

Abstract
Peripartum cardiomyopathy (PPCM) is an idiopathic form of pregnancy-induced heart failure associated with preeclampsia. Circulating factors in late pregnancy are thought to contribute to both diseases, suggesting a common underlying pathophysiological process. However, what drives this process remains unclear. Using serum proteomics, we identified the senescence-associated secretory phenotype (SASP), a marker of cellular senescence associated with biological aging, as the most highly up-regulated pathway in young women with PPCM or preeclampsia. Placentas from women with preeclampsia displayed multiple markers of amplified senescence and tissue aging, as well as overall increased gene expression of 28 circulating proteins that contributed to SASP pathway enrichment in serum samples from patients with preeclampsia or PPCM. The most highly expressed placental SASP factor, activin A, was associated with cardiac dysfunction or heart failure severity in women with preeclampsia or PPCM. In a murine model of PPCM induced by cardiomyocyte-specific deletion of the gene encoding peroxisome proliferator–activated receptor γ coactivator-1α, inhibiting activin A signaling in the early postpartum period with a monoclonal antibody to the activin type II receptor improved heart function. In addition, attenuating placental senescence with the senolytic compound fisetin in late pregnancy improved cardiac function in these animals. These findings link senescence biology to cardiac dysfunction in pregnancy and help to elucidate the pathogenesis underlying cardiovascular diseases of pregnancy.
INTRODUCTION
Cardiovascular diseases are a leading cause of maternal death worldwide (1, 2). Peripartum cardiomyopathy (PPCM), an idiopathic form of acute systolic heart failure (HF) that occurs during late pregnancy or early postpartum, is a major cause of maternal mortality (3). The incidence of PPCM is around 1 in 1000 in the United States, but as high as 1 in 100 in Nigeria, and is rising because of increasing prevalence of its risk factors, which include preeclampsia, advanced maternal age, and multiple gestation pregnancies (3). Since its formal recognition in 1971 by Demakis and Rahimtoola (4), the cause of PPCM has remained an enigma, limiting therapeutic development for this HF syndrome.
Accumulating evidence suggests a multifactor mechanism likely underlies PPCM pathophysiology, one in which a genetic predisposition to HF is unmasked by stresses extrinsic to the heart that occur with pregnancy (3, 5–9). Central to this pathophysiology is the role of circulating factors, such as the soluble vascular endothelial growth factor receptor, fms-like tyrosine kinase 1 (sFLT1), and cleaved prolactin, which increase during late pregnancy and can induce PPCM in animal models through antiangiogenic effects (5, 6). Many of the same circulating factors implicated in PPCM are also elevated in preeclampsia, a hypertensive disorder of pregnancy that is a leading risk factor for PPCM and postpartum HF (10–13). These observations have led to the hypothesis that cardiac dysfunction in preeclampsia and PPCM stems from a shared underlying mechanism (14). However, what drives this common pathophysiology remains unknown.
Cellular senescence, an intrinsic response to aging or stress, is a potential source of circulating factors that can have deleterious effects on the cardiovascular system (15). Secreted factors produced by senescent cells, collectively referred to as the senescence-associated secretory phenotype (SASP), have been shown to influence both local and remote tissue remodeling and function (15–18). Although senescence is most commonly associated with organismal aging and disease pathogenesis in older adults (19, 20), it can also occur in younger individuals (21). The placenta, a hybrid maternal-fetal organ unique to pregnancy, manifests markers of increased senescence by term (22), potentially reflecting an accelerated aging process in this organ relative to other maternal organs. However, the functional relevance of placental senescence and its potential contribution to disease has not previously been clear. We hypothesized that proteins circulating in young women with preeclampsia or PPCM might reflect a pathologic SASP derived from accelerated placental senescence. To test this hypothesis, we used an integrated approach incorporating serum proteomics–based human discovery with clinical and experimental validation, which collectively revealed an important role of placental senescence biology in the regulation of maternal cardiac function and HF in pregnancy.
RESULTS
Serum proteomics identifies the SASP as the most highly up-regulated pathway in both PPCM and preeclampsia
To gain insights into possible shared pathophysiology of PPCM and preeclampsia, we examined biospecimens (serum, placental, or heart tissue) from 281 individuals with PPCM (n = 98), preeclampsia (n = 56), gestational hypertension (n = 19), nonpregnant nonischemic cardiomyopathy (NICM, n = 13), uncomplicated pregnancies (n = 71), or nonpregnant controls (n = 24). We first performed parallel case-control serum proteomic studies in women presenting with incident PPCM or preeclampsia (table S1). Inflammation related pathways were up-regulated in both preeclampsia and PPCM, and vascular endothelial growth factor signaling was down-regulated in preeclampsia (Fig. 1), consistent with prior reports (23, 24). The SASP, a marker of biological aging, emerged as the most highly up-regulated pathway in both PPCM and preeclampsia, despite the relatively young age (32.5 ± 6.6 and 34.6 ± 5.9 years, respectively) of the affected women (Fig. 1, fig. S1, and data file S1). Twenty-eight distinct proteins contributed to SASP enrichment in the sera of women with PPCM or preeclampsia (data file S1).
Fig. 1. Senescence-associated secretory phenotype enrichment in PPCM and preeclampsia.
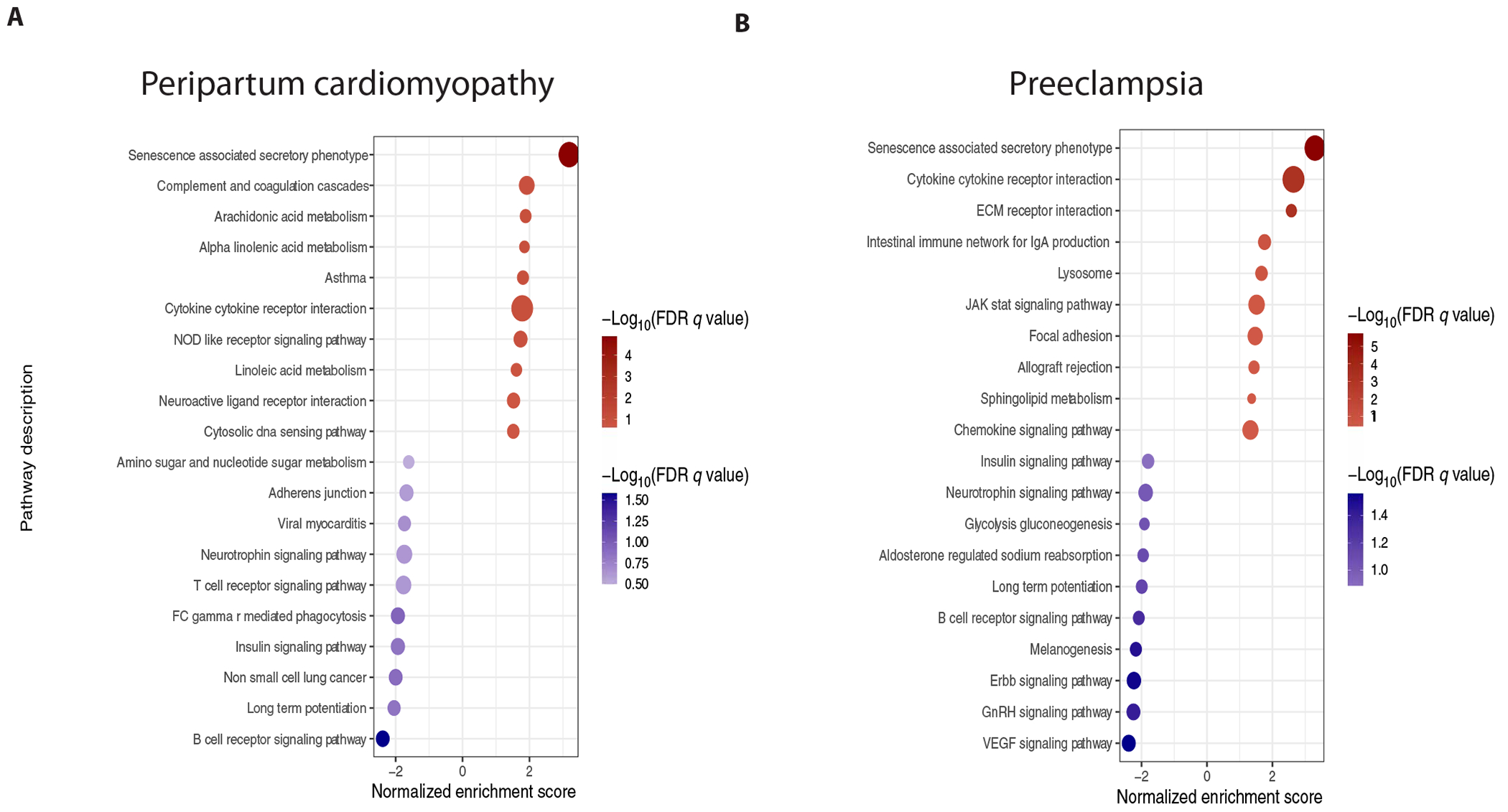
Pathway analysis on SOMAscan serum proteomic profiles from (A) women with PPCM (n = 13) versus healthy pregnant controls (n = 10) and (B) women with preeclampsia (n = 11) versus normotensive pregnant controls (n = 11). Gene set enrichment analysis using SASP protein set and KEGG pathway database. Only significant pathways [false discovery rate (FDR) <0.25] are displayed.
