

Abstract
Introduction
Infantile-onset Pompe disease (IOPD) is due to mutations in the GAA gene leading to profound deficiency of the lysosomal enzyme α-1,4-glucosidase. The disease is characterized by severe hypotonia, hypertrophic cardiomyopathy, macroglossia, and liver enlargement with onset in the first months of life. In the late-onset form (LOPD), muscle signs predominate with a clinical picture resembling muscle dystrophies. Enzyme replacement therapy with alglucosidase alfa (rhGAA) has been available since 2006 and patients treated with the enzyme show improved outcomes. Nevertheless, there is evidence that some patients have a suboptimal response or, after an initial improvement, reach a plateau with stabilization of the clinical picture. Thus, a new enzyme formulation, avalglucosidase alfa (neoGAA), with a higher degree of mannosylation, was developed.
Methods
We conducted a multicenter survey that collected data on four patients with IOPD, aged 6 to 16 years, who were switched to neoGAA thanks to a compassionate use program, after being treated for an average of 11.5 years with rhGAA. Follow-up data, including biochemical parameters and clinical features, were analyzed to determine clinical outcomes and the safety profile after a mean of 9 months.
Results
Patients with IOPD who were treated with neoGAA showed a positive change in biomarker levels. Moreover, the clinical picture revealed improved motor performance and cardiac parameters in patients who previously responded poorly.
Conclusion
This study highlights the improved efficacy of neoGAA, as a next generation enzyme replacement therapy, in 4 Italian patients with IOPD. Several clinical parameters showed a positive response to the new formulation suggesting that, if used at diagnosis, neoGAA may result in better outcomes for patients with IOPD.
1. Introduction
Pompe disease, glycogenosis type II, (OMIM 232300) is a glycogen storage disorder caused by deficiency of the lysosomal enzyme acid alpha-glucosidase, and mainly involves skeletal and cardiac muscles [1]. Mutations in the GAA gene on chromosome 17q25, causing severe enzyme deficiency, are associated with infantile-onset Pompe disease (IOPD), the earliest and most severe form. The clinical picture of IOPD is characterized by severe hypotonia, hypertrophic cardiomyopathy, macroglossia, and liver enlargement with onset in the first six months of life. In our experience, a variable degree of severity is present, with some patients showing hypertrophic cardiomyopathy and severe hypotonia after birth, and other manifesting clear signs in the first weeks or months of life. Mutations in GAA that allow some degree of enzyme synthesis and function cause later onset and slowly progressive clinical pictures (late onset Pompe disease, LOPD) which mimic other progressive myopathies (Limb-girdle dystrophy or Duchenne/Becker dystrophies) [[1], [2], [3]]. LOPD can also be diagnosed in the first year of life, in the absence of clear signs, through family history or newborn screening. Moreover, biochemical clues such as hypertransaminasemia or increased creatine kinase (CK) can be incidentally identified during routine investigations in infancy.
Since 2006 alglucosidase alfa (rhGAA) has been available for enzyme replacement therapy (ERT). Without treatment, patients with IOPD do not learn to walk and typically die within the first year of life, mainly because of progressive cardio-respiratory failure. ERT significantly changed the disease course, avoiding early death. Nevertheless, patients treated with ERT have variable outcomes that are mainly related to diagnostic delays. Some patients reach acceptable motor function, even if in the context of a variable myopathic clinical picture, while others never walk and need ventilatory assistance [[3], [4], [5], [6]]. Outcome-determining factors, at the time of initiation of ERT, are age and severity of the clinical picture [4,7]. Moreover, status of cross-reactive immunological material (CRIM) and antibody formation against rhGAA play a critical role in hampering the efficacy of ERT, necessitating an immunomodulation protocol at the start of treatment [7,8]. To date, variable dosages have been used to obtain the best possible results [4,9,10].
The uptake of exogenous lysosomal enzymes by receptors and their transfer into lysosomes improves with higher levels of mannose-6-phosphate (M6P) [11]. Thus, with the aim of improving the efficacy of rhGAA a new enzyme, avalglucosidase alfa (neoGAA) was glycoengineered, which is conjugated with bis-M6P to enhance its uptake in muscle tissue [12]. Positive results following treatment with neoGAA in patients with LOPD (COMET study) have been reported [[13], [14], [15]]. A Mini COMET study investigating the use of neoGAA was also carried out that enrolled 22 pediatric patients (aged 1 to 12 years) with IOPD in an open Phase 2 trial using escalating doses. Preliminary results have demonstrated a positive impact on motor and cardiac function as well as on biomarkers [16,17].
In 2022, neoGAA was made available in Italy by Sanofi through a compassionate use program in 4 Italian centers. Herein, we report on four patients with IOPD who switched to neoGAA after being treated for an average of 11.5 years with rhGAA.
2. Methods
2.1. Patients
In this observational cohort study, we collected data on four patients who switched to neoGAA in four different referral centers in Italy (Catania, Florence, Monza, and Padua) after obtaining informed consent from parents and approval by each respective local Ethics Committee. The treatment was made available by Sanofi through a compassionate use program. Inclusion criteria were the absence of other condition or illness or clinical worsening precluding safe participation in the trial. Exclusion criteria were pregnancy or breastfeeding, which was not the case in any of these pediatric patients.
They were 3 females and one male, with an age ranging from 6 to 16 years (mean age 12.25 years). Diagnosis of IOPD was reached through the demonstration of lack of GAA activity in fibroblasts and germline GAA mutations at an age ranging from 1 month to 11 months (mean age at diagnosis 4.5 months) as reported in Table 1.
Table 1.
