

SUMMARY
Infectious bacteria have both intrinsic and acquired mechanisms to combat harmful biocides that enter the cell. Through adaptive pressures, many of these pathogens have become resistant to many, if not all, of the current antibiotics used today to treat these often deadly infections. One prominent mechanism is the upregulation of efflux systems, especially the resistance-nodulation-cell division class of exporters. These tripartite systems consist of an inner membrane transporter coupled with a periplasmic adaptor protein and an outer membrane channel to efficiently transport a diverse array of substrates from inside the cell to the extracellular space. Detailed mechanistic insight into how these inner membrane transporters recognize and shuttle their substrates can ultimately inform both new antibiotic and efflux pump inhibitor design. This review examines the structural basis of substrate recognition of these pumps and the molecular mechanisms underlying multidrug extrusion, which in turn mediate antimicrobial resistance in bacterial pathogens.
KEYWORDS: efflux pumps, RND transporters, X-ray, cryo-EM
INTRODUCTION
One of the greatest threats to human health is the increasing prevalence of antibiotic-resistant microbes (ARM). Currently, around three million new ARM cases occur each year in the United States, with millions more throughout the world (1). Even this is likely a severe underestimation, as many cases go unreported and untreated, thereby rapidly increasing the global spread of these deadly bacteria. Often, our immune systems are unable to clear these infections without the aid of administered antibiotics. Potent drugs such as ciprofloxacin, erythromycin, metronidazole, and tetracycline have been used to effectively treat bacterial infections for decades, saving a countless number of lives. Unfortunately, the use of these antibiotics has devolved into their overuse. Bacteria have natural resistance mechanisms in place to help combat these types of biocides, and increased exposure through misuse (administered for viral and other non-bacterial infections) in both humans and animals has accelerated the timeline for acquired resistance (2–4). As a result, treatment regimens often require last-resort compounds administered for longer duration times than what was previously effective. In addition, antibiotic pollution into the environmental biota through water systems and contaminated human waste disposal leads to the transfer of antibiotic-resistant genes among many different bacterial species via horizontal gene transfer (5–7). The consequence of our antibiotic mismanagement is the evolution of new, potent ARM genotypes that cause increasingly difficult-to-treat infections, leading to higher morbidity and mortality rates.
The prevalence of ARM was further exacerbated throughout the COVID-19 pandemic. During this time, many antibiotic-resistant infections were undiagnosed due to limited resources. Hospitals and other medical centers repurposed their facilities, staff, and supplies for COVID-19 treatment and detection, and as a result, other types of infections were not immediately addressed. In fact, many ARM, such as vancomycin-resistant Enterococcus (14%), multidrug-resistant Pseudomonas aeruginosa (32%), extended-spectrum beta-lactamases-producing Enterobacterales (32%), methicillin-resistant Staphylococcus aureus (13%), and carbapenem-resistant Acinetobacter baumannii (CRAB) (78%) all had alarming increases of infection rates in patients during hospitalization (8). These hospital-borne pathogens, combined with carbapanem-resistant Enterobacterales, cost more than 4.6 billion annually in the United States (9). Clearly, along with increased stewardship of antibiotic use, the development and distribution of new antibiotics and other bactericides are paramount to combat these potent infections.
One of the most potent ways the bacterium overcomes exposure to harmful chemicals is through the expression of efflux systems. These systems are membrane-embedded machineries that shuttle their substrates through membrane lipid bilayers. The central protein in each system is an inner membrane transporter (or pump), which is responsible for substrate recognition and transport facilitated by the use of the energy generated from adenosine triphosphate (ATP) hydrolysis (primary transporter) or from ion gradients (secondary transporter). Based upon the classification of the Saier group (10–12), a total of six different families of multidrug transporters exist in Escherichia coli; the ATP-binding cassette superfamily (13), the major facilitator superfamily (14), the multidrug and toxic compound extrusion family (15), the resistance-nodulation-cell division family (16), the small multidrug resistance family (17, 18), and the proteobacterial antimicrobial compound extrusion family (19). Each family is distinct in its organization, assembly, and substrate specificity (17, 20).
Of special interest is the resistance-nodulation-cell division (RND) family of transporters. They are unique in that when compared to the other transporter families, their efflux machinery spans both the inner and outer membranes of Gram-negative bacteria. This allows them to recognize and transport substrate across the entire bacterial cell envelope. These efflux machineries are highly capable of removing substrates, particularly from the periplasm and the cytoplasmic membrane. In fact, RND transporters often coordinate with and take up substrates from other efflux families that only span the inner membrane and shuttle their substrates into the periplasmic space (21). For these reasons, RND efflux systems in general, and their inner membrane transporter in particular, hold great promise as targets for the development of inhibitors that render them nonfunctional, especially in conjunction with increasing the permeability of the outer membrane (22–24). Biocides introduced into the cell would not as easily be extruded, potentially allowing compounds previously impotent in antimicrobial-resistant (AMR) bacteria to regain their ability to treat these infectious microbes. Therefore, how RND transporters assemble within the cell, their substrate specificity, and their mechanism of action are critical components required as a basis for inhibitor development. This review outlines how protein structure guides mechanistic insight into RND transporters, with a specific focus on conformational flexibility during substrate transport.
THE RND TRANSPORT MACHINERY
In Escherichia coli, a Gram-negative bacterium, there exists six RND transporters that belong to the hydrophobe/amphiphile efflux 1 (HAE1) family (transporter classification database family 2.A.6.2). These transporters, acriflavine resistance protein B, AcrB (25, 26), acriflavine resistance protein D, AcrD (27–30), acriflavine resistance protein F, AcrF (28, 31), multidrug resistance protein B, MdtB (28, 32–34), multidrug resistance protein C, MdtC (28, 32–34), and multidrug resistance protein F, (MdtF or YhiV) (28, 35, 36), are important for the export of antibacterials and other toxic small molecules. A seventh RND transporter, cation efflux system protein A, CusA (28, 37–40), belongs to the heavy metal efflux (HME) RND family (41, 42) (transporter classification database family 2.A.6.2) and is critical for the export of Ag(I) and Cu(I) in order for the cell to maintain physiologic concentrations of these metal ions. The functional and phylogenic classifications of these and other membrane transport proteins can be viewed at www.tcdb.org.
The best-studied RND transporter in E. coli is AcrB, the inner membrane component of the AcrAB-TolC efflux system. AcrAB-TolC is a promiscuous system that recognizes multiple classes of antibiotics, dyes, bile salts, detergents, and organic compounds (17, 25, 43–52). It is the central efflux system in E. coli that removes toxic substances that have entered the cell via passive transport through the outer membrane or via porins and substrates deposited from the cytoplasm into the periplasmic space by single-component transporters. As a response to the challenge, increased expression of the acrB gene is a powerful tool that the bacterium uses to gain resistance and is often upregulated in clinical isolates (53, 54). All parts of the efflux system are required for efficient efflux, as the removal of any of the three components hinders the ability of the cell to confer resistance toward a variety of antibiotics (55, 56). The basic architecture of AcrAB-TolC, as well as the majority of RND efflux systems studied to date, consists of an inner membrane transporter (or inner membrane pump, IMP), AcrB, a soluble periplasmic adaptor protein (PAP), AcrA, and an outer membrane channel (OMC), TolC, to form a tripartite system (Fig. 1). A fully functioning system consists of IMP:PAP:OMC in a 3:6:3 ratio that spans both the inner and outer membranes of Gram-negative bacteria (57, 58). This assembly is prototypical for RND efflux systems of the HAE1 and HME classes, where IMP components are present in the form of a trimer. However, there are documented examples where the bacterial RND inner membrane pump exists as a dimer [HpnN from Burkholderia multivorans, HAE3 family (59), SecDF from E. coli, SecDF family (60), and as a monomer (MmpL3 from Mycobacterium species, HAE2 family (61–64)].
Fig 1.

Structure of the E. coli AcrAB-TolC efflux machinery (adapted from PDB ID: 5V5S). AcrAB-TolC consists of an inner membrane pump, AcrB (green), that spans the inner membrane and is responsible for substrate recognition and uptake. AcrA (red), the periplasmic adaptor protein provides stability to the complex and connects AcrB to the outer membrane channel TolC (blue), which forms a passage through the outer membrane to deposit their substrates into the extracellular space.
GENERAL STRUCTURE OF AN RND INNER MEMBRANE TRANSPORTER
To provide an overview of the general architecture of a canonical RND inner membrane transporter, the X-ray crystal structure for AcrB is used, as this protein is the RND transporter from the HAE-1 class that has been most extensively studied (Fig. 2A). The trimeric structure can effectively be divided into two regions: the transmembrane domain (~50 Å) located within the inner membrane and the periplasmic domain (~70 Å) situated in the periplasm. The periplasmic domain (PD) can be further split into the porter domain (~40 Å) and the docking domain (~30 Å) (65). Universally, the large PD is formed through external loops that extend between TM1–TM2 and TM7–TM8. Defining characteristics include 12 transmembrane helices (TM1–TM12) and six periplasmic subdomains (PC1, PC2, PN1, PN2, DC1, and DC2). This periplasmic domain is the main hub from which substrates are recognized and transported outside the cell. PC1 and PC2 are located near the periphery of the PD and are important for substrate recognition and binding. These two subdomains contribute to form a periplasmic cleft, which can be open or closed, and initiate passage through the PD for removal (Fig. 2A and B). PN1 is critical for protomer-protomer interactions and forming the central pore within the AcrB trimer, PN2 helps to shape the global orientation of the PD, and DC1 and DC2 provide the structural determinants for docking of the transporter to the outer membrane channel (in this instance, TolC). Each subdomain also helps to secure binding to periplasmic adaptor proteins (AcrA for AcrB) and is integral for the overall stability of the tripartite structure. As the size and shape of the PD can vary between members of the same class (HAE-1) and certainly between different classes (HAE-2, HAE-3, and HME), substrate recognition and transport can be unique for each RND pump. Therefore, it is worthwhile to critically examine each inner membrane transporter individually as well as the dynamics of the tripartite efflux system of which they are a part. In-depth details of substrate recognition and transport are detailed below for select RND pumps from the HAE-1 and HME classes of transporters.
Fig 2.

(A) Ribbon diagram of the E. coli AcrB IMP viewed in the membrane plane. AcrB assembles as a trimer (adapted from PDB ID: 2GIF). The three protomers of AcrB are colored gray (access protomer), light blue (binding protomer), and salmon (extrusion protomer). This color scheme is consistent throughout the review. Labeled are the subdomains DC and DN of the docking domain and PC1 and PC2 of the periplasmic domain. (B) Top view of the periplasmic domains of AcrB. The clefts formed between subdomains PC1 and PC2 are open in the access and binding protomers and closed in the extrusion protomer. (C) Ribbon diagram of the binding protomer of AcrB, with the flexible F-loop (yellow) and G-loop (orange) highlighted (full structure and inset—top). The red arrow highlights the substrate path starting at the entrance, E, and moving upward through the proximal binding site, PBS, and the distal binding site, DBS, toward the outer membrane channel (TolC). Full structure and inset—bottom—residues critical for proton uptake from the periplasm into the cell. The transmembrane domain is rotated 60° for better visualization of the residues important for proton translocation.
