

Abstract

A wide range of density functional methods and basis sets are available to derive the electronic structure and properties of molecules. Quantum mechanical calculations are too computationally intensive for routine simulation of molecules in the condensed phase, prompting the development of computationally efficient force fields based on quantum mechanical data. Parametrizing general force fields, which cover a vast chemical space, necessitates the generation of sizable quantum mechanical data sets with optimized geometries and torsion scans. To achieve this efficiently, choosing a quantum mechanical method that balances computational cost and accuracy is crucial. In this study, we seek to assess the accuracy of quantum mechanical theory for specific properties such as conformer energies and torsion energetics. To comprehensively evaluate various methods, we focus on a representative set of 59 diverse small molecules, comparing approximately 25 combinations of functional and basis sets against the reference level coupled cluster calculations at the complete basis set limit.
1. Introduction
General force fields offer a computationally efficient alternative to quantum mechanical (QM) calculations,1−13 in particular for pharmaceutical and biomolecular applications. Parametrizing general force fields or training machine learning (ML) potentials, for molecular mechanics (MM), requires extensive quantum chemistry data sets, with molecules covering a large chemical space, that inform the bonded parameters, typically bonds, angles, and torsions as well as nonbonded terms.14−21 The data sets include optimized chemical structures of different conformers of molecules and associated properties such as charges, bond orders, dipole moments, Hessian matrices, and torsion energy profiles for rotatable bonds, dimer interaction energies, among other data. A quantum mechanical method is needed that is globally accurate for different chemistries and properties, is computationally cost-effective, and can converge with a predefined set of hyperparameters (or a template with minimal changes) without too much human intervention, thus enabling automated generation of larger data sets essential for this effort. There are hundreds of density functionals, post Hartree–Fock methods, and basis sets to choose from, and it is cumbersome to pick one without a proper benchmark study.
Recent benchmark studies address the issue of how accurate a quantum mechanical method is with respect to a highly accurate gold standard such as coupled cluster with singles, doubles and perturbative triples in the complete basis set limit (CCSD(T)/CBS). Some studies are from the viewpoint of quantum chemistry method developers, where one would be concerned by a difference of even 0.1 kcal/mol in mean accuracy of absolute energies, with respect to the reference level of theory. However, from the perspective of practitioners engaged in force field development and general biochemical simulations, where relative energies such as conformer energetics and torsional profiles are of primary concern (as emphasized in Sellers et al.22), it is crucial that the quantum mechanical (QM) methods used to generate reference data for force field parametrization can reproduce torsion energy profiles with deviations of approximately 0.5 to 1.0 kcal/mol or smaller. This level of accuracy, as demonstrated in high-level QM calculations, is vital for ensuring the accuracy of trained force fields, and boosts the reliability of property predictions like protein–ligand binding affinities.22,23 By achieving accuracy within this specified range for the QM reference data employed in force field parametrization, and assuming the trained force field mimics the QM training data quite well, we can effectively capture the essential conformational behavior and energetic trends exhibited by the molecules under investigation. Our focus here is precisely in this area, conformational energetics, and not on modeling long-range electrostatic interactions, which may involve different considerations.24−27
Our primary focus is on accurately modeling small molecule organic compounds within pharmaceutically relevant regions of chemical space using force fields. As a result, we do not concern ourselves with evaluating the performance of quantum mechanical (QM) methods for compounds involving transition elements, lanthanides, noble gases, and other elements not directly implicated in the fundamental molecular interactions governing the binding and recognition of small molecule drugs. At the Open Force Field (OpenFF) Initiative,3,28 our attention lies on achieving performance benchmarks for organic molecules within the chemical space that is pertinent to small molecule drugs and that our force fields are currently capable of modeling. This chemical space encompasses C, H, O, N, P, S, F, Cl, Br, and I, as well as the monatomic ions Li+, Na+, K+, Rb+, F–, Cl–, Br–, and I–.
A variety of different method and basis set combinations in gas-phase calculations have been used in building various general force fields as well as ML potentials, depending on the force field and application. Some other common models were derived as follows:
GAFF: MP2/6-31G* was used as one of the sources of training bond and angle parameters. For the torsional parameters MP4/6-311G**//MP2/6-31G* was used.4
CHARMm: valence parameters were trained on QM data calculated at the MP2/6-31G* and MP2/6-31+G* levels.29
ANI-1: Training data generated at ωB97X/6-31G* level of theory. ANI-1ccx used CCSD(T)/CBS extrapolated structures.30
OPLS3e: M06-2X/cc-pVTZ(-f)//B3LYP/6-31G* level of theory was used for their training set.10
TorsionNet: a mix of data generated at B3LYP/6-31G**//HF/3-21G and B3LYP/6-31+G**//HF/3-21G.31
OrbNet Denali: ωB97X-D3/def2-TZVP level of theory.32
Open Force Field (OpenFF): B3LYP-D3(BJ)/DZVP was the default level of theory.2
Espaloma: A mix of SPICE16 data set and OpenFF data sets, all of the data generated at B3LYP-D3(BJ)/DZVP level of theory.18
MACE-OFF23: SPICE16 data set at ωB97M-D3(BJ)/def2-TZVPPD level of theory.21
Traditionally there are five rungs on “Jacob’s ladder” of chemical accuracy corresponding to the density functional theory (DFT) method used.33 Starting with the Local Density Approximation (LDA), we progress one level up the accuracy ladder each with Generalized Gradient Approximation (GGA), meta-GGA, hybrid functionals, and then double hybrid functionals. Although, all hybrid functionals do not necessarily include the ingredient added in “meta-GGA” which uses the second derivative of the density in the energy functional. For example, B3LYP does not have the meta-GGA term, but M06 does. Hybrid and double-hybrid functionals are distinguished by including a percentage of HF exchange, and a percentage of MP2 correlation, respectively. Therefore, double hybrid functionals are at least the cost of a MP2 calculation. Range separated functionals (RSF) are another very important development in DFT methods. In RSFs, such as ωB97 family, the percentage of HF exchange depends on the distance between the electrons. This accounts for the electron self-interaction error, and has become incorporated into several of the most modern functionals (i.e., the ones starting with LC or ω). DFT methods with dispersion corrections, which can be either ab initio,34−36 or empirical in nature,37 are notably more accurate than their standard forms as they improve the description of the noncovalent interactions. Grimme’s DFT-D3,38,39 along with Becke-Johnson (BJ) damping,40 is one of the most prominent dispersion corrections widely used in the field. On the other hand, wave function based methods (WFT) include Hartree–Fock, post-Hartree–Fock methods viz., Møller–Plesset perturbation theory of different orders (MP2, MP3, MP4), configuration interaction (CI), coupled cluster (CC), and multireference methods such as complete active space self-consistent field (CASSCF), in increasing order of complexity in describing electron correlation, and approaching higher accuracy.
Recent studies have utilized several benchmark data sets to assess the general quality of QM methods for specific molecular properties. Notable among these data sets are Database2015B,41 GMTKN3042 and its expanded version GMTKN55,43 MGCDB84,44 CHAL336,45 MPCONF196,46 YMPJ,47 S22,48 S66,49 HB375,50 TorsionNet500,31 Dakota Folmsbee’s set,51 XB18,52 CTB-279.53 The subsets of these data sets emphasize performance on predicting:
noncovalent interactions, both inter- and intramolecular
isomerization energies
chemical reaction barrier heights
relative conformer energies for various chemical moieties
torsional strain, and barriers
In our present study we want to address
the extent to which benchmark results of quantum mechanical methods obtained from larger basis sets, as documented in existing literature, are informative when deployed with smaller basis sets
how good OpenFF’s current choice of DZVP basis set is in describing conformational energetics for charged entities, and whether the error in relative energies of torsion profiles is within the desired range (0.5–1 kcal/mol) without addition of more diffuse functions to the basis set
how well different QM methods predict torsional energy profiles and whether the accuracy gain is significant enough that it warrants a change in QM reference for force field training
2. Motivation and Approach
2.1. Prior Studies and Selection of Molecules
Based on earlier benchmark studies,2,54 the OpenFF initiative initially chose B3LYP-D3(BJ)/DZVP as the method for generating QM training data to train OpenFF force fields, with the expectation that this choice might need to be revisited at a later date. Such a “revisit” is our focus here. The prior benchmarks2 included assessing accuracy of conformer energetics on the MPCONF196 set,46 which is a data set of conformers of smaller peptides and medium-sized macrocycles, that were all neutral, and of relative energies of torsional profiles on a curated set of 15 one-dimensional torsion scans. A good compromise between accuracy and cost on MPCONF196 and the smaller set of torsion scans led to the choice of the B3LYP-D3(BJ) functional along with Salahub’s double-ζ split-valence + polarization (DZVP) basis set55 for building OpenFF force fields.
