

Abstract
Background:
Primary progressive aphasia (PPA) defines a group of neurodegenerative disorders characterized by language decline. Three PPA variants correlate with distinct underlying pathologies: Semantic variant PPA (svPPA) with TDP-43 proteinopathy, agrammatic variant PPA (agPPA) with tau deposition, and logopenic variant PPA (lvPPA) with Alzheimer’s disease (AD). Our objectives were to differentiate PPA variants using clinical and neuroimaging features, assess progression, and evaluate structural MRI and a novel FDG-PET image decomposition machine-learning algorithm for neuropathology prediction.
Methods:
We analyzed 82 autopsied patients diagnosed with PPA from 1998–2022. Clinical histories, language characteristics, neuropsychological results and brain imaging were reviewed. A machine-learning framework using a k-nearest neighbors classifier assessed FDG-PET scans from 45 patients compared with a large reference database.
Results:
PPA variant distribution: 35 lvPPA (80% AD), 28 agPPA (89% tauopathy), and 18 svPPA(72% FTLD-TDP). Apraxia of speech was associated with 4R-tauopathy in agPPA, while pure agrammatic PPA without apraxia was linked to 3R-tauopathy. Longitudinal data-revealed language dysfunction remained the predominant deficit for lvPPA patients, agPPA evolved to corticobasal or progressive supranuclear palsy syndrome (64%), and svPPA progressed to bvFTD (44%). agPPA-4R-tauopathy exhibited limited pre-supplementary motor area atrophy, lvPPA-AD displayed temporal atrophy extending to the superior temporal sulcus, and svPPA-FTLD-TDP had severe temporal pole atrophy. The FDG-PET-based machine-learning algorithm accurately predicted clinical diagnoses and underlying pathologies.
Conclusions:
Distinguishing 3R- and 4R-tauopathy in agPPA may rely on apraxia of speech presence. Additional linguistic and clinical features can aid neuropathology prediction. Our data-driven brain metabolism decomposition approach effectively predicts underlying neuropathology.
Keywords: aphasia, apraxia of speech, MRI, FDG-PET, Alzheimer’s disease, frontotemporal lobar degeneration, tauopathy
INTRODUCTION
Primary progressive aphasia (PPA) encompasses neurodegenerative disorders characterized by gradual language deterioration [1 ,2]. Three major clinical variants include logopenic variant PPA (lvPPA); agrammatic variant PPA (agPPA); semantic variant PPA (svPPA). These variants align with three primary neuropathological classes: lvPPA with Alzheimer’s disease (AD) pathology, agPPA with tau deposition, and svPPA with transactive response DNA-binding protein of 43 kD (TDP-43) pathology [3–5]. Challenges include complex classification, clinicopathologic-group overlap, and variable neuropathology frequency.5 Identifying in-vivo features associated with neuropathology benefit patient counselling, symptom management, and increasingly, treatment.
Diagnostic criteria emphasize language deficits [2 ,6 ,7], but imaging biomarkers aid PPA-variant classification and neuropathology prediction. Studies reveal left-hemisphere atrophy, with distinct patterns of atrophy and hypometabolism by clinical variant [6 ,8 ,9]. Metabolic changes in FDG-PET precede atrophy and help identify PPA subtypes [10–12].
This study proposes a novel approach to analyzing FDG-PET data possibly offering insights into early-stage neuropathology prediction. Based on principal component decomposition of FDG-PET images, a multi-class, multi-label approach (StateViewer [13 ,14]) shows promise in classifying neurodegenerative syndromes given the relatively reliable correspondence between PPA phenotypes and their expected underlying pathology for neuropathology prediction.
Prognostic information includes understanding how the neurologic syndrome evolves. As underlying pathology progresses, additional symptoms emerge - but with limited longitudinal data on advanced PPA [15].
The study aimed to 1) identify neuropathology-based clinical, language, and neuroimaging features for PPA variants; 2) characterize clinical progression by variants, and 3) assess the clinical and neuropathological predictive utility of structural MRI and StateViewer analyses.. We hypothesized that 1) core language deficits correlate with expected/”typical” neuropathology, 2) progression corresponds to anticipated phenotypes based on underlying neuropathology, and 3) structural MRI and StateViewer analyses predict underlying neuropathology.
MATERIALS AND METHODS
Patient Consent
Participants provided research consent at their first clinical visit. The Mayo Clinic Institutional Review Board (IRB) approved the protocol.
Study Participants
We searched the Mayo Clinic autopsy database for patients meeting diagnostic criteria for PPA (n=82 [2 ,6–8], January 1998-December 2022).
Language evaluations
Language deficits were extracted from neurological evaluations for 82 patients; 66 (80.5%) underwent formal speech-pathology evaluation. The following deficits were defined according to PPA diagnostic criteria [2 ,8] and recorded as present or absent: agrammatism, impaired single-word retrieval in spontaneous speech, impaired sentence repetition, phonologic errors, dysarthria, AOS, impaired comprehension of syntactically complex sentences, impaired confrontation naming, impaired single-word comprehension, impaired object knowledge and surface dyslexia/dysgraphia. Evidence of binary terms reversal [16], such as yes/no.
Clinical Classification and Clinical Progression
Demographics and clinical data from neurological evaluations at presentation and late-stage were abstracted from the medical records, including age at symptom onset, age at death, memory impairment, prosopagnosia, verbal agnosia, limb apraxia, parkinsonism, motor-neuron signs, behavioral symptoms, instability/falls, visual hallucinations, and any additional symptoms or signs reported. Symptom duration was the difference between age at symptom onset and at death.
Patients without a previously assigned variant were subcategorized retrospectively based on language features and structural neuroimaging.
“Typical”/expected clinicopathological associations were considered lvPPA-AD, agPPA-4R tauopathy, and svPPA-FTLD-TDP. All other clinicopathologic associations were considered “atypical”/unexpected. The microtubule-associated protein tau can have 3 repeats (3R) or 4 repeats (4R) of the microtubule-binding domain due to tau mRNA alternative splicing [17]. We distinguished between 3R- and 4R-tauopathy based on the repeat numbers and associated pathology: Pick’s disease (PiD) with 3R-tau, whereas CBD, PSP, and globular glial tauopathy (GGT) are characterized by 4R-tau [17].
Patients were classified as bvFTD if they met consensus criteria [18]. The bvFTD diagnosis was made regardless of the time since language symptoms emerged. Published criteria [19 ,20] determined whether patients progressed to possible/probable corticobasal syndrome (CBS) or progressive supranuclear palsy (PSP). Core Lewy body features included REM-sleep behavioral disorder (RBD), cognitive fluctuations and visual hallucinations [21].
Neuropsychological evaluations
Sixty-four (78%) participants underwent formal neuropsychological testing including the Rey Auditory Verbal Learning Test (AVLT) for verbal encoding, 15-item Boston Naming Test (BNT) for word retrieval, category fluency for semantic networks, Short Test of Mental Status (STMS) and Mini-Mental State Examination (MMSE) for global cognition, and Trail Making Test A (TMTA) for psychomotor speed and attention. Participants were evaluated using the Neuropsychiatric Inventory (NPI-Q) questionnaire for neuropsychiatric symptoms.
Neuropathologic Assessment
Board-certified Mayo Clinic neuropathologists at (RR, D.W.D, A.T.N) conducted neuropathologic examinations to maintain uniformity. Neuropathologic diagnoses were based on consensus criteria for AD [22], FTLD [23], LBD [24], CBD [25], PSP [26], argyrophilic grain disease (AGD [27]), and aging-related tau astrogliopathy (ARTAG [28]). Patients were categorized by primary neuropathologic diagnosis and co-existing neuropathology recorded for each case.
Radiologic Assessment
Volumetric head MRI was performed on 70 patients. A standardized MRI protocol on either a 3.0 or 1.5 Tesla MRI (GE) scanner included a 3D T1-weighted volumetric sequence (magnetization-prepared rapid acquisition gradient echo [MPRAGE] at 3T or coronal spoiled gradient-recalled echo sequence at 1.5T). Gray- and white-matter atrophy patterns were assessed with voxel-based morphometry (VBM) and voxel-wise t-tests in SPM12 for statistical comparisons of pathology categories. The control group included 70 healthy, age- and sex-matched participants [29]. The Family-Wise Error (FWE) correction at P<0.05 corrected for multiple comparisons.