The TM domain consists of two TM bundles (TM1–TM6 and TM7–TM12) that confer pseudo-twofold symmetry to the overall structure. In general, the TMs do not extend extensively into the cytoplasm (Fig. 2A) and are not associated with any type of cellular signaling. The main functions of the TM domain are to provide the energetics for substrate transport and to anchor the protein in the inner membrane. RND pumps use the proton motive force (PMF) of the cell to catalyze the efflux of their substrates. Within the TM domain, there exists a proton relay network that generates the PMF needed to transport their substrates via an antiport mechanism. In the transmembrane region of AcrB, residues Asp 407, Asp 408, Lys 940, and Arg 971 have been found to be critical for H+ uptake and transport (Fig. 2C, bottom panel). Interestingly, the polar residue Thr 978 is also important for H+ transfer (66). Protons are acquired from the periplasm, and through a twisting of Lys 940 and Arg 971, they are released into the cytoplasm with concurrent propulsion of substrate toward the cell surface via a peristaltic motion during the export cycle. For select RND transporters, including E. coli AcrD (29, 30), E. coli CusA (37), and Ralstonia sp. CH4 CzcA (67), data suggest that substrates can be acquired from the cytoplasm directly. Thus, the TM domain is important for structural organization, housing the proton relay network required for substrate transport, and in some cases, providing an entryway for the uptake of substrates that originate in the cytoplasmic space. Below is a brief summary of how RND inner membrane pumps recognize and transport substrate highlighting the complexity of the transport cycle.
AcrB—EXPORT PATHWAY AND DYNAMICS
Crystallographic analyses of AcrB, both in the apo state and with various substrates such as ethidium (Et), rhodamine 6G, erythromycin A, and ciprofloxacin, show that it can assemble as either symmetric or asymmetric trimers. Three distinct conformations of the AcrB monomer have been elucidated, with each conformation distinct in its ability to recognize and bind substrates and the formation of unique tunnel systems in the PD that create a pathway for extrusion. For an excellent in-depth analysis on AcrB as well as the entire AcrAB-TolC system, please see references (17, 20, 65, 68).
In order for AcrB to function, it must go through consecutive transition states in the transport cycle. These have been designated as loose, or access state, tight, or binding state, and open, or extrusion state. For consistency in this review, these three conformers will be referred to as the access, binding, and extrusion states (Fig. 2A and B). The first crystal structure of AcrB solved in 2002 depicts a symmetric trimer without substrate present. In this conformer, the cleft between PC1 and PC2 is open; however, no ligand was detected at this proposed binding site. This is designated as the access state of the conformer with the assembled trimer being in the access:access:access conformation. In this state, there is a tunnel (tunnel 1) that starts at the PC1/PC2 interface and extends inward toward the central pore of the trimer, and substrate is posited to enter from the periplasm via this open cleft (Fig. 2C). Subsequent studies have elucidated additional structures showing that AcrB can also assemble as a variety of asymmetric trimers. The predominant form observed in these asymmetric assemblies is the access:binding:extrusion form (Fig. 2A and B), although other forms such as access:access:binding and access:binding:binding have been subsequently identified. To successfully complete the transport cycle, an AcrB monomer must sequentially transition from the access state to the binding state and then to the extrusion state to shuttle its substrate through the transport machinery powered by the PMF of the cell. Upon substrate binding, multiple allosteric changes occur in subdomains PN1/PC2 and PN2/PC1 to shift PC2 away from PC1 (Fig. 2B), thus opening a vertical cleft toward the periplasm. This and other structural rearrangements within the protomer transition it from the access state into the binding state. An additional tunnel is created with its entrance near the groove created by TM8 and TM9 near the outer leaflet of the inner membrane. This tunnel may be specific for hydrophobic substrates (as examples, ligands such as ethidium, N-phenylnaphthylamine, and penicillin-based antibiotics) that can partially embed into the membrane surface, but it has not been ruled out as a general entrance tunnel for all substrates (68). Additionally, in this conformer, this tunnel may also act as an additional pathway for substrates that enter via the PC1/PC2 cleft, or it may be used to extract poor substrates back into the periplasm. The binding conformer has three defined binding sites: the PC1/PC2 entrance site (E), the proximal binding site, and the distal binding site (Fig. 2C, upper panel). These sites are generally large and can accommodate a wide variety of structurally diverse compounds. Substrates move through the PD via a series of flexible loops (the F-loop 669PAIVELGT676 and the G-loop 614GFGFAGRG621 in AcrB (Fig. 2C, upper panel), which help transfer them sequentially from the periplasmic entrance site to the proximal and distal binding sites, and then through an exit tunnel toward the outer membrane channel TolC. The open cleft between PC1 and PC2 is closed, and an exit tunnel is created when the binding conformer transitions to the extrusion conformer. How this transition is precisely orchestrated is still unclear; however, remote conformational coupling between the docking domain and the transmembrane regions has been postulated (17). It has also been suggested that an additional substrate may need to bind to an adjacent protomer, thus forming the access:binding:binding or binding:binding:binding trimeric conformation as an intermediate in order for an extrusion conformer to exist within the access:binding:extrusion or binding:binding:extrusion asymmetric states (68–70). At this point, the adaptor protein, AcrA, helps to open the periplasmic channel toward TolC for substrate extrusion, and water molecules in the intramolecular substrate channel help to extrude hydrated substrates (65, 71).
Despite our considerable knowledge, precisely how AcrB, in particular, and all RND-inner membrane transporters, in general, function within their various trimeric conformations to efficiently export their substrates is still unresolved. To date, only the access:access:access, access:binding:extrusion, and binding:binding:binding forms of AcrB have been structurally characterized through X-ray crystallography. However, other forms have been suggested (69, 70). Cross-linking and mutagenesis studies point to each protomer within the intact trimer playing a role in substrate export via a functionally rotating mechanism, while thermodynamic calculations show that trimerization is necessary, as the formation of the trimer compensates for the entropy losses of each individual monomer during conformational switching (72–75). Through this rotating mechanism, the trimer would switch from access:binding:extrusion → binding:extrusion:access → extrusion:access:binding, and then finally → access:binding:extrusion to reset the cycle. It is proposed that adjacent protomers interact and help one another transition through the complete transport cycle and that trimerization is essential for the function of the fully assembled efflux system. However, analysis of several RND inner membrane transporters from different Gram-negative bacteria suggests that, in many cases, each protomer may act independently within the trimer and concurrently export their substrate. As each RND transport system is unique, it is possible that the mechanistic basis for substrate transport varies between species, Gram-positive and Gram-negative organisms, and perhaps even between RND pumps within the same bacteria.
Advances within the field of cryo-electron microscopy (cryo-EM) have allowed us to gain significant insight into the mechanistic basis of substrate binding and transport of inner membrane transporters (76, 77). Cryo-EM captures proteins in a frozen-hydrated state. From a single sample, it is possible to determine the structures of several different proteins/protein complexes, but importantly, it can also capture a single protein/complex in several distinct conformational states. These structures can then be coupled with various techniques, such as computational studies that analyze temperature, lipid composition, and solvent effects (71), molecular dynamics (MD) simulations (78–80), and experimental studies, such as kinetic measurements (78, 81), fluorescent efflux assays (82), and hydrogen/deuterium exchange mass spectrometry (83), to determine a complete picture of how inner membrane RND pump recognize and transport their substrates. Below are brief summaries of select RND transporters that have been studied through cryo-EM, X-ray crystallography, and other biophysical/computational techniques to help delineate the mechanistic basis of substrate recognition and transport.
CAMPYLOBACTER JEJUNI CmeB
Export of bactericides and other harmful agents through the Campylobacter multidrug efflux (Cme) tripartite system is a potent compensatory mechanism that C. jejuni uses to help maintain cell viability (84, 85). This system mirrors the AcrAB-TolC system in both its assembly and stoichiometry. The Cme locus contains three tandemly linked genes (cmeABC) that encode the periplasmic adaptor protein CmeA, the inner membrane pump CmeB, and the outer membrane channel protein CmeC (84–86). When assembled, CmeABC is able to export harmful substrates such as fluoroquinolones (ciprofloxacin), macrolides, tetracyclines, ethidium bromide, and chloramphenicol (86).
Via X-ray crystallography, two distinct structures of the CmeB transporter were successfully characterized (87). Both forms adopt the typical fold of an HAE-1 RND protein and assemble as a homotrimer, displaying a pseudo-threefold symmetrical axis perpendicular to the membrane surface (Fig. 3A). Each protomer contains a similar overall structure as described for AcrB: 12 transmembrane helices (TM1–TM12) and a large periplasmic domain separated into six subdomains (PC1, PC2, PN1, PN2, DC1, and DC2). This is consistent with each RND inner membrane pump reviewed here, and therefore, will not be explicitly detailed in the IMPs discussed below.
Fig 3.

(A) Ribbon diagram of Form II of the C. jejuni CmeB IMP viewed in the membrane plane (adapted from PDB ID: 5LQ3). The three protomers are colored light blue (binding protomer), salmon (extrusion protomer), and smudge (resting protomer). Lysine residues K781 and K843 are highlighted in the binding protomer as yellow spheres. (B) Top view of the periplasmic domain of Form II of CmeB. The clefts are closed in both the resting and extrusion protomers and open in the binding protomer. (C) Proposed model of drug efflux for CmeB. FRET experiments detailed four distinct states of the trimeric pump: Low (L), intermediate-1 (I1), intermediate-2 (I2), and high (H). Each protomer is thought to cycle through resting (R), binding (B), and extrusion (E) states independently. U, unlabeled protomer. (D) An overlay of the periplasmic binding pocket of RE-CmeB with various substrates—hydrolyzed ampicillin (Amp, green) (adapted from PDB ID: 8GJK), chloramphenicol (Chl, violet) (adapted from PDB ID: 8GK4), erythromycin (Ery, ruby) (adapted from PDB ID: 8GK0), and ciprofloxacin (Cip, orange) (adapted from PDB ID: 8GJL). Critical residues involved in ligand binding are highlighted.
The first X-ray structure (Form I) of CmeB is characterized as a nearly asymmetric trimer. Although similar, each protomer is slightly different yet all have their clefts between the subdomains PC1 and PC2 closed (87), comparable to the extrusion protomer of AcrB. A tunnel leading from the periplasmic domain toward the central cavity is present in each protomer, also closely mimicking the extrusion protomer of AcrB. Thus, this trimeric conformation can be categorized as the all-extrusion form (extrusion:extrusion:extrusion) (87).
The second X-ray structure (Form II) of CmeB is quite different. Form II also displays as an asymmetric trimer, but with two PC1/PC2 clefts closed and one cleft open (Fig. 3B) (87). The single protomer with the open cleft has an elongated tunnel spanning from the cleft entrance through the periplasmic domain toward the central cavity, similar to the binding form of AcrB. This CmeB protomer can be labeled as the binding form. Interestingly, the remaining two protomers with their periplasmic clefts closed yet are structurally not the same. One protomer can be classified as the extrusion form. The other cannot be categorized as being in either the binding or extrusion forms. No tunnel is apparent, and this protomer can potentially be considered an intermediate that the inner membrane pump transitions to after substrate export. No AcrB homologous structure is apparent; however, this CmeB protomer is similar to the resting state of the HME IMP of E. coli, CusA. Therefore, Form II is unique in that its overall assembly can be designated as the resting:binding:extrusion form (Fig. 3A and B) (87).
