

Abstract
The ability to site-selectively modify equivalent functional groups in a molecule has the potential to streamline syntheses and increase product yields by lowering step counts. Enzymes catalyze site-selective transformations throughout primary and secondary metabolism, but leveraging this capability for non-native substrates and reactions requires a detailed understanding of the potential and limitations of enzyme catalysis and how these bounds can be extended by protein engineering. In this review, we discuss representative examples of site-selective enzyme catalysis involving functional group manipulation and C─H bond functionalization. We include illustrative examples of native catalysis, but our focus is on cases involving non-native substrates and reactions often using engineered enzymes. We then discuss the use of these enzymes for chemoenzymatic transformations and target-oriented synthesis and conclude with a survey of tools and techniques that could expand the scope of non-native site-selective enzyme catalysis.
Graphical Abstract

1. INTRODUCTION
Controlling the selectivity of a chemical reaction is as central to synthetic organic chemistry as bond formation itself. From a practical perspective, forming a new bond is only useful if it provides a desired product with the correct connectivity between atoms (i.e., constitutional selectivity) and with the correct arrangement of those atoms in space (i.e., stereoselectivity).1 From a fundamental perspective, navigating the energy surfaces that govern selectivity constitutes a powerful test of our understanding of reactivity.2 This challenge can be simplified by exploiting the relative reactivity, or chemoselectivity,3,4 of reagents and catalysts toward different functional groups.5 If reactivity toward multiple functional groups is observed, protecting groups can be used to eliminate the chemoselectivity challenge, albeit at the expense of added operations and wasted material. Indeed, much of organic methodology involves generating a reactive intermediate and either blocking problematic functional groups or designing a synthesis so that they are not present to ensure that the intermediate reacts primarily with a desired functional group. This substrate-controlled approach reduces the problem of controlling constitutional selectivity if it remains a factor to controlling regioselectivity, that is, the “preferential making or breaking of bonds in one of two possible orientations”6 at that desired functional group (Scheme 1A).1
Scheme 1. (A) Definitions of Regioselectivity and Site Selectivity with Representative Examples of Each;a (B) Examples of Site-Selective Reactions in the Biosynthesis of Paclitaxelb 8.

aAdapted with permission from ref 1. Copyright 1996 Georg Thieme Verlag. bThroughout this review, crimson is used to highlight functional groups (A/B) or sites (B, filled circles) undergoing site-selective reactions and mint is used to highlight the functionalized sites.
But, what happens when the chemoselectivity simplification fails? As molecular complexity increases, the reactivities of different functional groups may be attenuated or exaggerated due to their molecular context such that they overlap. What if chemoselectivity is not relevant because one is dealing with identical functional groups? Carbon─hydrogen bonds can present a particularly challenging case in this regard since they are ubiquitous in organic molecules. The latter question poses the challenge of site selectivity, a subclassification of constitutional selectivity that “differentiates unrelated functional groups in a molecule which can undergo the same type of reaction” (Scheme 1A).1,5 While molecular context, including proximity to directing groups, may allow for differentiation of identical functional groups,7 this form of substrate control is limiting (what if you need to functionalize a less reactive or nondirected site?).
Site-selective catalysis emerged in nature long ago when enzymes evolved to achieve this capability presumably due to its benefit to organismal fitness. Site-selective oxidation, reduction, and phosphorylation are common throughout primary metabolism, and examples of site-selective hydroxylation in secondary metabolism are among the most-cited examples of the potential synthetic utility of site-selective catalysis. The extensive hydroxylation and acylation/benzoylation of taxadiene en route to paclitaxel, for example, involves discrimination of multiple secondary, tertiary, and allylic C─H bonds, olefins, and secondary alcohols, functionality that comprises nearly the entire molecule (Scheme 1B).8 Many of these enzymes catalyze the formation of reactive intermediates, but rather than requiring blocking/directing groups or circuitous synthetic routes, they use catalyst control to override inherent substrate reactivity. Critical to this capability is the extensive, redundant, and dynamic molecular recognition exerted by enzyme active sites: Extensive because enzymes use many different intermolecular forces to bind substrates, redundant because often one or more of these can be disrupted without completely ablating catalysis, and dynamic because enzymes can change conformations to suit different states along catalytic cycles. Chemists have long sought to mimic these properties.9 Early efforts used cyclodextrins that formed a hydrophobic pocket for substrate binding and that provided a scaffold on which catalysts could be appended to enable site-selective functionalization of the bound substrate.10,11 A variety of substrate binding and functionalization strategies have since been explored within this bifunctional catalyst framework.12
It is interesting that these “bioinspired” synthetic catalysts find themselves at a stage of development that wild-type enzymes appeared to occupy in what has been termed the “first wave” of biocatalysis,13 that is, the former are and the latter were known to catalyze site-selective functionalization of a relatively narrow range of substrates, a notable similarity given that substrate scope is often an advantage of synthetic catalysts over enzymes. We say that first-wave enzymes appeared to be limited in scope, however, because they have proven remarkably adept at site-selective catalysis when researchers decided to look for that reactivity outside the confines of the native substrate and reaction scope.14,15 Moreover, directed evolution has enabled rapid tuning of enzymes to enable site-selective reactions of diverse substrates.16,17 Will it be possible to design or otherwise engineer synthetic site-selective catalysts with molecular recognition abilities as versatile and adaptable as enzymes? How complex must such systems be to achieve these emergent properties? There is no doubt that learning the answers to these questions will be endlessly fascinating. Moreover, it is likely that both classes of catalysts, hybrids thereof,18 and others that have not yet been developed will be required to achieve the ultimate goal of site-selective catalysis: selective reaction at any site on any substrate.
With the goal of helping to frame the pursuit of this goal from the perspective of enzyme catalysis, we begin this review by discussing enzymes that control the site selectivity of functional group manipulation and C─H functionalization reactions. We then cover applications of these enzymes in the context of chemoenzymatic methods and target-oriented synthesis, and we conclude with a brief overview of site-selective modification of macromolecules. While we occasionally cover early studies involving native substrates, our focus is on non-native substrates, non-native reactions, and artificial enzymes that, collectively, illustrate the potential to use enzymes for site-selective catalysis in situations far beyond what they evolved for in nature. Because the definition of site selectivity provided above does not account for the fact that a given functional group can behave differently depending on its molecular context, we draw particular attention to cases involving groups in similar contexts or in which expected reactivity patterns are overridden. Also, while site selectivity is fundamentally different than stereoselectivity, we highlight systems that are site-selective and stereoselective since both are required to achieve the above-stated goal. We note that our focus differs from late-stage functionalization, which has been reviewed elsewhere,19 in that it often involves relatively simple substrates and excludes cases in which chemoselectivity alone is the key challenge. The basics of the techniques that were used to identify or engineer enzymes discussed in this review (e.g., directed evolution, genome mining, etc.) are also not extensively discussed since there are many reviews in this regard, but we do discuss key advances that are needed in these areas to advance the field of non-native site-selective enzyme catalysis.
2. FUNCTIONAL GROUP MANIPULATION
As noted above, functional groups lie at the heart of synthetic methodology, and while chemoselectivity can often be used to ensure reaction of one functional group over others, this is not always possible. As functional group reactivity and molecular context become more similar, addressing this challenge becomes harder so that one must use either protecting groups or an alternate synthetic strategy to avoid the chemoselectivity issue entirely. Enzymes capable of site-selectively manipulating similar or identical function groups using catalyst control provide an alternate strategy that enables otherwise impossible synthetic routes.
2.1. Carboxylic Acid Derivative Hydrolysis and Formation
2.1.1. Carboxylic Ester Hydrolysis.
Hydrolysis of carboxylic acid derivatives, including esters, amides, and nitriles, enables a range of subsequent reactions to elaborate compounds. Classical methods to hydrolyze these functional groups use strong acids or bases, but these reagents often lead to side reactions, making site-selective hydrolysis difficult. More recent methods correct this problem to varying extents,20-22 but the need for reactions that operate under mild conditions, including ambient temperature and neutral pH, have led to broad use of esterases and lipases as catalysts for carboxylic acid derivative hydrolysis. Indeed, these were among the first enzymes commonly used for organic synthesis.23 Particularly important in this regard are serine hydrolases, which do not require a cofactor and instead rely largely on a Ser─Asp/Glu─His catalytic triad and an H bonding by backbone amide N─H bonds in a so-called “oxyanion hole” to catalyze hydrolysis (Scheme 2).24 These enzymes often tolerate high concentrations of organic solvents,25 which allows for high substrate loadings in an aqueous solution. Extensive development of serine hydrolases has established that they can exhibit broad substrate scope while maintaining high site and stereoselectivity.
Scheme 2. Simplified Mechanism of Serine Hydrolases24.

The serine hydrolase C. antarctica lipase B (CALB) has been used in both fine and commodity chemical synthesis,26 and several examples of site-selective CALB catalysis have been reported. Hydrolysis of the terminal ester in 1 without hydrolysis of the more reactive phenolic ester linkage is challenging under conditions typically used for ester hydrolysis. Site-selective hydrolysis of the former was possible using Novozym 435, which is a resin-supported version of CALB,27 in 90/10 v/v t-BuOH/H2O at 20 °C to give pro-drug 2 in 84% yield (Scheme 3A).28 Novozym 435 has also been used for site-selective hydrolysis of triethylcitrate (3) to give 4 in 45% yield (Scheme 3B),29,30 and this compound can be converted to value-added products like rhizoferrin.30
Scheme 3. (A) Hydrolysis of 1 in 90/10 v/v t-BuOH/H2O Using Novozym 435 To Give 2;28 (B) Monohydrolysis of Triethylcitrate Using Novozym 435 To Give 430.

Pig liver esterase (PLE) has also been used for site-selective hydrolysis. For example, hydrolysis of methyl-(Z)-2-methyl-butenedioate 5 was achieved with high selectivity for the ester distal to the 2-methyl group (Scheme 4A).31 Castanospermine, an indolizidine alkaloid isolated from the seeds of Castanospermum austral and a potent inhibitor of dengue virus infection, bears four similarly reactive hydroxyl groups. To differentiate these groups, the natural product was selectively dibutyrylated to give 8.32 PLE preferentially hydrolyzes the butyryl group from the C-7 position of 8 to give 9, whereas a different serine hydrolase, subtilisin, hydrolyzes the ester bond at the C-1 position to give 10 (Scheme 4B).33 Finally, while poor site selectivity was observed for hydrolysis of (R)-aspartate dimethyl ester (11) using acid or base catalysis, PLE afforded 12a with a selectivity of 98:2 (Scheme 4C).34,35 This selectivity was maintained even for a gram-scale reaction, and both (R)- and (S)-dimethyl aspartate were viable substrates.
Scheme 4. (A) PLE-Catalyzed Hydrolysis of (Z)-2-Methylbutenedioic Acid Esters31 (B) PLE- and Subtilisin-Catalyzed Hydrolysis of 8;33 (C) Hydrolysis of (R)-Aspartate Dimethyl Ester 11 Using PLE35.

Several other hydrolases have also been used for site-selective catalysis involving polyacylated sugars. A lipase from Candida cylindracea was used to deacylate compound 13 to give the 6-OH derivative 14 in high yield (80–90%, Scheme 5A).36 This same group also examined lipase-catalyzed deacylation of other monosaccharides, including furanoses, pyranoses, and anomers of compounds in both classes.37 Lipases from Rhizopus japonics and Mucor sp., crude porcine pancreatic lipase, Aspergillus niger lipase (ANL), and C. cylindracea lipase (CCL) were used to site-selectively hydrolyze acylated furanoses. ANL and CCL provided the best results as shown in Scheme 5B, and hydrolysis of the pyranose series also proceeded via preferential hydrolysis at the primary positions.37 A separate study by the Crout group reported similar site-selective C-6 deacetylation of peracetylated α-d-hexopyranoses 17 (54–88% yield) and tetra-O-acetyl-α-d-hexopyranoses 19 (70–85% yield) using an esterase from Rhodosporidium toruloides (Scheme 5C).38 A more recent effort showed that C-1 of per-acylated glucose can be selectively hydrolyzed in preference to C-6 using porcine pancreatic lipase.39
Scheme 5. (A and B) Selective Hydrolysis of Peracylated Sugars from the Pyranose and Furanose Series by A. niger Lipase (ANL) and C. cylindracea Lipase (CCL); (C) R. toruloides Esterase-Catalyzed Hydrolysis of Peracetylated Compounds 17 and 1938,39.

2.1.2. Carboxylic Amide Formation.
Amide bond formation is among the most frequently performed organic transformation, and under typical chemical conditions, it requires the orchestration of different activating reagents and protecting group chemistries.40 Enzymatic amide bond formation is catalyzed by a variety of enzyme classes that activate carboxylic acid substrates under mild reaction conditions and without protecting groups. Many different classes of enzymes utilize ATP as a cofactor for activating carboxylic acids for amide bond formation (Scheme 6A),41 so application of these enzymes was initially limited by the requirement for superstoichiometric ATP, but efficient ATP recycling systems have alleviated this issue.42 ATP-dependent amide bond forming enzymes use different mechanisms to activate carboxylic acid substrates toward attack by amines. For example, acyl/aryl CoA synthetases form an acyl-AMP phosphoester that is attacked by CoASH to form the corresponding thioester. This acyl-CoA thioester is subsequently coupled to an amine via N-acyl/aryltransferases (NATs). Historically, the expense of the CoA cofactor and the relatively limited substrate scope of NATs limited applications of these enzymes for non-native amide bond formation. As with ATP recycling, CoA recycling allows for use of CoA and truncated derivatives in substoichiometric quantities for acylation of lysine residues in a histone-derived peptide with non-native amine donors.43
Scheme 6. (A) Activated Carboxylic Acid Intermediates; (B) Amidation of Diamine Substrate by CfaL and Related Enzymes; (C) PbCfaL-Catalyzed Coupling of Acids 25 and 2648.

Some amide bond forming enzymes catalyze the intermolecular reaction between two unprotected amino acids.44 The lack of homodimerization in these reactions highlights the inherent selectivity of these enzymes. ATP grasp enzymes are a class of amino acid ligases that release ADP to form an acyl─phosphate intermediate, which is attacked by an amine nucleophile to produce the amide. While ATP grasp enzymes have been engineered using site-directed mutagenesis,45 can have broad substrate scope,44 and have been used for the synthesis of macrocyclic peptides46 and natural product synthesis,47 no site-selective examples have been reported.
The enzyme CfaL belongs to a subset of amide bond synthetases that do not require an additional N-acyltransferase to affect amide bond formation. This enzyme and its homologues coupled a range of aliphatic and aromatic carboxylic acids to isoleucine and various amines to 3-methyl benzoic acid (Scheme 6B).48 These ligases also tolerate different amino acid coupling partners, including 2,4-diaminobutyrate (DAB) and l-ornithine, which underwent selective amide bond formation with m-methylbenzoic acid at the α-amino group using SsCfaL (Scheme 6B), obviating the need for side chain protecting groups. Notably, while PbCfaL gave a low yield and selectivity, it preferentially acylated the side chain amino group in DAB and l-ornithine, highlighting the catalyst-controlled site selectivity of this enzyme class. Improved activity of CfaL ligase variants generated via site-directed mutagenesis (i.e., Scheme 6C) also suggests that further protein engineering could be used to expand the scope and selectivity of these enzymes.
Increasing recognition of the importance of macrocyclic peptides49 has driven the use of thioesterases (TEs) to enable protecting group free macrocyclization via amide bond formation.50 The application of this enzyme class in vitro was historically limited by the requirement for a C-terminal N-acetylcysteamine (SNAC) peptide thioester to enable TE biocatalysis following solid-phase peptide synthesis.51 This penultimate coupling step introduces an additional expense by requiring coupling with N-acetylcysteamine, leads to epimerization of the α position, and complicates HPLC purification. Recently, it was found that the TE SurE tolerates peptide substrates with a simple C-terminal diol in place of the SNAC group required by most TE cyclases (Scheme 7). This modification simplified macrocycle synthesis and purification, and substrate profiling showed SurE accepted a variety of peptides with an l-amino acid at the AT-terminus and d-amino acid at the C-terminus. SurE G235L was found to fully cyclize a peptide substrate with a C-terminal glycine residue, eliminating the native requirement for the less common C-terminal d-amino acid. Other TE enzymes maintained high activity toward their native substrates upon incorporation of a C-terminal ethylene glycol group, showing the generality of this approach.
Scheme 7. Site-Selective Macrocyclization of Peptidic Substrate 28 to Surugamide 2951.

2.2. Alcohol and Amine Functionalization
2.2.1. Alcohol Acylation.
Many serine hydrolases also catalyze the acylation of alcohols with carboxylic acid derivatives to form esters. As in the hydrolysis reactions noted above, enzymes can provide these compounds with high selectivity and specificity relative to chemical methods.52,53 For example, polyhydroxylated steroids exhibit a wide range of biological activities, and while selective modification of the hydroxyl groups in these compounds is often desirable, their similar reactivity complicates this goal. Candida rugosa lipase was able to overcome this challenge to enable site-selective acylation of substrates 30 and 32 (Scheme 8A).54 Likewise, antisense oligonucleotides are used to selectively inhibit translation of disease-associated genes, but manipulating the hydroxyl groups in these compounds requires several protection/deprotection steps and with low yield.55 CALB-catalyzed transesterification, on the other hand, provides 5′-O-benzoyl-2′-deoxynucleosides in high yields (Scheme 8B).56 CALB has also been used for site-selective esterification in addition to transesterification. Naringin is a natural product with several similarly reactive alcohols on the sugar moiety and two phenol hydroxyl groups, but immobilized CALB allowed selective esterification of the primary hydroxyl group of the sugar (Scheme 8C).57
Scheme 8. (A) Lipase-Catalyzed Site-Selective Acylation of Vicinal Diols of Steroid Molecules;54 (B) Site-Selective Acylation of Deoxynucleosides by CALB;56 (C) Site-Selective Esterification of the Sugar Moiety of Naringin Using Immobilized Lipase from C. antarctica57.

Acyltransferases also catalyze site-selective acylation. For example, ArmB from Armillaria mellea is responsible for site-selective acylation of a single secondary hydroxyl group in the biosynthesis of melleolide (Scheme 9A).58 ArmB also catalyzes transesterification of orsellinic acid (OA) esters with various other alcohols (Scheme 9B). LovD is an acyltransferase found in Aspergillus terreus that converts the inactive monacolin J acid into lovastatin by the selective acylation of the hydroxy group.59 Wild-type LovD is inefficient and has poor stability, but directed evolution was used to engineer a LovD variant with activity 11-fold higher than the wild-type enzyme (Scheme 9C).59
Scheme 9. (A) ArmB-Catalyzed Site-Selective Acylation To Generate Melleolide;58 (B) Representative Orsellinic Acid Esters Found in Fungi;58 (C) Evolved Acyltransferase LovD Catalyzes the Acylation of Monacolin J Acid (MJA) To Generate Lovastatin60.

2.2.2. Alcohol Alkylation.
Selective alkylation of alcohols is critical to the function of natural products61 and pharmaceuticals.62 While a recent report describes a site-selective PLP-dependent O-alkylase capable of site-selective alkylation of a catechol moiety,63 this important transformation is typically catalyzed by S-adenosylmethionine (SAM)-dependent methyltransferases (MTases), which act by transferring a methyl group from the SAM cofactor to oxygen, carbon, sulfur, or nitrogen groups (Scheme 10A).64,65 O-Methyl transferases (OMTs) are particularly prevalent in natural product biosynthesis, as exemplified by the methylation of naringenin to the 7-O-methylated natural product sakuranetin 47 or 48 to give reticuline (Scheme 10B and 10C).66 The widespread presence of OMTs in biosynthetic pathways has facilitated the development of these enzymes for site-selective alkylation of hydroxyl groups on a variety of non-native substrates.
Scheme 10. (A) Mechanism for Methylation by the SAM Cofactor; (B) Methylation of Naringenin to Sakuranetin;66 (C) Methylation by OMT in Reticuline Biosynthesis67.

In one representative example of this capability, a catechol O-methyl transferase (COMT) and variants of an isoeugenol O-methyltransferase (IOMT) from C. breweri were used to selectively methylate either the para- or the meta-hydroxyl group of a series of substituted catechols (Scheme 11A and 11B).68 Although these enzymes share ~83% amino acid sequence identity, a triple mutant of IOMT (T133M/A134N/T135Q) obtained through site-directed mutagenesis could alter the site selectivity to provide para-methylation of caffeic acid. A different COMT from C. roseus (CaRo_MT) containing the same three mutations at the homologues residues provided the meta products (Scheme 11).68
Scheme 11. (A and B) Methylation of Phenols Catalyzed by OMTs to Give Meta (51) and Para (53) Products68.

OMTs can also catalyze selective methylation of the 2,4-dihydroxybenzophenone core in polyketide-derived natural products.69,70 A panel of these substrates was screened against wild-type and evolved variants of methyl transferases LtOMT and HsOMT, which were obtained from Lasiodiplodia theobromae and Hypomyces subiculosus. These enzymes methylated the ortho- and para-hydroxyl group (relative to the ketone moiety), respectively, with high site selectivity. These site preferences were maintained despite different substitution patterns on the benzophenone core. These same OMTs have also been used as models to understand and modify the site selectivity of the alkyl transfer reaction through protein engineering.71 Docking desmethyl lasiodiplodin into the active site of LtOMT and HsOMT suggested four residues that might affect substrate positioning relative to the SAM cofactor. Residues in LtOMT were mutated to the corresponding residues in HsOMT (Q384K, G386R, W387H, and Q388H, numbering taken from HsOMT), and the resulting OMT (M1) was capable of producing the para product as a minor byproduct in the reaction in Scheme 12A. A variant of M1 (HM1) constructed by installing segment 330–366 of HsOMT into M1 afforded exclusively the para-methylated product, indicating that this short region is critical for the site selectivity of the enzymes.71
Scheme 12. (A) Methylation of Novel Benzenediol Lactones Catalyzed by HsOMT and LtOMT Variants;71 (B) Selective Methylation of 60–62 by RnCOMT and MxSafC Using a SAM Regeneration System72.

As noted above, the high cost of SAM necessitates regeneration of this cofactor for efficient biocatalysis in alkyl transfer reactions.73-75 In one approach to this goal, an archaeal methionine adenosyltransferase (TkMAT) was used to catalyze the synthesis of SAM from l-methionine and ATP.76 This system was evaluated using OMTs with complementary site selectivity. Reaction of 3,4-dihydroxyphenethylamine (60), dihydrocaffeic acid (61), or 3,4-dihydroxybenzoic acid (62) catalyzed by RnCOMT from R. norvegicus77 or MxSafC from M. xanthus78 afforded complementary methylated products using substoichiometric SAM (Scheme 12B).72
Recent studies also show that MTases can accept synthetic SAM analogues, such as S-alkyl, -allyl, or -propargyl substituents, facilitating alkylation reactions beyond methylation.72,79-81 For example, RnCOMT accepts carboxy-S-adenosyl-l-methionine (cxSAM) to enable carboxymethylation of 3,4-dihydroxybenzaldehyde with roughly 4:3 m-/p-selectivity.82,83 The Y200L variant of this enzyme displayed improved m-/p-selectivity (64a′ 64% and 65a′ 3%), and similar selectivity trends were observed for 4-nitrocatechol. In a more recent study, an engineered halide methyltransferase (HMT) was evolved to accept ethyl iodide as the alkyl donor to enable ethylation reactions.74 A variant with improved kinetics for ethylation of S-adenosyl-l-homocysteine (SAH) was coupled with O-methyltransferases (IeOMT and COMT) to achieve site-selective ethylation of luteolin and allylation of 3,4-dihydroxybenzaldehyde (Scheme 13B).74 The ability of SAM cofactor regeneration systems to turn over SAH more than 1000 times and the more recent advent of thiomethyl-transferases for catalytic SAM production using sulfonate donors bode well for advances in biocatalytic alkylations.84
Scheme 13. (A) Site-Selective Carboxymethylation of 63 Using Carboxy-S-adenosyl-l-methionine (cxSAM) Catalyzed by COMT; (B) Regeneration of Alkyl-S-adenoxyl-l-methionines Using HMT, and Representative Ethyl and Allyl Products Generated by IOMT and COMT Variants, Respectively.

2.2.3. Amine Alkylation.
Selective alkylation of amines is a highly desirable yet challenging reaction.85 SAM-dependent enzymes also catalyze amine alkylation, but most reported substrates possess only a single amine substituent, so site selectivity is not an issue.86 Pyrazoles can be alkylated at either N-1 or N-2 as a result of tautomerization between these sites, making selective alkylation of these compounds is particularly challenging.87 Screening several promiscuous N-methyl transferases (NMTs) revealed that human nicotinamide NMT catalyzed the methylation of 3-methylpyrazole in 14% yield and roughly 2:1 N-1/N-2 site selectivity (Scheme 14A).86 Computational library design was used to generate a diverse panel of NMT active site variants, and several of these variants were found to give improved yields and complementary site selectivity toward several substituted pyrazoles (Scheme 14B). The engineered enzymes from this study were subsequently found to catalyze selective alkylation of a variety of other N-heterocycles (Scheme 14C).88
Scheme 14. (A) Site-Selective Alkylation Catalyzed by NMT Variant v36 and Haloalkanes; (B) Site-Selective Methylation by Variants Obtained through In Silico Mutational Studies;86 (C) Representative Examples of Site-Selective Alkylation Catalyzed by Variants of the NMTs88.

2.2.4. Hydroxyl Group Phosphorylation.
Phosphorylated molecules play critical roles in metabolism, signaling, and a wide array of other biological processes.89-91 Classes of phosphorylating enzymes include phosphotransferases, phosphorylases, phosphohydrolases, and phosphomutases.92 Phosphopentomutases (PPMs) catalyze the interconversion between ribose-5-phosphate and ribose-1-phosphate and are important in the bacterial nucleoside salvage pathway.93 Recently, a panel of PPMs was examined to identify an enzyme capable of converting 78 to 79 (Scheme 15) via transfer of a phosphate group from a primary hydroxyl to a more sterically encumbered secondary hydroxyl.94 A PPM from E. coli provided the highest activity on 78, and four rounds of directed evolution were used to improve the activity of this enzyme.95 This study also described an enzyme that catalyzes selective phosphorylation of the primary alcohol over the tertiary alcohol in substrate 80. Screening a range of kinases that naturally act on sugars and primary alcohols revealed that pantothenate kinase (PanK) from E. coli could accomplish this task, albeit with low activity. Directed evolution was used to improve this activity 100-fold to enable the desired transformation on the (R)-enantiomer of aldehyde 81. Directed evolution was also used to engineer a variant of 5-S-methylthioribose (MTR) kinase from Klebsiella sp. that could catalyze selective phosphorylation of the C-1 hydroxyl on 82 over the two other secondary alcohols on this substrate to give 83.94,96
Scheme 15. Evolved Variants of (A) E. coli Phosphopentomutase (PPM),94 (B) E. coli Pantothenate Kinase (PanK),94 and (C) Klebsiella sp. 5-S-Methylthioribose (MTR) Kinase Catalyze Site-Selective Phosphorylation of Different Non-Native Substrates96.

2.2.5. Hydroxyl Group Glycosylation.
Glycosylation of small molecules can also dramatically alter their biological function.97 Many natural glycosyl acceptors, including polyphenols, catechols, and sugars, often bear multiple similarly reactive hydroxyl groups, so site-selective glycosylation is required to ensure that only the biologically relevant isomer is formed.98,99 Glycosyl transferases (GTs) must also control the stereoselectivity at the anomeric carbon of the glycosyl donor. While this selectivity is typically accompanied by relatively high substrate specificity, researchers have found that some GTs and engineered variants can accept a range of glycosyl donors and acceptors to enable site-selective, non-native glycosylation.100-102 For example, while UGT71A15 was found to catalyze nonselective glycosylation of some polyphenolic acceptors, it catalyzes site-selective addition of glucose to 84 (Scheme 16A).103 In terms of donor variation, the antibiotic-modifying GT GtfE was shown to install a variety of monosaccharides onto vancomycin aglycone.104 The GTs OleD, OleI, and MGT accepted a variety of substrates from a panel of 64 acceptors and 18 sugar donors. Selective glycosylation of flavanols, coumarins, and 3,4-dichloroaniline was possible using these enzymes (Scheme 16B and 16C).105
Scheme 16. A) Glycosylation of 84 by Glycosyl Transferase UGT71A15;103 Representative Scope of (B) Glycosyl Acceptors and (C) Glycosyl Donors for Glycosylation of Oleandomycin with OleD, OleI, and MGTa.

aUDP = uridine diphosphate, GDP = guanosine diphosphate, and TDP = thymidine diphosphate glucose.105
Different protein engineering strategies have been used to expand the substrate scope and utility of GTs for site-selective biocatalysis. Directed evolution of a β-1,3-galactosyltransferase CgtB was successful in expanding the scope of acceptors the enzyme could functionalize (Scheme 17A).106 Fluorescence-activated cell sorting was used to screen large libraries of CgtB variants (from ~107 colonies) generated via error-prone PCR. This assay involved the use of fluorescently labeled glycosyl acceptors that are trapped within cells following GT-catalyzed reaction with a suitable donor. Improved sequences were subjected to DNA shuffling, and ~106 colonies were screened. This process provided variant S42 (Scheme 17B), which contained five amino acid mutations, provided significant improvements in activity toward different glycosyl acceptors and even UDP-Glu as a glycosyl donor, and maintained high site selectivity in all cases reported.
Scheme 17. Site-Selective Glycosylation by (A) β-1,3-Galactosyltransferase CgtB and (B) CgtB Variant S42; (C) Substrate Scope for Glycosylation of Non-Natural Sugar Acceptors and Uridine 5′-Diphosphogalactose (UDP Gal) Donors Catalyzed by LgtC.

Substrate engineering has also been used to expand the scope of site-selective glycosylation using GTs.107 For example, LgtC from Neisseria meningitidis is an α-1,4-galactosyltransferase that catalyzes the transfer of galactose from UDP Gal to lactose-containing acceptors (Scheme 17C).108 Different anomeric protecting groups were shown to enable glycosyl transfer involving several monosaccharides in place of the native disaccharide (lactose) substrate.107 Varying the protecting group could also be used to improve yields on a given monosaccharide while still maintaining high site selectivity.
Finally, site-selective C-glycosylation has been achieved using GT catalysis. For example, the CGT responsible for the biosynthesis of flavone-C-glycosides found in rice and wheat was shown to accept a range of non-natural substrates to generate the corresponding C-glycosylated products (Scheme 18A).109 Likewise, TcCGT1 catalyzes selective 8-C-glycosylation of several flavones and flavonoids.110 This enzyme could also be engineered through site-directed mutagenesis (I94E and G284 K) to switch the chemoselectivity from C- to O-glycosylation while maintaining high site selectivity for the latter reaction (Scheme 18B).
Scheme 18. (A) Glycosylation of the Open Chain Form of 2-Hydroxyflavanone (108) by C-Glucosyltransferase; (B) Site-Selective Catalytic Promiscuity of TcCGT1110.

Glycosidases, which natively catalyze the cleavage of glycosylated substrates,102 have also been engineered to catalyze site-selective transglycosylation, but these reactions are thermodynamically disfavored, and the reaction products are susceptible to the native reverse reaction.111 Withers found that mutating the active site Glu/Asp of several different glycosidases to alanine ablated the undesired hydrolysis reaction to enable site-selective glycosylation of substrates using glycosyl fluoride acceptors.111,112 Representative glycosidases on which this approach has proven successful are T. thermophilus β-glycosidase,113 Thermotoga maritima β-glucuronidase,114 and β-endoglycosidase from Rhodococcus sp.115 One example of site-selective catalysis by such a system involved the E197S mutant of Humicola insolens Cel7B.116 This enzyme was found to catalyze site-selective glycosylation of different flavonoids using the glycosyl acceptor LacF (Scheme 19B).
Scheme 19. Site-Selective Glycosidase Catalysis: (A) Synthesis of Glycoflavonoids Catalyzed by Cel7B–E197S with Disaccharide Donor Lactosyl Fluoride (LacF);116 (B) Structures of Saponins QS-21, QS-18, and QS-17.

