

Summary
The N 6‐methyladenosine (m6A) mRNA modification is crucial for plant development and stress responses. In rice, the male sterility resulting from the deficiency of OsFIP37, a core component of m6A methyltransferase complex, emphasizes the significant role of m6A in male fertility. m6A is reversible and can be removed by m6A demethylases. However, whether mRNA m6A demethylase regulates male fertility in rice has remained unknown. Here, we identify the mRNA m6A demethylase OsALKBH9 and demonstrate its involvement in male fertility regulation. Knockout of OsALKBH9 causes male sterility, dependent on its m6A demethylation activity. Cytological analysis reveals defective tapetal programmed cell death (PCD) and excessive accumulation of microspores exine in Osalkbh9‐1. Transcriptome analysis of anthers shows up‐regulation of genes involved in tapetum development, sporopollenin synthesis, and transport pathways in Osalkbh9‐1. Additionally, we demonstrate that OsALKBH9 demethylates the m6A modification in TDR and GAMYB transcripts, which affects the stability of these mRNAs and ultimately leads to excessive accumulation of pollen exine. Our findings highlight the precise control of mRNA m6A modification and reveal the pivotal roles played by OsALKBH9‐mediated m6A demethylation in tapetal PCD and pollen exine accumulation in rice.
Keywords: m6A, demethylase, tapetal PCD, exine, OsALKBH9, rice
Introduction
Rice (Oryza sativa) is a prominent crop globally and serves as a monocot model. The development of pollen in anthers is a highly regulated process, which involves the formation of male meiocytes, gametophytes and gametes (Zhang and Yang, 2014). Tapetum, the innermost layer of the anther wall, supports gametogenesis and undergoes programmed cell death (PCD) after meiosis (Zhang and Yang, 2014). Abnormal development and/or delayed or premature degradation of the tapetum cells results in male sterility. In rice, the development and PCD of tapetum cells are controlled by a highly conserved complex network of transcription factors (TFs): OsUDT1 (UNDEVELOPED TAPETUM 1), OsTDF1 (DEFECTIVE in TAPETAL DEVELOPMENT and FUNCTION1), OsTDR (TAPETUM DEGENERATION RETARDATION), OsMS188 (Male‐sterile 188) and OsPTC1 (PERSISTENT TAPETAL CELL1) (Yao et al., 2022). Any genetic mutations in these five genes can lead to male sterility and abnormalities in tapetal development and/or PCD. Additionally, OsEAT1 (ETERNAL TAPETUM 1), another bHLH TF, plays a crucial role in tapetal PCD by directly regulating the transcription of two aspartic protease encoding genes, OsAP25 and OsAP27 (Niu et al., 2013). Furthermore, OsGAMYB, a MYB TF regulated by gibberellin (GA), also contributes to tapetal development and PCD (Aya et al., 2009).
The pollen wall serves a critical function in safeguarding male gametes from multiple environmental stresses. It is composed of two layers, intine and exine, which consist primarily of polysaccharides and lipidic sporopollenin, respectively (Shi et al., 2015). Sporopollenin is synthesized within the tapetum cells and subsequently transported to the surface of microspores for exine formation (Shi et al., 2015). In Arabidopsis, sporopollenin biosynthesis and exine accumulation are regulated by a series of TFs which form a regulatory cascade (AtDYT1‐AtTDF1‐AtAMS‐AtMS188‐AtMS1) activating transcription of their downstream TFs and target genes (Yao et al., 2022). In rice, cascade (OsUDT1‐OsTDF1‐OsTDR‐OsMS188‐OsPTC1) also regulates exine formation (Yao et al., 2022). OsMS188 and OsGAMYB operate several genes involved in sporopollenin synthesis or transport, enabling them to directly activate gene expression, such as PTC1, CYP703A3, CYP704B2, ABCG15 and PKS1 (Aya et al., 2009; Jin et al., 2022).
N 6‐methyladenosine (m6A) is the most prevalent chemical modification in mRNA, and it regulates gene expression at both transcriptional and post‐transcriptional levels (Tang et al., 2023). m6A is installed, removed and recognized by methyltransferases (writers), demethylases (erasers) and m6A‐binding proteins (readers), respectively (Tang et al., 2023). In rice, male fertility is significantly impacted by mRNA m6A writers (Cheng et al., 2022; Zhang et al., 2019). Knocking out OsFIP37, a core component of m6A methyltransferase complex, leads to abnormal meiosis and early degeneration of microspores during the vacuolated pollen stage (Cheng et al., 2022; Zhang et al., 2019). Moreover, the absence of OsEDM2L (ENHANCED DOWNY MILDEW 2‐LIKE), a protein containing an N 6‐adenine methyltransferase‐like domain vital for proper mRNA m6A modification in anthers, causes defective pollen development and delayed tapetal PCD (Ma et al., 2021). m6A is a reversible mRNA modification that can be demethylated by m6A demethylases. Several m6A demethylases have been identified in Arabidopsis and tomato, including ALKBH10B (Duan et al., 2017), ALKBH9B (Martinez‐Perez et al., 2017) and SlALKBH2 (Zhou et al., 2019). These demethylases are involved in various biological processes such as floral transition (Duan et al., 2017), abiotic stress response (ALKBH10B) (Shoaib et al., 2021; Tang et al., 2021), alfalfa mosaic virus (AMV) infection (Martinez‐Perez et al., 2017) and abscisic acid (ABA) response (ALKBH9B) (Tang et al., 2022), and tomato fruit ripening (SlALKBH2) (Zhou et al., 2019). However, the biological functions of demethylase in rice development remain unknown, despite its crucial role.
Here, we identified OsALKBH9 as a rice mRNA m6A demethylase. Loss of function of OsALKBH9 results in male sterility, delayed tapetal degradation, and excessive accumulation of pollen exine. OsALKBH9 is highly expressed in anthers and regulates the expression of genes involved in tapetal development and pollen exine accumulation. Our findings demonstrate that OsALKBH9 mediates the m6A demethylation of TDR and GAMYB mRNAs, leading to decreased stability and repressing of downstream gene expression, such as OsMS188, PTC1, CYP703A3, CYP704A4, PKS2 and ABCG15. These insights provide valuable information on the vital roles of m6A in controlling rice fertility and offer male sterile materials that can aid in hybrid breeding.
Results
Knock out of OsALKBH9 caused male sterility in rice
To identify mRNA m6A demethylases and explore their functions in rice, we searched for homologues of mammalian m6A demethylase ALKBH5. We designated two proteins predicted as putative m6A demethylases (LOC_Os06g04660 and LOC_Os05g33310) as OsALKBH9 and OsALKBH10, respectively (Figure S1). Using clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR‐associated nuclease 9 (Cas9) mediated gene editing in the Nipponbare cultivar background (Oryza sativa ssp. Japonica), we obtained two mutants of OsALKBH9 with predicted early termination of translation, which were named Osalkbh9‐1 and Osalkbh9‐2 (Figure 1a). Although both mutants exhibited normal vegetative growth, they failed to produce seeds (Figure 1b, c, f). By CRISPR‐Cas9 technology, we also obtained two mutants of OsALKBH10, which caused predicted early termination of translation (Figure S2). However, we did not find obvious development phenotypes in the Osalkbh10 mutants. So, we focused on the research of OsALKBH9. Reciprocal cross‐pollination between Osalkbh9‐1 and the wild‐type (WT) plants showed that all WT ovules were unable to be fertilized by Osalkbh9‐1 pollen grains, whereas the ovules of Osalkbh9‐1 were fertilized by WT pollen grains (Figure 1f), indicating that the lack of seeds in Osalkbh9 mutant is due to the defective pollen grains. Compared to the WT, the anthers of both mutants did not fill up and became pale (Figure 1d), and the pollen grains were shrunken and lacked starch inclusions (Figure 1e), indicating male sterile of Osalkbh9 mutants. To test the genetic action of the Osalkbh9 mutation, we calculated the ratio of sterile plants to fertile plants and determined the genotype of the inbred offspring of heterozygous mutants (CRISPR‐Cas9 T‐DNA free). The three genotypes, OsALKBH9/OsALKBH9 (WT): OsALKBH9/Osalkbh9‐1 (heterozygote) and Osalkbh9‐1/Osalkbh9‐1 (homozygote), segregated at a ratio of 1:2:1, and the phenotypic ratio of male fertility to male sterility fit a 3:1 ratio, with co‐segregation between the genotypes and phenotypes (Table S1). These results indicate that the Osalkbh9 mutation acted in a recessive sporophytic manner.
Figure 1.