Interestingly, a snapshot of all known bacterial RND structures determined to date shows a myriad of assemblies, ranging from symmetric trimers with their PC1/PC2 clefts either all open or all closed to asymmetric trimers with one or two clefts open. This substantiates the notion that the transport dynamics of these efflux systems are complicated. Can each protomer function independently or is there a coordinated action between protomers that is requisite for substrate transport? As has been postulated, after a substrate binds to AcrB, a second substrate that binds to a neighboring protomer can help assist the transition from the binding state to the extrusion state (88). Therefore, this binding to extrusion transition is thought to be aided by both bi-site activation with a second substrate as well as the utilization of the cellular PMF. It has also been suggested that not all trimeric assemblies are likely, as steric clashes can dissuade certain configurations. It is clear that the static depictions of these inner membrane pumps are not sufficient to completely delineate the mechanistic basis of the transport cycle. To help answer this, total internal reflection single molecule-fluorescence resonance energy transter (sm-FRET) imaging was used to discern specifically how each CmeB protomer functions within its trimeric assembly (87).
Transport dynamics of CmeB
Wild-type CmeB contains three cysteine amino acids located within the transmembrane region, and replacement of these residues to serine does not affect its overall function (87). A single cysteine was then introduced at position K843. This lysine is located near subdomain PC2 facing the periplasm, an area readily exposed to solvent (Fig. 3A). This K843C mutant on the cys-less background was found to be fully functional via in vitro proton transport and in vivo susceptibility assays (87). Alexa Fluor 546 (donor) and Alexa Fluor 647 (acceptor) dyes were introduced, and only CmeB molecules with one donor and one acceptor dye per trimer were used for analysis. As a control, a cysteine introduced at K781, located near the top of the funnel region within subdomain DC1 (Fig. 3A), was used. This area of the transporter is predicted to have little movement regardless of protomeric form. These proteins were reconstituted into proteoliposomes, and the single molecule-level transport dynamics were analyzed with and without substrate via FRET. Presumably, a rotating mechanism would give rise to three distinct signals, mimicking the access:binding:extrusion trimeric form observed in the asymmetric assembly of known AcrB X-ray structures. However, the FRET analysis indicated at least four distinct states that K843C cys-less CmeB could adopt upon the addition of taurodeoxycholate, a known substrate of this transporter. These results suggested that there are more than three different conformational states that the trimeric CmeB pump goes through during the transport cycle. These states were designated low (L), intermediate-1 (I1), intermediate-2 (I2), and high (H). This, combined with the symmetrical nature of the density plots, suggests that each CmeB protomer likely functions independently of each other rather than in a tightly coordinated fashion. Little change was seen with the K781C cys-less CmeB, confirming that the funnel region of the trimer undergoes little movement upon substrate transport and substantiates the reliability of the FRET system. Of note, the removal of the PMF by abolishing the proton-relay network within CmeB eliminated almost all dynamic movement, suggesting that the transition between different states of the IMP is most likely energy dependent. Taken together, structural and functional analyses of CmeB have helped to uncover the mechanism of drug export by an RND transporter. Upon substrate binding, each protomer can independently advance through the transport cycle by coupling with the proton relay network to effectively remove substrates from the cell (Fig. 3C). Whether the binding of substrate to more than one protomer sequentially aids in this transition is still unclear. Further study is also required to determine a complete picture of all favored trimeric states that CmeB can adopt during active efflux.
The resistance-enhancing CmeB multidrug efflux pump
In 2015, a clinical isolate of Salmonella Typhimurium from a patient who failed ciprofloxacin therapy was subjected to genome sequencing. Sequencing results identified a point mutation, G288D, within the distal binding site of the AcrB, and computational modeling suggests that this mutation lowered the drug-binding affinity at this site (89). In 2016, a “super” efflux pump variant was identified that shifts the minimum inhibitory concentration distribution of various antibiotics to the higher range among clinical C. jejuni isolates (90). Using gene replacement, it was discovered that amino acid changes in both the promoter region and within the cmeABC operon were associated with elevated levels of multidrug resistance. It should be noted that this cmeABC genetic variant was originally identified from the Campylobacter coli DH161 isolate, where the resistant gene is capable of horizontally transferring to C. jejuni (90). This CmeABC variant was designated as resistance-enhancing CmeABC (RE-CmeABC) (90). RE-CmeABC represents an emerging multidrug resistance mechanism utilized by Campylobacter for adaptation to antibiotic selection pressure and is a second example of sequence variation as a significant mechanism to enhance the function of an RND multidrug resistance transport system against antimicrobials. In 2018, a “super drug-resistant” strain of Neisseria gonorrhoeae was identified in the United Kingdom (91). In the same year, it was found that gonococci bearing mosaic-like sequences within the gene mtrD, encoding the RND-type MtrD multidrug efflux pump, is able to elevate drug resistance and enhance the transport function of the MtrD pump (see the MtrD section) (92). Thus, it is likely that additional drug-resistant isolates with mutations within RND transporters from these and other pathogenic strains of bacteria will be discovered. In the case of C. jejuni, there have been 198 amino acid differences between wild-type and variant CmeB transporters identified, with 22 of these located in the putative drug-binding cavity (90). Docking studies suggest that wild-type and variant transporters utilize different sets of residues to anchor drugs (specifically fluorfenicol and ciprofloxacin), with the RE-CmeB variant displaying enhanced drug binding via predicted binding energy calculations (90). However, exactly how this is achieved structurally was unclear.
To elucidate how this resistance-enhancing variant elevates drug resistance and enhances transport function, cryo-EM structures of the C. jejuni RE-CmeB membrane protein were solved, both in the absence and presence of the antibiotics, ciprofloxacin (Cip), chloramphenicol (Chl), and erythromycin (Ery) (93). These structures detail unique binding modes the pump utilizes to efficiently recognize a variety of antibiotics.
The structure of RE-CmeB in the absence of added drugs (apo-RE-CmeB) (93) adopts the overall architecture of RND-type proteins and forms a homotrimer (25, 37, 45, 57, 87, 94–97). Similar to the structures of asymmetric AcrB (25, 46), MtrDCR103 (98, 99), and AdeJ (100, 101), the cryo-EM structure of apo-RE-CmeB presents an asymmetric trimer with the access:binding:extrusion conformational state. This structure, however, is quite distinct from those of the X-ray structures of CmeB, where the membrane protein displays either a symmetric extrusion:extrusion:extrusion form or an asymmetric resting:binding:extrusion form (87).
In addition to images that led to the apo-RE-CmeB structure, a distinct class of images of RE-CmeB that contains densities of a ligand within the pump was also observed. This ligand was identified as a hydrolyzed ampicillin (Amp) antibiotic with an open form of the four-member β-lactam ring (93). The structure of RE-CmeB-Amp also presents an asymmetric trimer with the access:binding:extrusion conformational state identical to that of apo-RE-CmeB. The bound Amp ligand was found within the binding protomer of RE-CmeB. Surprisingly, the binding mode for this ligand is completely different from those drug binding modes observed before in other multidrug efflux pumps, including AcrB, AdeB, AdeJ, and MtrDCR103. The cryo-EM structure indicates that the bound Amp drug is located directly below the G-loop of the pump, where the drug spans both the proximal and distal drug binding sites of the “binding” protomer of RE-CmeB (Fig. 3D). Within 4 Å of bound Amp, binding residues from both the proximal and distal sites of RE-CmeB anchor this drug. All of these residues are hydrophobic in nature, underscoring that drug recognition by RE-CmeB is mainly governed by hydrophobic interactions.
Additionally, cryo-EM structures of RE-CmeB bound with Cip, Ery, and Chl were determined to high resolutions to elucidate how RE-CmeB recognizes fluoroquinolone, macrolide, and chloramphenicol antibiotics (93). All of these RE-CmeB complexes present more or less identical access:binding:extrusion conformations, similar to that found in apo-RE-CmeB and RE-CmeB-Amp, with all three drugs located within the binding protomer of the RE-CmeB trimer (Fig. 3D). Cip is primarily bound at the distal drug binding site, but Ery and Chl span both the proximal and distal sites. This binding mode of spanning two multidrug-binding sites is quite novel and very distinct from those seen in AcrB (45, 48), MtrDCR103 (98), and AdeJ (100, 101). This binding mechanism should empower the pump to recognize a large collection of different antimicrobials and drugs. It is also observed that each drug uses a slightly different subset of RE-CmeB residues to secure the binding, thus greatly potentiating the range of drug-pump interactions. However, residues M570, L612, F625, and L662 are consistent for Amp, Cip, Ery, and Chl binding (90).
Intriguingly, these drug-bound cryo-EM structures of RE-CmeB may represent a snapshot of the intermediate states of RE-CmeB during substrate transport, where the drug molecules are in the process of being shuttled from the proximal to distal sites during the extrusion process. The fact that some of these drugs are partially bound at the proximal site, as well as the distal site, may also indicate that there is no real boundary between these two sites. Thus, RE-CmeB can employ a large collection of residues from these two sites to optimize binding.
ESCHERICHIA COLI AcrD
AcrD is unique in that it has the capability to export physiologic substrates such as amphipathic bile acids as well as aminoglycoside-based drugs (27, 102). It appears to be more selective in the type of ligands it binds than some of the prototypical RND transporters, as their multidrug-binding sites allow them to recognize a wide range of chemical scaffolds. To further understand the substrate selectivity and transport function of this RND pump, structures of AcrD, both in the presence and absence of the aminoglycoside-based drug, gentamicin (Gen), were determined by cryo-EM (30). This work provided the first structural model as to how an RND transporter binds aminoglycoside drugs, as structural information was previously unavailable for any aminoglycoside-specific IMP. Both apo-AcrD and Gen-bound AcrD (AcrD-Gen) assemble as trimers, similar to other known structures of the RND HAE-1 transporter class (Fig. 4A). Consistent with CusA but somewhat distinct from AcrB, both TM2 and TM8 form continuous helices and extend into the periplasm, contributing to the global orientation of the PD (Fig. 4A). These two structures display a conformational state with one periplasmic cleft open and two clefts closed, with the open cleft protomer representing the binding form, while the two protomers with their periplasmic clefts closed representing the resting and extrusion forms (Fig. 4B). Global structural analysis determined that this particular asymmetric trimer is in the binding:resting:extrusion conformation, distinct from known structures of E. coli AcrB, yet similar to Form II of C. jejuni CmeB (87). This suggests that the binding:resting:extrusion trimeric conformation may be common across select RND transporters.
Fig 4.