Patient characteristics before therapy with neoGAA. HCM, hypertrophic cardiomyopathy; PEG, percutaneous endoscopic gastrostomy.
| Case #/sex | Gene 1 | Gene 2 | CRIM status |
Age at diagnosis and ERT initiation | ERT duration before switch | Clinical outcome | Anti-rhGAA antibody titer |
Adverse events while on rhGAA |
|---|---|---|---|---|---|---|---|---|
| #1 M | c.693–2 A > C (IVS3–2 A > C) | c.368G > A (p.Gly123Glu) c.1288G > A (p.Glu430Lys) |
Negative | 3 months | 6 years | Severe hypotonia, HCM, never walking, PEG-tracheostomy | 120,000 | Rash, laryngospasm to anaphylaxis |
| #2 F | c.236_246DEL11 | c.1927G > A | Positive | 11 months | 11 years | PEG, never walking, sensorineural deafness at 5 years | 52,100 | Rash, nausea, epigastric pain |
| #3 F | c.1933G > A | c.1564C > G | Positive | 3 months | 16 years | Sensorineural deafness at 12 years, walking |
400 | None |
| #4 F | c.1933G > A (p.D645N) | c.1933G > A (p.D645N) | Positive | 1 month | 13 years | Scoliosis, ptosis, walking with aid | 0 | None |
2.2. Clinical and laboratory assessments
Data were collected on cardiac involvement, motor and cognitive performance, blood biomarkers and swallowing tolerance. Blood samples were drawn every 4 weeks for complete blood count (CBC), liver function tests, and biomarkers (transaminases, CK). Urine tetrasaccharides (uGlc4) were tested in 3 of 4 patients following the indications of Pirauda et al. [18]. Antibodies against rhGAA and neoGAA were tested by LabCorp, California, USA.
All patients underwent pulmonary function testing, and a six-minute walking test (6MWT) was possible in 2 of 4 patients. Neurocognitive function was assessed by the Weschler Preschool and Primary Scale of Intelligence, 4th edition (WPPSI-IV) and WAIS-IV according to age. Instrumental control visits were performed every two months. Liver volume was assessed using abdominal ultrasonography (US). Heart electrocardiogram and US were performed in each center by skilled Pediatric Cardiologists with experience in metabolic diseases.
3. Results
3.1. Case #1
This boy was born in 2015 to healthy, unrelated parents. At the age of 3 months, he was evaluated because of progressive hypotonia. Clinical and instrumental investigations showed hypertrophic cardiomyopathy (HCM) and high levels of CK [655 IU/L; normal value (nv) <50 IU/L] and transaminases (ALT 157; AST 304 U/L; nv <35 U/L). Diagnosis of IOPD was reached through the acid maltase assay in fibroblasts, which showed no activity. Molecular analysis of the GAA gene confirmed a splicing mutation c.693–2 A > C IVS3–2 A > C and two exonic changes c.368G > Ap. (Gly123Glu) and c.1288G > A p. (Glu430Lys). His CRIM status was negative, but at that time no immunotolerance protocol was known. Soon after diagnosis, he started ERT with rhGAA 20 mg/kg every other week. Four months later, at 7 months of age, he was admitted for a cardio-respiratory crisis following infection with Cytomegalovirus. He was treated with antibiotics, cortisone, salbutamol, and O2. There was a sudden worsening of cardiac function with sporadic peaks of hypertension, the left ventricular ejection fraction (LVEF) was 57%, a reduction of myocardial kinesis and left ventricle function with inversion of mitral valve E/A ratio (0.7), and an increase of the ventricular contraction system (VCS) which was emptying at 125 cm/s. These features prompted us to increase the dosage of ERT 20 mg/kg/week and two weeks later to 40 mg/kg/week. Captopril and furosemide were also started. At that time, phoniatric evaluation revealed dysphagia and considering the high risk of tracheal inhalation, a nasogastric tube (NGT) was used for feeding. One month later, the heart was still enlarged with hypertrophic and hyperechoic lateral ventricles and interventricular septum (IV). The left ventricular mass index (LVMI) was 143 g/m2 and LVEF was increased to 68%, while the mitral valve E/A ratio was 1.3 although moderate mitral valve patency persisted. Thanks to logopedic treatment, the NGT was removed.
At the age of 11 months, at the 45th infusion, he developed macular rash all over his body that was successfully treated with an antihistamine and corticosteroids. The antibody titers were very high (120,000 IgG), and thus he underwent 3 plasma exchanges and an immunosuppression protocol with rituximab and pooled intravenous immunoglobulins (IVIg). At follow-up, heart echocardiogram showed worsening mitral valve insufficiency and the LVMI was 98 g/m2 with a LVEF of 75%.
During the 57th infusion, 20 min after initiation, a new episode of reaction was observed with recurrent cough and respiratory insufficiency. This forced us to reduce the dosage to 20 mg/kg every other week, according to the advice of our Ethic Committee. The patient had no further reactions up to the age of 30 months, when recurrent respiratory infections appeared that were followed by further worsening of respiratory ability. Moreover, the persistent increase in antibodies against rhGAA required a new cycle of plasma-exchange. The dosage was again increased to 20 mg/kg/week and he was pre-treated with hydrocortisone and antihistamine. No reaction was observed.
At follow-up, two months later, he was not able to stand, his chronic respiratory failure required nocturnal non-invasive ventilation (NIV), cardiomyopathy was severe, and NGT was applied to allow nutrition. Despite these aids, he had two cardiac arrests and was transferred to the intensive care unit where he underwent tracheotomy. We thus decided to increase the dosage to 40 mg/kg/week. Since then, his general conditions and respiratory function both improved. Nevertheless, 15 min after the start of each ERT infusion he consistently presented desaturation, tachycardia, and dyspnea that were controlled with salbutamol, hydrocortisone, and 02.