Although very insightful, this prior benchmark study did not include charged molecules, and was limited to [C, H, N, O] chemical space. Because of these limitations, here we focused on running a new benchmark which would more adequately represent the pharmaceutically relevant chemical space that our force field needs to describe accurately. Thus, to expand the scope of the prior benchmark we selected 59 torsions from molecules in OpenFF’s Roche (general, and tautomer+protomer data set) and Coverage molecule sets.
Molecule selection followed these criteria:
ensuring a comprehensive representation of chemical diversity
prioritizing size similarity among molecules to mitigate the influence of intrinsic size-related effects on electronic properties
incorporating variations in central bonds, formal charges, element compositions, and intramolecular interactions
-
and, inclusion of molecules that added additional complexity beyond those in the prior set, such as those with
nonzero formal charges
strong internal interactions
conjugated central rotatable bonds (>10 kcal/mol rotational barrier)
halogens
The extracted charged molecules were further subdivided into groups based on the type of charged functional group. Here are the SMARTS patterns of the subgroups which include,
-
functional groups with a charge of −1
phenolate: c[O-]
carboxamide: C(=O)[N-]
aromatic carbons connected to a nitrogen with a negative charge: c[N-]c
sulfonamide: S(=O)(=O)[N-]
sulfinamide: S(=[N-])(=O)
-
functional groups with a charge of +1:
protonated amine with carbon bond: [NH+,nH+](=,:[C,c])[C,c]
protonated amine: [NH+]([*])[*]
protonated primary amine: [NH2+]([*])[*]
protonated ammonium: [NH3+][*]
We then performed one-dimensional torsion scans on one selected molecule per cluster. We selected one molecule per cluster by picking the centroid using the MACCS keys fingerprint method.56 Following selection, we performed one-dimensional torsion scans by rotating around the central bond in the chosen dihedral. In total, 59 torsions were scanned in 36 unique molecules, shown in Figure 1 with the dihedral atoms highlighted, in this benchmark study.
Figure 1.


Molecules used in this benchmark study. The atoms and central bonds engaged in the driven torsions are highlighted.
2.2. Computational Details
Torsion scans were performed with a 15-degree grid spacing, using Torsiondrive,57 resulting in a total of 24 points on the grid within the range of [-180°, 180°]. More details on the compute infrastructure were provided in SI section 1.2.
2.2.1. Choice of Reference Theory
The benchmark geometries were the final geometries from the torsion scan at MP2/heavy-aug-cc-pVTZ level of theory. Whereas, the benchmark relative energies were obtained using coupled cluster with single, double, and perturbative triple excitations at the complete basis set limit, CCSD(T)/CBS. It is to be noted that MP2 is a wave function-based method that treats electron correlation explicitly, and does not rely on the same approximations as DFT functionals. Hence, MP2 geometries offer a more neutral and consistent basis for benchmarking devoid of the specific biases inherent to individual DFT functionals. For the complete basis set calculation, Helgaker’s 2-point extrapolation scheme was used as implemented in Psi4.58−60 Helgaker extrapolation scheme here includes a reference total energy from Hartree–Fock, correlation correction which includes correlation effects beyond HF with the MP2 method, and a delta correction, which gives a highly accurate correlation calculation with CCSD(T), accounting for the error in MP2. The difference between CCSD(T) and MP2 converges quickly with increase in basis functions and hence a smaller basis set can be used for this part of calculation. Psi4 performs these calculations in stages and the treatment follows this equation:60
| 1 |
where 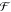 is an energy or energy extrapolation scheme.
And, in our case this translates to
is an energy or energy extrapolation scheme.
And, in our case this translates to
| 2 |
Although heavy-aug-cc-pVDZ (or haDZ) is computationally affordable it may fall short of the gold standard reference level of theory often used in the community.61,62 It is expected that the delta correction error with haDZ would fall somewhere in between 0.1 and 0.25 kcal/mol, the errors observed with aug-cc-pVDZ and cc-pVDZ on either end of the range.63,64 To assess how good our choice of reference theory level is we have performed energy calculations with the gold standard reference theory level for a subset of 7 molecules from the benchmark set61 at CCSD(T)/CBS, where for the extrapolation to CBS the correlation basis is aug-cc-pV[TQ]Z, and delta basis is aug-cc-pVTZ. The gold standard energy is calculated as,60
| 3 |
Our reference theory level employed
a computationally efficient
smaller basis set (haDZ) for delta correction when compared to a larger
basis (aTZ) used in the gold standard reference. And, the RMSE in
relative energies for the 7 molecule subset (7 × 24 grid points)
of our reference theory with respect to the gold standard was 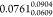 kcal/mol (the subscript and superscript
are the 95% confidence intervals). So, the difference with respect
to the gold standard was one tenths of a kcal/mol for our choice of
reference theory level, which was accurate enough and quite affordable
for our study.
kcal/mol (the subscript and superscript
are the 95% confidence intervals). So, the difference with respect
to the gold standard was one tenths of a kcal/mol for our choice of
reference theory level, which was accurate enough and quite affordable
for our study.
2.2.2. Choice of Density Functionals to Benchmark
We have chosen a smaller pool of density functional approximations (DFA) that are cost-effective from the get go and we are not looking into a comprehensive evaluation of all available DFAs. The choice of DFAs include those commonly used in developing force fields and charge models, and better performing ones from other benchmark studies. From our prior studies DZVP has been proven cost friendly and all of the DFAs were tested with this basis set. And, within our current choice of DFA used in developing OpenFF force fields, B3LYP-D3BJ, we tested Ahlrichs def2 basis sets incrementing them systematically in size, and the Pople basis sets in increasing size as they are commonly used in practice in industry. Also, one of the purposes of the study was to determine which DFAs are useful at medium sized basis sets. The rankings and differences might vary from the benchmarks at larger basis sets. This selection represents different rungs on the Jacob’s ladder of chemical accuracy.
B97 (GGA), with D3(BJ) dispersion correction and with def2-TZVP basis set
-
B3LYP hybrid family (hybrid-GGA), with65
-
Berkeley range separated functionals (meta-GGA),77 with DZVP basis and dispersion corrections,
ωB97M-V, ωB97M-D3(BJ)
ωB97X-D3(BJ)
-
Minnesota functionals (meta-GGA),78−81 with DZVP basis and dispersion corrections
M05-2X-D3
M06-2X-D3
M08-HX-D3
-
PW6B95 (meta-GGA),82 with DZVP and dispersion corrections
D3(BJ)
D3
Double hybrid DSD-BLYP-D3(BJ)83 with haTZ basis
3. Results and Discussion
3.1. Comparison of Different Levels of Theory
In the context of force field fitting, apart from conformer energies and optimized geometries, torsion energy profiles are another important source of molecular geometries/energetics data. The energy barriers observed in torsion energy profiles are a measure of the strength of steric hindrance or strong intramolecular interactions that prevent certain conformations. Thus, accurately capturing torsion profiles is relevant for understanding molecular recognition, binding, and other interactions that occur in complex systems. Our aim here is to pick an accurate and computationally efficient QM level of theory to train the valence parameters in a general small molecule force field. For this purpose, single point energies were evaluated at different levels of theory for comparison at the benchmark geometries, and the RMSE in relative energies was tabulated. Single point energies were evaluated at the same geometry to ensure parity between the methods since performing a geometry optimization with each of the methods will result in minor differences in final geometries, and sometimes TorsionDrive may push them to a completely different minima. SI Table S1 lists the RMSE in torsion profile energies for each of the molecules considered in this benchmark set with respect to the reference level of theory, CCSD(T)/[haTQZ; δ:haDZ].
The relative energies were calculated by subtracting the minimum energy for each of the methods. And, the RMSE was calculated with respect to the reference theory level.
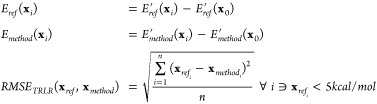 |
4 |
where E’ represents the absolute energies, x0 represents the minimum energy point. And, the RMSE and MUE were evaluated with the relative energies. For calculating the RMSE of thermally relevant low-energy region (TRLR), only the relative energies below 5 kcal/mol on the reference energy surface were chosen. The cutoff of 5 kcal/mol was chosen to favor low-energy regions in state space.3,28,84
From Table 1 we can see that RMSE values of relative energies for most of the functional and basis set combinations studied here fall in the range of 0.4–0.7 kcal/mol with respect to CCSD(T)/[haTQZ; δ:haDZ], the reference level of theory. ωB97M-D3(BJ)/DZVP is the most accurate among those tested here with an RMSE in torsion profile energies of 0.41 kcal/mol. This range-separated hybrid functional has been one of the top performers in several other recent studies that were done with larger basis sets.43,44 Our current choice of theory, B3LYP-D3(BJ)/DZVP, has an RMSE of 0.52 kcal/mol and it closely trails by a difference of 0.1 kcal/mol when compared to the best functional in this study. Almost half of the of the other tested levels of theory were within 0.1 kcal/mol of the best functional. No method is consistently the most accurate across all of the subsets, highlighting the importance of considering a large and diverse benchmark set.