(A) Ribbon diagram of the E. coli AcrD IMP viewed in the membrane plane (adapted from PDB ID: 8F4R). The three protomers of AcrD are colored smudge (resting protomer), light blue (binding protomer), and salmon (extrusion protomer). TM2 and TM8 are colored wheat in the binding protomer. Gen is represented as colored spheres. (B) Upper panel: top view of the periplasmic domain of AcrD, displaying two closed PC1/PC2 clefts and one open PC1/PC2 cleft. Lower panel: magnified view of the drug binding site. Interacting residues from each protomer are depicted as red and white sticks.
Within this same sample, both the dimeric form and the monomeric form of AcrD were detected. The resolution of monomeric AcrD was quite low so the overall structure was not refined. The dimeric configuration, however, was resolved to a resolution of 2.95 Å (30). This dimer displayed the extrusion:extrusion conformation, with both periplasmic clefts closed. Superimposition of the extrusion protomer of dimeric AcrD onto the extrusion form of trimeric AcrD gave rise to a root mean square deviation of 0.9 Å, suggesting that these two are similar. The main observation taken from this dimeric assembly was that the AcrD protomer:protomer interface was much tighter when compared with the interface between protomers within the fully assembled trimer. Overall, the dimeric form of AcrD likely represents an intermediate form toward the complete assembly of fully functioning AcrD (30).
AcrD bound with aminoglycoside
Several cryo-EM structures of AcrD were also determined with bound Gen (30). While all three oligomerization states were observed in the sample (trimer, dimer, and monomer), this drug was only detected within the trimeric form. AcrD-Gen also assembles as an asymmetric trimer, with the three protomers displaying as binding:resting:extrusion (Fig. 4A), the identical configuration seen in the apo-AcrD form without bound substrate. Perhaps unexpectedly, Gen was neither seen at the open PC1/PC2 periplasmic cleft in the binding protomer nor at the membrane interface. Rather, this drug was detected at the ceiling of the central cavity formed by the trimer (Fig. 4B, inset). At this position, Gen was anchored by the charged residues D99, D101, and E102 to secure binding. Interestingly, the corresponding position in AcrB has also been reported to contribute to binding drugs, where the charged residues D99 and D101 participate in anchoring drug molecules (97, 103). Additional charged residues R130, K131, D174, and D176 line this vertical tunnel, which ends at the bottom of the periplasmic funnel created by subdomains DN and DC. Mutagenesis experiments indicate that these residues are critical for E. coli cells to grow in the presence of aminoglycosides (30). Presumably, these charged residues propel the drug toward the outer membrane channel TolC for extrusion. Finally, the substrate exits the extrusion tunnel aided by the AcrD residues Q125 and Y756. Indeed, both mutagenesis and molecular dynamics simulations point to these residues as being critical for the function of this transporter (30). Interestingly, in AcrD, the D99, D101, and E102 amino acid residues are located at the beginning of the tunnel near the inner membrane surface. This extrusion tunnel is unique from those in other multidrug transporters in that aminoglycosides are transported from the central cavity vertically through the trimer for extrusion and not through the canonical periplasmic entrance, proximal binding site, distal binding site, and exit pathway.
The structural work detailed here has substantiated the ability of AcrD to bind aminoglycosides within the central cavity of the trimer. Other studies have shown that AcrD can pick up the substrate from the periplasm, likely via the open PC1/PC2 cleft displayed in the access and binding conformers, as well as from the cytoplasm (29). Interestingly, antibiotic recognition within the periplasmic domain appears to depend upon residues within the proximal binding site, as β-lactam specificity of AcrD can be transferred to AcrB with the substitution of three residues, Q569R, I626R, and E673G (104). Additionally, based on AcrB binding data (105), there is also the potential for AcrD to transport substrates from the vestibule that forms at the interface between two protomers near the membrane surface. This area is near the cationic triad D99, D101, and E102 located at the ceiling of the central cavity. Therefore, it appears that AcrD is efficient in recruiting its substrates from both the cytoplasm as well as the periplasmic space via multiple entrance sites. Driven by the PMF, it can then be funneled into the central cavity through interactions with charged residues that line the export pathway. Despite this substantial data, the complete mechanism of substrate binding and export is still incomplete. How exactly does each protomer cycle through the resting, access, binding, and extrusion states? Thus far, only the binding:resting:extrusion trimeric conformation has been elucidated in AcrD. It is clear from other work on RND transporters that there are several different transient states the trimer can adopt as it binds and exports its ligand. Is it possible that the binding of an additional ligand may help advance the transport cycle, as postulated for AcrB (17)? Characterization of AcrD with additional bound substrates will significantly help to fully understand this important aminoglycoside transporter.
KLEBSIELLA PNEUMONIAE AcrB
The Gram-negative pathogen Klebsiella pneumoniae is an opportunistic bacterium able to initiate different types of healthcare-associated infections, including pneumonia, septicemia, wound or surgical site infections, and meningitis (106). It has emerged as one of the most problematic and highly antibiotic-resistant pathogens worldwide. Like A. baumannii, K. pneumoniae is one of the most common causative agents involved in secondary bacterial infection in COVID-19 patients (107). Together with other ESKAPE bacterial pathogens, such as Enterococcus faecium, Staphylococcus aureus, and Pseudomonas aeruginosa, K. pneumoniae and A. baumannii are leading causes of nosocomial infections and present one of the greatest challenges in modern medicine. In particular, most of these bacteria have evolved to become resistant to multiple antimicrobials (108). Recently, Klebsiella pneumoniae carbapenemase (KPC)-producing K. pneumoniae has drawn wide attention, as the mortality rate for this infection can be as high as 71% (109). As a result, KPC-producing K. pneumoniae is listed as a first-priority pathogen for the research and development of new antibiotics (110).
In K. pneumoniae, the best characterized multidrug efflux system is the prevalent AcrB multidrug efflux pump (111–114). This efflux system is capable of mediating resistance to a broad spectrum of clinically relevant antimicrobial agents, including quinolones, β-lactams, tetracyclines, macrolides, aminoglycosides, and chloramphenicol (114). The K. pneumoniae AcrB (KpAcrB) multidrug efflux pump is homologous to E. coli AcrB and assembles with the KpAcrA periplasmic adaptor protein and the KpTolC outer membrane channel to form a tripartite efflux complex that spans the entire cell envelope to directly extrude drugs out of K. pneumoniae cells (114). Like E. coli AcrB, KpAcrB is a proton-motive-force-dependent efflux pump, where it provides energy to the entire K. pneumoniae AcrAB-TolC system to actively export drug molecules.
Cryo-EM structure of K. pneumoniae AcrB
To elucidate the molecular mechanisms of drug recognition and extrusion of the KpAcrB multidrug efflux pump, cryo-EM structures of this membrane protein embedded in lipidic nanodiscs, were solved, both in the absence and presence of the antibiotic Ery (Fig. 5) (115).
Fig 5.

(A) tTop view of the periplasmic domain of apo-KpAcrB (adapted from PDB ID: 8FFK), displaying two open PC1/PC2 clefts and one closed PC1/PC2 cleft. In the apo form, access:binding:extrusion is depicted in a clockwise direction. (B) Top view of the periplasmic domain of Ery-bound KpAcrB (adapted from PDB ID: 8FFS). In the substrate-bound form, access:binding:extrusion is depicted in a counterclockwise direction. The access state is colored grey, the binding state colored light blue and the extrusion state colored salmon. (C) Magnified view of Ery (green sticks) within the periplasmic binding pocket of the binding protomer, located between the flexible F- (yellow) and G-loops (orange).
KpAcrB forms an asymmetric trimer, where the overall structures of the three protomers display the access:binding:extrusion configuration (Fig. 5A). This familiar asymmetric trimeric conformational state has been detected in several RND pumps, including E. coli AcrB (45, 48), N. gonorrhoeae MtrD (98, 99), and A. baumannii AdeJ (100, 101). Each KpAcrB subunit shows distinct entrance, proximal, and distal drug-binding sites, similar to those seen in E. coli AcrB (45, 48), C. jejuni CmeB (87, 93), A. baumannii AdeB (96, 116, 117), and A. baumannii AdeJ (100, 101).
Structure of K. pneumoniae AcrB bound with erythromycin
The cryo-EM structure of KpAcrB bound with the macrolide antibiotic Ery was determined, as KpAcrB is capable of mediating resistance to this class of drugs (114). Microscale thermophoresis (118) was first used to quantify the binding affinity of KpAcrB to Ery. The equilibrium dissociation constant (KD) for KpAcrB and Ery interaction was measured to be 14.4 ± 2.6 µM (115). After confirming the specific interaction between KpAcrB and Ery, the cryo-EM structure of the KpAcrB-Ery complex was solved.
The overall structure of KpAcrB-Ery is similar to that of apo-KpAcrB, in which these two structures display an asymmetric trimeric assembly. However, a detailed inspection reveals that the conformations of these asymmetric trimers are very distinct. For instance, a top-down view of the apo-KpAcrB trimer indicates that the three protomers display conformations in the form of access:binding:extrusion as observed in a clockwise direction. However, this top-down view shows that the KpAcrB-Ery trimeric complex is arranged in the form of access:binding:extrusion in an anticlockwise direction (Fig. 5A and B) (115). Therefore, the access:binding:extrusion form of KpAcrB-Ery is observed with both clockwise and anticlockwise directionality within the trimer, distinct from the postulated rotating mechanism for AcrB and other RND IMPs that require a clockwise progression from access to binding to extrusion states. This uncoordinated directionality may also have deeper implications for its molecular mechanism, where the three KpAcrB protomers may not need to be synchronized and coordinated to export substrates. This observation is indeed in good agreement with the single-molecule FRET data of C. jejuni CmeB (87) and cryo-EM data of E. coli CusA (39), where the transport mechanisms of these efflux pump protomers function uncoordinatedly and independently of each other within the trimer.
In the cryo-EM structure of the KpAcrB pump, the Ery molecule binds deep inside the distal drug-binding site of the binding protomer (Fig. 5C). This binding mode is quite different from that was found in the crystal structure of E. coli AcrB, where the bound Ery molecule was anchored in the proximal drug-binding site, closer to the entrance of the opening of the periplasmic cleft of the E. coli AcrB pump (48). Based on the structural information, drug binding by KpAcrB seems to be mainly governed by hydrophobic interactions. This observation is indeed supported by docking calculations, where the hydrophobic residues F136, F178, and F627 are critical for binding a variety of antibiotics (115).
ACINETOBACTER BAUMANNII EFFLUX SYSTEMS
Similar to E. coli, A. baumannii contains several RND efflux systems (119), with AdeABC being the highest expressed. AdeABC and AdeIJK are the two most potent efflux systems used to eliminate a broad range of clinically relevant antibiotics from the cell (120–128). This is, in part, a central reason why multidrug-resistant A. baumannii is considered a global threat, with CRAB a first-priority pathogen (1, 129), which necessitates the development of novel antibiotic compounds and A. baumannii-specific efflux pump inhibitors to help eliminate these infections. Thus, determination of both the mode of substrate binding and the mechanistic basis of substrate transport will greatly aid in the identification of new drugs.
