016/j.jmb.2011.12.013 [DOI] [PubMed] [Google Scholar]
- 5.Zou, T., Risso, V. A., Gavira, J. A., Sanchez-Ruiz, J. M. & Ozkan, S. B. Evolution of Conformational Dynamics Determines the Conversion of a Promiscuous Generalist into a Specialist Enzyme. Mol. Biol. Evol.32, 132–143 (2015). 10.1093/molbev/msu281 [DOI] [PubMed] [Google Scholar]
- 6.Nam, H. et al. Network context and selection in the evolution to enzyme specificity. Science337, 1101–1104 (2012). 10.1126/science.1216861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaltenbach, M., Emond, S., Hollfelder, F. & Tokuriki, N. Functional Trade-Offs in Promiscuous Enzymes Cannot Be Explained by Intrinsic Mutational Robustness of the Native Activity. PLOS Genet.12, e1006305 (2016). 10.1371/journal.pgen.1006305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaltenbach, M. & Tokuriki, N. Dynamics and constraints of enzyme evolution. J. Exp. Zool. Part B: Mol. Dev. Evol.322, 468–487 (2014). 10.1002/jez.b.22562 [DOI] [PubMed] [Google Scholar]
- 9.Loo, et al. Balancing Specificity and Promiscuity in Enzyme Evolution: Multidimensional Activity Transitions in the Alkaline Phosphatase Superfamily. Journal of the American Chemical Society10.1021/jacs.8b10290 (2018) [DOI] [PubMed]
- 10.Miton, C. M. et al. Evolutionary repurposing of a sulfatase: A new Michaelis complex leads to efficient transition state charge offset. Proc. Natl Acad. Sci.115, E7293–E7302 (2018). 10.1073/pnas.1607817115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khanal, A., Yu McLoughlin, S., Kershner, J. P. & Copley, S. D. Differential Effects of a Mutation on the Normal and Promiscuous Activities of Orthologs: Implications for Natural and Directed Evolution. Mol. Biol. Evol.32, 100–108 (2015). 10.1093/molbev/msu271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blount, Z. D., Lenski, R. E. & Losos, J. B. Contingency and determinism in evolution: Replaying life’s tape. Science362, eaam5979 (2018). 10.1126/science.aam5979 [DOI] [PubMed] [Google Scholar]
- 13.Dickinson, B. C., Leconte, A. M., Allen, B., Esvelt, K. M. & Liu, D. R. Experimental interrogation of the path dependence and stochasticity of protein evolution using phage-assisted continuous evolution. Proc. Natl Acad. Sci.110, 9007–9012 (2013). 10.1073/pnas.1220670110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaltenbach, M., Jackson, C. J., Campbell, E. C., Hollfelder, F. & Tokuriki, N. Reverse evolution leads to genotypic incompatibility despite functional and active site convergence. eLife4, e06492 (2015). 10.7554/eLife.06492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson, D. W., Baier, F., Yang, G. & Tokuriki, N. The adaptive landscape of a metallo-enzyme is shaped by environment-dependent epistasis. Nat. Commun.12, 3867 (2021). 10.1038/s41467-021-23943-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poelwijk, F. J., Tănase-Nicola, S., Kiviet, D. J. & Tans, S. J. Reciprocal sign epistasis is a necessary condition for multi-peaked fitness landscapes. J. Theor. Biol.272, 141–144 (2011). 10.1016/j.jtbi.2010.12.015 [DOI] [PubMed] [Google Scholar]
- 17.Poelwijk, F. J., Socolich, M. & Ranganathan, R. Learning the pattern of epistasis linking genotype and phenotype in a protein. Nat. Commun.10, 4213 (2019). 10.1038/s41467-019-12130-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miton, C. M., Buda, K. & Tokuriki, N. Epistasis and intramolecular networks in protein evolution. Curr. Opin. Struct. Biol.69, 160–168 (2021). 10.1016/j.sbi.2021.04.007 [DOI] [PubMed] [Google Scholar]
- 19.Noor, S. et al. Intramolecular Epistasis and the Evolution of a New Enzymatic Function. PLOS ONE7, e39822 (2012). 10.1371/journal.pone.0039822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Triplett, T. A. et al. Reversal of indoleamine 2,3-dioxygenase-mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat. Biotechnol.36, 758–764 (2018). 10.1038/nbt.4180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lima, S., Khristoforov, R., Momany, C. & Phillips, R. S. Crystal structure of Homo sapiens kynureninase. Biochemistry46, 2735–2744 (2007). 10.1021/bi0616697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karamitros, C. S. et al. Conformational Dynamics Contribute to Substrate Selectivity and Catalysis in Human Kynureninase. ACS Chem. Biol.15, 3159–3166 (2020). 10.1021/acschembio.0c00676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blazeck, J. et al. Bypassing evolutionary dead ends and switching the rate-limiting step of a human immunotherapeutic enzyme. Nat. Catal.5, 952–967 (2022). 10.1038/s41929-022-00856-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karamitros, C. S. et al. Leveraging intrinsic flexibility to engineer enhanced enzyme catalytic activity. Proc. Natl Acad. Sci. USA119, e2118979119 (2022). 10.1073/pnas.2118979119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Phillips, R. S. Structure and mechanism of kynureninase. Arch. Biochem. Biophys.544, 69–74 (2014). 10.1016/j.abb.2013.10.020 [DOI] [PubMed] [Google Scholar]
- 26.Momany, C., Levdikov, V., Blagova, L., Lima, S. & Phillips, R. S. Three-dimensional structure of kynureninase from Pseudomonas fluorescens. Biochemistry43, 1193–1203 (2004). 10.1021/bi035744e [DOI] [PubMed] [Google Scholar]
- 27.Koushik, S. V., Moore, J. A., Sundararaju, B. & Phillips, R. S. The catalytic mechanism of kynureninase from Pseudomonas fluorescens: insights from the effects of pH and isotopic substitution on steady-state and pre-steady-state kinetics. Biochemistry37, 1376–1382 (1998). 10.1021/bi971130w [DOI] [PubMed] [Google Scholar]
- 28.Johnson, K. A. 1 Transient-State Kinetic Analysis of Enzyme Reaction Pathways. in The Enzymes (ed. Sigman, D. S.) 20 1–61 (Academic Press, 1992).