Placental senescence is the likely source of circulating SASP in preeclampsia and PPCM
Given previous reports indicating progressive placental senescence throughout pregnancy (22), we hypothesized that amplification of this process might explain the increased circulating SASP profile detected in women with PPCM or preeclampsia. Given the rarity of PPCM, we analyzed placentas from preeclamptic women. Compared with placentas from maternal- and gestational-age–matched women with normotensive pregnancies (table S2), preeclamptic placentas displayed multiple markers of amplified senescence and tissue aging, including increased senescence-associated β-galactosidase (SA-βgal), cyclin-dependent kinase inhibitor 1A and 2A (CDKN1A and CDKN2A) expression, and a nonsignificant decrease in global DNA methylation (P = 0.07) (fig. S2) (25, 26). Overall gene expression of the 28 SASP proteins was increased in preeclamptic placentas (Fig. 2A), indicating that the shared SASP in preeclampsia and PPCM is associated with increased placental senescence.
Fig. 2. Activin A is a dominant placental SASP protein in preeclampsia.
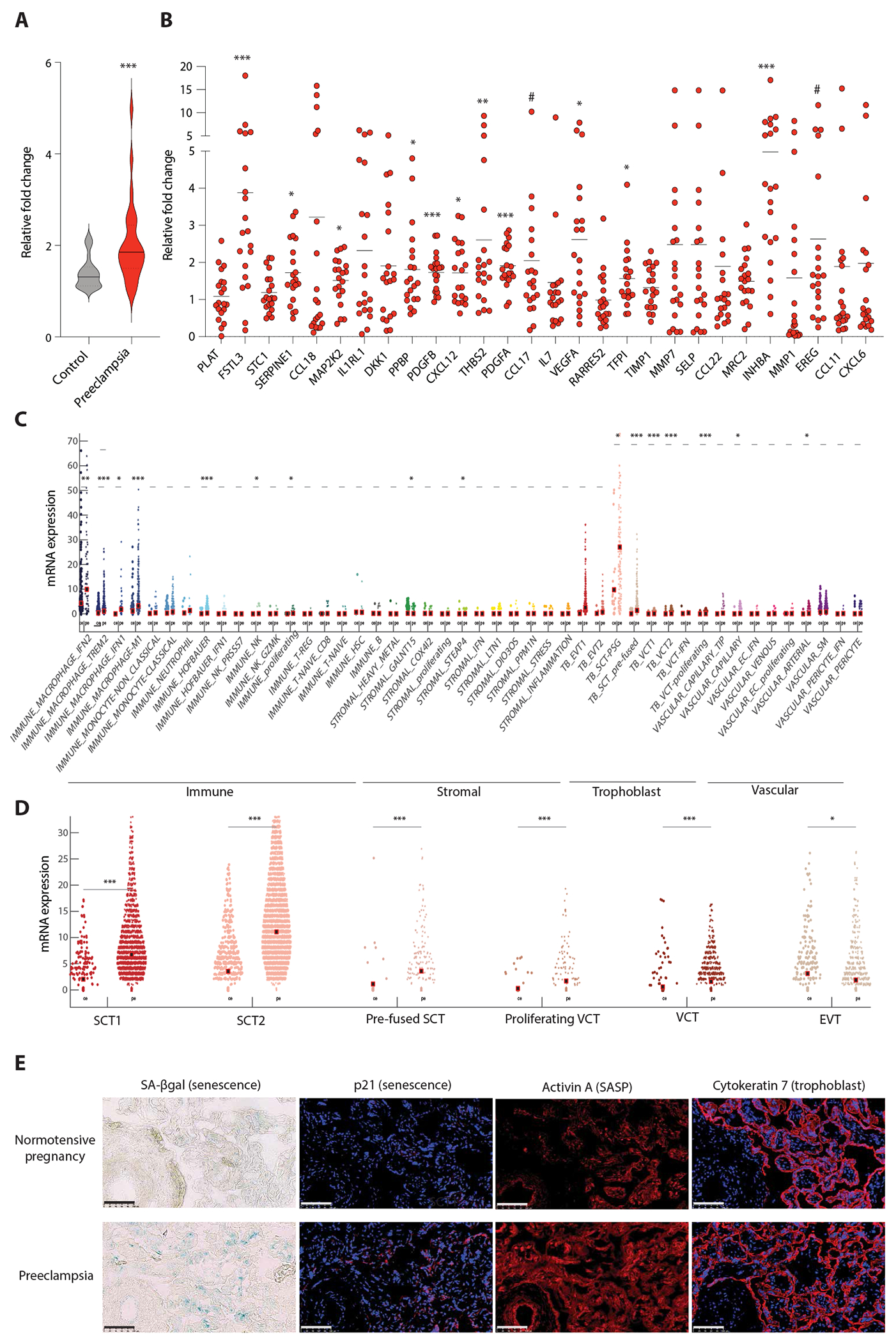
(A) Violin plot of the composite fold change in relative gene expression of the 28 SASP proteins in placental tissue from preeclamptic versus normotensive pregnancy. n = 20 per group. Solid line is median. Dashed line is quartiles. Mann-Whitney test. P = 0.0004. (B) Relative gene expression of individual SASP proteins. Mean fold change (black bar) and individual data points from the 20 individuals with preeclampsia are displayed. See table S2 for individual data. Student’s t test or Mann-Whitney test. #P ≤ 0.1, *P < 0.05, **P < 0.01, ***P < 0.001. (C) Violin plot of gene expression of INHBA (gene encoding activin A) from placental scRNAseq of early preeclampsia (pe, n = 10) versus uncomplicated preterm delivery (ce, n = 3). Wilcoxon or Mann-Whitney rank sum test. *P < 0.01, **P < 0.001, ***P < 0.0001. (D) Violin plot of INHBA expression from snRNAseq from five distinct trophoblast subtypes (SCT, syncytiotrophoblast; VCT, villous cytotrophoblast; EVT, extravillous trophoblast). Wilcoxon or Mann-Whitney rank sum test. *P < 0.01, **P < 0.001, ***P < 0.0001. Analysis for (C) and (D) was done on datasets acquired by Admati and colleagues (27). Dots represent single cells (c) or single nuclei (D). Black square is mean. (E) SA-βgal (blue, senescence) and immunofluorescence images of p21 (senescence), activin A (SASP), and cytokeratin-7 (trophoblast) in placenta from a patient with preeclampsia versus a patient with normotensive pregnancy. Scale bar, 100 μm. Blue, DAPI (nuclei); red, target antigen.
Association of the placental SASP protein, activin A, with cardiac dysfunction and HF severity in preeclampsia and PPCM
Among the 28 shared SASP proteins, activin A was the most highly up-regulated in preeclamptic placenta (gene ID: INHBA, 5.0 fold, P = 0.0008; Fig. 2B). Moreover, established downstream targets of activin A signaling, including follistatin-like 3 (FSTL3) and serpin family E member 1 (SERPINE1), were the next two most highly up-regulated SASP genes (Fig. 2B), suggesting a role for activin biology in these pregnancy-induced cardiovascular diseases. Post hoc analysis of publicly available placental single-cell and single-nucleus RNA sequencing datasets (27) confirmed that placental gene expression of INHBA, FSTL3, and SERPINE1 all increased in preeclampsia, with senescent trophoblasts being the predominant placental fetal cells responsible for this activin A–related SASP signal (Fig. 2, C to E, and figs. S3 and S4).
Although activin A, a member of the transforming growth factor–β superfamily, has previously been reported as increased in hypertensive disorders of pregnancy and to have an important role in HF among older adults (28–31), its functional contribution in pregnancy-induced HF is unclear. To investigate this further, we used two independent cohorts that included women with incident preeclampsia or PPCM for validation. The preeclampsia cohort was generated from a racially diverse maternal population in the United States, which included women with preeclampsia, gestational hypertension, or normotensive pregnancies (table S3). The PPCM validation cohort was derived from the Investigations of Pregnancy-Associated Cardiomyopathy (IPAC) Study, the largest prospective PPCM cohort in North America, and included patients with PPCM or uncomplicated pregnancies, along with nonpregnant women with NICM or no HF (table S4) (32, 33). In both cohorts, serum activin A concentrations were not only higher in patients with preeclampsia or PPCM, compared with matched pregnant and nonpregnant controls (Fig. 3, A and B), but also associated with worsening cardiac function and/or HF severity (Fig. 3, D to F). Evidence of increased activin A signaling was also detected in the hearts of women with advanced PPCM compared with individuals without HF (Fig. 3C) (34). Moreover, plasma from patients with preeclampsia versus matched normotensive pregnant controls directly induced transcriptional profiles of activin A signaling and nonsignificantly increased pathologic gene expression profiles (NPPB and MYH7, P = 0.1) in human induced pluripotent stem cell–derived cardiomyocytes (iPSC-CMs), independent of hemodynamic influences (fig. S5). Together, these data raised the possibility that placental-derived SASP proteins, specifically activin A, could contribute to the development of acute cardiac dysfunction in pregnancy-induced HF syndromes.
Fig. 3. Association of SASP protein, activin A, with cardiac dysfunction and HF severity in human preeclampsia and PPCM.

(A) Violin plot of third trimester serum activin A concentration in women with preeclampsia (n = 21), compared with maternal- and gestational-age–matched individuals with gestational hypertension (n = 19) or normotensive pregnancy (n = 18). Bold line is median. Dashed line is quartile. See table S3 for individual data. Pairwise comparisons with Wilcoxon rank sum test. Normotensive control versus gestational hypertension (P = 0.01), normotensive control versus preeclampsia (P < 0.001). (B) Violin plot of postpartum serum activin A concentration in women with PPCM (n = 82) compared with matched healthy pregnant (n = 8), healthy nonpregnant (n = 11), and nonpregnant nonischemic cardiomyopathy (NICM; n = 13) controls. See table S4 for individual data. Pairwise comparisons with Wilcoxon rank sum test. Healthy pregnant versus PPCM (P < 0.001), healthy nonpregnant versus PPCM (P < 0.001), and NiCM versus PPCM (P = 0.08). (C) Cardiac gene expression of downstream activin A targets in individuals with severe PPCM (n = 3) versus control individuals without HF (n = 13). Bar graphs show means ± SD with individual data points displayed. Mann-Whitney or Student’s t test. FSTL3 (P = 0.005), SERPINE1 (P = 0.04), CCN2 (P = 0.0009). (D and E) Pearson correlations of serum activin A with left ventricle (LV) systolic function metrics. Global longitudinal strain (GLS) in the preeclampsia cohort (n = 57), r = 0.6, P < 0.001 (D), and PPCM cohort (n = 94), r = 0.5, P < 0.001 (E), cohorts. More negative GLS indicates better systolic function. Correlation of LV ejection fraction in PPCM cohort (n = 111), r = −0.5; P < 0.001 (E). (F) Association of serum activin A with HF severity metrics in patients with PPCM. Brain natriuretic protein (BNP), Pearson correlation, r = 0.3; P = 0.04, n = 46. New York Heart Association (NYHA) functional class, (I, n = 11; II, n = 35; III, n = 23; IV, n = 13), displayed as violin plot (bold line, median; dashed line, quartiles). One-way analysis of variance (ANOVA) with post hoc Tukey’s test. #P ≤ 0.1, *P < 0.05, **P < 0.01, ***P < 0.001.