StateViewer framework
45 patients underwent 18F fluorodeoxyglucose (FDG)-PET on a PET/CT scanner (GE Healthcare). FDG-PET images were processed using an MRI-free pipeline, which involved registration to the MCALT space (available online at https://www.nitrc.org/projects/mcalt/) using non-linear symmetric diffeomorphic registration [30], standardization of the FDG-PET signal to the pons for standard uptake ratio values, and smoothing with a 6-mm full-width at half-maximum kernel.
FDG-PET images were analyzed using a multi-class, multi-label, unsupervised machine-learning framework (StateViewer). This involves the projection of the patients onto a latent space based on singular-value decomposition of an independent, non-overlapping set of 1100 FDG-PET images [14]. This generated eigenvalues for each participant, which were used in a k-nearest neighbors algorithm to assess the similarity between an individual’s FDG-PET scan and a database of >1500 scans from cognitively unimpaired individuals and those diagnosed with ≥1 neurodegenerative dementia syndrome (e.g., probable AD, lvPPA, bvFTD, PSP, agPPA, sematic dementia (SD), dominant AOS, PPAOS, DLB, PCA, dysexecutive AD). Similarity or dissimilarity between a patient’s metabolic pattern and the classes of neurodegenerative disorders in StateViewer was determined by enrichment or depletion of those classes among the k-nearest neighbors. This approach involves Haldane-Anscombe-corrected log odds-ratio of a 2×2 table, considering the presence or absence of labels inside and outside the patient neighborhood. Unlike the conventional k-nearest neighbor formulation, the decision function is based on the raw neighborhood fraction and better accounts for the differences in labels prevalence. Odds ratios extracted for lvPPA, agPPA, and SD for each patient indicated the patients’ metabolism-pattern similarity or dissimilarity to an independent cohort of SD, lvPPA, and/or agPPA patients. To prevent circularity, we computed odds ratio for each patient by excluding their images from the database. We then evaluated the predictive power of these log-transformed odd ratios for underlying neuropathology using linear discriminant analysis (LDA). LDA is a supervised machine-learning classifier that determines the optimal number of linear discriminants to predict predetermined class (i.e., primary pathological diagnosis) based on input (i.e., StateViewer log-transformed odds ratios).
Statistical Analyses
Statistical analyses utilized SPSS software (IBM Corp., Version 28.0.1.1 Armonk, NY) with significance established at P<0.05. Demographic and clinical characteristics were summarized using descriptive statistics. Kruskal-Wallis tests determined differences in demographics, neuropathologic findings, and cognitive scores. Fisher’s exact tests assessed language and clinical-feature differences across pathologic categories. Voxel-wise t-tests in SPM12 compared structural MRI differences for each disease category and controls, with age, sex, and field strength as covariates. LDA was performed using Python version 3.7.12.
RESULTS
Patient characteristics
Table 1 summarizes demographic and neuropathologic features. The cohort consisted of 82 PPA patients, 31 (38%) women. The mean ± SD symptom duration was 9.3 ± 3.7 years. The mean ± SD age at death was 71.9 ± 8.5 years. Follow-up duration averaged 4.25 years (range: 1 month to 14 years). Seven (8.5%) study participants were left-handed but had a more severely affected left hemisphere.
Table 1.
Disease Characteristics and Neuropathology Features
| PPA Variant | Age at onset | Age at death | Duration (yr.) | Femalea (total) | Education (yr.) | ApoE4 carrier (n=68) | Braak NFT stage (n=72) | Thal amyloid phase (n=44) | CERAD score (n=77) | Brain weight (g) (n=56) |
|---|---|---|---|---|---|---|---|---|---|---|
|
Logopenic (n=35) |
63 (13) | 74 (12) | 9 (4.1) | 12 (34) | 16 (4) | 17/27 (63) | VI (1) | 5 (1.5) | 3 (0) | 1101 (301) |
|
Agrammatic (n=28) |
64 (14) | 72.6 (14) | 8.2 (3.8) | 12 (43) | 14 (4) | 3/25 (12) | II (1.5) | 0 (2) | 0 (0) | 1094 (277) |
|
Semantic (n=18) |
61 (11) | 71 (9) | 10 (5) | 7 (39) | 15.5 (2) | 6/15 (40) | 1 (2) | 0.5 (3) | 0 (0.5) | 1046 (118) |
| p. value | ns | ns | ns | ns | ns | <0.001 a | <0.001 | <0.001 | <0.001 | ns |
Median (IQR); for sex, ApoE4 n (%).
P. values are based on Kruskal-Wallis H test. One unclassifiable-PPA patient was excluded from analyses.
For sex, ApoE4 p. value is based on Fisher’s Exact.
P < 0.05.
PPA: Primary Progressive Aphasia.
Distribution of clinical PPA variants was as follows: 35 (43%) logopenic, 28 (34%) agrammatic, 18 (22%) semantic. One patient was categorized as “unclassifiable” due to a mixed deficit pattern.
Neuropathology Findings
Primary neuropathologic diagnoses according to clinical variants appear in Figure 1A. Among lvPPA patients, 29 (80%) had advanced AD, 5 (14%) had diffuse/neocortical-predominant LBD, 1 (3%) had CBD, and 1 (3%) had FTLD-TDP43. Considering any AD co-pathology, the number of lvPPA with AD increased to 32 (91%). Among agPPA patients, 11 (39%) had CBD, 9 (32%) had PSP, 5 (18%) had PiD, and 3 (11%) had FTLD-TDP. Among svPPA patients, 13 (72%) had FTLD-TDP, 2 (11%) had GGT, 1 (6%) had FTLD-tau with MAPT mutation, 1 (6%) had AD, and 1 (6%) had PiD. One patient was “unclassifiable” and had PiD at autopsy.
Figure 1. Primary Progressive Aphasia Clinical Variants according to Primary Neuropathologic Diagnosis.