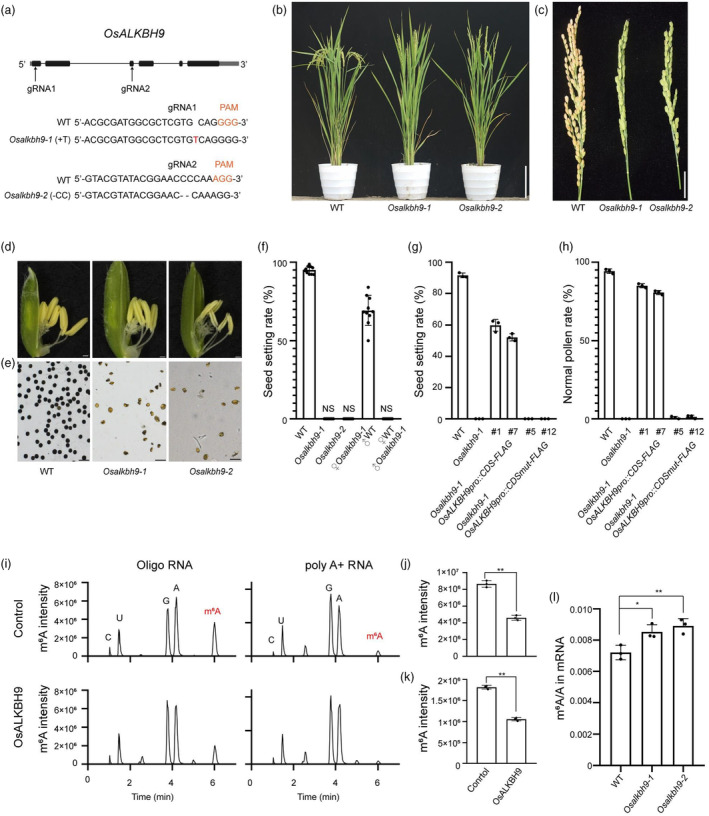
The regulation of OsALKBH9 in male fertility depends on its m6A demethylase activity. (a) CRISPR/Cas9‐mediated target mutagenesis of OsALKBH9. The upper panel shows the OsALKBH9 genomic region and the two CRISPR/Cas9 target sites indicated by arrows. Exons and other sequences are indicated by black boxes and lines, respectively. The lower panel shows alignment of wild‐type (WT), Osalkbh9‐1 and Osalkbh9‐2 sequences containing the CRISPR/Cas9 target sites. Osalkbh9‐1 and Osalkbh9‐2 contain a 1‐bp insertion of T (red) and a 2‐bp deletion of C (dash), respectively. (b) The phenotype of WT, Osalkbh9‐1 and Osalkbh9‐2 mutant plants after heading. Scale bar, 20 cm. (c) Comparison of WT, Osalkbh9‐1 and Osalkbh9‐2 panicles at the mature stage. Scale bar, 2 cm. (d) Spikelets of WT, Osalkbh9‐1 and Osalkbh9‐2 plants. Scale bars, 0.5 mm. (e) Pollen grains of WT, Osalkbh9‐1 and Osalkbh9‐2 stained by I2/KI solution. Scale bars, 50 μm. (f) The seed setting rates of the mutants crossed with WT plants (n = 10 plants). (g‐h) The seed setting rates (g) and pollen grains I2/KI solution (h) of genetically complementary plants and demethylase inactive complementary plants (n = 3 plants). The results of two representative lines of genetically complementary plants and demethylase inactive complementary plants were present, individually. (i–k) Recombinant OsALKBH9 protein demethylates the m6A modification in m6A‐containing ssRNA and poly A+ RNA extracted from rice in vitro. n = 3 biological replicates. (i) LC–MS/MS chromatograms of digested substrates of in vitro demethylase activity reaction. (j, k) m6A changes using ssRNA (j) and poly A+ RNA (k) as substrates after in vitro demethylase activity reaction. (l) m6A percentage relative to adenosine (m6A/A ratio) determined by LC–MS/MS in mRNA purified from 14‐day‐old shoots of WT, Osalkbh9‐1 and Osalkbh9‐2. n = 3 biological replicates. Data are means ± SD for three biological replicates. Student's t test: *(P < 0.05), **(P < 0.01).
We conducted genetic complementation experiments to further confirm the function of OsALKBH9 in pollen development. We transformed Osalkbh9‐1/Osalkbh9‐1 calli with the construct of OsALKBH9pro::OsALKBH9CDS‐FLAG, which contains a 2.1 kb native promoter and a 1.9 kb coding sequence fused in‐frame with a 3 × FLAG tag. In the T0 generation of complementation lines, the seed setting rate and pollen activity were largely restored (Figure 1g, h; Figure S3), confirming the male‐sterile phenotype of Osalkbh9 mutants is caused by the loss of function of OsALKBH9.
The regulation of OsALKBH9 in male fertility depends on its m6A demethylase activity
To verify if OsALKBH9 functions as an mRNA m6A demethylase, we expressed full‐length OsALKBH9 with a His‐tag in Escherichia coli as fusion proteins. Demethylation assays were performed with the purified proteins by incubating overnight with synthetic 14‐mer m6A‐modified RNA or the full‐length mRNA isolated from rice seedlings. The nucleosides that were digested from the reaction products were analysed by Liquid chromatography–tandem mass spectrometry (LC–MS/MS). The results indicated that approximately 50% of the m6A present in either synthetic ssRNA or rice mRNA was removed by OsALKBH9 in vitro (Figure 1i–k), clearly demonstrating its RNA m6A demethylation activity in vitro. Furthermore, we observed an increase in the level of mRNA m6A modification in 14‐day‐old shoots of both Osalkbh9‐1 and Osalkbh9‐2 compared to WT (Figure 1l), verifying the in vivo m6A demethylation activity of OsALKBH9 in rice.
To determine whether the regulation of male fertility by OsALKBH9 depends on its putative RNA demethylase activity, we conducted complementation experiments using the OsALKBH9pro::OsALKBH9CDSmut‐FLAG (H324A/D326A) construct which consists of the catalytically inactive OsALKBH9 coding sequence (Duan et al., 2017). We obtained 20 independent transgenic lines, but none of them showed any restoration of the seed‐setting rate or pollen fertility (Figure 1g, h). These results suggest that the male sterile phenotype of Osalkbh9 mutant is directly linked to the RNA demethylase activity of OsALKBH9 being compromised.
OsALKBH9 is required for the tapetal PCD and pollen exine accumulation
To investigate the cellular abnormality of Osalkbh9‐1 during male reproductive development, anther transverse sections were examined at different developmental stages. From stage 7 to stage 8 (as labelled by (Zhang et al., 2011)), no obvious differences were observed between anthers from WT and Osalkbh9‐1 (Figure 2a). At stage 9, the tapetum of WT was condensed, forming a thin cell layer, while that of Osalkbh9‐1 remained large (Figure 2a). At stage 10, the tapetum in WT became more concentrated, degraded and thinned, and the microspores underwent vacuolation (Figure 2a). Conversely, in Osalkbh9‐1, the degradation of tapetal cells was abnormal, with some cells remaining undegraded, and the microspores appeared irregularly shaped (Figure 2a). At stage 11, only a small amount of degradation residue was left in the tapetum of WT anthers, and the microspore vacuoles enlarged (Figure 2a). However, there were still undegraded tapetal cells, and all the microspores were aborted in Osalkbh9‐1 (Figure 2a).
Figure 2.
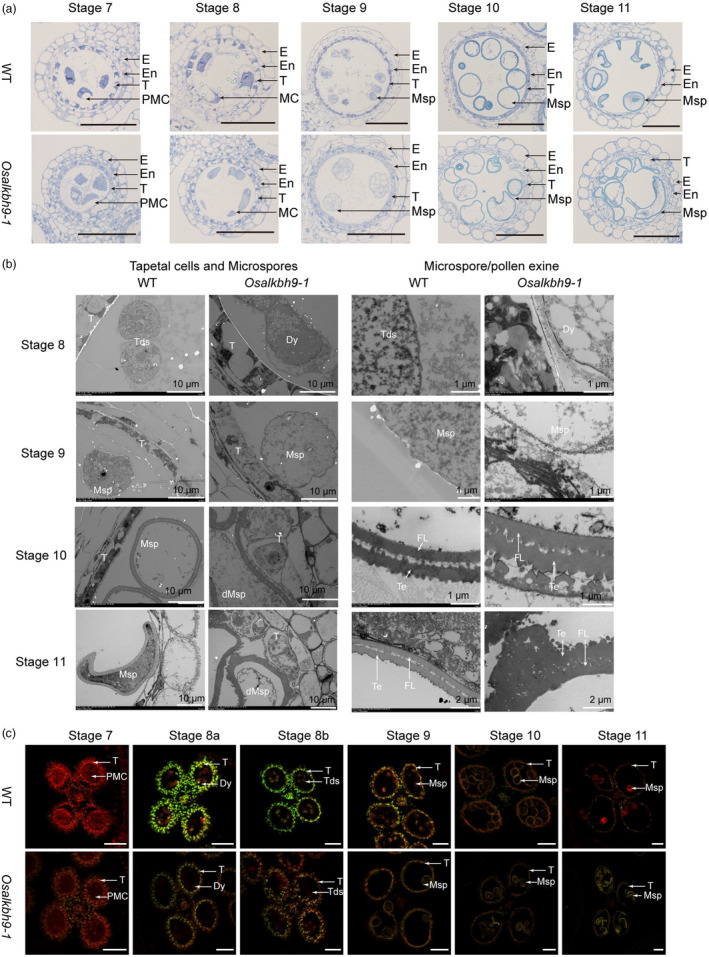
Knockout of OsALKBH9 caused delayed degradation of tapetal cells and abnormal pollen wall patterning. (a) Semi‐thin sections of anthers at stage 7 to stage 11 in the wild type and Osalkbh9‐1 mutant. The images show a single locule of an anther. Scale bars, 50 μm. (b) Transmission electron micrographs of the wild‐type and Osalkbh9‐1 anthers from stage 8 to stage 11. (c) TUNEL analyses of WT and Osalkbh9‐1 anthers at stage 7 to stage 11. Propidium iodide staining is indicated by red fluorescence and TUNEL‐positive staining is indicated by yellow to green fluorescence. Scale bars, 50 μm. E, epidermis; En, endothecium; T, tapetum; PMC, pollen mother cell; MC, meiotic cell; Msp, microspore; dMsp, aborted microspore; Tds, tetrads; Dy, dyad cell; FL, foot layer; Te, tectum.