Senolytics attenuate placental senescence and improve cardiac function in a murine model of PPCM
To assess the functional role of placental senescence in PPCM, we used a murine model of PPCM in which the peroxisome proliferator–activated receptor γ coactivator-1α (Ppargc1a) gene, which encodes the PGC1α protein, is knocked out (KO) specifically in cardiomyocytes. For simplicity, these mice will be referred to as PGC1α KO mice here forth. Similar to many patients with PPCM, PGC1α KO mice harbor a genetic predisposition to HF and develop progressively more severe cardiac dysfunction with serial pregnancies (6). We found that even a single pregnancy decreased cardiac systolic function, as measured by left ventricular (LV) fractional shortening (FS), by ~43% (table S5), in association with a pathological gene expression profile and evidence of increased cardiac activin A signaling (figs. S6 and S7). The increase in activin A signaling in the late pregnant PGC1α KO heart occurred in the absence of increased cardiac activin A expression, suggesting that it is likely being driven by an extracardiac source of activin A, such as the placenta.
Placental senescence and a similar placental SASP profile also occurred in mice (fig. S8), supporting the use of this species for studying placental senescence biology. In accordance with clinical PPCM, which presents in late pregnancy, we found that both placental senescence and SASP expression increased progressively throughout mid to late gestation (fig. S9). Compared with other maternal organs, SASP expression was generally higher in placenta, supporting it as the primary source of most of the circulating SASP in late pregnancy (fig. S10). Activin A gene expression and protein secretion were both >80-fold higher in the late gestational placenta compared with other maternal organs (figs. S10 and S11).
In the PGC1α KO PPCM model, we found that placental expression of p16, p21, and activin A were further increased (Fig. 4A). Similar to our human preeclampsia samples, immunofluorescence characterization of placentas confirmed the presence of these senescence markers and activin A expression in the trophoblast-enriched labyrinth area (fig. S12). The smaller size of the murine placenta also afforded us the ability to simultaneously analyze the decidua, junctional zone, and labyrinth. This revealed that the decidua, which primarily consists of maternal-derived stromal cells, displayed higher expression of p16, p21, and activin A compared with the fetal portions of the placenta, suggesting that it likely also contributes to the senescence pathobiology in preeclampsia and PPCM (fig. S13).
Fig. 4. Fisetin attenuates placental senescence and activin A expression in a mouse model of PPCM.

During their first pregnancy, PGC1α KO mice were treated with the senolytic fisetin (20 mg/kg per day) versus DMSO vehicle from gestational day 13 (GD13) to GD17. n = 6 per group. Pregnant floxed (fl/fl) littermate controls (n = 7) treated with DMSO vehicle from GD13 to GD17 were also included to assess for placental differences between healthy and PPCM mice. (A) Representative Western blot images and quantification of senescence markers (p16, p21, and p53) and activin family SASP proteins (activin A, FSTL3, and PAI-1) in placental tissue at GD18. Protein amounts normalized to vinculin. One-way ANOVA with post hoc Tukey’s test. #P ≤ 0.1, *P < 0.05, **P < 0.01, ***P < 0.001. (B) Representative images of SA-βgal and immunofluorescence stains of trophoblast (cytokeratin 7) and stromal cell (vimentin) markers in placental tissue at GD18. Placental decidua (D), labyrinth (L), and junctional zone (J) displayed. Scale bar, 100 μm. For SA-βgal: blue, senescence. For immunofluorescence: blue, DAPi (nuclei); red, target antigen. (C) Relative quantification of SA-βgal intensity per total placenta area or specific anatomical areas. One-way ANOVA with post hoc Tukey’s test. #P ≤ 0.1, *P < 0.05, **P < 0.01. Bar graphs show means ± SD with individual data points displayed.
To determine whether attenuating placental senescence can reverse cardiac dysfunction and activin A signaling in PPCM, we treated pregnant PGC1α KO mice with the senolytic fisetin (Figs. 4 and 5). Fisetin is a flavonoid that selectively eliminates senescent cells in aged mice (35) and has been shown to be safe in pregnant rodents at low doses (36). Five consecutive days of orally administered fisetin (20 mg/kg per day), spanning mid to late pregnancy, effectively attenuated placental senescence, as indicated by reductions in p16 and p21 protein expression (Fig. 4A) and SA-βgal intensity in the junctional zone of the placenta (Fig. 4, B and C). In addition, both latent and active forms of activin A were decreased in the placentas from fisetin-treated mice (Fig. 4A).
Fig. 5. Fisetin partially rescues cardiac dysfunction in a mouse model of PPCM.

(A) Schematic of fisetin treatment study in PGC1α KO mice during their first pregnancy. DMSO = vehicle control. n = 6 per group. (B) Representative echocardiography images of the LV before treatment and after treatment. Quantification of LV FS (metric of systolic function). Two-way ANOVA with post hoc Šidák’s test. Posttreatment LV FS, DMSO versus fisetin, P = 0.009. (C) Heart and lung weights normalized to body weight. Student’s t test. Lung weight to body weight, P = 0.09. (D) Pathologic gene expression profile in cardiac tissue. Student’s t test. Nppa (P = 0.04) and Myh7 (P = 0.06). (E) Representative Western blot images and quantification of activin A and ActRii downstream targets (FSTL3, PAI-1, and CTGF) in cardiac tissue at GD18. Student’s t test. FSTL3 (P = 0.07), CTGF (P = 0.008), activin A (P = 0.02). Bar graphs show means ± SD with individual data points displayed. #P ≤ 0.1, *P < 0.05, **P < 0.01, ***P < 0.001.
Compared with pregnant PGC1α KO mice treated with dimethyl sulfoxide (DMSO) vehicle control, fisetin treatment resulted in a substantive improvement in cardiac function reflected in an LV FS that was 34% higher than vehicle control–treated KOs (Fig. 5B and table S6). We observed a nonsignificant decrease in lung weight in fisetin-treated animals (P = 0.09) (Fig. 5C) in addition to reduction in some of the pathological markers associated with HF and downstream targets of activin A (Fig. 5, D and E). As previously reported (36), treatment with this relatively low dose of fisetin during pregnancy appeared safe without adverse effects on gestational duration, placental weight, placental resorption, placental/fetal weight ratios, litter size, or congenital malformations (fig. S14) (37–41). In addition, no differences in Cdkn1a, Cdkn2a, or Tp53 gene expression were detected in the heart, suggesting that fisetin’s effects on cardiac function in this model were likely being mediated more through its attenuation of placental senescence, as opposed to direct cardiac effects (fig. S15).
Postpartum inhibition of the SASP factor activin A improves cardiac function in a murine model of severe PPCM
Although attenuating placental senescence with fisetin during late gestation improved cardiac function in a murine model of PPCM, clinically, PPCM most often presents after delivery, typically in the first month postpartum. Because our serum proteomic profiling in patients with PPCM suggested that SASP proteins were still increased in the maternal circulation after delivery (Figs. 1 and 3), we hypothesized that residual placental-derived SASP likely still contributed to maternal cardiac dysfunction in PPCM, even after expulsion of the placenta. To test whether inhibiting SASP factors in the early postpartum period could yield benefits similar to attenuating placental senescence directly, we treated PGC1α KO mice for 2 weeks postpartum after their second pregnancy with a monoclonal antibody (CDD866, Novartis Pharmaceuticals) that blocks the activin A receptors activin type IIA and IIB (collectively referred to as ActRIIA/B) (Fig. 6A). Because the cardiomyopathy becomes more severe with repeated pregnancy in this model (6), these mice had even more severe cardiac dysfunction than the mice treated with fisetin during their first pregnancy (fig. S16). Postpartum treatment with CDD866 resulted in a marked improvement in cardiac function reflected in a 77% higher LV FS compared with isotype control-treated animals (Fig. 6B and fig. S17). The isotype control–treated animals had increased heart and lung weights, indicative of HF with pulmonary congestion, both of which were reversed in CDD866-treated animals (Fig. 6C and fig. S16). Compared with fisetin treatment, CDD866 treatment resulted in a more uniform and marked reduction in pathological markers associated with HF, along with more consistent and extensive inhibition of activin A signaling, reducing expression of its downstream targets, FSTL3, plasminogen activator inhibitor-1 (PAI-1), and connective tissue growth factor (CTGF) (Fig. 6, D and E). Postpartum ActRIIA/B inhibition did not affect myocardial microvascular remodeling, the primary mechanism by which previously identified circulating factors, such as sFLT1 and cleaved prolactin, contribute to PPCM pathophysiology (fig. S18) (5, 6). However, consistent with prior studies (30, 42), ActRIIA/B inhibition increased protein abundance of sarcoplasmic reticulum Ca2+ adenosine triphosphatase 2, a critical regulator of cardiomyocyte contractility (fig. S18). Collectively, these data provide additional evidence supporting a causal role of the placental-derived SASP factor, activin A, in PPCM, and show that inhibition of its binding receptors, ActRIIA/B, in the early postpartum period improves cardiac function through different mechanisms than antagonizing sFLT1 and cleaved prolactin pathobiology.