The adeABC locus encodes for the canonical proteins that comprise most RND efflux pump machineries: the AdeA periplasmic adaptor protein, the AdeB inner membrane pump, and the AdeC outer membrane channel. This system mediates resistance to a broad spectrum of clinically relevant antimicrobial agents, including β-lactams, tetracyclines, macrolides, fluoroquinolones, chloramphenicol, and trimethoprim (122–125). Of note, not all Acinetobacter strains carry the adeC gene (130), as it is carried by major clonal lineages of A. baumannii but few other strains (131). This suggests that AdeB alone or AdeAB together may be sufficient to export substrates, or that other components could aid in the export process. Indeed, evidence suggests that AdeK can cooperate with AdeAB to form a functional tripartite complex, albeit at a decreased efficiency when compared to the canonical AdeABC complex (132). The presence of AdeC likely increases the efficiency of this process, as it has been shown that the outer membrane channel increases the resistance levels of A. baumannii to several different antibiotics (130).
Similarly, the AdeIJK locus encodes for AdeI, AdeJ, and AdeK, the periplasmic adaptor protein, the inner membrane pump, and the outer membrane channel, respectively, together forming the AdeIJK tripartite efflux system that powerfully confers broad-spectrum resistance to β-lactams, tetracyclines, fluoroquinolones, erythromycin, and novobiocin (132–139).
ACINETOBACTER BAUMANNII AdeB
Cryo-EM performed on purified AdeB embedded in nanodiscs led to the characterization of six related but distinct trimeric structures (140), three in the apo form and three bound with one or more molecules of the AdeB substrate ethidium. These structures shed light on trimer orientation and flexibility as well as substrate binding and transport, leading to a semi-comprehensive understanding of this IMP.
Apo-AdeB shows interprotomeric plasticity
All three forms (Forms I-III) of trimeric apo-AdeB show each protomer in the extrusion form (Fig. 6A through C). Each cleft between PC1/PC2 is closed, and there is an extrusion tunnel in the periplasmic domain leading up through the exit site at the docking domain, consistent with the extrusion forms of other RND-HAE1 class of transporters (45, 51). The main difference between the three apo-AdeB forms is how each protomer packs within its trimeric orientation. Form I (AdeB-I) is packed in a similar way to the prototypical RND transporter. Each protomer tightly interacts with each other, and this appears to be facilitated by phosphatidylethanolamine (PE) lipids (Fig. 6A). Within each protomer, there is one PE located at the interior of the large central cavity, while a second is located at the protomer-protomer interface. It is likely that these PE molecules help to stabilize the overall structure by providing tight binding contacts at the subunit interface.
Fig 6.

(A–C) Ribbon diagrams of apo forms of A. baumannii AdeB IMP viewed in the membrane plane (adapted from PDB IDs: 7KGD (A), 7KGE (B), and 7KGF (C). Individual protomers are colored red, purple, and yellow. The protomer labeled with a* is designated as the central protomer and provides a reference to show orientation when the trimer is rotated 60°clockwise. PE molecules are depicted as blue and red spheres. The arrows from the central protomer show how the TM domain of the adjacent protomer swings out to form dimer + monomer and monomer + monomer + monomer orientations. IM, inner membrane and PD, periplasmic domain.
This notion is further corroborated when examining Form II and Form III of apo-AdeB. In Form II (AdeB-II), there are only three PE molecules, two in the central cavity of adjacent protomers and one at the protomer-protomer interface (Fig. 6B). The other protomer is PE deficient and is packed in a way that it appears to swing out from the other two, thus this trimeric orientation can be considered as dimer + monomer. Assembly of the pump is maintained through the docking domain. A long-arm feature formed by subdomain DN strongly coordinates with a hook-like feature of subdomain DC of the adjacent protomer, thus securely anchoring this monomeric protomer to the dimer. Within the PD, there also exists a hinge region, allowing considerable movement of the TM domains of each protomer; this generates inherent flexibility within the system. Form III (AdeB-III) can be classified as a monomer + monomer + monomer configuration (Fig. 6C). No PE is present in any of these protomers, and similar to Form II, their association is guided by interprotomeric associations originating from the PD. The three configurations, trimer, dimer + monomer, and monomer + monomer + monomer, demonstrate the dynamic nature of this transporter, and it is postulated that these forms can influence AdeA binding (140) to facilitate substrate binding and transport.
AdeB-Et structures provide insight into the transport dynamics of RND HAE-1 transporters
Three distinct structures of AdeB were determined in the presence of ethidium (AdeB-Et-I–III). Each one is unique in that while multiple ethidium molecules are present, their location and distribution within each trimer vary. This suggests that AdeB can bind and transport multiple Et molecules simultaneously within an individual protomer as well as concurrently with other protomers in the same complex.
The structure of form AdeB-Et-I depicts a trimer that has one cleft open (assigned the binding form) and two clefts closed (assigned the extrusion form). Thus, this structure is considered to represent the binding:extrusion:extrusion configuration. Within the binding conformer, there exists three Et molecules, one located at the entrance of the periplasmic cleft, one located at the distal drug binding site located approximately 20 Å above the membrane surface, and the third located directly above the second bound Et near the hydrophobic patch of the distal drug binding site (Fig. 7A) (96). The visualization of three separate Et ligands within the same channel clearly delineates a path for drug extrusion. Indeed, molecular dynamics simulations using this Et-bound form of AdeB substantiate this pathway (140). MD simulations show that drug enters through the PC1/PC2 periplasmic cleft and is shuttled through the tunnel through the proximal and distal binding sites via the highly flexible F- and G-loops, similar to what is observed in E. coli AcrB (141). Hydrophobic and aromatic interactions guide the substrate through the periplasmic space with the energetics for transport provided by the PMF of the cell. This unique form also demonstrates the plasticity of the individual protomers within the trimer, as it can tolerate three Et ligands simultaneously. It also suggests that a second ligand-bound protomer is not requisite for substrate transport, although it may help the efficiency of the transport process.
Fig 7.
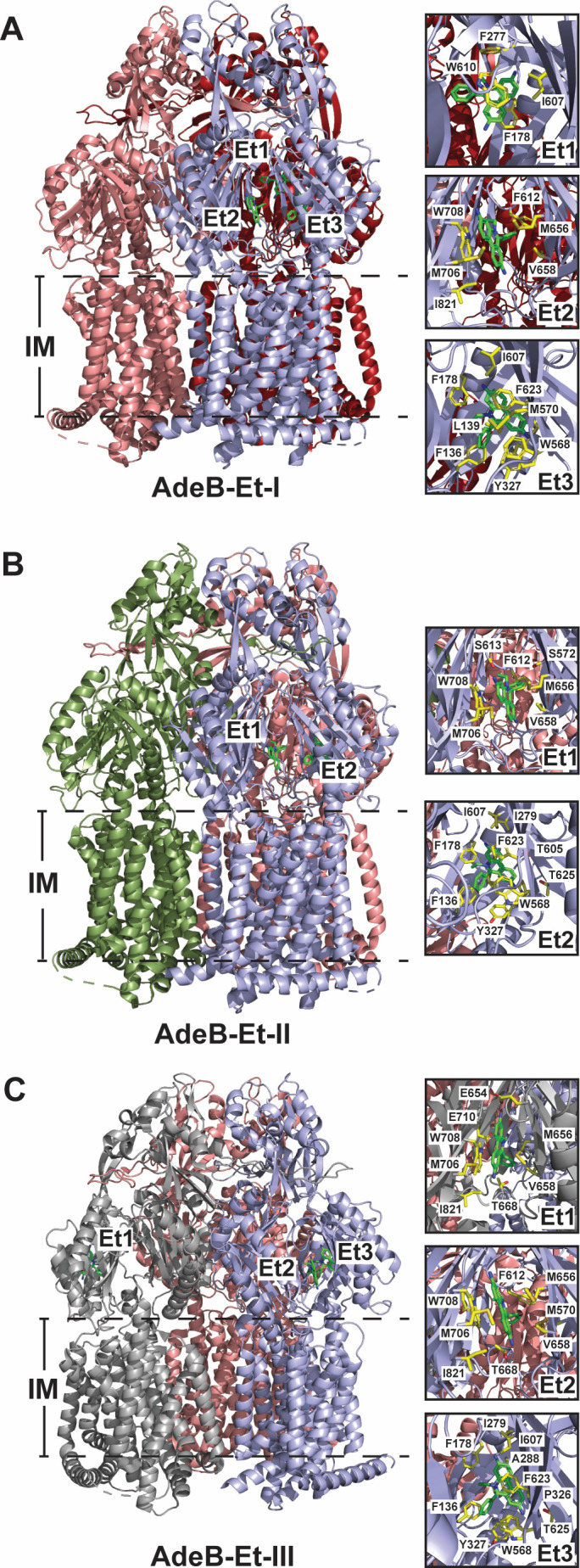
(A–C) Ribbon diagrams of Et-bound forms of A. baumannii AdeB IMP viewed in the membrane plane (adapted from PDB IDs: 7KGG (A), 7KGH (B), and 7KGI (C). For AdeB-Et-I, the individual protomers are colored in shades of red for the two extrusion forms and light blue for the binding form. AdeB-Et-II is colored smudge, light blue, and salmon to depict the resting, binding, and extrusion protomeric forms, respectively. AdeB-Et-III is colored gray, light blue, and salmon to depict the access, binding, and extrusion protomeric forms with Et ligands found in both the access and binding protomers. Et molecules are shown as green sticks located within the access and binding protomers. Right panels: residues important for Et binding at each location are highlighted as yellow sticks.
The structure of form AdeB-Et-II shows one cleft open and two clefts closed, the same as seen in form AdeB-Et-I. The main difference between the two forms is that in one of the closed cleft protomers, there is no discernable tunnel; thus, this protomer is designated as being in the resting state. The trimeric configuration of this form is binding:resting:extrusion, a configuration observed in C. jejuni CmeB (87, 93) and E. coli AcrD (30, 87). In the extrusion protomer, two Et molecules were identified, however, in slightly different locations when compared to the Et molecules in form AdeB-Et-I (Fig. 7B). Both Et molecules are bound approximately 2 Å higher with respect to the membrane surface (140). This suggests that channel flexibility will allow AdeB to accommodate larger and potentially diverse substrates, adding to the overall promiscuity of this transporter.
The third ethidium-bound form of AdeB, form AdeB-Et-III, is completely distinct from other known RND HAE-1 structures. This form has two clefts open and one cleft closed, presenting as the access:binding:extrusion trimer. However, it is unique in that Et molecules are present in two separate protomers within the trimer (Fig. 7C). In the access conformer, the Et molecule binds at the entrance drug binding site. In the adjacent binding conformer, one Et molecule is located at the entrance site, while the second Et molecule is positioned within the deep distal binding site.
Interestingly, a unique trimeric form of AdeB has recently been identified, the access*:extrusion:extrusion form. The access* protomer has a wider cleft between the PC1 and PC2 subdomains than previously observed with other AdeB structures (116). This access* form is postulated to be a conformation required for initial substrate binding or it may help to transition from the access to the binding conformer and allow the substrate to reach the deep distal binding site (116).