In 2022, at the age of 6 years, he was enrolled in the Avalglucosidase compassionate use program and received 40 mg/kg every other week. After 3 months, we observed a surprising regression of HCM with a ventricular mass of 53 g/m2 (Fig. 1). Six months later, he showed improved lifting of arms and grasping objects and a progressive improvement in speech, which was previously almost understandable due to weak and coarse voice.
Fig. 1.
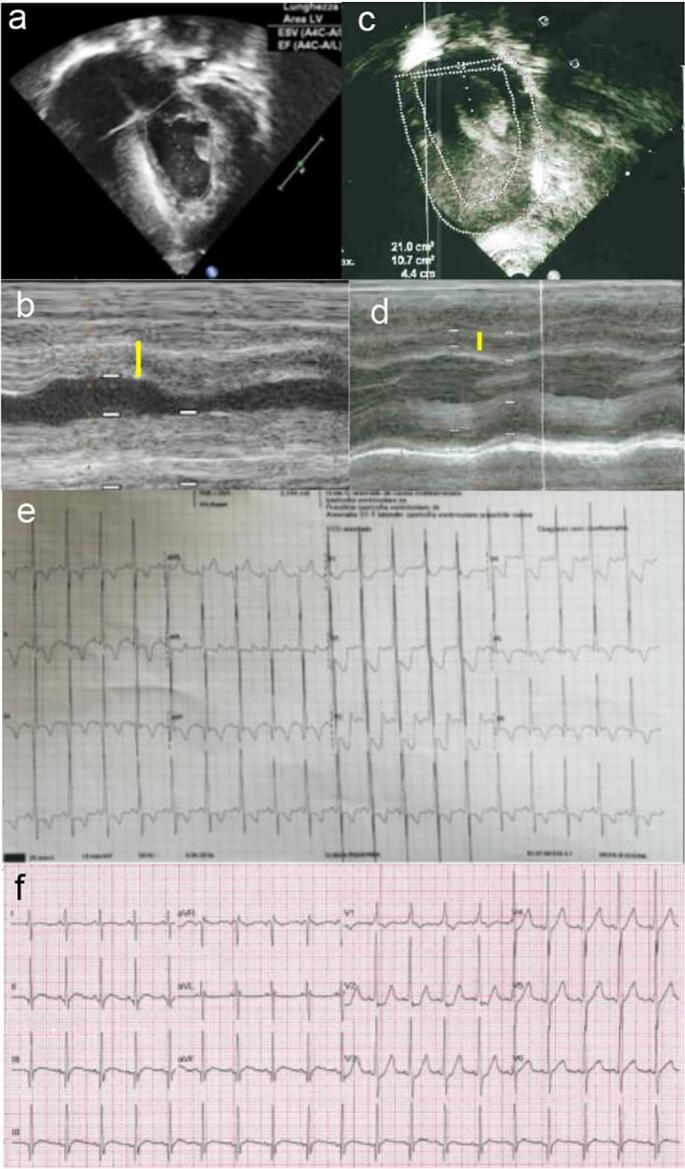
Patient #1. Color Doppler echocardiogram at baseline before the switch to neoGAA (a-b) showing marked myocardial hypertrophy (white and yellow lines), reduced in T3, after 9 months of ERT with neoGAA (c-d). Electrocardiogram at baseline (e) showing biventricular overload with high narrow QRS, Q waves, ST-segment elevation, and negative T in all leads; at T3 (f), the biventricular overload is markedly reduced (reduced QRS height RV > Vsn), the ST segment is no longer elevated, and negative T waves in the left leads disappeared.
3.2. Case #2
Born in 2012, this girl is the first born of unrelated parents, after two spontaneous abortions in the first trimester. Since the first months of life, she had tongue protrusion with swallowing difficulties when sucking milk. She sat at the age of 7 months and stood up with aid at 11 months. Thereafter, the mother referred arrest of psychomotor development and loss of previously acquired motor skills because of progressive severe hypotonia. At the time of first evaluation, at 10 months, thyroid hormones were normal but she had hypertransaminasemia (AST 399 IU/L, ALT 205 IU/L; nv <35), LDH 1658 IU/L (nv 60–160), CK 639 U/L (nv 30–170), CK_MB 18.82 IU/L (nv 0–25), troponin HS 79 ng/L (nv <15). Heart US control showed severe left ventricle hypertrophy, increased IV septum (15 mm), reduced kinesis, and mitral insufficiency. IOPD was diagnosed by lack of acid maltase activity in lymphocytes (0 nM/mg/h; nv 11–32) and confirmed by molecular investigation that revealed C.236_246DEL11 and C.1927G > A.
ERT with rhGAA was started at the age of 11 months with 20 mg/kg every other week passing to 20 mg/kg/week and then to 30 mg/kg/week over the years. A gradual reduction until normalization of HCM was observed. At the 11th infusion, a rash appeared that was successfully treated with antihistamine and anti-rhGAA Ab titers were increased to 12,800. Three years later she was admitted to the intensive care unit for severe respiratory distress and NIV has been needed ever since. A few months later, for recurrence of episodes of food inhalation with pulmonary involvement, she also needed a percutaneous endoscopic gastrostomy (PEG) to ensure adequate caloric intake. The patient recovered well and regained oral feeding thanks to logopedic treatment. Sensorineural hypoacusia was diagnosed at 5 years.