Very recently, researchers at GSK used a combination of genome mining and directed evolution to engineer glycosidases for site-selective cleavage of sugars from a complex mixture of saponins obtained from bark extracts of the evergreen tree Quillaja saponaria Molina (Scheme 19B).117 Saponins QS-17, QS-18, and QS-21 from Quillaja extracts are used as adjuvants, but they must be purified by tedious reversed phase HPLC; removal of Q-18 is particularly important since it possesses high toxicity in mice. QS-21 can be obtained from QS-18 via loss of a specific β-d-glucose, and QS-18 can be obtained from QS-17 via loss of a specific rhamnose. Researchers envisioned that they could improve the yield of QS-21, eliminate toxic QS-18, and simplify the saponin mixture to facilitate isolation of QS-21 by identifying enzymes that could achieve these transformations. An extensive genome mining effort led to the identification of a glucosidase and a rhamnosidase with activity on Q-18 and Q-17, respectively. Concurrent directed evolution of these enzymes led to site-selective variants with improved activity that achieved the desired enrichment of QS-21 from Quillaja extracts when the engineered enzymes were used together.
2.2.6. Hydroxyl Group Epimerization.
In addition to enabling site-selective functionalization of hydroxyl groups via phosphorylation and glycosylation, enzymes can also epimerize hydroxyl groups. This is an especially desirable application in carbohydrate chemistry in that it enables converting readily available sugars to rare ones.118,119 Rare sugars can serve as low-calorie sweeteners to replace more common sugars, influence cell wall biosynthesis,120 and attenuate or increase immune response.121 While chemical methods for site-selective epimerization have been reported,122,123 enzymes from several different classes of epimerases often offer superior selectivity.124 Many enzymes in this family exhibit distinct site-selectivity for native reactions, but only those that have been applied to non-natural catalysis are highlighted below.
Uracil diphosphate (UDP) hexose-4-epimerases are NAD-dependent enzymes that epimerize specific hydroxyl groups on substrates, as dictated by the orientation of the substrate relative to the oxidized cofactor.125,126 Hydride transfer from the carbohydrate results in ketone formation, and rotation of the sugar within the active site of the epimerase repositions the ketone such that hydride transfer from the reduced cofactor produces the C-4 epimer of the original compound. A few examples of evolving UDP hexose epimerases with improved activity or expanded scope have been reported.118,119 The requirement for a UDP group on the substrates of these enzymes limits their synthetic utility, but GalE was found to catalyze site-selective 4-epimerization of free sugars.127 Site-saturation mutagenesis of residues predicted to interact with nucleoside portion of UDP–galactose in this enzyme showed that GalE N179S displayed improved activity for epimerization of the 4 position of free fructose and tagatose (i.e., lacking a UDP group). Further improvement of this activity could significantly improve the synthetic utility of these enzymes.
d-Tagatose epimerases have been extensively explored for non-native catalysis.128-131 These enzymes enable C-3 epimerization of d/l-tagatose, -fructose, -psicose, and -sorbose. Both C-3 and C-4 epimerization involves both Lewis acid catalysis by an M2+ ion and general acid/base catalysis involving a glutamic or aspartic acid residue. In the case of C-3 epimerization,132 these species catalyze formation of a cis-enediolate, and subsequent protonation from the opposite face of the enolate by a second aspartic acid residue leads to epimerization. C-4 epimerization133 proceeds via a retro-aldol reaction of the keto-sugar substrate involving a M2+ ion and a glutamic acid residue in the epimerase active site. Rotation of the resulting C-4 aldehyde presents the opposite face of the carbonyl to the intermediate M2+-bound enolate, so that the aldol reaction gives the opposite stereoisomer at C-4.
By exploiting a thermostabilized variant of the d-tagatose 3-epimerase from P. cichorii,134 divergent evolutions were carried out for epimerization of l-sorbose and d-fructose to l-tagatose and d-psicose, respectively.128 By coupling production of the product sugar to a NADH-dependent dehydrogenase, a high-throughput assay for detection of cofactor oxidation was developed that ultimately allowed for efficient production l-tagatose in 6 rounds of evolution and d-psicose in 8134 (Scheme 20A). Crystal structure analysis of the two final variants showed minor impacts on the overall structure and indicated that the mutations seemed to influence the charge at the surface of the protein where the substrate entered the active site.
Scheme 20. Epimerization of Abundant Carbohydrates To Furnish Rare Sugars: (A) Evolved d-Fructose Epimerases for d-Psicose and l-Tagatose Production; (B) Wild-Type C-3 d-Frucuronate Epimerase and Evolved C-4 d-Fructose Epimerase.

While use of d-tagatose-3-epimerases for production of non-native sugars is common, methods to produce d-tagatose from commodity sugars are underdeveloped. d-Tagatose is a low-calorie alternative to more abundant natural sugars but is rarely present in nature, making scalable isolation a challenge, and natural enzymes capable of epimerizing d-fructose to d-tagatose have proven elusive.133 An enzyme that catalyzes C-3 epimerization of a related substrate135 was engineered to achieve this goal using several rounds of structure-guided site-directed mutagenesis (Scheme 20B). The final variant 5V was capable of producing d-tagatose at 200 g/L titers and had reduced activity on the native d-tagaturonate substrate compared to the wild-type variant, indicating a significant modification of the substrate scope.
2.2.7. Alcohol Oxidation.
Alcohol oxidation is common in biosynthetic pathways, and several classes of enzymes, including alcohol oxidases, have been used to catalyze oxidation of primary alcohols over secondary alcohols.136 Other enzymes, including NAD(P)(H)-dependent hydroxysteroid dehydrogenases (HSDHs), are capable of discriminating between secondary alcohols in steroids and bile acids.137,138 HSDHs from Bacteroides fragilis and Clostridium absonum DSM 599, for example, were used to develop a chemoenzymatic method for converting cholic acid to ursodeoxycholic acid, which is used to treat gallstones.139 A one-pot strategy for this transformation used 7α- and 12α-HSDHs to oxidize the hydroxyl groups at positions C-7 and C-12 of cholic acid; a 7β-HSDH was then used to reduce the ketone at position C-7 to a β-hydroxyl group (Scheme 21). A more recent study focused on the epimerization of chenodeoxycholic acid to produce ursodeoxycholic acid in another one-pot experiment where a NAD(H)-dependent 7α-HSDH and a NADP(H)-dependent 7β-HSDH were employed.140 In this case, the oxidative step was coupled to a flavin reductase to oxidize NADH to NAD+, and the reaction was driven toward the production of ursodeoxycholic acid thanks to the auto-oxidation of FMN under aerobic conditions.
Scheme 21. One-Pot Synthesis of 12-Ketoursodeoxycholic Acid from Cholic Acid Using HSDHs139.

2.3. Carbonyl Transformations
2.3.1. Carbonyl Reduction.
In nature, enzymes from the aldo-keto reductase (AKR) superfamily catalyze the interconversion of a wide range of carbonyl compounds and the corresponding alcohols.141 Representative AKRs include aldose reductases, aldehyde reductases, hydroxysteroid dehydrogenases, ketoreductases (KREDs), and alcohol dehydrogenases (ADHs).142 KREDs and ADHs have been widely used and engineered to synthesize chiral alcohols.143 One example of site-selective KRED biocatalysis involves reduction of triketone 127 to the corresponding dihydro iso-alpha acid 128 (Scheme 22A). Directed evolution was used to engineer a site-selective KRED for this transformation.144 Compound 129 presents an interesting challenge for site-selective ketone reduction since reduction of the trifluoromethyl ketone would be expected based on electronic considerations (Scheme 22B). Indeed, chemoselective reduction of 129 to give racemic 131 has been reported.145 Several commercially available KREDs (KRED-112, KRED-129, KRED-131) were found to catalyze this same reaction with high enantioselectivity (>99% ee). Notably, an alcohol dehydrogenase from Candida parapsilosis (ADH-CP) and KRED-A1p demonstrated enantioselectivity (98%) and chemoselectivity (100% and 87%, respectively) toward the reduction of the methyl carbonyl, producing the methyl hydroxyketone 130.146
Scheme 22. Site-Selective Reduction of (A) Sterically and Electronically Similar Ketones in 127 and (B) Either Ketone in 129 Using Engineered KREDs.

The commercial availability of KREDs and ADHs has facilitated several additional examples of site-selective ketone reduction. Screening 20 commercially available KREDs for the reduction of α-alkyl-1,3-diketones, for example, led to the site-selective reduction 3-methyl-2,4-hexadione with 100% yield after 24 h and >99% diastereomeric ratio (Scheme 23A).147 Similarly, more than 400 KREDs, both proprietary and commercial, were screened for the reduction of 5-androstene-3,17-dione (5-AD) to dehydroepiandrosterone (DHEA).148 This study identified a KRED from Sphingomonas wittichii capable of fully converting 5-AD to DHEA with excellent site selectivity and diastereoselectivity (>99% de, Scheme 23B). Additionally, a more recent study evaluated more than 500 commercial KREDs for the reduction of diketone 136 as part of the development of a chemoenzymatic strategy for the synthesis of navoximod.149 KRED ADH 430 selectively reduced the cyclohexanone over the acyclic ketone in this substrate with a 95% yield and >99% selectivity for the trans isomer 137 (Scheme 23C). Finally, ADHs from different origins showed high site selectivity toward the reduction of tert-butyl 4-methyl-3,5-dioxohexanoate to tert-butyl 5-hydroxy-4-methyl-3-oxohexanoates.150,151 ADHs from Lactobacillus brevis (LBADH) and Saccharomyces cerevisiae (YGL157w) both showed activity on the (S)-enantiomer with complementary diastereoselectivities, while ADH from Rhodococcus sp. (RS 1-ADH) was able to reduce the (R)-enantiomer (Scheme 23D), providing a platform to access most stereoisomers of this synthetically attractive building block via enzymatic dynamic kinetic resolution.
Scheme 23. Site-Selective Carbonyl Reductions: (A) Reduction of 3-Methyl-2,4-hexadione;147 (B) Formation of Dehydroepiandrosterone (DHEA) by a KRED from S. wittichii;148 (C) Reduction of a Diketone as Part of the Chemoenzymatic Synthesis of Navoximod;149 (D) Kinetic Resolution of tert-Butyl 4-Methyl-3,5-dioxohexanoate with Alcohol Dehydrogenases150,151.

2.3.2. Carbonyl Reductive Amination.
Biocatalytic methods for amine formation have been widely adopted in both industry and academia. This capability was first facilitated by the development of transaminases (TAs), which catalyze the formation of primary amines from ketones, as exemplified at industrial scale by the biocatalytic synthesis of sitagliptin.152 More recently, reductive aminases (RedAMs) and imine reductases (IREDS) have expanded this capability significantly to allow for the formation and/or enantioselective reduction of secondary and tertiary amines.153,154 Both TAs and IREDs are now available as commercial enzyme panels to facilitate easy screening. TAs use an amine donor and the pyridoxal phosphate (PLP) cofactor, while IREDs require a reducing equivalent provided by NAD(P)H to reduce an imine to the corresponding amine.154 These enzymes typically have restricted active sites formed upon dimerization of the enzymes in solution. This active site structure has been exploited to enable site-selective catalysis in cases where two ketones are distinguished by their relative steric environments (Scheme 24A). Even for examples where sterically similar groups, like methyl and n-propyl, are used, a panel of (S)- and (R)-selective TAs was nearly entirely 99:1 selective for the formation of the less hindered amine (Scheme 24B).155
Scheme 24. ω-Transaminase-Catalyzed Cyclization of Diketones: (A) General Scheme for Cyclization with TAs; (B) Cyclization of Nonane-2,6-dione with a Panel of ω-TAs.

ω-TAs have also been used for the asymmetric synthesis of 2-methyl-5-phenylpyrrolidines via enzymatic and chemoenzymatic cascades. In the latter approach, a ω-TA was used to site selectively aminate the 4 position of 1-phenylpentane-1,4-dione, and the resulting amine underwent spontaneous cyclization to form an imine (Scheme 25A).156 This intermediate was nonspecifically reduced to the racemic pyrrolidine by ammonia borane, and the trans-pyrrolidine was selectively reoxidized to the imine by a monoamine oxidase, leading to enrichment of the cis isomer. By replacing the ammonia borane and MAO-N with an imine reductase (Scheme 25B), this cascade was rendered fully enzymatic with up to 68% yield and >98% dr and er.157 This predictable preference for amination of less hindered ketones also extends beyond diketones, as shown in the synthesis of the bicyclic alkaloid xenovenine.158 In this study, the first key asymmetric step was accomplished by screening a panel of wild-type (R)- and (S)-selective transaminases. The best variants provided the desired products with perfect site selectivity for the less hindered ketone and 99% enantioselectivity, enabling the most efficient synthesis of the xenovenine enantiomers at the time.
Scheme 25. (A) Coupling of ω-TA with Nonselective Ammonia Borane Reduction and trans-Pyrrolidine-Selective MAO-N Oxidation To Accumulate Product 154; (B) ω-TA-Catalyzed Amination Coupled to IRED-Catalyzed Reduction of Simple Diketone 155.

2.4. Alkene Transformations
2.4.1. Alkene Hydration.
Alkenes are substrates for a wide range of catalytic asymmetric transformations like hydrogenation, hydroformylation, dihydroxylation, and epoxidation. While differentiating the prochiral faces of a given olefin is often possible in many of these reactions, site-selective reaction when multiple olefins are present remains challenging. This task is particularly difficult for olefin hydration to produce the corresponding alcohols since this reaction is traditionally carried out using catalysts like mineral acids, metal oxides, and zeolites under harsh conditions.159 Several hydratases have been discovered that can activate unactivated alkenes to achieve stereoselective hydration.159-161 While site-selective hydration of non-native olefin substrates remains rare, linalool dehydratase isomerase (LinD)162 from β-protobacterium Castellaniella defragrans highlights the potential utility of this transformation in biocatalysis. The native substrate of this enzyme, β-myrcene, is a monoterpene that is produced by a variety of plants, and it is a key ingredient in the flavor and food industries. LinD catalyzes site-selective hydration of the 1,1-disubstituted olefin in this compound to generate (S)-linalool in 24% conversion after 24 h (Scheme 26).163 Preliminary analysis of LinD variants confirmed the importance of active site residues believed to play a role in water activation and altered product distributions in the reverse reaction of geraniol to give myrcene and linalool. These efforts suggest that LinD may be a viable platform for biocatalytic olefin hydration.
Scheme 26. Reversible (De)hydration of the Tertiary Alcohol (S)-Linalool to β-Myrcene and Its Isomerization to the Primary Alcohol Geraniol Catalyzed by LinD163.

2.4.2. Alkene Epoxidation.
While site-selective olefin epoxidation can be achieved using directing groups or electronic bias,164 methods to achieve this transformation on diverse substrates remain highly desirable due to its synthetic utility. Cytochromes P450 have been found to be especially useful for catalyzing this reaction due to the reactivity of the intermediate compound I, a ferryl porphyrin cation radical species (vide infra), toward different types of olefins.165 The high reactivity of olefins toward this species relative to C─H bonds enables chemoselective epoxidation of substrates that might otherwise undergo C─H hydroxylation by these enzymes. For example, the antitumor sesquiterpene lactone parthenolide, which contains two alkenes and multiple C─H bonds that could potentially be hydroxylated, gave a mixture of epoxidation and allylic hydroxylation using P450-BM3 variant FL#62.166 Three active site mutations were found to provide an enzyme that is 90% selective for formation of epoxide product 164 (Scheme 27A) with only minor formation of the hydroxylated side reactions and nearly 5-fold improved TTN. A variant of BM3 also catalyzed site-selective epoxidation of the terpenoid β-cembrenediol, albeit as a side product in a reaction that produced many oxidized products.167
Scheme 27. Alkene Epoxidation by P450 Enzymes: (A) Epoxidation of Parthenolide with Evolved BM3 Variant III-D4; (B) Epoxidation of a Terminal Alkene by a TamI Variant; (C) Selective Epoxidation of Alkenes Appended to Theobromine by CYP 3A4.

Several other P450s have also been used for site-selective epoxidation. For example, TamI oxidizes its native substrate 165 in a cascade of reactions to form the tirandamycins, including epoxide formation of a bicyclic core in the presence of several similarly reactive alkenes (Scheme 27B).168 Active site mutations showed that the order and degree of TamI-catalyzed oxidations could be modified, as exemplified by TamI L101A_L295I, which catalyzes the epoxidation as the first reaction before later showing improved hydroxylation of the adjacent C─H bond and decreased hydroxylation at other sites usually transformed during the native oxidative cascade. Theobromine is a substrate of the human P450 enzyme CYP 3A4.169 Using this known enzyme─substrate pair, epoxidation of various polyolefins appended to N3 in the theobromine core (Scheme 27C) was studied. In the cases of substrates 167 and 171, the epoxidation was perfectly selective for the distal alkene. Site selectivity for 1,3-hexadiene 169 was measured at 53%, although the enantioselectivity for the major product was higher than that observed for the corresponding 2,4-hexadiene 167. This study not only shows an effective site and enantioselective epoxidation reaction but also cleverly uses a known substrate for P450s to anchor the modified compounds in the active site, spurring the promiscuous activity.
2.5. Miscellaneous Functional Group Transformations
2.5.1. Nitrile Hydration.
Hydration of nitriles to the corresponding amide bond is a valuable transformation, and nitrile hydratases (NHases) have been used to catalyze this transformation under mild conditions even for commodity chemicals like acrylamide.170,171 NHases are metalloenzymes that typically contain an active site Co or Fe ion, although an exact mechanism has not been fully described for this enzyme class.172,173 NHase reactions are also frequently performed using whole cells to reduce the cost associated with biocatalyst preparation. The sequestration of the NHase enzyme within cells also allows for the incorporation of NHases into chemoenzymatic cascades where the free enzyme may be inhibited by other cofactors or cocatalysts in solution.174 This was demonstrated in the synthesis of receptor agonist compound 174,175 which was performed in a two-step, one-pot reaction wherein nitrile bond hydration was catalyzed by the Co-dependent CGA009 NHase (Scheme 28). The enzyme was highly selective for the less hindered nitrile, which allowed for the formation of the receptor agonist in 49% yield, providing a much shorter and higher yielding synthesis than the best previously reported effort. In addition to this site-selective example, the same work also describes several enantioselective kinetic resolutions, showing the wide utility of this highly active class of biocatalysts.
Scheme 28. Chemoenzymatic Cascade to Compound 174 Enabled by Site-Selective Hydration of Polynitrile Precursor.

2.5.2. Nucleophilic Demethylation.
Demethylation of methyl phenyl ethers is an important transformation in synthetic organic chemistry,176,177 but traditional synthetic methods rely on harsh reagents or/and reaction conditions that limit site selectivity.178,179 Multicomponent B12-dependent methyl transferases (MTs) catalyze this reaction with concomitant methylation of an acceptor substrate.180 While the native methyl donors for these enzymes include substrates like CH3─H4folate, methanol, methylamine, and dimethylsulfide,180-182 some can also accept various methyl phenyl ethers as substrates. Demethylation proceeds via attack of the methyl group by the nucleophilic cob(I)alamin form of a cobalamin binding protein (e.g., vdmB) catalyzed by a suitable carrier protein (e.g., dhaf4611) to form the methyl cob(III)alamin form of the protein and the demethylated substrate. The reverse reaction then occurs using an acceptor substrate to drive the reaction in the desired direction.181 For example, site-selective monodemethylation of papaverine (175) to form 176 was achieved (Scheme 29) using a veratrol-O-demethylase from A. dehalogenans (vdmB) in combination with the cobalamin carrier protein dhaf4611 from D. hafniense.183 This same system also enabled selective monodemethylation of 177 to give 178 (6% conversion), and several other aromatic substrates with methyl ether substituents provided preferences in the site selectivity of demethylation.
Scheme 29. Site-Selective Demethylation of Papaverine and rac-Yatein by Cobalamin-Dependent Methyltransferase MT-vdmB.

3. C─H FUNCTIONALIZATION
Site-selective functionalization of C─H bonds constitutes a long-standing challenge in synthetic chemistry since such transformations could eliminate the need for prefunctionalized starting materials and enable new synthetic routes.184,185 The same difficulties associated with site-selective functional group manipulation apply to C─H bonds, but at least at first glance, the challenge appears greater since C─H bonds are ubiquitous in organic molecules. The molecular context of a given C─H bond, however, leads to differences in proximity to blocking or directing groups, acidity, bond dissociation energy, and stereoelectronic properties that can be differentiated by reagents and catalysts.186 Indeed, the reactivity of C─H bonds is so diverse and approaches to functionalize them so varied that Dyker once recounted in an early review of the field that “C─H functionalization begins just below one’s own results”.187 Nonetheless, there are many cases where similar C─H bond reactivity leads to poor site selectivity using small molecule catalysts. Just as importantly, selective functionalization of a single C─H bond on a given substrate is just the start of the challenge since, ideally, one would be able to functionalize any C─H bond on that substrate. Only enzymes have been able to achieve this feat,188,189 enabling sequential functionalization of C─H bonds in natural product biosyntheses, for example,190 that far surpasses current synthetic methods. Leveraging this power for synthetic chemistry provides a means to complement our ability to exploit differences in C─H bond reactivity with differences in substrate binding and molecular recognition to expand the scope of site-selective C─H functionalization.
3.1. C─H Hydroxylation
Several classes of enzymes catalyze C─H hydroxylation due to the broad importance of hydroxylation for the function and metabolism of natural products and xenobiotic compounds.191 As noted above, alcohols are also important for the function of pharmaceuticals, natural products, and agrochemicals, and they serve as useful intermediates and building blocks. The diversity of enzymes that catalyze this transformation has led to several options for site-selective hydroxylation of different substrate classes, so these are grouped below based on enzyme class.
3.1.1. Cytochromes P450.
Cytochrome P450 monooxygenases (P450s) catalyze a variety of oxidative reactions, including C─H hydroxylation.192 These enzymes possess a heme cofactor with a cysteine thiolate acting as a ligand to the iron center on the proximal face, leaving the distal face free for O2 activation. P450-catalyzed hydroxylation is initiated by substrate binding, which promotes electron transfer from a NAD(P)H cofactor through a P450 reductase or ferredoxin to the iron center of the heme cofactor (Scheme 30).192,193 Heme reduction is followed by O2 binding to the distal face of heme, transfer of a second electron by a P450 reductase or ferredoxin to produce an Fe(III)─peroxo species, and protonolysis of this intermediate to give a highly reactive Fe(IV)─oxo complex known as “compound I”. This intermediate can abstract a hydrogen atom from primary, secondary, or tertiary C─H bonds (or react with olefins as noted in section 2.4.2). C─H abstraction is followed by a radical rebound step between the substrate radical and the newly formed Fe(III)─hydroxo species, resulting in the hydroxylated product. Perhaps not surprisingly, given their ability to react with such a broad range of C─H bonds in diverse substrates, protein engineering has proven central to the utility of P450s for site-selective catalysis. Because of the extensive effort that this field has attracted, the examples below are meant to highlight early studies in the field and recent examples of the diversity of substrates that can now be site-selectively functionalized. Reviews on P450 biocatalysis should be consulted for a more exhaustive coverage.194-196
Scheme 30. Simplified Scheme of the Catalytic Cycle for the Hydroxylation of a Substrate R─H by a Cytochrome P450.

Many P450s have been used for site-selective hydroxylation,197,198 but cytochrome P450BM3 (BM3), a fatty acid hydroxylase from Bacillus megaterium, has attracted particular attention because it has a fused reductase domain that leads to high reaction rates and obviates the need for a separate reductase.199 This architecture also simplifies protein engineering, and several groups have developed BM3 variants that provide high yields and site selectivity on a range of different substrates. Early studies by Arnold used directed evolution to develop BM3 variants with activity on n-alkanes,200,201 and subsequent mutagenesis of these variants led to enzymes with high site selectivity on these substrates, including 86% selectivity for 2-nonanol with 1-12G and 67% selectivity for 4-nonanol with 9-10A A82L (Scheme 31A).202 Further evolution of variant 139-3 led to the development of propane monooxygenase (PMO) P450PMOR2, capable of carrying out the challenging hydroxylation of propane with native-like efficiency with a 9:1 2-propanol:1-propanol product ratio.203
Scheme 31. (A) Site-Selective Hydroxylation of n-Alkanes by P450BM3 Variants;202 (B) Demethylation of Protected Monosaccharides by P450BM3 Variants Shown as the Major Product for Each Reaction204.

The P450 variants obtained via these evolution efforts proved to be remarkably versatile for site-selective hydroxylation of different compounds, including drugs,205 natural products,206 and other small molecules. In a particularly notable example that illustrated the ability to tune site selectivity to different C─H bonds using a combination of protein engineering and substrate engineering, P450BM3 was used for site-selective demethylation of permethylated monosaccharides (Scheme 31B). This capability enabled chemoenzymatic functionalization of different sugars via subsequent manipulation of the single deprotected hydroxyl group.204 Screening previously engineered P450BM3 variants to identify hits for different sugars followed by further directed evolution yielded a panel of enzymes with high selectivity (~50–100% in terms of product distribution) for the demethylation of protected hexoses (182–184).
Hydroxylation of steroid substrates is of particular interest for pharmaceutical applications, but this goal requires distinguishing several nonactivated C─H bonds. Additionally, while various eukaryotic P450s can perform these transformations,207 these are often membrane proteins, which makes them unattractive as biocatalysts. The previously engineered P450BM3 variant 9-10A F87V TS was therefore further evolved using combinatorial alanine scanning and random mutagenesis to enable selective C-2 hydroxylation of 11α-hydroxyprogesterone (185) and demethylation of the alkaloids thebaine and dextromethorphan (Scheme 32A).208 Site-selective hydroxylation of a range of steroids using different BM3 variants was subsequently reported by other groups.209-212
Scheme 32. Site Selectivity Expressed as Percent in the Product Distribution in the Hydroxylation of Steroids by Engineered P450 Enzymesa.

aRemaining products consist of hydroxylation at different sites and other oxidation products. Reactions shown include (A) 11α-hydroxyprogesterone (185) with P450BM3 F1,208 (B) testosterone (186) with P450BM3 variants KSA-1 and KSA-14,213 and (C) progesterone (187) with CYP106A2 T89N/A395I.214
Wong also conducted early studies focusing on the hydroxylation of compounds containing aromatic moieties, and these efforts led to the development of BM3 variant KT5.215 While hydroxylation of propylbenzene (188) with wild-type P450BM3 resulted in a product distribution of 99% 1-phenyl-1-propanol, variant KT5 yielded 78% 1-phenyl-2-propanol and 20% 1-phenyl-1-propanol (Scheme 33A). This effect was even more pronounced when using toluene as substrate, resulting in 95% benzyl alcohol and 5% o-cresol, while the product distribution for BM3 consisted of 98% o-cresol. Further computational studies on KT5 linked the change in site selectivity in the hydroxylation of toluene to changes in the active site caused by mutations F87A/A330P, which allow for toluene to bind in previously inaccessible orientations.216
Scheme 33. Site-Selective Hydroxylation Reactions Carried out with Engineered P450BM3 Enzymes and (A) Propylbenzene (188),215 (B) Cyclohexene-1-carboxylic Acid Methyl Ester (189), and Cyclopentene-1-carboxylic Acid Methyl Ester (191)217a.

aProduct distribution is presented as the percentage of the major product in each reaction.
Reetz and co-workers used P450BM3 to catalyze site-selective hydroxylation of cyclohexenes such as cyclohexene-1-carboxylic acid methyl ester (189).217 Wild-type P450BM3 provided a product distribution comprising 84% (R)-3-hydroxycyclohexene-1-carboxylic acid methyl ester (34% ee) and 16% isomeric alcohols and other oxidation products. Targeted mutagenesis of the active site resulted in variants with comparable conversion levels and improved selectivities, including one (F87V/A328N) that provided the desired (R)-3-hydroxycyclohexene product with 93% site selectivity and 96% ee. A second variant (I263G/A328S) gave the corresponding S-enantiomer with 97% site selectivity and 94% ee (Scheme 33B). The variants engineered for the reaction of the model cyclohexene 189 were successfully tested on other substrates, with cyclopentene-1-carboxylic acid methyl ester (191) showing significant improvements in site selectivity, from 58% of side products with wild-type P450BM3 to 13% and 17% with the (R)- and (S)-selective variants, respectively.
Several P450BM3 biocatalysts have been developed for the site-selective hydroxylation of testosterone (187, Scheme 32C). BM3 variant F87A was used in two different studies to engineer variants KSA-1 with 97% selectivity toward 2β-hydroxylation, KSA-14 with 96% selectivity toward 15β-hydroxylation, LIFI-WQM with 98% selectivity toward 6α-hydroxylation, and WWV-M with 92% selectivity toward 16β-hydroxylation.209,213 Additionally, the triple-mutant P450BM3 F87G/A328G/A330W was evolved for hydroxylation at the C-7 position of 186, with final variant LG-23 having an improved site selectivity of 90% 7β-hydroxytestosterone.210
Finally, Fasan has engineered BM3 variants that catalyze site-selective and enantioselective hydroxylation of artemisinin (193) using the promiscuous variant FL#62 as a starting point.218,219 While the parent enzyme showed a product distribution of 83% 7(S)-hydroxyartemisinin, 10% 7(R)-hydroxyartemisinin, and 7% 6α-hydroxyartemisinin, highly selective variants were obtained resulting in IV-H4 producing 100% 7(S)-hydroxyartemisinin, II-H10 producing 100% 7(R)-hydroxyartemisinin, and X-E12 producing 94% 6α-hydroxyartemisinin (Scheme 34A). FL#62 variants that exhibit site-selective hydroxylation of parthenolide analogs were also developed, showcasing the value of promiscuous variants that can be further engineered into selective biocatalysts.166,220,221
Scheme 34. (A) Selectivity in the Product Distribution for the Hydroxylation of Artemisinin (193) by Engineered P450BM3 Variants;219 (B) Site Selectivity of Hydroxylation Shown as the Ratio of C-10:C-12 Hydroxylated Products for YC-17 Analogues with Varying Anchoring Groups with PikC226.

A variety of other prokaryotic P450s have also been the focus of extensive engineering efforts. For instance, CYP106A2 has been developed as a platform for selective steroid hydroxylation with mutant T89N/A395I showcasing the potential of this enzyme by having its site selectivity switched to 80.9% 11α-hydroxyprogesterone from wild-type 27.7% (Scheme 32C).214,222 Additionally, the P450 PikC, from the pikromycin biosynthetic pathway,223 has been engineered into PikCD50N, a single mutant with increased activity on the native macrolide substrates YC-17 and narbomycin.224,225 This variant was also used to show that the native site selectivity between C-10 and C-12 hydroxylation could be modulated by modifying the desosamine sugar group with a site selectivity C-10:C-12 of 1:1 and >20:1 for YC-17 and an analogue with a benzylic amine as anchoring group, respectively (Scheme 34B).226
While aliphatic hydroxylation occurs via abstraction of the C─H bond, aromatic hydroxylation by P450s is believed to take place via the formation of an arene oxide or electrophilic attack of compound I on the aromatic substrate.227-229 Site-selective aromatic hydroxylation by engineered P450BM3 variant M2 (P450BM3 R47S/Y51W/I401W)230 was shown to have high selectivity for o-hydroxylation of monosubstituted benzenes, including substrates that were not accepted by the wild-type enzyme (Scheme 35A).231 A more recent study showed that CYP199A4 from Rhodopseudomonas palustris could be engineered to hydroxylate 4-phenylbenzoic acid (202) to 4-(2′-hydroxyphenyl)benzoic acid (203) with a 83% site selectivity, a reaction that was not catalyzed by wild-type CYP199A4 (Scheme 35B).232
Scheme 35. (A) Product Distribution in the Hydroxylation of Monosubstituted Benzenes by P450BM3 M2;231 (B) Selective Hydroxylation of 4-Phenylbenzoic Acid (202) by Variant CYP199A4 F182L with Percentages Depicting the Product Distribution232.

3.1.2. Fe(II)- and α-Ketoglutarate-Dependent Oxygenases.
A second class of enzymes that hydroxylate C─H bonds site selectively is the Fe(II)-/α-ketoglutarate-dependent oxygenases (FeDOs). The consensus mechanism of FeDO-catalyzed C─H hydroxylation is initiated via bidentate binding of α-ketoglutarate to a Fe(II) ion that is coordinated by a conserved His─Asp/Glu─His facial triad and three water ligands, resulting in the displacement of two water molecules (Scheme 36).233 Substrate binding causes loss of the last water ligand and creates an open coordination site for O2 for binding and activation to form a Fe(III)─superoxo intermediate. Oxidative decarboxylation of α-ketoglutarate by the superoxo species leads to formation of a Fe(IV)─oxo intermediate, succinate, and CO2. As in P450s, the Fe(IV)─oxo species is responsible for abstracting a hydrogen atom from the primary substrate to generate a Fe(III)─OH intermediate and a substrate radical, which react via radical rebound to form the hydroxylated product and regenerate the Fe(II)(H2O)3 center.
Scheme 36. Simplified Scheme of the Catalytic Cycle for the Hydroxylation of Substrate R–H by a FeDO.