Taken together, the structural and computational data obtained for AdeB have significantly added to our knowledge of RND assembly and transport dynamics. AdeB is flexible. This inherent flexibility may influence assembly with the AdeA and how these two work together to allow access to the substrate at the periplasmic cleft entrance. Et-bound forms show that this transporter can bind multiple ligands simultaneously within the same protomer as well as in multiple protomers of the same trimer. This suggests that each protomer can independently bind and transport substrate simultaneously. Future studies on AdeB and others within the HAE-1 class of RND transporters are still needed to substantiate the cooperativity vs independence of each protomer within the fully assembled tripartite efflux system.
ACINETOBACTER BAUMANNII AdeJ
Tetracyclines are an important class of antibiotics because of their ability to directly bind to the bacterial ribosome (142), the complex responsible for protein synthesis inside the cell (143). Unfortunately, in A. baumannii, resistance to these compounds can be gained through the expression of the AdeIJK drug efflux system, the most prominent system responsible for conferring tetracycline resistance (20, 100, 132, 136–138). AdeIJK is a multipartite system that consists of the periplasmic adaptor protein, AdeI, the inner membrane transporter, AdeJ, and the outer membrane channel, AdeK. This complex spans both the inner and outer membranes of A. baumannii and actively removes substrates through the energetics provided by the proton motive force of the cell (138).
AdeJ uniquely binds tetracycline-based compounds
Cryo-EM structures of AdeJ, both in the absence and presence of bound drugs, have been determined (100, 101). Apo-AdeJ displays as the resting:binding:extrusion trimeric form, with global features similar to what has been found for the prototypical apo forms of RND transporters, including KpAcrB (115) and E. coli AcrB (140, 141). Notably, the entrance of the periplasmic cleft is surrounded by hydrophobic residues that likely influence substrate specificity, and four conserved residues, F178, F277, V613, and F616, form a hydrophobic patch within the distal site of AdeJ that are hypothesized to be critical residues for drug binding and export, similar to what is observed for AcrB (100).
Structure of the eravacycline-bound AdeJ transporter
The eravacycline-bound AdeJ transporter (AdeJ-Era) displays as the access:binding:extrusion form. Within the binding protomer, a large extra density was found located at the distal multidrug-binding site of this pump deep inside the periplasmic cleft (Fig. 8A). The binding of Era to AdeJ is extensive, guided by hydrophobic residues that form π-alkyl and π-π stacking interactions with the A, C, and the D rings of Era, respectively. Additionally, alkyl-alkyl and π-alkyl interactions are formed between V139, A326, and F611 and the dimethylamine group of Era while F277 stabilizes its pyrrolidinoacetamido group. This is in direct contrast to Era bound to the ribosome, which is mainly guided by electrostatic interactions (100), and thus provides the framework for structure-based drug modification to exploit these very different modes of binding.
Fig 8.

A. baumannii AdeJ bound with eravacycline (adapted from PDB ID: 7M4P). (A) Left: trimeric form viewed in the membrane plane. Individual protomers are colored gray (access), light blue (binding), and salmon (extrusion). Middle: magnified view of Era in the periplasmic binding pocket. Era, green. G-loop, orange. Important residues involved in Era binding are labeled. Right: 2D diagram of the interactions between AdeJ and Era. Amino acid residues are colored cyan. (B) A. baumannii AdeJ bound with TP-6076 (adapted from PDB ID: 7RY3). Left: trimeric form viewed in the membrane plane. Individual protomers are colored gray (access), light blue (binding), and salmon (extrusion). Middle: magnified view of TP-6076 in the periplasmic binding pocket. TP-6076 (TP), green. F-loop, yellow. G-loop, orange. Important residues involved in Era binding are labeled. Right: 2D diagram of the interactions between AdeJ and TP-6076. Amino acid residues are colored cyan.
Structure of the AdeJ transporter bound to the novel fluorocycline TP-6076
TP-6076 is a fully synthetic antibiotic initially developed by TetraPhase Pharmaceuticals and is classified as a fluorinated-hydrocarbon antibacterial (101). It inhibits protein synthesis by directly binding to the 70S ribosome and has been shown to have enhanced antibacterial activity against hard-to-treat Gram-negative and Gram-positive pathogens compared to other tetracycline-based antibiotics (117, 144). Unfortunately, the potency of TP-6076 in A. baumannii may be diminished due to the interaction with the AdeJ efflux transporter. Cryo-EM studies of AdeJ with the bound TP-6076 fluorocycline reveal that this compound also binds within the distal site of the binding promoter in the access:binding:extrusion trimeric assembly (Fig. 8B) (101). As with AdeJ-Era, binding is mainly guided by aromatic and hydrophobic interactions within the deep binding pocket. All hydrophobic residues within this pocket participate in the stabilization of both Era and TP-6076. However, TP6076 is flipped in relation to Era in the binding pocket. This allows additional electrostatic interactions between two glutamines (Q87 and Q176) and the carboxamide group at position 2 of ring A to provide additional stabilization of this compound (Fig. 8B). This also highlights the flexibility of the binding pocket that allows for the stabilization and successive transport of a variety of structurally unique hydrophobic compounds.
NEISSERIA GONORRHOEAE MtrD
Neisseria gonorrhoeae is a Gram-negative diplococcus that infects humans causing the sexually transmitted infection gonorrhea. Gonorrhea is one of the oldest described diseases, yet it remains a significant global problem, with approximately 87 million cases reported annually worldwide (145). AMR is a major concern for the treatment of gonorrhea threatening future clinical treatment regimens, especially if new antibiotics are not brought into clinical practice (146, 147). Unfortunately, the emergence of a “super drug-resistant” strain of N. gonorrhoeae has been identified (91). Infections caused by this strain are refractory to treatment by azithromycin (Azi) and ceftriaxone (Cro), two first-choice antibiotics recommended for dual treatment of gonorrhea.
In N. gonorrhoeae, the best characterized and most clinically important efflux system that mediates multidrug resistance is the multiple transferrable resistance (Mtr)CDE tripartite efflux pump (95, 148–154). This system recognizes and confers resistance to a variety of antimicrobial agents, including macrolides, β-lactams, cationic antimicrobial peptides, bile salts, dyes, and detergents (155). The mtrCDE locus consists of three tandemly linked genes encoding MtrC, MtrD, and MtrE, where all three components are absolutely required for substrate extrusion and antimicrobial resistance. This tripartite system comprises the MtrD inner membrane multidrug efflux pump, which belongs to the RND superfamily of transport proteins (42) and constitutes substrate-binding sites and a proton-relay network to generate the PMF. MtrD works in conjunction with the MtrC periplasmic adaptor protein and the MtrE outer membrane channel to actively export antimicrobials (95, 148–154). Importantly, it has been shown that overexpression of the MtrCDE multidrug efflux pump contributes significantly to clinically relevant levels of resistance to β-lactams and macrolides (155).
X-RAY STRUCTURE OF MtrD
To elucidate the mechanism used by the MtrCDE efflux system for multidrug recognition and extrusion, the crystal structure of the MtrD efflux pump from N. gonorrhoeae strain PID332, designated MtrDPID332, was determined (95).
MtrDPID332 assembles as a homotrimer (95). The three MtrDPID332 protomers are identical in structure and each cleft is open within the trimer. In comparison with the structures of AcrB, the conformation of MtrDPID332 closely resembles that of the “access” protomer of AcrB. Therefore, each MtrDPID332 protomer should represent the “access” transient state of the pump, and the MtrDPID332 trimer displays the access:access:access configuration (95). Perhaps the most distinct secondary structural feature of MtrDPID332 is TM9 when compared with other RND efflux pump structures. TM9 of MtrDPID332 contains an extended region that protrudes from the transmembrane into the periplasm. This extended helix (residues 917–927) contributes to the periplasmic domain. Protein sequence alignment suggests that these extra residues are only found in MtrD and not in other homologous RND proteins. This extra elongated helix may help the pump to transport substrates more effectively from the outer leaflet of the inner membrane to the periplasmic multidrug binding sites.
CRYO-EM STRUCTURES OF MtrD
An increasing amount of evidence suggests that transfer of DNA from commensal Neisseria spp. into the mtr locus results in multiple nucleotide changes, which escalates gonococcal resistance to clinically important antibiotics, such as Azi and Cro (155, 156). Recently, it was identified that gonococci bearing mosaic-like sequences within the mtrD gene exhibit strong linkage disequilibrium and epistatic effects capable of enhancing the activity of the pump (92). To elucidate how MtrD carrying these mosaic-like sequences elevates drug resistance and enhances transport function, cryo-EM structures of these mutated efflux pumps were determined, with the focus on full-length MtrD from the gonococcal strain CR.103, designated MtrDCR103, as this multidrug efflux pump has been shown to decrease the level of susceptibility for several antimicrobials. Two cryo-EM structures of the N. gonorrhoeae MtrDCR103 multidrug efflux pump bound with hydrolyzed Amp and Ery were solved to high resolutions (98). These structures allowed the identification of important drug-binding residues as well as modes of MtrDCR103-drug interactions.
MtrD bound with antibiotics
Unlike the X-ray structure of MtrDPID332, the cryo-EM structure of MtrDCR103 indicates that this pump forms an asymmetric trimer with the three protomers displaying three different conformational states, with the trimer adopting the access:binding:extrusion configuration (98). An extra density was found within the distal drug-binding site of the “binding” protomer of MtrDCR103, indicating that the structure of MtrDCR103 is bound by a ligand identified as a hydrolyzed Amp molecule with its four-member β-lactam ring open (Fig. 9A). Based on the structural information, the nature of protein-substrate interaction is mostly hydrophobic, where residues such as F610 and F612 of the F-loop provide hydrophobic interactions at the distal site to stabilize substrate binding. In addition, N135, S275, and T277 contribute electrostatic interactions to anchor this inactive drug. Interestingly, the positively charged residue R174 is also found within the vicinity of the Amp binding site. It is likely that the cationic side chain of this residue makes additional contacts to strengthen substrate binding.
Fig 9.

(A–C) Magnified views of the periplasmic binding pocket of N. gonorrhoeae MtrD with (A) bound Amp (adapted from PDB ID: 6VKS), (B) bound CASP peptide (CASP) (adapted from PDB ID: 8DEU), and (C) bound LL peptide (adapted from PDB ID: 8DEW). The F-loops are colored yellow, and the G-Loops are colored orange. Each ligand is depicted as green sticks (Amp and CASP) and a green helix (LL peptide). The phenylalanine side chains within the F-loop, F610, and F612 are colored orange. Residues important for substrate binding are highlighted.
The cryo-EM structure of MtrDCR103 bound with the macrolide antibiotic erythromycin was also determined. The overall structure of the MtrDCR103-Ery complex is almost identical to that of MtrDCR103-Amp, where the MtrDCR103 trimer displays the access:binding:extrusion form (98). In the binding protomer of MtrDCR103, the cryo-EM images depict that an additional large density corresponding to the bound Ery drug was located at the distal multidrug-binding site. This binding mode is very distinct from that found in the crystal structure of the E. coli AcrB-Ery complex (48). The binding of Ery in MtrDCR103 is quite extensive with residues participating in aromatic stacking and hydrophobic and electrostatic interactions to facilitate Ery binding. Many of these interaction residues are shared between the binding of Amp and Ery. However, MtrDCR103 also utilizes slightly different subsets of amino acids to anchor each individual antibiotic. This binding versatility suggests that the MtrDCR103 pump may be able to recognize a broad spectrum of antimicrobial compounds.