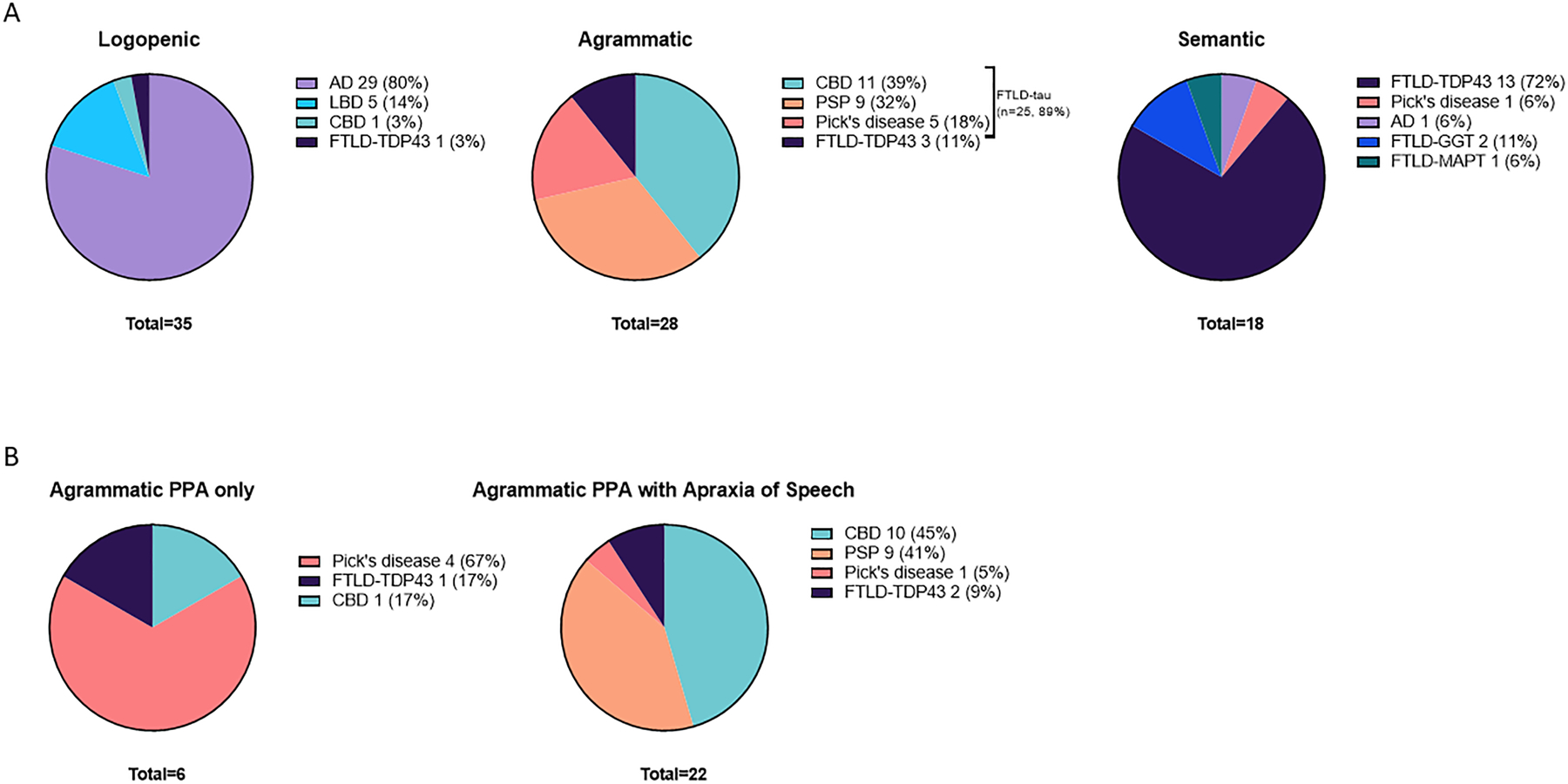
AD: Alzheimer’s Disease; LBD: Lewy body disease; CBD: corticobasal degeneration; FTLD-TDP43: Frontotemporal lobar degeneration-TAR DNA-binding protein 43; PSP: progressive supranuclear gaze palsy; PiD: Pick’s disease; FTLD-GGT Frontotemporal lobar degeneration-Globular glial inclusions; FTLD-MAPT Frontotemporal lobar degeneration- with MAPT mutation.
Among agPPA patients (Figure 1B), those with “pure” aphasia without apraxia of speech were primarily associated with PiD pathology (4/6 [67%]). In comparison, among 22 patients with both apraxia of speech and aphasia, the majority had underlying 4R-tauopathy: 10 (45%) had CBD, 9 (41%) had PSP, 2 (9%) had FTLD-TDP, and 1 (5%) had PiD.
Co-pathology
Of patients with a primary neuropathologic diagnosis of AD, 5/29 (17%) had co-existing LBD pathology. In patients with diffuse/neocortical-predominant LBD, 4/5 had concurrent AD pathology. Of 12 patients (15%) primarily diagnosed with CBD, 5 (42%) had co-existing AGD. Seven patients had a primary diagnosis of PiD (8.5%), one had concurrent ARTAG. Among the 17 (21%) patients primarily diagnosed with FTLD-TDP, 12 (71%) had additional pathologic findings, mostly AD changes. Co-pathology of patients diagnosed with PSP was mainly AD.
Differences in disease characteristics and neuropathology across clinical variants
The three PPA clinical variants did not differ in age at symptom onset, age at death, sex, years of education, or symptoms duration (Table 1). Frequency of APOE4 carriership, Braak neurofibrillary-tangle stage, Thal amyloid phase, and CERAD neuritic plaque score were all higher in lvPPA patients compared to the other variants (P < 0.001).
Table 2 summarized neurocognitive performance across clinical variants. svPPA patients exhibited lower scores on STMS (P < 0.001), MMSE (P = 0.046), AVLT (P = 0.027), and BNT (P < 0.001) compared to other variants. Scores on the verbal fluency test were also lower for svPPA patients without reaching significance (P = 0.063). Sum of positive neuropsychiatric symptoms as measured by the NPI-Q was highest among svPPA patients (5 ± 3), followed by lvPPA (4 ± 3), and lowest for agPPA patients (2 ± 2).
Table 2.
Cognitive and Neuropsychiatric Features
| PPA Variant | STMSa score (n=67) | MMSE score (n=41) | AVLT sum of 5 trials (n=52) | Category Fluency (animals, n=48) | Boston Naming Test (n=53) | Trail Making Test A (seconds, n=56) | NPI-Qb (sum of symptoms, n=48): |
|---|---|---|---|---|---|---|---|
|
Logopenic (n=35) |
23.5 (8) | 22 (5) | 24 (15) | 9 (5) | 5 (4) | 56 (31) | 4 (3) |
|
Agrammatic (n=28) |
34 (3) | 25 (6) | 31 (11) | 11 (4) | 11.5 (3) | 56 (23) | 2 (2) |
|
Semantic (n=18) |
21 (8) | 17 (8) | 21 (9) | 5.5 (3) | 1 (0.7) | 49 (14) | 5 (3) |
| p. value | <0.001 | <0.05 | 0.027 | ns | <0.001 | ns | ns |
| Mean (SD) |
P. values are based on Kruskal-Wallis H test; for NPI subscales, Fisher’s Exact. One unclassifiable-PPA patient was excluded from analyses.
Cognition Scores converted to Short Test of Mental Status scores.
NPI-Q: presence of each of the following symptoms was considered as 1 point: delusions, hallucination, agitation/aggression, depression/dysphoria, anxiety, elation/euphoria, apathy/indifference, disinhibition, irritability/lability, motor disturbance, nighttime behaviors, changes in appetite/eating.
P < 0.05.
STMS: Short Test of Mental Status; MMSE: Mini-Mental State Examination; AVLT: Rey Auditory Verbal Learning Test; NPI-Q: Neuropsychiatric Inventory Questionnaire.
Language deficits according to clinical variant and neuropathology
Comparing language deficits detected in patients with “typical” versus “atypical” clinicopathologic associations revealed no significant differences (Supplementary Tables 1–3), except for apraxia of speech. In agPPA patients, apraxia of speech was more frequent in cases with 4R-tauopathy (PSP/CBD) compared to those with non-4R-tauopathy (P = 0.038, Supplementary Table 3). Categorizing by neuropathologic diagnosis revealed an association between language deficits and neuropathologic primary diagnosis (Figure 2, Supplementary Table 4). Language deficits consistent with root criteria for agPPA appear in Figure 2A. Apraxia of speech (P<0.001) was more frequent in patients with 4R-tauopathy, and agrammatism (P<0.001) in AgPPA patients with either 4R- or 3R-tauopathy. Although not included in the diagnostic criteria, binary reversals were more frequent in 4R-tauopathy (P = 0.013). Patients with FTLD-TDP neuropathology were more likely to exhibit svPPA-consistent language deficits, (Figure 2B), including single-word comprehension difficulties (P<0.001) and object knowledge loss (P=0.023), along with associative agnosia (verbal agnosia and prosopagnosia, P=0.045). Surface dyslexia did not differ between neuropathology categories (P = 0.239). Language deficits included in lvPPA criteria, including single-word retrieval in spontaneous speech (P=0.006), impaired repetition (P=0.034), and phonologic errors (P<0.001), were more frequent in patients with AD and LBD neuropathologic diagnoses (Figure 2C). Additionally, memory impairment at presentation was more frequent in AD- and LBD-diagnosed patients (P<0.001).
Figure 2. Application of Criteria for Variants of Primary Progressive Aphasia Across Neuropathology.