Fig. 6. Postpartum activin type II receptor inhibition improves cardiac dysfunction in severe PPCM.
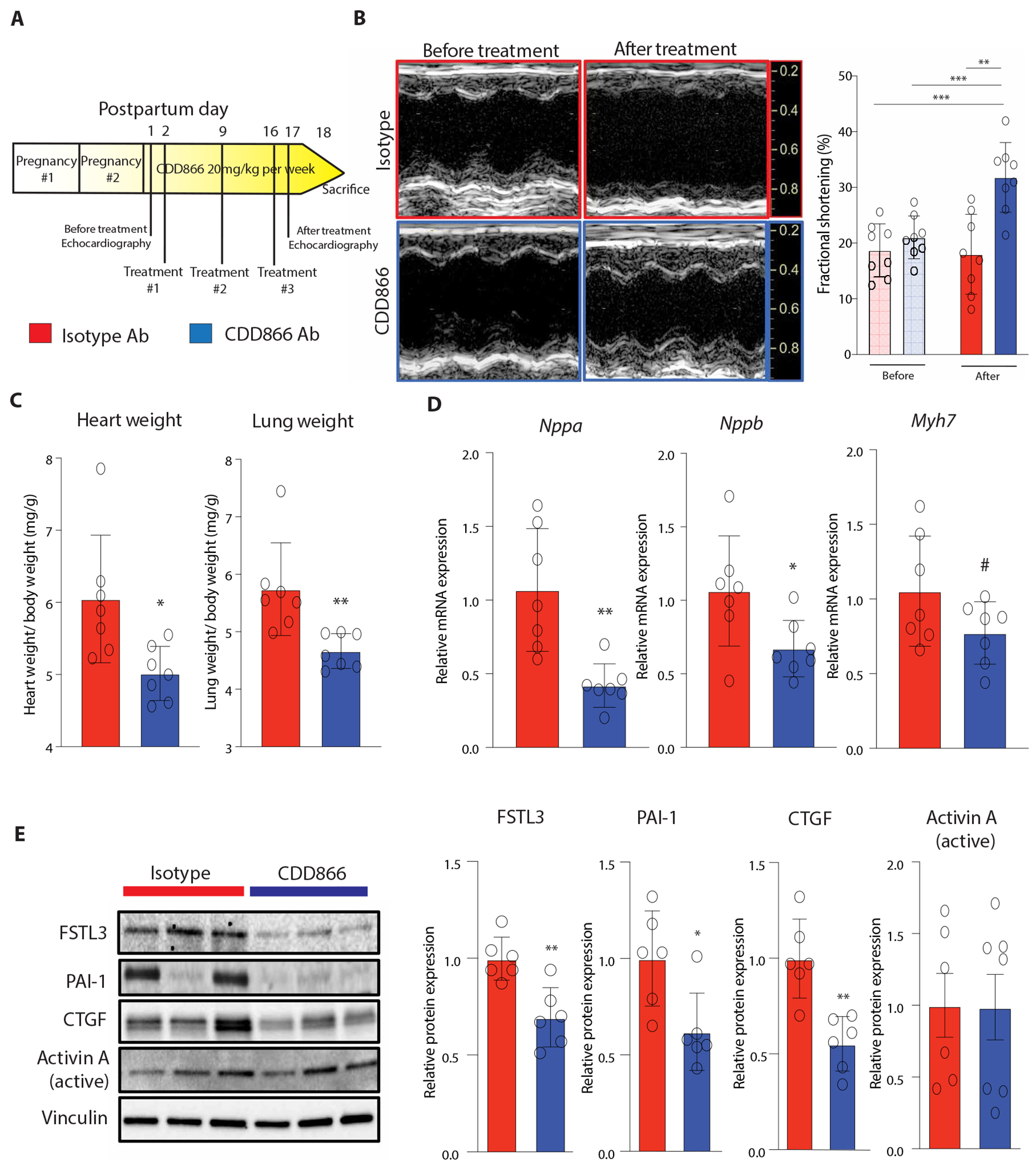
(A) Schematic of CDD866 treatment study. PGC1α KO mice were subjected to two serial pregnancies to induce severe PPCM, after which they were treated with weekly injections of CDD866 (ActRIIA/B blocking Ab) versus isotype control Ab for 2 weeks (n = 8 per group). One mouse died in each group before the final time point for tissue collection. (B) Representative echocardiography images and quantification of LV FS before and after treatment. Two-way ANOVA with post hoc Šidák’s test. **P < 0.01, ***P < 0.001. (C) Heart and lung weights normalized to body weight. Student’s t test used for heart weight (P = 0.01). Mann-Whitney test used for lung weight (P = 0.001). (D) Pathologic gene expression profile in hearts. Student’s t test. Nppa (P = 0.002), Nppb (P = 0.03), and Myh7 (P = 0.1). (E) Representative Western blot images and quantification of activin A and downstream ActRII targets. Student’s t test or Mann-Whitney test. FSTL3 (P = 0.003), PAI-1 (P = 0.02), and CTGF (P = 0.001). Bar graphs show means ± SD with individual data points displayed. #P ≤ 0.1, *P < 0.05, **P < 0.01, ***P < 0.001.
DISCUSSION
Our study identified amplified placental senescence as a core process underlying the pathophysiology shared between PPCM and preeclampsia. Although placental senescence was found over 50 years ago and has been associated with adverse pregnancy outcomes (22, 43), its roles in disease pathogenesis and remote organ dysfunction have remained unclear.
Here, we present evidence demonstrating a functional role of placental senescence in pregnancy-induced HF, paradoxically linking fundamental aging biology to cardiac dysfunction in relatively young pregnant women. The inflammatory properties of senescence and its associated SASP have been implicated in the pathogenesis of chronic diseases in older adults (19, 20), but its role in cardiovascular diseases of pregnancy, conditions inherently linked to younger individuals at reproductive age, was unexpected. Emerging epidemiological data in women with hypertensive disorders of pregnancy, however, support this unique relationship, suggesting that an accelerated form of cardiovascular aging occurs in these women (44). Whether this long-term accelerated aging phenotype is initially triggered by increased placental senescence at the time of incident disease or reflects a preexisting propensity remains to be determined. However, the ability of SASP, including activin A, to induce irreversible paracrine senescence (45) raises the possibility that amplified placental senescence during pregnancy may initiate a cascade that leads to systemic and progressive cardiovascular aging or senescence in these women (44, 46). We found that the circulating SASP in women with preeclampsia persists well into the postpartum period (data file S1).
In a broader context, the identification of a functional role of placental senescence in regulating maternal cardiac function establishes a framework for investigating disease pathogenesis and therapeutic development for pregnancy-related cardiac dysfunction. Dynamic fluid shifts and hemodynamic changes throughout gestation were previously considered the primary drivers of cardiac remodeling in pregnancy (47). However, our data challenge this historical paradigm. This study not only adds to a growing body of evidence implicating a causal role of circulating factors in pregnancy-induced cardiac dysfunction (5, 6, 48) but also identifies a mechanism responsible for this secretory process, which should accelerate understanding of how and why the maternal heart remodels in pregnancy.
Our studies also leverage these insights to provide proof-of-concept support for two therapeutic approaches to PPCM: a senolytic, fisetin, delivered during pregnancy and an antibody that inhibits the receptors for activin A, one of the SASP proteins, delivered postpartum. Both were effective in an animal model of PPCM and warrant further investigation given the limited treatments available for this condition. Although activin A has been previously shown to increase in preeclampsia and contribute to age-related HF (28–30), our data link it to placental senescence biology and its functional effects in pregnancy-induced HF. Consistent with our data, prior reports have noted genetic associations between the ActRIIA locus and preeclampsia (49). Moreover, because inhibitors of this pathway have been approved for other clinical indications (50), these findings could lead to the development of much needed disease-specific treatments for PPCM and preeclampsia.
Whereas our data provide mechanistic insights into the shared pathophysiology of preeclampsia and PPCM with important therapeutic implications, several limitations are worth noting. First, the SASP profile identified in preeclampsia and PPCM was limited by the coverage of the proteomics platform used and previously identified SASP proteins, both of which continue to expand. Our normotensive control group for our preeclampsia studies also included women with early (< 34 weeks) delivery. Although some of these women may not represent normal pregnancy, we felt that gestational age was an important variable to control for given its association with placental maturation and senescence. Second, although extensive clinical and experimental validation of our top SASP candidate, activin A, was performed, other placental-derived SASP factors may also play important roles and should be further investigated. Third, although we provide proof-of-concept evidence showing that attenuating placental senescence improves cardiac function in an animal model of PPCM, the safety and efficacy of this approach needs to be rigorously assessed in clinical studies. Last, the cause of increased placental senescence in preeclampsia and PPCM remains unclear. Placental senescence is likely a normal part of pregnancy in preparing the placenta for delivery (51). Consistent with this, we saw evidence of placental senescence in normal murine pregnancy (figs. S8 to S10) (52). In the setting of genetic predisposition, as in patients with cardiomyopathy mutations (7–9) or the cardiac-specific PGC1a KO mice, even the typical SASP protein abundances likely suffice to induce cardiac dysfunction. However, the higher SASP observed in PPCM and preeclampsia may indicate a predisposition to SASP that could tip the balance even with milder or absent genetic predisposition (fig. S19). Accumulating evidence suggests that various triggers, such as DNA damage, oxidative stress, ischemia, and other environmental stresses, can exacerbate placental senescence (43, 51, 52). Thus, targeting these upstream placental stressors could represent an additional approach to treating pregnancy-associated cardiovascular disease.