During the following years, she had recurrent pulmonary infections due to impaired deglutition. At 8 years, she showed a reduced range of motion of hips and knees and dorso-lumbar kyphoscoliosis with pelvis dysmetria. Gait was possible with extra-rotation of the right foot. X-ray investigations detected bilateral coxo-femoral subluxation. Spirometry results were compatible with a restrictive respiration pattern. The Weschler Intelligence Scale for Children revealed an IQ of 61. Heart US showed normal parameters with normal ventricle volume and IV septum.
At 9 years, during ERT, the girl began to complain of epigastric pain, nausea, and rash. Following this, these reactions occurred at each infusion, despite pre-medication with hydrocortisone and antihistamine. Infusion velocity was lowered but without success, and thus a desensitization protocol was applied with partial control of reactions.
Antibodies against rhGAA increased to 12,800–51,200, and an immunotolerance protocol was started with rituximab (375 mg/m2/week for 4 weeks, and a maintenance dose after 3 months), IVIg (440 mg/kg/month) followed, after 7 weeks, by methotrexate at 0.5 mg/kg/week. During this protocol she developed malar rash, pale and cold skin, vomiting, and tremors. Considering the high recurrence of hypersensitivity reactions, she was enrolled in the compassionate use program for neoGAA and started treatment with 40 mg/kg every other week, continuing her immunosuppressive protocol.
Before switching to neoGAA, she had stable respiratory impairment, oxygen saturation 98%, vital capacity (VC) 0.46 L (20% of predicted, same as 6 moths earlier), and Peak Cough Expiratory Flow (PcEF) of 104 L/min (>5th percentile for age). A cough assist machine and NIV were regularly used, while motor evaluation (by Quick Motor Function Test and Pediatric Evaluation of Disability Inventory (PEDI), and Short Form Health Survey 36 (SF36) all revealed a clear worsening of motor abilities. Heart US was normal.
After 8 months on neoGAA, the VC was 21% of predicted (z-score − 7.44). She can maintain a sitting position, with evident kyphoscoliosis, and lower limbs are held flexed and extra-rotated. Vertical head control is good. She is now able to make mild rotation movements of the trunk and is able to lift arms and grasp small objects like a pen.
3.3. Case #3
This patient was diagnosed with IOPD at the age of 3 months for cardiomyopathy and started rhGAA 20 mg/kg every other week soon after (age 5 months). She had a good response to ERT and walked at the age of 13 months. At age 6 years, asymmetry of hips and dorso-lumbar kyphoscoliosis were evident. Bilateral sensorineural deafness was diagnosed at 12 years. Brain MRI revealed T2 and FLAIR hyperintensity of supratentorial white matter involving capsulae and partial involvement of U fibers. Moreover, spirometry was performed and revealed moderate-severe respiratory insufficiency. Cardiologic follow-up with electrocardiogram, US, and heart MRI showed a stable picture without significant issues. In contrast, her scoliosis worsened with appearance of a dorsal gibbus.
At 14 years, her physical examination showed dysarthria, muscle hypotropia, and right scoliosis, with uncertain autonomous walk. No dysphagia or worsening of the respiratory pattern was observed. At 15 years, while on rhGAA at 40 mg/kg/week, she experienced a sudden worsening of motor function. She was able to complete 100 m during her 6MWT. Thus, she was enrolled in neoGAA compassionate use program.
Before the switch to neoGAA, muscle MRI showed adipose substitution in all muscles with atrophy in T1 and T2 sequences. Heart evaluation was considered normal, although mild repolarization anomalies were apparent. There was objective worsening of respiratory function, but spirometry results were stable.
Three months after switching to neoGAA, motor function and 6MWT improved as well as quality of life. After 8 months of neoGAA therapy, her 6MWT was 190 m. The anti-rhGAA antibody titer was 400 U/L and CK-MB was 17.8 U/L (nv <4.88). Her physical examination showed a hypomimic, asthenic face with ogival palate. The liver was palpable at the costal rime. She walks unaided, although with a waddling gait and a hyperlordotic spine. She can stand up from a sitting position without aid, but Gowers maneuver is positive, and she can lift her arms but with suboptimal strength of upper girdle. Anti-neoGAA antibodies were repeatedly negative. Unfortunately, there are no data on uGlc4 before the switch to neoGAA. After 15 months on neoGAA, uGlc4 was 9.51 (nv 0.08–1.37) which decreased 7.56 after 18 months.
3.4. Case #4
This patient's detailed clinical history was described in 2010 [19]. Born by Cesarian section because of intrauterine bradycardia, at birth she presented mild hypotonia that progressively worsened during the following two weeks when macroglossia was also evident. Heart US confirmed bilateral ventricular hypertrophy. Routine blood exams revealed moderate hypertransaminasemia (AST 200 U/L, ALT 99 U/L) and elevation of CK (731 U/L, nv 0–295). Electromyography (EMG) showed a myopathic pattern. Increased urine excretion of uGlc4 and absence of GAA activity in white blood cells led to the diagnosis of Pompe disease, which was confirmed by the presence of a homozygous missense mutation: c.1933 G > A, p.Asp645Asn (GAA exon 14).
She started ERT with rhGAA in the first month of life (20 days of age) at 20 mg/kg every other week for 26 months. After the 3rd month of treatment, heart US showed improvement of cardiomyopathy up to normalization at 24 months of follow-up. At baseline, her LVMI was 171 g/m2 (Z-score = 4.3), while the LVEF was normal. After 2 years of ERT, LVMI decreased to 35 g/m2 (Z-score = −2.1). Antibodies against rhGAA were tested every 3 months and no positive result was found. She had normal physical and motor growth and walked unaided at 12 months, but had a slight delay in speech.