Table 1. Overall RMSE and MUE (in kcal/mol) in Torsion Profile Energies of the Molecule Set with Respect to the Reference Level CCSD(T)/[haTQZ; δ:haDZ]//MP2/heavy-aug-cc-pVTZ Level of Theorya.
| metric (kcal/mol) |
||||||
|---|---|---|---|---|---|---|
| specification | RMSE | MUE | RMSE-TRLR | |||
| B97-D3(BJ)/def2-TZVP | ||||||
| M05-2X-D3/DZVP | ||||||
| M06-2X-D3/DZVP | ||||||
| M08-HX-D3/DZVP | ||||||
| ωB97X-D3BJ/DZVP | ||||||
| ωB97M-D3BJ/DZVP | ||||||
| ωB97M-V/DZVP | ||||||
| PW6B95-D3(BJ)/DZVP | ||||||
| PW6B95-D3/DZVP | ||||||
| MP2/aug-cc-pVTZ | ||||||
| MP2/heavy-aug-cc-pVTZ | ||||||
| DSD-BLYP-D3(BJ)/heavy-aug-cc-pVTZ | ||||||
| B3LYP-D3(BJ)/DZVP | ||||||
| B3LYP-D3MBJ/DZVP | ||||||
| B3LYP-NL/DZVP | ||||||
| B3LYP-D3(BJ)/def2-SV(P) | ||||||
| B3LYP-D3(BJ)/def2-SVP | ||||||
| B3LYP-D3(BJ)/def2-TZVP | ||||||
| B3LYP-D3(BJ)/def2-TZVPD | ||||||
| B3LYP-D3(BJ)/def2-TZVPP | ||||||
| B3LYP-D3(BJ)/def2-TZVPPD | ||||||
| B3LYP-D3(BJ)/def2-QZVP | ||||||
| B3LYP-D3(BJ)/6-31G* | ||||||
| B3LYP-D3(BJ)/6-31+G** | ||||||
| B3LYP-D3(BJ)/6-311+G** | ||||||
The 95% confidence intervals, calculated with cinnabar, are presented as subscripts and superscripts. Furthermore, the last column includes the RMSE within the thermally relevant low-energy region (TRLR) with energies of <5 kcal/mol, averaged over all of the molecules, which serves as a metric for assessing accuracy in low-energy regions. The best performer on this set of molecules is ωB97M-D3(BJ)/DZVP, and our current choice of theory level, B3LYP-D3(BJ)/DZVP, lags behind it by only 0.1 kcal/mol in RMSE.
The RMSEs on the whole set and the subsets of neutral and charged molecule sets were depicted in Figure 3 and tabulated in Table 2. We can see from Figure 3(f), and from Table 2, that the accuracy of B3LYP-D3(BJ) functional increases for charged molecules with addition of polarization and diffuse functions from DZVP to def2-TZVP and higher. Within B3LYP-D3(BJ) it is to be noted that the high quality DZVP basis set yields comparable results to the larger triple-ζ level basis set, def2-TZVP. PW6B95-D3(BJ)/DZVP is the best among the tested methods for neutral molecules with an RMSE of 0.43 kcal/mol. ωB97M-D3(BJ)/DZVP is also on the same level with statistically insignificant difference. And, B3LYP-D3(BJ)/DZVP, has an RMSE of 0.45 kcal/mol and it closely trails these two functionals by a negligible difference of 0.02 kcal/mol for neutral molecules.
Figure 3.
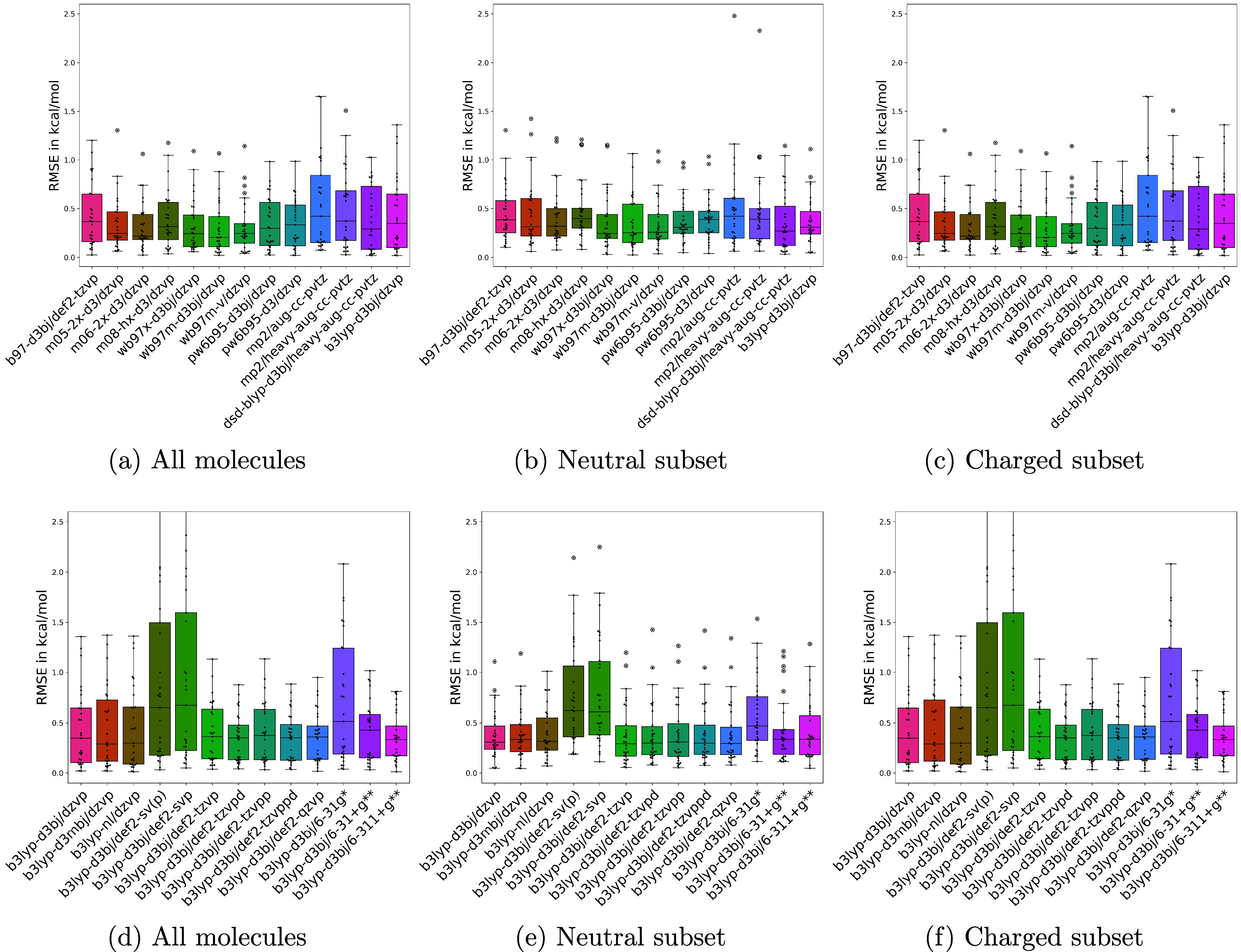
RMSE on the entire benchmark data set and on subsets of neutral and charged molecules. The top row shows the RMSE in torsion profile energies with different functional and basis set combinations. The bottom row shows the RMSE within the B3LYP functional.
Table 2. Comparison between Neutral and Charged Molecule Subsetsa.
| RMSE (kcal/mol) |
||||||
|---|---|---|---|---|---|---|
| specification | whole set | neutral subset | charged subset | |||
| B97-D3(BJ)/def2-TZVP | ||||||
| M05-2X-D3/DZVP | ||||||
| M06-2X-D3/DZVP | ||||||
| M08-HX-D3/DZVP | ||||||
| ωB97X-D3(BJ)/DZVP | ||||||
| ωB97M-D3BJ/DZVP | ||||||
| ωB97M-V/DZVP | ||||||
| PW6B95-D3(BJ)/DZVP | ||||||
| PW6B95-D3/DZVP | ||||||
| MP2/aug-cc-pVTZ | ||||||
| MP2/heavy-aug-cc-pVTZ | ||||||
| DSD-BLYP-D3(BJ)/heavy-aug-cc-pVTZ | ||||||
| B3LYP-D3(BJ)/DZVP | ||||||
| B3LYP-D3MBJ/DZVP | ||||||
| B3LYP-NL/DZVP | ||||||
| B3LYP-D3(BJ)/def2-SV(P) | ||||||
| B3LYP-D3(BJ)/def2-SVP | ||||||
| B3LYP-D3(BJ)/def2-TZVP | ||||||
| B3LYP-D3(BJ)/def2-TZVPD | ||||||
| B3LYP-D3(BJ)/def2-TZVPP | ||||||
| B3LYP-D3(BJ)/def2-TZVPPD | ||||||
| B3LYP-D3(BJ)/def2-QZVP | ||||||
| B3LYP-D3(BJ)/6-31G* | ||||||
| B3LYP-D3(BJ)/6-31+G** | ||||||
| B3LYP-D3(BJ)/6-311+G** | ||||||
The overall RMSE in torsion profile energies of the molecule set with respect to the reference level CCSD(T)/[haTQZ; δ:haDZ]//MP2/heavy-aug-cc-pVTZ level of theory for the whole benchmark set and subsets of neutral and charged molecules. The 95% confidence intervals, calculated with cinnabar, are presented as subscripts and superscripts. The RMSE of neutral molecules for OpenFF’s default theory level, B3LYP-D3(BJ)/DZVP, is comparable to the best functional.