Transmission electron microscopy (TEM) was utilized to investigate the developmental defects in the tapetum and microspores of Osalkbh9‐1. While the tapetum of WT continued to degrade until complete degradation from stage 9 to stage 11, intact tapetal cells were still evident in Osalkbh9‐1 at stage 11, which corroborates with the transverse sections (Figure 2b). From stage 10 to stage 11, both Osalkbh9‐1 and WT microspores were enclosed by a double‐layered exine consisting of tectum and nexine (Figure 2b); however, the exine of Osalkbh9‐1 was thicker than that of WT (Figure 2b). At stage 10, the exine structure of Osalkbh9‐1 resembled that of WT (Figure 2b); but the exine pattern of Osalkbh9‐1 at stage 11 became irregular due to abnormal tectum accumulation (Figure 2b). These results indicate that OsALKBH9 is necessary for both pollen exine accumulation and appropriate patterning.
To investigate the disturbed PCD in Osalkbh9‐1 anthers, the terminal deoxynucleotidyl transferase‐mediated dUTP‐biotin nick end labelling (TUNEL) assay was performed due to the delayed tapetum degradation in Osalkbh9‐1. At stage 7, no TUNEL signals were observed in the anthers of either WT or Osalkbh9‐1 mutant (Figure 2c). However, strong TUNEL signals were observed in the tapetum of the WT at stage 8a and continued until stage 9, while TUNEL signals in Osalkbh9‐1 were detected at stage 8a and stage 8b, but were weaker than those of the WT (Figure 2c), indicating inadequate degradation of tapetum cells in Osalkbh9‐1 (Figure 2c). Additionally, the microspores from stage 9 to stage 11 in Osalkbh9‐1 mutant but not in WT displayed TUNEL signals (Figure 2c). These results suggest abnormal tapetal PCD and microspores development in Osalkbh9‐1, further confirming the critical roles of OsALKBH9 in tapetal PCD, microspores development and microspores/pollen exine accumulation and patterning.
OsALKBH9 is highly expressed in anthers and mainly localized in cytoplasm
To investigate the expression pattern of OsALKBH9, we measured its transcript levels in different tissues by qRT‐PCR. The results showed that OsALKBH9 was highly expressed in anthers at stage 9 to stage 10 and stage 11 to stage 12 (Figure 3a). We further investigated the expression pattern of OsALKBH9 during anthers development using OsALKBH9pro::GUS (β‐glucuronidase) transgenic plants. GUS staining revealed that OsALKBH9 was highly expressed in anthers from stage 8 to stage 10 (Figure 3b). We also examined the OsALKBH9 expression pattern by RNA in situ hybridization. The results showed that OsALKBH9 transcripts are highly expressed in tapetal cells (S8a, S8b and S9), meiotic cells (S8a and S8b) and microspore (S9) (Figure 3d).
Figure 3.
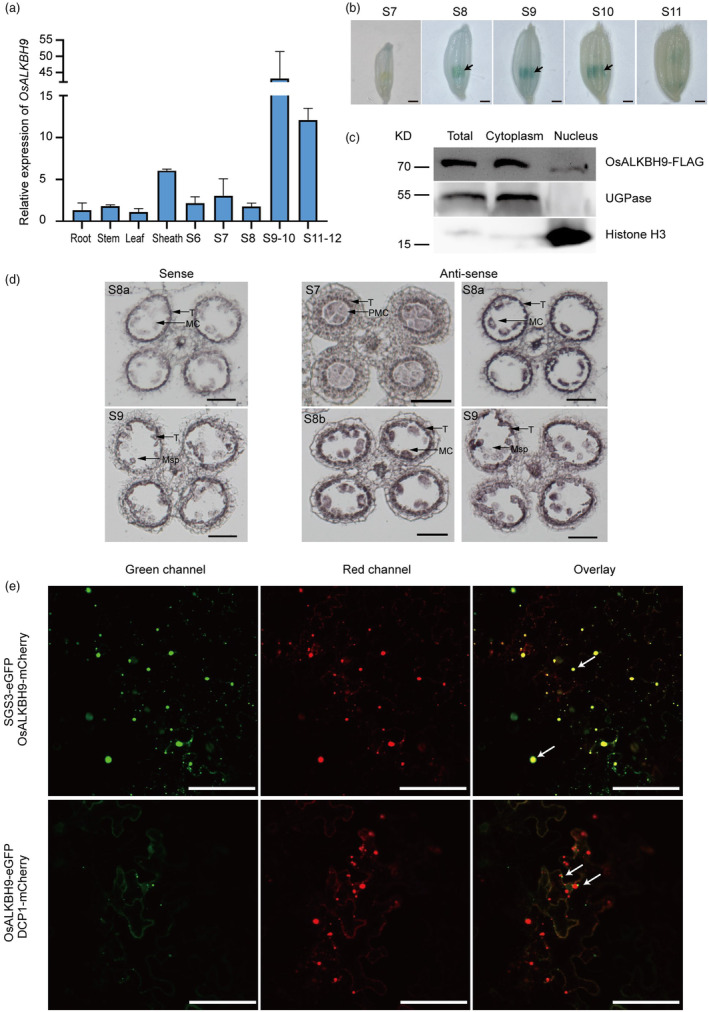
OsALKBH9 is highly expressed in anthers and OsALKBH9 is mainly localized in cytoplasm. (a) Expression analysis of OsALKBH9 in different tissues by qRT‐PCR. Anthers were collected at developmental stage 6 to stage 12. Other tissues were harvested from plants at the flowering stage. S6, stage 6; S7, stage 7; S8, stage 8; S9–10, stage 9 to stage 10; S11–12, stage 11 to stage 12. (b) GUS staining of transgenic anthers containing OsALKBH9pro::GUS. Scale bars, 1 mm. Arrows pointing anthers with strong GUS signals. S7, stage 7; S8, stage 8; S9, stage 9; S10, stage 10; S11, stage 11. (c) Subcellular fraction and immunoblot assay. Total protein, cytoplasm, and nuclei‐enriched fractions from Osalkbh9‐1 OsALKBH9pro::OsALKBH9CDS‐FLAG transformants are subject to SDS‐PAGE. UGPase and Histone H3 are used as cytoplasmic and nuclear markers, respectively. (d) In situ hybridization of OsALKBH9 transcripts in WT anthers. S7, stage 7; S8a, stage 8a; S8b, stage 8b; S9, stage 9. T, tapetum; PMC, pollen mother cell; MC, meiotic cell; Msp, microspore. Scale bars, 50 μm. (e) Subcellular localization of OsALKBH9 in N. benthamiana leaves epidermal cells. Arrows pointing co‐localized foci of OsALKBH9‐mCherry and SGS3‐eGFP (the upper panel), and non‐co‐localized foci of OsALKBH9‐eGFP and DCP1‐mCherry (the lower panel). Scale bars, 100 μm.
To determine the subcellular localization of OsALKBH9, we transiently expressed OsALKBH9‐GFP via agroinfiltration in N. benthamiana leaves. We found that OsALKBH9‐GFP exhibited a dotted cytoplasm distribution pattern (Figure S4), similar to the localization pattern of its homologues (AtALKBH9B) in Arabidopsis thaliana (Martinez‐Perez et al., 2017). We further confirmed the cytoplasmic localization of OsALKBH9 by isolating the cytoplasmic and nuclear components of Osalkbh9‐1 OsALKBH9pro::OsALKBH9CDS‐FLAG and determining its location through western blotting. The outcome demonstrated that almost all OsALKBH9 proteins are localized in the cytoplasm (Figure 3c). Because AtALKBH9B was founded co‐localized with DCP1 and SGS3 (Martinez‐Perez et al., 2017), which are marker proteins of P‐bodies and siRNA‐bodies. So, we checked whether OsALKBH9 co‐localized with DCP1 and SGS3, and the results showed that OsALKBH9 co‐localized with SGS3 but not with DCP1 (Figure 3e). Since the siRNA bodies commonly co‐localized with stress granules (SGs) (Jouannet et al., 2012; Kakutani et al., 2012), it suggests that the function of OsALKBH9 might involve in siRNA‐bodies and/or stress granules.