In 2016, 8 years after starting ERT, frequent respiratory infections and worsening of motor performance were observed. She climbed stairs with difficulty and her gait was unstable. Thus, the dosage of rhGAA was increased to 40 mg/kg every other week. In the following 5 years, spirometry showed moderate-severe restrictive ventilation and the dosage of rhGAA was increased to 40 mg/kg/week. In November 2021, at the age of 13 years, she was enrolled in the neoGAA compassionate use program.
At baseline, she had a rich language although speech was sometimes dyslalic. She had a hypomimic face, palpebral ptosis, and increased lumbar lordosis. Cranial nerve exam was normal. She was able to pass from lying to sitting position and from sitting to standing. Muscle hypotrophy was more evident at lower limbs and she had a waddling gait with wide base and extra rotation of feet. She completed 170 m during a 6MWT.
Heart US showed no hypertrophy and the LVEF was 60%. Spirometry confirmed a restrictive ventilatory pattern without further progression compared to previous exam. Brain MRI showed periventricular white matter involvement that remained stable after 9 months of neoGAA therapy. She could walk with braces and the 6MWT showed no worsening, progressively passing from 175 m to 180 m and then 240 m. Spirometry showed severe restrictive ventilatory dysfunction in the standing position (VC 1.30 L, FEV1 1.13 L, 45% of predicted), which was reduced by 20% in the supine position, that became stable after neoGAA therapy (at previous follow-up it was worsening).
3.5. Clinical and laboratory assessments
The clinical picture at onset was characterized by hypotonia and cardiomyopathy in all cases; liver enlargement was present in all but one patient (case #2). Laboratory exams revealed high CK levels (572–3000 U/L); and elevated transaminases [ALT (68–241 U/L) AST (118–477 U/L)] and LDH (1203–1935 IU/L).
After assessing CRIM status (positive in 3 and negative in the male patient), all patients started ERT with rhGAA at a dosage of 20 mg/kg every other week, which was later increased to 40 mg/kg/week in cases #1 and #4. The dosage was later increased to 40 mg/kg/week in case #3. One of the CRIM-positive females (case #2) underwent an immunosuppressive protocol with rituximab, methotrexate, and IVIg because of the development of high antibody titers (52,100 IgG). The male patient underwent plasmapheresis at 20 months because of the very high Ab titer (120,000 IgG), and immunosuppression with rituximab and IVIg. In our patients, rhGAA had been administered for long periods (6 to 16 years).
Two patients attained walking abilities, while two never walked (cases #1 and #2). Both these patients needed a PEG to allow adequate nutritional intake, even if case #2 maintained a partial ability to feed orally. Moreover, in case #1 tracheostomy was necessary and he is still receiving NIV.
Heart involvement was present in all cases at the time of diagnosis, described as HCM with increased left ventricle mass or both left and right ventricle enlargement), while the LVEF ranged between 60% and 79%. Clear HCM normalization was obtained in the 3 female patients while on rhGAA after 3 to 10 months of treatment. Modifications in clinical features following treatment with neoGAA are reported in Table 2.
Table 2.
Outcomes after treatment with neoGAA. eow, every other week; HCM, hypertrophic cardiomyopathy; 6MWT, 6 min walk test; PEG, percutaneous endoscopic gastrostomy.
| Case #/sex/age at switch | neoGAA dosage and duration | Heart involvement on rhGAA | Heart involvement on neoGAA (months of treatment) |
Motor performanceon rhGAA | Motor performance on neoGAA (months of treatment) | Other clinical outcomes before and after neoGAA |
Anti-rhGAA titer |
|---|---|---|---|---|---|---|---|
| #1 M 6 years |
40 mg/kg/eow 9 months |
Biventricular hypertrophy with main left involvement, ventricular mass 105 g/m2 and LVEF 74% | Clear HCM regression with a ventricular mass of 53 g/m2 (3 months) |
Profound hypotonia, never walked | Improved arms lifting and grasping objects (6 months) | PEG, tracheostomy already performed, improved speech | 51,200 |
| #2 F 11 years |
40 mg/kg/eow 8 months |
HCM normalized after 3 months | None | Never walked | Improved arms lifting and grasping objects (3 months) |
PEG already present. Improved swallowing and fatigue tolerance |
400 |
| #3 F 15 years |
40 mg/kg/eow 18 months |
HCM normalized after 10 months | None | Walking at 1 year, sudden loss of motor abilities at 15 years | 6MWT improvement (3 months) | Improved quality of life | 0 |
| #4 F 13 years |
40 mg/kg/eow 20 months |
HCM normalized after 6 months | None | Hypotonia, ptosis, walking with braces | Walking unaided, 6MWT improvement (9 months) | Improvement of spirometry results |
0 |
At the time of data collection (October 2022), patients had been on neoGAA for a mean period of 11 months (range 8–20 months), with an mean of 19 infusions. All patients were adherent to therapy and no infusion was missed. In cases #3 and #4 the initial dosage was 20 mg/kg every other week, passing to 40 mg/kg every other week after the 8th to 10th infusions; the other two cases received 40 mg/kg every other week from the start of therapy because of the severe clinical picture.
neoGAA was well tolerated in all patients in the absence of adverse events, including the two patients who had high anti-rhGAA antibodies. Moreover, a significant reduction in CK (mean 47%), AST (48%), ALT (46%), and LDH (32%) was observed (Fig. 2).
Fig. 2.

ALT, AST, CK, and LDH after initiation of neoGAA therapy in the four patients.