MtrD bound with antimicrobial peptides
In addition to mediating resistance to a variety of antibiotics, the MtrD contributes to the resistance of host-derived antimicrobial peptides, such as cathelicidins, and peptide-based antibiotics, such as polymyxins (157–161). Although there are several structures regarding how multidrug efflux pumps specifically interact with drug molecules, little information is found in relation to how RND pumps recognize and interact with antimicrobial peptides. To elucidate the molecular mechanisms of efflux pump-antimicrobial peptide interactions, cryo-EM structures of MtrDCR103 bound with three different peptides were solved.
Initially, the PhD-C7C Phage Display combinatorial peptide library (162–167) was used to select for heptapeptides that specifically bind to the purified N. gonorrhoeae MtrD protein (strain FA19). Based on this screening approach coupled with cell growth experiments, a cationic antibiotic-sensitizing peptide (CASP) that directly interacts with the MtrD multidrug efflux pump was identified. Potentially, this CASP could serve as an adjuvant to enhance the efficiency of antibiotic treatments to treat bacterial infections. The cryo-EM structure of the MtrDCR103-CASP complex was then determined in order to understand how this cyclic peptide interacts with the pump (Fig. 9B). Additionally, cryo-EM structures of MtrDCR103 bound with the cyclic antimicrobial peptide colistin (Col) (168) and the α-helical, linear peptide LL (Fig. 9C) (168) (a peptide that contains residues 17–32 of human cathelicidin LL-37) were also elucidated.
In the MtrDCR103-CASP structure (168), the MtrDCR103 trimer displays the typical access:binding:extrusion organization. An extra density corresponding to the CASP molecule is observed to anchor at the distal drug-binding site of the “binding” protomer. As many as 20 residues, including those within the entrance drug-binding site, F-loop, proximal drug-binding site, G-loop, and distal drug-binding site, participate in anchoring this peptide. Particularly, the side chains of R174 and T277 are involved in forming two hydrogen bonds with bound CASP to further secure peptide binding (Fig. 9B).
Similar to MtrDCR103-CASP, the structure of MtrDCR103-Col reveals an asymmetric trimer where the MtrDCR103 trimer exhibits the access:binding:extrusion configuration (168). Within the “binding” protomer of MtrDCR103, an additional large density corresponding to the bound Col peptide was observed. This Col-binding site is found deep inside the distal site of the pump and overlaps with that of CASP. However, it appears that MtrDCR103 utilizes a slightly different subset of residues to attach this peptide. Again, the nature of this protein-peptide interaction is mostly hydrophobic. Within 4.5 Å of this bound peptide, there are 26 residues that participate in Col binding.
As the MtrD multidrug efflux pump recognizes and confers resistance to the human antimicrobial peptide LL-37 (157), the cryo-EM structure of MtrDCR103 bound by LL that contains 16 amino acids of LL-37 (residues 17–32) was determined. LL has been shown to be sufficient to kill N. gonorrhoeae FA19 cells (169). Similar to MtrDCR103-CASP and MtrDCR103-Col, the MtrDCR103-LL complex indicates that the MtrDCR103 trimer shows the typical access:binding:extrusion conformation. Again, the bound α-helical LL peptide is found within the “binding” protomer of MtrDCR103.
On comparing the binding sites of LL, Col, and CASP, it is observed that MtrDCR103 binds Col and CASP in a very similar manner. The majority of these two cyclic peptides are located at the distal drug-binding site. However, the binding mode of the linear human cathelicidin LL peptide is quite distinct from those of Col and CASP. LL is found to span both the distal and proximal drug-binding sites, as well as part of the entrance site (Fig. 9C) (168). Binding of LL is accomplished by a collaborative effort among residues creating the entrance, proximal, distal, and hydrophobic-patched drug-binding sites. This binding is expansive and involves at least 33 residues spanning different drug-binding sites. In particular, residues D83 and S87 form two hydrogen bonds with the bound LL peptide for additional stabilization (Fig. 9C). These observations indeed highlight the cabability of MtrDCR103 to utilize different binding sites and different subsets of residues to accommodate for the binding of a variety of antimicrobial agents, which include antibiotics and antimicrobial peptides.
SUBSTRATE BINDING CHARACTERISTICS OF HAE-1 RND TRANSPORTERS
In addition to those described above, several structures, both in their apo and substrate-bound forms, of the HAE-1 class of transporters have been solved, including MexB from Pseudomonas aeruginosa (94) and BpeB from Burkholderia pseudomallei (170), further delineating how these transporters are able to recognize and bind a diverse array of substrates. E. coli AcrB has been shown to bind several different antibiotics at its proximal binding site (such as rifampicin and erythromycin) and also at its distal binding site (doxorubicin) (48), highlighting its promiscuity. Interestingly, binding sites within the transmembrane domain have also been characterized (88). Fusidic acid can bind in the PD of E. coli AcrB but has also been found in multiple sites within the transmembrane domain, including between TM1 and TM2, between TM7 and TM8, and in the deep transmembrane binding pocket (88), supporting the notion that substrate uptake can occur from the cytoplasm in addition to directly from the periplasm.
These studies and others have substantiated that the HAE-1 class of RND transporters recognizes a wide array of chemically diverse compounds and can transport them through multiple pathways. Since these efflux pumps are integral to the inherent and acquired resistance of the cell to many different antibiotics, they will continue to be extensively studied, with the overall goal of developing new inhibitors to help combat their potent resistance capabilities.
ESCHERICHIA COLI CusA—A HEAVY METAL EFFLUX RND TRANSPORTER
The HME class, however, has not been thoroughly investigated. To date, only a few structures have been reported, namely CusA from E. coli (37, 39) and ZneA from Cupriavidus metallidurans (171).
The crystal structure of E. coli CusA (37) represents the first structure of the HME class of efflux pumps. The main function of CusA is to export toxic levels of copper and silver ions from the cell (41). Although CusA is globally similar structurally to transporters of the HAE-1 class, its substrate specificity and method of transport are markedly different. This is not surprising, as its sequence homology to other HAE-1 transporters is quite low. For example, CusA and AcrB only share 19% sequence identity. Here, we describe the overall structure of CusA and its method of metal ion transport, highlighting the differences between the two classes of transporters. Several reviews are available for a more detailed structural analysis of the CusA IMP (20, 172).
As with most, if not all, trimeric RND transporters, CusA pairs with a periplasmic adaptor protein, CusB (173, 174), and an outer membrane channel, CusC (173, 174), in a ratio of 3:6:3 (57) to form the complete efflux machinery. X-ray structures depict CusA as a homotrimer. Identical to the structures of the HAE-1 transporters described above, each protomer of this HME transporter has 12 transmembrane helices and a large periplasmic domain formed by extensive loops between TM1–TM2 and TM7–TM8. The PD is also divided into six subdomains, PC1, PC2, PN1, PN2, DC1, and DC2 (Fig. 10A, left). Different from the HAE-1 class of transporters, in CusA, TM4, TM5, TM10, and TM11 extend into the cytoplasm, forming a cytoplasmic domain, while similar to what has been previously observed, TM2 and TM8 extend into the periplasmic domain (Fig. 10A) (37).
Fig 10.

(A) Ribbon diagrams of single protomers of E. coli CusA viewed in the membrane place. Apo-CusA (adapted from PDB ID: 3K07) (left) and copper ion-bound (adapted from PDB ID: 3KSS) (right). Apo CusA is in the access form and is colored gray. Cu(I)-CusA is in the binding form and is colored slate. TM2 and TM8 are colored teal to highlight their extension into the periplasmic space. Inset: amino acid residues involved in the binding of Cu(I). The methionine triad is depicted as yellow sticks. The glutamic acid residue 625 is depicted as red and white sticks. Copper ion is depicted as an orange sphere. IM, inner membrane. HH, horizontal helix. (B) Stick diagram of E. coli CusA (adapted from PDB ID: 3K07) viewed in the membrane place. The methionine pairs and the methionine triad involved in metal ion transport are labeled and depicted as colored spheres. Dotted lines represent the pathways metal ion can enter and shuttle through the transporter. IM, inner membrane.
Metal binding and transport through CusA
The X-ray structures of CusA can be classified as either the apo form with each PC1/PC2 cleft closed or the metal ion-bound form with each PC1/PC2 cleft open. The apo form represents the resting state of the transporter and the trimer can be categorized as the resting:resting:resting configuration. The ion-bound form shows either Cu(I) or Ag(I) bound to three proximal methionines, M573, M623, and M672, with additional stabilization provided by E625. This methionine triad is located directly above a horizontal helix residues 665–675 located at the bottom of the cleft (Fig. 10A, middle and right inset). This helix is absent from other transporters such as AcrB and likely helps to guide substrate specificity (39). During uptake from the periplasm, the resting state of CusA is converted to the binding state in order to accommodate its metal substrate. The PC2 subdomain significantly swings out by as much as 30°, shifting it away from the PC1 subdomain, allowing substrate entry. The horizontal helix also tilts upward by approximately 21°. This positions M672 closer to M573 and M623, forming the transient binding site (175). Since one metal ion was found in each protomer of the homotrimer, this is classified as the binding:binding:binding configuration.
Interestingly, based on the structural information, it is possible that Cu(I) or Ag(I) can also be acquired directly through the cytoplasm. Indeed, in vitro transport assays substantiate that CusA is capable of picking up ions from the cytoplasm (37). This observation is also in good agreement with previous studies with both E. coli AcrD (30) and Ralstonia sp. CH4 CzcA (67). Within the transmembrane region, there are three methionine pairs, M410-M501 (cytoplasmic side of the inner membrane), M403-M486 (middle), and M391-M1009 (periplasmic side of the inner membrane), which are somewhat evenly spaced within this TM domain (Fig. 10B). Each methionine pair is roughly 15 Å apart from each other. Cytoplasmic Cu(I) or Ag(I) can bind one of these three methionine pairs (likely M410-M501) and pass through the membrane into the periplasmic domain aided by the PMF of the cell. There, a fourth methionine pair, M271-M755, can help guide the metal ion through the central pore to the outer membrane channel CusC. Substrate entering from the periplasm through the open PC1/PC2 cleft and binding to the methionine triad can also be propelled through the central pore through this fourth methionine pair. Thus, these structures provide a mechanism for substrate export from multiple entry points. Indeed, mutagenesis studies, molecular docking, stopped-flow transport, and silver tolerance assays all lend evidence in support of this transport mechanism (39).
Cryo-EM structures of CusA help to elucidate a mechanism of metal ion export
Symmetric CusA
As observed with AdeB, cryo-EM was able to capture multiple trimeric conformations of CusA from a single sample preparation. Four distinct cryo-EM structures were determined; two symmetric structures with all PC1/PC2 clefts either open or all three closed and two novel asymmetric structures (39). The cryo-EM CusA symmetric trimer with all clefts open is nearly identical to that of the comparable X-ray structure of CusA bound with Cu(I). Therefore, this trimeric form can be classified as the binding:binding:binding configuration. In both structures, a single Cu(I) ion was found to bind to the methionine triad located within the periplasmic domain. The cryo-EM CusA symmetric trimer with three closed clefts is distinct from what was observed in the X-ray structure of apo-CusA with three closed PC1/PC2 clefts, in that an extrusion tunnel was found to extend vertically through the PD of each protomer perpendicular to the membrane surface. This homotrimer is designated as being in the extrusion:extrusion:extrusion configuration. The superimposition of an extrusion protomer onto the resting protomer resolved via X-ray crystallography gives rise to a root mean square deviation of 1.5 Å, which suggests these two protomers are quite different.