In summary, with pregnancy-induced cardiovascular diseases representing a growing and leading cause of maternal mortality (1, 2), elucidating the pathobiology underlying these complications is critical to improving maternal health. We identify placental senescence as a causal factor driving the shared secretory pathophysiology in PPCM and preeclampsia. This paradigm, linking aging biology to the regulation of maternal cardiac function, not only provides mechanistic insights into cardiovascular diseases of pregnancy but also opens the door to new diagnostic and therapeutic approaches for these important conditions.
MATERIALS AND METHODS
Study design
This study was designed to investigate mechanisms underlying the shared secretory pathophysiology in PPCM and preeclampsia. With prior evidence suggesting similar circulating factors contribute to both diseases, we started by performing parallel case-control serum proteomics studies in patients with PPCM or preeclampsia compared with healthy postpartum or normotensive pregnant controls. The primary objectives were to identify a broader scope of shared circulating factors in these closely related diseases and to gain insights into the mechanisms driving this similar secretory phenomenon. With senescence biology emerging as the most highly up-regulated biological process in both diseases, we next performed extensive validation and senescence characterization in placental samples from patients with preeclampsia versus patients with normotensive pregnancy and mice with experimental PPCM versus healthy pregnant controls. Moreover, we validated the most highly up-regulated placental-derived SASP candidate in independent cohorts of patients with preeclampsia or PPCM, which included relevant controls and cardiac function and HF phenotyping. To then assess the functional role of placental senescence in regulating maternal cardiac function, we tested both a broad senolytic approach and a targeted SASP intervention in a murine model of PPCM. Given the case-control design of the study, randomization was not done. No statistical methods were used to predetermine sample sizes (n), which are indicated in the figures and legends. All human and animal studies were approved by the appropriate institutional review boards at the participating centers and, for human studies, conformed to the Declaration of Helsinki principles with all patients providing written informed consent.
Human specimens
Biospecimens (serum, placental, or heart tissue) from 281 human patients were included in this study: preeclampsia (n = 56), gestational hypertension (n = 19), PPCM (n = 98), nonpregnant NICM (n = 13), normotensive pregnancy without cardiac failure (n = 71), and non-pregnant controls (n = 24). Patients with preeclampsia or gestational hypertension were recruited from Beth Israel Deaconess Medical Center (Boston, MA), University of Chicago Medicine (Chicago, IL), and Brigham and Women’s Hospital (Boston, MA). Patients with PPCM were from a prospective registry in Germany (n = 16) or the multicenter IPAC study (n = 82) (32). Patients with NICM were from the Intervention in Myocarditis and Acute Cardiomyopathy registry. Pregnant and nonpregnant controls were matched as closely as possible to cases for maternal age and gestational age based on the study design (tables S1 to S4). Echocardiography analyses were performed by S.S. for the preeclampsia cohorts and by core labs for the German registry and IPAC PPCM cohorts (33). Biospecimens were collected at the time of study entry and stored at −80°C until analyses.
Serum proteomics
The 1.1k (1129 proteins) and 1.3k (1305 proteins) SOMAscan platforms were used for the preeclampsia and PPCM cohorts, respectively, using previously published methods (21, 30). Quality control was done using 12 hybridization control sequences for each serum sample with median normalization performed to remove sample or assay biases. Relative fluorescence units were quantified using oligonucleotide microarrays (Agilent Technologies).
Single-cell and single-nucleus RNA sequencing
Post hoc analysis was performed on single-cell and single-nucleus RNA sequencing datasets generated by Admati and colleagues (27). Detailed methods are outlined in the original manuscript. Briefly, placenta cotyledons were dissected immediately after delivery from 10 women with early-onset preeclampsia (<34 weeks of gestation) and three women with idiopathic preterm delivery (spontaneous delivery) before 34 weeks with no placental pathology, fetal malformations, or maternal chronic diseases. Maternal decidua was cleared from all samples before tissue processing. Isolated cells (~5000 to 6000) or nuclei (~8000 to 10,000) were then processed on the 10X Genomics Chromium platform. Sequencing was done using Illumina NextSeq or NovaSeq, targeting a depth of ~35,000 reads per cell.
Mice
Cardiomyocyte-specific PGC1α knockout mice (referred to for simplicity as PGC1α KO mice) were generated by crossing Ppargc1a floxed mice (Jackson Laboratory, #009666) with Myh6 cre mice (Jackson Laboratory, #011038) to induce cardiomyocyte-specific deletion of PGC1α. Cardiomyocyte-specific KO of the target Ppargc1a gene was confirmed by gene expression in whole-heart lysates. All mice used were on a C57BL/6 J background. Flox/flox littermates and C57BL/6 J mice were used as controls for the various experiments. All mouse experiments were approved by the MGH IACUC (institutional animal care and use committee).
Ex vivo maternal organ culture
Pregnant mice were anesthetized with ketamine and xylazine on gestational day 18 (GD18). Organs, including heart, gastrocnemius, liver, kidney, abdominal visceral fat, and placentas, were explanted and rinsed in sterile phosphate-buffered saline (PBS). Organs were immediately cultured for 24 hours in 5 ml of high glucose, serum-free Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1% penicillin/streptomycin, at 37°C, 5% CO2. The medium from cultured organs was collected and frozen at −80°C until ready for analysis.
Senolytic mouse study
PPCM was induced in 10- to 12-week-old female PGC1α KO mice with a first pregnancy. Pregnancy was confirmed by the presence of a vaginal plug the day after mating, and mice were followed serially to assess for anticipated weight gain associated with pregnancy. The senolytic fisetin (Selleck Chemicals; S2298) was diluted in water and 0.1% DMSO and administered at 20 mg/kg per day by oral gavage from GD13 to G17 of the first pregnancy versus vehicle (0.1% DMSO). Cardiac function was assessed with serial echocardiography done on GD11 (before treatment) and GD17 (after treatment). At GD18, mice were euthanized, and hearts and placenta were collected, weighed, and stored for future analysis.
Mouse fetuses were also isolated on GD18, rinsed in PBS, dried on absorbent tissue, and then weighed. Assessment of fetal development and malformations was done by macroscopic observations using reference anatomical pictures of normal and pathological fetal development (37–41). Fetal development assessment included the following: body weight, curvature of the caudal body axis, craniofacial shape, nose and midfacial cleft shape, eyelids and eye shape, ear shape, amount of interdigital webbing, shape, length and curvature of the tail, fragile embryo (skin, limbs, join), and presence or absence of edema and subcutaneous hemorrhage.
ActRII inhibition mouse study
Monoclonal antibody–mediated blockade of the activin type II receptors ActRIIA and ActRIIB was performed using previously published reagents and methods (30). Briefly, PGC1α KO mice were first subjected to two serial pregnancies to induce a severe form of PPCM (6). Immediately after the second pregnancy, mice were subcutaneously administered a dose (20 mg/kg per week) of CDD866, a murinized version of bimagrumab (Novartis Pharmaceuticals) versus an equivalent dose of isotype control antibody for 2 weeks. Mice received a total of three doses during the study period. Echocardiography was done after second pregnancy (before treatment), 9 to 10 days after second pregnancy (1-week treatment), and 16 to 17 days after second pregnancy (after treatment). Mice were euthanized ~24 hours after the third dose of antibody treatment.
Senescence and SASP analysis
The Senescence Cells Histochemical Staining kit (Sigma-Aldrich) was used to detect SA-βgal activity, a marker of senescent cells, in unfixed frozen sections of placental tissue. Placenta sections were incubated at 37°C without CO2 for 5 days (for human sections) and 72 hours (for mouse sections), after which slides were scanned using NanoZoomer 2.0RS (Hamamatsu). To quantify mean SA-βgal intensity per placenta, additional images were acquired with a 20× objective using a Leica DMi8 microscope equipped with a DFC7000T camera. To ensure representative quantification, two sections per placenta were used. For analysis of human samples, which only included the labyrinth area, three images were acquired for each section. For the mouse samples, because the entire placenta was able to be sectioned on a single slide, we took three images in each of the following anatomical areas (labyrinth, junctional zone, and decidua), yielding a total of nine images per section. The SA-βgal blue intensity for each image was measured with ImageJ software [National Institutes of Health (NIH)], which were then averaged to represent the mean SA-βgal blue intensity per placenta. For the mouse, additional quantification was performed for each of the specific anatomical areas.
To assess epigenetic DNA methylation in frozen placental samples, we used the MethylFlash Global DNA Methylation ELISA Easy Kit (Epigentek) to quantify amounts of 5-methylcytosine. Given the heterogeneity of human placental samples, we pooled five random tissue sections from each placenta together to represent a composite sample from each study individual.
Gene and protein expression of senescence markers and candidate SASP were measured using standard methods for real-time quantitative polymerase chain reactions, Western blotting, and immunofluorescence staining. Methods, primer sequences, and antibodies used are included in the supplementary materials.
Mouse echocardiography
Echocardiography was performed on unanesthetized mice using a Vivid E90 system (GE Healthcare) equipped with a high-frequency L8-18i-D linear array probe. Parasternal short-axis images at the papillary muscle level were acquired at a depth of 10 mm using M-mode and 2D imaging. Image analyses were performed off-line using EchoPACS software (GE Healthcare, version 201). LV FS was calculated as [(LVEDD – LVESD)/LVEDD] × 100, where LVEDD is the left ventricular end-diastolic dimension and LVESD is the left ventricular end-systolic dimension. Heart rate was averaged over nine beats. LV mass (LVM) was calculated using a modified cubed formula: LVM = 1.05 × [(AWT + PWT + LVEDD)3 – LVEDD3), where AWT is the anterior wall thickness (at end diastole) and PWT is the posterior wall thickness (at end diastole). Image acquisition and analysis were done in a blinded fashion.