OsALKBH9 deficiency affects the expression of genes related to male reproductive development and exine accumulation
As OsALKBH9 is highly expressed in anthers during stage 8 to stage 10, we conducted RNA‐seq analysis to evaluate its effect on male reproductive development. We compared gene expression profiles of stage 7 to stage 8 (Table S3) and stage 9 to stage 10 (Table S4) anthers between Osalkbh9‐1 and the WT. Clustering analysis confirmed the robustness of our RNA‐seq data (Figures S5 and S6). Our analysis revealed differential expression of 2606 genes in stage 7 to stage 8 anthers (1195 upregulated, 1411 downregulated) and 6316 genes in stage 9 to stage 10 anthers (3860 upregulated, 2456 downregulated) (with the threshold criteria | Log2[fold‐change] | ≥0.585 and FDR <0.05) (Figure S7a,c). We performed Gene Ontology (GO) analysis and found that the upregulated genes in stage 7 to stage 8 anthers were primarily associated with lipid metabolism, fatty acid biosynthesis, pollen and sporopollenin development, and photosynthesis (Figure S7b), while those in stage 9 to stage 10 anthers were linked to photosynthesis, flower development, sporopollenin biosynthesis and pollen exine formation (Figure S7d). Downregulated genes were mainly involved in ribosome, translation, symporter activity and auxin response in stage 7 to stage 8 anthers (Figure S7b), whereas DNA replication and cell cycle were affected in stage 9 to stage 10 anthers (Figure S7d). We compared co‐upregulated and co‐downregulated genes in stages 7 to stage 8 and stage 9 to stage 10, revealing 985 co‐differentially expressed genes (655 upregulated and 330 downregulated) (Figure 4a). GO analysis showed that the upregulated genes were primarily associated with sporopollenin biosynthesis, pollen development and pollen exine formation, while the downregulated genes were linked to carbohydrate metabolic and auxin response (Figure 4b). We further utilized qRT‐PCR to detect gene expression related to tapetum development and sporopollenin synthesis/transport pathways (Figure 4c; Table S2). The results demonstrated upregulation of TFs TDF1, TDR, EAT1, MS188 and PTC1 at both stage 7 to stage 8 and stage 9 to stage 10 and GAMYB at stage 9 to stage 10 in Osalkbh9‐1 (Figure 4c). Furthermore, the sporopollenin synthesis and transport genes CYP703A3, CYP704A4, PKS2 and ABCG15 were also upregulated at both stages (Figure 4c). These results suggest that the increased expression of genes involved in microspores sporopollenin accumulation in Osalkbh9‐1 could account for the excessive accumulation of pollen exine in the mutant.
Figure 4.
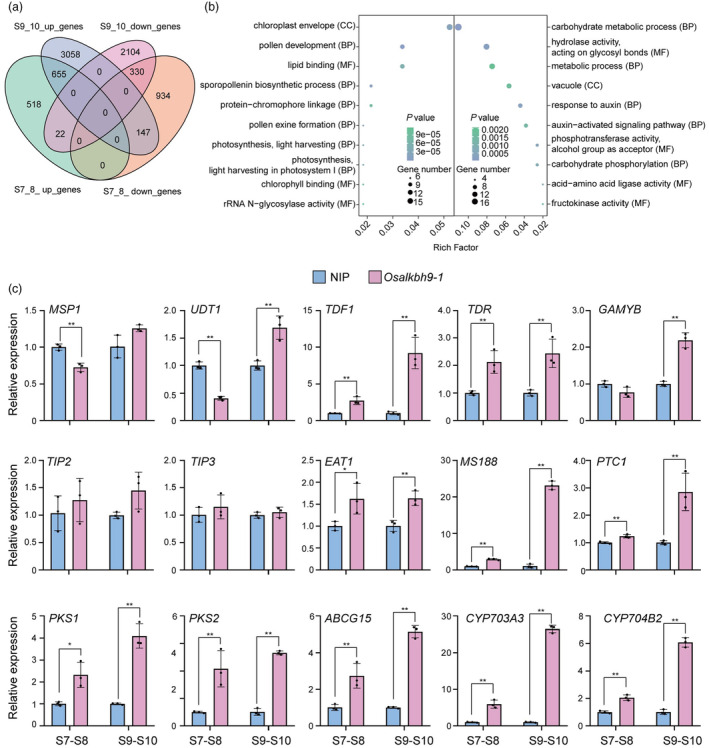
OsALKBH9 affects the expression of genes related to male reproductive development and exine accumulation. (a) Venn diagram depicting the differentially expressed genes (DEGs, Osalkbh9‐1/WT) at stage 7 to stage 8 and stage 9 to stage 10. (b) Gene ontology analysis of the co‐upregulated and co‐downregulated genes at stage 7 to stage 8 and stage 9 to stage 10. Left part indicates the co‐upregulated genes, while right part indicates the co‐downregulated genes. CC, cell component; BP, biological process; MF, molecular function. (c) qRT‐PCR detection of genes required for tapetum development and pollen wall accumulation. UBQ was used as an internal control. Data are means ± SD for three biological replicates. Student's t test: *(P < 0.05), **(P < 0.01).
Disruption of OsALKBH9 leads to transcriptome‐wide m6A hypermethylation
We conducted m6A‐seq analysis on anthers at stage 9 to stage 10 from both Osalkbh9‐1 and WT to investigate changes in m6A methylation patterns (Table S5). Our analysis revealed 6551 hyper‐methylated m6A peaks corresponding to 5968 genes and 563 hypo‐methylated peaks corresponding to 548 genes in the Osalkbh9‐1 mutant (Figure 5a). Comparing m6A enrichment between WT and Osalkbh9‐1, we found significantly higher m6A enrichment in Osalkbh9‐1 than in WT (Figure 5b), suggesting that disruption of OsALKBH9 leads to hyper‐methylation of m6A. Analysis of the metagene profiles revealed highly enriched hyper‐methylated m6A peaks in the 3′ untranslated region (3′ UTR) of transcripts (Figure 5c). Further analysis of hyper‐methylated m6A peaks within five non‐overlapping transcript regions showed that they were primarily enriched in the 3′ UTR (>75%) (Figure 5c). The enriched m6A motifs (UGHAC and GWAACU) observed in hyper‐methylated m6A peaks were consistent with the previously reported RRACH motif (Figure 5f).
Figure 5.
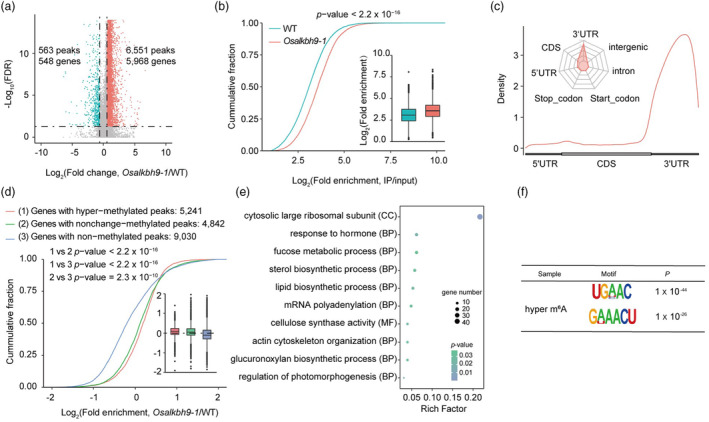
Loss of function of OsALKBH9 caused transcriptome‐wide m6A hypermethylation. (a) The result of the volcano plot shows the changes in m6A modification between Osalkbh9‐1 and WT. (b) The results of cumulative fraction and boxplot show the log2(fold enrichment) of m6A peaks in Osalkbh9‐1 and WT. The Kolmogorov–Smirnov test revealed a P‐value of less than 2.2 × 10−16. (c) The results of metaplot and radar plot represent the distribution of identified m6A hyper‐methylated peaks in Osalkbh9‐1 across the indicated mRNA segments. (d) The cumulative fraction and boxplots illustrate the gene expression patterns of hyper‐methylated, non‐change methylated and hypo‐methylated peaks in Osalkbh9‐1/WT The Kolmogorov–Smirnov test revealed a P‐value of less than 2.2 × 10−16. (e) Gene ontology analysis was performed on the genes with hyper‐methylated peaks. (f) OsALKBH9‐dependent motifs were identified by using HOMER based on the top 1000 peaks.