The use of FeDOs for site-selective hydroxylation has primarily focused on amino acid substrates. These enzymes exhibit relatively high substrate specificity compared to P450s, so they tend to be used on compounds similar to their native substrates. For example, bacterial and fungal l-proline hydroxylases can selectively produce cis-3-, cis-4-, trans-3-, and trans-4-hydroxy-l-proline (Scheme 37A).234 These enzymes also accept other cyclic amino acids, including l-pipecolic acids. However, native pipecolic acid hydroxylases, such as GetF and PiFa, have been shown to have poor activity on l-proline, with efforts to increase substrate promiscuity via protein engineering being unsuccessful.235,236 A patent by Merck & Co. reported the evolution of a proline hydroxylase over 12 rounds of evolution for the production of (2S,5S)-hydroxypipecolic acid at 180 g/L substrate loading.237 The evolution approach included several rounds aimed at mitigating self-deactivation of the enzyme by preincubating it with reaction components before adding substrate into the bioconversion. Additionally, a FeDO from the same gene cluster as GetF, GetI, was characterized as a citrulline hydroxylase and engineered to selectively hydroxylate arginine to 4-hydroxyarginine with an activity high enough to allow for incorporation in the synthesis of novel dipeptides.238,239
Scheme 37. Examples of Site-Selective Hydroxylation of Amino Acids with FeDOs: (A) Proline and Pipecolic Acid Hydroxylases;236 (B) Lysine Hydroxylases;240 (C) Engineered SadA Variantsa.

aPercentages correspond to relative amounts in the product distribution.
Genome mining has been used to discover new FeDOs with novel selectivities. A series of l-lysine (KDO1–3) and l-ornithine (ODO) hydroxylases obtained via such a strategy resulted in biocatalysts with the less common C-4 site selectivity (KDO2 and KDO3) as opposed to the more frequent C-3 hydroxylation of polar amino acids like lysine (210, Scheme 37B).240 This capability allowed for the synthesis of dihydroxylated l-lysine via the sequential use of KDO1 and KDO2/3. These lysine hydroxylases have been used in combination with a lysine decarboxylase to form an enzymatic cascade for the synthesis of chiral amino alcohols from l-lysine.241 Furthermore, the lysine hydroxylase GlbB from the glidobactin biosynthetic cluster in Polyangium brachysporum was shown to hydroxylate C-4 of both l-lysine and l-leucine.242
Finally, a recent study by our group showed that the site selectivity of the FeDO SadA can be altered.243 Variants SadX (MBP-fused SadA D157G) and SadXL (SadX F152L) lack the native glutamic acid in their facial triad, allowing for exogenous anions to bind to the Fe(II) center and rescue native activity. The F152L mutation in SadXL produced a distinct change in the site selectivity of the hydroxylation of N-succinyl-l-leucine (213), from 98% β-hydroxylation with SadX to 80% γ-hydroxylation when CsF is added to a reaction. The identity of the anion also has a significant impact, with the product distribution of SadXL being 57% γ-hydroxylation in the presence of sodium formate (Scheme 37C).
3.1.3. Other Site-Selective Metalloenzyme Oxygenases and Dioxygenases.
Heme peroxidases are widely distributed enzymes that carry out oxidative transformations by reducing peroxide (H2O2) through their heme prosthetic group.244,245 The promiscuous unspecific peroxidase (UPO) is an aromatic peroxygenase known to perform oxyfunctionalization of C─H bonds that has been extensively studied and engineered.244 For example, a fungal UPO from Agrocybeaegerita (AaeUPO) was engineered to enable selective hydroxylation of naphthalene to give 1-naphthol.246 This effort resulted in a 2-fold increase in activity from the parent enzyme and a final ratio of 1-naphthol:2-naphthol of 97:3. AaeUPO also catalyzes site-selective hydroxylation of short alkanes such as propane and n-butane to the corresponding 2-alcohols and flavonoids like apigenin and luteolin to the corresponding 6-hydroxyflavonoids, showcasing the broad substrate scope of this enzyme (Scheme 38A).247,248 Finally, engineered AaeUPO variant AaeUPO Fett was recently reported to show improved selectivity for the functionalization of subterminal positions of fatty acid substrates.249 The crystal structure of the Fett variant showed a narrower channel into the active site, restricting the access of bulkier substrates and resulting in a higher site selectivity toward the ω position of lauric (218), myristic (219), palmitic (220), and stearic (221) acids. While the ω-1 selectivity of AaeUPO ranged from 20% to 56%, the Fett variant provided >92% selectivity in all cases (Scheme 38B).
Scheme 38. (A) Flavonoid Site-Selective Aromatic Hydroxylation by Heme Peroxidase AaeUPO;248 (B) Hydroxylation of Saturated Fatty Acids by AaeUPO Variantsa.

aProducts are shown as percentages of the total product pool, with remaining products being ω-hydroxylated fatty acids and overoxidation products.249
Rieske nonheme iron-dependent oxygenases are iron─sulfur cluster-containing enzymes involved in the degradation of aromatic compounds by bacteria via cis-dihydroxylation reactions.250 The iron─sulfur cluster transfers electrons from a reductase to the catalytic nonheme iron center, allowing for O2 reduction and the formation of a high-valent iron intermediate. This species is responsible for oxidizing the substrate via a series of radical intermediates. A set of Rieske dioxygenases from Pseudomonas strains was shown to oxidize alkenes and aromatic compounds with complementary selectivity; naphthalene dioxygenase (NDO) catalyzed dihydroxylation of the olefin in styrene exclusively, while cumene dioxygenase (CDO) provided >99% selectivity for the corresponding arene-1,2-dihydrodiol (Scheme 39A).251 Additionally, a single-point mutation was found to switch the selectivity of CDO from 0.3% to 92% for olefin dihydroxylation, highlighting the potential for these enzymes to be engineered for non-native selectivities. More recent studies focused on characterizing enzymes from the saxitoxin biosynthetic pathway showed that Rieske monooxygenases SxtT and GxtA give different hydroxylation patterns on a series of tricyclic natural products derived from saxitoxin (Scheme 39B).252,253
Scheme 39. (A) Site Selectivity in Hydroxylation Expressed as Percentage of Major Product in the Product Distribution for Rieske Dioxygenases NDO and CDO;251 (B) Site-Selective Monooxygenation of Saxitoxin-Derived Natural Products by Rieske Monooxygenases SxtT and GxtA252.

Bacterial multicomponent monooxygenases (BMMs) have also been used for site-selective hydroxylation. These enzymes comprise three or four components: a carboxylate-bridged diiron(III) center-containing hydroxylase enzyme, a NADH reductase, an effector protein required for the coupling of electron consumption and substrate oxidation, and, in some cases, a Rieske-type [2Fe-2S] ferredoxin. Variants of the BMM toluene 4-monooxygenase (T4MO), which natively hydroxylates toluene (227) to p-cresol (228), were engineered to provide different site selectivity toward aromatic substrates (Scheme 40A).254,255 One variant, T4MO G103L, catalyzes toluene hydroxylation to give O-cresol (229) as the major product. This superfamily of enzymes also includes methane monooxygenases (MMOs) from methanotrophic bacteria. Soluble MMO (sMMO) from Methylococcus capsulatus, for example, is capable of hydroxylating aliphatic C─H bonds selectively (Scheme 40B), and this enzyme catalyzes other oxidative reactions on alkanes, alkenes, and aromatic substrates.256,257 While sMMOs are produced only by some methanotrophs, nearly all methanotrophic bacteria produce particulate MMO (pMMO).258 These membrane-bound, copper-dependent enzymes possess a narrower substrate scope than sMMO, hydroxylating mostly linear alkanes (Scheme 40B).259 Whereas the mechanism of sMMOs has been studied extensively, with most of its intermediates being identified,260 characterization of the catalytic cycle of pMMOs is actively under study.261
Scheme 40. Major Products in Hydroxylation as Percentage in the Product Pool for (A) Bacterial Multicomponent Monooxygenase T4MO255 and (B) Methane Monooxygenases sMMO256,262 and pMMO259.

3.1.4. Flavin-Dependent Monooxygenases.
Flavin-dependent monooxygenases (FMOs) are part of the FAD-oxidoreductase family and catalyze among other reactions hydroxylation of aromatic sp2 C─H bonds.263,264 These enzymes can be classified as either single-component FMOs that tightly bind FAD as a prosthetic group or two-component FMOs that require a second reductase enzyme to provide the reduced flavin as a cosubstrate. The mechanism of single-component FMOs involves the reduction of FAD by NAD(P)H in the enzyme followed by reaction with O2 to generate the reactive hydroperoxyflavin intermediate. This species can react with electron-rich aromatic substrates via electrophilic aromatic substitution to generate the hydroxylated arene and hydroxyflavin, which undergoes elimination to form oxidized FAD. Depending on the class of single-component FMO, substrate binding may be required for the reduction of FAD (Scheme 41) or it may happen after formation of the hydroperoxyflavin intermediate. In the case of two-component FMOs, the flavin is reduced in the reductase by NAD(P)H and transferred to the oxygenase, where reaction with O2 occurs to form the hydroperoxyflavin intermediate. The mechanism then follows a similar path to the one described for single-component FMOs.
Scheme 41. Catalytic Cycle for Hydroxylation of a Phenolic Compound by a Single-Component FMO Triggered by Substrate Binding.

The native activity and substrates of many FMOs have been thoroughly characterized,263 and this has facilitated efforts to explore site-selective non-native FMO catalysis. For example, the single-component 4-hydroxybenzoate 3-hydroxylase (PHBH), which natively converts 4-hydroxybenzoate into 3,4-dihydroxybenzoate, can also hydroxylate a limited scope of phenolic acids. A study focused on PHBH enzymes from Rhodococcus rhodnii 135 and Rhodococcus opacus 557 showed that while both enzymes hydroxylate 2,4-dihydroxybenzoate at C-3, they hydroxylate 2-chloro-4-hydroxybenzoate with C-3:C-5 ratios of 40:60 and 77:23, respectively.265 2-Hydroxybiphenyl 3-monooxygenase (HbpA), which natively hydroxylates 2-hydroxybiphenyl (238) at C-3, was engineered to hydroxylate tert-butyl-2-hydroxybiphenyl.266 A more recent study showed that HbpA M321A catalyzes selective C-4 hydroxylation of 3-hydroxybiphenyl (240), a substrate not accepted by wild-type HbpA (Scheme 42A).267
Scheme 42. Hydroxylase Activity of Native and Engineered FMOs with Strict Site Selectivity: (A) Mutation M321A in HbpA Increases Substrate Scope;267 (B) Site-Selective Hydroxylation by Engineered HpaB Variants in Non-Native Substrates;a 273 (C) SorbC Catalyzed Oxidative Dearomatization277.

aHydroxylation sites are denoted with arrows.
The 4-hydroxyphenylacetate 3-hydroxylases (HPAHs) are two-component FMOs that natively catalyze hydroxylation of 4-hydroxyphenylacetate to 3,4-dihydroxyphenylacetate and accept a range of additional substrates.268 The S146A variant of HPAH from Acinetobacter baumannii provides improved activity on 4-aminophenylacetic acid to give 3-hydroxy-4-aminophenylacetic acid when compared to wild type.269 HpaBC from Pseudomonas aeruginosa catalyzes site-selective hydroxylation of cinnamic acid derivatives such as ferulic acid, p-coumaric acid, and caffeic acid.270 HpaBC was also used to synthesize the natural antioxidant piceatannol from resveratrol.271 More recent studies on HpaB from E. coli272 showed that this enzyme could be engineered to site selectively hydroxylate bulkier molecules like naringenin (242, Scheme 42B).273
Some FMOs, such as TropB, AzaH, and SorbC, natively catalyze the oxidative dearomatization of resorcinol derivatives to quinol products via site-selective hydroxylation.274-276 These enzymes showed complementary substrate scope and orthogonal site selectivity toward a panel of resorcinol substrates with varying steric and electronic features. While TropB and AzaH preferentially hydroxylate the C-3 position, SorbC was found to preferentially hydroxylate the C-5 position (Scheme 42C).277 Further study of SorbC showed that the substituent at the C-1 position of its native substrate, sorbicillin, plays an important role in placing the substrate in a productive pose. Replacing this substituent with a crotyl ester led to expanded substrate scope, and the crotyl group could be removed to yield the corresponding carboxylic acid.278
3.2. C─H Halogenation
Just as different classes of hydroxylases evolved to act on different types of C─H bonds, so too did different classes of halogenases emerge in nature. Aromatic and aliphatic C─H halogenations proceed via electrophilic aromatic substitution and radical rebound mechanisms, respectively, and unique mechanisms for generating the relevant halogenating species evolved to accommodate these pathways.
3.2.1. Aromatic C─H Halogenation.
Early studies on chloroperoxidase (CPO) from C. fumago showed that this enzyme could promote aromatic C─H halogenation,279 but the selectivity of CPO280 and the later characterized vanadium-dependent haloperoxidases281 is substrate controlled. This reactivity was found to result from peroxidase-catalyzed oxidation of halide ions to generate free hypohalous acid (HOX, X = Cl, Br, I), which was responsible for the observed halogenation reactions. In the early 2000s, however, it was established that site-selective chlorination of the 7 position of tryptophan is catalyzed by PrnA (Scheme 44A),282 the first characterized flavin-dependent halogenase (FDH). Following this discovery, FDHs capable of halogenating different sites of tryptophan283,284 and various indole-285,286 pyrrole-,287-289 and phenol-containing290-295 natural products were characterized, showing that these enzymes are responsible for site-selective halogenation of aromatic C─H bonds on a variety of substrates in nature, and many subsequent examples further illustrate this point.296-299 These enzymes bind reduced flavin,300 which reacts with oxygen to form a hydroperoxyflavin intermediate analogous to that formed in FMOs (Scheme 41). Rather than reacting with an organic substrate, however, this species reacts with a halide ion (X = I, Br, or Cl) bound proximal to the hydroperoxyflavin to generate the corresponding hypohalous acid (Scheme 43B).301 Unlike the case for peroxidases, this species then travels through a tunnel within the enzyme to access the active site.302 Recent computational evidence supports the original suggestion302,303 that HOX is activated toward electrophilic attack via general acid catalysis by an active site lysine residue.304,305 Structural studies show that substrate positioning relative to this activated species then controls the site of halogenation (Scheme 43B).306,307
Scheme 44. (A) Biocatalysis with Wild-Type RebH and Rdc2 for Halogenation of Non-Native Substrates;a(B) Directed Evolution of a Thermostable RebH Variant for Halogenation of Biologically Active Molecules; (C) Site-Selective and Atroposelective Catalysis with FDHs.

aNumber refers to HPLC yield when provided.
Scheme 43. (A) Simplified FDH Mechanism; (B) PrnA and RebH Natively Chlorinate L-Tryptophan Site Selectively at C7 of the Indole Ring.

Early studies showed that the native site selectivity of tryptophan halogenases could be used to access various halogenated natural product derivatives by incorporating these enzymes into organisms expressing genes for different metabolic pathways.308,309 Halogenation of substrates other than tryptophan by PrnA was reported to proceed with substrate-controlled selectivity,310 but RebH catalyzed 7-halogenation of tryptamine,311 showing that at least certain FDHs could exert catalyst-controlled selectivity toward non-native substrates. It was then established that RebH,312 Rdc2,313 and later several additional FDHs314-317 exhibit catalyst-controlled selectivity toward a number of non-native substrates (Scheme 44A).
Early efforts to expand FDH reactivity beyond that offered by native enzymes showed that site-directed mutagenesis of active site residues could be used to change RebH substrate preference to favor tryptamine over tryptophan.311 Directed evolution was used to evolve a thermostable RebH variant,318 and this enzyme was further evolved to expand the substrate scope to encompass larger, biologically active compounds (Scheme 44B).319 This effort involved a substrate walking approach in which iterative rounds of mutagenesis and screening were conducted on progressively larger and more complex substrates. Variant 3-SS possessed 65-fold improved activity for the tetrahydrocarbazole derivatives tryptoline and eleagnine relative to RebH, and variant 4-V exhibited 40-fold improved chlorination for yohimbine. Variants 3-SS, 4-V, and related active site mutants were later shown to catalyze site-selective and enantioselective halogenation of methylene dianilines substrates.320 Inspired by the reports of peptide-catalyzed atroposelective halogenation,321 directed evolution of a RebH variant also enabled site-selective and atroposelective halogenation of 3-aryl-4(3H)-quinazolinones (Scheme 44C).322 While most FDH variants examined in a preliminary screening halogenated probe substrates at the less sterically hindered C4 site, one RebH variant 6-TLP gave the desired C6-brominated product with high enantioselectivity. Three rounds of evolution were required to evolve this enzyme for improved site selectivity and yield, and the final variant 3-T demonstrated excellent site selectivity and atroposelectivity for a panel of related quinazolinones.
In addition to expanding the FDH substrate scope, researchers have sought to alter the site selectivity of FDHs on both native and non-native substrates.323 For example, incorporating arginine and lysine residues in the active site of PrnA was used to reshape the size and charge distribution of the active site to modify the regioselectivity and improve the activity for halogenation of anthranilic acid derivatives.324 In a separate study, the similarity of the structures for the 5- and 6-tryptophan halogenases PyrH and SttH was exploited to engineer a 5-selective SttH variant by mutating three active site residues in SttH to the homologous residues in PyrH.325 While wild-type SttH halogenated tryptophan and 3-indolepropionic acid with 6 selectivities of >99% and 90%, respectively, the triple mutant gave the 5-chlorinated compounds in 32% and 75% selectivity, respectively. In a similar fashion, the crystal structures of tryptophan 6- and 7-halogenases Thal and RebH were used to identify key residues for positioning the substrate in the active site.326 A total of 5 residues in the active site chosen based upon their interactions with substrate were switched to produce variant Thal-RebH-5, which generated the 7-chlorinated tryptophan product in 30% yield and with a 7:6 selectivity of 19:1, representing the most complete switch in regioselectivity for an FDH for a structure-guided approach (Scheme 45A). Structure-based mutagenesis was also used to alter the site selectivity of MalA.327
Scheme 45. (A) Use of a Deuterated Tryptamine Probe–Substrate Allows for MALDI Screening Based on m/z Values; (B) Directed Evolution of RebH into 5- and 6-Selective Chlorinases; (C) Using the Crystal Structures of Thal and RebH Substitution of Key Active Site Residues from RebH into Thal Results in Modified Site Selectivity.

These structure-guided approaches show that key active site residues identified in crystal structures of FDHs with different selectivity toward a cocrystallized substrate can be mutated to achieve the corresponding switch in site selectivity. To avoid the need for crystal structures and to provide a means to switch site selectivity to any desired site, a directed evolution approach to altering FDH site selectivity was developed.328 A MALDI-based screen using deuterated tryptamine probe substrates enabled direct evaluation FDH variant site selectivity (Scheme 45B). Several rounds of evolution led to the generation of 7-selective variant 0S, which possessed an increased rate relative to WT and 5- and 6-selective halogenases 8F and 10S that provided their respective products (relative to all others) with 90:10 and 95:5 site selectivity (Scheme 45C). Variants from this lineage were later used in conjunction with the native 6-halogenase Thal and two fungal phenol halogenases to compare a panel of substrates for chemical and biocatalytic halogenation.329 Characterization of products generated by biocatalytic halogenation using RebH variants 0S, 8F, and 10S showed that mutations designed to modify site selectivity for tryptamine carried over to other substrates as well. In many cases the site selectivity of the FDH-catalyzed halogenation differed from the chemical NCS reaction, showing the ability of the enzyme scaffold to override the inherent reactivity of the substrate. Crystal structures of these variants coupled with reversion mutations and MD simulations were used to rationalize the observed changes in site selectivity and to develop a computational model to predict FDH site selectivity.305
Genome mining has often been used to identify individual FDHs with novel properties from diverse sources.295,315,330 Our group pursued a family-wide genome mining approach to build a panel of FDHs with novel site selectivity toward diverse non-native substrates.331 A sequence-similarity network (SSN) was constructed, and the substrate preferences for known FDHs was mapped onto the SSN. FDH sequences with like substrate preferences were found to cluster, providing a means to probe FDH sequence space for novel function. Ultimately, 80 new FDHs were obtained from this analysis, and the chlorinase and brominase activity of these enzymes on a panel of 12 probe substrates was examined using a mass spectrometry-based assay. Several of the active enzymes identified from this analysis catalyze site-complementary bromination of diverse substrates (Scheme 46).
Scheme 46. Genome Mining for FDHs Enables Site-Selective Catalysis on Biologically Active Compoundsa.

aNumbers represent isolated yields.
The FDHs discussed above require a separate flavin reductase (FRED) to supply the FADH2 required for the mechanism shown in Scheme 43. This requirement complicates FDH biocatalysis since these enzymes are not widely available, and any FDH evolution done to improve process compatibility is ultimately limited by the FRED unless it is also engineered. The FDH Bmp5 is notable in that it is a single-component FRED/FDH, but its reported activity is limited to substrate-controlled site selectivity on phenols and aryl ethers.332 This contrasts with the recently described single-component FRED/FDH AetF,333 which natively catalyzes site-selective 5-/7-dibromination of l-tryptophan.334 This novel site selectivity led to a recent effort to evaluate the activity and selectivity of AetF toward a diverse range of substrates.335 AetF accepted not only indoles and phenols analogous to those halogenated by many conventional FDHs but also heterocycles and less electronically activated substrates that provide low or no yield using other FDHs (Scheme 47A). Remarkably, AetF also catalyzed site-selective iodination of the benzene ring of l-tryptophan and 6-fluorotryptamine with high yield. Previous reports of iodination reactions involving FDHs have typically involved substrate-controlled site selectivity,315,332,336 and further analysis of the iodination activity has shown that FREDs can release hydrogen peroxide into solution that oxidizes I− to HOI, leading to substrate-controlled iodination selectivity.337 The broad substrate and halide scope of AetF coupled with its catalyst-controlled site selectivity therefore makes this and related single-component FRED/FDHs promising tools for biocatalytic halogenation.
Scheme 47. Substrate Scope of AetF for Site-Selective (A) Bromination and (B) Iodination.

3.2.2. Aliphatic C─H Halogenation.
Even in comparison to the relatively recent development of FDH biocatalysis, biocatalytic halogenation of aliphatic C─H bonds is in its infancy. To date, only Fe(II)-/α-ketoglutarate-dependent halogenases (FeDHs) can achieve this challenging reaction. These enzymes are similar to the analogous FeDOs discussed above, but their Fe(II) center lacks a conserved Asp/Glu residue found in FeDOs (Scheme 36). The resulting open coordination site allows FeDHs to bind halide ions (X = Cl, Br). FeDH proceeds in analogy to FeDO catalysis in that substrate binding leads to O2 activation and formation of a reactive Fe(IV)(X)(oxo) intermediate capable of abstracting unactivated C─H bonds to generate a Fe(III)(X)(OH) intermediate and a carbon-centered radical. The presence of the X ligand leads to the possibility for X• rebound to generate the halogenated product, which proceeds with high specificity in FeDHs.
The first FeDHs characterized acted on substrates linked to a carrier protein,339-341 so it was unclear whether this class of enzymes would be useful for biocatalysis involving small molecule substrates. Eventually, however, it was established that two FeDHs, WelO5 and AmBO5, were responsible for site-selective installation of the chlorine substituents in the natural products welwitindolinone342 and ambiguine,343 respectively. These enzymes could accept fisherindole, hapalindole, and ambiguine substrates related to their native substrates (Scheme 48A), and WelO5 catalyzed selective bromination of its native substrate in the presence of excess bromide.344
Scheme 48. Site-Selective Halogenation of Fisherindole (285), Hapalindole (287 and 288), and Ambiguine (289) Substrates by WelO5 and AmbO5; (B) WelO5* and Evolved Variants Catalyze Halogenation of a Martinelline-Derived Substrate;338 (C) Site-Selective Halogenation of Soraphen A by Evolved WelO5* Variants.

WelO5 was the subject of the first efforts to engineer FeDH variants with substrate scope beyond close analogues of the natural fisherindole substrates. Buller used site-saturation mutagenesis of residues suspected to be involved in substrate binding to generate variants CA2 and CB2, which provided different chlorinated isomers of martinelline-derived substrate 290 (Scheme 48B).338 On the other hand, CA2 provided only 6% yield of the chlorinated product, and the reaction was dominated by hydroxylation (36% yield of two diastereomers). CB2 gave 30% of a chlorinated product, but the site selectivity had changed to an adjacent tertiary position. This same group later screened libraries of WelO5* variants for activity on the macrolide soraphen A to identify the initial hit WelO5*-GAP.345 Subsequent mutagenesis of active site residues established that a triple mutant dubbed SLP (WelO5*-GAP V81S, A88L, I161P) possessed 13-fold activity for formation of a single-product isomer relative to the parent enzyme (Scheme 48C). Using the data from this initial library, machine learning was applied to predict functional sequences, which were then characterized experimentally. It was found that all seven ML-derived sequences were active with four outperforming the previous best SLP enzyme, and one ML-derived sequence also showed altered site selectivity, with enzyme variant WelO5*-AHG being highly selective for chlorination of C16 in soraphen A, whereas the parent enzyme GAP was largely a C14-selective halogenase.
In an effort to expand the substrate scope of WelO5, a directed evolution campaign was conducted via screening for halogenation on compounds that contained a ketone rather than the typical isonitrile group present in the natural products accepted by these enzymes.348 Although the wild-type enzyme had no activity for the substrate with the ketone replacing the isonitrile, a library constructed by an active site scan of 7 residues deemed likely to influence substrate binding resulted in a variant capable of 30% halogenation. The activity was further improved by three additional rounds of evolution incorporating mutations found to be beneficial as single mutants, ultimately providing a panel of variants capable of preparative halogenation for a panel of related ketone-containing haplaindole compounds with high site selectivity, chemoselectivity, and enantioselectivity.
The lysine halogenase BesD (Scheme 49A) has been successfully used as a template for the study and identification of FeDHs.346 While the substrate scope of BesD proved limited in a full screen of l-amino acids, this enzyme was used as a template gene sequence in a BLAST search to identify active halogenases with activity on small aliphatic amino acids (Scheme 49B). Further exploration of the gene clusters identified in this study led to the identification of two closely related enzymes dubbed Hal (WP_122981682) and Hydrox (WP_107105619) which had over 70% sequence identity, and both functionalized lysine with the same site selectivity, favoring either hydroxylation or halogenation (Scheme 49C).347 The native alkyne forming pathway of BesABCD was used to enable a high-throughput screen for lysine chlorination, which led to the identification of residues crucial for favoring halogenation over hydroxylation. By comparing the location of the conserved residues in halogenase variants, two adjacent beta loops were identified as being crucial for maintaining both high chemoselectivity and activity. Such structural insights into this enzyme class may inform future engineering efforts.
Scheme 49. (A) Natural Product Pathway of BesD Results in Alkyne Formation;346 (B) Homologues of BesD Site Selectively Halogenate Amino Acid Substrates; (C) Fluorogenic Click Assay Developed for High-Throughput Screening of Chimera Hydox/Hal Libraries347.

3.3. C─H Amination, Azidation, and Amidation
Remarkably, several anionic species can bind to the Fe(II) center in FeDH and FeDO facial triad mutants, and the resulting X-type ligand can undergo rebound with the substrate radical.349 Transformations such as nitration, azidation, and cyanation have been demonstrated, although thus far only azidation has been successfully demonstrated for functionalization of small molecules.243 The ability of FeDHs to accept peusdo-halides varies among the enzymes studied to date. Most FeDHs which are natively known for chlorination accept non-native anions but do so only in low yield or with poor chemoselectivity in the presence of chloride in the media.350
The FeDH SyrB2 was the first enzyme of this family to successfully carry out C─H functionalization with nitrogenous groups, catalyzing both nitration and azidation reactions.351 The engineered FeDO SadX appears to be relatively broadly accepting of anions, demonstrating site-selective and chemoselective rebound for chloride, bromide, azide, and isocyanate.243,352 SadX was also used as the starting point in a directed evolution effort to evolve site-selective and chemoselective azidation biocatalysts.353 Over four rounds of evolution, the chemoselectivity increased 7.5-fold over the parent enzyme and the total yield of the azidated product increased 6.7-fold. In keeping with the need for precise substrate positioning relative to the reactive iron center, the substrate scope for individual mutants from this lineage was narrow. Instead, a panel of enzymes from the lineage was used to site-selectively azidate a variety of N-succinylated amino acids (Scheme 50), which were then readily functionalized to the reduced amine and the desuccinylated products to produce modified amino acids. Azidation chemistry in FeDHs has also been accessed through an anaerobic abiological radical relay reaction as well. While no site-selective examples were presented, a key development from this work was the synthesis and implementation of a pro-fluorogenic alkyne probe which enabled rapid, high-throughput screening of azidase libraries through the copper-catalyzed azide─alkyne cycloaddition.354 Given the substrate-agnostic nature of the alkyne probe, this high-throughput screening platform should lay a foundation for future azidase evolution.
Scheme 50. Site-Selective Azidation by Engineered SadX Variants353.

Heme-containing proteins have also been used for site-selective C─H functionalization via C─N bond formation. For example, a BM3 variant (P411BM3-CIS-T438S) containing key mutations that reduce monooxygenation activity (T268A) and alter the primary coordination sphere of the heme cofactor (C400S) displayed robust nitrene transfer activity.355 Site saturation mutagenesis of active site residues in P411BM3-CIS-T438S was then used to engineer variants with orthogonal site selectivity toward functionalization of benzylic and homobenzylic bonds, both with high enantioselectivity (Scheme 51A).356 The variants developed in this study maintained the desired site selectivity for a small panel of related compounds. A similar reaction was catalyzed by an artificial metalloenzyme generated from the CYP119 enzyme from the thermophilic S. solfataricus.357 In the absence of the protein scaffold, the free iridium cofactor generated the 5- (317) and 6-membered (318) rings in a 60:40 ratio, presumably dominated by the lower BDE of the benzylic C─H bond. When the Ir(Me)─PIX cofactor (PIX = protoporphyrin IX) was encapsulated within the CYP119 variant, this selectivity switched to 20:80 toward the less favored 6-membered ring 318 with a 84:16 er (Scheme 51B).
Scheme 51. (A) P411 Enzymes Evolved for Regiodivergent Intramolecular Sulfamidation; (B) Sulfamidation Catalyzed by Iridium–Heme Bearing Artificial Metalloenzymes.

Intermolecular C─H amination reactions have also been developed. For example, site saturation mutagenesis of a P411BM3 variant led to variant P411BPA, which catalyzes site-selective amination of both m- and p-ethylmethylbenzene (among many other substrates) using hydroxylamine ester as a nitrene source (Scheme 52A).358 A second evolution effort was used to engineer an allylic C─H aminase. The resulting P411APA enzyme demonstrated remarkable site selectivity for allylic C─H bonds with up to 20:1 site selectivity observed for substrate bearing benzylic C─H bonds. Similarly high selectivity was obtained for α-terpinene, although usually P411APA had somewhat limited selectivity in the presence of multiple primary alkene C─H bonds. Intermolecular amidation of benzylic C─H bonds using P411 enzymes was also demonstrated,43 and constitutional isomers were not detected in examples bearing both secondary and primary C─H bonds (Scheme 52B).
Scheme 52. (A) P411BPA and (B) uAMD9 Expressed in Whole Cells Are Capable of Site-Selective Functionalization Amination and Amidation of Benzyl C─H Bonds.
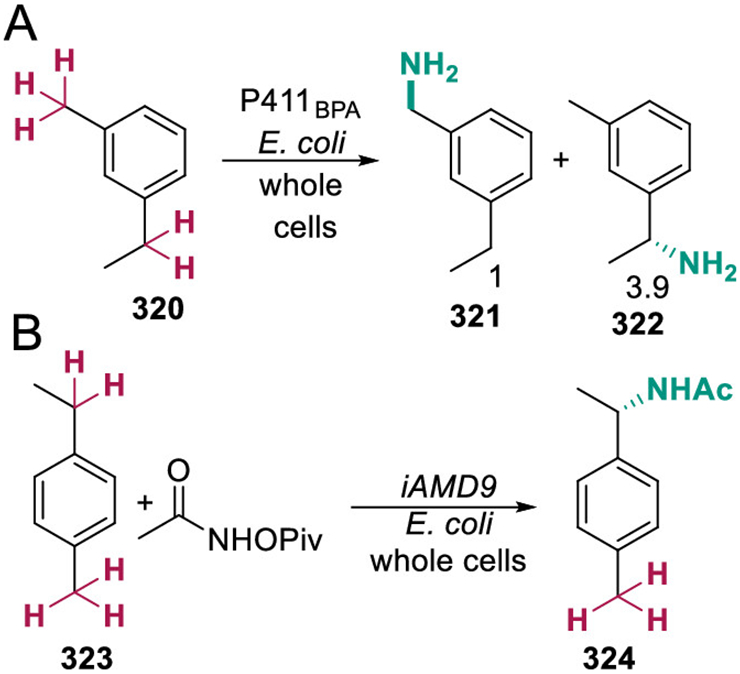
Perhaps most impressively, BM3 variants capable of functionalizing unactivated C─H bonds via amidation or amination were recently reported.359 The latter reaction constitutes the nitrogen analogue of the native P450 reaction. By screening variants previously developed for nitrene insertion chemistry, researchers from the Arnold lab identified two variants with low activity for 3-amidation and 2-amination of methylcyclohexanone. Over 9 rounds of evolution, the activity and selectivity of these enzymes were improved to ultimately provide two variants uPA9 and uAMD9, which were 86% and 91% site selective for the 3 and 2 position, respectively (Scheme 53). These enzymes showed high site selectivity toward several cyclic and acyclic compounds, and promising activity (though low selectivity) was observed on many additional substrates. Through the kinetic isotope effect and computational efforts, site selectivity was found to be largely determined by the steric environment of the protein active site, whereas the stereoselectivity was determined by both an irreversible HAT and the subsequent radical rebound.
Scheme 53. Evolution of P450 Variants for Site-Selective Amidation and Amination of Unactivated C─H Bonds.

3.4. C─H Deuteration
Deuteration is a nearly traceless way to modify a compound that has both biochemical and therapeutic applications. Recently, the isoleucine epimerization enzyme DsaD and the partner protein DsaE were used to enable selective α,β-, α-, and β-deuteration of amino acids via selective epimerization.360 Reaction of the amino acid with DsaD in D2O resulted in selective α-deuteration, whereas reactions with DsaD and excess DsaE led to deuteration of both the α and the β positions (Scheme 54A). Incubating the α- and β-trideuterated compound with DsaD in protic water led to selective α-protonation, resulting in the selectively β-deuterated product (Scheme 54B). The DsaD/DsaE system showed efficient α-deuteration for nonpolar amino acids, although the trends for DsaE-assisted β-deuteration were less clear. Derivatization of the resulting deuterated amino acids with Marfey’s reagent revealed that in most cases, perfect stereoretention was observed regardless of whether the α-, β-, or α,β-deuterated product was obtained. Given the increasing importance of peptide-based drugs, the ability to site-selectively deuterate these building blocks could prove useful for future drug development.
Scheme 54. Biocatalytic Deuteration of Amino Acids: (A) General Site-Selective Deuteration by DsaD and DsaE; (B) Synthetic Outline for Production of α-, β-, and αβ-Deuterated Amino Acid Reaction Outcomes Are Formatted as % Recovery,% Deuteration (Site), and $ ee360.