Data on uGLC4 pre- and post-neoGAA were available for 3 of 4 patients. In Case #2, uGLC4 before neoGAA was 77 mmol/mol Creatinine (Cr) (range 49–122; nv 0.08–1.37 mmol/mol Cr); after 3 months of treatment uGLC4 excretion was 2.78 mmol/mol Cr. Case #3 had an uGlc4 of 9.51 mmol/mol Cr after 15 months of neoGAA, passing to 7.56 mmol/mol Cr after 18 months. In case #4, uGLC4 decreased by 10% after 9 months of therapy (from 28.9 mmol/mol Cr to 25.5 mmol/mol Cr.
Although a good response on HCM was observed in 3 of 4 patients on rhGAA, a surprisingly positive effect was registered in case #1 after switching to neoGAA. He had a significantly progressive HCM despite treatment with rhGAA, as shown by heart echography which showed biventricular hypertrophy with main left involvement, LVM of 105 g/m2, and LVEF of 74% (Fig. 1 a,b). After 3 months on neoGAA, clear signs of regression were documented with a LVMI of 53 g/m2 (Fig. 1 c,d). Tricuspid annular plane excursion was 1.74 (nv >15) when starting neoGAA that increased to 2.57 after 6 months.
In 2 of 4 patients there was antibody positivity (Fig. 3) In case #1, Anti-rhGAA antibodies were high and still detectable when starting neoGAA, while anti-neoGAA antibodies appeared after 6 months of treatment, with a slight decrease in the following months; in case #2, these antibodies were also present at baseline, perhaps due to cross-reactivity. Anti-neoGAA antibodies progressively decreased along with anti-rhGAA antibodies.
Fig. 3.

Changes in anti-rhGAA and anti-neoGAA antibodies after starting treatment with neoGAA in cases #1 (a) and #2, (b).
4. Discussion
>15 years after the introduction of ERT, good clinical results in patients with Pompe disease have been described in several studies. Dornelles et al. [20] carried out a large systematic review and meta-analysis of published experiences in LOPD. It was concluded that there is evidence that rhGAA increases endurance, as shown by 6MWT, and is associated with improved quality of life and decreased time on ventilation support. They also underlined the importance of starting treatment in a timely fashion, although some studies pointed to a possible decline of efficacy after 3–5 years of treatment [21].
Overall, patients with IOPD treated with a high dose (40 mg/kg/week or every other week) of rhGAA showed prolonged overall survival and invasive ventilation-free survival [22]. Improvements in cardiomyopathy and motor skills are also observed [23,24]. In 2018, van Capelle et al. [25] investigated the effects of ERT on cardiac features and found that LVMI normalized after 30 weeks of treatment and was maintained for 4.8 years. More recently, Scheffers et al. [26] reported on 27 patients treated with ERT who were followed for a median of 9.9 years. It was reported that cardiac function, measured using myocardial deformation analysis, normalized within 1 year after the start of ERT and was stable over time. Nevertheless, despite early diagnosis and treatment, and reaching older ages, when compared to the natural history of the untreated disease, a subset of patients with severe IOPD do not gain motor skills and may also demonstrate significant heart involvement.
The availability of ERT has represented an important therapeutic opportunity for patients with Pompe disease, given the significant impact on the clinical course of disease. However, as the treated cohort becomes larger and the patients become older, new challenges are being unveiled. Involvement of the central nervous system has been described in surviving patients [27]. Hsu et al. collected data on 19 patients from Taiwan who started ERT very early (within the first month of life). These patients were followed for 17 years by regular MRI investigation. White matter abnormalities were estimated using a scoring system for metachromatic leukodystrophy. The authors reported that all patients showed progressive white matter anomalies along with possible neuroaxonal injury for the correlation found with neurofilament levels in plasma. Indeed, there is evidence that levels of neurofilament protein in cerebrospinal fluid and blood are a sensitive, although unspecific, marker of axonal injury and neuronal damage [28].
Unfortunately, our patients #1 and #2 could not be investigated by imaging as the parents denied their consent, being fearful of the need for general anesthesia. Nevertheless, neither patient had clear neurological signs. Patients #3 and #4 had periventricular white matter changes, which remained stable on neoGAA.
Another issue is hearing impairment, which was also present in our cases #2 and #3. The most commonly described type of hearing impairment is sensorineural. Kishnani et al. described inner ear involvement in patients with IOPD in a retrospective, multicenter study on the natural history of the disease in 2006 [29]. In a recent survey on IOPD patients in Taiwan, identified through newborn screening and treated early with rhGAA ERT, Hsueh et al. documented the occurrence of hearing impairment in 6 of 11 cases and 5 were of the sensorineural type. They stressed the positive role of early ERT thanks to identification by newborn screening [30]. Nevertheless, as shown by our two patients, despite early and long-term treatment, hearing failure may still occur and represents another concern for these patients.
The need to improve outcomes has pushed researchers to focus on the possible enhanced capture by lysosomes and deriving increased effect of ERT through the association with chaperons [31] or the synthesis of new recombinant enzymes enriched in M6P [12]. In this regard, neoGAA was developed and the first studies on the late onset forms confirmed its higher efficacy compared to rhGAA. The phase 3 multicenter/multinational study (COMET), designed to evaluate the safety, tolerability, pharmacokinetics, pharmacodynamics, and efficacy of neoGAA in LOPD in patients ≥18 years, showed significant benefits in terms of stabilization and improvement of respiratory and motor functions without significant adverse events [13].