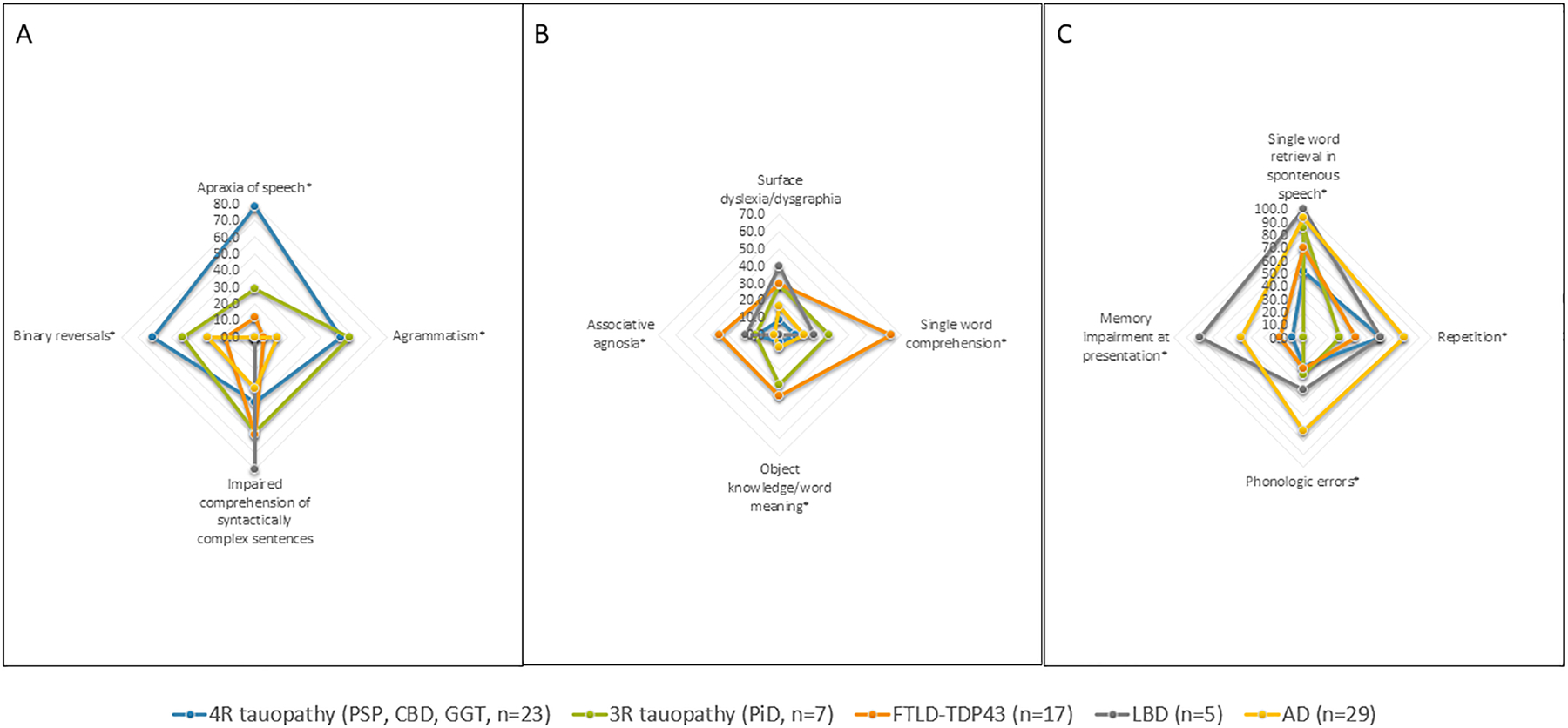
Axes present percentage. Language deficits that significantly differed between the groups are marked with *.
PSP: progressive supranuclear gaze palsy; CBD: corticobasal degeneration; GGT - Globular glial inclusions; FTLD-TDP43: Frontotemporal lobar degeneration-TAR DNA-binding protein 43; LBD: Lewy body disease; AD: Alzheimer’s Disease.
For agPPA, having more core features (apraxia of speech, agrammatism) did not differentiate between 4R-tauopathy (“typical”) and non-4R tauopathy (“atypical”, P=0.194). However, more supportive features (impaired comprehension of syntactically complex sentences, spared single word comprehension and spared object knowledge) correlated with 4R-tauopathy (P=0.032). In lvPPA and svPPA, having more core or supportive features did not associate with “typical” clinicopathologic association (i.e., lvPPA-AD or svPPA-FTLD-TDP). “Atypical” clinicopathologic associations occurred at similar frequencies among patients with agPPA (8/28, 29%), svPPA (5/18, 28%), and lvPPA (7/35, 20%; P = 0.753).
Clinical Progression across PPA variants
Figure 3 displays clinical progression in PPA variants. Although global cognitive impairment or additional symptoms like limb apraxia or visuospatial deficits emerged with illness progression, language dysfunction remained the predominant deficit for most lvPPA patients (n = 29, 83%, symptom duration mean ±SD: 9±3 years). Among the five lvPPA patients with neocortical LBD, three developed Lewy body features later in the disease (RBD/fluctuations/visual hallucinations ± parkinsonism). Agrammatic PPA patients were more likely to develop CBS/PSP/CBS-PSP hybrid syndrome (n = 18, 64%, symptom duration 9±2 years). Nearly half the svPPA patients developed bvFTD (n=8, 44%, symptom duration 10±3 years), while the other half showed predominantly speech-language progression (n = 10, 56%, symptom duration 10±4.5 years).
Figure 3. Clinical course in PPA variants. (A) Logopenic PPA. (B) Agrammatic PPA. (C) Semantic PPA.
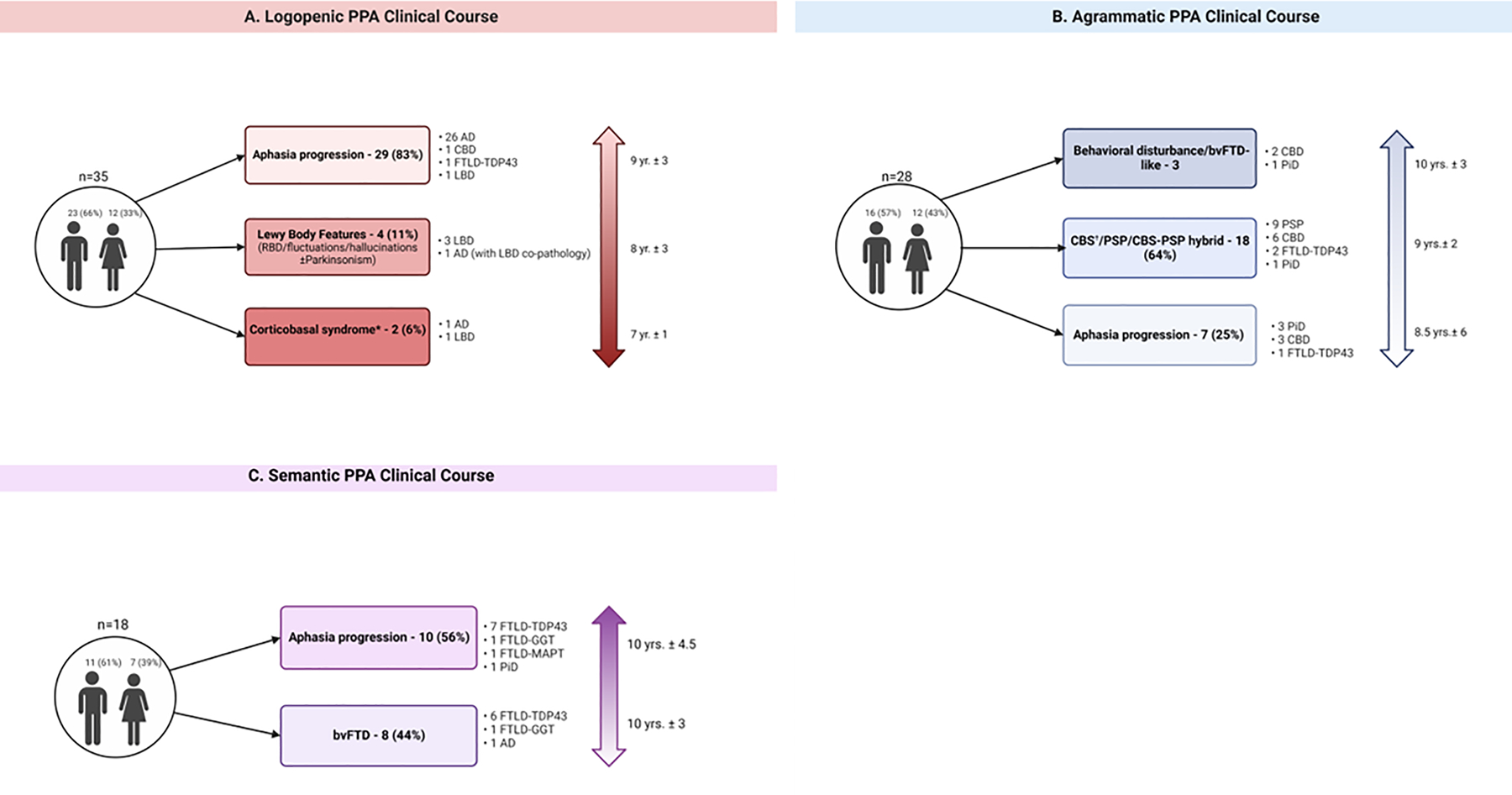
Mean ± SD symptom duration from symptom onset to death shown for each progression group next to the vertical arrows on the right. Created with BioRender.com
*One patient met criteria for probable CBS; One patient met criteria for possible CBS.