We next investigated whether altered m6A levels resulting from OsALKBH9 deficiency affect gene expression. Analysis of transcript accumulation in genes with non‐methylated, unchanged‐methylated and hyper‐methylated peaks indicated that genes with hyper‐methylated peaks were more likely to be upregulated than those without (Figure 5d). GO analysis revealed that hyper‐methylated genes were mainly associated with hormone response, regulation of photomorphogenesis, lipid biosynthetic and mRNA polyadenylation (Figure 5e). Joint analysis of m6A‐seq and RNA‐seq data identified 865 upregulated and 499 downregulated genes containing hyper‐methylated peaks, as well as 35 upregulated and 131 downregulated genes with hypo‐m6A methylation (Figure S8a). GO analysis demonstrated that hyper‐methylated and up‐regulated genes were enriched in pathways such as positive regulation of transcription, signal transduction and transcription coactivator activity, while hyper‐methylated and down‐regulated genes were enriched in carbohydrate metabolic processes and carbohydrate phosphorylation (Figure S8b). Overall, our study demonstrates that disruption of OsALKBH9 results in transcriptome‐wide m6A hypermethylation.
Loss of function of OsALKBH9 caused m6A hypermethylation in TDR and GAMYB transcripts
We observed higher m6A peaks in the exon of TDR and 3′ UTR of GAMYB in Osalkbh9‐1, both of which are involved in tapetum and pollen exine development (Figure 6a). We also performed m6A‐immunoprecipitation followed by quantitative PCR (m6A‐IP‐qPCR) using anthers at stage 9 to stage 10 to confirm that TDR and GAMYB mRNA are indeed m6A‐hypermethylated in Osalkbh9‐1 compared to WT (Figure 6b). To validate TDR and GAMYB mRNA as direct targets of OsALKBH9, we performed RNA immunoprecipitation followed by RT‐qPCR (RIP‐RT‐qPCR) with an anti‐FLAG antibody in the anthers from Osalkbh9‐1 OsALKBH9pro::OsALKBH9‐FLAG. The results confirmed direct binding of OsALKBH9 to TDR and GAMYB mRNAs (Figure 6c). Note that we have found the higher expressed TDR and GAMYB mRNAs in Osalkbh9‐1 (Figure 4c). As the mRNA m6A modification has been shown to promote mRNA stabilization in Arabidopsis and human, we assessed the mRNA stability of the TDR and GAMYB transcripts using actinomycin D in anthers. Our results showed that TDR and GAMYB transcripts are more stable in Osalkbh9‐1 than in WT (Figure 6d), confirming that m6A modification mediates mRNA stabilization in rice. Collectively, our results demonstrate that disruption of OsALKBH9 increases m6A levels in TDR and GAMYB transcripts, which promotes mRNA stabilization and activates downstream gene expression of tapetum development, sporopollenin synthesis and transport, thereby leading to excessive accumulation of pollen exine (Figure 6e).
Figure 6.
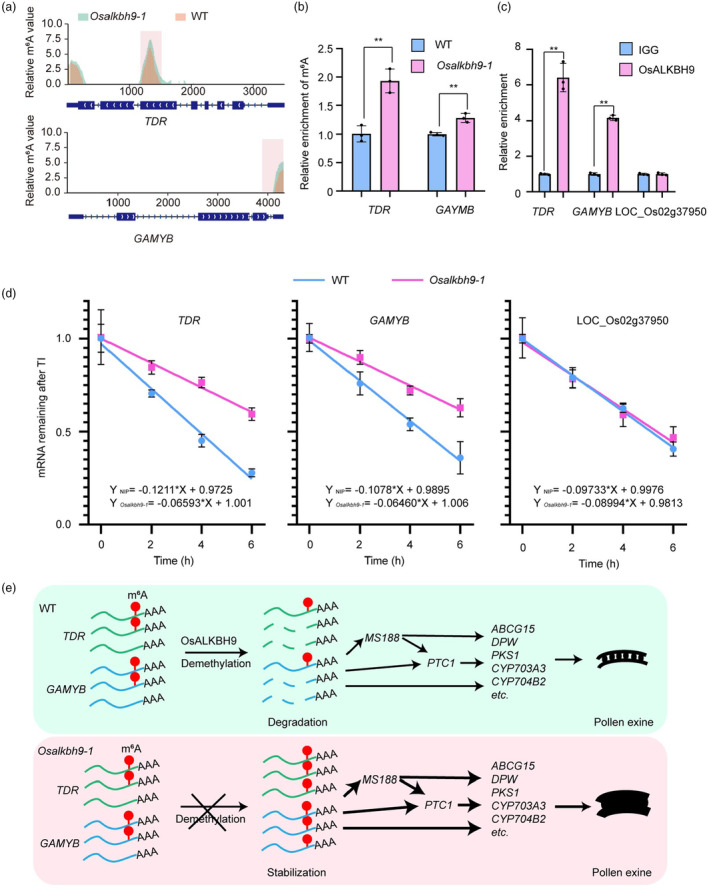
OsALKBH9‐dependent m6A demethylation regulates pollen exine accumulation. (a) The relative m6A value of TDR and GAMYB. The relative m6A value of TDR and GAMYB is determined using log2 (Osalkbh9‐1/NIP, reads count (IP/input)). (b) m6A‐IP‐qPCR results showing the relative m6A levels in stage 9 to stage 10 anthers of WT and Osalkbh9‐1. (c) RIP‐qPCR assays in Osalkbh9‐1 OsALKBH9pro::OsALKBH9CDS‐FLAG spikelets (anthers at stage 7 to stage 12) showing that OsALKBH9 directly binds to the TDR and GAMYB transcripts. (d) mRNA lifetimes of TDR and GAMYB in WT and Osalkbh9‐1 spikelets (anthers at stage 7 to stage 12). LOC_Os02g37950 was used as the negative control. 18S RNA was used as a reference. TI, transcription inhibition. Data are means ± SD for three biological replicates. Student's t test: *(P < 0.05), **(P < 0.01). (e) A proposed working model for how OsALKBH9 modulates pollen exine accumulation. In wild type, OsALKBH9 removes m6A on TDR and GAMYB transcripts, reducing their mRNA stability and ensuring appropriate accumulation of pollen exine. In Osalkbh9‐1, TDR and GAMYB transcripts are m6A hypermethylated, promoting the stability of their mRNA, activating the expression of genes involved in pollen exine accumulation, leading to excessive accumulation of pollen exine and abnormal exine patterning.
Discussion
As the most abundant internal chemical modification found in eukaryotic mRNA, m6A modification plays vital roles in plants, including development, biotic and abiotic stress responses, as well as crop trait improvement (Tang et al., 2023). This reversible RNA modification can be removed by m6A demethylase. In mammals, two identified mRNA m6A demethylases are fat mass and obesity‐associated protein (FTO) (Jia et al., 2011) and alkylated DNA repair protein AlkB homologue 5 (ALKBH5) (Zheng et al., 2013). Arabidopsis has five homologues (ALKBH9A, ALKBH9B, ALKBH9C, ALKBH10A and ALKBH10B) of mammalian ALKBH5, but rice only has two homologues (OsALKBH9 and OsALKBH10) (Figure S1), suggesting low redundancy of m6A demethylases. To deeply explore and understand the roles of m6A demethylases in plants, it is necessary to study the functions of demethylases in rice, which serves as a model for monocotyledonous plants.
In this work, we identified OsALKBH9 as an mRNA m6A demethylase that plays a vital role in regulating male fertility in rice. Knockout of OsALKBH9 resulted in delayed tapetal cell degradation and abnormal accumulation of pollen exine, ultimately leading to pollen abortion. The tapetum, which is the innermost cell layer of the anther, is essential for the development and maturation of microspores. During pollen development, the tapetum undergoes PCD. Early or delayed PCD of the tapetum causes abnormal microspore development. In the Osalkbh9‐1 mutant, tapetum formation was normal, but its degradation was inhibited. The initiation time of tapetal PCD in the Osalkbh9‐1 mutant was similar to that of the WT, starting from stage 8a. However, as measured by the TUNEL assays (Figure 2c), the tapetum degradation of Osalkbh9‐1 was weaker than that of the WT, causing delayed tapetum degradation. In rice, tapetal PCD delayed mutants can be classified into two groups: tapetum expanded mutants, including udt1 (Jung et al., 2005), tdr (Li et al., 2006), gamyb (Aya et al., 2009) and tip2 (Fu et al., 2014), which exhibit resistance to tapetum degradation and little or no exine accumulation; and tapetum persistent mutants, including ptc1 (Li et al., 2011), eat1 (Niu et al., 2013), ptc2 (Uzair et al., 2020) and post (Che et al., 2021), which have normal tapetum cell size and metabolic activity and these genes are highly expressed at post‐meiotic stages. While both ptc1 and ptc2 mutants accumulate less exine, eat1 exhibited much thicker exine than that of WT. The Osalkbh9‐1 male sterile mutant delayed degradation of tapetal cells and persistence at late stages, indicating that it is a tapetum‐persistent mutant. Interestingly, unlike most tapetal PCD delayed mutants, Osalkbh9‐1 accumulated more exine on the pollen surface than WT (Figure 2b), suggesting excessive activation of pollen exine synthesis and/or transport pathways in Osalkbh9‐1. Indeed, we found OsALKBH9 can directly remove m6A in TDR and GAMYB mRNA, which reduces their stability and promotes accurate expression of downstream genes. In the mutant, m6A levels in TDR and GAMYB transcripts increased, leading to mRNA stabilization and activation of OsMS188, PTC1, ABCG15, DPW, PKS1, CYP703A3 and CYP704B2 expression, ultimately causing excessive accumulation of pollen exine. A recent study in Maize is similar to our results. In Msms1 mutant, the tapetal PCD is delayed and the exine is aberrantly thickened (Hou et al., 2023). By map‐based clone, they identified a male‐sterility mutant gene, ZmMS1, encoding a tapetum‐specific lateral organ boundaries domain transcription factor ZmLBD30. Further study found that several tapetal development and pollen exine formation genes are upregulated in the mutant, causing an aberrantly thickened exine. Furthermore, ZmMS1/LBD30 serves as a repressor to shut down ZmbHLH51‐ZmMYB84‐ZmMS7 cascade to ensure timely tapetal degeneration and the proper level of exine (Hou et al., 2023).