Younger patients were enrolled in Mini-COMET, a phase 2, open label, ascending dose study. A total of 22 patients with IOPD aged 1 to 12 years, previously treated with rhGAA, were selected based on either clinical decline or a suboptimal response to treatment. Preliminary results have indicated a rapid improvement in ptosis [32]. More recent results reported improvement in motor function as measured by GMFM-88, QMFT and POMPE Pedi (Pompe specific Pediatric Evaluation of Disability Inventory) Functional Skills Scale. Biomarkers of muscle storage (CK, AST, ALT, and uGlc4) also decreased to nearly normal levels over time [16,17].
Our data, although limited to 4 patients, represents the first “real-world” experience in Italy of the use of neoGAA in patients with IOPD. Patients were selected based on their age and the inability to achieve optimal results with rhGAA even if it was administered early and for many years. However, due to the real-world setting, the outcomes reported in our four patients varied, even if our findings suggest a significant positive effect of neoGAA, given that a constant and rapid decrease in biomarkers was observed and motor performances appeared to increase. Even heart involvement, which was stable or worsening over the past years, showed a surprising improvement in case #1. In this patient, the high antibody titer might have been responsible for the poor response to rhGAA. While on neoGAA this benefit was detected in the first months of treatment, suggesting a rapid response to the drug. Moreover, no adverse events were registered in our sample, even in patients who had developed high antibody titers against rhGAA.
The need for early diagnosis and treatment of IOPD remains an important issue. In Italy, newborn screening for lysosomal disorders is already active in selected centers and possible expansion in the entire country is under consideration. This would allow identification of patients at birth and prompt, early treatment with neoGAA in the first weeks, through which even better results can be expected.
Declaration of competing interest
None.
Data availability
Data will be made available on request.
References
- 1.Reuser A.J., Hirschhorn R., Kroos M.A. McGraw Hill: The Online Metabolic & Molecular Bases of Inherited Disease (OMMBID); 2018. Pompe Disease: Glycogen Storage Disease type II, Acid Alpha-Glucosidase (Acid Maltase) Deficiency.https://ommbid.mhmedical.com [Google Scholar]
- 2.Gungor D., Reuser A.J. How to describe the clinical spectrum in Pompe disease? Am. J. Med. Genet. A. 2013;161 A(2):399–400. doi: 10.1002/ajmg.a.35662. [DOI] [PubMed] [Google Scholar]
- 3.Duong T., Kishnan K.S., An Haackc K., et al. Motor responses in pediatric Pompe disease in the ADVANCE participant cohort. J. Neuromusc. Dis. 2022;9:713–730. doi: 10.3233/JND-210784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Gelder C.M., Poelman E., Plug I., et al. Effects of a higher dose of alglucosidase alfa on ventilator-free survival and motor outcome in classic infantile Pompe disease: an open-label single-center study. J. Inherit. Metab. Dis. 2016;39:383–390. doi: 10.1007/s10545-015-9912-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broomfield A., Fletcher J., Davison J., et al. Response of 33 UK patients with infantile-onset Pompe disease to enzyme replacement therapy. J. Inherit. Metab. Dis. 2016;39(2):261–271. doi: 10.1007/s10545-015-9898-5. Epub 2015 Oct 26.PMID: 26497565. [DOI] [PubMed] [Google Scholar]
- 6.Hahn A., Schänzer A. Long-term outcome and unmet needs in infantile-onset Pompe disease. Ann. Transl. Med. 2019;7(13):283. doi: 10.21037/atm.2019.04.70.PMID. 31392195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li C., Desai A.K., Gupta P., et al. l. Transforming the clinical outcome in CRIM-negative infantile Pompe disease identified via newborn screening: the benefit of early treatment with enzyme replacement therapy and immune tolerance induction. Genet. Med. 2021;23(5):845–855. doi: 10.1038/s41436-020-01080-y. Epub 2021 Jan 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gragnaniello V., Deodato F., Gasperini S., et al. Immune response to alglucosidase in infantile Pompe disease: reccomendations from an Italian pediatric expert panel. Ital. J. Pediatr. 2022;48(1):41. doi: 10.1186/s13052-022-01219-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khan A.A., Case L.E., Herbert M., et al. Higher dosing of alglucosidase alfa improves outcomes in children with Pompe disease: a clinical study and review of the literature. Genet.Med. 2020;22:898–907. doi: 10.1038/s41436-019-0738-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Case L.E., Bjartmar C., Morgan C., et al. Safety and efficacy of alternative alglucosidase alfa regimens in Pompe disease. Neuromuscul. Disord. 2015;25:321–332. doi: 10.1016/j.nmd.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 11.Seo J., Oh D.B. Mannose-6-phosphate glycan for lysosomal targeting: various applications from enzyme replacement therapy to lysosome-targeting chimeras. Animal Cells System. 2022;26(3):84–91. doi: 10.1080/19768354.2022.2079719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Y., Jiang J.L., Gumlaw N.K., et al. Glycoengineered acid alpha-glucosidase with improved efficacy at correcting the metabolic aberrations and motor function deficits in a mouse model of Pompe disease. Mol. Ther. 2009;17:954–963. doi: 10.1038/mt.2009.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diaz-Manera J., Kishnani P.S., Kushlaf H., et al. Safety and efficacy of avalglucosidase alfa versus alglucosidase alfa in patients with late- onset Pompe disease (COMET): a phase 3, randomised, multicentre trial. Lancet Neurol. 2021;12:1012–1026. doi: 10.1016/S1474-4422(21)00241-6. [DOI] [PubMed] [Google Scholar]