†14 patients met criteria for probable CBS, 1 for possible CBS.
RBD: probable REM sleep behavioral disorder; CBS: corticobasal syndrome; AD: Alzheimer’s Disease; CBD: corticobasal degeneration; LBD: Lewy body disease; FTLD-TDP43: Frontotemporal lobar degeneration-TAR DNA-binding protein 43; PSP: progressive supranuclear gaze palsy; PiD Pick’s disease; FTLD-GGT Frontotemporal lobar degeneration-Globular glial inclusions; FTLD-MAPT Frontotemporal lobar degeneration- with MAPT mutation; bvFTD behavioral variant frontotemporal dementia.
Structural MRI findings
Figure 4 displays regions of gray-matter volume loss according to clinical variant and neuropathology. The expected asymmetry involving left-greater-than-right hemisphere was apparent. Compared to controls, patients with agPPA with 4R-tauopathy (e.g., Agrammatic-Tau) had minimal frontal pre-supplementary motor-area gray-matter loss, while agPPA patients with PiD or FTLD-TDP neuropathology (e.g., Agrammatic-Other) had prefrontal, ventrolateral, dorsolateral, and premotor cortex involvement, with additional involvement of the inferior temporal lobe. The Agrammatic-Other group showed greater loss throughout the left prefrontal cortex and anterior temporal lobe compared to the Agrammatic-Tau group. lvPPA patients with underlying AD pathology had temporal-lobe volume loss extending into the parietal lobe compared to controls, and gray-matter loss around the posterior part of superior temporal sulcus compared to lvPPA-Other. No regions of gray-matter loss survived correction for multiple comparisons in the Logopenic-Other group compared to controls. Comparison of Logopenic-Other and Logopenic-AD revealed greater asymmetry involving the left anteromedial region in Logopenic-Other patients. For svPPA patients with underlying FTLD-TDP pathology (e.g., Semantic-TDP), severe temporal-pole volume loss occurred medially and laterally, while Semantic-Other showed more medial temporal lobe loss. The two semantic groups displayed no differences in direct comparison.
Figure 4. Regions of volume loss in PPA patients according to pathology category.
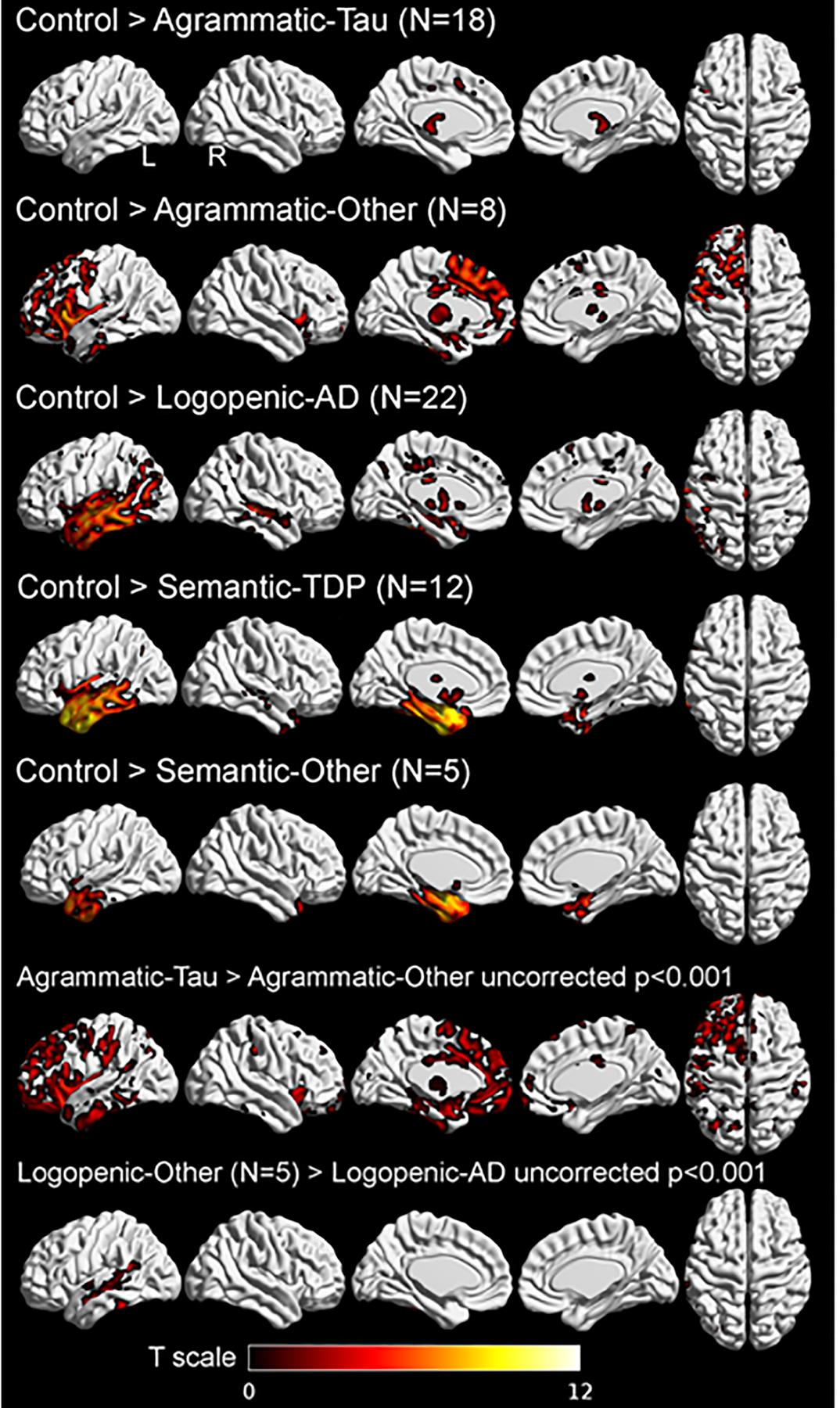
SPM maps of grey matter volume loss in agPPA, lvPPA and svPPA compared to controls and within group comparisons according to neuropathology for agPPA and lvPPA. Lateral, medial, and superior view of grey matter volume loss in all cohorts, compared to age- and sex-matched controls. Results from comparisons with control subjects are FWE corrected for multiple comparisons, p<0.05, extent threshold = 20. Legends show t-scores with brighter colors representing higher t-score.
AD: Alzheimer’s Disease; TDP: TAR DNA-binding protein 43.
FDG-PET findings
Figure 5 depicts the LDA aimed at predicting underlying primary pathological diagnosis based on odds ratios for agPPA, lvPPA and SD predictions from the StateViewer algorithm along with the confusion matrix between true and predicted pathological diagnosis. The optimal solution to classify the patient sample, stratified according to pathological diagnosis based on their StateViewer predictions. included three linear discriminants. The overall balanced accuracy of primary pathology, regardless of class, was 75.6% with overall sensitivity of 83.3% and specificity of 92.6%. The breakdown per primary pathology was the following: AD (89.5% sensitivity, 70.8% specificity, 80.0% accuracy); Tau (83.3% sensitivity, 90.5% specificity, 88.9% accuracy); FTLD-TDP (66.7% sensitivity, 97.0% specificity, 95.6% accuracy); LBD (0% sensitivity, 97.1% specificity, 86.7% accuracy). Qualitatively, hypometabolic patterns consistent with agPPA were typically associated with underlying tauopathy (red dots) and less likely with AD pathology (green triangles) or FTLD-TDP pathology (pink stars). LBD pathology (blue squares) resembled clinical lvPPA patients on FDG-PET. Patients with underlying AD pathology (green triangles) had the highest likelihood of hypometabolism patterns consistent with lvPPA. Among the three patients with FTLD-TDP pathology (pink stars), an agPPA hypometabolism pattern was unlikely; two exhibited a higher likelihood of svPPA phenotype, but one leaned toward lvPPA phenotype.