3.5. C─C Bond Forming Reactions
3.5.1. Oxidative Coupling.
Cross-coupling reactions are widely used to form biaryl compounds from prefunctionalized coupling partners like aryl halides and aryl boronic acids.361 A wide range of C─H functionalization methods has been developed to form biaryl compounds while avoiding the need for functionalization of one or both coupling partners.362,363 One approach to accomplish the latter involves oxidative coupling, in which the biaryl bond is forged with formal loss of H2 from the reacting aromatic coupling partners. Chemical methods for oxidative coupling are often limited by poor site selectivity.364,365 Biaryl bond formation in nature often proceeds via oxidative coupling, but enzymes enforce remarkable site selectivity, chemoselectivity, and enantioselectivity on these reactions. Extending the scope of these reactions to enable site-selective non-native oxidative coupling is therefore highly desirable.
One of the most well-studied examples of intramolecular biaryl bond formation via oxidative coupling in natural product biosynthesis is the OxyC-catalyzed coupling of the A and B rings in vancomycin.366 OxyC and other P450s367 involved in the formation of vancomycin have been studied for biocatalytic synthesis of related compounds, but the need for a PCP-tethered substrate limits the practical application of these enzymes. It was recently discovered that the enzyme AryC accomplishes a similar intramolecular C─C coupling reaction using only a lipophilic chain instead of a PCP tether to anchor the substrate within the enzyme active site.368 Recently, a P450 from Streptomyces sp. MG-AR, originally identified for its ability to hydroxylate testosterone, was engineered to enable gram-scale oxidative coupling of the arylomycin core, demonstrating scalable production of this medicinally relevant motif (Scheme 55A).369 This study constitutes the first example of a native hydroxylase being engineered for oxidative C─C bond formation, suggesting that other P450s originally studied for oxygenation could be used for site-selective oxidative coupling.
Scheme 55. (A) Percent of Product Pool in the Synthesis of the Arylomycin Core by P450 MG-AR Variant;369 (B) Intermolecular Cross-Coupling of Nonidentical Monomers by KtnC;370 (C) Reactions of 7-Demethylsiderin (347) with Enzymes KtnC and DesC371.

Most efforts aimed at understanding P450-catalyzed oxidative couplings focus on identifying enzymes that natively catalyze these reactions. For example, the enzyme DesC from E. desertorum was found to catalyze site-selective and atroposelective oxidative homocoupling of 7-demethylsiderin (347) to produce the unsymmetrical 6,8′-dimer 348, showing strong catalyst control of selectivity.371 Similar catalyst-controlled site selectivity was observed for the enzymes AunB and BfoB, which produce 10,7′- and 10,10′-coupled products of fonsecin B and rubrofusarin B.372 A related P450, KtnC, was shown to catalyze site-selective and atroposelective intermolecular coupling of 347 to produce the natural product P-orlandin via 8–8′ coupling (349, Scheme 55C), but related substrates were not successfully coupled to the corresponding dimers.371 More recently, directed evolution targeting residues in the active site of KtnC was used to improve the activity and site selectivity for the 8,1′-cross-coupled product 346, albeit at the expense of enantioselectivity (Scheme 55B).370 Further evolution led to variant LxC7 with improved site selectivity and enantioselectivity comparable to the parent enzyme (80:20 er wild-type KtnC, 77:23 er LxC7), but this variant had lower total activity than previous enzymes in the lineage for the substrate used in the evolution. Genome mining of the sequence space around KtnC identified several additional P450s capable of intermolecular coupling, including several examples where KtnC had no activity.
3.5.2. Prenylation.
Prenyl-, gerenyl-, and farnesyltransferases catalyze nucleophilic substitution of the respective organic phosphates by electronically activated aromatic compounds or heteroatom substituents. The former reactivity is typified by the site-selective functionalization of tryptophan (350) by a class of prenyltransferases known as dimethylallyl-tryptophan synthases (DMATSs, Scheme 56A). The enzymes DmaW373 and FgaPT2374 have also been used to produce 4-prenyl-l-tryptophan, and shortly after its discovery, the substrate scope of FgaPT2 was elucidated by modifying both the alkyl and the aryl substituents.375 While these substrates were typically prenylated in low yield, this enzyme was later found to be competent for the site-selective prenylation of cyclo-l-Trp-l-Tyr and other diketopiperazines, albeit at higher enzyme loading.376 A related 4-DMATS enzyme from the organism A. japonicus was recently found to accept a variety of tryptophan derivatives with exceptional site selectivity in high yields even at relatively high substrate loadings.377
Scheme 56. (A) General Scheme of the Native Prenyltransferase Reaction by Dimethylallyltryptophan Synthases (DMATSs) and Representative Non-Native Products for DMATSs with Different Site Selectivity;375,383 (B) Geranylation Reactions on Genistein (359) by AtaPT Single Mutants384.

The 4-DMATS enzymes were the first prenyltransferases to be characterized, and shortly thereafter, the 7-DMATS analogue from A. fumigatus was discovered.378 This enzyme has a broader substrate scope than FgaPT2.379 The first 6-DMATS enzyme characterized was IptA, which selectively prenylates the C-6 position of l-tryptophan and various indole compounds.380 These C-6 prenyltransferases were present in many organisms, and two homologues of these enzymes from S. ambofaciens (6-DMATSSa) and S. violaceusniger (6-DMATSSv) were used as catalysts for prenylation of a variety of hydroxynaphthalenes and tryptophan derivatives.381 Unlike previously described DMATS enzymes, 6-DMATSSa was also capable of using GPP as an alkyl donor, though it did not accept FPP. A unique 6-DMATS designated PriB was found to accept a variety of prenyl acceptors, including phenazine-2,3-diamine, 1,4-diaminoanthraquinone, and pindolol and even different alkyl donors, including geranyl diphosphate, farnesyl diphosphate, and a variety of farnesyl diphosphate analogs with phenoxy, anilino, phenylsulfanyl, and benzyl substitutions.382
DMATS-catalyzed functionalization of the remaining position on the benzene ring of indoles was enabled by the discovery of the C-5 prenyltransferase from A. clavatus.385 This enzyme and the later discovered C-5 prenyltransferase 5-DMATSSc can also transfer non-native alkyl chains, including benzyl and 2-pentenyl groups.386 Yields for these reactions were typically 5–15% of those involving the native DMAPP donor, but they provide viable starting points for further engineering. While 5-DMATS remains relatively rare, the 4-DMATS FgaPT2 catalyzes C-5 alkylation when non-native alkyl donors like 2-pentenyl, 1-methyl, and benzyl phosphate are used.387 The ability to control site selectivity was also shown in an effort to functionalize different sites on the four diastereomers of a fumiquinazoline substrate with a DMATS panel.383 This effort also highlighted the stereoselectivity of DMATS catalysis. For example, perfect site selectivity was observed for C-7 prenylation of the (11S,14S) diastereomer using a 7-DMATS, but a 2.5:1 C-6:C-7 ratio was observed for the (11R,14R) diastereomer.
DMATS-catalyzed alkylation of substrates other than those containing indole cores has also been demonstrated. The activity of a prenyl transferase from A. terreus (AtaPT) was examined on over 100 substrates, including lignanoids, quinoline alkaloids, xanthones, coumarins, and flavonoid glycosides. Of these substrates, 46 were prenylated in over 5% conversion, and 26 additional substrates provided measurable levels of product.384 While AtaPT generally prenylated alcohols, activated aromatic sites on several flavonoid compounds and umbelliferone were also prenylated. This enzyme also showed both evolvability and a broader than usual acceptance for prenyl donors as the single mutants W397A and E91A were capable of selectively geranylating genistein (359) whereas the wild-type enzyme digeranylated the compound (Scheme 56B).
3.5.3. Radical Coupling Reactions.
Radical SAM enzymes catalyze a wide range of reactions, including site-selective sp3 C─C bond formation,388 as exemplified by the sequential methylation of carbapenam scaffold 362 by Tokk to forge an isopropyl group (Scheme 57A).389 While some radical SAM enzymes, including TsrM, which was actually found to employ a polar mechanism for methylation,390 have been explored as biocatalysts, such applications remain rare. To facilitate radical SAM biocatalysis by eliminating the need for stoichiometric SAM in these reactions, a SAM regeneration system described above (Scheme 12B)72 was further developed and used for selective alkylation of a glutamine residue in a 24-mer polypeptide (366 Scheme 57B).391 This one-pot system involves ribophosphorylation of adenine and two subsequent phosphorylations to produce ATP, which is used by a methionine adenosyltransferase to produce SAM. This regeneration system was used with cobalamin-dependent glutamine C-methyltransferase (QCMT) to alkylate 366, showing the ability of this approach to generate SAM analogues by replacing the addition of methionine by ethionine (Scheme 57B).
Scheme 57. (A) Sequential Methylation of Carbapenem Substrate 362 by SAM-Dependent Methylase;389 (B) Alkylation of the Glutamine Residue in a 24-Mer Polypeptide Substrate Employing a One-Pot SAM Regeneration System391a.

aSAE: S-adenosylethionine. DOA: 5′-deoxyadenosine. SAH: S-adenosylhomocysteine. Met: l-methionine. Eth: l-ethionine.
Although they lack catalytic activity themselves, dirigent proteins (DIRs) also mediate the site selectivity and enantioselectivity of radical C─C coupling reactions.392 DIRs are involved in lignan biosynthesis by binding and controlling the reactivity of free radical lignol intermediates that would otherwise result in a mixture of products.393 The DIR from Podophyllum hexandrum (PhDIR) was used in a study that focused on the heterocoupling of coniferyl alcohol analogues when used with the fungal laccase TvLac.394 The addition of PhDIR resulted in an increase of the 8–8′ isomer for both the homo- and the heterodimer products when compared to the 8–5′ isomer (Scheme 58A). This study showcased not only the control that DIRs can exert on radical coupling reactions but also the synthetic relevance of PhDIR due to its substrate flexibility.
Scheme 58. (A) Radical Heterocoupling of Coniferyl Alcohol Analogues by Laccase TvLac and the Effect of Dirigent Protein PhDIR in the Product Ratio;a 394 (B) Non-Native Heteroarene Alkylation by Ene-Reductasesb 395.

aPercentages represent the amount of 8–8′ heterodimer product compared to the 5–8′ heterodimer product. bSite selectivity expressed as the percentage of the shown compound in the total product pool.
In recent years, flavin-dependent ene-reductases (EREDs) have been engineered to carry out non-native light-promoted C─C bond forming reactions.396-398 A recent study used a previously optimized variant of the ERED from Gluconobacter oxidans, GluER T36A/Y343F, as the parent enzyme for a directed evolution campaign aimed at developing a biocatalyst for the selective alkylation of heteroarene substrates.395 The final enzyme, PagER, accumulated five additional mutations and was able to override the initial preference for alkylation of indole at the C-2 and C-3 positions in favor of C-4 with a ratio of 9:1 for C-4:(C-2 + C-3) (Scheme 58B). PagER and other GluER variants derived from its lineage were shown to have high site selectivity on a panel of both electron-rich and electron-poor arenes.
3.5.4. Carbene and Nitrene Insertion into C─H Bonds.
The utility of cytochromes P450 for site-selective C─H hydroxylation and olefin epoxidation were outlined above, but these enzymes have more recently been explored as catalysts for non-native carbene and nitrene insertion reactions.399 Early studies toward this end established that cytochrome P450BM3 variant P411, whose native cysteine axial ligand was substituted for serine as noted above, provide selective olefin cyclopropanation over the native hydroxylation chemistry.400 Several rounds of directed evolution and removal of the P411 FAD domain led to variant P411-CHF, which catalyzes carbene insertion into sp3 C─H bonds at benzylic, allylic, propargylic, and α-amino sites.401
Most importantly for this discussion, P411-CHF catalyzes site-selective alkylation of N-methyltetrahydroquinoline with a selectivity of 9:1 for alkylation of the 2 position of the tetrahydroquinoline moiety over alkylation of the methyl group (Scheme 59A). A variant from the same lineage, P411-gen5, showed superior selectivity (>50:1) and formed the opposite enantiomer, demonstrating the utility in developing a panel of enzymes for a given transformation. A similar lineage of P411 enzymes was later developed and found to catalyze alkylation of a simple N-methylpyrrole with orthogonal site selectivity depending on the variant used.402 Four rounds of evolution of P411–C10 generated variant L9, which catalyzes site-selective functionalization of N-ethyl-N-methylaniline (379) with a preference of up to 99:1 for the methyl carbon over the ethyl carbon (Scheme 59B).403 A second variant developed from this lineage, P411-L10, had a significantly lower selectivity of 71:29 for the methyl carbon but showed high diastereoselectivity and enantioselectivity for the minor product, suggesting this enzyme could be further evolved for this minor product if desired.
Scheme 59. (A) Alkylation of the 2 Position of the Tetrahydroquinoline Moiety of N-Methyltetrahydroquinoline (376) with the Remaining Product Pool Being the Alkylation of the Methyl Group;401 (B) P411 Variants Selective for Methyl and Ethyl Alkylation of N-Ethyl-N-methylaniline (379)403.

Another approach to site-selective enzymatic carbene insertion involves replacing the native heme cofactor with metal-substituted protoporphyrin IX derivatives.404,405 A recent report showed that directed evolution of an iridium-containing P450 artificial metalloenzyme enabled site-selective alkylation of substituted benzofurans and demonstrated alkylation with divergent selectivities for meta and para positions to various substituents with an intermediate variant from the lineage showing high enantioselectivities in a subset of select reactions (up to 97:3 er, Scheme 60A).406 Streptavidin-based artificial metalloenzymes have also been used for site-selective carbene insertion. Specifically, a streptavidin variant containing a biotinylated copper(I) heteroscorpionate complex catalyzed cyclization of aryl amides resulting in the formation of β- and γ-lactams. Site saturation mutagenesis of hotspots within the streptavidin scaffold resulted in two variants for selective formation of the 4- and 5-membered lactam (Scheme 60B).407
Scheme 60. (A) Distribution of Para and Meta Products in the Alkylation of Several Benzofurans by Artificial Iridium-Containing P450s;406 (B) Lactam Formation via Intramolecular Cyclization by Engineered Artificial Copper–Streptavidin Metalloenzymes with Major Products Displayed with the Corresponding Percent of the Product Pool407.

4. APPLICATIONS OF SITE-SELECTIVE ENZYME CATALYSIS
The ability to insert functional groups into molecules at will opens up a variety of subsequent bond forming capabilities. When paired with the vast arsenal of chemoselective methods available to synthetic chemists, site-selective catalysis enables a range of chemoenzymatic methods and novel synthetic disconnections for target-oriented synthesis.
4.1. Chemoenzymatic Methods
Nearly all transformations discussed above result in functionality that can be used in subsequent transformations. Some, including hydroxylation, halogenation, and azidation, are particularly notable in that they have been paired with chemical transformations to enable bond disconnections that are not known for any enzymes. For example, P450-catalyzed C─H hydroxylation has been coupled with deoxyfluorination to enable chemoenzymatic fluorination of unactivated C─H bonds (Scheme 61A).204,206,219 While fluorinases are known, these enzymes act on the adenoside fragment of their SAM cofactor via an SN2 reaction that defines their site selectivity, so the chemoenzymatic approach provides far greater generality.408 P450-catalyzed demethylation has also been paired with either Barton–McCombie deoxygenation or glycosylation to enable site-selective sugar modification.204 Site-selective azidation catalyzed by the engineered Fe(II)/KG-dependent oxygenase SadX (Scheme 50) has also been combined with both reduction and click chemistry to enable access to amines and triazoles.353
Scheme 61. Representative Examples of (A) Site-Selective Chemoenzymatic Hydroxylation/Deoxy Fluorination and (B) Halogenation/Cross-Coupling.

Given the importance of halogenation in a wide range of transformations, particularly transition-metal-catalyzed cross-coupling, it is perhaps not surprising that enzymatic halogenation has been paired with a variety of reactions to enable site-selective formation of carbon─carbon and carbon─heteroatom bonds. Early efforts toward this end focused on using wild-type FDHs to access natural product derivatives with halide substitution that enabled subsequent Suzuki─Miyaura coupling to access biaryl derivatives (Scheme 61B).409,410 The use of engineered FDHs allowed for extension of this approach to diverse substrates and different cross-coupling reactions,411 and the use of membrane-divided reactors allowed for the halogenation and cross-coupling steps to be conducted in a two-phase “one-pot” fashion.412 Concurrent halogenation/cross-coupling was achieved using a whole cell biocatalyst and a Pd catalyst in the culture media.413 This transformation was also used to enable a high-throughput screen for FDH activity,414 and cross-linked enzyme aggregates comprised of different FDHs, a FRED, and an alcohol dehydrogenase were shown to be effective catalysts.413 Extending this approach even further, enzymatic halogenation and Pd-catalyzed cyanation has been followed by either nitrile hydratase or nitrilase catalysis to enable site-selective installation of nitrile, amide, or carboxylic acid groups.174
4.2. Target-Oriented Synthesis
Site-selective enzyme catalysis has also been used to increase the efficiency of synthetic routes to natural products and other compounds. Importantly, however, these efforts have been limited relative to chemoselective enzyme catalysis due to the fundamental challenge of identifying enzymes with the correct site selectivity and substrate scope, since the native activity of many enzymes often provides little information regarding their non-native catalytic potential and scope. This is particularly challenging for enzymes like cytochromes P450 and flavin-dependent halogenases, both of which have been shown to functionalize a broad range of substrates at different sites.16,329,331,415 In methodology efforts, this capability is a highlight since it shows the versatility of these enzymes, but target-oriented synthesis demands specific site selectivity that might prove elusive using known enzymes. Despite these difficulties, several promising examples of target-oriented synthesis enabled by enzymatic site-selective functionalization show the potential of this approach.
To date, target-oriented site-selective enzyme catalysis has largely focused on C─H oxyfunctionalization reactions catalyzed by enzymes such as P450s and Fe(II)-dependent hydroxylases. For P450s, these efforts are facilitated by decades of research that has revealed key residues responsible for selectivity and engineered variants that provide excellent starting points for site-selective catalysis even on relatively complex scaffolds. For example, based on previous data regarding the substrate scope of BM3 variant IL-H8,218 which bears 15 mutations from wild-type BM3, a panel of BM3 variants with mutations similar to IL-H8 was screened to identify variants with activity on sclareolide.416 Alanine scanning of the top hit from this panel provided BM3 MERO1, which hydroxylated C-3 of sclareolide in 60–70% isolated yield on gram scale, enabling the total synthesis of phenylpropene C. A related BM3 variant dubbed KSA15 exhibited the same site selectivity for episclareolide, providing a precursor for the synthesis of N-acetyl-polveoline (Scheme 62A).
Scheme 62. (A) BM3 MERO1 and KSA 15 Are Both Highly Selective for the C3 Hydroxylation of Sclareolide and Episclareolide, Respectively; (B) Sequential Gram-Scale Oxidation of Sclareol with BM3 Variants LG-23 and MERO1 Provide a Precursor for the Core of (+)-Pallavincin.

BM3 variant LG-23 was similarly found to enable hydroxylation of C-6 of sclareol.210,213 This enzyme was further improved through two rounds of directed evolution.417 The final variant contained two additional mutations and allowed for the gram-scale production of the 6-hydroxylated compound 401 coupled to oxidative chemistry. Using the complementary hydroxylase BM3 MERO1 then afforded product 402 (Scheme 62B). These sequential oxidation reactions allowed for the efficient conversion of this compound into product 402, a precursor to the core moiety found in (+)-pallavincin. Demonstrating incorporation into longer synthetic sequences, MERO1 L437A was used to produce 3-hydroxylated sclareolide in the first total synthesis of gedunin, accomplished in 13 steps in 5.5% yield.418
Complementing the utility of engineered P450 BM3 variants, the diversity of enzymes that catalyze C─H hydroxylation provides a means to rapidly identify enzymes that might be better suited to different targets. To demonstrate this, BM3 MERO1, a fusion of the P450 PtmO5 with the reductase RHFRed, and the FeDO PtmO6 were used to enable site-selective oxidation of stevioside aglycone 403 at three different sites (Scheme 63).419 These selectively oxidized compounds provided functional handles for further diversification through traditional organic chemistry and ultimately provided access to nine natural products. The reported syntheses often included sequential biocatalytic oxidations using evolved enzymes and shortened reported syntheses for these compounds significantly. While bacterial P450s such as PtmO5 and BM3 are often easier to handle than enzymes from eukaryotic sources, enzymes sourced from higher organisms can yield catalysts bearing new function. This was demonstrated in the synthesis of several cardiotonic steroids using a fungal P450 in C. lunata,420 capable of producing more than 5 g of 14α-hydroxy-androst-4-ene-3,17-dione. This hydroxylated intermediate was used to produce bufogenin B and digitoxigenin in 11 and 12 steps, respectively.
Scheme 63. Site-Selective Oxidation of a Complex Diterpenoid Using P450 and FeDO Enzymes.

Unnatural amino acids are often used as building blocks for synthesis of both natural products and pharmaceuticals, so methods to access these compounds from proteinogenic amino acids and related compounds are highly sought after. FeDOs and FeDHs have proven particularly useful in this regard as their native activity often involves site-selective amino acid functionalization. The FeDO GetF was used in the synthesis of (2S,3-R)-3-hydroxy-3-methylproline, a key monomer in several peptide-derived natural products.421 By coupling GetF in a cascade with FeDO UcsF, sequential site-selective C─H hydroxylations enabled the isolation of the Boc-protected amino acid 410 in 28% yield over 3 steps, providing 300 mg of the product (Scheme 64A). Similarly, the FeDO GriE was found to accept additional bulk at the gamma position of the native substrate l-leucine. Consequently, hydroxylation of an azidated l-leucine derivative provided a quick way to access the lactone core of manzacidin C in significantly fewer steps than some previous reports (Scheme 64B).
Scheme 64. (A) Hydroxylation with UscF and GetF Is Used To Construct a Core Unnatural Amino Acid Fragment in Polyoxpeptin A; (B) GriE Hydroxylation of an Azidated Substrate; (C) KDO1 Catalyzed the Hydroxylation of Lysine in the Total Synthesis of Tambromycin.

While some applications of FeDOs in target-oriented synthesis utilize non-native substrates, most take advantage of native enzyme─substrate pairs. For example, the aforementioned hydroxylation of leucine by GriE was also used to produce (2S,4R)-4-methylproline en route to the total synthesis of cavinafungin B.422 The enzymatically produced compound was used as a monomer in solid-phase peptide synthesis as the penultimate coupling partner before hydrolysis of the product from the resin, providing the target compound entirely from building blocks derived from proteinogenic amino acids. A similar approach was used to access the pyrrolidine-containing amino acid tambroline in tambromycin.423 The native reactivity of a 3-selective lysine hydroxylase KDO1 was used to provide 3-hydroxylysine on gram scale with perfect site selectivity and stereoselectivity (Scheme 64C).240 This enabled gram-scale production of N-Boc-protected tambroline (416) and ultimately the efficient synthesis of tambromycin in 11 total steps, demonstrating an efficient use of biocatalytic retrosynthesis.
The native substrate scope of new FeDOs can be leveraged to diversify natural products in programmable ways. For example, the biosynthetic gene cluster of the terpenoid natural product GE81112 contains two FeDOs, GetI and GetF, that, respectively, catalyze site-selective and enantioselective hydroxylation of l-citrulline and l-pipecolic acid.424 These enzymatically produced unnatural amino acids could then be used as monomers in the chemical synthesis of the final GE81112 product. To expand this approach beyond total synthesis of GE81112, the native reactivity of other FeDOs was used to produce diverse unnatural lysine monomers. These were incorporated into the GE81112 backbone to produce novel analogues, identifying modifications that could be tolerated without ablating antimicrobial activity. A similar approach was recently used for profiling the antimicrobial activity of cepafungin I, wherein retrosynthetic analysis identified 4-hydroxylysine as a potential synthon for macrolactam 417 (Scheme 65).425 A panel of lysine hydroxylases with complementary selectivities could be used to provide diversified northern fragments of the macrocyclic core. Furthermore, SadA in combination with the desuccinylase LasA were used to provide a modified center amino acid fragment, further proving the utility of biocatalysts for product diversification.
Scheme 65. Chemoenzymatic Synthesis of Cepafungin Ia.

aA lysine-derived fragment can be modified with various lysine hydroxylases to study the impact of modification. For KDO1- and KDO3-derived peptides, yield refers to the Boc-protected monomer. Two-step enzymatic synthesis of 422 combining SadA hydroxylation with LasA decuccinylation; yield for 422 refers to the Fmoc-protected monomer.
In a similar way to how GetF and GetI were identified from analyzing biosynthetic gene clusters, BscD and Bsc9 were found to be involved in the formation of a tertiary alcohol late in cotylenol biosynthesis.426 These enzymes were initially investigated for in vitro late-stage hydroxylation of compound 423 but could not be solubly expressed in E. coli. Screening of related Bsc9 homologues led to the identification of MoBsc9, which was found to catalyze the desired transformation with roughly 1:1 selectivity for the desired alcohol 424 and a ketone byproduct. Site saturation mutagenesis of active site residues was used to generate MoBsc9 L110A, Y112R, which provided the desired tertiary alcohol and allowed for synthesis of over 100 mg of brassicicene I 426 in 10 total steps (Scheme 66 A). A second variant with the mutation Y112 M maintained high activity and selectivity for hydroxylation of an analog of this compound bearing different oxygenation patterns on the cyclooctene fragment (Scheme 66B). This example shows how bioinformatic approaches coupled with protein engineering in a total synthesis context can enable powerful transformations.
Scheme 66. Hydroxylation of Cotylenol and Brassicicene I Precursors with Evolved Variants of MoBsc9, a Homologue of the Native Hydroxylase.

The FeDO 2-ODD-PH natively catalyzes the C─C bond forming cyclization of the C ring in (−)-podophyllotoxin, which serves as a precursor for the chemotherapeutic compound etoposide.427 Substrate profiling of 2-ODD-PH showed that this enzyme accepted modifications of the native substrate, especially in the “A” ring of the compound, although in the absence of electron-donating groups the enzyme affects a C─H hydroxylation reaction at the benzylic position. When an electron-donating moiety was present, enantioselective C─C bond formation was accomplished instead, providing homologues of podophyllotoxin. Later exploration of 2-ODD-PH reactivity showed that product outcomes with 2-ODD-PH were highly susceptible to minor modifications of the substrate as shown by the difference between the selectivity observed for the methylenedioxy substrate 427 and the corresponding dimethoxy compound 429 (Scheme 67A and 67B).428 Ultimately, by utilizing 2-ODD-PH for the key cyclization step in a chemoenzymatic synthesis, (−)-podophyllotoxin was produced in 28% yield over 5 steps.
Scheme 67. Oxidative Coupling of Etoposide Precursors with FeDOs: (A) Native Reaction; (B) Closely Related Non-Native Substrate Shows Altered Selectivity for the Ring Closure.

Flavin-dependent enzymes have also been used for target-oriented synthesis. For example, the site-selective chlorination of napthacemycin B1 by flavin-dependent halogenase FasV provided fasamycin A 432 in 5% isolated yield as the final step in a chemoenzymatic approach starting from prenol and a simple phenol.429 Confirming the activity of FasV for chlorination of napthacemycin B1 both supports this enzyme as being responsible for late-stage functionalization in its native context and enables a competitive route to this product stepwise compared to the previous synthesis. Other than this example of target-oriented synthesis with halogenases, most flavin-dependent enzymes used for synthetic purposes catalyze oxidative chemistry. As noted above, the flavin-dependent oxidase SorbC catalyzes a site-selective and enantioselective hydroxylation reaction to produce sorbicillinol in biosynthesis of sorbicillactone A (Scheme 68B).277 This sorbicillinol intermediate has been used to synthesize several natural products,430,431 for example, by cyclization with a bisacyl urea to produce natural product sorbacatechol in 30% yield over 2 steps, the first total synthesis of this natural product (Scheme 68B).430
Scheme 68. (A) FasV Was Used as the Final Step in the Total Synthesis of Fasamycin A To Affect Site-Selective Halogenation of Napthacemycin B1; (B) SorbC Was Used in the First Total Synthesis of Sorbicillactone A.

5. SITE-SELECTIVE FUNCTIONALIZATION OF MACROMOLECULES
The use of protein-based pharmaceuticals has increased dramatically in recent years, leading to a commensurate increase in the need for generalizable methods for site-selective modification of these macromolecules.432 Site-selective modification of proteins is particularly challenging given that they are of course comprised of only 20 amino acid building blocks. Most enzymes that catalyze site-selective protein functionalization, including glycosyltransferases, lipoic acid ligases, sortases, and transpeptidases, therefore rely on the presence of specific amino acid sequence motifs that are recognized by the biocatalyst, a form of substrate control. While the respective motifs for these enzymes can often be recognized in different proteins, this requires modification of the original protein, which might not be desirable, and the context of that motif can have a significant impact on its modification efficiency (just as with directing groups in small molecule substrates). Overcoming this limitation with sequence-independent, site-selective protein modification is therefore a major challenge to this area. Here, we also note that DNA base-editing technologies such as the Crispr–Cas9 system have shown a remarkable ability to effect site-selective transformations both in vitro and in vivo, but since the site selectivity of these methods is dictated by a guide-RNA, it is not relevant to other biopolymers, and we refer readers to other excellent reviews of this area.433,434
5.1. Hydrolases for Peptide Modification
Insulin is a peptidic natural product with inter- and intramolecular bonds between the peptide fragments. The enormous clinical relevance of this protein makes it a prime target for the development of site-selective methods for biomolecule modification. In addition to the two free amines in the N-terminal glycine and phenylalanine residues (labeled A1 and B1, respectively, Scheme 69) a reactive lysine residue must also be protected before chemical modification, presenting a challenge for selective modification. Early studies showed that the penicillin G acylase from E. coli (EcPGA) could be used to deprotect the B29 lysine residue with a phenylacetyl protecting group on an insulin-derived peptide without disrupting the peptide structure.435 This finding was exploited by researchers at Merck to develop a comprehensive chemoenzymatic strategy for site-selective protection and deprotection of the insulin peptide. A penicillin G acylase enzyme from K. cryocrescens (KcPGA) was found to exhibit differential rates of hydrolysis for a triacylated insulin peptide and could be modified to express well in E. coli. After divergent directed evolution campaigns resulted in selective B29 and A1 deacylating enzymes, the reverse acylating reaction starting from unprotected insulin was developed to provide a route to A1-PAc in high selectivity. Ultimately, five distinct variants of this enzyme were obtained which in conjunction with the wild-type PGA from Achromobacter sp. allowed for the site-selective modification of both N-terminal residues and the internal lysine side chain. For global protection and deprotection, further evolution of the starting enzyme produced a variant capable of efficient nonselective modification of each exposed amine group, finishing a fully biocatalytic approach to mono-, di-, and triprotected insulin (Scheme 69).436
Scheme 69. Toolbox for Insulin Modification with Penicillin Acylases.