- 14.Dimachkie M.M., Barohn R.J., Byrne B., et al. Long-term safety and efficacy of Avalglucosidase alfa in patients with late-onset Pompe disease. Neurology. 2022;99(5):e536–e548. doi: 10.1212/WNL.0000000000200746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kishnani Priya S., Diaz-Manera Jordi, Toscano Antonio, et al. Efficacy and safety of Avalglucosidase alfa in patients with late-onset Pompe disease after 97WeeksA phase 3 randomized clinical trial. JAMA Neurol. 2023;apr 10 doi: 10.1001/jamaneurol.2023.0552. (Online ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kronn D., Davison J., Brassier A., et al. Mini-COMET: safety and efficacy of ≥97 weeks’ avalglucosidase alfa in infantile-onset Pompe disease participants previously treated with alglucosidase alfa. Genet. Med. 2022;24:S348–S354. [Google Scholar]
- 17.Kishnani P.S., Kronn D., Brassier A., et al. Safety and efficacy of avalglucosidase alfa in individuals with infantile-onset Pompe disease enrolled in the phase 2, open-label Mini-COMET study: the 6-month primary analysis report. Genet. Med. 2022 doi: 10.1016/j.gim.2022.10.010. (Online ahead of print.PMID: 36542086) [DOI] [PubMed] [Google Scholar]
- 18.Pirauda M., Pettazzonia M., de Antoniob M., et al. Urine glucose tetrasaccharide: a good biomarker for glycogenoses type II and III? A study of the French cohort. Mol. Genet. Metabol. Reports. 2020;23 doi: 10.1016/j.ymgmr.2020.100583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Del Rizzo M., Fanin M., Cerutti A., et al. Long-term follow-up results in enzyme replacement therapy for Pompe disease: a case report. J. Inherit. Metab. Dis. 2010;33(Suppl. 3):S389–S393. doi: 10.1007/s10545-010-9195-2. [DOI] [PubMed] [Google Scholar]
- 20.Dornelles A.D., Pedroso Junges A.P., Pereira T.V., et al. A systematic review and meta-analysis of enzyme replacement therapy in late-onset Pompe disease. J. Clin. Med. 2021;10:4828. doi: 10.3390/jcm10214828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harlaar L., Hogrel J.Y., Perniconi B., et al. Large variation in effects during 10 years of enzyme therapy in adults with Pompe disease. Neurology. 2019;93:e1756–e1767. doi: 10.1212/WNL.0000000000008441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.IAMs Ditter, Huidekoper H.H., Kruijshaar M.E., et al. Effect of alglucosidase alfa dosage on survival and walking ability in patients with classic infantile Pompe disease: a multicentre observational cohort study from the European Pompe consortium. Lancet Child Adolesc. Health. 2022;6:28–37. doi: 10.1016/S2352-4642(21)00308-4. [DOI] [PubMed] [Google Scholar]
- 23.Nicolino M., Byrne B.J., Wraith E., et al. Clinical outcomes after long-term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet. Med. 2009;11(3):210–219. doi: 10.1097/GIM.0b013e31819d0996. [DOI] [PubMed] [Google Scholar]
- 24.Parini R., De Lorenzo P., Dardis A., et al. Long term clinical history of an Italian cohort of infantile onset Pompe disease treated with enzyme replacement therapy. Orphanet J. Rare Dis. 2018;13(1):32. doi: 10.1186/s13023-018-0771-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Capelle C.I., Poelman E., Frohn-Mulder I.M., et al. Cardiac outcome in classic infantile Pompe disease after 13 years of treatment with recombinant human acid alpha-glucosidase. Int. J. Cardiol. 2018;269:104–110. doi: 10.1016/j.ijcard.2018.07.091. [DOI] [PubMed] [Google Scholar]
- 26.Scheffers L.E., Kok R., van der Berg L.E., et al. Effects of enzyme replacement therapy on cardiac function in classic infantile Pompe disease. Int. J. Cardiol. 2023;380:65–71. doi: 10.1016/j.ijcard.2023.03.010. [DOI] [PubMed] [Google Scholar]
- 27.Hsu Y.K., Chien Y.H., Shinn-Forng Peng S., et al. Evaluating brain white matter hyperintensity, IQ scores, and plasma neurofilament light chain concentration in early-treated patients with infantile-onset Pompe disease. Genet. Med. 2023;25:27–36. doi: 10.1016/j.gim.2022.10.005. [DOI] [PubMed] [Google Scholar]
- 28.Dardis A., Pavan E., Fabris M., et al. Plasma Neurofilament light (NfL) in patients affected by Niemann-pick type C diseases (NPCD) J. Clin. Med. 2021;10(20) doi: 10.3390/jcm10204796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kishnani P.S., Hwu W.-L., Mandel H., et al. A retrospective, multinational, multicenter study on the natural history of infantile-onset Pompe disease. J. Pediatr. 2006;148(5):671–676. doi: 10.1016/j.jpeds.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 30.Hsueh C.Y., Huang C.Y., Yang C.F., et al. Hearing characteristics of infantile-onset Pompe disease after early enzyme-replacement therapy. Orphanet J. Rare Dis. 2021;16:348. doi: 10.1186/s13023-021-01817-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parenti G., Fecarotta S., la Marca G., et al. A chaperon enhances blood a glucosidase activity in Pompe disease patients treated with enzyme replacement therapy. Mol. Ther. 2014;22(11):2004–2012. doi: 10.1038/mt.2014.138. (Epub 2014 Jul 23) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davison J., Brassier A., Broomfield A., et al. Effects of repeat avalglucosidase dosing on ptosis in participants with infantile onset Pompe disease (IOPD) who were previously treated with alglucosidase alfa. Mol. Genet. Metab. 2022;132:S13–S116. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available on request.