Myocardial capillary density quantification
To quantify myocardial capillary density in murine hearts, midventricular sections were stained with rabbit anti-mouse CD31 (1:50; 77699, Cell Signaling Technology). Twelve randomly selected images were taken of each heart. The number of capillaries (CD31-positive cells) was measured in each image and normalized to the area. Quantification of all 12 images was then averaged to represent a single sample. Image quantification was done using ImageJ software (NIH) in a blinded fashion.
iPSC-cardiomyocyte study
Human iPSC-CMs were used for this study. Stem cells were maintained in StemFlex media (Life Technologies) and passaged using ReLeSR (STEMCELL Technologies). At 80% confluency, cultures were dissociated into single-cell suspension using Accutase (STEMCELL Technologies). After 3 days, single cells grew into uniformly sized spheroids, and ventricular cardiomyocyte differentiation was then launched by manipulating Wnt/β catenin signaling. On day 17, beating spheroids were dissociated and cultured on fibronectin-coated flasks. Cells were then subjected to glucose starvation for a 6-day period in a lactate purification media comprising DMEM with no glucose, no pyruvate, 1 M lactate, 200 μM ascorbic acid, and sodium selenite (14 ng/ml). After purification, iPSC-CMs were cultured for 24 hours at 37°C, 5% CO2 in serum free media (RPMI 1640 supplemented with B27 insulin) supplemented with 2% plasma from either patients with preeclampsia versus matched patients with normotensive pregnancies.
Statistical analysis
Proteomics analysis was done using median normalized relative amounts of serum protein analytes that were imported into R, and differential protein expression was assessed using limma. To adjust for multiple hypothesis testing, a Bonferroni correction was used with Padj < 0.05 considered significant. Protein pathway analysis was performed using gene set enrichment analysis (Broad Institute) with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. Because the SASP is not formally included in KEGG, we generated a SASP protein set from proteins documented as secreted by senescent cells in PubMed as of January 2019. The search term “SASP” was used in conjunction with an English language filter yielding 984 publications, which were reviewed. Only proteins derived from a senescent cell type and directly measured in the media were included in the protein set, resulting in a final list of 250 SASP proteins (data file S1). Pathways with a false discovery rate < 0.25 were considered significant. To validate SASP pathway enrichment, additional independent enrichment analysis was performed using more contemporary SASP protein sets (18, 21).
For the human validation cohorts, given the non-normal distribution of activin A serum concentrations, pairwise comparisons with Wilcoxon rank sum tests were used to assess differences in circulating activin A. Fisher’s exact test was used for categorical variables and a two-sided t test for continuous variables in the patient cohorts. Pearson correlations or Kruskal-Wallis tests were performed to assess activin A associations with cardiac function and HF metrics. For animal and cardiomyocyte studies, unpaired Student’s t test, Mann-Whitney test, Fisher’s exact test, and one-way or two-way analysis of variance followed by post hoc multiple comparison testing were used as outlined in the figure legends.
Supplementary Material
Acknowledgments:
We would like to thank Novartis Pharmaceuticals for the donation of the CDD866 reagent.
Funding:
Funding for this work was provided by the NIH (K76AG064328 and R01HL170058 to J.D.R.; R01AG061034 and R35HL155318 to A.R.; K08HL146963, R01HL163234, and R03HL162756 to K.J.G.; K08HL166687 to M.C.H.; R37AG013925 and R33AG061456 to J.L.K.; R01HL126797 and R01HL152446 to Z.A.; R01HL092577, R01HL157635, and R01HL139731 to P.T.E.; R01HL102429 to D.M.M.; T32HL007208 and K01AG0880077 to P.X.; R01HL148191-01A1 to S.S.; and R56HL157579-01 to S.R.); MGH Transformative Scholars Award (to J.D.R.); Fred and Ines Yeatts Fund for Innovative Research (to J.D.R.); Hassenfeld Scholars Award (to J.D.R.); American Heart Association (940166 and 979465 to M.C.H.; 18SFRN34110082 to P.T.E.; and AHA MERIT Award to A.R.); Department of Defense (W81XWH18 to Z.A.); European Union (MAESTRIA 965286 to P.T.E.); Deutsche Forschungsgemeinschaft KFO 311 (HI 842/10-2 to D.H.-K. and M.R.-H., RI2531/2-2 to M.R.-H., and RI2531/4-1 to M.R.-H.); Foundation Leducq (19CVD02 to D.H.-K.); Lower Saxony Ministry of Science and Culture REBIRTH I/II (ZN3400 to D.H.-K. and M.R.-H.); Connor Fund (to J.L.K.); Robert J and Theresa Ryan Fund (to J.L.K.); and the Noaber Foundation (to J.L.K.).
Competing interests:
J.D.R. and A.R. are coinventors on patents (WO-2018175460-A1; Methods for Preventing and Treating Heart Disease and 11834508; Method of treating structural and/or functional cardiac abnormalities by administering an anti-ActRII receptor antibody). J.D.R. reports research support from Amgen, Keros, and Genentech. A.R. reports consulting fees from Keros and Versanis as well as serving as a scientific cofounder and equity owner of Thryv Therapeutics. M.C.H. reports research support from Genentech, consulting fees from CRISPR Therapeutics, and advisory board service for Miga Health. S.R. reports serving as a consultant to Roche and Thermo Fisher Scientific and has received funding from Roche and Siemens for studies related to the use of angiogenic factors in pregnancy, which is unrelated to the present work. J.B. reports research support from Zoll, CVRx, Abiomed, Norgine, and Roche and honoraria for lectures/consulting from Novartis, Vifor, Bayer, Pfizer, Boehringer Ingelheim, AstraZeneca, Cardior, CVRx, BMS, Amgen, Corvia, Norgine, Edwards, and Roche. P.T.E. reports research support from Bayer AG, Novo-Nordisk, Bristol Myers Squibb and Pfizer and has served on advisory boards or consulted for Bayer AG and MyoKardia. K.J.G. has served as a consultant to Illumina, Aetion, Roche, and BillionToOne. S.A.K. reports research support from Thermo Fisher Scientific, Roche, Siemens, and Beckman Coulter and serves as a consultant for Thermo Fisher Scientific, Roche, and Siemens. S.A.K. has financial interest in Aggamin Pharmaceuticals and Comanche Biopharma and has multiple patents on angiogenic biomarkers that have been outlicensed to multiple companies. All reported research support and consulting are unrelated to this work.
The IPAC Investigators
In addition to IPAC members who are authors (D.M.M., K.H.-Y., J.G., J.D., and M.S.), the following IPAC members are collaborators who have contributed to study design, data analysis, and interpretation:
James D. Fett17, Jessica Pisarcik17, Charles McTiernan17, Erik Schelbert17, Rami Alharethi20, Kismet Rasmusson20, Kim Brunisholz20, Amy Butler20, Deborah Budge20, A.G. Kfoury20, Benjamin Horne20, Joe Tuinei20, Heather Brown20, Allen J. Naftilan16, Jill Russell16, Darla Freehardt16, Eileen Hsich21, Cynthia Oblak21, Greg Ewald22, Donna Whitehead22, Jean Flanagan22, Anne Platts22, Uri Elkayam23, Jorge Caro23, Stephanie Mullin23, Michael M. Givertz24, M. Susan Anello24, Navin Rajagopalan25, David Booth25, Tiffany Sandlin25, Wendy Wijesiri25, Leslie T. Cooper26, Lori A. Blauwet26, Joann Brunner26, Mary Phelps26, Ruth Kempf26, Kalgi Modi27, Tracy Norwood27, Joan Briller28, Decebal Sorin Griza28, G. Michael Felker29, Robb Kociol29, Patricia Adams29, Gretchen Wells30, Vinay Thohan30, Deborah Wesley-Farrington30, Sandra Soots30, Richard Sheppard31, Caroline Michel31, Nathalie Lapointe31, Heather Nathaniel31, Angela Kealey32, Marc Semigran1, Maureen Daher1, John Boehmer14, David Silber14, Eric Popjes14, Patricia Frey14, Todd Nicklas14, Jeffrey Alexis33, Lori Caufield33, John W. Thornton III34, Mindy Gentry34, Vincent J.B. Robinson34, Gyanendra K. Sharma34, Joan Holloway34, Maria Powell34, David Markham35, Mark Drazner35, Lynn Fernandez35, Mark Zucker36, David A. Baran36, Martin L. Gimovsky36, Natalia Hochbaum36, Bharati Patel36, Laura Adams36, Gautam Ramani37, Stephen Gottlieb37, Shawn Robinson37, Stacy Fisher37, Joanne Marshall37, Jennifer Haythe38, Donna Mancini38, Rachel Bijou38, Maryjane Farr38, Marybeth Marks38, Henry Arango38, Biykem Bozkurt39, Mariana Bolos39, Paul Mather40, Sharon Rubin40, Raphael Bonita40, Susan Eberwine40, Hal Skopicki41, Kathleen Stergiopoulos41, Ellen McCathy-Santoro41, Jennifer Intravaia41, Elizabeth Maas41, Jordan Safirstein42, Audrey Kleet42, Nancy Martinez42, Christine Corpoin42, Donna Hesari42, Sandra Chaparro43, Laura J. Hudson43, Jalal K. Ghali44, Zora Injic44, Ilan S. Wittstein45.