Some of the larger errors were seen in the subset of charged molecules, when compared to the neutral molecules, among the whole set of molecules used in this benchmark. ωB97M-D3(BJ)/DZVP is the best among the tested methods for the charged subset of molecules. And, OpenFF’s default level of theory, B3LYP-D3(BJ)/DZVP, has an RMSE error 0.18 kcal/mol worse than the best method, with an overall RMSE of 0.58 kcal/mol. The difference in RMSE between charged molecules and the neutral molecules is slightly higher for B3LYP-D3(BJ)/DZVP level of theory, and addition of basis functions helped drive this error down. The addition of basis functions from DZVP to def2-TZVPD basis set shows a significant improvement, with B3LYP-D3(BJ) functional, and can be a choice for fine-tuning the force field with more accurate data.
Despite observing higher errors in certain molecules, the methodology used to construct the torsion profile target data, which prioritizes the match to low energy regions, has the potential to mitigate some of these discrepancies. During the training of OpenFF force fields we construct a torsion profile target and optimize the force field using ForceBalance,85,86 and the objective function in terms of relative energies is defined as follows:3
| 5 |
where the primes indicate the absolute energies at each grid point and the weighted differences in relative energy profiles serve as the loss function to minimize:
| 6 |
where xi represents the coordinates of ith conformer, the 0th conformer is the minimum energy conformer in respective potential energy landscapes, θ is the force field parameter set at that iteration, and OptMM(xi, θ) corresponds to the MM energy obtained via constrained minimization and dE = 1 kcal/mol is a conversion factor to make the sum over deviations dimensionless. The applied weights w(EQM) in eq 6 prioritize matching the torsion profile at energy minima since Boltzmann sampling favors low-energy regions in state space.84
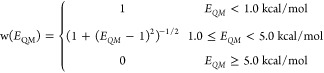 |
7 |
In the context of fitting OpenFF force fields, the torsion profile energy loss function defined above in eq 6 may further mitigate the differences between the methods since we applied a hard cutoff of 5 kcal/mol to exclude the higher energy regions from torsion fits for the OpenFF Parsley and Sage line of force fields.3,28
The RMSE on thermally relevant low-energy region (TRLR), only considering the energies less than 5 kcal/mol with respect to the minima on the torsion profile, were tabulated in the last column of Table 1. We can see that ωB97M-D3(BJ)/DZVP still holds its place as the most accurate functional with torsion target score as well, and the differences between various methods drop drastically in the low energy regions. In a sense, the modeling of low-energy, thermally relevant regions was quite accurate, while it was in the high-energy regions where the influence of stereoelectronic and steric effects became prominent, leading to discernible differences between various methods.
The error in the subset of neutral molecules (30 data points) is small for OpenFF’s default of B3LYP-D3(BJ) and close to the best functional, ωB97M-D3(BJ). However, the error is larger for the charged subset of molecules (29 data points) with OpenFF’s default, compared to the best functional, and yet remains accurate in low-energy regions. Most of the large deviations come from high energy regions which were (and usually are) excluded in fitting to torsion profile energies as they are thermally irrelevant. In this sense, our default method may remain appealing given its low computational cost and relatively low error in key regions of torsion profiles. For charged molecules, addition of more basis functions would help as seen in Figure 3, subplot (f).
Referring to SI Table S1, when we examine the molecule with the largest error using OpenFF’s default, B3LYP-D3(BJ)/DZVP, which exhibits an RMSE in relative energies of 1.36 kcal/mol, we observe that the majority of discrepancies arise from the high-energy region exceeding 5 kcal/mol. The torsion profile for this molecule with respect to different basis sets was shown in Figure 2. It is worth reiterating that we exclude this high-energy region during the training process.
Figure 2.
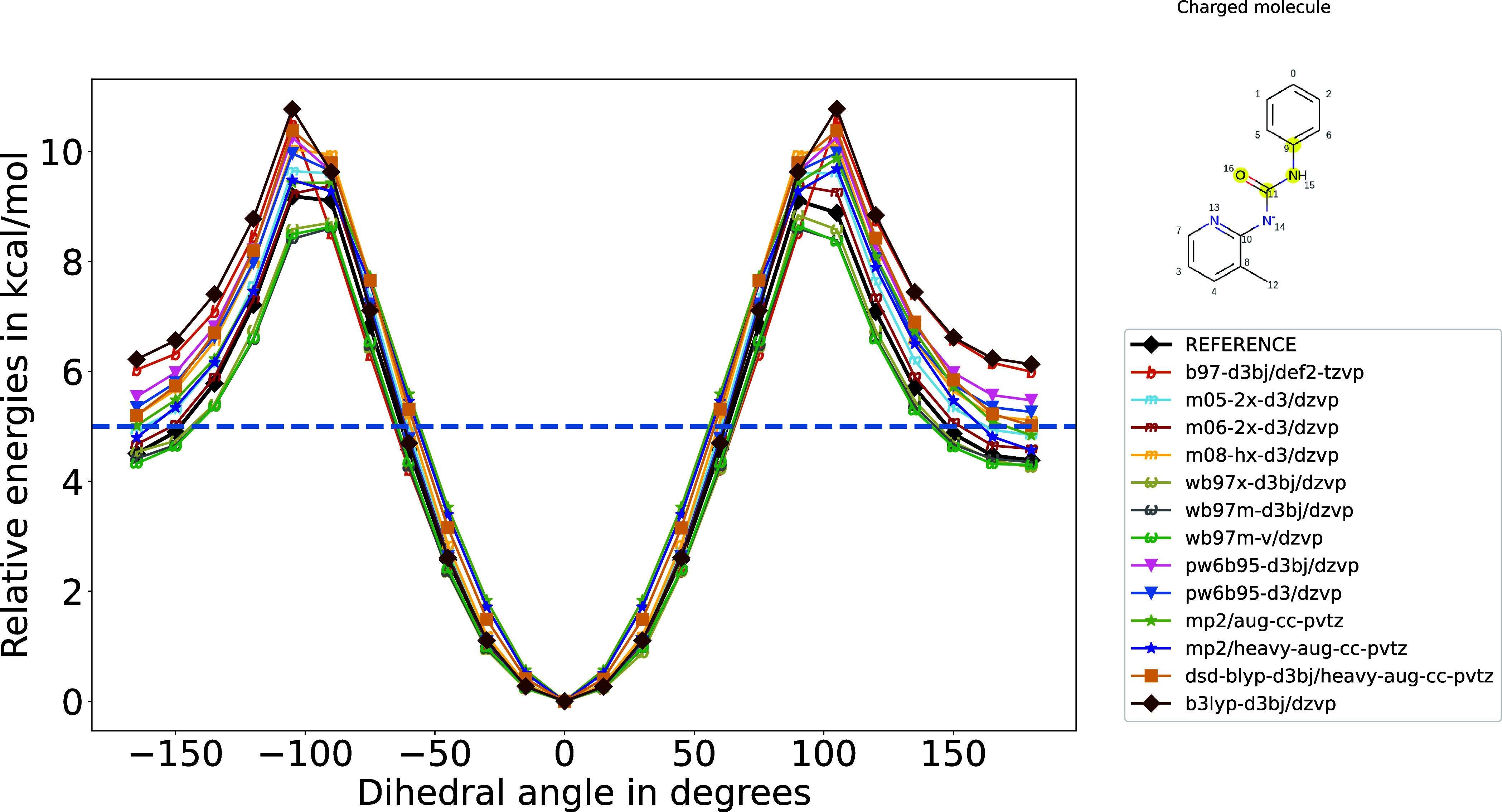
Torsion profile that corresponds to a molecule with a high RMSE at relative energies of >1.0 kcal/mol for most of the methods (row 23 from Table S1). The horizontal dashed line represents 5 kcal/mol, and if we truncate the energies above it, we can see that the low-energy regions were captured better than were the high-energy regions.