In mammalians, both ALKBH5 and FTO are nuclear localization protein (Jia et al., 2011; Zheng et al., 2013), however, several mRNA m6A demethylase in plants, such as AtALKBH9B (Martinez‐Perez et al., 2017), SlALKBH2 (Zhou et al., 2019) and OsALKBH9, are localized in the cytoplasm, suggesting different functions of demethylases may exist in plants. AtALKBH9B was found to be localized with SGS3, a marker of stress granule (SG) and cytoplasmic siRNA body, implying a functional association with siRNA‐mediated RNA decay, RNA storage or mRNA translation. A recent work found that AtALKBH9B could demethylase the m6A of a heat‐activated long terminal repeat retrotransposon RNA (Onsen) in SG, releasing Onsen RNA out of SG (Fan et al., 2023). In this work, we also found OsALKBH9 co‐localized with SG, suggesting OsALKBH9 may involve SG related functions.
The precise regulation of mRNA m6A modification is crucial for rice fertility. In addition to OsALKBH9, other studies have also highlighted the significant roles of mRNA m6A modifications in rice male fertility. Knocking out of OsFIP37, a core component of m6A methyltransferase, leads to abnormal meiosis and early degeneration of microspores during the vacuolated pollen stage (Cheng et al., 2022; Zhang et al., 2019). Further studies demonstrated that OsFIP37 is recruited by OsFAP1 to mediate m6A modification on an auxin biosynthesis gene OsYUCCA3 during microsporogenesis (Cheng et al., 2022). Deficiency of ENHANCED DOWNY MILDEW 2‐LIKE (OsEDM2L), an N 6‐adenine methyltransferase‐like domain‐containing protein essential for proper mRNA m6A modification in anthers, results in delayed tapetal PCD and defective pollen development in rice (Ma et al., 2021).
Methods
Plant materials and growth conditions
The rice mutants Osalkbh9‐1 and Osalkbh9‐2 were created by a clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 genome editing method in the background of Japonica rice ‘Nipponbare’. Osalkbh9‐1 was obtained from Baige Gene Technology (Jiangsu, China) by CRISPR/Cas9 technology with target ‘ACGCGATGGCGCTCGTGCAGGGG’. Osalkbh9‐2 was obtained from BIORUN (Wuhan, China) by CRISPR/Cas9 technology with target ‘GTACGTATACGGAACCCCAAAGG’. The Osalkbh10 mutants were obtained from Baige Gene Technology (Jiangsu, China) by CRISPR/Cas9 technology with target ‘CCCTATCTGCTCTGATCACGAGG’ and ‘CACAACTTCAGAGCTCATGAAGG’. The CRISPR‐Cas9 T‐DNA free Osalkbh9‐1 plants were obtained by screening plants without hygromycin bands through PCR, the primers are listed in Table S6. All plant materials were grown in the fields located in Beijing, China, and maintained with routine management practices.
Phenotypic characterization and cytological observation
Pollen grains were stained by 1% (w/v) I2‐KI and observed by a lab microscope. The anther semi‐section, TEM analysis, TUNEL assay and the RNA in situ hybridization were performed as described previously (Bai et al., 2019). GUS activity was measured by staining spikelets at different stages of T1 transgenic lines as described previously (Bai et al., 2019).
Plasmid construction and plant transformation
To generate OsALKBH9pro::OsALKBH9CDS‐FLAG construct. Firstly, we cloned the coding sequence (CDS) of OsALKBH9 into vector pCAMBIA1300‐35S‐3 × Flag by KpnI and SalI, and we obtained pCAMBIA1300‐35S‐OsALKBH9‐3 × Flag construct. Then the 35S promotor of pCAMBIA1300‐35S‐OsALKBH9‐3 × Flag was replaced by a 2068‐bp fragment upstream ATG start codon of OsALKBH9 by HindIII and KpnI. We created the catalytically inactive mutant, OsALKBH9pro::OsALKBH9CDSmut‐FLAG, by introducing the OsALKBH9 H324A/D326A mutation using the Mut Express II Fast Mutagenesis Kit V2 (Vazyme). Both constructs were transformed into Osalkbh9‐1/Osalkbh9‐1 calli, respectively. To construct OsALKBH9pro::GUS, we cloned the promoter fragments into pCAMBIA1300‐GUS‐3U (Tang et al., 2020) by XbaI and BamHI, and transformed them into Nipponbare. All primers used for vector construction were listed in Table S6.
LC‐MS/MS for m6A quantification
The m6A quantification by LC–MS/MS was performed as described previously (Wang et al., 2022). Poly A+ RNA (200 ng) was digested with 1 U Nuclease P1 in 40 μL of buffer containing 10% 0.1 M ammonium acetate NH4AC (pH 5.3) at 42 °C for 4 h, followed by the addition of 2 U Shrimp Alkaline Phosphatase (NEB, USA) and 10× CutSmart buffer (NEB). The mixture was incubated at 37 °C for 3 h, and the resulting aqueous phase was injected into an LC–MS/MS system. Nucleosides were separated using a UPLC pump (Shimadzu, Japan) with a ZORBAX SB‐Aq column (Agilent, USA) and analysed by MS/MS using a Triple QuadTM 5500 (AB SCIEX, USA) mass spectrometer running in positive ion mode and the multiple reaction‐monitoring (MRM) feature.
Protein purification and in vitro demethylation assays
The coding sequence of OsALKBH9 was amplified and cloned into pET28a+. The resulting construct, Pet28a+‐OsALKBH9 was transformed into E. coli strain BL‐21 Gold competent cells, and induced by 0.5 mM IPTG at 18 °C overnight. The protein purification process followed the procedures outlined in the Ni‐NTA Spin Kit handbook (QIAGEN).
Demethylation activity assays were performed with limited modifications as previously reported (Zheng et al., 2013). Briefly, reaction mixtures contained the following components: 0.5 μL oligo RNA with m6A (AUUGUCA(m6A) CAGCAGC) or 500 ng full‐length rice mRNA, OsALKBH9, KCl (100 μM), MgCl2 (2 mM), RNasin (0.2 U μL −1, Thermofisher), l‐ascorbic acid (200 μM), a‐ketoglutarate (300 μM), (NH4)2Fe (SO4)2 (150 μM) and 50 mM of HEPES buffer (PH 7.0). The reaction was incubated at room temperature overnight, the quenched by adding 5 mM EDTA followed by heating at 95 °C for 10 min, and analysed by LC–MS/MS.
RNA extraction and qRT‐PCR
Total RNA was extracted from anthers at stage 7 to stage 8 and stage 9 to stage 10 using Quick‐RNA™ Plant Miniprep kit (Zymo research). One microgram of total RNA was used for reverse transcription by PrimeScript RT reagent kit with gDNA Eraser (Takara). qRT‐PCR was performed using SYBR Green Master Mix (YEASEN) on a ViiA 7 Dx (Applied Biosystems). The 2−ΔΔCT method was used to calculate the gene expression levels. All primers were designed by multiPrime at http://www.multiprime.cn (Xia et al), and listed in (Table S6).
RNA seq and data analysis
Ribosomal RNA was depleted from total RNA samples extracted from stage 7 to stage 8 and stage 9 to stage 10 using riboPOOL (siTOOLs Biotech). The remaining rRNA‐depleted RNA was used to generate libraries with a NEBNext Ultra II RNA Library Prep Kit, followed by paired‐end sequencing on an Illumina HiSeq X Ten instrument with 150 bp per read (Genewiz). Two biological replicates were performed for each sample.