20Heart Institute, Intermountain Medical Center, Murray, UT 84157, USA. 21Robert and Suzanne Tomsich Department of Cardiovascular Medicine, Cleveland Clinic, Cleveland, OH 44195, USA. 22Division of Cardiology, Washington University School of Medicine, St Louis, MO 63110, USA. 23Division of Cardiovascular Medicine, University of Southern California Keck School of Medicine, Los Angeles, CA 90033, USA. 24Division of Cardiovascular Medicine, Brigham and Women’s Hospital, Boston, MA 02115, USA. 25Division of Cardiovascular Medicine, University of Kentucky College of Medicine, Lexington, KY 40536, USA. 26Department of Cardiovascular Medicine, Mayo Clinic, Rochester, MN 55905, USA. 27Division of Cardiology, Louisiana State University Health Science Center, Shreveport, LA 71103, USA. 28Division of Cardiology, University of Illinois College of Medicine, Chicago, IL 60612, USA. 29Division of Cardiology, Duke University School of Medicine, Durham, NC 27705, USA. 30Department of Cardiovascular Medicine, Wake Forest University School of Medicine, Winston-Salem, NC 27157, USA 31Division of Cardiology, Jewish General Hospital, Montreal H3T 1E2, Canada. 32Division of Cardiology, Libin Cardiovascular Institute, University of Calgary, Calgary T2N 1 N4, Canada. 33Department of Cardiology, University of Rochester Medical Center, Rochester, NY 14642, USA. 34Division of Cardiology, Medical College of Georgia, Augusta, GA 30912, USA. 35Division of Cardiology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA. 36Division of Cardiology, Newark Beth Israel Medical Center, Newark, NJ 07112, USA. 37Division of Cardiovascular Medicine, University of Maryland School of Medicine, Baltimore, MD 21201, USA. 38Division of Cardiology, Columbia University Irving Medical Center, New York, NY 10032, USA. 39Section of Cardiology, Baylor College of Medicine, Houston, TX 77030, USA. 40Division of Cardiology, Thomas Jefferson University Sidney Kimmel Medical College, Philadelphia, PA 19107, USA. 41Division of Cardiology, Stony Brook University Renaissance School of Medicine, Stony Brook, NY 11794, USA. 42Gagnon Cardiovascular Institute, Morristown Medical Center, Morristown, NJ 07960, USA. 43Division of Cardiovascular Medicine, University of Miami Miller School of Medicine, Miami, FL 33136, USA. 44Division of Cardiology, Harper University Hospital, Detroit, MI 48201, USA. 45Division of Cardiology, Johns Hopkins School of Medicine, Baltimore, MD 21287, USA.
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. The PGC1α KO mice can be obtained by contacting the corresponding author through a material transfer agreement. CDD866 is a proprietary material of Novartis and may be shared under material transfer agreement under restricted industry standard conditions. All the raw proteomics data are included in the supplementary file. The codes used for proteomics analysis are available at https://zenodo.org/records/10669226. Links to the raw data in figshare and code in GitHub for the placenta scRNAseq are available in the original manuscript by Admati and colleagues (27).
REFERENCES AND NOTES
- 1.Say L, Chou D, Gemmill A, Tuncalp O, Moller AB, Daniels J, Gulmezoglu AM, Temmerman M, Alkema L, Global causes of maternal death: A WHO systematic analysis. Lancet Glob. Health 2, e323–e333 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Mehta LS, Warnes CA, Bradley E, Burton T, Economy K, Mehran R, Safdar B, Sharma G, Wood M, Valente AM, Volgman AS, American Heart Association Council on Clinical Cardiology, Council on Arteriosclerosis, Thrombosis and Vascular Biology, Council on Cardiovascular and Stroke Nursing; and Stroke Council, Cardiovascular Considerations in Caring for Pregnant Patients: A Scientific Statement From the American Heart Association. Circulation 141, e884–e903 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Hoes MF, Arany Z, Bauersachs J, Hilfiker-Kleiner D, Petrie MC, Sliwa K, van der Meer P, Pathophysiology and risk factors of peripartum cardiomyopathy. Nat. Rev. Cardiol 19, 555–565 (2022). [DOI] [PubMed] [Google Scholar]
- 4.Demakis JG, Rahimtoola SH, Peripartum cardiomyopathy. Circulation 44, 964–968 (1971). [DOI] [PubMed] [Google Scholar]
- 5.Hilfiker-Kleiner D, Kaminski K, Podewski E, Bonda T, Schaefer A, Sliwa K, Forster O, Quint A, Landmesser U, Doerries C, Luchtefeld M, Poli V, Schneider MD, Balligand JL, Desjardins F, Ansari A, Struman I, Nguyen NQ, Zschemisch NH, Klein G, Heusch G, Schulz R, Hilfiker A, Drexler H, A cathepsin D-cleaved 16 kDa form of prolactin mediates postpartum cardiomyopathy. Cell 128, 589–600 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Patten IS, Rana S, Shahul S, Rowe GC, Jang C, Liu L, Hacker MR, Rhee JS, Mitchell J, Mahmood F, Hess P, Farrell C, Koulisis N, Khankin EV, Burke SD, Tudorache I, Bauersachs J, del Monte F, Hilfiker-Kleiner D, Karumanchi SA, Arany Z, Cardiac angiogenic imbalance leads to peripartum cardiomyopathy. Nature 485, 333–338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ware JS, Li J, Mazaika E, Yasso CM, De Souza T, Cappola TP, Tsai EJ, Hilfiker-Kleiner D, Kamiya CA, Mazzarotto F, Cook SA, Halder I, Prasad SK, Pisarcik J, Hanley-Yanez K, Alharethi R, Damp J, Hsich E, Elkayam U, Sheppard R, Kealey A, Alexis J, Ramani G, Safirstein J, Boehmer J, Pauly DF, Wittstein IS, Thohan V, Zucker MJ, Liu P, Gorcsan J III, Namara DMM, Seidman CE, Seidman JG, Arany Z, IMAC-, IPAC Investigators, Shared Genetic Predisposition in Peripartum and Dilated Cardiomyopathies. N. Engl. J. Med 374, 233–241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Spaendonck-Zwarts KY, Posafalvi A, van den Berg MP, Hilfiker-Kleiner D, Bollen IA, Sliwa K, Alders M, Almomani R, van Langen IM, van der Meer P, Sinke RJ, van der Velden J, Van Veldhuisen DJ, van Tintelen JP, Jongbloed JD, Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. Eur. Heart J 35, 2165–2173 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Goli R, Li J, Brandimarto J, Levine LD, Riis V, McAfee Q, DePalma S, Haghighi A, Seidman JG, Seidman CE, Jacoby D, Macones G, Judge DP, Rana S, Margulies KB, Cappola TP, Alharethi R, Damp J, Hsich E, Elkayam U, Sheppard R, Alexis JD, Boehmer J, Kamiya C, Gustafsson F, Damm P, Ersboll AS, Goland S, Hilfiker-Kleiner D, McNamara DM, Imac I, Z. A. Investigators, Genetic and Phenotypic Landscape of Peripartum Cardiomyopathy. Circulation 143, 1852–1862 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA, Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J. Clin. Invest 111, 649–658 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, Schisterman EF, Thadhani R, Sachs BP, Epstein FH, Sibai BM, Sukhatme VP, Karumanchi SA, Circulating angiogenic factors and the risk of preeclampsia. N. Engl. J. Med 350, 672–683 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Gonzalez C, Parra A, Ramirez-Peredo J, Garcia C, Rivera JC, Macotela Y, Aranda J, Lemini M, Arias J, Ibarguengoitia F, de la Escalera GM, Clapp C, Elevated vasoinhibins may contribute to endothelial cell dysfunction and low birth weight in preeclampsia. Lab. Invest 87, 1009–1017 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Wu P, Haththotuwa R, Kwok CS, Babu A, Kotronias RA, Rushton C, Zaman A, Fryer AA, Kadam U, Chew-Graham CA, Mamas MA, Preeclampsia and Future Cardiovascular Health. Circ. Cardiovasc. Qual. Outcomes 10, e003497 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Bello N, Rendon ISH, Arany Z, The relationship between pre-eclampsia and peripartum cardiomyopathy: A systematic review and meta-analysis. J. Am. Coll. Cardiol 62, 1715–1723 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehdizadeh M, Aguilar M, Thorin E, Ferbeyre G, Nattel S, The role of cellular senescence in cardiac disease: Basic biology and clinical relevance. Nat. Rev. Cardiol 19, 250–264 (2022). [DOI] [PubMed] [Google Scholar]
- 16.Coppe JP, Desprez PY, Krtolica A, Campisi J, The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol 5, 99–118 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stojanovic SD, Fiedler J, Bauersachs J, Thum T, Sedding DG, Senescence-induced inflammation: An important player and key therapeutic target in atherosclerosis. Eur. Heart J 41 , 2983–2996 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, Holtz A, Shah S, Sharma V, Ferrucci L, Campisi J, Schilling B, A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLOS Biol. 18, e3000599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schafer MJ, Zhang X, Kumar A, Atkinson EJ, Zhu Y, Jachim S, Mazula DL, Brown AK, Berning M, Aversa Z, Kotajarvi B, Bruce CJ, Greason KL, Suri RM, Tracy RP, Cummings SR, White TA, LeBrasseur NK, The senescence-associated secretome as an indicator of age and medical risk. JCI Insight 5, e133668 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chaib S, Tchkonia T, Kirkland JL, Cellular senescence and senolytics: The path to the clinic. Nat. Med 28, 1556–1568 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roh JD, Kitchen RR, Guseh JS, McNeill JN, Aid M, Martinot AJ, Yu A, Platt C, Rhee J, Weber B, Trager LE, Hastings MH, Ducat S, Xia P, Castro C, Singh A, Atlason B, Churchill TW, Di Carli MF, Ellinor PT, Barouch DH, Ho JE, Rosenzweig A, Plasma Proteomics of COVID-19-Associated Cardiovascular Complications: Implications for Pathophysiology and Therapeutics. JACC Basic Transl. Sci 7, 425–441 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fox H, Senescence of placental villi. J. Obstet. Gynaecol. Br. Commonw 74, 881–885 (1967). [DOI] [PubMed] [Google Scholar]