3.2. Comparison of Timings and Accuracy
The computation time of a single point (energy + gradient) calculation provides a rough approximation of the method’s cost for a torsion scan or geometry optimization. When examining the OpenFF QM data sets of small molecules, it is observed that a geometry optimization typically requires a median of 42 gradient calculations (based on data from approximately 8000 geometry optimizations). Additionally, a 1D torsion scan with 24 grid points generally costs around 788 gradient calculations (from data on roughly 4000 torsion scans). As a rule of thumb, a geometry optimization calculation is 40x costlier than a single point energy and gradient calculation, and a 1D torsion scan is 800x costlier if executed serially, or nearly 72-fold for parallelized torsion scans (considering the median of maximum number of optimization steps taken among all grid points). So, the differences scale up pretty quickly with the type of calculation. To provide a reference point, we present timing data for a molecule containing 16 heavy atoms. The timings were normalized with the time for a B3LYP-D3(BJ)/DZVP calculation. The cost factor versus RMSE plot is shown in Figure 4, and the most accurate functional is almost twice the cost of a B3LYP-D3(BJ)/DZVP calculation, despite only a modest accuracy benefit. It is to be noted that Psi4, as of v1.4.1, does not yet have analytic gradients for NL and VV10 dispersion terms, and also for DSD-BLYP method, so for these methods only the costs of an energy calculation were reported and scaled with respect to the cost of a B3LYP-D3(BJ) energy calculation (Table 3).
Figure 4.

Cost factor of a single point energy and gradient calculations within each method, averaged over 10 runs of different conformations of the same molecule (with 16 heavy atoms) for all of the methods. All of the run times of each method were normalized with the time taken for a B3LYP-D3(BJ)/DZVP calculation. All of the calculations were run on an Intel(R) Xeon(R) Gold 6148 CPU @ 2.40 GHz CPU using 16 threads and 48 GB of memory. The best functional, ωB97M-D3(BJ)/DZVP, is indicated with an ω, while OpenFF’s default, B3LYP-D3(BJ)/DZVP, is indicated with a ◆.
Table 3. Cost Factors of a Single Energy + Gradient Calculation of Various Methods with Respect to a B3LYP-D3(BJ) Calculationa.
| specification | mean wall time ± standard deviation (s) | cost factor |
|---|---|---|
| B97-D3(BJ)/def2-TZVP | 49.41 ± 1.10 | 2.33 |
| M05-2X-D3/DZVP | 27.05 ± 0.42 | 1.28 |
| M06-2X-D3/DZVP | 27.26 ± 0.35 | 1.29 |
| M08-HX-D3/DZVP | 27.06 ± 0.49 | 1.28 |
| ωB97X-D3(BJ)/DZVP | 37.49 ± 0.84 | 1.77 |
| ωB97M-D3(BJ)/DZVP | 43.17 ± 0.82 | 2.04 |
| PW6B95-D3(BJ)/DZVP | 27.11 ± 0.39 | 1.28 |
| PW6B95-D3/DZVP | 26.98 ± 0.33 | 1.27 |
| MP2/aug-cc-pVTZ | 397.87 ± 15.91 | 18.78 |
| MP2/heavy-aug-cc-pVTZ | 312.62 ± 12.16 | 14.75 |
| B3LYP-D3(BJ)/DZVP | 21.19 ± 0.24 | 1.00 |
| B3LYP-D3MBJ/DZVP | 21.21 ± 0.21 | 1.00 |
| B3LYP-D3(BJ)/def2-SV(P) | 18.21 ± 0.36 | 0.86 |
| B3LYP-D3(BJ)/def2-SVP | 19.89 ± 0.17 | 0.94 |
| B3LYP-D3(BJ)/def2-TZVP | 51.80 ± 0.63 | 2.44 |
| B3LYP-D3(BJ)/def2-TZVPD | 80.60 ± 0.63 | 3.80 |
| B3LYP-D3(BJ)/def2-TZVPP | 60.27 ± 0.95 | 2.84 |
| B3LYP-D3(BJ)/def2-TZVPPD | 92.33 ± 1.18 | 4.36 |
| B3LYP-D3(BJ)/def2-QZVP | 203.17 ± 3.71 | 9.59 |
| B3LYP-D3(BJ)/6-31G* | 19.41 ± 0.32 | 0.92 |
| B3LYP-D3(BJ)/6-31+G** | 30.22 ± 0.49 | 1.43 |
| B3LYP-D3(BJ)/6-311+G** | 37.98 ± 0.46 | 1.79 |
| Energy Calculation Only | ||
| ωB97M-V/DZVP | 66.97 ± 0.61 | 6.21 |
| B3LYP-NL/DZVP | 64.02 ± 0.47 | 5.94 |
| DSD-BLYP-D3(BJ)/heavy-aug-cc-pVTZ | 87.43 ± 1.13 | 8.11 |
| B3LYP-D3(BJ)/DZVP | 10.78 ± 0.17 | 1.00 |
All of the wall times were averaged over 10 different runs of 10 different conformers of the same molecule, molecule 1 in Table 1, which contains 16 heavy atoms. For a few methods for which timings for an energy calculation were reported, they were scaled with respect to the cost of a B3LYP-D3(BJ) energy calculation.
B3LYP-D3(BJ) with def2-SVP and def2-SV(P) were lower in accuracy but cheaper, and by adding more basis functions to improve accuracy the cost increases, and it can be seen from Figure 4 that Salahub’s DZVP offers a perfect balance in cost and accuracy for this functional. In the basis set study using B3LYP, 6-311+G** is already very close to the basis set limit (def2-QZVP) in terms of accuracy, in fact it is closer than def2-TZVP and def2-TZVPP, yet it is much cheaper than all of those. When comparing the Pople basis sets, perhaps the largest improvement in accuracy for B3LYP came from adding the first set of diffuse functions (from 6-31g* to 6-31+g*). In the Minnesota functionals, M06-2X-D3 performs better than M05-2X-D3 and M08-HX-D3. In both the B3LYP and ωB97 M tests, the ab initio nonlocal dispersion correction actually gives larger RMSEs than the empirical dispersion.
Psi4’s efficient implementation results in quadratic scaling of DFT calculation time with an increase in molecule size.87 Both the quadratic and linear fits of the data yielded a similar R2, which prompted considering the Akaike information criterion (AIC) and the Bayesian information criterion (BIC) to differentiate the models better. Higher R2, lower AIC, and lower BIC values indicate a better fit to the quadratic model. From Figure 5 we can see that with an increase in number of atoms represented by an increase in number of basis functions and the scaling of computation cost for a B3LYP-D3(BJ)/DZVP single point energy plus gradient calculation.
Figure 5.

Scaling of B3LYP-D3(BJ)/DZVP single point energy plus gradient calculation with an increase in the number of basis functions, which in turn represents an increase in the number of atoms. The time of calculation for the dispersion correction term is not included as it is negligible, on the order of a fraction of a second. The quadratic (orange) and linear (blue) fits to the data are shown along with the metrics R2, Akaike information criterion (AIC), and the Bayesian information criterion (BIC). The quadratic scaling model seems appropriate from these metrics.
3.3. Faster Parametrization with SQM/ML Potentials
Creating bespoke force fields on-the-fly adds a lot of new torsion parameters, which in turn requires generation of new QM reference data for training them.88 Generation of new QM data is time-consuming, and a possible alternative for faster parametrization, without too much loss in accuracy, can be a semiempirical method, or a couterpoise corrected method with minimal basis, or machine learning potentials.21,89−96 Here we checked the performance of the semiempirical method GFN2-XTB,95 Grimme’s 3-corrected Hartree–Fock method (HF-3c),93 and two recent machine learning potentials, AIMNET296 and MACE-OFF23,21 the ML potentials which have demonstrated accuracy very close to the level of DFT that they were trained on. Only single point energies were evaluated here, as in the comparisons above.
Table 4 summarizes the performance of the four methods across the whole benchmark set, and also broken down into neutral and charged subsets (see also Table 2). GFN2-XTB shows reasonable accuracy, but is not competitive with the DFT methods. AIMNet2 shows very good accuracy, close to DFT, consistently across both neutral and charged species. MACE-OFF23 shows remarkable accuracy for the neutral subset, in fact more accurate than any of the DFT methods studied here. Note that MACE-OFF23 was not trained on charged species, and hence the error is much higher for this subset, but this will be addressed in future models. HF-3c does not perform well here. Thus, machine learning potentials such as these are a reasonable alternative to DFT, particularly for high-throughput, bespoke parametrization work.88
Table 4. Comparison of SQM, DFT-3c, and ML Methods on This Benchmark Seta.
| RMSE (kcal/mol) |
||||||
|---|---|---|---|---|---|---|
| specification | whole set | neutral subset | charged subset | |||
| GFN2-XTB | ||||||
| AIMNET2 | ||||||
| MACE-OFF23 | ||||||
| HF-3c | ||||||
The overall RMSE in torsion profile energies of the molecule set with respect to the reference level CCSD(T)/[haTQZ; δ:haDZ]//MP2/heavy-aug-cc-pVTZ level of theory for the whole benchmark set and subsets of neutral and charged molecules. The 95% confidence intervals, calculated with cinnabar, are presented as subscripts and superscripts.