Figure 5. Clinical variant predictions according to FDG-PET hypometabolism, stratified by neuropathology.
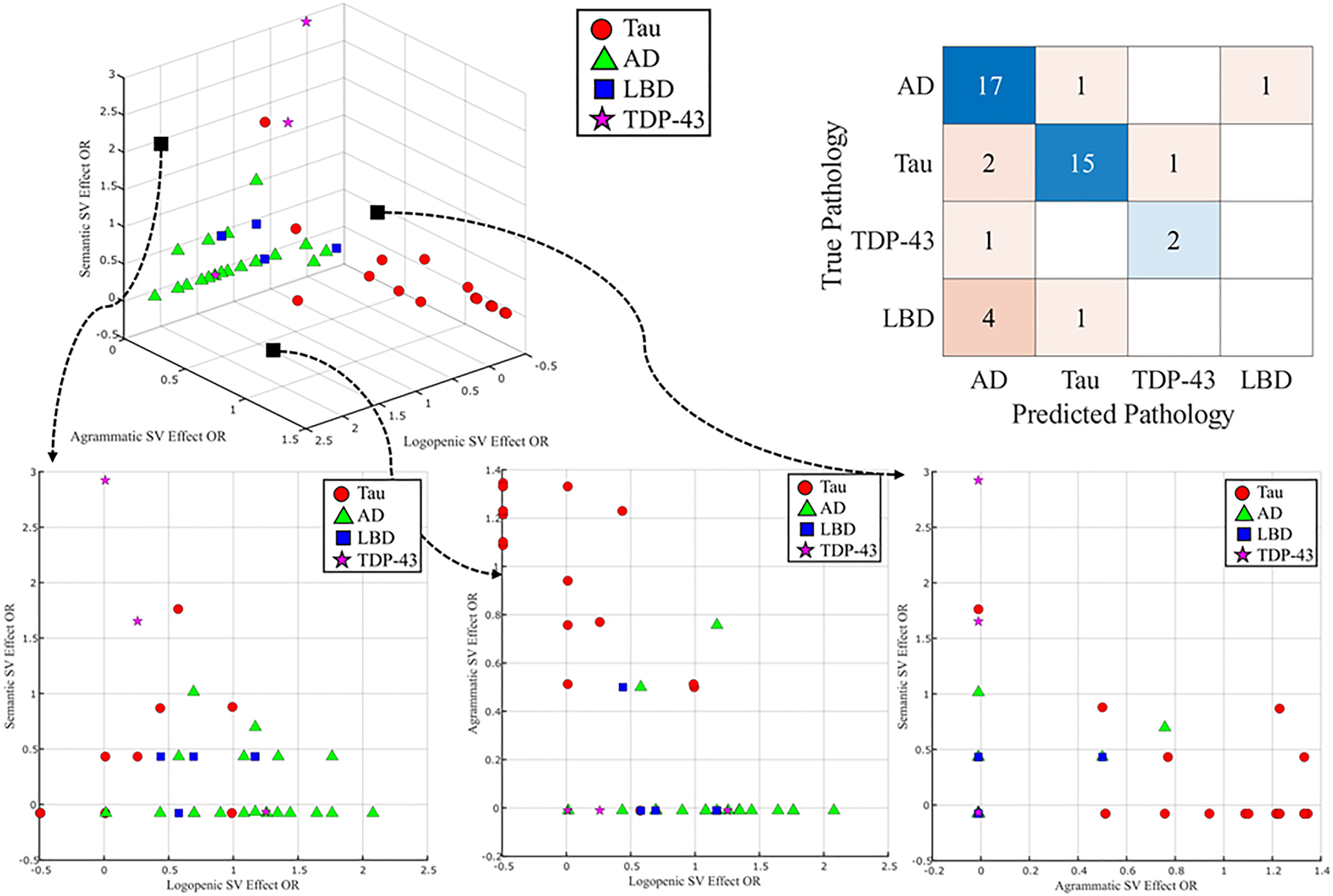
A subset of 45 patients who underwent FDG-PET are presented on the 3D model; Color and shape are representative of neuropathology primary diagnosis. X, Y, Z axes are odds-ratio representative of the likelihood of specific pathology according to FDG-PET. Linear discriminant analysis is shown in the upper right corner.
Tau: primary neuropathology diagnosis of 4R- or 3R- tauopathy.
SV: StateViewer. AD: Alzheimer’s Disease; LBD: Lewy body disease; FTLD-TDP43: Frontotemporal lobar degeneration-TAR DNA-binding protein 43.
DISCUSSION
We found associations between PPA clinical variants and underlying neuropathologies, effectively captured by clinical, linguistic features, and neuroimaging biomarkers including areas of atrophy and patterns of hypometabolism. Moreover, we provide early evidence supportive of data-driven approaches, specifically the decomposition of FDG-PET images to enhance in-vivo predictive accuracy of underlying proteinopathy. We suggest further granulation of 3R- versus 4R-tauopathy by considering the presence/absence of apraxia of speech. We emphasize other linguistic and clinical features to facilitate neuropathology prediction including binary reversals, memory impairment, associative agnosia, and prosopagnosia.
Consistent with prior cohorts [3 ,4], PPA diagnosis correlated with FTLD-tau in 38%, AD in 35%, FTLD-TDP in 21%, and neocortical-predominant LBD in 6% of patients. Each clinical variant aligned with “typical” neuropathology: lvPPA mostly associated with AD (80%), agPPA with tauopathy (89%), and svPPA with FTLD-TDP (72%).
Most agPPA patients with apraxia of speech had 4R-tauopathy (PSP/CBD,86%), while “pure” agrammatic PPA without apraxia of speech correlated with 3R-tauopathy (PiD, 67%). PiD tauopathy typically links to bvFTD, with PPA occurring less frequently [31]. Existing descriptions of PPA with PiD include both agPPA and svPPA variants [31]. As tauopathies are known to underlie agPPA more frequently than other neuropathologies [32], we suggest further differentiation of 3R- versus 4R-tauopathy based on presence/absence of apraxia of speech.
Our results suggest that while language deficits considered in the diagnostic criteria predict neuropathology accurately, recognition of “atypical” cases could be improved. Despite not being the predominant symptom, detection of memory impairment at initial presentation was linked to AD and to neocortical LBD pathology, warranting focused testing in lvPPA. In agPPA, apraxia of speech may indicate a “typical” clinicopathologic association (e.g., agPPA-4R-tauopathy). Likewise, detection of binary reversals or associative agnosia may indicate “typical” clinicopathologic association in agPPA and svPPA, respectively. Incorporating these clinical features into the diagnostic criteria may refine clinical-pathologic correlations. Surface dyslexia/dysgraphia was not associated with FTLD-TDP pathology in svPPA, possibly due to infrequent assessment or detection challenges. Alternatively, this criterion may not closely align with FTLD-TDP pathology.
While impaired communication at onset signals a diagnosis of PPA, patients and their families often inquire about prognosis and disease progression [15]. Longitudinal follow-up revealed a relatively extended disease course. Although global cognitive impairment or symptoms such as limb apraxia or visuospatial deficits emerged with disease progression, language dysfunction remained the predominant deficiency for most lvPPA patients, further highlighting the importance of communication-preservation strategies in this group. Patients with underlying LBD presented with lvPPA, consistent with existing literature [33], and accounted for 14% of lvPPA patients. We advise monitoring nonverbal features by clinical variant: motor manifestations in agPPA, profound behavioural changes in svPPA, and LBD features in lvPPA.