- 23.Sliwa K, Forster O, Libhaber E, Fett JD, Sundstrom JB, Hilfiker-Kleiner D, Ansari AA, Peripartum cardiomyopathy: inflammatory markers as predictors of outcome in 100 prospectively studied patients. Eur. Heart J 27, 441–446 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Borzychowski AM, Sargent IL, Redman CW, Inflammation and pre-eclampsia. Semin. Fetal Neonatal Med 11,309–316 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, Gil J, Hara E, Krizhanovsky V, Jurk D, Maier AB, Narita M, Niedernhofer L, Passos JF, Robbins PD, Schmitt CA, Sedivy J, Vougas K, von Zglinicki T, Zhou D, Serrano M, Demaria M, Cellular senescence: Defining a path forward. Cell 179, 813–827 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Wilson VL, Jones PA, DNA methylation decreases in aging but not in immortal cells. Science 220, 1055–1057 (1983). [DOI] [PubMed] [Google Scholar]
- 27.Admati I, Skarbianskis N, Hochgerner H, Ophir O, Weiner Z, Yagel S, Solt I, Zeisel A, Two distinct molecular faces of preeclampsia revealed by single-cell transcriptomics. Med 4, 687–709.e7 (2023). [DOI] [PubMed] [Google Scholar]
- 28.Muttukrishna S, Knight PG, Groome NP, Redman CW, Ledger WL, Activin A and inhibin A as possible endocrine markers for pre-eclampsia. Lancet 349, 1285–1288 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Shahul S, Ramadan H, Nizamuddin J, Mueller A, Patel V, Dreixler J, Tung A, Lang RM, Weinert L, Nasim R, Chinthala S, Rana S, Activin a and late postpartum cardiac dysfunction among women with hypertensive disorders of pregnancy. Hypertension 72, 188–193 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Roh JD, Hobson R, Chaudhari V, Quintero P, Yeri A, Benson M, Xiao C, Zlotoff D, Bezzerides V, Houstis N, Platt C, Damilano F, Lindman BR, Elmariah S, Biersmith M, Lee SJ, Seidman CE, Seidman JG, Gerszten RE, Lach-Trifilieff E, Glass DJ, Rosenzweig A, Activin type II receptor signaling in cardiac aging and heart failure. Sci. Transl. Med 11 , eaau8680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yndestad A, Ueland T, Oie E, Florholmen G, Halvorsen B, Attramadal H, Simonsen S, Froland SS, Gullestad L, Christensen G, Damas JK, Aukrust P, Elevated levels of activin A in heart failure: Potential role in myocardial remodeling. Circulation 109, 1379–1385 (2004). [DOI] [PubMed] [Google Scholar]
- 32.McNamara DM, Elkayam U, Alharethi R, Damp J, Hsich E, Ewald G, Modi K, Alexis JD, Ramani GV, Semigran MJ, Haythe J, Markham DW, Marek J, Gorcsan J III, Wu WC, Lin Y, Halder I, Pisarcik J, Cooper LT, Fett JD, Investigators I, Clinical Outcomes for Peripartum Cardiomyopathy in North America: Results of the IPAC Study (Investigations of Pregnancy-Associated Cardiomyopathy). J. Am. Coll. Cardiol 66, 905–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugahara M, Kagiyama N, Hasselberg NE, Blauwet LA, Briller J, Cooper L, Fett JD, Hsich E, Wells G, McNamara D, Gorcsan J III, Investigators I, Global Left Ventricular Strain at Presentation Is Associated with Subsequent Recovery in Patients with Peripartum Cardiomyopathy. J. Am. Soc. Echocardiogr 32, 1565–1573 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ricke-Hoch M, Hoes MF, Pfeffer TJ, Schlothauer S, Nonhoff J, Haidari S, Bomer N, Scherr M, Stapel B, Stelling E, Kiyan Y, Falk C, Haghikia A, Binah O, Arany Z, Thum T, Bauersachs J, van der Meer P, Hilfiker-Kleiner D, In peripartum cardiomyopathy plasminogen activator inhibitor-1 is a potential new biomarker with controversial roles. Cardiovasc. Res 116, 1875–1886 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL, McGuckian C, Wade EA, Kato JI, Grassi D, Wentworth M, Burd CE, Arriaga EA, Ladiges WL, Tchkonia T, Kirkland JL, Robbins PD, Niedernhofer LJ, Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacob S, Thangarajan S, Effect of gestational intake of fisetin (3,3′,4′,7-Tetrahydroxyflavone) on developmental methyl mercury neurotoxicity in F1 generation rats. Biol. Trace Elem. Res 177, 297–315 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Hsu CW, Wong L, Rasmussen TL, Kalaga S, McElwee ML, Keith LC, Bohat R, Seavitt JR, Beaudet AL, Dickinson ME, Three-dimensional microCT imaging of mouse development from early post-implantation to early postnatal stages. Dev. Biol 419, 229–236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peeters MC, Hekking WM, Vainas T, Drukker J, van Straaten HW, Spatio-temporal curvature pattern of the caudal body axis for non-mutant and curly tail mouse embryos during the period of caudal neural tube closure. Anat. Embryol 195, 259–266 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Petiet AE, Kaufman MH, Goddeeris MM, Brandenburg J, Elmore SA, Johnson GA, High-resolution magnetic resonance histology of the embryonic and neonatal mouse: A 4D atlas and morphologic database. Proc. Natl. Acad. Sci. U.S.A 105, 12331–12336 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill MA. Embryology Mouse Stages. 2022; https://embryology.med.unsw.edu.au/embryology/index.php/Mouse_Stages. [Google Scholar]
- 41.Hill MA. Embryology Mouse Timeline Detailed. 2022; https://embryology.med.unsw.edu.au/embryology/index.php/Mouse_Timeline_Detailed. [Google Scholar]
- 42.Castillero E, Akashi H, Najjar M, Ji R, Brandstetter LM, Wang C, Liao X, Zhang X, Sperry A, Gailes M, Guaman K, Recht A, Schlosberg I, Sweeney HL, Ali ZA, Homma S, Colombo PC, Ferrari G, Schulze PC, George I, Activin type II receptor ligand signaling inhibition after experimental ischemic heart failure attenuates cardiac remodeling and prevents fibrosis. Am. J. Physiol. Heart Circ. Physiol 318, H378–H390 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sultana Z, Maiti K, Dedman L, Smith R, Is there a role for placental senescence in the genesis of obstetric complications and fetal growth restriction? Am. J. Obstet. Gynecol 218, S762–S773 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Honigberg MC, Zekavat SM, Aragam K, Klarin D, Bhatt DL, Scott NS, Peloso GM, Natarajan P, Long-term cardiovascular risk in women with hypertension during pregnancy. J. Am. Coll. Cardiol 74, 2743–2754 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, Jin H, Dharmalingam G, Snijders AP, Carroll T, Capper D, Pritchard C, Inman GJ, Longerich T, Sansom OJ, Benitah SA, Zender L, Gil J, A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol 15, 978–990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suvakov S, Ghamrawi R, Cubro H, Tu H, White WM, Tobah YSB, Milic NM, Grande JP, Cunningham JM, Chebib FT, Prata L, Zhu Y, Tchkonia T, Kirkland JL, Nath KA, Milosavljevic A, Garovic VD, Epigenetic and senescence markers indicate an accelerated ageing-like state in women with preeclamptic pregnancies. EBioMedicine 70, 103536 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung E, Leinwand LA, Pregnancy as a cardiac stress model. Cardiovasc. Res 101, 561–570 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Damp J, Givertz MM, Semigran M, Alharethi R, Ewald G, Felker GM, Bozkurt B, Boehmer J, Haythe J, Skopicki H, Hanley-Yanez K, Pisarcik J, Halder I, Gorcsan J III, Rana S, Arany Z, Fett JD, McNamara DM, IPAC Investigators, Relaxin-2 and Soluble Flt1 Levels in Peripartum Cardiomyopathy: Results of the Multicenter IPAC Study. JACC Heart Fail 4, 380–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roten LT, Johnson MP, Forsmo S, Fitzpatrick E, Dyer TD, Brennecke SP, Blangero J, Moses EK, Austgulen R, Association between the candidate susceptibility gene ACVR2A on chromosome 2q22 and pre-eclampsia in a large Norwegian population-based study (the HUNT study). Eur. J. Hum. Genet 17, 250–257 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fenaux P, Platzbecker U, Mufti GJ, Garcia-Manero G, Buckstein R, Santini V, Diez-Campelo M, Finelli C, Cazzola M, Ilhan O, Sekeres MA, Falantes JF, Arrizabalaga B, Salvi F, Giai V, Vyas P, Bowen D, Selleslag D, DeZern AE, Jurcic JG, Germing U, Gotze KS, Quesnel B, Beyne-Rauzy O, Cluzeau T, Voso MT, Mazure D, Vellenga E, Greenberg PL, Hellstrom-Lindberg E, Zeidan AM, Ades L, Verma A, Savona MR, Laadem A, Benzohra A, Zhang J, Rampersad A, Dunshee DR, Linde PG, Sherman ML, Komrokji RS, List AF, Luspatercept in patients with lower-risk myelodysplastic syndromes. N. Engl. J. Med 382, 140–151 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Gal H, Lysenko M, Stroganov S, Vadai E, Youssef SA, Tzadikevitch-Geffen K, Rotkopf R, Biron-Shental T, de Bruin A, Neeman M, Krizhanovsky V, Molecular pathways of senescence regulate placental structure and function. EMBO J. 38, e100849 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ciampa EJ, Flahardy P, Srinivasan H, Jacobs C, Tsai L, Karumanchi SA, Parikh SM, Hypoxia-inducible factor 1 signaling drives placental aging and can provoke preterm labor. eLife 12, RP85597 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. The PGC1α KO mice can be obtained by contacting the corresponding author through a material transfer agreement. CDD866 is a proprietary material of Novartis and may be shared under material transfer agreement under restricted industry standard conditions. All the raw proteomics data are included in the supplementary file. The codes used for proteomics analysis are available at https://zenodo.org/records/10669226. Links to the raw data in figshare and code in GitHub for the placenta scRNAseq are available in the original manuscript by Admati and colleagues (27).