4. Conclusion
We conducted a benchmark of QM levels of theory that strike a balance between accuracy and computational efficiency for generating large QM data sets with diverse chemistries to train the valence parameters in a general small molecule force field. The benchmark set of molecules included charged molecules, biaryls, complex hypervalent sulfur chemistry, and complex nitrogen chemistry. This benchmark study is an extension to an earlier work2 on benchmarking conformer energies, which suggested B3LYP-D3(BJ)/DZVP as the level of theory to generate OpenFF force field training and validation data. In the context of force field development, aside from conformer energies and optimized geometries, torsion energy profiles represent another indispensable source of molecular interaction data. Achieving accuracy in torsion profile energies relative to a highly accurate QM level of theory reflects in the trained force field. For this purpose, relative energies were compared against CCSD(T)/[haTQZ; δ:haDZ]//MP2/haTZ level of theory for different functional and basis set combinations. And, among the tested methods, ωB97M-D3(BJ) outperforms the others even within a smaller basis set of DZVP, boasting an RMSE in torsion profile energies of just 0.41 kcal/mol. This range-separated hybrid functional has consistently ranked among the top performers in various recent studies43,44 which were done with a larger basis set. OpenFF’s choice of B3LYP-D3(BJ)/DZVP closely follows it with an RMSE in relative energies of 0.52 kcal/mol. And, the computational cost of B3LYP-D3(BJ)/DZVP for a single gradient is only half of the best functional, but depending on the type of data set, geometry optimization or torsion scans, the cost would scale up with the number of steps taken during the calculation. Within a subset of neutral molecules the RMSE in relative energies with B3LYP-D3(BJ)/DZVP is comparable to the most accurate method. And, the larger errors appear to originate from molecules with charges. However, in practice, the distinctions between levels of theory become evident in the high-energy regions, which are typically excluded during force field training with torsion profiles as they are thermally irrelevant.
Acknowledgments
P.K.B. appreciates the financial support from National Institute of General Medical Sciences Grant R01GM132386. D.L.M. appreciates the financial support from the National Institutes of Health (R35GM148236 and R01GM132386). D.J.C. and J.T.H. acknowledge support from a UKRI Future Leaders Fellowship (Grant MR/T019654/1). L.-P.W. and H.J. acknowledge support from ACS PRF 58158-DNI6. For the purpose of open access, the author has applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising.
Data Availability Statement
The scripts to download the structure data from QCArchive and the downloaded energy and dipole data are available at https://github.com/MobleyLab/qm-theory-benchmark/tree/master.
Supporting Information Available
Supporting Information includes tables on individual errors in each molecule, details on compute infrastructure used, dipole moments comparison, correlation between energy and delta energy difference wrt reference, error estimates in barriers. The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.4c03167.
(PDF)
The authors declare the following competing financial interest(s): D.L.M. serves on the scientific advisory boards of Anagenex and OpenEye Scientific Software, Cadence Molecular Sciences, and is an Open Science Fellow with Psivant.
Supplementary Material
References
- Jorgensen W. L.; Tirado-Rives J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- Wang L. P.Parameterization perspective I: Parameter optimization methodology. 2019. https://zenodo.org/records/3405539 (last accessed 2024-02-12).
- Boothroyd S.; Behara P. K.; Madin O. C.; Hahn D. F.; Jang H.; Gapsys V.; Wagner J. R.; Horton J. T.; Dotson D. L.; Thompson M. W.; Maat J.; Gokey T.; Wang L.-P.; Cole D. J.; Gilson M. K.; Chodera J. D.; Bayly C. I.; Shirts M. R.; Mobley D. L. Development and Benchmarking of Open Force Field 2.0.0: The Sage Small Molecule Force Field. J. Chem. Theory Comput. 2023, 19, 3251–3275. 10.1021/acs.jctc.3c00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; De Vries A. H. The MARTINI Force Field: Coarse Grained Model for Biomolecular Simulations. J. Phys. Chem. B 2007, 111, 7812–7824. 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- Maerzke K. A.; Schultz N. E.; Ross R. B.; Siepmann J. I. TraPPE-UA Force Field for Acrylates and Monte Carlo Simulations for Their Mixtures with Alkanes and Alcohols. J. Phys. Chem. B 2009, 113, 6415–6425. 10.1021/jp810558v. [DOI] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; Mackerell A. D. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayly C. I.; McKay D.; Truchon J.-F.. An Informal AMBER Small Molecule Force Field: parm@Frosst. http://www.ccl.net/cca/data/parm_at_Frosst/ (last accessed 2024-01-04).
- Wang L.-P.; McKiernan K. A.; Gomes J.; Beauchamp K. A.; Head-Gordon T.; Rice J. E.; Swope W. C.; Martínez T. J.; Pande V. S. Building a More Predictive Protein Force Field: A Systematic and Reproducible Route to AMBER-FB15. J. Phys. Chem. B 2017, 121, 4023–4039. 10.1021/acs.jpcb.7b02320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos K.; Wu C.; Damm W.; Reboul M.; Stevenson J. M.; Lu C.; Dahlgren M. K.; Mondal S.; Chen W.; Wang L.; Abel R.; Friesner R. A.; Harder E. D. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. 10.1021/acs.jctc.8b01026. [DOI] [PubMed] [Google Scholar]
- Oostenbrink C.; Villa A.; Mark A. E.; Van Gunsteren W. F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- Schmid N.; Eichenberger A. P.; Choutko A.; Riniker S.; Winger M.; Mark A. E.; Van Gunsteren W. F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. 10.1007/s00249-011-0700-9. [DOI] [PubMed] [Google Scholar]
- Lu C.; Wu C.; Ghoreishi D.; Chen W.; Wang L.; Damm W.; Ross G. A.; Dahlgren M. K.; Russell E.; Von Bargen C. D.; Abel R.; Friesner R. A.; Harder E. D. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. 10.1021/acs.jctc.1c00302. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Zubatyuk R.; Nebgen B.; Lubbers N.; Barros K.; Roitberg A. E.; Isayev O.; Tretiak S. The ANI-1ccx and ANI-1x data sets, coupled-cluster and density functional theory properties for molecules. Sci. Data 2020, 7, 134. 10.1038/s41597-020-0473-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan R.; Dral P. O.; Rupp M.; von Lilienfeld O. A. Quantum chemistry structures and properties of 134 kilo molecules. Sci. Data 2014, 1, 140022. 10.1038/sdata.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman P.; Behara P. K.; Dotson D. L.; Galvelis R.; Herr J. E.; Horton J. T.; Mao Y.; Chodera J. D.; Pritchard B. P.; Wang Y.; De Fabritiis G.; Markland T. E. SPICE, A Dataset of Drug-like Molecules and Peptides for Training Machine Learning Potentials. Sci. Data 2023, 10, 11. 10.1038/s41597-022-01882-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donchev A. G.; Taube A. G.; Decolvenaere E.; Hargus C.; McGibbon R. T.; Law K.-H.; Gregersen B. A.; Li J.-L.; Palmo K.; Siva K.; Bergdorf M.; Klepeis J. L.; Shaw D. E. Quantum chemical benchmark databases of gold-standard dimer interaction energies. Sci. Data 2021, 8, 55. 10.1038/s41597-021-00833-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaba K.; Pulido I.; Behara P. K.; Cavender C. E.; Friedman A. J.; Henry M. M.; Opeskin H. M.; Iacovella C. R.; Nagle A. M.; Payne A. M.; Shirts M. R.; Mobley D. L.; Chodera J. D.; Wang Y. Machine-learned molecular mechanics force field for the simulation of protein-ligand systems and beyond. arXiv 2023, 10.48550/arXiv.2307.07085. [DOI] [Google Scholar]
- Lehner M. T.; Katzberger P.; Maeder N.; Schiebroek C. C.; Teetz J.; Landrum G. A.; Riniker S. DASH: Dynamic Attention-Based Substructure Hierarchy for Partial Charge Assignment. J. Chem. Inf. Model. 2023, 63, 6014–6028. 10.1021/acs.jcim.3c00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isert C.; Atz K.; Jiménez-Luna J.; Schneider G. QMugs, quantum mechanical properties of drug-like molecules. Sci. Data 2022, 9, 273. 10.1038/s41597-022-01390-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovács D. P.; Moore J. H.; Browning N. J.; Batatia I.; Horton J. T.; Kapil V.; Witt W. C.; Magdău I.-B.; Cole D. J.; Csányi G. MACE-OFF23: Transferable Machine Learning Force Fields for Organic Molecules. arXiv 2023, 10.48550/arXiv.2312.15211. [DOI] [Google Scholar]