The obtained sequencing reads were preprocessed using Cutadapt (v1.18) to remove adapter sequences and low‐quality bases. High‐quality reads were subsequently aligned to the Oryza_sativa IRGSP‐1.0 reference genome using HISAT2 (v2.1.0), while PCR duplicates were removed with the Picard Toolkit. Differentially expressed genes were identified with the R package DESeq2, and Gene Ontology (GO) enrichment analysis was conducted using the plant‐regulomics (http://bioinfo.sibs.ac.cn/plant‐regulomics/).
m6A‐seq and data analysis
Poly A+ RNA was enriched from total RNA using Oligo(dT)25 Dynabeads (Thermo Fisher Scientific). Subsequently, 500 ng Poly A+ RNA was fragmented into 100 nt fragments using a Magnesium RNA fragmentation module (NEB). Immunoprecipitation (IP) of m6A was conducted with an EpiMark N 6‐Methyladenosine enrichment kit (NEB), and the RNA eluted from the m6A‐IP and input RNA were used to prepare libraries with the NEBNext Ultra II RNA Library Prep Kit. Paired‐end sequencing was performed on an Illumina HiSeq X Ten platform with 150 bp per read (Genewiz).
Cutadapt (v1.18) was used to preprocess the raw sequencing reads and remove adapter sequences and low‐quality bases. The resulting high‐quality reads were aligned to the Oryza_sativa IRGSP‐1.0 reference genome using HISAT2 (v2.1.0). The resulting mapping reads were utilized to identify m6A peaks with the R package EXOMEPEAK, and MeTDiff was employed to detect differential m6A peaks based on criteria of fold change >2 and FDR < 0.05. Peak annotation was performed using Bedtools and custom python scripts available at https://github.com/joybio/m6A‐seq/tree/main/feature_annotation/. Gene Ontology (GO) functional annotations were conducted using the plant‐regulomics database (http://bioinfo.sibs.ac.cn/plant‐regulomics/), while motifs were identified with HOMER.
m6A‐IP‐qPCR
The procedure utilized in this study was based on the previously described m6A‐seq method (Dominissini et al., 2013). Briefly, 50 μg total RNA was fragmented to 100–150 nt using RNA Fragmentation Reagents (Invitrogen), followed by ethanol precipitation. Subsequently, 10% of the eluted RNA was saved as input sample, while the remaining RNA was incubated with 5 μg m6A antibody (#202203; Synaptic Systems) in 500 μL IP buffer 750 mM NaCl, 0.5% Igepal CA‐630, 50 mM Tris–HCl (pH 7.4), 200 U RiboLock RNase Inhibitor (Thermo Fisher) at 4 °C for 2 h. The m6A‐containing fragments were pulled down with Dynabeads Protein A and eluted with RLT buffer, followed by ethanol precipitation. Both input and IP samples were analysed by qRT‐PCR using primers listed in Table S6. The relative enrichment of m6A in each sample was calculated by normalizing the value of the amplification cycle (Cq) of the m6A‐IP portion to the Cq of the corresponding input portion.
RIP‐qPCR
The RNA immunoprecipitation was carried out according to previously described method (Koster and Staiger, 2014). Briefly, spikelets (anthers at stage 7 to stage 12) of Osalkbh9‐1 and NIP were harvested and fixed in 1% formaldehyde under vacuum for 15 min. Subsequently, they were terminated with 150 mM glycine for 10 min. Two grams of fixed material was homogenized in 2 mL of lysis buffer (50 mM HEPES, pH 7.5, 150 mM KCl, 2 mM EDTA, 0.5% Igepal CA‐630, 0.5 mM DTT, 1× Roche Protease inhibitor cocktail and 40 U/mL Ribolock RNase inhibitor). The extract was centrifuged at 13,000 rpm for 10 min at 4 °C. A part of the lysate was taken as input samples, while the rest was divided into two equal volumes and subsequently immunoprecipitated with either anti‐Flag M2 magnetic beads (Sigma‐Aldrich, USA) or normal rabbit IgG (Cell Signaling Technology, USA) bounded to Dynabeads Protein A. After washing and ethanol precipitation, the recovered RNA fractions were used for qRT‐PCR. LOC_Os02g37950, which was devoid of an m6A peak from m6A profiling data, was employed as the internal control.
mRNA stability assay
The procedure was based on the previously described method (Duan et al., 2017). Briefly, spikelets (anthers at stage 7 to stage 12) of Osalkbh9‐1 and NIP were harvested and transferred to 1/2 MS liquid medium. A final concentration of 100 μM actinomycin D was added to the medium, followed by infiltration for 1 h. The spikelets were then collected and considered as time 0 controls, while subsequent samples were collected every 2 h. qRT‐PCR was used to determine the remaining mRNA levels, with 18S rRNA serving as a reference.
Author contributions
G.J., J.W., S.Z. and J.T. conceived the project; J.T. and D.L. performed the experiments with the help of S.C., X.W., X.H., S.Z. and Z.C.; J.Y. analysed the sequencing data; J.T., D.L., J.Y., G.J., J.W. and S.Z. designed the experiments, interpreted the results, and wrote the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare no competing interests.
Supporting information
Figure S1 Sequence alignment of the AlkB domain proteins.
Figure S2 CRISPR/Cas9‐mediated target mutagenesis of OsALKBH10.
Figure S3 Pollen grains of genetically complementary plants and demethylase inactive complementary plants.
Figure S4 Subcellular localization of OsALKBH9‐eGFP in N. benthamiana leaves epidermal cells.
Figure S5 Correlation of RNA‐seq data.
Figure S6 PCA analysis of RNA‐seq data.
Figure S7 Hierarchical clustering and Gene Ontology analysis of RNA‐seq data.
Figure S8 Joint analysis of m6A‐seq and RNA‐seq.
Table S1 The genetic segregation ratios of genotypes and phenotypes from heterozygotes.
Table S2 List of RNA‐seq data of genes corresponding to Figure 4C.
Table S3 RNA‐seq of anthers at S7‐8.
Table S4 RNA‐seq of anthers at S9‐10.
Table S5 m6A hypermethylated peaks.
Table S6 Primers used in this study.
Acknowledgements
This work was supported by the National Key R&D Program of China (2023ZD04073), the National Natural Science Foundation of China (nos. 22225704 and 22321005), and the National Basic Research Program of China (2019YFA0802201). Jun Tang was supported in part by the Postdoctoral Fellowship of Peking‐Tsinghua Center for Life Sciences.
[Correction added on 9 May 2024, after first online publication: The affiliation of Shanshan Zhu is corrected in this version.]
Contributor Information
Shanshan Zhu, Email: zhushanshan@caas.cn.
Jianmin Wan, Email: wanjm@njau.edu.cn.
Guifang Jia, Email: guifangjia@pku.edu.cn.
Data availability statement
The raw sequencing data reported in this paper have been deposited in the Genome Sequence Archive in the National Genomics Data Center (NGDC), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA010684) which is publicly accessible at https://ngdc.cncb.ac.cn/gsa.