This so-called insulin toolbox of PGA enzymes greatly streamlined the existing syntheses for several clinical candidates and provided the starting point for evolving an enzyme capable of modifying the insulin dimer MK-5160 in up to twice the yield of the previous chemical synthesis with high purity.437 The PGA enzymes evolved for acylation of insulin were investigated for their promiscuous activity on other peptides and in several cases were found to be highly selective for modification of N-terminal residues. In the case of the small peptide somatostatin which contains several lysine residues in addition to the N-terminal alanine, evolved acylase enzymes were capable of differential acylation reminiscent of the differential hydrolysis that inspired selection of KcPGA as the template. Taken together, this study is a significant step forward in the development of site-selective peptide modifications and demonstrates a marked improvement over existing pKa-based strategies exploited by chemical synthesis.438
5.2. Microbial Transglutaminases
Microbial transglutaminases (mTGs) catalyze the intermolecular formation of an amide bond between the γ-amine in glutamine and the primary amine from lysine or a similar amine donor. A cysteine residue in the active site of the mTG forms an acyl enzyme─substrate intermediate, which is then attacked by the primary amine of the amine donor, resulting in the release of ammonia and the formation of a cross-linking bond. Unlike transpeptidases, mTGs can recognize glutamine residues in unique primary and secondary structures without a consistent motif. Furthermore, studies focusing on incorporation of an acyl donor containing a fluorescent probe revealed that mTG was competent for forming amide bonds on a variety of secondary structures including α-helices, β-sheets, and unstructured portions when glutamine residues were genetically incorporated into Protein G.439 This combined site-selective ligation without absolute motif or structural requirements underpins the potential of transglutaminases, but while this can provide a benefit by obviating the need to genetically incorporate a specific motif into the substrate, it also results in site selectivity that must often be determined experimentally and is largely not programmable. This difficulty in predicting and screening for site selectivity in biomolecule modification means that even in instances where directed evolution of mTGs is attempted, the targets are largely activity or thermoselectivity.440,441
Many non-naturally occurring amines including aromatic and aliphatic compounds can act as acyl acceptors in mTG-catalyzed reactions,442 although mTGs exhibit a distinct preference for less sterically hindered primary amines. Early studies with mTGs showed that even for proteins containing multiple surface-exposed glutamine residues, addition of Boc-PEG-NH2 or PEG-NH2 resulted in only mono- or di-PEGylation of the target protein sequences.443,444 Analysis of the glutamine residues pegylated by mTG showed only glutamine residues located on flexible portions of the target protein surface were functionalized with high selectivity compared to glutamine residues on more rigid portions of the surface. A critical study found that for humanized, aglycosylated antibodies an mTG enzyme was highly site selective for Q295 in the heavy chain portion of the protein. This high selectivity has been used to produce antibody–drug conjugates using primary amines connected to a payload via a defined linker which usually contains a site designed to be readily hydrolyzed when uptaken into the target cells.445 Incorporation of a linker connected to both azide and tetrazine moieties enabled the production of bifunctionally labeled ADCs, which proved advantageous for the reduction of hetereogeneous tumors, demonstrating benefits over the monolabeled antibody treatment.446
6. OUTLOOK
The examples outlined above highlight the current state of the art in our ability to use and engineer enzymes for non-native site-selective enzyme catalysis. With this information in mind, we can consider what types of advances might have the potential to expand the substrate and reaction scope of these enzymes and thus applications for which they could be used. As for biocatalysis and protein engineering more broadly,447 tight integration of tools and techniques from disparate research areas will be required, but the added challenge of site selectivity will provide an interesting context for their development.
Identifying new enzymes is central to biocatalysis. This task has historically been accomplished by characterizing enzymes involved in natural product biosynthesis.13 Looking at the diversity of natural product structures, we can infer that enzymes with remarkable biosynthetic capabilities exist in nature.448 The unique reactivity of enzymes involved in anaerobic secondary metabolism is notable in this regard, particularly in light of the dearth of studies examining their utility for biocatalysis.449 Even relatively common enzymatic activities like sp3 C─H hydroxylation and aromatic halogenation can belie the fact that they may involve novel enzymes with untapped potential, like recently reported Mn/Fe-dependent hydroxylases450 and single-component flavin reductase/halogenases.333,334 For these enzymes to be broadly useful, however, it is equally important to establish whether these capabilities extend to non-native substrates and reactions. Metagenomic and family-wide genome mining have also been used to identify new enzymes,451 and these efforts allow for direct analysis of non-native activity on probe substrates331,452 to identify enzymes with broad substrate scope. Advances in de novo enzyme design continue to emerge, but these remain limited to relatively simple chemical processes.453 Efforts to enable the design of enzymes that catalyze complex multistep reactions involving cofactors, including the metal cofactors responsible for much of the reactivity outlined above, are needed. For all of these systems, chemical and mechanistic intuition is critical to judge whether a given enzyme might reasonably catalyze a desired transformation and how probe substrates might be designed to identify novel activity and site selectivity.
Artificial metalloenzymes (ArMs)18 represent a particularly interesting source of enzymes for site-selective catalysis since they could be used to impart site selectivity to synthetic catalysts, thus avoiding the blocking and protecting group strategies noted in the Introduction. To date, there have been relatively few examples of this capability,406,407 and these involved the generation of carbene intermediates. The ArM scaffold is primarily involved in binding and orienting substrates relative to these intermediates to impart selectivity, which mimics the mechanisms of P450s, FeDOs, FDHs, and other enzymes that generate intermediates capable of reacting with a range of functional groups and C─H bonds in organic substrates (e.g., Schemes 30 and 36). This approach simplifies ArM (and natural enzyme) evolution since one need not worry about mutations disrupting complex networks of amino acids involved in substrate activation, and any compound reactive toward the intermediate could potentially serve as a substrate. On the other hand, it requires robust methods to orient the catalyst within the scaffold and to avoid diffusion of reactive intermediates since both could lead to nonselective reactions. Native substrate activation modes in ArM scaffolds mitigate these issues and can also be used to enable non-native catalysis,454,455 but this approach is inherently limited to substrates that contain suitable functionality for activation, such as a carbonyl compound in the case of serine hydrolases (Scheme 2). Beyond the significant technical challenges to ArM engineering,456-458 perhaps the biggest limitation to broadening their utility is the fact that the vast majority of transition-metal catalysis is incompatible with water. The benefits to developing transition-metal-catalyzed reactions that tolerate water are manifold, and the potential to incorporate water-tolerant catalysts into ArMs and to interface them with biocatalysts more broadly should certainly be counted among these.459
Identifying an enzyme with novel activity and/or selectivity is a necessary but not sufficient requirement for biocatalysis since extensive directed evolution is often needed for synthetic utility and essentially always required for commercial applications.460,461 The successful examples of directed evolution outlined above should not distract from the fact that enzymes are not equally evolvable462 and that technical hurdles can derail evolution efforts. Moreover, the field has now moved on from evolving individual enzymes to evolving entire synthetic pathways,447 so the scope and speed of directed evolution must be increased. Tools like laboratory automation, in vitro transcription/translation,463 and in vivo continuous evolution can increase the library size and ease of generation. Importantly, however, improved screening throughput is required to realize the potential of these tools, and screening for site selectivity is often challenging since isomeric products are formed. These products are difficult to resolve using chromatographic methods, and while microfluidic ESI-MS464,465 and DESI-MS466 can increase screening throughput, particularly using pooled reactions,331 site selectivity can only be distinguished if the products have different ionization patterns. Selectively deuterated probe substrates can be used to directly screen for site selectivity using mass spectrometry, but synthesis of such probes can be difficult, and the selectivity analysis must account for possible deuterium kinetic isotope effects.328 Biosensors, including ligand-dependent transcription factors, can provide a direct readout of formation of a specific product isomer, but these must be engineered to achieve the desired substrate and site specificity.467 New, rapid methods for assaying the site selectivity of chemical reactions are therefore needed.
Computational tools also have the potential to enable de novo design of enzymes468,469 and to accelerate directed evolution and genome mining,470 though the use of these tools to engineer enzymes for site-selective catalysis remains rare. In the context of site-selective catalysis, methods to rapidly model471 enzyme structures and analyze differential substrate binding in enzyme active sites can be used to guide enzyme selection or library design. Molecular dynamics simulations have been used to rationalize size selectivity in cases where docking simulations fail to provide useful insights,305 so decreasing the computational cost of these simulations or finding ways to use machine learning to avoid them entirely would be beneficial. Machine learning-directed evolution472 could also prove particularly useful for site-selective catalysis since deep learning methods are adept at handling complex data,473 such as the selectivity data associated with substrates that can undergo reaction at multiple sites with variable stereoselectivity. This approach also requires efficient methods to pair sequence information for entire enzyme libraries,473 not just improved variants, with enzyme activities from high-throughput assays such as those noted above.
Given the scope and limitation of these tools and the diversity of molecules to which they could be applied, it is difficult at this time to envision a “general” approach to generating enzymes for biocatalysis and non-native site-selective enzyme catalysis in particular. Careful consideration of parent enzymes and protein scaffolds, target reactivity, selectivity, and process compatibility, and methods for library generation and analysis will likely be required for some time. Site-selective catalysis is therefore not just a challenging goal for organic synthesis; it is a driving force for fundamental developments in protein engineering.
ACKNOWLEDGMENTS
We thank the David and Lucile Packard Foundation and the Searle Scholars Program for supporting my group’s studies on site-selective catalysis at an early phase. We also thank the NIH (R00GM087551, R01GM115665), the NSF (CHE-1351991, CHE-2154726, and the Center for Chemical Innovation Center for Selective C─H Functionalization, CHE-1700982), and the ARO (W911NF-14-1-0334, W911NF-19-1-0074, and W911NF-22-1-0118) for continued support of our studies aimed at engineering flavin-dependent halogenases, Fe(II)/α-ketoglutarate-dependent hydroxylases and halogenases, and artificial metalloenzymes for site-selective catalysis. D.M. was supported by the Precision Health Initiative of Indiana University.
Biographies
Dibyendu Mondal was born in West Bengal, India. He received his M.Sc. degree in Organic Chemistry from the University of Pune. Following graduation, he spent a few years in the drug discovery division of Takeda and Daiichi Sankyo Pharmaceuticals. In 2012, he began his graduate studies at the University of Iowa (Amnon Koehn), where his research focused on understanding the mechanism of flavin-dependent thymidylate synthase. In 2018, he took up a postdoctoral fellowship at the Indiana University (Jared C. Lewis) and spent 3 years engineering flavin-dependent halogenase for selective catalysis. In 2021, he joined the discovery group of Kalsec Inc. as a Lead Scientist.
Harrison M. Snodgrass is a native of Audubon, PA. He obtained his B.S. degree in Chemistry from West Chester University of Pennsylvania, where he worked with Felix Goodson and Kurt Kolasinski. After starting graduate school in 2016, he earned his Ph.D. degree from the University of Chicago (2022, Jared Lewis), studying biocatalysis with a focus on flavin-dependent halogenases. He is currently a protein engineer at Merck & Co., working at Merck Research Laboratories.
Christian A. Gomez was born and raised in Buenos Aires, Argentina. He obtained his B.S. degree in Chemistry from the University of Buenos Aires, where he worked with Marta B. Mazzetti. In 2016, he began his graduate studies at the University of Chicago with Jared C. Lewis and moved to Indiana University in 2018 to continue his work on developing Fe(II)- and α-ketoglutarate-dependent enzymes as selective biocatalysts for C─H functionalization reactions.
Jared C. Lewis was born and raised in Effingham, IL. He obtained his B.S. degree in Chemistry from the University of Illinois (2002, Eric Oldfield), earned his Ph.D. degree in Chemistry from the University of California, Berkeley (2007, Jonathan Ellman and Robert Bergman), and conducted postdoctoral studies at Caltech (2010, Frances Arnold). He started his independent career at the University of Chicago in 2011 and moved to Indiana University as an associate professor in 2018. His group engineers enzymes and develops new protein engineering tools to enable selective chemical transformations.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Helmchen G. In Stereoselective Synthesis; Helmchen G, Hoffmann RW, Mulzer J, Schaumann E, Eds.; Methods of Organic Chemistry (Houben-Weyl); Georg Thieme Verlag: New York, 1996; Vol. Workbench ed. E21 Vol. 1, pp 45–63. [Google Scholar]
- (2).Mahatthananchai J; Dumas AM; Bode JW Catalytic Selective Synthesis. Angew. Chem., Int. Ed 2012, 51, 10954–10990. [DOI] [PubMed] [Google Scholar]
- (3).Trost BM; Salzmann TN; Hiroi K New Synthetic Reactions. Sulfenylations and Dehydrosulfenylations of Esters and Ketones. J. Am. Chem. Soc 1976, 98, 4887–4902. [Google Scholar]
- (4).Trost BM Selectivity: A Key to Synthetic Efficiency. Science. 1983, 219, 245–250. [DOI] [PubMed] [Google Scholar]
- (5).Afagh NA; Yudin AK Chemoselectivity and the Curious Reactivity Preferences of Functional Groups. Angew. Chem., Int. Ed 2010, 49, 262–310. [DOI] [PubMed] [Google Scholar]
- (6).Hassner A. Correction. Regiospecificity. A Useful Terminology in Addition and Elimination Reactions. J. Org. Chem 1970, 35, 4004–4004. [Google Scholar]
- (7).Brown CJ; Toste FD; Bergman RG; Raymond KN Supramolecular Catalysis in Metal-Ligand Cluster Hosts. Chem. Rev 2015, 115, 3012–3035. [DOI] [PubMed] [Google Scholar]
- (8).Ishihara Y; Baran PS Two-Phase Terpene Total Synthesis: Historical Perspective and Application to the Taxol® Problem. Synlett 2010, 2010, 1733–1745. [Google Scholar]
- (9).Breslow R. Biomimetic Chemistry and Artificial Enzymes: Catalysis by Design. Acc. Chem. Res 1995, 28, 146–153. [Google Scholar]
- (10).Breslow R; Yang J; Yan J Biomimetic Hydroxylation of Saturated Carbons with Artificial Cytochrome P-450 Enzymes-Liberating Chemistry from the Tyranny of Functional Groups. Tetrahedron 2002, 58, 653–659. [Google Scholar]
- (11).Yang J; Gabriele B; Belvedere S; Huang Y; Breslow R Catalytic Oxidations of Steroid Substrates by Artificial Cytochrome P-450 Enzymes. J. Org. Chem 2002, 67, 5057–5067. [DOI] [PubMed] [Google Scholar]
- (12).Toste FD; Sigman MS; Miller SJ Pursuit of Noncovalent Interactions for Strategic Site-Selective Catalysis. Acc. Chem. Res 2017, 50, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bornscheuer UT; Huisman GW; Kazlauskas RJ; Lutz S; Moore JC; Robins K Engineering the Third Wave of Biocatalysis. Nature. 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- (14).Khersonsky O; Tawfik DS Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annu. Rev. Biochem 2010, 79, 471–505. [DOI] [PubMed] [Google Scholar]
- (15).Prier CK; Arnold FH Chemomimetic Biocatalysis: Exploiting the Synthetic Potential of Cofactor-Dependent Enzymes To Create New Catalysts. J. Am. Chem. Soc 2015, 137, 13992–14006. [DOI] [PubMed] [Google Scholar]
- (16).Yang Y; Arnold FH Navigating the Unnatural Reaction Space: Directed Evolution of Heme Proteins for Selective Carbene and Nitrene Transfer. Acc. Chem. Res 2021, 54, 1209–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Andorfer MC; Lewis JC Understanding and Improving the Activity of Flavin-Dependent Halogenases via Random and Targeted Mutagenesis. Annu. Rev. Biochem 2018, 87, 159–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Schwizer F; Okamoto Y; Heinisch T; Gu Y; Pellizzoni MM; Lebrun V; Reuter R; Köhler V; Lewis JC; Ward TR Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev 2018, 118, 142–231. [DOI] [PubMed] [Google Scholar]
- (19).Romero E; Jones BS; Hogg BN; Rué Casamajo A; Hayes MA; Flitsch SL; Turner NJ; Schnepel C Enzymatic Late-Stage Modifications: Better Late Than Never. Angew. Chem., Int. Ed 2021, 60, 16824–16855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Niwayama S. Highly Efficient Selective Monohydrolysis of Symmetric Diesters. J. Org. Chem 2000, 65, 5834–5836. [DOI] [PubMed] [Google Scholar]
- (21).Niwayama S; Cho H; Lin C Highly Efficient Selective Monohydrolysis of Dialkyl Malonates and their Derivatives. Tetrahedron Lett. 2008, 49, 4434–4436. [Google Scholar]
- (22).Nicolaou KC; Estrada AA; Zak M; Lee SH; Safina BS A Mild and Selective Method for the Hydrolysis of Esters with Trimethyltin Hydroxide. Angew. Chem., Int. Ed 2005, 44, 1378–1382. [DOI] [PubMed] [Google Scholar]
- (23).Domínguez de María P; García-Burgos CA; Bargeman G; van Gemert RW Pig Liver Esterase (PLE) as Biocatalyst in Organic Synthesis: From Nature to Cloning and to Practical Applications. Synthesis 2007, 2007, 1439–1452. [Google Scholar]
- (24).Hedstrom L. Serine Protease Mechanism and Specificity. Chem. Rev 2002, 102, 4501–4524. [DOI] [PubMed] [Google Scholar]
- (25).Bornscheuer UT Microbial Carboxyl Esterases: Classification, Properties and Application in Biocatalysis. FEMS Microbiol. Rev 2002, 26, 73–81. [DOI] [PubMed] [Google Scholar]
- (26).Chandra P; Enespa; Singh R; Arora PK Microbial Lipases and their Industrial Applications: a Comprehensive Review. Microb. Cell Fact 2020, 19, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ortiz C; Ferreira ML; Barbosa O; dos Santos JCS; Rodrigues RC; Berenguer-Murcia A; Briand LE; Fernandez-Lafuente R Novozym 435: the “Perfect” Lipase Immobilized Biocatalyst? Catal. Sci. Technol 2019, 9, 2380–2420. [Google Scholar]
- (28).Carey JS; McCann E Lipase-Catalyzed Regioselective Ester Hydrolysis as a Key Step in an Alternative Synthesis of a Buprenorphine Pro-Drug. Org. Process Res. Dev 2019, 23, 771–774. [Google Scholar]
- (29).Chênevert R; Ngatcha BT; Rose YS; Goupil D Regio- and Enantioselectivity of the Enzyme-catalyzed Hydrolysis of Citric Acid Derivatives. Tetrahedron: Asymmetry 1998, 9, 4325–4329. [Google Scholar]
- (30).Serbian I; Wiese J; Raschke A; Deising HB; Csuk R Streamlined Synthesis of (R, R)-Rhizoferrin, (S, S)-Rhizoferrin and (R, S, R)-Staphyloferrin A. Phytochem. Lett 2019, 33, 64–69. [Google Scholar]
- (31).Schmid R; Partali V; Anthonsen T; Anthonsen HW; Kvittingen L Regioselective Hydrolysis of Diesters of (Z)- and (E)-2-Methyl-butenedioic Acids by PLE. Tetrahedron Lett. 2001, 42, 8543–8545. [Google Scholar]
- (32).Zaks A; Dodds DR Application of Biocatalysis and Biotransformations to the Synthesis of Pharmaceuticals. Drug Discovery Today 1997, 2, 513–531. [Google Scholar]
- (33).Margolin AL; Delinck DL Enzymic Synthesis of Biologically Active Compounds: Synthesis of Castanospermine Derivatives. Biotechnol. Prog 1990, 6, 203–204. [Google Scholar]
- (34).Gmeiner P; Feldman PL; Chu-Moyer MY; Rapoport H An Efficient and Practical Total Synthesis of (+)-Vincamine from L-Aspartic Acid. J. Org. Chem 1990, 55, 3068–3074. [Google Scholar]
- (35).Stein KA; Toogood PL Enzyme-catalyzed Regioselective Hydrolysis of Aspartate Diesters. J. Org. Chem 1995, 60, 8110–8112. [Google Scholar]
- (36).Sweers HM; Wong CH Enzyme-catalyzed Regioselective Deacylation of Protected Sugars in Carbohydrate Synthesis. J. Am. Chem. Soc 1986, 108, 6421–6422. [Google Scholar]
- (37).Hennen WJ; Sweers HM; Wang YF; Wong CH Enzymes in Carbohydrate Synthesis. Lipase-catalyzed Selective Acylation and Deacylation of Furanose and Pyranose Derivatives. J. Org. Chem 1988, 53, 4939–4945. [Google Scholar]
- (38).Horrobin T; Tran CH; Crout D Esterase-catalysed Regioselective 6-deacylation of Hexopyranose Per-acetates, Acid-catalysed Rearrangement to the 4-deprotected Products and Conversions of these into Hexose 4- and 6-sulfates. J. Chem. Soc., Perkin Trans 1 1998, 1069–1080. [Google Scholar]
- (39).Wang J.-b.; Li G; Reetz MT Enzymatic Site-selectivity Enabled by Structure-guided Directed Evolution. Chem. Commun 2017, 53, 3916–3928. [DOI] [PubMed] [Google Scholar]
- (40).Massolo E; Pirola M; Benaglia M Amide Bond Formation Strategies: Latest Advances on a Dateless Transformation. Eur. J. Org. Chem 2020, 2020, 4641–4651. [Google Scholar]
- (41).Winn M; Richardson SM; Campopiano DJ; Micklefield J Harnessing and Engineering Amide Bond Forming Ligases for the Synthesis of Amides. Curr. Opin. Chem. Biol 2020, 55, 77–85. [DOI] [PubMed] [Google Scholar]
- (42).Andexer JN; Richter M Emerging Enzymes for ATP Regeneration in Biocatalytic Processes. ChemBioChem. 2015, 16, 380–386. [DOI] [PubMed] [Google Scholar]
- (43).Schnepel C; Pérez LR; Yu Y; Angelastro A; Heath RS; Lubberink M; Falcioni F; Mulholland K; Hayes MA; Turner NJ; et al. Thioester-mediated Biocatalytic Amide Bond Synthesis with in situ Thiol Recycling. Nat. Catal 2023, 6, 89–99. [Google Scholar]
- (44).Arai T; Arimura Y; Ishikura S; Kino K L-Amino Acid Ligase from Pseudomonas syringae Producing Tabtoxin Can Be Used for Enzymatic Synthesis of Various Functional Peptides. Appl. Environ. Microbiol 2013, 79, 5023–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Tsuda T; Asami M; Koguchi Y; Kojima S Single Mutation Alters the Substrate Specificity of l-Amino Acid Ligase. Biochem. 2014, 53, 2650–2660. [DOI] [PubMed] [Google Scholar]
- (46).Lee H; Choi M; Park J-U; Roh H; Kim S Genome Mining Reveals High Topological Diversity of ω-Ester-Containing Peptides and Divergent Evolution of ATP-Grasp Macrocyclases. J. Am. Chem. Soc 2020, 142, 3013–3023. [DOI] [PubMed] [Google Scholar]
- (47).Petchey MR; Rowlinson B; Lloyd RC; Fairlamb IJS; Grogan G Biocatalytic Synthesis of Moclobemide Using the Amide Bond Synthetase McbA Coupled with an ATP Recycling System. ACS Catal. 2020, 10, 4659–4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Winn M; Rowlinson M; Wang F; Bering L; Francis D; Levy C; Micklefield J Discovery, Characterization and Engineering of Ligases for Amide Synthesis. Nature. 2021, 593, 391–398. [DOI] [PubMed] [Google Scholar]
- (49).Vinogradov AA; Yin Y; Suga H Macrocyclic Peptides as Drug Candidates: Recent Progress and Remaining Challenges. J. Am. Chem. Soc 2019, 141, 4167–4181. [DOI] [PubMed] [Google Scholar]
- (50).Kobayashi M; Fujita K; Matsuda K; Wakimoto T Streamlined Chemoenzymatic Synthesis of Cyclic Peptides by Nonribosomal Peptide Cyclases. J. Am. Chem. Soc 2023, 145, 3270–3275. [DOI] [PubMed] [Google Scholar]
- (51).Trauger JW; Kohli RM; Mootz HD; Marahiel MA; Walsh CT Peptide Cyclization Catalysed by the Thioesterase Domain of Tyrocidine Synthetase. Nature. 2000, 407, 215–218. [DOI] [PubMed] [Google Scholar]
- (52).Stergiou P-Y; Foukis A; Filippou M; Koukouritaki M; Parapouli M; Theodorou LG; Hatziloukas E; Afendra A; Pandey A; Papamichael EM Advances in Lipase-catalyzed Esterification Reactions. Biotechnol. Adv 2013, 31, 1846–1859. [DOI] [PubMed] [Google Scholar]
- (53).Vilas Bôas RN; de Castro HF A Review of Synthesis of Esters with Aromatic, Emulsifying, and Lubricant Properties by Biotransformation Using Lipases. Biotechnol. Bioeng 2022, 119, 725–742. [DOI] [PubMed] [Google Scholar]
- (54).Cruz Silva MM; Riva S; Saé Melo ML Regioselective Enzymatic Acylation of Vicinal Diols of Steroids. Tetrahedron 2005, 61, 3065–3073. [Google Scholar]
- (55).Mitsunobu O; Kimura J; Fujisawa Y Studies on Nucleosides and Nucleotides. II. Selective Acylation of 5′-Hydroxyl Group of Thymidine. Bull. Chem. Soc. Jpn 1972, 45, 245–247. [Google Scholar]
- (56).Garcıa J; Fernández S; Ferrero M; Sanghvi YS; Gotor V A Mild, Efficient and Regioselective Enzymatic Procedure for 5′-O-Benzoylation of 2′-deoxynucleosides. Tetrahedron Lett. 2004, 45, 1709–1712. [Google Scholar]
- (57).Almeida VM; Branco CRC; Assis SA; Vieira IJC; Braz-Filho R; Branco A Synthesis of Naringin 6″-ricinoleate Using Immobilized Lipase. Chem. Cent. J 2012, 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Lackner G; Bohnert M; Wick J; Hoffmeister D Assembly of Melleolide Antibiotics Involves a Polyketide Synthase with Cross-Coupling Activity. Chem. Biol 2013, 20, 1101–1106. [DOI] [PubMed] [Google Scholar]
- (59).Gao X; Xie X; Pashkov I; Sawaya MR; Laidman J; Zhang W; Cacho R; Yeates TO; Tang Y Directed Evolution and Structural Characterization of a Simvastatin Synthase. Chem. Biol 2009, 16, 1064–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Antonopoulou I; Varriale S; Topakas E; Rova U; Christakopoulos P; Faraco V Enzymatic Synthesis of Bioactive Compounds with High Potential for Cosmeceutical Application. Appl. Microbiol. Biotechnol 2016, 100, 6519–6543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Zhang Q; van der Donk WA Catalytic Promiscuity of a Bacterial α-N-methyltransferase. FEBS Lett. 2012, 586, 3391–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Cha B; Jho E-H Protein Arginine Methyltransferases (PRMTs) as Therapeutic Targets. Expert Opin. Ther. Targets 2012, 16, 651–664. [DOI] [PubMed] [Google Scholar]
- (63).Yee DA; Niwa K; Perlatti B; Chen M; Li Y; Tang Y Genome Mining for Unknown-Unknown Natural Products. Nat. Chem. Biol 2023.19633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Liao C; Seebeck FP S-adenosylhomocysteine as a Methyl Transfer Catalyst in Biocatalytic Methylation Reactions. Nat. Catal 2019, 2, 696–701. [Google Scholar]
- (65).Abdelraheem E; Thair B; Varela RF; Jockmann E; Popadić D; Hailes HC; Ward JM; Iribarren AM; Lewkowicz ES; Andexer JN; et al. Methyltransferases: Functions and Applications. ChemBioChem. 2022, 23, No. e202200212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Shimizu T; Lin F; Hasegawa M; Okada K; Nojiri H; Yamane H Purification and Identification of Naringenin 7-O-Methyltransferase, a Key Enzyme in Biosynthesis of Flavonoid Phytoalexin Sakuranetin in Rice. J. Biol. Chem 2012, 287, 19315–19325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Minami H; Kim J-S; Ikezawa N; Takemura T; Katayama T; Kumagai H; Sato F Microbial Production of Plant Benzylisoquinoline Alkaloids. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 7393–7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Tang Q; Bornscheuer UT; Pavlidis IV Specific Residues Expand the Substrate Scope and Enhance the Regioselectivity of a Plant O-Methyltransferase. ChemCatChem. 2019, 11, 3227–3233. [Google Scholar]
- (69).Reeves CD; Hu Z; Reid R; Kealey JT Genes for the Biosynthesis of the Fungal Polyketides Hypothemycin from Hypomyces subiculosus and Radicicol from Pochonia chlamydosporia. Appl. Environ. Microbiol 2008, 74, 5121–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Xu Y; Zhou T; Espinosa-Artiles P; Tang Y; Zhan J; Molnár I Insights into the Biosynthesis of 12-Membered Resorcylic Acid Lactones from Heterologous Production in Saccharomyces cerevisiae. ACS Chem. Biol 2014, 9, 1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Wang X; Wang C; Duan L; Zhang L; Liu H; Xu Y.-m.; Liu Q; Mao T; Zhang W; Chen M; et al. Rational Reprogramming of O-Methylation Regioselectivity for Combinatorial Biosynthetic Tailoring of Benzenediol Lactone Scaffolds. J. Am. Chem. Soc 2019, 141, 4355–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Mordhorst S; Siegrist J; Müller M; Richter M; Andexer JN Catalytic Alkylation Using a Cyclic S-Adenosylmethionine Regeneration System. Angew. Chem., Int. Ed 2017, 56, 4037–4041. [DOI] [PubMed] [Google Scholar]
- (73).Struck A-W; Thompson ML; Wong LS; Micklefield J S-Adenosyl-Methionine-Dependent Methyltransferases: Highly Versatile Enzymes in Biocatalysis, Biosynthesis and Other Biotechnological Applications. ChemBioChem. 2012, 13, 2642–2655. [DOI] [PubMed] [Google Scholar]
- (74).Tang Q; Grathwol CW; Aslan-Üzel AS; Wu S; Link A; Pavlidis IV; Badenhorst CPS; Bornscheuer UT Directed Evolution of a Halide Methyltransferase Enables Biocatalytic Synthesis of Diverse SAM Analogs. Angew. Chem., Int. Ed 2021, 60, 1524–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Popadić D; Mhaindarkar D; Dang Thai MHN; Hailes HC; Mordhorst S; Andexer JN A Bicyclic S-adenosylmethionine Regeneration System Applicable with Different Nucleosides or Nucleotides as Cofactor Building Blocks. RSC chem. biol 2021, 2, 883–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Siegrist J; Aschwanden S; Mordhorst S; Thöny-Meyer L; Richter M; Andexer JN Regiocomplementary O-Methylation of Catechols by Using Three-Enzyme Cascades. ChemBioChem. 2015, 16, 2576–2579. [DOI] [PubMed] [Google Scholar]