- Sellers B. D.; James N. C.; Gobbi A. A Comparison of Quantum and Molecular Mechanical Methods to Estimate Strain Energy in Druglike Fragments. J. Chem. Inf. Model. 2017, 57, 1265–1275. 10.1021/acs.jcim.6b00614. [DOI] [PubMed] [Google Scholar]
- Ehrlich S.; Göller A. H.; Grimme S. Towards full Quantum-Mechanics-based Protein–Ligand Binding Affinities. ChemPhysChem 2017, 18, 898–905. 10.1002/cphc.201700082. [DOI] [PubMed] [Google Scholar]
- Hait D.; Head-Gordon M. How Accurate Is Density Functional Theory at Predicting Dipole Moments? An Assessment Using a New Database of 200 Benchmark Values. J. Chem. Theory Comput. 2018, 14, 1969–1981. 10.1021/acs.jctc.7b01252. [DOI] [PubMed] [Google Scholar]
- Lehtola S.; Dimitrova M.; Fliegl H.; Sundholm D. Benchmarking Magnetizabilities with Recent Density Functionals. J. Chem. Theory Comput. 2021, 17, 1457–1468. 10.1021/acs.jctc.0c01190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou A.; Schauperl M.; Nerenberg P. S. Benchmarking Electronic Structure Methods for Accurate Fixed-Charge Electrostatic Models. J. Chem. Inf. Model. 2020, 60, 249–258. 10.1021/acs.jcim.9b00962. [DOI] [PubMed] [Google Scholar]
- Hickey A. L.; Rowley C. N. Benchmarking Quantum Chemical Methods for the Calculation of Molecular Dipole Moments and Polarizabilities. J. Phys. Chem. A 2014, 118, 3678–3687. 10.1021/jp502475e. [DOI] [PubMed] [Google Scholar]
- Qiu Y.; Smith D. G. A.; Boothroyd S.; Jang H.; Hahn D. F.; Wagner J.; Bannan C. C.; Gokey T.; Lim V. T.; Stern C. D.; Rizzi A.; Tjanaka B.; Tresadern G.; Lucas X.; Shirts M. R.; Gilson M. K.; Chodera J. D.; Bayly C. I.; Mobley D. L.; Wang L.-P. Development and Benchmarking of Open Force Field v1.0.0—the Parsley Small-Molecule Force Field. J. Chem. Theory Comput. 2021, 17, 6262–6280. 10.1021/acs.jctc.1c00571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Raman E. P.; MacKerell A. D. J. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. 10.1021/ci3003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. S.; Isayev O.; Roitberg A. E. ANI-1, A data set of 20 million calculated off-equilibrium conformations for organic molecules. Sci. Data 2017, 4, 170193. 10.1038/sdata.2017.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai B. K.; Sresht V.; Yang Q.; Unwalla R.; Tu M.; Mathiowetz A. M.; Bakken G. A. TorsionNet: A Deep Neural Network to Rapidly Predict Small-Molecule Torsional Energy Profiles with the Accuracy of Quantum Mechanics. J. Chem. Inf. Model. 2022, 62, 785–800. 10.1021/acs.jcim.1c01346. [DOI] [PubMed] [Google Scholar]
- Christensen A. S.; Sirumalla S. K.; Qiao Z.; O’Connor M. B.; Smith D. G. A.; Ding F.; Bygrave P. J.; Anandkumar A.; Welborn M.; Manby F. R.; Miller T. F. OrbNet Denali: A machine learning potential for biological and organic chemistry with semi-empirical cost and DFT accuracy. J. Chem. Phys. 2021, 155, 204103. 10.1063/5.0061990. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Schmidt K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 2001, 577, 1–20. 10.1063/1.1390175. [DOI] [Google Scholar]
- Dion M.; Rydberg H.; Schröder E.; Langreth D. C.; Lundqvist B. I. Van der Waals Density Functional for General Geometries. Phys. Rev. Lett. 2004, 92, 246401. 10.1103/PhysRevLett.92.246401. [DOI] [PubMed] [Google Scholar]
- Lee K.; Murray D.; Kong L.; Lundqvist B. I.; Langreth D. C. Higher-accuracy van der Waals density functional. Phys. Rev. B 2010, 82, 081101. 10.1103/PhysRevB.82.081101. [DOI] [Google Scholar]
- Vydrov O. A.; Van Voorhis T. Nonlocal van der Waals density functional: The simpler the better. J. Chem. Phys. 2010, 133, 244103. 10.1063/1.3521275. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Hansen A.; Brandenburg J. G.; Bannwarth C. Dispersion-Corrected Mean-Field Electronic Structure Methods. Chem. Rev. 2016, 116, 5105–5154. 10.1021/acs.chemrev.5b00533. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Becke A. D.; Johnson E. R. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 123, 154101. 10.1063/1.2065267. [DOI] [PubMed] [Google Scholar]
- Yu H. S.; He X.; Li S. L.; Truhlar D. G. MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions. Chem. Sci. 2016, 7, 5032–5051. 10.1039/C6SC00705H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goerigk L.; Grimme S. Efficient and Accurate Double-Hybrid-Meta-GGA Density Functionals—Evaluation with the Extended GMTKN30 Database for General Main Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2011, 7, 291–309. 10.1021/ct100466k. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Hansen A.; Bauer C.; Ehrlich S.; Najibi A.; Grimme S. A look at the density functional theory zoo with the advanced GMTKN55 database for general main group thermochemistry, kinetics and noncovalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 32184–32215. 10.1039/C7CP04913G. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. Thirty years of density functional theory in computational chemistry: an overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. 10.1080/00268976.2017.1333644. [DOI] [Google Scholar]
- Mehta N.; Fellowes T.; White J. M.; Goerigk L. CHAL336 Benchmark Set: How Well Do Quantum-Chemical Methods Describe Chalcogen-Bonding Interactions?. J. Chem. Theory Comput. 2021, 17, 2783–2806. 10.1021/acs.jctc.1c00006. [DOI] [PubMed] [Google Scholar]
- ŘezáČ J.; Bím D.; Gutten O.; RulíŠek L. Toward Accurate Conformational Energies of Smaller Peptides and Medium-Sized Macrocycles: MPCONF196 Benchmark Energy Data Set. J. Chem. Theory Comput. 2018, 14, 1254–1266. 10.1021/acs.jctc.7b01074. [DOI] [PubMed] [Google Scholar]
- Yuan Y.; Mills M. J. L.; Popelier P. L. A.; Jensen F. Comprehensive Analysis of Energy Minima of the 20 Natural Amino Acids. J. Phys. Chem. A 2014, 118, 7876–7891. 10.1021/jp503460m. [DOI] [PubMed] [Google Scholar]
- Jurečka P.; Šponer J.; Černý J.; Hobza P. Benchmark database of accurate (MP2 and CCSD(T) complete basis set limit) interaction energies of small model complexes, DNA base pairs, and amino acid pairs. Phys. Chem. Chem. Phys. 2006, 8, 1985–1993. 10.1039/B600027D. [DOI] [PubMed] [Google Scholar]
- Řezáč J.; Riley K. E.; Hobza P. S66: A Well-balanced Database of Benchmark Interaction Energies Relevant to Biomolecular Structures. J. Chem. Theory Comput. 2011, 7, 2427–2438. 10.1021/ct2002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Řezáč J. Non-Covalent Interactions Atlas Benchmark Data Sets: Hydrogen Bonding. J. Chem. Theory Comput. 2020, 16, 2355–2368. 10.1021/acs.jctc.9b01265. [DOI] [PubMed] [Google Scholar]
- Folmsbee D.; Hutchison G. Assessing conformer energies using electronic structure and machine learning methods. Int. J. Quantum Chem. 2021, 121, e26381 10.1002/qua.26381. [DOI] [Google Scholar]
- Siiskonen A.; Priimagi A. Benchmarking DFT methods with small basis sets for the calculation of halogen-bond strengths. J. Mol. Model. 2017, 23, 50. 10.1007/s00894-017-3212-4. [DOI] [PubMed] [Google Scholar]
- Nam S.; Cho E.; Sim E.; Burke K. Explaining and Fixing DFT Failures for Torsional Barriers. J. Phys. Chem. Lett. 2021, 12, 2796–2804. 10.1021/acs.jpclett.1c00426. [DOI] [PubMed] [Google Scholar]
- Kesharwani M. K.; Karton A.; Martin J. M. L. Benchmark ab Initio Conformational Energies for the Proteinogenic Amino Acids through Explicitly Correlated Methods. Assessment of Density Functional Methods. J. Chem. Theory Comput. 2016, 12, 444–454. 10.1021/acs.jctc.5b01066. [DOI] [PubMed] [Google Scholar]
- Godbout N.; Salahub D. R.; Andzelm J.; Wimmer E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. 10.1139/v92-079. [DOI] [Google Scholar]
- Durant J. L.; Leland B. A.; Henry D. R.; Nourse J. G. Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- Qiu Y.; Smith D. G. A.; Stern C. D.; Feng M.; Jang H.; Wang L.-P. Driving torsion scans with wavefront propagation. J. Chem. Phys. 2020, 152, 244116. 10.1063/5.0009232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgaker T.; Klopper W.; Koch H.; Noga J. Basis-set convergence of correlated calculations on water. J. Chem. Phys. 1997, 106, 9639–9646. 10.1063/1.473863. [DOI] [Google Scholar]