Our cohort featured unique patients, including two with svPPA-GGT, a rare 4R-tauopathy with widespread globular glial inclusions. Previous reports of GGT clinical phenotypes include bvFTD, PSP, CBS, primary lateral sclerosis, and combinations of dementia, parkinsonism, and motor neuron disease [34]. The second-most common cause of svPPA after TDP-43 is tauopathy; most reported cases are due to PiD, a 3R-tauopathy [35]. Our cohort included a MAPT-positive case, manifesting as svPPA years prior to behavioral manifestation. MAPT gene mutations are associated with a svPPA-like presentation [36].
Unlike a recent clinicopathological study [3], we compared structural, clinical variant-based imaging and the distinction between “typical” and “atypical” neuropathology, rather than organizing it by neuropathological categories. Our approach prioritized the clinical context and the sequence of events encountered in clinical practice. In our cohort, lvPPA-AD patients showed more localized temporal atrophy than in Mesulam et al. [3], without frontal or extensive parietal involvement. Yet compared to lvPPA-Other patients, lvPPA-AD had greater posterior superior temporal sulcus involvement. Consistent with a previous study looking at PiB-negative lvPPA [37], lvPPA-Other had increased asymmetry involving the left anteromedial region; this could facilitate predicting “atypical” lvPPA patients. Our findings align with previous research demonstrating very focal atrophy at presentation in patients with underlying 4R-tauopathy [3 ,38]. Absent prefrontal atrophy at presentation in agPPA patients suggests underlying 4R-tauopathy. svPPA showed a distinct left temporal-pole involvement, regardless of underlying pathology.
The subtle nature of PPA structural findings [39] requires additional diagnostic-imaging tools. FDG-PET may offer higher diagnostic accuracy than MRI [10]. Results from StateViewer suggest that hypometabolism topographic patterns capture PPA diagnoses and their relationship to neuropathology. This underscores the value of data-driven approaches, leveraging an imaging modality widely used in clinical practice to perform in-vivo predictions of pathology in PPA patients. This carries important implications for clinical endeavors, including patient care, diagnosis, and prognosis counseling, especially because different neuropathologies across PPA syndromes may lead to distinct phenotypic evolutions over time, necessitating different approaches to risk reduction and symptomatic management.
These findings could enhance therapeutic strategies aimed at molecular diagnosis by improving the identification of patients with specific proteinopathy for enrollment in clinical trials of disease-modifying therapies. Objective quantification of diagnostic PET patterns can unearth subtle patterns in data that might elude visual reads alone. Incorporation of objective PET analysis may eventually complement established visual reading methods. Overall, LDA model accuracy was poor in most classes, primarily due to the low performance in patients with LBD pathology and the paucity of patients in this groupbut was expected because LBD patients presenting with lvPPA-like syndromes are considered “atypical” [40]. Consequently, the high odd ratios observed for the lvPPA category in these patients were expected, but this lowered the overall model performance. Although this study involves a large cohort of PPA patients, the multiple clinical and pathologic groupings resulted in small groups limiting the power of some comparisons.
This study includes several novel findings. First, it offers clinical insights to aid in neuropathology prediction. Second, our data-driven approach based on brain metabolism decomposition allows in-vivo prediction of underlying proteinopathy. Lastly, increased data regarding the late stages of PPA can improve clinical management.
Supplementary Material
Key Messages.
1. What is already known on this topic
Primary progressive aphasia (PPA) includes a group of neurodegenerative disorders characterized by language decline. Three major clinical variants are tied to distinct underlying pathologies: Semantic variant PPA (svPPA) with TDP-43 proteinopathy, agrammatic variant PPA (agPPA) with tau deposition, and logopenic variant PPA (lvPPA) with Alzheimer’s disease (AD).
2. What this study adds
We suggest linguistic and clinical features for neuropathology prediction and underscore the potential of data-driven brain metabolism decomposition for pathology prediction.
3. How might this study affect research, practice, or policy?
. Our study offers clinical insights that could aid in neuropathology prediction. Results from the novel machine-learning FDG-PET framework suggest that clinical diagnoses of PPA and their relationship to neuropathology are captured by topographic patterns of hypometabolism.
ACKNOWLEDGEMENTS
We thank the patients and their families for participating in this study.
FUNDING
This study was supported by the following resources:
NRG SLD (R01-DC010367, R01-DC12519, R01-DC14942, R01-AG50603)
ADRC (P30AG062677)
ALLFTD (U19 AG63911)
MCSA (U01 AG006786)
Footnotes
Data Sharing
The data that support the findings of this study are available from the corresponding author, upon request.
COMPETING INTERESTS
D.S. Knopman serves on a Data Safety Monitoring Board for the DIAN study. He served on a Data Safety monitoring Board for a tau therapeutic for Biogen but receives no personal compensation. He is a site investigator in the Biogen aducanumab trials. He is an investigator in a clinical trial sponsored by Lilly Pharmaceuticals and the University of Southern California. He serves as a consultant for Samus Therapeutics, Roche and Alzeca Biosciences but receives no personal compensation. R.C. Petersen serves as a consultant for Roche, Inc., Genentech Inc., Nestle, Inc., Eli Lilly and Co., and Eisai, Inc., receives publishing royalties from Mild Cognitive Impairment (Oxford University Press, 2003), and UpToDate. B.F. Boeve receives honoraria for SAB activities for the Tau Consortium; is a site investigator for clinical trials sponsored by Alector, Biogen, and Transposon; and receives research support from NIH. V. J. Lowe consults for Bayer Schering Pharma, Eli Lilly and Co., Piramal Life Sciences, Life Molecular Imaging, Eisai Inc., AVID Radiopharmaceuticals, and Merck Research and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals and the NIH (NIA, NCI, NINDS). G.S. Day serves as a consultant for Parabon Nanolabs Inc, as a Topic Editor (Dementia) for DynaMed (EBSCO), and as the Clinical Director of the Anti-NMDA Receptor Encephalitis Foundation (Inc, Canada; uncompensated). He is the co-Project PI for a clinical trial in anti-NMDAR encephalitis, which receives support from Horizon Pharmaceuticals. He has developed educational materials for PeerView Media, Inc, and Continuing Education Inc. He owns stock in ANI pharmaceuticals. Dr. Day’s institution has received support from Eli Lilly for Dr. Day’s development and participation in an educational event promoting early diagnosis of symptomatic Alzheimer disease. W.K. Kremers supported in part by NIH funding. D.W. Dickson, D.T. Jones, K.A. Josephs and J.L. Whitwell received research funding from the NIH and declare no competing financial interests. M.E. Murray is a consultant for AVID Radiopharmaceuticals. She receives support from the NIH/NIA and Eli Lilly. N.R. Graff-Radford receives royalties from UpToDate, has participated in multi-center therapy studies sponsored by Biogen,TauRx, AbbVie, Novartis, and Lilly, and he receives research support from NIH. J.A. Fields is on the OSMB for the SWAN-Aging Study, serves as a consultant for Medtronic, Inc., and received NIH funding. J. Graff-Radford serves on the DSMB for STROKENET and receives research support from the NIH. The other authors declare no financial or other conflict of interest.