- 29.Johnson, K. A. Fitting enzyme kinetic data with KinTek Global Kinetic Explorer. Meth. Enzymol.467, 601–626 (2009). 10.1016/S0076-6879(09)67023-3 [DOI] [PubMed] [Google Scholar]
- 30.Johnson, K. A., Simpson, Z. B. & Blom, T. FitSpace explorer: an algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem.387, 30–41 (2009). 10.1016/j.ab.2008.12.025 [DOI] [PubMed] [Google Scholar]
- 31.Lima, S., Kumar, S., Gawandi, V., Momany, C. & Phillips, R. S. Crystal structure of the Homo sapiens kynureninase-3-hydroxyhippuric acid inhibitor complex: insights into the molecular basis of kynureninase substrate specificity. J. Med. Chem.52, 389–396 (2009). 10.1021/jm8010806 [DOI] [PubMed] [Google Scholar]
- 32.Krieger, E. et al. Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8. Proteins: Struct., Funct., Bioinforma.77, 114–122 (2009). 10.1002/prot.22570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput Chem.31, 455–461 (2010). 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Konermann, L., Pan, J. & Liu, Y.-H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev.40, 1224–1234 (2011). 10.1039/C0CS00113A [DOI] [PubMed] [Google Scholar]
- 35.Zheng, J., Strutzenberg, T., Pascal, B. D. & Griffin, P. R. Protein dynamics and conformational changes explored by hydrogen/deuterium exchange mass spectrometry. Curr. Opin. Struct. Biol.58, 305–313 (2019). 10.1016/j.sbi.2019.06.007 [DOI] [PubMed] [Google Scholar]
- 36.Storz, J. F. Causes of molecular convergence and parallelism in protein evolution. Nat. Rev. Genet17, 239–250 (2016). 10.1038/nrg.2016.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soskine, M. & Tawfik, D. S. Mutational effects and the evolution of new protein functions. Nat. Rev. Genet11, 572–582 (2010). 10.1038/nrg2808 [DOI] [PubMed] [Google Scholar]
- 38.Otten, R. et al. Rescue of conformational dynamics in enzyme catalysis by directed evolution. Nat. Commun.9, 1314 (2018). 10.1038/s41467-018-03562-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee, J. & Goodey, N. M. Catalytic Contributions from Remote Regions of Enzyme Structure. Chem. Rev.111, 7595–7624 (2011). 10.1021/cr100042n [DOI] [PubMed] [Google Scholar]
- 40.Conserved conformational dynamics determine enzyme activity | Science Advances. https://www-science-org.ezproxy.lib.utexas.edu/doi/10.1126/sciadv.abo5546. [DOI] [PMC free article] [PubMed]
- 41.Karamitros, C. S. & Konrad, M. Fluorescence-Activated Cell Sorting of Human l-asparaginase Mutant Libraries for Detecting Enzyme Variants with Enhanced Activity. ACS Chem. Biol.11, 2596–2607 (2016). 10.1021/acschembio.6b00283 [DOI] [PubMed] [Google Scholar]
- 42.Joern, J. M. DNA Shuffling. In Directed Evolution Library Creation: Methods and Protocols (eds. Arnold, F. H. & Georgiou, G.) 85–89 (Humana Press, Totowa, NJ, 2003). 10.1385/1-59259-395-X:85.
- 43.Krieger, E. & Vriend, G. New ways to boost molecular dynamics simulations. J. Comput Chem.36, 996–1007 (2015). 10.1002/jcc.23899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang, N., Yu, X., Zhang, X. & D’Arcy, S. HD-eXplosion: visualization of hydrogen-deuterium exchange data as chiclet and volcano plots with statistical filtering. Bioinformatics37, 1926–1927 (2021). 10.1093/bioinformatics/btaa892 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The data that support the findings of this study are available in the Supplementary Information. All in-silico datasets have been deposited in the Zenodo.org database and can be accessed by using the following 10.5281/zenodo.12600974. PDB accession entries for the structures that were used in this work can be found here 3E9K, and here 7S3V. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD053893. Source data are provided as a Source Data file.