- Halkier A.; Helgaker T.; Jørgensen P.; Klopper W.; Olsen J. Basis-set convergence of the energy in molecular Hartree–Fock calculations. Chem. Phys. Lett. 1999, 302, 437–446. 10.1016/S0009-2614(99)00179-7. [DOI] [Google Scholar]
- Burns L. A.; Smith D. G. A.; Kraus P.. Complete Basis Set. 2023. https://psicode.org/psi4manual/master/cbs.html. [Google Scholar]
- Burns L. A.; Marshall M. S.; Sherrill C. D. Appointing silver and bronze standards for noncovalent interactions: A comparison of spin-component-scaled (SCS), explicitly correlated (F12), and specialized wavefunction approaches. J. Chem. Phys. 2014, 141, 234111. 10.1063/1.4903765. [DOI] [PubMed] [Google Scholar]
- Kodrycka M.; Patkowski K. Platinum, gold, and silver standards of intermolecular interaction energy calculations. J. Chem. Phys. 2019, 151, 070901. 10.1063/1.5116151. [DOI] [PubMed] [Google Scholar]
- Marshall M. S.; Burns L. A.; Sherrill C. D. Basis set convergence of the coupled-cluster correction, δ(MP2)(CCSD(T)): Best practices for benchmarking non-covalent interactions and the attendant revision of the S22, NBC10, HBC6, and HSG databases. J. Chem. Phys. 2011, 135, 194102. 10.1063/1.3659142. [DOI] [PubMed] [Google Scholar]
- Marshall M. S.; Sears J. S.; Burns L. A.; Brédas J.-L.; Sherrill C. D. An Error and Efficiency Analysis of Approximations to Møller Plesset Perturbation Theory. J. Chem. Theory Comput. 2010, 6, 3681–3687. 10.1021/ct100468f. [DOI] [Google Scholar]
- Becke A. D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. 10.1063/1.464304. [DOI] [Google Scholar]
- Rassolov V. A.; Ratner M. A.; Pople J. A.; Redfern P. C.; Curtiss L. A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. 10.1002/jcc.1058. [DOI] [Google Scholar]
- Francl M. M.; Pietro W. J.; Hehre W. J.; Binkley J. S.; Gordon M. S.; DeFrees D. J.; Pople J. A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. 10.1063/1.444267. [DOI] [Google Scholar]
- Dill J. D.; Pople J. A. Self-consistent molecular orbital methods. XV. Extended Gaussian-type basis sets for lithium, beryllium, and boron. J. Chem. Phys. 1975, 62, 2921–2923. 10.1063/1.430801. [DOI] [Google Scholar]
- Hehre W. J.; Ditchfield R.; Pople J. A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. 10.1063/1.1677527. [DOI] [Google Scholar]
- Hellweg A.; Rappoport D. Development of new auxiliary basis functions of the Karlsruhe segmented contracted basis sets including diffuse basis functions (def2-SVPD, def2-TZVPPD, and def2-QVPPD) for RI-MP2 and RI-CC calculations. Phys. Chem. Chem. Phys. 2015, 17, 1010–1017. 10.1039/C4CP04286G. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Dunning T. H. Jr Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Wilson A. K.; Woon D. E.; Peterson K. A.; Dunning T. H. Jr Gaussian basis sets for use in correlated molecular calculations. IX. The atoms gallium through krypton. J. Chem. Phys. 1999, 110, 7667–7676. 10.1063/1.478678. [DOI] [Google Scholar]
- Weigend F.; Furche F.; Ahlrichs R. Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr. J. Chem. Phys. 2003, 119, 12753–12762. 10.1063/1.1627293. [DOI] [Google Scholar]
- Pritchard B. P.; Altarawy D.; Didier B.; Gibson T. D.; Windus T. L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. 10.1021/acs.jcim.9b00725. [DOI] [PubMed] [Google Scholar]
- Smith D. G. A.; Burns L. A.; Patkowski K.; Sherrill C. D. Revised Damping Parameters for the D3 Dispersion Correction to Density Functional Theory. J. Phys. Chem. Lett. 2016, 7, 2197–2203. 10.1021/acs.jpclett.6b00780. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. B97M-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation. J. Chem. Phys. 2016, 144, 214110. 10.1063/1.4952647. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Schultz N. E.; Truhlar D. G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. 10.1021/ct0502763. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Schultz N. E.; Truhlar D. G. Exchange-correlation functional with broad accuracy for metallic and nonmetallic compounds, kinetics, and noncovalent interactions. J. Chem. Phys. 2005, 123, 161103. 10.1063/1.2126975. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. Exploring the Limit of Accuracy of the Global Hybrid Meta Density Functional for Main-Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2008, 4, 1849–1868. 10.1021/ct800246v. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Zhao Y.; Truhlar D. G. Design of Density Functionals That Are Broadly Accurate for Thermochemistry, Thermochemical Kinetics, and Nonbonded Interactions. J. Phys. Chem. A 2005, 109, 5656–5667. 10.1021/jp050536c. [DOI] [PubMed] [Google Scholar]
- Kozuch S.; Gruzman D.; Martin J. M. L. DSD-BLYP: A General Purpose Double Hybrid Density Functional Including Spin Component Scaling and Dispersion Correction. J. Phys. Chem. C 2010, 114, 20801–20808. 10.1021/jp1070852. [DOI] [Google Scholar]
- Robertson M. J.; Tirado-Rives J.; Jorgensen W. L. Improved Peptide and Protein Torsional Energetics with the OPLS-AA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. 10.1021/acs.jctc.5b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.-P.; Martinez T. J.; Pande V. S. Building Force Fields: An Automatic, Systematic, and Reproducible Approach. J. Phys. Chem. Lett. 2014, 5, 1885–1891. 10.1021/jz500737m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.-P.; Chen J.; Van Voorhis T. Systematic Parametrization of Polarizable Force Fields from Quantum Chemistry Data. J. Chem. Theory Comput. 2013, 9, 452–460. 10.1021/ct300826t. [DOI] [PubMed] [Google Scholar]
- Smith D. G. A.; Burns L. A.; Simmonett A. C.; Parrish R. M.; Schieber M. C.; Galvelis R.; Kraus P.; Kruse H.; Di Remigio R.; Alenaizan A.; James A. M.; Lehtola S.; Misiewicz J. P.; Scheurer M.; Shaw R. A.; Schriber J. B.; Xie Y.; Glick Z. L.; Sirianni D. A.; O’Brien J. S.; Waldrop J. M.; Kumar A.; Hohenstein E. G.; Pritchard B. P.; Brooks B. R.; Schaefer H. F. III; Sokolov A. Y.; Patkowski K.; DePrince A. E. III; Bozkaya U.; King R. A.; Evangelista F. A.; Turney J. M.; Crawford T. D.; Sherrill C. D. PSI4 1.4: Open-source software for high-throughput quantum chemistry. J. Chem. Phys. 2020, 152, 184108. 10.1063/5.0006002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horton J. T.; Boothroyd S.; Wagner J.; Mitchell J. A.; Gokey T.; Dotson D. L.; Behara P. K.; Ramaswamy V. K.; Mackey M.; Chodera J. D.; Anwar J.; Mobley D. L.; Cole D. J. Open Force Field BespokeFit: Automating Bespoke Torsion Parametrization at Scale. J. Chem. Inf. Model. 2022, 62, 5622–5633. 10.1021/acs.jcim.2c01153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar M. J. S.; Zoebisch E. G.; Healy E. F.; Stewart J. J. P. Development and use of quantum mechanical molecular models. 76. AM1: a new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. 10.1021/ja00299a024. [DOI] [Google Scholar]
- Stewart J. J. P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. 10.1007/s00894-007-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J. J. P. Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. 10.1007/s00894-012-1667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte J.; Neaton J. B.; Head-Gordon M. Effective empirical corrections for basis set superposition error in the def2-SVPD basis: gCP and DFT-C. J. Chem. Phys. 2017, 146, 234105. 10.1063/1.4986962. [DOI] [PubMed] [Google Scholar]
- Sure R.; Grimme S. Corrected small basis set Hartree-Fock method for large systems. J. Comput. Chem. 2013, 34, 1672–1685. 10.1002/jcc.23317. [DOI] [PubMed] [Google Scholar]
- Müller M.; Hansen A.; Grimme S. ωB97X-3c: A composite range-separated hybrid DFT method with a molecule-optimized polarized valence double- basis set. J. Chem. Phys. 2023, 158, 014103. 10.1063/5.0133026. [DOI] [PubMed] [Google Scholar]
- Bannwarth C.; Ehlert S.; Grimme S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. 10.1021/acs.jctc.8b01176. [DOI] [PubMed] [Google Scholar]
- Anstine D.; Zubatyuk R.; Isayev O. AIMNet2: A Neural Network Potential to Meet your Neutral, Charged, Organic, and Elemental-Organic Needs. chemRxiv 2023, 10.26434/chemrxiv-2023-296ch. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scripts to download the structure data from QCArchive and the downloaded energy and dipole data are available at https://github.com/MobleyLab/qm-theory-benchmark/tree/master.