REFERENCES
- 1.Mesulam MM. Primary progressive aphasia--a language-based dementia. N Engl J Med 2003;349(16):1535–42. doi: 10.1056/NEJMra022435 [DOI] [PubMed] [Google Scholar]
- 2.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76(11):1006–14. doi: 10.1212/WNL.0b013e31821103e6 [published Online First: 20110216] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mesulam MM, Coventry CA, Bigio EH, et al. Neuropathological fingerprints of survival, atrophy and language in primary progressive aphasia. Brain 2022;145(6):2133–48. doi: 10.1093/brain/awab410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grossman M Primary progressive aphasia: clinicopathological correlations. Nat Rev Neurol 2010;6(2):88–97. doi: 10.1038/nrneurol.2009.216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Josephs KA, Hodges JR, Snowden JS, et al. Neuropathological background of phenotypical variability in frontotemporal dementia. Acta Neuropathol 2011;122(2):137–53. doi: 10.1007/s00401-011-0839-6 [published Online First: 20110526] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mesulam MM. Primary Progressive Aphasia and the Left Hemisphere Language Network. Dement Neurocogn Disord 2016;15(4):93–102. doi: 10.12779/dnd.2016.15.4.93 [published Online First: 20161231] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Botha H, Josephs K. Primary Progressive Aphasias and Apraxia of Speech. CONTINUUM: Lifelong Learning in Neurology 2019;25:101–27. doi: 10.1212/CON.0000000000000699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Botha H, Duffy JR, Whitwell JL, et al. Classification and clinicoradiologic features of primary progressive aphasia (PPA) and apraxia of speech. Cortex 2015;69:220–36. doi: 10.1016/j.cortex.2015.05.013 [published Online First: 20150527] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tippett DC. Classification of primary progressive aphasia: challenges and complexities. F1000Res 2020;9 doi: 10.12688/f1000research.21184.1 [published Online First: 20200130] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Josephs KA, Martin PR, Botha H, et al. [(18) F]AV-1451 tau-PET and primary progressive aphasia. Ann Neurol 2018;83(3):599–611. doi: 10.1002/ana.25183 [published Online First: 20180313] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Josephs KA, Duffy JR, Fossett TR, et al. Fluorodeoxyglucose F18 positron emission tomography in progressive apraxia of speech and primary progressive aphasia variants. Arch Neurol 2010;67(5):596–605. doi: 10.1001/archneurol.2010.78 [DOI] [PubMed] [Google Scholar]
- 12.Minoshima S, Mosci K, Cross D, et al. Brain [F-18]FDG PET for Clinical Dementia Workup: Differential Diagnosis of Alzheimer’s Disease and Other Types of Dementing Disorders. Semin Nucl Med 2021;51(3):230–40. doi: 10.1053/j.semnuclmed.2021.01.002 [published Online First: 20210202] [DOI] [PubMed] [Google Scholar]
- 13.Jones D, Lowe V, Graff-Radford J, et al. A computational model of neurodegeneration in Alzheimer’s disease. Nat Commun 2022;13(1):1643. doi: 10.1038/s41467-022-29047-4 [published Online First: 20220328] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barnard LR, Botha H, Corriveau-Lecavalier N, et al. Latent space projection of brain FDG-PET creates a powerful classifier for neurodegenerative diseases. Alzheimer’s & Dementia 2022;18(S1):e066614. doi: 10.1002/alz.066614 [DOI] [Google Scholar]
- 15.de la Sablonnière J, Tastevin M, Lavoie M, et al. Longitudinal Changes in Cognition, Behaviours, and Functional Abilities in the Three Main Variants of Primary Progressive Aphasia: A Literature Review. Brain Sci 2021;11(9) doi: 10.3390/brainsci11091209 [published Online First: 20210914] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frattali C, Duffy JR, Litvan I, et al. Yes/no reversals as neurobehavioral sequela: a disorder of language, praxis, or inhibitory control? Eur J Neurol 2003;10(1):103–6. doi: 10.1046/j.1468-1331.2003.00545.x [DOI] [PubMed] [Google Scholar]
- 17.Arendt T, Stieler JT, Holzer M. Tau and tauopathies. Brain Res Bull 2016;126(Pt 3):238–92. doi: 10.1016/j.brainresbull.2016.08.018 [published Online First: 20160909] [DOI] [PubMed] [Google Scholar]
- 18.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134(Pt 9):2456–77. doi: 10.1093/brain/awr179 [published Online First: 20110802] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80(5):496–503. doi: 10.1212/WNL.0b013e31827f0fd1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov Disord 2017;32(6):853–64. doi: 10.1002/mds.26987 [published Online First: 20170503] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017;89(1):88–100. doi: 10.1212/WNL.0000000000004058 [published Online First: 20170607] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging 1997;18(4 Suppl):S1–2. [PubMed] [Google Scholar]
- 23.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 2010;119(1):1–4. doi: 10.1007/s00401-009-0612-2 [published Online First: 20091119] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology 1996;47(5):1113–24. doi: 10.1212/wnl.47.5.1113 [DOI] [PubMed] [Google Scholar]
- 25.Dickson DW, Bergeron C, Chin SS, et al. Office of Rare Diseases neuropathologic criteria for corticobasal degeneration. J Neuropathol Exp Neurol 2002;61(11):935–46. doi: 10.1093/jnen/61.11.935 [DOI] [PubMed] [Google Scholar]
- 26.Roemer SF, Grinberg LT, Crary JF, et al. Rainwater Charitable Foundation criteria for the neuropathologic diagnosis of progressive supranuclear palsy. Acta Neuropathol 2022;144(4):603–14. doi: 10.1007/s00401-022-02479-4 [published Online First: 20220810] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez RD, Suemoto CK, Molina M, et al. Argyrophilic Grain Disease: Demographics, Clinical, and Neuropathological Features From a Large Autopsy Study. J Neuropathol Exp Neurol 2016;75(7):628–35. doi: 10.1093/jnen/nlw034 [published Online First: 20160609] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kovacs GG, Ferrer I, Grinberg LT, et al. Aging-related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 2016;131(1):87–102. doi: 10.1007/s00401-015-1509-x [published Online First: 20151210] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts RO, Geda YE, Knopman DS, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 2008;30(1):58–69. doi: 10.1159/000115751 [published Online First: 20080207] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Avants BB, Tustison NJ, Song G, et al. A reproducible evaluation of ANTs similarity metric performance in brain image registration. Neuroimage 2011;54(3):2033–44. doi: 10.1016/j.neuroimage.2010.09.025 [published Online First: 20100917] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whitwell JL, Tosakulwong N, Schwarz CC, et al. Longitudinal anatomic, functional, and molecular characterization of Pick disease phenotypes. Neurology 2020;95(24):e3190–e202. doi: 10.1212/WNL.0000000000010948 [published Online First: 20200928] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Josephs KA, Duffy JR, Strand EA, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain 2006;129(Pt 6):1385–98. doi: 10.1093/brain/awl078 [published Online First: 20060413] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kakinuma K, Narita W, Baba T, et al. “Semantic variant primary progressive aphasia” due to comorbidity of Lewy body disease and a previous cerebral venous infarction in the left anterior temporal lobe: A case report. eNeurologicalSci 2021;22:100318. doi: 10.1016/j.ensci.2021.100318 [published Online First: 20210119] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buciuc M, Pham NTT, Duffy J, et al. Unraveling globular glial tauopathy: a clinical and neuroimaging study. Neurology 2022;98 [published Online First: 04/01/2022]35300622 [Google Scholar]
- 35.Graff-Radford J, Josephs KA, Parisi JE, et al. Globular Glial Tauopathy Presenting as Semantic Variant Primary Progressive Aphasia. JAMA Neurol 2016;73(1):123–5. doi: 10.1001/jamaneurol.2015.2711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pickering-Brown SM, Richardson AM, Snowden JS, et al. Inherited frontotemporal dementia in nine British families associated with intronic mutations in the tau gene. Brain 2002;125(Pt 4):732–51. doi: 10.1093/brain/awf069 [DOI] [PubMed] [Google Scholar]
- 37.Whitwell JL, Duffy JR, Strand EA, et al. Clinical and neuroimaging biomarkers of amyloid-negative logopenic primary progressive aphasia. Brain Lang 2015;142:45–53. doi: 10.1016/j.bandl.2015.01.009 [published Online First: 20150203] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Josephs KA, Duffy JR, Clark HM, et al. A molecular pathology, neurobiology, biochemical, genetic and neuroimaging study of progressive apraxia of speech. Nat Commun 2021;12(1):3452. doi: 10.1038/s41467-021-23687-8 [published Online First: 20210608] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sajjadi SA, Sheikh-Bahaei N, Cross J, et al. Can MRI Visual Assessment Differentiate the Variants of Primary-Progressive Aphasia? AJNR Am J Neuroradiol 2017;38(5):954–60. doi: 10.3174/ajnr.A5126 [published Online First: 20170324] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buciuc M, Whitwell JL, Kasanuki K, et al. Lewy Body Disease is a Contributor to Logopenic Progressive Aphasia Phenotype. Ann Neurol 2021;89(3):520–33. doi: 10.1002/ana.25979 [published Online First: 20201217] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.