- (77).Vidgren J; Svensson LA; Liljas A Crystal Structure of Catechol O-Methyltransferase. Nature. 1994, 368, 354–358. [DOI] [PubMed] [Google Scholar]
- (78).Nelson JT; Lee J; Sims JW; Schmidt EW Characterization of SafC, a Catechol 4-O-Methyltransferase Involved in Saframycin Biosynthesis. Appl. Environ. Microbiol 2007, 73, 3575–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Dalhoff C; Lukinavicius G; Klimasauskas S; Weinhold E Direct transfer of Extended Groups from Synthetic Cofactors by DNA Methyltransferases. Nat. Chem. Biol 2006, 2, 31–32. [DOI] [PubMed] [Google Scholar]
- (80).Wang R; Zheng W; Yu H; Deng H; Luo M Labeling Substrates of Protein Arginine Methyltransferase with Engineered Enzymes and Matched S-Adenosyl-l-methionine Analogues. J. Am. Chem. Soc 2011, 133, 7648–7651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Singh S; Zhang J; Huber TD; Sunkara M; Hurley K; Goff RD; Wang G; Zhang W; Liu C; Rohr J; et al. Facile Chemoenzymatic Strategies for the Synthesis and Utilization of S-Adenosyl-L-Methionine Analogues. Angew. Chem., Int. Ed 2014, 53, 3965–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Herbert AJ; Shepherd SA; Cronin VA; Bennett MR; Sung R; Micklefield J Engineering Orthogonal Methyltransferases to Create Alternative Bioalkylation Pathways. Angew. Chem., Int. Ed 2020, 59, 14950–14956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Law BJC; Bennett MR; Thompson ML; Levy C; Shepherd SA; Leys D; Micklefield J Effects of Active-Site Modification and Quaternary Structure on the Regioselectivity of Catechol-O-Methyltransferase. Angew. Chem., Int. Ed 2016, 55, 2683–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Wen X; Leisinger F; Leopold V; Seebeck FP Synthetic Reagents for Enzyme-Catalyzed Methylation. Angew. Chem., Int. Ed 2022, 61, No. e202208746. [DOI] [PubMed] [Google Scholar]
- (85).Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nat. Chem 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- (86).Bengel LL; Aberle B; Egler-Kemmerer A-N; Kienzle S; Hauer B; Hammer SC Engineered Enzymes Enable Selective N-Alkylation of Pyrazoles With Simple Haloalkanes. Angew. Chem., Int. Ed 2021, 60, 5554–5560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Faheem M; Rathaur A; Pandey A; Kumar Singh V; Tiwari AK A Review on the Modern Synthetic Approach of Benzimidazole Candidate. ChemistrySelect. 2020, 5, 3981–3994. [Google Scholar]
- (88).Ospina F; Schülke KH; Soler J; Klein A; Prosenc B; Garcia-Borràs M; Hammer SC Selective Biocatalytic N-Methylation of Unsaturated Heterocycles. Angew. Chem., Int. Ed 2022, 61, No. e202213056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Manning G; Whyte DB; Martinez R; Hunter T; Sudarsanam S The Protein Kinase Complement of the Human Genome. Science. 2002, 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- (90).Saier MH Protein Phosphorylation and Allosteric Control of Inducer Exclusion and Catabolite Repression by the Bacterial Phosphoenolpyruvate: Sugar Phosphotransferase System. Microbiol. Rev 1989, 53, 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Westheimer FH Why Nature Chose Phosphates. Science. 1987, 235, 1173–1178. [DOI] [PubMed] [Google Scholar]
- (92).Wohlgemuth R. The Power of Biocatalysts for Highly Selective and Efficient Phosphorylation Reactions. Catalysts 2022, 12, 1436. [Google Scholar]
- (93).Tozzi MG; Camici M; Mascia L; Sgarrella F; Ipata PL Pentose Phosphates in Nucleoside Interconversion and Catabolism. FEBS J. 2006, 273, 1089–1101. [DOI] [PubMed] [Google Scholar]
- (94).Huffman MA; Fryszkowska A; Alvizo O; Borra-Garske M; Campos KR; Canada KA; Devine PN; Duan D; Forstater JH; Grosser ST; et al. Design of an in vitro Biocatalytic Cascade for the Manufacture of Islatravir. Science. 2019, 366, 1255–1259. [DOI] [PubMed] [Google Scholar]
- (95).Kilstrup M; Hammer K; Ruhdal Jensen P; Martinussen J Nucleotide Metabolism and its Control in Lactic Acid Bacteria. FEMS Microbiol. Rev 2005, 29, 555–590. [DOI] [PubMed] [Google Scholar]
- (96).McIntosh JA; Benkovics T; Silverman SM; Huffman MA; Kong J; Maligres PE; Itoh T; Yang H; Verma D; Pan W; et al. Engineered Ribosyl-1-Kinase Enables Concise Synthesis of Molnupiravir, an Antiviral for COVID-19. ACS Cent. Sci 2021, 7, 1980–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Reily C; Stewart TJ; Renfrow MB; Novak J Glycosylation in Health and Disease. Nat. Rev. Nephrol 2019, 15, 346–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Ouadhi S; López DMV; Mohideen FI; Kwan DH Engineering the Enzyme Toolbox to Tailor Glycosylation in Small Molecule Natural Products and Protein Biologics. Protein Eng. Des. Sel 2023, 36. DOI: 10.1093/protein/gzac010 [DOI] [PubMed] [Google Scholar]
- (99).Danieli B; Falcone L; Monti D; Riva S; Gebhardt S; Schubert-Zsilavecz M Regioselective Enzymatic Glycosylation of Natural Polyhydroxylated Compounds: Galactosylation and Glucosylation of Protopanaxatriol Ginsenosides1. J. Org. Chem 2001, 66, 262–269. [DOI] [PubMed] [Google Scholar]
- (100).He B; Bai X; Tan Y; Xie W; Feng Y; Yang G-Y Glycosyltransferases: Mining, Engineering and Applications in Biosynthesis of Glycosylated Plant Natural Products. Synth. Syst. Biotechnol 2022, 7, 602–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).McArthur JB; Chen X Glycosyltransferase Engineering for Carbohydrate Synthesis. Biochem. Soc. Trans 2016, 44, 129–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Shaikh FA; Withers SG Teaching Old Enzymes New Tricks: Engineering and Evolution of Glycosidases and Glycosyl Transferases for Improved Glycoside Synthesis. Biochem.Cell Biol 2008, 86, 169–177. [DOI] [PubMed] [Google Scholar]
- (103).Gutmann A; Schiller M; Gruber-Khadjawi M; Nidetzky B An Ortho C-methylation/O-glycosylation Motif on a Hydroxy-Coumarin Scaffold, Selectively Installed by Biocatalysis. Org. Biomol. Chem 2017, 15, 7917–7924. [DOI] [PubMed] [Google Scholar]
- (104).Losey HC; Jiang J; Biggins JB; Oberthür M; Ye X-Y; Dong SD; Kahne D; Thorson JS; Walsh CT Incorporation of Glucose Analogs by GtfE and GtfD from the Vancomycin Biosynthetic Pathway to Generate Variant Glycopeptides. Chem. Biol 2002, 9, 1305–1314. [DOI] [PubMed] [Google Scholar]
- (105).Yang M; Proctor MR; Bolam DN; Errey JC; Field RA; Gilbert HJ; Davis BG Probing the Breadth of Macrolide Glycosyltransferases: In Vitro Remodeling of a Polyketide Antibiotic Creates Active Bacterial Uptake and Enhances Potency. J. Am. Chem. Soc 2005, 127, 9336–9337. [DOI] [PubMed] [Google Scholar]
- (106).Yang G; Rich JR; Gilbert M; Wakarchuk WW; Feng Y; Withers SG Fluorescence Activated Cell Sorting as a General Ultra-High-Throughput Screening Method for Directed Evolution of Glycosyltransferases. J. Am. Chem. Soc 2010, 132, 10570–10577. [DOI] [PubMed] [Google Scholar]
- (107).Lairson LL; Watts AG; Wakarchuk WW; Withers SG Using Substrate Engineering to Harness Enzymatic Promiscuity and Expand Biological Catalysis. Nat. Chem. Biol 2006, 2, 724–728. [DOI] [PubMed] [Google Scholar]
- (108).Ly HD; Lougheed B; Wakarchuk WW; Withers SG Mechanistic Studies of a Retaining α-Galactosyltransferase from Neisseria meningitidis. Biochem. 2002, 41, 5075–5085. [DOI] [PubMed] [Google Scholar]
- (109).Brazier-Hicks M; Evans KM; Gershater MC; Puschmann H; Steel PG; Edwards R The C-Glycosylation of Flavonoids in Cereals. J. Biol. Chem 2009, 284, 17926–17934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).He J-B; Zhao P; Hu Z-M; Liu S; Kuang Y; Zhang M; Li B; Yun C-H; Qiao X; Ye M Molecular and Structural Characterization of a Promiscuous C-Glycosyltransferase from Trollius chinensis. Angew. Chem., Int. Ed 2019, 58, 11513–11520. [DOI] [PubMed] [Google Scholar]
- (111).Hancock SM; Vaughan MD; Withers SG Engineering of Glycosidases and Glycosyltransferases. Curr. Opin. Chem. Biol 2006, 10, 509–519. [DOI] [PubMed] [Google Scholar]
- (112).Mackenzie LF; Wang Q; Warren RAJ; Withers SG Glycosynthases: Mutant Glycosidases for Oligosaccharide Synthesis. J. Am. Chem. Soc 1998, 120, 5583–5584. [Google Scholar]
- (113).Drone J; Feng H.-y.; Tellier C; Hoffmann L; Tran V; Rabiller C; Dion M Thermus thermophilus Glycosynthases for the Efficient Synthesis of Galactosyl and Glucosyl β-(1→3)-Glycosides. Eur. J. Org. Chem 2005, 2005, 1977–1983. [Google Scholar]
- (114).Müllegger J; Chen H-M; Chan WY; Reid SP; Jahn M; Warren RAJ; Salleh HM; Withers SG Thermostable Glycosynthases and Thioglycoligases Derived from Thermotoga maritima β-Glucuronidase. ChemBioChem. 2006, 7, 1028–1030. [DOI] [PubMed] [Google Scholar]
- (115).Vaughan MD; Johnson K; DeFrees S; Tang X; Warren RAJ; Withers SG Glycosynthase-Mediated Synthesis of Glycosphingolipids. J. Am. Chem. Soc 2006, 128, 6300–6301. [DOI] [PubMed] [Google Scholar]
- (116).Yang M; Davies GJ; Davis BG A Glycosynthase Catalyst for the Synthesis of Flavonoid Glycosides. Angew. Chem., Int. Ed 2007, 46, 3885–3888. [DOI] [PubMed] [Google Scholar]
- (117).Brown M; Chapman E; Collis A; Fuerst D; Hosford J; Macdermaid C; Morrison J Saponins. WO2022122830A2; Dec 8, 2021. [Google Scholar]
- (118).Beerens K; Desmet T; Soetaert W Enzymes for the Biocatalytic Production of Rare Sugars. J. Ind. Microbiol. Biotechnol 2012, 39, 823–834. [DOI] [PubMed] [Google Scholar]
- (119).Izumori K Izumoring: A Strategy for Bioproduction of all Hexoses. J. Biotechnol 2006, 124, 717–722. [DOI] [PubMed] [Google Scholar]
- (120).Lafite P; Daniellou R Rare and Unusual Glycosylation of Peptides and Proteins. Nat. Prod. Rep 2012, 29, 729–738. [DOI] [PubMed] [Google Scholar]
- (121).Nishimura S-I; Hato M; Hyugaji S; Feng F; Amano M Glycomics for Drug Discovery: Metabolic Perturbation in Androgen-Independent Prostate Cancer Cells Induced by Unnatural Hexosamine Mimics. Angew. Chem., Int. Ed 2012, 51, 3386–3390. [DOI] [PubMed] [Google Scholar]
- (122).Wang Y; Carder HM; Wendlandt AE Synthesis of Rare Sugar Isomers Through Site-selective Epimerization. Nature. 2020, 578, 403–408. [DOI] [PubMed] [Google Scholar]
- (123).Angyal SJ The Lobry de Bruyn-Alberda van Ekenstein Transformation and Related Reactions. In Glycoscience: Epimerisation, Isomerisation and Rearrangement Reactions of Carbohydrates; Stütz AE, Ed.; Springer Berlin Heidelberg, 2001; pp 1–14. [Google Scholar]
- (124).Allard STM; Giraud MF; Naismith JH Epimerases: Structure, Function and Mechanism. Cell. Mol. Life Sci 2001, 58, 1650–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Fushinobu S. Molecular Evolution and Functional Divergence of UDP-hexose 4-epimerases. Curr. Opin. Chem. Biol 2021, 61, 53–62. [DOI] [PubMed] [Google Scholar]
- (126).Beerens K; Soetaert W; Desmet T UDP-Hexose 4-Epimerases: A View on Structure, Mechanism and Substrate Specificity. Carbohydr. Res 2015, 414, 8–14. [DOI] [PubMed] [Google Scholar]
- (127).Kim H-J; Kang SY; Park JJ; Kim P Novel Activity of UDP-Galactose-4-Epimerase for Free Monosaccharide and Activity Improvement by Active Site-Saturation Mutagenesis. Appl. Biochem. Biotechnol 2011, 163, 444–451. [DOI] [PubMed] [Google Scholar]
- (128).Bosshart A; Hee CS; Bechtold M; Schirmer T; Panke S Directed Divergent Evolution of a Thermostable D-Tagatose Epimerase towards Improved Activity for Two Hexose Substrates. ChemBioChem. 2015, 16, 592–601. [DOI] [PubMed] [Google Scholar]
- (129).Bosshart A; Wagner N; Lei L; Panke S; Bechtold M Highly Efficient Production of Rare Sugars D-psicose and L-tagatose by Two Engineered D-tagatose Epimerases. Biotechnol. Bioeng 2016, 113, 349–358. [DOI] [PubMed] [Google Scholar]
- (130).Zhu Z; Gao D; Li C; Chen Y; Zhu M; Liu X; Tanokura M; Qin H-M; Lu F Redesign of a Novel d-allulose 3-Epimerase from Staphylococcus aureus for Thermostability and Efficient Biocatalytic Production of d-Allulose. Microb. Cell Fact 2019, 18, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Shompoosang S; Yoshihara A; Uechi K; Asada Y; Morimoto K Enzymatic Production of Three 6-deoxy-Aldohexoses from L-Rhamnose. Biosci. Biotechnol. Biochem 2014, 78, 317–325. [DOI] [PubMed] [Google Scholar]
- (132).Yoshida H; Yamada M; Nishitani T; Takada G; Izumori K; Kamitori S Crystal Structures of d-Tagatose 3-Epimerase from Pseudomonas cichorii and Its Complexes with d-Tagatose and d-Fructose. J. Mol. Biol 2007, 374, 443–453. [DOI] [PubMed] [Google Scholar]
- (133).Shin K-C; Lee T-E; Seo M-J; Kim DW; Kang L-W; Oh D-K Development of Tagaturonate 3-Epimerase into Tagatose 4-Epimerase with a Biocatalytic Route from Fructose to Tagatose. ACS Catal. 2020, 10, 12212–12222. [Google Scholar]
- (134).Bosshart A; Panke S; Bechtold M Systematic Optimization of Interface Interactions Increases the Thermostability of a Multimeric Enzyme. Angew. Chem., Int. Ed 2013, 52, 9673–9676. [DOI] [PubMed] [Google Scholar]
- (135).Rodionova IA; Scott DA; Grishin NV; Osterman AL; Rodionov DA Tagaturonate-fructuronate epimerase UxaE, a novel enzyme in the hexuronate catabolic network in Thermotoga maritima. Environ. Microbiol 2012, 14, 2920–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Heath RS; Turner NJ Recent Advances in Oxidase Biocatalysts: Enzyme Discovery, Cascade Reactions and Scale up. Curr. Opin. Green Sustain. Chem 2022, 38, 100693. [Google Scholar]
- (137).Bianchini E; Chinaglia N; Dean M; Giovannini PP; Medici A; Pedrini P; Poli S Regiospecific Oxidoreductions Catalyzed by a New Pseudomonas Paucimobilis Hydroxysteroid Dehydrogenase. Tetrahedron 1999, 55, 1391–1398. [Google Scholar]
- (138).Secundo F; Carrea G; De Amici M; Joppolo di Ventimiglia S; Dordick JS A Combinatorial Biocatalysis Approach to an Array of Cholic Acid Derivatives. Biotechnol. Bioeng 2003, 81, 391–396. [DOI] [PubMed] [Google Scholar]
- (139).Monti D; Ferrandi EE; Zanellato I; Hua L; Polentini F; Carrea G; Riva S One-Pot Multienzymatic Synthesis of 12-Ketoursodeoxycholic Acid: Subtle Cofactor Specificities Rule the Reaction Equilibria of Five Biocatalysts Working in a Row. Adv. Synth. Catal 2009, 351, 1303–1311. [Google Scholar]
- (140).Chen X; Cui Y; Feng J; Wang Y; Liu X; Wu Q; Zhu D; Ma Y Flavin Oxidoreductase-Mediated Regeneration of Nicotinamide Adenine Dinucleotide with Dioxygen and Catalytic Amount of Flavin Mononucleotide for One-Pot Multi-Enzymatic Preparation of Ursodeoxycholic Acid. Adv. Synth. Catal 2019, 361, 2497–2504. [Google Scholar]
- (141).Barski OA; Tipparaju SM; Bhatnagar A The Aldo-Keto Reductase Superfamily and its Role in Drug Metabolism and Detoxification. Drug Metab. Rev 2008, 40, 553–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Hyndman D; Bauman DR; Heredia VV; Penning TM The Aldo-keto Reductase Superfamily Homepage. Chem. Biol. Interact 2003, 143–144, 621–631. [DOI] [PubMed] [Google Scholar]
- (143).France SP; Lewis RD; Martinez CA The Evolving Nature of Biocatalysis in Pharmaceutical Research and Development. JACS Au. 2023.3715735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Whalen K; Berdahl DR; Patrick B; Jones MB; Williams K; WO2020069139A2. Enzymatic Process for Production of Modified Hop Products. USA April 2, 2020. [Google Scholar]
- (145).Sasaki S; Yamauchi T; Kubo H; Kanai M; Ishii A; Higashiyama K Chemoselective Reduction of Ketones: Trifluoromethylketones Versus Methylketones. Tetrahedron Lett. 2005, 46, 1497–1500. [Google Scholar]
- (146).Grau BT; Devine PN; DiMichele LN; Kosjek B Chemo- and Enantioselective Routes to Chiral Fluorinated Hydroxyketones Using Ketoreductases. Org. Lett 2007, 9, 4951–4954. [DOI] [PubMed] [Google Scholar]
- (147).Kalaitzakis D; Rozzell JD; Kambourakis S; Smonou I Highly Stereoselective Reductions of α-Alkyl-1,3-diketones and α-Alkyl-β-keto Esters Catalyzed by Isolated NADPH-Dependent Ketoreductases. Org. Lett 2005, 7, 4799–4801. [DOI] [PubMed] [Google Scholar]
- (148).Fryszkowska A; Peterson J; Davies NL; Dewar C; Evans G; Bycroft M; Triggs N; Fleming T; Gorantla SSC; Hoge G; et al. Development of a Chemoenzymatic Process for Dehydroepiandrosterone Acetate Synthesis. Org. Process Res. Dev 2016, 20, 1520–1528. [Google Scholar]
- (149).St-Jean F; Angelaud R; Bachmann S; Carrera DE; Remarchuk T; Piechowicz KA; Niedermann K; Iding H; Meier R; Hou H; et al. Stereoselective Synthesis of the IDO Inhibitor Navoximod. J. Org. Chem 2022, 87, 4955–4960. [DOI] [PubMed] [Google Scholar]
- (150).Ji A; Wolberg M; Hummel W; Wandrey C; Müller M Dynamic Kinetic Resolution of tert-butyl 4-methyl-3,5-Dioxohexanoate Through Enzymatic Reduction. Chem. Commun. 2001, 57–58. [Google Scholar]
- (151).Lüdeke S; Richter M; Müller M Stereoselective Synthesis of Three Isomers of tert-Butyl 5-Hydroxy-4-methyl-3-oxohexanoate through Alcohol Dehydrogenase-Catalyzed Dynamic Kinetic Resolution. Adv. Synth. Catal 2009, 351, 253–259. [Google Scholar]
- (152).Savile CK; Janey JM; Mundorff EC; Moore JC; Tam S; Jarvis WR; Colbeck JC; Krebber A; Fleitz FJ; Brands J; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science. 2010, 329, 305–309. [DOI] [PubMed] [Google Scholar]
- (153).Aleku GA; France SP; Man H; Mangas-Sanchez J; Montgomery SL; Sharma M; Leipold F; Hussain S; Grogan G; Turner NJ A Reductive Aminase from Aspergillus Oryzae. Nat. Chem 2017, 9, 961–969. [DOI] [PubMed] [Google Scholar]
- (154).Gilio AK; Thorpe TW; Turner N; Grogan G Reductive Aminations by Imine Reductases: From Milligrams to Tons. Chem. Sci 2022, 13, 4697–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Simon RC; Zepeck F; Kroutil W Chemoenzymatic Synthesis of All Four Diastereomers of 2,6-Disubstituted Piperidines through Stereoselective Monoamination of 1,5-Diketones. Chem. Eur. J 2013, 19, 2859–2865. [DOI] [PubMed] [Google Scholar]
- (156).O’Reilly E; Iglesias C; Ghislieri D; Hopwood J; Galman JL; Lloyd RC; Turner NJ A Regio- and Stereoselective ω-Transaminase/Monoamine Oxidase Cascade for the Synthesis of Chiral 2,5-Disubstituted Pyrrolidines. Angew. Chem., Int. Ed 2014, 53, 2447–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).France SP; Hussain S; Hill AM; Hepworth LJ; Howard RM; Mulholland KR; Flitsch SL; Turner NJ One-Pot Cascade Synthesis of Mono- and Disubstituted Piperidines and Pyrrolidines using Carboxylic Acid Reductase (CAR), ω-Transaminase (ω-TA), and Imine Reductase (IRED) Biocatalysts. ACS Catal. 2016, 6, 3753–3759. [Google Scholar]
- (158).Payer SE; Schrittwieser JH; Grischek B; Simon RC; Kroutil W Regio- and Stereoselective Biocatalytic Monoamination of a Triketone Enables Asymmetric Synthesis of Both Enantiomers of the Pyrrolizidine Alkaloid Xenovenine Employing Transaminases. Adv. Synth. Catal 2016, 358, 444–451. [Google Scholar]
- (159).Resch V; Hanefeld U The Selective Addition of Water. Catal. Sci. Technol 2015, 5, 1385–1399. [Google Scholar]
- (160).Hiseni A; Arends IWCE; Otten LG New Cofactor-Independent Hydration Biocatalysts: Structural, Biochemical, and Biocatalytic Characteristics of Carotenoid and Oleate Hydratases. ChemCatChem. 2015, 7, 29–37. [Google Scholar]
- (161).Demming RM; Fischer M-P; Schmid J; Hauer B (De)hydratases—Recent Developments and Future Perspectives. Curr. Opin. Chem. Biol 2018, 43, 43–50. [DOI] [PubMed] [Google Scholar]
- (162).Brodkorb D; Gottschall M; Marmulla R; Luddeke F; Harder J Linalool Dehydratase-Isomerase, a Bifunctional Enzyme in the Anaerobic Degradation of Monoterpenes. J. Biol. Chem 2010, 285, 30436–30442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Cuetos A; Iglesias-Fernández J; Danesh-Azari H-R; Zukic E; Dowle A; Osuna S; Grogan G Mutational Analysis of Linalool Dehydratase Isomerase Suggests That Alcohol and Alkene Transformations Are Catalyzed Using Noncovalent Mechanisms. ACS Catal. 2020, 10, 11136–11146. [Google Scholar]
- (164).Katsuki T; Martin V Asymmetric Epoxidation of Allylic Alcohols: the Katsuki-Sharpless Epoxidation Reaction. Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, 1996; pp 1299 [Google Scholar]
- (165).Coleman T; Kirk AM; Chao RR; Podgorski MN; Harbort JS; Churchman LR; Bruning JB; Bernhardt PV; Harmer JR; Krenske EH; et al. Understanding the Mechanistic Requirements for Efficient and Stereoselective Alkene Epoxidation by a Cytochrome P450 Enzyme. ACS Catal 2021, 11, 1995–2010. [Google Scholar]
- (166).Kolev JN; O’Dwyer KM; Jordan CT; Fasan R Discovery of Potent Parthenolide-Based Antileukemic Agents Enabled by Late-Stage P450-Mediated C─H Functionalization. ACS Chem. Biol 2014, 9, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Le-Huu P; Heidt T; Claasen B; Laschat S; Urlacher VB Chemo-, Regio-, and Stereoselective Oxidation of the Monocyclic Diterpenoid β-Cembrenediol by P450 BM3. ACS Catal. 2015, 5, 1772–1780. [Google Scholar]
- (168).Espinoza RV; Maskeri MA; Turlik A; Nangia A; Khatri Y; Montgomery J; Houk KN; Sherman DH Epoxidation and Late-Stage C-H Functionalization by P450 TamI Are Mediated by Variant Heme-Iron Oxidizing Species. ACS Catal. 2022, 12, 3731–3742. [Google Scholar]
- (169).Polic V; Cheong KJ; Hammerer F; Auclair K Regioselective Epoxidations by Cytochrome P450 3A4 Using a Theobromine Chemical Auxiliary to Predictably Produce N-Protected or β- or γ-Amino Epoxides. Adv. Synth. Catal 2017, 359, 3983–3989. [Google Scholar]
- (170).Kobayashi M; Nagasawa T; Yamada H Enzymatic Synthesis of Acrylamide: a Success Story Not Yet Over. Trends Biotechnol. 1992, 10, 402–408. [DOI] [PubMed] [Google Scholar]
- (171).Yamada H; Kobayashi M Nitrile Hydratase and Its Application to Industrial Production of Acrylamide. Biosci. Biotechnol. Biochem 1996, 60, 1391–1400. [DOI] [PubMed] [Google Scholar]
- (172).Kobayashi M; Shimizu S Metalloenzyme Nitrile Hydratase: Structure, Regulation, and Application to Biotechnology. Nat. Biotechnol 1998, 16, 733–736. [DOI] [PubMed] [Google Scholar]
- (173).Cheng Z; Xia Y; Zhou Z Recent Advances and Promises in Nitrile Hydratase: From Mechanism to Industrial Applications. Front. Bioeng. Biotechnol 2020, 8, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Craven EJ; Latham J; Shepherd SA; Khan I; Diaz-Rodriguez A; Greaney MF; Micklefield J Programmable Late-stage C-H bond Functionalization Enabled by Integration of Enzymes with Chemocatalysis. Nat. Catal 2021, 4, 385–394. [Google Scholar]
- (175).Bering L; Craven EJ; Sowerby Thomas SA; Shepherd SA; Micklefield J Merging Enzymes with Chemocatalysis for Amide Bond Synthesis. Nat. Commun 2022, 13, 380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Farnberger JE; Richter N; Hiebler K; Bierbaumer S; Pickl M; Skibar W; Zepeck F; Kroutil W Biocatalytic Methylation and Demethylation via a Shuttle Catalysis Concept Involving Corrinoid Proteins. Commun. Chem 2018, 1, 82. [Google Scholar]
- (177).Wuts PGM; Greene TW Greene’s Protective Groups in OrganicSynthesis; John Wiley & Sons, Inc., 2006; pp 367–430. [Google Scholar]
- (178).Selva M; Perosa A Green Chemistry Metrics: a Comparative Evaluation of Dimethyl Carbonate, Methyl Iodide, Dimethyl Sulfate and Methanol as Methylating Agents. Green Chem. 2008, 10, 457–464. [Google Scholar]
- (179).Zhang M-X; Hu X-H; Xu Y-H; Loh T-P Selective Dealkylation of Alkyl Aryl Ethers. Asian J. Org. Chem 2015, 4, 1047–1049. [Google Scholar]
- (180).Banerjee R; Ragsdale SW The Many Faces of Vitamin B12: Catalysis by Cobalamin-Dependent Enzymes. Annu. Rev. Biochem 2003, 72, 209–247. [DOI] [PubMed] [Google Scholar]
- (181).Richter N; Zepeck F; Kroutil W Cobalamin-dependent Enzymatic O-, N-, and S-demethylation. Trends Biotechnol. 2015, 33, 371–373. [DOI] [PubMed] [Google Scholar]
- (182).Pompei S; Grimm C; Schiller C; Schober L; Kroutil W Thiols Act as Methyl Traps in the Biocatalytic Demethylation of Guaiacol Derivatives. Angew. Chem., Int. Ed 2021, 60, 16906–16910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Grimm C; Lazzarotto M; Pompei S; Schichler J; Richter N; Farnberger JE; Fuchs M; Kroutil W Oxygen-Free Regioselective Biocatalytic Demethylation of Methyl-phenyl Ethers via Methyltransfer Employing Veratrol-O-demethylase. ACS Catal. 2020, 10, 10375–10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Davies HML; Morton D Guiding Principles for Site Selective and Stereoselective Intermolecular C-H Functionalization by Donor/Acceptor Rhodium Carbenes. Chem. Soc. Rev 2011, 40, 1857–1869. [DOI] [PubMed] [Google Scholar]
- (185).White MC Chemistry. Adding aliphatic C-H bond Oxidations to Synthesis. Science. 2012, 335, 807–809. [DOI] [PubMed] [Google Scholar]
- (186).Newhouse T; Baran PS If C-H Bonds Could Talk: Selective C-H Bond Oxidation. Angew. Chem., Int. Ed 2011, 50, 3362–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Dyker G. Transition Metal Catalyzed Coupling Reactions under C-H Activation. Angew. Chem, Int. Ed 1999, 38, 1698–1712. [DOI] [PubMed] [Google Scholar]
- (188).Lewis JC; Coelho PS; Arnold FH Enzymatic Functionalization of Carbon-hydrogen Bonds. Chem. Soc. Rev 2011, 40, 2003–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Cochrane RVK; Vederas JC Highly Selective but Multifunctional Oxygenases in Secondary Metabolism. Acc. Chem. Res 2014, 47, 3148–3161. [DOI] [PubMed] [Google Scholar]
- (190).Li F; Zhang X; Renata H Enzymatic CH Functionalizations for Natural Product Synthesis. Curr. Opin. Chem. Biol 2019, 49, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Nozaki M. Oxygenases and dioxygenases. Top Curr. Chem 1979, 78, 145–186. [DOI] [PubMed] [Google Scholar]
- (192).Denisov IG; Makris TM; Sligar SG; Schlichting I Structure and Chemistry of Cytochrome P450. Chem. Rev 2005, 105, 2253–2278. [DOI] [PubMed] [Google Scholar]
- (193).Groves JT Models and Mechanisms of Cytochrome P450 Action. In Cytochrome P450: Structure, Mechanism, and Biochemistry; Ortiz de Montellano PR, Ed.; Springer US, 2005; pp 1–43. [Google Scholar]
- (194).McIntosh JA; Farwell CC; Arnold FH Expanding P450 Catalytic Reaction Space Through Evolution and Engineering. Curr. Opin. Chem. Biol 2014, 19, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Wei Y; Ang EL; Zhao H Recent Developments in the Application of P450 Based Biocatalysts. Curr. Opin. Chem. Biol 2018, 43, 1–7. [DOI] [PubMed] [Google Scholar]
- (196).Urlacher VB; Girhard M Cytochrome P450 Monooxygenases in Biotechnology and Synthetic Biology. Trends Biotechnol. 2019. 37, 882–897. [DOI] [PubMed] [Google Scholar]
- (197).Bernhardt R. Cytochromes P450 as Versatile Biocatalysts. J. Biotechnol 2006, 124, 128–145. [DOI] [PubMed] [Google Scholar]
- (198).Ortiz de Montellano PR Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Warman AJ; Roitel O; Neeli R; Girvan HM; Seward HE; Murray SA; McLean KJ; Joyce MG; Toogood H; Holt RA; et al. Flavocytochrome P450 BM3: an Update on Structure and Mechanism of a Biotechnologically Important Enzyme. Biochem. Soc. Trans 2005, 33, 747–753. [DOI] [PubMed] [Google Scholar]
- (200).Farinas ET; Schwaneberg U; Glieder A; Arnold FH Directed Evolution of a Cytochrome P450 Monooxygenase for Alkane Oxidation. Adv. Synth. Catal 2001, 343, 601–606. [Google Scholar]
- (201).Glieder A; Farinas ET; Arnold FH Laboratory Evolution of a Soluble, Self-sufficient, Highly Active Alkane Hydroxylase. Nat. Biotechnol 2002, 20, 1135–1139. [DOI] [PubMed] [Google Scholar]
- (202).Peters MW; Meinhold P; Glieder A; Arnold FH Regio- and Enantioselective Alkane Hydroxylation with Engineered Cytochromes P450 BM-3. J. Am. Chem. Soc 2003, 125, 13442–13450. [DOI] [PubMed] [Google Scholar]
- (203).Fasan R; Chen MM; Crook NC; Arnold FH Engineered Alkane-Hydroxylating Cytochrome P450BM3 Exhibiting Nativelike Catalytic Properties. Angew. Chem., Int. Ed 2007, 46, 8414–8418. [DOI] [PubMed] [Google Scholar]
- (204).Lewis JC; Bastian S; Bennett CS; Fu Y; Mitsuda Y; Chen MM; Greenberg WA; Wong C-H; Arnold FH Chemoenzymatic Elaboration of Monosaccharides Using Engineered Cytochrome P450BM3 Demethylases. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 16550–16555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Sawayama AM; Chen MMY; Kulanthaivel P; Kuo M-S; Hemmerle H; Arnold FH A Panel of Cytochrome P450 BM3 Variants to Produce Drug Metabolites and Diversify Lead Compounds. Chem. Eur. J 2009, 15, 11723–11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Rentmeister A; Arnold FH; Fasan R Chemo-enzymatic Fluorination of Unactivated Organic Compounds. Nat. Chem. Biol 2009, 5, 26–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Wrighton SA; Stevens JC The Human Hepatic Cytochromes P450 Involved in Drug Metabolism. Crit. Rev. Toxicol 1992, 22, 1–21. [DOI] [PubMed] [Google Scholar]
- (208).Lewis JC; Mantovani SM; Fu Y; Snow CD; Komor RS; Wong C-H; Arnold FH Combinatorial Alanine Substitution Enables Rapid Optimization of Cytochrome P450BM3 for Selective Hydroxylation of Large Substrates. ChemBioChem. 2010, 11, 2502–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Acevedo-Rocha CG; Gamble CG; Lonsdale R; Li A; Nett N; Hoebenreich S; Lingnau JB; Wirtz C; Fares C; Hinrichs H; et al. P450-Catalyzed Regio- and Diastereoselective Steroid Hydroxylation: Efficient Directed Evolution Enabled by Mutability Landscaping. ACS Catal. 2018, 8, 3395–3410. [Google Scholar]
- (210).Li A; Acevedo-Rocha CG; D’Amore L; Chen J; Peng Y; Garcia-Borràs M; Gao C; Zhu J; Rickerby H; Osuna S; et al. Regio- and Stereoselective Steroid Hydroxylation at C7 by Cytochrome P450 Monooxygenase Mutants. Angew. Chem., Int. Ed 59, 12499–12505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Peng Y; Gao C; Zhang Z; Wu S; Zhao J; Li A A Chemoenzymatic Strategy for the Synthesis of Steroid Drugs Enabled by P450 Monooxygenase-Mediated Steroidal Core Modification. ACS Catal. 2022, 12, 2907–2914. [Google Scholar]
- (212).Zhang X; Shen P; Zhao J; Chen Y; Li X; Huang J-W; Zhang L; Li Q; Gao C; Xing Q; et al. Rationally Controlling Selective Steroid Hydroxylation via Scaffold Sampling of a P450 Family. ACS Catal. 2023, 13, 1280–1289. [Google Scholar]
- (213).Kille S; Zilly FE; Acevedo JP; Reetz MT Regio- and Stereoselectivity of P450-Catalysed Hydroxylation of Steroids Controlled by Laboratory Evolution. Nat. Chem 2011, 3, 738–743. [DOI] [PubMed] [Google Scholar]
- (214).Nguyen KT; Virus C; Gunnewich N; Hannemann F; Bernhardt R Changing the Regioselectivity of a P450 from C15 to C11 Hydroxylation of Progesterone. ChemBioChem. 2012, 13, 1161–1166. [DOI] [PubMed] [Google Scholar]
- (215).Whitehouse CJC; Bell SG; Tufton HG; Kenny RJP; Ogilvie LCI; Wong L-L Evolved CYP102A1 (P450BM3) Variants Oxidise a Range of Non-natural Substrates and Offer New Selectivity Options. Chem. Commun 2008, 966–968. [DOI] [PubMed] [Google Scholar]