References
- Aya, K. , Ueguchi‐Tanaka, M. , Kondo, M. , Hamada, K. , Yano, K. , Nishimura, M. and Matsuoka, M. (2009) Gibberellin modulates anther development in rice via the transcriptional regulation of GAMYB. Plant Cell, 21, 1453–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, W. , Wang, P. , Hong, J. , Kong, W. , Xiao, Y. , Yu, X. , Zheng, H. et al. (2019) Earlier degraded tapetum1 (EDT1) encodes an ATP‐citrate lyase required for tapetum programmed cell death. Plant Physiol. 181, 1223–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Che, R. , Hu, B. , Wang, W. , Xiao, Y. , Liu, D. , Yin, W. , Tong, H. et al. (2021) POLLEN STERILITY, a novel suppressor of cell division, is required for timely tapetal programmed cell death in rice. Sci. China Life Sci. 65, 1235–1247. [DOI] [PubMed] [Google Scholar]
- Cheng, P. , Bao, S. , Li, C. , Tong, J. , Shen, L. and Yu, H. (2022) RNA N 6‐methyladenosine modification promotes auxin biosynthesis required for male meiosis in rice. Dev. Cell 57, 246–259.e4. [DOI] [PubMed] [Google Scholar]
- Dominissini, D. , Moshitch‐Moshkovitz, S. , Salmon‐Divon, M. , Amariglio, N. and Rechavi, G. (2013) Transcriptome‐wide mapping of N 6‐methyladenosine by m6A‐seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 8, 176–189. [DOI] [PubMed] [Google Scholar]
- Duan, H.C. , Wei, L.H. , Zhang, C. , Wang, Y. , Chen, L. , Lu, Z. , Chen, P.R. et al. (2017) ALKBH10B is an RNA N 6‐methyladenosine demethylase affecting Arabidopsis floral transition. Plant Cell, 29, 2995–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan, W. , Wang, L. , Lei, Z. , Li, H. , Chu, J. , Yan, M. , Wang, Y. et al. (2023) m6A RNA demethylase AtALKBH9B promotes mobilization of a heat‐activated long terminal repeat retrotransposon in Arabidopsis. Sci. Adv. 9, eadf3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Z. , Yu, J. , Cheng, X. , Zong, X. , Xu, J. , Chen, M. , Li, Z. et al. (2014) The rice basic helix‐loop‐helix transcription factor TDR INTERACTING PROTEIN2 is a central switch in early anther development. Plant Cell, 26, 1512–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou, Q. , An, X. , Ma, B. , Wu, S. , Wei, X. , Yan, T. , Zhou, Y. et al. (2023) ZmMS1/ZmLBD30‐orchestrated transcriptional regulatory networks precisely control pollen exine development. Mol. Plant, 16, 1321–1338. [DOI] [PubMed] [Google Scholar]
- Jia, G. , Fu, Y. , Zhao, X. , Dai, Q. , Zheng, G. , Yang, Y. , Yi, C. et al. (2011) N 6‐methyladenosine in nuclear RNA is a major substrate of the obesity‐associated FTO. Nat. Chem. Biol. 7, 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, Y. , Song, X. , Chang, H. , Zhao, Y. , Cao, C. , Qiu, X. , Zhu, J. et al. (2022) The GA–DELLA–OsMS188 module controls male reproductive development in rice. New Phytol. 233, 2629–2642. [DOI] [PubMed] [Google Scholar]
- Jouannet, V. , Moreno, A.B. , Elmayan, T. , Vaucheret, H. , Crespi, M.D. and Maizel, A. (2012) Cytoplasmic Arabidopsis AGO7 accumulates in membrane‐associated siRNA bodies and is required for ta‐siRNA biogenesis. EMBO J. 31, 1704–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, K.H. , Han, M.J. , Lee, Y.S. , Kim, Y.W. , Hwang, I. , Kim, M.J. , Kim, Y.K. et al. (2005) Rice Undeveloped Tapetum1 is a major regulator of early tapetum development. Plant Cell, 17, 2705–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakutani, T. , McCue, A.D. , Nuthikattu, S. , Reeder, S.H. and Slotkin, R.K. (2012) Gene expression and stress response mediated by the epigenetic regulation of a transposable element small RNA. PLoS Genet. 8, e1002474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster, T. and Staiger, D. (2014) RNA‐binding protein immunoprecipitation from whole‐cell extracts. Methods Mol. Biol. 1062, 679–695. [DOI] [PubMed] [Google Scholar]
- Li, N. , Zhang, D.S. , Liu, H.S. , Yin, C.S. , Li, X.x. , Liang, W.q. , Yuan, Z. et al. (2006) The rice tapetum degeneration retardation gene is required for tapetum degradation and anther development. Plant Cell, 18, 2999–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Yuan, Z. , Vizcay‐Barrena, G. , Yang, C. , Liang, W. , Zong, J. , Wilson, Z.A. et al. (2011) PERSISTENT TAPETAL CELL1 encodes a PHD‐finger protein that is required for tapetal cell death and pollen development in rice. Plant Physiol. 156, 615–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, K. , Han, J. , Zhang, Z. , Li, H. , Zhao, Y. , Zhu, Q. , Xie, Y. et al. (2021) OsEDM2L mediates m6A of EAT1 transcript for proper alternative splicing and polyadenylation regulating rice tapetal degradation. J. Integr. Plant Biol. 63, 1982–1994. [DOI] [PubMed] [Google Scholar]
- Martinez‐Perez, M. , Aparicio, F. , Lopez‐Gresa, M.P. , Belles, J.M. , Sanchez‐Navarro, J.A. and Pallas, V. (2017) Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc. Natl. Acad. Sci. 114, 10755–10760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu, N. , Liang, W. , Yang, X. , Jin, W. , Wilson, Z.A. , Hu, J. and Zhang, D. (2013) EAT1 promotes tapetal cell death by regulating aspartic proteases during male reproductive development in rice. Nat. Commun. 4, 1445. [DOI] [PubMed] [Google Scholar]
- Shi, J. , Cui, M. , Yang, L. , Kim, Y.‐J. and Zhang, D. (2015) Genetic and biochemical mechanisms of pollen wall development. Trends Plant Sci. 20, 741–753. [DOI] [PubMed] [Google Scholar]
- Shoaib, Y. , Hu, J. , Manduzio, S. and Kang, H. (2021) Alpha‐ketoglutarate‐dependent dioxygenase homolog 10B, an N 6‐methyladenosine mRNA demethylase, plays a role in salt stress and abscisic acid responses in Arabidopsis thaliana . Physiol. Plant, 173, 1078–1089. [DOI] [PubMed] [Google Scholar]
- Tang, J. , Jia, P. , Xin, P. , Chu, J. , Shi, D.‐Q. , Yang, W.‐C. and Bozhkov, P. (2020) The Arabidopsis TRM61/TRM6 complex is a bona fide tRNA N 1‐methyladenosine methyltransferase. J. Exp. Bot. 71, 3024–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, J. , Yang, J. , Duan, H. and Jia, G. (2021) ALKBH10B, an mRNA m6A demethylase, modulates ABA response during seed germination in Arabidopsis. Front. Plant Sci. 12, 712713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, J. , Yang, J. , Lu, Q. , Tang, Q. , Chen, S. and Jia, G. (2022) The RNA N 6‐methyladenosine demethylase ALKBH9B modulates ABA responses in Arabidopsis. J. Integr. Plant Biol. 64, 2361–2373. [DOI] [PubMed] [Google Scholar]
- Tang, J. , Chen, S. and Jia, G. (2023) Detection, regulation, and functions of RNA N 6‐methyladenosine modification in plants. Plant Commun. 4, 100546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzair, M. , Xu, D. , Schreiber, L. , Shi, J. , Liang, W. , Jung, K.‐H. , Chen, M. et al. (2020) PERSISTENT TAPETAL CELL2 is required for normal tapetal programmed cell death and pollen wall patterning. Plant Physiol. 182, 962–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. , Yang, J. , Song, P. , Zhang, W. , Lu, Q. , Yu, Q. and Jia, G. (2022) FIONA1 is an RNA N 6‐methyladenosine methyltransferase affecting Arabidopsis photomorphogenesis and flowering. Genome Biol. 23, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, X. , Hu, W. and Yang, Z.N. (2022) The contributions of sporophytic tapetum to pollen formation. Seed Biol. 1, 1–13. [Google Scholar]
- Zhang, D. and Yang, L. (2014) Specification of tapetum and microsporocyte cells within the anther. Curr. Opin. Plant Biol. 17, 49–55. [DOI] [PubMed] [Google Scholar]
- Zhang, D. , Luo, X. and Zhu, L. (2011) Cytological analysis and genetic control of rice anther development. J. Genet. Genomics, 38, 379–390. [DOI] [PubMed] [Google Scholar]
- Zhang, F. , Zhang, Y.C. , Liao, J.Y. , Yu, Y. , Zhou, Y.F. , Feng, Y.Z. , Yang, Y.W. et al. (2019) The subunit of RNA N 6‐methyladenosine methyltransferase OsFIP regulates early degeneration of microspores in rice. PLoS Genet. 15, e1008120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, G. , Dahl, J.A. , Niu, Y. , Fedorcsak, P. , Huang, C.‐M. , Li, C.J. , Vågbø, C.B. et al. (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell, 49, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, L. , Tian, S. and Qin, G. (2019) RNA methylomes reveal the m6A‐mediated regulation of DNA demethylase gene SlDML2 in tomato fruit ripening. Genome Biol. 20, 156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Sequence alignment of the AlkB domain proteins.
Figure S2 CRISPR/Cas9‐mediated target mutagenesis of OsALKBH10.
Figure S3 Pollen grains of genetically complementary plants and demethylase inactive complementary plants.
Figure S4 Subcellular localization of OsALKBH9‐eGFP in N. benthamiana leaves epidermal cells.
Figure S5 Correlation of RNA‐seq data.
Figure S6 PCA analysis of RNA‐seq data.
Figure S7 Hierarchical clustering and Gene Ontology analysis of RNA‐seq data.
Figure S8 Joint analysis of m6A‐seq and RNA‐seq.
Table S1 The genetic segregation ratios of genotypes and phenotypes from heterozygotes.
Table S2 List of RNA‐seq data of genes corresponding to Figure 4C.
Table S3 RNA‐seq of anthers at S7‐8.
Table S4 RNA‐seq of anthers at S9‐10.
Table S5 m6A hypermethylated peaks.
Table S6 Primers used in this study.
Data Availability Statement
The raw sequencing data reported in this paper have been deposited in the Genome Sequence Archive in the National Genomics Data Center (NGDC), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA010684) which is publicly accessible at https://ngdc.cncb.ac.cn/gsa.