- (216).Wu X; Chen Y; Wang X; Wei W; Liang Y Origin of Site Selectivity in Toluene Hydroxylation by Cytochrome P450 Enzymes. J. Org. Chem 2021, 86, 13768–13773. [DOI] [PubMed] [Google Scholar]
- (217).Agudo R; Roiban G-D; Reetz MT Achieving Regio- and Enantioselectivity of P450-Catalyzed Oxidative CH Activation of Small Functionalized Molecules by Structure-Guided Directed Evolution. ChemBioChem. 2012, 13, 1465–1473. [DOI] [PubMed] [Google Scholar]
- (218).Zhang K; El Damaty S; Fasan R P450 Fingerprinting Method for Rapid Discovery of Terpene Hydroxylating P450 Catalysts with Diversified Regioselectivity. J. Am. Chem. Soc 2011, 133, 3242–3245. [DOI] [PubMed] [Google Scholar]
- (219).Zhang K; Shafer BM; Demars MD II; Stern HA; Fasan R Controlled Oxidation of Remote sp3 C-H Bonds in Artemisinin via P450 Catalysts with Fine-Tuned Regio- and Stereoselectivity. J.Am. Chem. Soc 2012, 134, 18695–18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Tyagi V; Alwaseem H; O’Dwyer KM; Ponder J; Li QY; Jordan CT; Fasan R Chemoenzymatic Synthesis and Antileukemic Activity of Novel C9- and C14-functionalized Parthenolide Analogs. Bioorg. Med. Chem 2016, 24, 3876–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Alwaseem H; Frisch BJ; Fasan R Anticancer Activity Profiling of Parthenolide Analogs Generated via P450-mediated Chemoenzymatic Synthesis. Bioorg. Med. Chem 2018, 26, 1365–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (222).Schmitz D; Zapp J; Bernhardt R Steroid Conversion with CYP106A2 - Production of Pharmaceutically Interesting DHEA Metabolites. Microb. Cell Fact 2014, 13, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Xue Y; Zhao L; Liu H.-w.; Sherman DH A Gene Cluster for Macrolide Antibiotic Biosynthesis in Streptomyces Venezuelae: Architecture of Metabolic Diversity. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 12111–12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Sherman DH; Li S; Yermalitskaya LV; Kim Y; Smith JA; Waterman MR; Podust LM The Structural Basis for Substrate Anchoring, Active Site Selectivity, and Product Formation by P450 PikC from Streptomyces venezuelae. J. Biol. Chem 2006, 281, 26289–26297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Li S; Ouellet H; Sherman DH; Podust LM Analysis of Transient and Catalytic Desosamine-binding Pockets in Cytochrome P-450 PikC from Streptomyces venezuelae. J. Biol. Chem 2009, 284, 5723–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Negretti S; Narayan ARH; Chiou KC; Kells PM; Stachowski JL; Hansen DA; Podust LM; Montgomery J; Sherman DH Directing Group-Controlled Regioselectivity in an Enzymatic C-H Bond Oxygenation. J. Am. Chem. Soc 2014, 136, 4901–4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).de Visser SP; Shaik S A Proton-Shuttle Mechanism Mediated by the Porphyrin in Benzene Hydroxylation by Cytochrome P450 Enzymes. J. Am. Chem. Soc 2003, 125, 7413–7424. [DOI] [PubMed] [Google Scholar]
- (228).Asaka M; Fujii H Participation of Electron Transfer Process in Rate-Limiting Step of Aromatic Hydroxylation Reactions by Compound I Models of Heme Enzymes. J. Am. Chem. Soc 2016, 138, 8048–8051. [DOI] [PubMed] [Google Scholar]
- (229).Stok JE; Chow S; Krenske EH; Farfan Soto C; Matyas C; Poirier RA; Williams CM; De Voss JJ Direct Observation of an Oxepin from a Bacterial Cytochrome P450-Catalyzed Oxidation. Chem. Eur. J 2016, 22, 4408–4412. [DOI] [PubMed] [Google Scholar]
- (230).Dennig A; Marienhagen J; Ruff AJ; Guddat L; Schwaneberg U Directed Evolution of P 450 BM 3 into a p-Xylene Hydroxylase. ChemCatChem. 2012, 4, 771–773. [Google Scholar]
- (231).Dennig A; Lulsdorf N; Liu H; Schwaneberg U Regioselective o-Hydroxylation of Monosubstituted Benzenes by P450 BM3. Angew. Chem., Int. Ed 2013, 52, 8459–8462. [DOI] [PubMed] [Google Scholar]
- (232).Coleman T; Lee JZH; Kirk AM; Doherty DZ; Podgorski MN; Pinidiya DK; Bruning JB; De Voss JJ; Krenske EH; Bell SG Enabling Aromatic Hydroxylation in a Cytochrome P450 Monooxygenase Enzyme through Protein Engineering. Chem. Eur. J 2022, 28, No. e202201895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Martinez S; Hausinger RP Catalytic Mechanisms of Fe(II)- and 2-Oxoglutarate-dependent Oxygenases. J. Biol. Chem 2015, 290, 20702–20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Klein C; Hüttel W A Simple Procedure for Selective Hydroxylation of L-Proline and L-Pipecolic Acid with Recombinantly Expressed Proline Hydroxylases. Adv. Synth. Catal 2011, 353, 1375–1383. [Google Scholar]
- (235).Hibi M; Mori R; Miyake R; Kawabata H; Kozono S; Takahashi S; Ogawa J Novel Enzyme Family Found in Filamentous Fungi Catalyzing trans-4-Hydroxylation of L-Pipecolic Acid. Appl. Environ. Microbiol 2016, 82, 2070–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Mattay J; Huttel W Pipecolic Acid Hydroxylases: A Monophyletic Clade Among cis-Selective Bacterial Proline Hydroxylases that Discriminates l-Proline. ChemBioChem. 2017, 18, 1523–1528. [DOI] [PubMed] [Google Scholar]
- (237).Chen HB; Bong YK; Cabirol FL; Gohel Prafulchandra A; Li T; Moore JC; Quintanar-Audelo M; Hong Y; Collier SJ; Smith D Biocatalysts and Methods for Hydroxylation of Chemical Compounds. US Patent 2015/0118719A1, 2015.
- (238).Binz TM; Maffioli SI; Sosio M; Donadio S; Müller R Insights into an Unusual Nonribosomal Peptide Synthetase Biosynthesis: Insights into an Unusual Nonribosomal Peptide Synthetase. J. Biol. Chem 2010, 285, 32710–32719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Zwick III CR; Sosa MB; Renata H Characterization of a Citrulline 4-Hydroxylase from Nonribosomal Peptide GE81112 Biosynthesis and Engineering of Its Substrate Specificity for the Chemoenzymatic Synthesis of Enduracididine. Angew. Chem., Int. Ed 2019, 58, 18854–18858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Baud D; Saaidi P-L; Monfleur A; Harari M; Cuccaro J; Fossey A; Besnard M; Debard A; Mariage A; Pellouin V; et al. Synthesis of Mono- and Dihydroxylated Amino Acids with New α-Ketoglutarate-Dependent Dioxygenases: Biocatalytic Oxidation of C-H Bonds. ChemCatChem. 2014, 6, 3012–3017. [Google Scholar]
- (241).Baud D; Peruch O; Saaidi P-L; Fossey A; Mariage A; Petit J-L; Salanoubat M; Vergne-Vaxelaire C; de Berardinis V; Zaparucha A Biocatalytic Approaches towards the Synthesis of Chiral Amino Alcohols from Lysine: Cascade Reactions Combining alpha-Keto Acid Oxygenase Hydroxylation with Pyridoxal Phosphate-Dependent Decarboxylation. Adv. Synth. Catal 2017, 359, 1563–1569. [Google Scholar]
- (242).Amatuni A; Renata H Identification of a Lysine 4-Hydroxylase from the Glidobactin Biosynthesis and Evaluation of its Biocatalytic Potential. Org. Biomol. Chem 2019, 17, 1736–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Chan NH; Gomez CA; Vennelakanti V; Du Q; Kulik HJ; Lewis JC Non-Native Anionic Ligand Binding and Reactivity in Engineered Variants of the Fe(II)- and α-Ketoglutarate-Dependent Oxygenase, SadA. Inorg. Chem 2022, 61, 14477–14485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Beltrán-Nogal A; Sánchez-Moreno I; Mández-Sánchez D; Gómez de Santos P; Hollmann F; Alcalde M Surfing the Wave of Oxyfunctionalization Chemistry by Engineering Fungal Unspecific Peroxygenases. Curr. Opin. Struct. Biol 2022, 73, 102342. [DOI] [PubMed] [Google Scholar]
- (245).Hobisch M; Holtmann D; Gomez de Santos P; Alcalde M; Hollmann F; Kara S Recent Developments in the Use of Peroxygenases - Exploring their High Potential in Selective Oxyfunctionalisations. Biotechnol. Adv 2021, 51, 107615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Molina-Espeja P; Cañellas M; Plou FJ; Hofrichter M; Lucas F; Guallar V; Alcalde M Synthesis of 1-Naphthol by a Natural Peroxygenase Engineered by Directed Evolution ChemBioChem. 2016, 17, 341–349. [DOI] [PubMed] [Google Scholar]
- (247).Peter S; Kinne M; Wang X; Ullrich R; Kayser G; Groves JT; Hofrichter M Selective Hydroxylation of Alkanes by an Extracellular Fungal Peroxygenase. FEBS J. 2011, 278, 3667–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Barková K; Kinne M; Ullrich R; Hennig L; Fuchs A; Hofrichter M Regioselective Hydroxylation of Diverse Flavonoids by an Aromatic Peroxygenase. Tetrahedron 2011, 67, 4874–4878. [Google Scholar]
- (249).Gomez de Santos P; González-Benjumea A; Fernandez-Garcia A; Aranda C; Wu Y; But A; Molina-Espeja P; Maté DM; Gonzalez-Perez D; Zhang W; et al. Engineering a Highly Regioselective Fungal Peroxygenase for the Synthesis of Hydroxy Fatty Acids. Angew. Chem., Int. Ed 2023, 62, No. e202217372. [DOI] [PubMed] [Google Scholar]
- (250).Barry SM; Challis GL Mechanism and Catalytic Diversity of Rieske Non-Heme Iron-Dependent Oxygenases. ACS Catal. 2013, 3, 2362–2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Gally C; Nestl BM; Hauer B Engineering Rieske Non-Heme Iron Oxygenases for the Asymmetric Dihydroxylation of Alkenes. Angew. Chem., Int. Ed 2015, 54, 12952–12956. [DOI] [PubMed] [Google Scholar]
- (252).Lukowski AL; Ellinwood DC; Hinze ME; DeLuca RJ; Du Bois J; Hall S; Narayan ARH C-H Hydroxylation in Paralytic Shellfish Toxin Biosynthesis. J. Am. Chem. Soc 2018, 140, 11863–11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Lukowski AL; Liu J; Bridwell-Rabb J; Narayan ARH Structural Basis for Divergent C-H Hydroxylation Selectivity in two Rieske Oxygenases. Nat. Commun 2020, 11, 2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Lippard SJ Hydroxylation of C-H Bonds at Carboxylate-bridged Diiron Centres. Philos. trans., Math. phys. eng. sci 2005, 363, 861–877. [DOI] [PubMed] [Google Scholar]
- (255).Mitchell KH; Studts JM; Fox BG Combined Participation of Hydroxylase Active Site Residues and Effector Protein Binding in a Para to Ortho Modulation of Toluene 4-Monooxygenase Regiospecificity. Biochem. 2002, 41 , 3176–3188. [DOI] [PubMed] [Google Scholar]
- (256).Colby J; Stirling DI; Dalton H The Soluble Methane Mono-oxygenase of Methylococcus capsulatus (Bath). Its ability to Oxygenate n-Alkanes, n-Alkenes, Ethers, and Alicyclic, Aromatic and Heterocyclic Compounds. Biochem. J 1977, 165, 395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Green J; Dalton H Substrate Specificity of Soluble Methane Monooxygenase. Mechanistic Implications. J. Biol. Chem 1989, 264, 17698–17703. [PubMed] [Google Scholar]
- (258).Ross MO; Rosenzweig AC A tale of Two Methane Monooxygenases. J. Biol. Inorg. Chem 2017, 22, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Elliott SJ; Zhu M; Tso L; Nguyen HHT; Yip JHK; Chan SI Regio- and Stereoselectivity of Particulate Methane Monooxygenase from Methylococcus capsulatus (Bath). J. Am. Chem. Soc 1997, 119, 9949–9955. [Google Scholar]
- (260).Banerjee R; Jones JC; Lipscomb JD Soluble Methane Monooxygenase. Annu. Rev. Biochem 2019, 88, 409–431. [DOI] [PubMed] [Google Scholar]
- (261).Peng W; Qu X; Shaik S; Wang B Deciphering the Oxygen Activation Mechanism at the CuC Site of Particulate Methane Monooxygenase. Nat. Catal 2021, 4, 266–273. [Google Scholar]
- (262).Green J; Dalton H Substrate specificity of soluble methane monooxygenase: Mechanistic implications. J. Biol. Chem 1989, 264, 17698–17703. [PubMed] [Google Scholar]
- (263).Chenprakhon P; Wongnate T; Chaiyen P Monooxygenation of Aromatic Compounds by Flavin-dependent Monooxygenases. Protein Sci. 2019, 28, 8–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Reis RAG; Li H; Johnson M; Sobrado P New Frontiers in Flavin-dependent Monooxygenases. Arch. Biochem. Biophys 2021, 699, 108765. [DOI] [PubMed] [Google Scholar]
- (265).Jadan AP; Moonen MJH; Boeren S; Golovleva LA; Rietjens IMCM; van Berkel WJH Biocatalytic Potential of p-Hydroxybenzoate Hydroxylase from Rhodococcus rhodnii 135 and Rhodococcus opacus 557. Adv. Synth. Catal 2004, 346, 367–375. [Google Scholar]
- (266).Meyer A; Schmid A; Held M; Westphal AH; Rothlisberger M; Kohler H-PE; van Berkel WJH; Witholt, B. Changing the Substrate Reactivity of 2-Hydroxybiphenyl 3-Monooxygenase from Pseudomonas azelaica HBP1 by Directed Evolution. J. Biol. Chem 2002, 277, 5575–5582. [DOI] [PubMed] [Google Scholar]
- (267).Bregman-Cohen A; Deri B; Maimon S; Pazy Y; Fishman A Altering 2-Hydroxybiphenyl 3-Monooxygenase Regioselectivity by Protein Engineering for the Production of a New Antioxidant. ChemBioChem. 2018, 19, 583–590. [DOI] [PubMed] [Google Scholar]
- (268).Chaiyen P; Suadee C; Wilairat P A Novel two-protein Component Flavoprotein Hydroxylase. Eur. J. Biochem 2001, 268, 5550–5561. [DOI] [PubMed] [Google Scholar]
- (269).Dhammaraj T; Pinthong C; Visitsatthawong S; Tongsook C; Surawatanawong P; Chaiyen P A Single-Site Mutation at Ser146 Expands the Reactivity of the Oxygenase Component of p-Hydroxyphenylacetate 3-Hydroxylase. ACS Chem. Biol 2016, 11, 2889–2896. [DOI] [PubMed] [Google Scholar]
- (270).Furuya T; Kino K Catalytic Activity of the Two-component Flavin-dependent Monooxygenase from Pseudomonas Aeruginosa Toward Cinnamic Acid Derivatives. Appl. Microbiol. Biotechnol 2014, 98, 1145–1154. [DOI] [PubMed] [Google Scholar]
- (271).Furuya T; Kino K Regioselective Synthesis of Piceatannol from Resveratrol: Catalysis by Two-component Flavin-dependent Monooxygenase HpaBC in Whole Cells. Tetrahedron Lett. 2014, 55, 2853–2855. [Google Scholar]
- (272).Deng Y; Faivre B; Back O; Lombard M; Pecqueur L; Fontecave M Structural and Functional Characterization of 4-Hydroxyphenylacetate 3-Hydroxylase from Escherichia coli. ChemBioChem. 2020, 21, 163–170. [DOI] [PubMed] [Google Scholar]
- (273).Herrmann S; Dippe M; Pecher P; Funke E; Pietzsch M; Wessjohann LA Engineered Bacterial Flavin-Dependent Monooxygenases for the Regiospecific Hydroxylation of Polycyclic Phenols. ChemBioChem. 2022, 23, No. e202100480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Abood A; Al-Fahad A; Scott A; Hosny AE-DMS; Hashem AM; Fattah AMA; Race PR; Simpson TJ; Cox RJ Kinetic characterisation of the FAD Dependent Monooxygenase TropB and Investigation of its Biotransformation Potential. RSC Adv. 2015, 5, 49987–49995. [Google Scholar]
- (275).Zabala AO; Xu W; Chooi Y-H; Tang Y Characterization of a Silent Azaphilone Gene Cluster from Aspergillus niger ATCC 1015 Reveals a Hydroxylation-Mediated Pyran-Ring Formation. Chem. Biol 2012, 19, 1049–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Fahad A. a.; Abood A; Fisch KM; Osipow A; Davison J; Avramović M; Butts CP; Piel J; Simpson TJ; Cox RJ Oxidative Dearomatisation: the Key Step of Sorbicillinoid Biosynthesis. Chem. Sci 2014, 5, 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Baker Dockrey SA; Lukowski AL; Becker MR; Narayan ARH Biocatalytic Site- and Enantioselective Oxidative Dearomatization of Phenols. Nat. Chem 2018, 10, 119–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Dockrey SAB; Suh CE; Benítez AR; Wymore T; Brooks CL III; Narayan ARH Positioning-Group-Enabled Biocatalytic Oxidative Dearomatization. ACS Cent. Sci 2019, 5, 1010–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Hager LP; Morris DR; Brown FS; Eberwein H Chloroperoxidase. II. Utilization of Halogen Anions. J. Biol. Chem 1966, 241, 1769–1777. [PubMed] [Google Scholar]
- (280).Getrey L; Krieg T; Hollmann F; Schrader J; Holtmann D Enzymatic Halogenation of The Phenolic Monoterpenes Thymol and Carvacrol with Chloroperoxidase. Green Chem. 2014, 16, 1104–1108. [Google Scholar]
- (281).Fernández-Fueyo E; van Wingerden M; Renirie R; Wever R; Ni Y; Holtmann D; Hollmann F Chemoenzymatic Halogenation of Phenols by using the Haloperoxidase from Curvularia inaequalis. ChemCatChem. 2015, 7, 4035–4038. [Google Scholar]
- (282).Keller S; Wage T; Hohaus K; Hölzer M; Eichhorn E; van Pée K-H Purification and Partial Characterization of Tryptophan 7-Halogenase (PrnA) from Pseudomonas fluorescens. Angew. Chem., Int. Ed 2000, 39, 2300–2302. [DOI] [PubMed] [Google Scholar]
- (283).Seibold C; Schnerr H; Rumpf J; Kunzendorf A; Hatscher C; Wage T; Ernyei AJ; Dong C; Naismith JH; Van Pée K-H A Flavin-dependent Tryptophan 6-halogenase and its Use in Modification of Pyrrolnitrin Biosynthesis. Biocatal. Biotransformation 2006, 24, 401–408. [Google Scholar]
- (284).Heemstra JR Jr; Walsh CT Tandem Action of the O2- and FADH2-Dependent Halogenases KtzQ and KtzR Produce 6,7-Dichlorotryptophan for Kutzneride Assembly. J. Am. Chem. Soc 2008, 130, 14024–14025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Zehner S; Kotzsch A; Bister B; Süssmuth RD; Méndez C; Salas JA; van Pee K-H A Regioselective Tryptophan 5-Halogenase is Involved in Pyrroindomycin Biosynthesis in Streptomyces rugosporus LL-42D005. Chem. Biol 2005, 12, 445–452. [DOI] [PubMed] [Google Scholar]
- (286).Foulston LC; Bibb MJ Microbisporicin Gene Cluster Reveals Unusual Features of lantibiotic Biosynthesis in Actinomycetes. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 13461–13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Wynands I; van Pee K-H A Novel Halogenase Gene from the Pentachloropseudilin Producer Actinoplanes sp. ATCC 33002 and Detection of in vitro Halogenase Activity. FEMS Microbiol. Lett 2004, 237, 363–367. [DOI] [PubMed] [Google Scholar]
- (288).Dorrestein PC; Yeh E; Garneau-Tsodikova S; Kelleher NL; Walsh CT Dichlorination of a Pyrrolyl-S-carrier Protein by FADH2-dependent Halogenase PltA During Pyoluteorin Biosynthesis. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 13843–13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Heide L; Westrich L; Anderle C; Gust B; Kammerer B; Piel J Use of a Halogenase of Hormaomycin Biosynthesis for Formation of New Clorobiocin Analogues with 5-Chloropyrrole Moieties. ChemBioChem. 2008, 9, 1992–1999. [DOI] [PubMed] [Google Scholar]
- (290).Yu T-W; Bai L; Clade D; Hoffmann D; Toelzer S; Trinh KQ; Xu J; Moss SJ; Leistner E; Floss HG The Biosynthetic Gene Cluster of the Maytansinoid Antitumor Agent Ansamitocin from Actinosynnema Pretiosum. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 7968–7973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Lin S; Van Lanen SG; Shen B Regiospecific Chlorination of (S)-β-Tyrosyl-S-Carrier Protein Catalyzed by SgcC3 in the Biosynthesis of the Enediyne Antitumor Antibiotic C-1027. J. Am. Chem. Soc 2007, 129, 12432–12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Wang S; Xu Y; Maine EA; Wijeratne EMK; Espinosa-Artiles P; Gunatilaka AAL; Molnár I Functional Characterization of the Biosynthesis of Radicicol, an Hsp90 Inhibitor Resorcylic Acid Lactone from Chaetomium chiversii. Chem. Biol 2008, 15, 1328–1338. [DOI] [PubMed] [Google Scholar]
- (293).Buedenbender S; Rachid S; Müller R; Schulz GE Structure and Action of the Myxobacterial Chondrochloren Halogenase CndH: A New Variant of FAD-dependent Halogenases. J. Mol. Biol 2009, 385, 520–530. [DOI] [PubMed] [Google Scholar]
- (294).Podzelinska K; Latimer R; Bhattacharya A; Vining LC; Zechel DL; Jia Z Chloramphenicol Biosynthesis: The Structure of CmlS, a Flavin-Dependent Halogenase Showing a Covalent Flavin-Aspartate Bond. J. Mol. Biol 2010, 397, 316–331. [DOI] [PubMed] [Google Scholar]
- (295).Agarwal V; Blanton JM; Podell S; Taton A; Schorn MA; Busch J; Lin Z; Schmidt EW; Jensen PR; Paul VJ; et al. Metagenomic Discovery of Polybrominated Diphenyl Ether Biosynthesis by Marine Sponges. Nat. Chem. Biol 2017, 13, 537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Agarwal V; Miles ZD; Winter JM; Eustáquio AS; El Gamal AA; Moore BS Enzymatic Halogenation and Dehalogenation Reactions: Pervasive and Mechanistically Diverse. Chem. Rev 2017, 117, 5619–5674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Latham J; Brandenburger E; Shepherd SA; Menon BRK; Micklefield J Development of Halogenase Enzymes for Use in Synthesis. Chem. Rev 2018, 118, 232–269. [DOI] [PubMed] [Google Scholar]
- (298).Gkotsi DS; Dhaliwal J; McLachlan MMW; Mulholand KR; Goss RJM Halogenases: Powerful Tools for Biocatalysis (Mechanisms Applications and Scope). Curr. Opin. Chem. Biol 2018, 43, 119–126. [DOI] [PubMed] [Google Scholar]
- (299).Weichold V; Milbredt D; van Pée K-H Specific Enzymatic Halogenation—From the Discovery of Halogenated Enzymes to Their Applications In Vitro and In Vivo. Angew. Chem., Int. Ed 2016, 55, 6374–6389. [DOI] [PubMed] [Google Scholar]
- (300).Yeh E; Cole LJ; Barr EW; Bollinger JM; Ballou DP; Walsh CT Flavin Redox Chemistry Precedes Substrate Chlorination during the Reaction of the Flavin-Dependent Halogenase RebH. Biochemistry 2006, 45, 7904–7912. [DOI] [PubMed] [Google Scholar]
- (301).Yeh E; Garneau S; Walsh CT Robust in vitro Activity of RebF and RebH, a Two-component Reductase/Halogenase, Generating 7-Chlorotryptophan During Rebeccamycin Biosynthesis. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 3960–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Dong C; Flecks S; Unversucht S; Haupt C; van Pée K-H; Naismith JH Tryptophan 7-Halogenase (PrnA) Structure Suggests a Mechanism for Regioselective Chlorination. Science. 2005, 309, 2216–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (303).Flecks S; Patallo EP; Zhu X; Ernyei AJ; Seifert G; Schneider A; Dong C; Naismith JH; van Pée K-H New Insights into the Mechanism of Enzymatic Chlorination of Tryptophan. Angew. Chem., Int. Ed 2008, 47, 9533–9536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (304).Karabencheva-Christova TG; Torras J; Mulholland AJ; Lodola A; Christov CZ Mechanistic Insights into the Reaction of Chlorination of Tryptophan Catalyzed by Tryptophan 7-Halogenase. Sci. Rep 2017, 7, 17395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Andorfer MC; Evans D; Yang S; He CQ; Girlich AM; Vergara-Coll J; Sukumar N; Houk KN; Lewis JC Analysis of Laboratory-evolved Flavin-dependent Halogenases Affords a Computational Model for Predicting Halogenase Site Selectivity. Chem. Catal 2022, 2, 2658–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Zhu X; De Laurentis W; Leang K; Herrmann J; Ihlefeld K; van Pée K-H; Naismith JH Structural Insights into Regioselectivity in the Enzymatic Chlorination of Tryptophan. J. Mol. Biol 2009, 391, 74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Phintha A; Prakinee K; Chaiyen P Structures, Mechanisms and Applications of Flavin-dependent Halogenases. In The Enzymes; Chaiyen P, Tamanoi F, Eds.; Academic Press, 2020; Vol. 47, Chapter 11, pp 327–364. [DOI] [PubMed] [Google Scholar]
- (308).Sánchez C; Zhu L; Braña AF; Salas AP; Rohr J; Méndez C; Salas JA Combinatorial Biosynthesis of Antitumor Indolocarbazole Compounds. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (309).Runguphan W; Qu X; O’Connor SE Integrating Carbonhalogen Bond Formation into Medicinal Plant Metabolism. Nature. 2010, 468, 461–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (310).Gray KA; Richardson TH; Kretz K; Short JM; Bartnek F; Knowles R; Kan L; Swanson PE; Robertson DE Rapid Evolution of Reversible Denaturation and Elevated Melting Temperature in a Microbial Haloalkane Dehalogenase. Adv. Synth. Catal 2001, 343, 607–617. [Google Scholar]
- (311).Glenn WS; Nims E; O’Connor SE Reengineering a Tryptophan Halogenase To Preferentially Chlorinate a Direct Alkaloid Precursor. J. Am. Chem. Soc 2011, 133, 19346–19349. [DOI] [PubMed] [Google Scholar]
- (312).Payne JT; Andorfer MC; Lewis JC Regioselective Arene Halogenation using the FAD-Dependent Halogenase RebH. Angew. Chem., Int. Ed 2013, 52, 5271–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (313).Zeng J; Lytle AK; Gage D; Johnson SJ; Zhan J Specific Chlorination of Isoquinolines by a Fungal Flavin-dependent Halogenase. Bioorg. Med. Chem. Lett 2013, 23, 1001–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (314).Frese M; Guzowska PH; Voß H; Sewald N Regioselective Enzymatic Halogenation of Substituted Tryptophan Derivatives Using the FAD-Dependent Halogenase RebH. ChemCatChem. 2014, 6, 1270–1276. [Google Scholar]
- (315).Gkotsi DS; Ludewig H; Sharma SV; Connolly JA; Dhaliwal J; Wang Y; Unsworth WP; Taylor RJK; McLachlan MMW; Shanahan S; et al. A Marine Viral Halogenase that Iodinates Diverse Substrates. Nat. Chem 2019, 11, 1091–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Smith DRM; Uria AR; Helfrich EJN; Milbredt D; van Pée K-H; Piel J; Goss RJM An Unusual Flavin-Dependent Halogenase from the Metagenome of the Marine Sponge Theonella swinhoei WA. ACS Chem. Biol 2017, 12, 1281–1287. [DOI] [PubMed] [Google Scholar]
- (317).Patallo EP; Walter A; Milbredt D; Thomas M; Neumann M; Caputi L; O’Connor S; Ludwig-Müller J; van Pée K-H Strategies to Produce Chlorinated Indole-3-Acetic Acid and Indole-3-Acetic Acid Intermediates. ChemistrySelect. 2017, 2, 11148–11153. [Google Scholar]
- (318).Poor CB; Andorfer MC; Lewis JC Improving the Stability and Catalyst Lifetime of the Halogenase RebH By Directed Evolution. ChemBioChem. 2014, 15, 1286–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (319).Payne JT; Poor CB; Lewis JC Directed Evolution of RebH for Site-Selective Halogenation of Large Biologically Active Molecules. Angew. Chem., Int. Ed 2015, 54, 4226–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Payne JT; Butkovich PH; Gu Y; Kunze KN; Park HJ; Wang D-S; Lewis JC Enantioselective Desymmetrization of Methylenedianilines via Enzyme-Catalyzed Remote Halogenation. J. Am. Chem. Soc 2018, 140, 546–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Metrano AJ; Miller SJ Peptide-Based Catalysts Reach the Outer Sphere through Remote Desymmetrization and Atroposelectivity. Acc. Chem. Res 2019, 52, 199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Snodgrass HM; Mondal D; Lewis JC Directed Evolution of Flavin-Dependent Halogenases for Site- and Atroposelective Halogenation of 3-Aryl-4(3H)-Quinazolinones via Kinetic or Dynamic Kinetic Resolution. J. Am. Chem. Soc 2022, 144, 16676–16682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Lang A; Polnick S; Nicke T; William P; Patallo EP; Naismith JH; van Pee K-H Changing the Regioselectivity of the Tryptophan 7-Halogenase PrnA by Site-Directed Mutagenesis. Angew. Chem., Int. Ed 2011, 50, 2951–2953. [DOI] [PubMed] [Google Scholar]
- (324).Shepherd SA; Karthikeyan C; Latham J; Struck A-W; Thompson ML; Menon BRK; Styles MQ; Levy C; Leys D; Micklefield J Extending the Biocatalytic Scope of Regiocomplementary Flavin-dependent Halogenase Enzymes. Chem. Sci 2015, 6, 3454–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Shepherd SA; Menon BRK; Fisk H; Struck A-W; Levy C; Leys D; Micklefield J A Structure-Guided Switch in the Regioselectivity of a Tryptophan Halogenase. ChemBioChem. 2016, 17, 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (326).Moritzer A-C; Minges H; Prior T; Frese M; Sewald N; Niemann HH Structure-based Switch of Regioselectivity in the Flavin-Dependent Tryptophan 6-halogenase Thal. J. Biol. Chem 2019, 294, 2529–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (327).Fraley AE; Garcia-Borràs M; Tripathi A; Khare D; Mercado-Marin EV; Tran H; Dan Q; Webb GP; Watts KR; Crews P; et al. Function and Structure of MalA/MalA′, Iterative Halogenases for Late-Stage C-H Functionalization of Indole Alkaloids. J. Am. Chem. Soc. 2017, 139, 12060–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (328).Andorfer MC; Park HJ; Vergara-Coll J; Lewis JC Directed Evolution of RebH for Catalyst-controlled Halogenation of Indole C-H Bonds. Chem. Sci 2016, 7, 3720–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (329).Andorfer MC; Grob JE; Hajdin CE; Chael JR; Siuti P; Lilly J; Tan KL; Lewis JC Understanding Flavin-Dependent Halogenase Reactivity via Substrate Activity Profiling. ACS Catal. 2017, 7, 1897–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (330).Neubauer PR; Widmann C; Wibberg D; Schröder L; Frese M; Kottke T; Kalinowski J; Niemann HH; Sewald N A Flavin-dependent Halogenase from Metagenomic Analysis Prefers Bromination Over Chlorination. PLoS One. 2018, 13, No. e0196797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (331).Fisher BF; Snodgrass HM; Jones KA; Andorfer MC; Lewis JC Site-Selective C-H Halogenation Using Flavin-Dependent Halogenases Identified via Family-Wide Activity Profiling. ACS Cent. Sci 2019, 5, 1844–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Agarwal V; El Gamal AA; Yamanaka K; Poth D; Kersten RD; Schorn M; Allen EE; Moore BS Biosynthesis of Polybrominated Aromatic Organic Compounds by Marine Bacteria. Nat. Chem. Biol 2014, 10, 640–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (333).Breinlinger S; Phillips TJ; Haram BN; Mareš J; Martínez Yerena JA; Hrouzek P; Sobotka R; Henderson WM; Schmieder P; Williams SM; et al. Hunting the Eagle Killer: A Cyanobacterial Neurotoxin Causes Vacuolar Myelinopathy. Science 2021, 371, No. eaax9050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (334).Adak S; Lukowski AL; Schäfer RJB; Moore BS From Tryptophan to Toxin: Nature’s Convergent Biosynthetic Strategy to Aetokthonotoxin. J. Am. Chem. Soc 2022, 144, 2861–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (335).Jiang Y; Snodgrass HM; Zubi YS; Roof CV; Guan Y; Mondal D; Honeycutt NH; Lee JW; Lewis RD; Martinez CA; et al. The Single-Component Flavin Reductase/Flavin-Dependent Halogenase AetF is a Versatile Catalyst for Selective Bromination and Iodination of Arenes and Olefins. Angew. Chem., Int. Ed 2022, 61, No. e202214610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (336).Mori S; Pang AH; Thamban Chandrika N; Garneau-Tsodikova S; Tsodikov OV Unusual Substrate and Halide Versatility of Phenolic Halogenase PltM. Nat. Commun 2019, 10, 1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (337).Zhang Y; Chen L; Chen H; Huang T; Shi Q; Wang X; Wang Y; Tang M-C; Zhou N-Y; Lin S Aryl C-H Iodination: are There Actual Flavin-dependent Iodinases in Nature? Sci. China Chem 2021, 64, 1730–1735. [Google Scholar]
- (338).Hayashi T; Ligibel M; Sager E; Voss M; Hunziker J; Schroer K; Snajdrova R; Buller R Evolved Aliphatic Halogenases Enable Regiocomplementary C-H Functionalization of a Pharmaceutically Relevant Compound. Angew. Chem., Int. Ed 2019, 58, 18535–18539. [DOI] [PubMed] [Google Scholar]
- (339).Ueki M; Galonić DP; Vaillancourt FH; Garneau-Tsodikova S; Yeh E; Vosburg DA; Schroeder FC; Osada H; Walsh CT Enzymatic Generation of the Antimetabolite γ,γ-Dichloroaminobutyrate by NRPS and Mononuclear Iron Halogenase Action in a Streptomycete. Chem. Biol 2006, 13, 1183–1191. [DOI] [PubMed] [Google Scholar]
- (340).Vaillancourt FH; Yin J; Walsh CT SyrB2 in Syringomycin E Biosynthesis is a Nonheme FeII Alpha-Ketoglutarate- and O2-Dependent Halogenase. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 10111–10116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (341).Vaillancourt FH; Yeh E; Vosburg DA; O’Connor SE; Walsh CT Cryptic Chlorination by a Non-haem Iron Enzyme During Cyclopropyl Amino acid Biosynthesis. Nature. 2005, 436, 1191–1194. [DOI] [PubMed] [Google Scholar]
- (342).Hillwig ML; Liu X A New Family of Iron-Dependent Halogenases Acts on Freestanding Substrates. Nat. Chem. Biol 2014, 10, 921–923. [DOI] [PubMed] [Google Scholar]
- (343).Hillwig ML; Zhu Q; Ittiamornkul K; Liu X Discovery of a Promiscuous Non-Heme Iron Halogenase in Ambiguine Alkaloid Biogenesis: Implication for an Evolvable Enzyme Family for Late-Stage Halogenation of Aliphatic Carbons in Small Molecules. Angew. Chem., Int. Ed 2016, 55, 5780–5784. [DOI] [PubMed] [Google Scholar]
- (344).Zhu Q; Hillwig ML; Doi Y; Liu X Aliphatic Halogenase Enables Late-Stage C-H Functionalization: Selective Synthesis of a Brominated Fischerindole Alkaloid with Enhanced Antibacterial Activity. ChemBioChem. 2016, 17, 466–470. [DOI] [PubMed] [Google Scholar]
- (345).Büchler J; Malca SH; Patsch D; Voss M; Turner NJ; Bornscheuer UT; Allemann O; Le Chapelain C; Lumbroso A; Loiseleur O; et al. Algorithm-aided Engineering of Aliphatic Halogenase WelO5* for the Asymmetric Late-stage Functionalization of Soraphens. Nat. Commun 2022, 13, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (346).Neugebauer ME; Sumida KH; Pelton JG; McMurry JL; Marchand JA; Chang MCY A Family of Radical Halogenases for the Engineering of Amino-acid-based Products. Nat. Chem. Biol 2019, 15, 1009–1016. [DOI] [PubMed] [Google Scholar]
- (347).Neugebauer ME; Kissman EN; Marchand JA; Pelton JG; Sambold NA; Millar DC; Chang MCY Reaction Pathway Engineering Converts a Radical Hydroxylase into a Halogenase. Nat. Chem. Biol 2022, 18, 171–179. [DOI] [PubMed] [Google Scholar]
- (348).Duewel S; Schmermund L; Faber T; Harms K; Srinivasan V; Meggers E; Hoebenreich S Directed Evolution of an FeII-Dependent Halogenase for Asymmetric C(sp3)-H Chlorination. ACS Catal. 2020, 10, 1272–1277. [Google Scholar]
- (349).Matthews ML; Chang W.-c.; Layne AP; Miles LA; Krebs C; Bollinger JM Direct Nitration and Azidation of Aliphatic Carbons by an Iron-Dependent Halogenase. Nat. Chem. Biol 2014, 10, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (350).Kim CY; Mitchell AJ; Glinkerman CM; Li F-S; Pluskal T; Weng J-K The Chloroalkaloid (−)-acutumine is Biosynthesized via a Fe(II)- and 2-Oxoglutarate-dependent Halogenase in Menispermaceae Plants. Nat. Commun 2020, 11, 1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Matthews ML; Chang W.-c.; Layne AP; Miles LA; Krebs C; Bollinger JM Direct nitration and azidation of aliphatic carbons by an iron-dependent halogenase. Nat. Chem. Biol 2014, 10, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Mitchell AJ; Dunham NP; Bergman JA; Wang B; Zhu Q; Chang W.-c.; Liu X; Boal AK Structure-Guided Reprogramming of a Hydroxylase To Halogenate Its Small Molecule Substrate. Biochem. 2017, 56, 441–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (353).Gomez CA; Mondal D; Du Q; Chan N; Lewis JC Directed Evolution of an Iron(II)- and α-Ketoglutarate-Dependent Dioxygenase for Site-Selective Azidation of Unactivated Aliphatic C-H Bonds. Angew. Chem., Int. Ed 2023, 135, e202301370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (354).Rui J; Zhao Q; Huls AJ; Soler J; Paris JC; Chen Z; Reshetnikov V; Yang Y; Guo Y; Garcia-Borràs M; et al. Directed Evolution of Nonheme Iron Enzymes to Access Abiological Radical-relay C(sp3)-H Azidation. Science. 2022, 376, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (355).McIntosh JA; Coelho PS; Farwell CC; Wang ZJ; Lewis JC; Brown TR; Arnold FH Enantioselective Intramolecular C-H Amination Catalyzed by Engineered Cytochrome P450 Enzymes In Vitro and In Vivo. Angew. Chem., Int. Ed 2013, 52, 9309–9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Hyster TK; Farwell CC; Buller AR; McIntosh JA; Arnold FH Enzyme-Controlled Nitrogen-Atom Transfer Enables Regiodivergent C-H Amination. J. Am. Chem. Soc 2014, 136, 15505–15508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (357).Dydio P; Key HM; Hayashi H; Clark DS; Hartwig JF Chemoselective, Enzymatic C-H Bond Amination Catalyzed by a Cytochrome P450 Containing an Ir(Me)-PIX Cofactor. J. Am. Chem. Soc 2017, 139, 1750–1753. [DOI] [PubMed] [Google Scholar]
- (358).Jia Z-J; Gao S; Arnold FH Enzymatic Primary Amination of Benzylic and Allylic C(sp3)-H Bonds. J. Am. Chem. Soc 2020, 142, 10279–10283. [DOI] [PubMed] [Google Scholar]
- (359).Athavale SV; Gao S; Das A; Mallojjala SC; Alfonzo E; Long Y; Hirschi JS; Arnold FH Enzymatic Nitrogen Insertion into Unactivated C-H Bonds. J. Am. Chem. Soc 2022, 144, 19097–19105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (360).Doyon TJ; Buller AR Site-Selective Deuteration of Amino Acids through Dual-Protein Catalysis. J. Am. Chem. Soc 2022, 144, 7327–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Kohei T; Miyaura N Introduction to Cross-Coupling Reactions. In Cross-Coupling Reactions: A Practical Guide; Miyaura N, Ed.; Springer Berlin Heidelberg, 2002; pp 1–9. [Google Scholar]
- (362).Kuhl N; Hopkinson MN; Wencel-Delord J; Glorius F Beyond Directing Groups: Transition-Metal-Catalyzed C-H Activation of Simple Arenes. Angew. Chem, Int. Ed 2012, 51, 10236–10254. [DOI] [PubMed] [Google Scholar]
- (363).Ackermann L. Carboxylate-Assisted Transition-Metal-Catalyzed C-H Bond Functionalizations: Mechanism and Scope. Chem. Rev 2011, 111, 1315–1345. [DOI] [PubMed] [Google Scholar]
- (364).Kozlowski MC Oxidative Coupling in Complexity Building Transforms. Acc. Chem. Res 2017, 50, 638–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).Shalit H; Dyadyuk A; Pappo D Selective Oxidative Phenol Coupling by Iron Catalysis. J. Org. Chem 2019, 84, 1677–1686. [DOI] [PubMed] [Google Scholar]
- (366).Forneris CC; Seyedsayamdost MR In Vitro Reconstitution of OxyC Activity Enables Total Chemoenzymatic Syntheses of Vancomycin Aglycone Variants. Angew. Chem., Int. Ed 2018, 57, 8048–8052. [DOI] [PubMed] [Google Scholar]
- (367).Pylypenko O; Vitali F; Zerbe K; Robinson JA; Schlichting I Crystal Structure of OxyC, a Cytochrome P450 Implicated in an Oxidative C-C Coupling Reaction during Vancomycin Biosynthesis. J. Biol. Chem 2003, 278, 46727–46733. [DOI] [PubMed] [Google Scholar]
- (368).Aldemir H; Shu S; Schaefers F; Hong H; Richarz R; Harteis S; Einsiedler M; Milzarek TM; Schneider S; Gulder TAM Carrier Protein-Free Enzymatic Biaryl Coupling in Arylomycin A2 Assembly and Structure of the Cytochrome P450 AryC**. Chem. Eur. J 2022, 28, No. e202103389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (369).Molinaro C; Kawasaki Y; Wanyoike G; Nishioka T; Yamamoto T; Snedecor B; Robinson SJ; Gosselin F Engineered Cytochrome P450-Catalyzed Oxidative Biaryl Coupling Reaction Provides a Scalable Entry into Arylomycin Antibiotics. J. Am. Chem. Soc 2022, 144, 14838–14845. [DOI] [PubMed] [Google Scholar]
- (370).Zetzsche LE; Yazarians JA; Chakrabarty S; Hinze ME; Murray LAM; Lukowski AL; Joyce LA; Narayan ARH Biocatalytic Oxidative Cross-coupling Reactions for Biaryl Bond Formation. Nature. 2022, 603, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (371).Mazzaferro LS; Hüttel W; Fries A; Müller M Cytochrome P450-Catalyzed Regio- and Stereoselective Phenol Coupling of Fungal Natural Products. J. Am. Chem. Soc 2015, 137, 12289–12295. [DOI] [PubMed] [Google Scholar]
- (372).Obermaier S; Müller M Biaryl-Forming Enzymes from Aspergilli Exhibit Substrate-Dependent Stereoselectivity. Biochem. 2019, 58, 2589–2593. [DOI] [PubMed] [Google Scholar]
- (373).Coyle CM; Panaccione DG An Ergot Alkaloid Biosynthesis Gene and Clustered Hypothetical Genes from Aspergillus fumigatus. Appl. Environ. Microbiol 2005, 71, 3112–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (374).Unsöld IA; Li S-M Overproduction, Purification and Characterization of FgaPT2, a Dimethylallyltryptophan Synthase from Aspergillus Fumigatus. Microbiol. 2005, 151, 1499–1505. [DOI] [PubMed] [Google Scholar]
- (375).Steffan N; Unsöld IA; Li S-M Chemoenzymatic Synthesis of Prenylated Indole Derivatives by Using a 4-Dimethylallyltryptophan Synthase from Aspergillus fumigatus. ChemBioChem. 2007, 8, 1298–1307. [DOI] [PubMed] [Google Scholar]
- (376).Steffan N; Li S-M Increasing Structure Diversity of Prenylated Diketopiperazine Derivatives by using a 4-Dimethylallyltryptophan Synthase. Arch. Microbiol 2009, 191, 461–466. [DOI] [PubMed] [Google Scholar]
- (377).Eggbauer B; Schrittwieser JH; Kerschbaumer B; Macheroux P; Kroutil W Regioselective Biocatalytic C4-Prenylation of Unprotected Tryptophan Derivatives. ChemBioChem. 2022, 23, No. e202200311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378).Kremer A; Westrich L; Li S-M A 7-dimethylallyltryptophan Synthase from Aspergillus Fumigatus: Overproduction, Purification and Biochemical Characterization. Microbiol. 2007, 153, 3409–3416. [DOI] [PubMed] [Google Scholar]
- (379).Kremer A; Li S-M Potential of a 7-dimethylallyltryptophan Synthase as a Tool for Production of Prenylated Indole Derivatives. Appl. Microbiol. Biotechnol 2008, 79, 951–961. [DOI] [PubMed] [Google Scholar]
- (380).Takahashi S; Takagi H; Toyoda A; Uramoto M; Nogawa T; Ueki M; Sakaki Y; Osada H Biochemical Characterization of a Novel Indole Prenyltransferase from Streptomyces sp. SN-593. J. Bacteriol 2010, 192, 2839–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (381).Winkelblech J; Li S-M Biochemical Investigations of Two 6-DMATS Enzymes from Streptomyces Reveal New Features of L-Tryptophan Prenyltransferases. ChemBioChem. 2014, 15, 1030–1039. [DOI] [PubMed] [Google Scholar]
- (382).Elshahawi SI; Cao H; Shaaban KA; Ponomareva LV; Subramanian T; Farman ML; Spielmann HP; Phillips GN; Thorson JS; Singh S Structure and Specificity of a Permissive Bacterial C-Prenyltransferase. Nat. Chem. Biol 2017, 13, 366–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Mai P; Zocher G; Ludwig L; Stehle T; Li S-M Actions of Tryptophan Prenyltransferases Toward Fumiquinazolines and their Potential Application for the Generation of Prenylated Derivatives by Combining Chemical and Chemoenzymatic Syntheses. Adv. Synth. Catal 2016, 358, 1639–1653. [Google Scholar]
- (384).Chen R; Gao B; Liu X; Ruan F; Zhang Y; Lou J; Feng K; Wunsch C; Li S-M; Dai J; et al. Molecular Insights into the Enzyme Promiscuity of an Aromatic Prenyltransferase. Nat. Chem. Biol 2017, 13, 226–234. [DOI] [PubMed] [Google Scholar]
- (385).Yu X; Liu Y; Xie X; Zheng X-D; Li S-M Biochemical Characterization of Indole Prenyltransferases: Filling The Last Gap of Prenylation Positions by a 5-Dimethylallyltryptophan Synthase from Aspergillus Clavatus. J. Biol. Chem 2012, 287, 1371–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (386).Winkelblech J; Liebhold M; Gunera J; Xie X; Kolb P; Li S-M Tryptophan C5-, C6- and C7-Prenylating Enzymes Displaying a Preference for C-6 of the Indole Ring in the Presence of Unnatural Dimethylallyl Diphosphate Analogues. Adv. Synth. Catal 2015, 357, 975–986. [Google Scholar]
- (387).Liebhold M; Li S-M Regiospecific Benzylation of Tryptophan and Derivatives Catalyzed by a Fungal Dimethylallyl Transferase. Org. Lett 2013, 15, 5834–5837. [DOI] [PubMed] [Google Scholar]
- (388).Yokoyama K; Lilla EA C-C Bond Forming Radical SAM Enzymes Involved in the Construction of Carbon Skeletons of Cofactors and Natural Products. Nat. Prod. Rep 2018, 35, 660–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (389).Knox HL; Sinner EK; Townsend CA; Boal AK; Booker SJ Structure of a B12-dependent Radical SAM Enzyme in Carbapenem Biosynthesis. Nature. 2022, 602, 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (390).Soualmia F; Guillot A; Sabat N; Brewee C; Kubiak X; Haumann M; Guinchard X; Benjdia A; Berteau O Exploring the Biosynthetic Potential of TsrM, a B12-dependent Radical SAM Methyltransferase Catalyzing Non-radical Reactions. Chem. Eur. J 2022, 28, No. e202200627. [DOI] [PubMed] [Google Scholar]
- (391).Gericke L; Mhaindarkar D; Karst LC; Jahn S; Kuge M; Mohr MKF; Gagsteiger J; Cornelissen NV; Wen X; Mordhorst S Biomimetic S-Adenosylmethionine Regeneration Starting from Multiple Byproducts Enables Biocatalytic Alkylation with Radical SAM Enzymes. ChemBioChem. 2023, 24, e202300133. [DOI] [PubMed] [Google Scholar]
- (392).Pickel B; Schaller A Dirigent Proteins: Molecular Characteristics and Potential Biotechnological Applications. Appl. Microbiol. Biotechnol 2013, 97, 8427–8438. [DOI] [PubMed] [Google Scholar]
- (393).Davin LB; Lewis NG Dirigent Proteins and Dirigent Sites Explain the Mystery of Specificity of Radical Precursor Coupling in Lignan and Lignin Biosynthesis1. Plant Physiol. 2000, 123, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Kim SS; Sattely ES Dirigent Proteins Guide Asymmetric Heterocoupling for the Synthesis of Complex Natural Product Analogues. J. Am. Chem. Soc 2021, 143, 5011–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (395).Page CG; Cao J; Oblinsky DG; MacMillan SN; Dahagam S; Lloyd RM; Charnock SJ; Scholes GD; Hyster TK Regioselective Radical Alkylation of Arenes Using Evolved Photoenzymes. J. Am. Chem. Soc 2023, 145, 11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (396).Biegasiewicz KF; Cooper SJ; Gao X; Oblinsky DG; Kim JH; Garfinkle SE; Joyce LA; Sandoval BA; Scholes GD; Hyster TK Photoexcitation of Flavoenzymes Enables a Stereoselective Radical Cyclization. Science. 2019, 364, 1166–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (397).Clayman PD; Hyster TK Photoenzymatic Generation of Unstabilized Alkyl Radicals: An Asymmetric Reductive Cyclization. J. Am. Chem. Soc 2020, 142, 15673–15677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (398).Nicholls BT; Oblinsky DG; Kurtoic SI; Grosheva D; Ye Y; Scholes GD; Hyster TK Engineering a Non-Natural Photoenzyme for Improved Photon Efficiency**. Angew. Chem., Int. Ed 2022, 61, No. e202113842. [DOI] [PubMed] [Google Scholar]
- (399).Brandenberg OF; Fasan R; Arnold FH Exploiting and Engineering Hemoproteins for Abiological Carbene and Nitrene Transfer Reactions. Curr. Opin. Biotechnol 2017, 47, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (400).Coelho PS; Wang ZJ; Ener ME; Baril SA; Kannan A; Arnold FH; Brustad EM A Serine-substituted P450 Catalyzes Highly Efficient Carbene Transfer to Olefins in Vivo. Nat. Chem. Biol 2013, 9, 485–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (401).Zhang RK; Chen K; Huang X; Wohlschlager L; Renata H; Arnold FH Enzymatic Assembly of Carbon-Carbon Bonds via Iron-catalysed sp3 C-H Functionalization. Nature. 2019, 565, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (402).Brandenberg OF; Chen K; Arnold FH Directed Evolution of a Cytochrome P450 Carbene Transferase for Selective Functionalization of Cyclic Compounds. J. Am. Chem. Soc 2019, 141, 8989–8995. [DOI] [PubMed] [Google Scholar]
- (403).Zhou AZ; Chen K; Arnold FH Enzymatic Lactone-Carbene C-H Insertion to Build Contiguous Chiral Centers. ACS Catal. 2020, 10, 5393–5398. [Google Scholar]
- (404).Dydio P; Key HM; Nazarenko A; Rha JY-E; Seyedkazemi V; Clark DS; Hartwig JF An Artificial Metalloenzyme With the Kinetics of Native Enzymes. Science. 2016, 354, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (405).Natoli SN; Hartwig JF Noble-Metal Substitution in Hemoproteins: An Emerging Strategy for Abiological Catalysis. Acc. Chem. Res 2019, 52, 326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (406).Gu Y; Natoli SN; Liu Z; Clark DS; Hartwig JF Site-Selective Functionalization of (sp3)C-H Bonds Catalyzed by Artificial Metalloenzymes Containing an Iridium-Porphyrin Cofactor. Angew. Chem., Int. Ed 2019, 58, 13954–13960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (407).Rumo C; Stein A; Klehr J; Tachibana R; Prescimone A; Häussinger D; Ward TR An Artificial Metalloenzyme Based on a Copper Heteroscorpionate Enables sp3 C-H Functionalization via Intramolecular Carbene Insertion. J. Am. Chem. Soc 2022, 144, 11676–11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (408).O’Hagan D; Deng H Enzymatic Fluorination and Biotechnological Developments of the Fluorinase. Chem. Rev 2015, 115, 634–649. [DOI] [PubMed] [Google Scholar]
- (409).Roy AD; Gruschow S; Cairns N; Goss RJM Gene Expression Enabling Synthetic Diversification of Natural Products: Chemogenetic Generation of Pacidamycin Analogs. J. Am. Chem. Soc 2010, 132, 12243–12245. [DOI] [PubMed] [Google Scholar]
- (410).Runguphan W; O’Connor SE Diversification of Monoterpene Indole Alkaloid Analogs through Cross-Coupling. Org. Lett 2013, 15, 2850–2853. [DOI] [PubMed] [Google Scholar]
- (411).Durak LJ; Payne JT; Lewis JC Late-Stage Diversification of Biologically Active Molecules via Chemoenzymatic C-H Functionalization. ACS Catal. 2016, 6, 1451–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Latham J; Henry J-M; Sharif HH; Menon BRK; Shepherd SA; Greaney MF; Micklefield J Integrated Catalysis Opens New Arylation Pathways via Regiodivergent Enzymatic C-H Activation. Nat. Commun 2016, 7, 11873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (413).Frese M; Schnepel C; Minges H; Voß H; Feiner R; Sewald N Modular Combination of Enzymatic Halogenation of Tryptophan with Suzuki-Miyaura Cross-Coupling Reactions. ChemCatChem. 2016, 8, 1799–1803. [Google Scholar]
- (414).Schnepel C; Minges H; Frese M; Sewald N A High-Throughput Fluorescence Assay to Determine the Activity of Tryptophan Halogenases. Angew. Chem., Int. Ed 2016, 55, 14159–14163. [DOI] [PubMed] [Google Scholar]
- (415).Lewis JC; Arnold FH Catalysts on Demand: Selective Oxidations by Laboratory-Evolved Cytochrome P450 BM3. Chimia. 2009, 63, 309. [Google Scholar]
- (416).Li J; Li F; King-Smith E; Renata H Merging Chemoenzymatic and Radical-based Retrosynthetic Logic for Rapid and Modular Synthesis of Oxidized Meroterpenoids. Nat. Chem 2020, 12, 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (417).Li F; Deng H; Renata H Remote B-Ring Oxidation of Sclareol with an Engineered P450 Facilitates Divergent Access to Complex Terpenoids. J. Am. Chem. Soc 2022, 144, 7616–7621. [DOI] [PubMed] [Google Scholar]
- (418).Li J; Chen F; Renata H Concise Chemoenzymatic Synthesis of Gedunin. J. Am. Chem. Soc 2022, 144, 19238–19242. [DOI] [PubMed] [Google Scholar]
- (419).Zhang X; King-Smith E; Dong L-B; Yang L-C; Rudolf JD; Shen B; Renata H Divergent Synthesis of Complex Diterpenes Through a Hybrid Oxidative Approach. Science. 2020, 369, 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (420).Zhao Y; Zhang B; Sun ZQ; Zhang H; Wang W; Wang ZR; Guo ZK; Yu S; Tan RX; Ge HM Biocatalytic C14-Hydroxylation on Androstenedione Enabled Modular Synthesis of Cardiotonic Steroids. ACS Catal. 2022, 12, 9839–9845. [Google Scholar]
- (421).Zhang X; Renata H Efficient Chemoenzymatic Synthesis of (2S,3R)-3-hydroxy-3-methylproline, a Key Fragment in Polyoxypeptin A and FR225659. Tetrahedron 2019, 75, 3253–3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (422).Zwick CR; Renata H A one-pot Chemoenzymatic Synthesis of (2S, 4R)-4-methylproline Enables the First Total Synthesis of Antiviral Lipopeptide Cavinafungin B. Tetrahedron 2018, 74, 6469–6473. [Google Scholar]
- (423).Zhang X; King-Smith E; Renata H Total Synthesis of Tambromycin by Combining Chemocatalytic and Biocatalytic C-H Functionalization. Angew. Chem., Int. Ed 2018, 57, 5037–5041. [DOI] [PubMed] [Google Scholar]
- (424).Zwick CR III; Sosa MB; Renata H Modular Chemoenzymatic Synthesis of GE81112 B1 and Related Analogues Enables Elucidation of Its Key Pharmacophores. J. Am. Chem. Soc 2021, 143, 1673–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (425).Amatuni A; Shuster A; Abegg D; Adibekian A; Renata H Comprehensive Structure-Activity Relationship Studies of Cepafungin Enabled by Biocatalytic C-H Oxidations. ACS Cent. Sci 2023, 9, 239–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Jiang Y; Renata H Eleven-Step Chemoenzymatic Synthesis of Cotylenol. ChemRxiv 2022, DOI: 10.26434/chemrxiv-2022-7nvn3. [DOI] [Google Scholar]
- (427).Lazzarotto M; Hammerer L; Hetmann M; Borg A; Schmermund L; Steiner L; Hartmann P; Belaj F; Kroutil W; Gruber K; et al. Chemoenzymatic Total Synthesis of Deoxy-, epi-, and Podophyllotoxin and a Biocatalytic Kinetic Resolution of Dibenzylbutyrolactones. Angew. Chem., Int. Ed 2019, 58, 8226–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Li J; Zhang X; Renata H Asymmetric Chemoenzymatic Synthesis of (−)-Podophyllotoxin and Related Aryltetralin Lignans. Angew. Chem., Int. Ed 2019, 58, 11657–11660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Li J; Renata H Concise Chemoenzymatic Synthesis of Fasamycin A. J. Org. Chem 2021, 86, 11206–11211. [DOI] [PubMed] [Google Scholar]
- (430).Sib A; Gulder TAM Chemo-enzymatic Total Synthesis of Oxosorbicillinol, Sorrentanone, Rezishanones B and C, Sorbicatechol A, Bisvertinolone, and (+)-Epoxysorbicillinol. Angew. Chem., Int. Ed 2018, 57, 14650–14653. [DOI] [PubMed] [Google Scholar]
- (431).Sib A; Gulder TAM Stereoselective Total Synthesis of Bisorbicillinoid Natural Products by Enzymatic Oxidative Dearomatization/Dimerization. Angew. Chem., Int. Ed 2017, 56, 12888–12891. [DOI] [PubMed] [Google Scholar]
- (432).Hoyt EA; Cal PMSD; Oliveira BL; Bernardes GJL Contemporary Approaches to Site-Selective Protein Modification. Nat. Rev. Chem 2019, 3, 147–171. [Google Scholar]
- (433).Rees HA; Liu DR Base Editing: Precision Chemistry on the Genome and Transcriptome of Living Cells. Nat. Rev. Genet 2018, 19, 770–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (434).Porto EM; Komor AC; Slaymaker IM; Yeo GW Base Editing: Advances and Therapeutic Opportunities. Nat. Rev. Drug Discovery 2020, 19, 839–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (435).Žáková L; Zyka D; Ježek J; Hančlová I; Šanda M; Brzozowski AM; Jiráček J The use of Fmoc-Lys(Pac)-OH and Penicillin G Acylase in the Preparation of Novel Semisynthetic Insulin Analogs. J. Pept. Sci 2007, 13, 334–341. [DOI] [PubMed] [Google Scholar]
- (436).Fryszkowska A; An C; Alvizo O; Banerjee G; Canada KA; Cao Y; DeMong D; Devine PN; Duan D; Elgart DM; et al. A Chemoenzymatic Strategy for Site-Selective Functionalization of Native Peptides and Proteins. Science. 2022, 376, 1321–1327. [DOI] [PubMed] [Google Scholar]
- (437).Pissarnitski DA; Kekec A; Yan L; Zhu Y; Feng DD; Huo P; Madsen-Duggan C; Moyes CR; Nargund RP; Kelly T; et al. Discovery of Insulin Receptor Partial Agonists MK-5160 and MK-1092 as Novel Basal Insulins with Potential to Improve Therapeutic Index. J. Med. Chem 2022, 65, 5593–5605. [DOI] [PubMed] [Google Scholar]
- (438).Wu M; Carballo-Jane E; Zhou H; Zafian P; Dai G; Liu M; Lao J; Kelly T; Shao D; Gorski J; et al. Functionally Selective Signaling and Broad Metabolic Benefits by Novel Insulin Receptor Partial Agonists. Nat. Commun 2022, 13, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (439).Rachel NM; Quaglia D; Levesque E; Charette AB; Pelletier JN Engineered, Highly Reactive Substrates of Microbial Transglutaminase Enable Protein Labeling Within Various Secondary Structure Elements. Protein Sci. 2017, 26, 2268–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (440).Marx CK; Hertel TC; Pietzsch M Random Mutagenesis of a Recombinant Microbial Transglutaminase for the Generation of Thermostable and Heat-sensitive Variants. J. Biotechnol 2008, 136, 156–162. [DOI] [PubMed] [Google Scholar]
- (441).Deweid L; Neureiter L; Englert S; Schneider H; Deweid J; Yanakieva D; Sturm J; Bitsch S; Christmann A; Avrutina O; et al. Directed Evolution of a Bond-Forming Enzyme: Ultrahigh-Throughput Screening of Microbial Transglutaminase Using Yeast Surface Display. Chem. Eur. J 2018, 24, 15195–15200. [DOI] [PubMed] [Google Scholar]
- (442).Zhou L; Kooy-Winkelaar YMC; Cordfunke RA; Dragan I; Thompson A; Drijfhout JW; van Veelen PA; Chen H; Koning F Abrogation of Immunogenic Properties of Gliadin Peptides through Transamidation by Microbial Transglutaminase Is Acyl-Acceptor Dependent. J. Agric. Food Chem 2017, 65, 7542–7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (443).Fontana A; Spolaore B; Mero A; Veronese FM Site-specific modification and PEGylation of pharmaceutical proteins mediated by transglutaminase. Adv. Drug Delivery Rev 2008, 60, 13–28. [DOI] [PubMed] [Google Scholar]
- (444).Mero A; Spolaore B; Veronese FM; Fontana A Transglutaminase-Mediated PEGylation of Proteins: Direct Identification of the Sites of Protein Modification by Mass Spectrometry using a Novel Monodisperse PEG. Bioconjugate Chem. 2009, 20, 384–389. [DOI] [PubMed] [Google Scholar]
- (445).Anami Y; Yamazaki CM; Xiong W; Gui X; Zhang N; An Z; Tsuchikama K Glutamic acid-valine-citrulline Linkers Ensure Stability and Efficacy of Antibody-drug Conjugates in Mice. Nat. Commun 2018, 9, 2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (446).Yamazaki CM; Yamaguchi A; Anami Y; Xiong W; Otani Y; Lee J; Ueno NT; Zhang N; An Z; Tsuchikama K Antibody-drug Conjugates with Dual Payloads for Combating Breast Tumor Heterogeneity and Drug Resistance. Nat. Commun 2021, 12, 3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (447).Bell EL; Finnigan W; France SP; Green AP; Hayes MA; Hepworth LJ; Lovelock SL; Niikura H; Osuna S; Romero E; et al. Biocatalysis. Nat. Rev. Methods Primers 2021, 1 , 46. [Google Scholar]
- (448).van der Donk WA Introduction: Unusual Enzymology in Natural Product Synthesis. Chem. Rev 2017, 117, 5223–5225. [DOI] [PubMed] [Google Scholar]
- (449).Booker SJ Anaerobic Functionalization of Unactivated C-H Bonds. Curr. Opin. Chem. Biol 2009, 13, 58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (450).Powell MM; Rao G; Britt RD; Rittle J Enzymatic Hydroxylation of Aliphatic C-H Bonds by a Mn/Fe Cofactor. bioRxiv 2023, DOI: 10.1101/2023.03.10.532131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (451).Ziemert N; Alanjary M; Weber T The Evolution of Genome Mining in Microbes - a Review. Nat. Prod. Rep 2016, 33, 988–1005. [DOI] [PubMed] [Google Scholar]
- (452).Marshall JR; Yao P; Montgomery SL; Finnigan JD; Thorpe TW; Palmer RB; Mangas-Sanchez J; Duncan RAM; Heath RS; Graham KM; et al. Screening and Characterization of a Diverse Panel of Metagenomic Imine Reductases for Biocatalytic Reductive Amination. Nat. Chem 2021, 13, 140–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (453).Vaissier Welborn V; Head-Gordon T Computational Design of Synthetic Enzymes. Chem. Rev 2019, 119, 6613–6630. [DOI] [PubMed] [Google Scholar]
- (454).Biegasiewicz KF; Cooper SJ; Emmanuel MA; Miller DC; Hyster TK Catalytic Promiscuity Enabled by Photoredox Catalysis in Nicotinamide-Dependent Oxidoreductases. Nat. Chem 2018, 10, 770–775. [DOI] [PubMed] [Google Scholar]
- (455).Devamani T; Rauwerdink AM; Lunzer M; Jones BJ; Mooney JL; Tan MAO; Zhang Z-J; Xu J-H; Dean AM; Kazlauskas RJ Catalytic Promiscuity of Ancestral Esterases and Hydroxynitrile Lyases. J. Am. Chem. Soc 2016, 138, 1046–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (456).Yang H; Swartz AM; Park HJ; Srivastava P; Ellis-Guardiola K; Upp DM; Lee G; Belsare K; Gu Y; Zhang C; et al. Evolving Artificial Metalloenzymes via Random Mutagenesis. Nat. Chem 2018, 10, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (457).Jeschek M; Reuter R; Heinisch T; Trindler C; Klehr J; Panke S; Ward TR Directed Evolution of Artificial Metalloenzymes for in vivo Metathesis. Nature. 2016, 537, 661–665. [DOI] [PubMed] [Google Scholar]
- (458).Markel U; Sauer DF; Schiffels J; Okuda J; Schwaneberg U Towards the Evolution of Artificial Metalloenzymes—A Protein Engineer’s Perspective. Angew. Chem., Int. Ed 2019, 58, 4454–4464. [DOI] [PubMed] [Google Scholar]
- (459).Gröger H; Gallou F; Lipshutz BH Where Chemocatalysis Meets Biocatalysis: In Water. Chem. Rev 2023, 123, 5262–5296. [DOI] [PubMed] [Google Scholar]
- (460).Hughes G; Lewis JC Introduction: Biocatalysis in Industry. Chem. Rev 2018, 118, 1–3. [DOI] [PubMed] [Google Scholar]
- (461).Wu S; Snajdrova R; Moore JC; Baldenius K; Bornscheuer UT Biocatalysis: Enzymatic Synthesis for Industrial Applications. Angew. Chem., Int. Ed 2021, 60, 88–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (462).Romero PA; Arnold FH Exploring Protein Fitness Landscapes by Directed Evolution. Nat. Rev. Mol. Cell Biol 2009, 10, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (463).Quertinmont LT; Orru R; Lutz S RApid Parallel Protein EvaluatoR (RAPPER), from Gene to Enzyme Function in One Day. Chem. Commun 2015, 51, 122–124. [DOI] [PubMed] [Google Scholar]
- (464).Holland-Moritz DA; Wismer MK; Mann BF; Farasat I; Devine P; Guetschow ED; Mangion I; Welch CJ; Moore JC; Sun S; et al. Mass Activated Droplet Sorting (MADS) Enables High-Throughput Screening of Enzymatic Reactions at Nanoliter Scale. Angew. Chem., Int. Ed 2020, 59, 4470–4477. [DOI] [PubMed] [Google Scholar]
- (465).Diefenbach XW; Farasat I; Guetschow ED; Welch CJ; Kennedy RT; Sun S; Moore JC Enabling Biocatalysis by High-Throughput Protein Engineering Using Droplet Microfluidics Coupled to Mass Spectrometry. ACS Omega 2018, 3, 1498–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Wleklinski M; Loren BP; Ferreira CR; Jaman Z; Avramova L; Sobreira TJP; Thompson DH; Cooks RG High Throughput Reaction Screening Using Desorption Electrospray Ionization Mass Spectrometry. Chem. Sci 2018, 9, 1647–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (467).Mahr R; Frunzke J Transcription Factor-based Biosensors in Biotechnology: Current State and Future Prospects. Appl. Microbiol. Biotechnol 2016, 100, 79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (468).Kries H; Blomberg R; Hilvert D De Novo Enzymes by Computational Design. Curr. Opin. Chem. Biol 2013, 17, 221–228. [DOI] [PubMed] [Google Scholar]
- (469).Yeh AH-W; Norn C; Kipnis Y; Tischer D; Pellock SJ; Evans D; Ma P; Lee GR; Zhang JZ; Anishchenko I; et al. De Novo Design of Luciferases Using Deep Learning. Nature. 2023, 614, 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (470).Derat E; Kamerlin SCL Computational Advances in Protein Engineering and Enzyme Design. J. Phys. Chem. B 2022, 126, 2449–2451. [DOI] [PubMed] [Google Scholar]
- (471).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature. 2021, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (472).Yang KK; Wu Z; Arnold FH Machine-learning-guided Directed Evolution for Protein Engineering. Nat. Methods 2019, 16, 687–694. [DOI] [PubMed] [Google Scholar]
- (473).Freschlin CR; Fahlberg SA; Romero PA Machine Learning to Navigate Fitness Landscapes for Protein Engineering. Curr. Opin. Biotechnol 2022, 75, 102713. [DOI] [PMC free article] [PubMed] [Google Scholar]