Asymmetric CusA
Interestingly, the CusA structures that were resolved from the two most abundant classes of cryo-EM particles were asymmetric trimers. The most abundant form represents the transient state with two PC1/PC2 clefts closed and one PC1/PC2 cleft open (Fig. 11A). The two protomers with their clefts closed represent the extrusion state, while the protomer with an open cleft represents the binding state. This binding protomer has one Cu(I) ion bound to the M573-M623-M672 methionine triad (Fig. 11A, inset), similar to what was seen with the comparable Cu(I)- and Ag(I)-bound CusA X-ray structures. Therefore, this asymmetric trimer represents the extrusion:extrusion:binding configuration of the transporter. The second asymmetric trimer that was observed has one PC1/PC2 cleft closed and two PC1/PC2 clefts open, with a Cu(I) ion found in the periplasmic binding pocket in these two protomers (Fig. 11B). This form can be viewed as the extrusion:binding:binding configuration. These asymmetric trimeric forms of CusA are unique to the HME class of transporters and help to determine the possible states each protomer can cycle through in order to bind, transport, and extrude substrate (Fig. 11C).
Fig 11.

(A) Top view of the periplasmic domain of the extrusion:extrusion:binding trimeric form of CusA (adapted from PDB ID: 7KF7). The extrusion protomers are colored in shades of red and the binding protomer is colored slate. Inset: magnified view of Cu(I) bound to the methionine triad. Copper ion is depicted as an orange sphere. (B) Top view of the periplasmic domain of the extrusion:binding:binding trimeric form of CusA (PDB ID: 7KF8). The extrusion protomer is colored in red and the binding protomers are colored in shades of blue. Copper ions are depicted as orange spheres. (C) Proposed efflux mechanism for CusA. B, binding protomer and E, extrusion protomer.
Mechanistic insights into CusA substrate transport
The cryo-EM structures, combined with MD simulations and previous mutagenesis studies, outline a clear mechanism of how CusA transports substrates through structural conversion of the binding conformer into the extrusion conformer. After Cu(I) or Ag(I) binds, there is considerable rearrangement in both the transmembrane helices (specifically, TM4, TM6, TM8, TM9, TM10, TM11, and TM12), as well as in the periplasmic domain. This results in the closing of the periplasmic cleft and creation of the extrusion channel required for metal ion export. In fact, MD simulations performed with Cu(I) point to this ion as being the driving force for protomer conversion. As a result of these structural changes, a reorientation of several amino acids critical for proton transport occurs. Specifically, the proton sweeper K984 is repositioned to effectively transfer a proton within the transport chain. Point mutations of the conserved residues, D405, E939, and K984, have established the importance of these three amino acids within the proton relay network, with each one vital for the transport of metal ions across the membrane. TM4 was found to have enhanced importance as it participates in both the proton relay network as well as the methionine relay network.
MD simulations also suggest that each monomer works independently; however, cooperativity may not be dismissed. Root mean square fluctuation measurements between the binding and extrusion conformers are similar in both the trimeric and monomeric simulations, suggesting that each protomer can effectively function independently (39).
Each protomer of the HME class of transporters may have a simpler transport cycle (Fig. 11C) when compared to the HAE-1 class, as there has only been one periplasmic binding site determined (the methionine triad) for CusA vs the entrance, proximal, and distal binding sites detailed for AcrB, CmeB, AcrD, and many others. The method of substrate transport may be quite different between these two RND classes of transporters. CusA, with the help of the CusB periplasmic adaptor protein, shuttles Cu(I) or Ag(I) through a methionine relay channel toward the outer membrane channel CusC powered by the PMF of the cell. In contrast, substrates that bind through the open PC1/PC2 periplasmic cleft in HAE-1 transporters stepwise move through the periplasmic tunnel through a series of conserved loops. The F-loop is part of the proximal binding site where it helps to pass substrate through the PD toward the G-loop, which compartmentalizes the proximal and distal binding sites. The substrate is then further guided to the outer membrane channel for extrusion. These loops are absent in CusA, and presumably in most, if not all HME transporters. Instead, a horizontal helix replaces the F-loop and is thought to help guide substrate specificity.
CONCLUDING REMARKS
Significant progress has been made in determining the structures and understanding the functional dynamics of RND transport systems. Through X-ray crystallography, and more recently cryo-EM, many detailed structures of IMPs alone and bound with substrates/cofactors/adaptor proteins have been solved at atomic or near atomic resolution. We are now able to understand and characterize the binding sites of antibiotics and other substrates, such as peptides and efflux pump inhibitors, and outline their path from entry to export in detail. In many cases, in the HAE-1 class of transporters such as AcrB, binding is governed by a loose but cooperative effort of both hydrophobic and polar amino acids. This, along with the relative flexibility of the PD, allows promiscuity in the chemical scaffolds that can be recognized, thus making these transport systems a powerful tool against cellular insult. In contrast, the HME class of transporters is much more selective. For CusA, only metal ions Cu(I) and Ag(I) are able to bind and be transported out of the cell. Substrate recognition is methionine-based instead of through hydrophobic and aromatic residues. Therefore, in the development of binding-site and/or proton relay network inhibitors, structure-guided design is certainly paramount.
It has been postulated that the development of physiologically safe, effective RND inhibitors may be useful as an adjunct therapy to help increase the effectiveness of commonly administered antibiotics, especially in patients with multidrug-resistant infections. However, the promiscuity of RND IMPs as well as the existence of compensatory efflux mechanisms in the bacterium makes successful inhibition a challenge. By knowing intricate details of the recognition and binding of the substrate at multiple sites (entrance site, proximal site, and distal site), through computational methods such as docking studies, MD simulations, and experimental design, compounds can be formulated with increased affinity for a particular IMP. This would allow potent inhibition of the desired target with less affinities for other export systems. In addition, it may be possible to develop a single compound that can dually function as both an IMP inhibitor and a bactericide. An example is the novel fluorocycline TP-6076 that can bind to both the ribosome as well as the RND transporter AdeJ in A. baumannii (101)
Our understanding of the functional dynamics of RND inner membrane transporters in part and the entire tripartite system as a whole is less complete. Structural studies of HAE-1 RND transporters have identified many unique trimer conformations that they can adopt as they bind and extrude substrate. Several studies performed with AcrB point to a functionally rotating mechanism. Notably, locking one protomer prohibits another from completing its access:binding:extrusion cycle, suggesting a certain synchronization must occur between each protomer within the trimer. However, FRET experiments with CmeB point to more than three transition states and show that each protomer can function independently. Both AdeB and CusA support the notion of independent protomeric function through cryo-EM studies. In addition, the directionality of the arrangement of the access, binding, and extrusion protomers can be reversed within the K. pneumoniae AcrB trimer, thus disagreeing with the presumed prerequisite of the unidirectionality of the rotating mechanism. This bidirectionality is also in good agreement with the symmetrical nature of the FRET data from the CmeB pump, implying that the protomers can rotate clockwise or anticlockwise with equal probability. Finally, not all RND transporters are trimers. HpnN from Burkholderia multivorans can assemble as a dimer (59) and MmpL3 from Mycobacterium species seem to be able to function as a monomer (61, 62), although there is a possibility that MmpL3 also functions as a trimer (176). Therefore, it is quite possible that there are mechanistic differences between species based on evolution and environmental pressures. There is still much to be discovered with these efflux systems, as their mechanistic ambiguity highlights their complexity.
ACKNOWLEDGMENTS
We would like to thank Dr. Zhemin Zhang (Assistant Professor, Department of Pharmacology, Case Western Reserve University, Cleveland, OH, USA) for his contribution to Fig. 8.
Cited work from our laboratory and the preparation of this review were supported by grants from the National Institutes of Health (R01AI145069, R01AI140669, R01AI154860, R01AI1572080, and R01AI021150).
Biographies

Philip A. Klenotic received his Ph.D. in Chemistry from The Ohio State University studying the structure and function of E. coli Signal Peptidase I. He has since held research positions in Ophthalmology and Cell Biology at the Cleveland Clinic, and Dermatology, Cardiovascular Medicine, and Pharmacology at Case Western Reserve University. His current focus as a researcher in the Yu lab is how efflux systems recognize and transport their substrates from inside the cell into the extracellular space. A better understanding of this process will help in the design of more effective inhibitors to help treat antibiotic resistance in bacteria and other infectious pathogens.

Edward W. Yu is a Full Professor of Pharmacology at Case Western Reserve University School of Medicine. He is also Co-Leader of the Molecular Mechanisms of Resistance Research Group of the Case VA CARES Center. His research program broadly focuses on the structure, assembly, and mechanisms of the resistance-nodulation-cell division (RND)-superfamily of efflux pumps as well as their regulation. These efflux systems are key components for Gram-negative pathogens to ensure their survival in toxic environments by extruding a variety of antimicrobial agents from bacterial cells. His lab has elucidated the structures of several of these important RND-type membrane proteins with the overall goal to discover novel ways to effectively inhibit their export functions.
Contributor Information
Edward W. Yu, Email: edward.w.yu@case.edu.
Corrella S. Detweiler, University of Colorado Boulder, Boulder, Colorado, USA
REFERENCES
- 1. Antibiotic resistance threats in the United States. 2019. [PubMed]
- 2. Cabello FC. 2006. Heavy use of prophylactic antibiotics in aquaculture: a growing problem for human and animal health and for the environment. Environ Microbiol 8:1137–1144. doi: 10.1111/j.1462-2920.2006.01054.x [DOI] [PubMed] [Google Scholar]
- 3. Sarmah AK, Meyer MT, Boxall ABA. 2006. A global perspective on the use, sales, exposure pathways, occurrence, fate and effects of veterinary antibiotics (VAs) in the environment. Chemosphere 65:725–759. doi: 10.1016/j.chemosphere.2006.03.026 [DOI] [PubMed] [Google Scholar]
- 4. Segura PA, François M, Gagnon C, Sauvé S. 2009. Review of the occurrence of anti-infectives in contaminated wastewaters and natural and drinking waters. Environ Health Perspect 117:675–684. doi: 10.1289/ehp.11776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Larsson DGJ, Flach CF. 2022. Antibiotic resistance in the environment. Nat Rev Microbiol 20:257–269. doi: 10.1038/s41579-021-00649-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Witte W. 1998. Medical consequences of antibiotics use in agriculture. Science 279:996–997. doi: 10.1126/science.279.5353.996 [DOI] [PubMed] [Google Scholar]
- 7. Aarestrup FM. 2005. Veterinary drug usage and antimicrobial resistance in bacteria of animal origin. Basic Clin Pharmacol Toxicol 96:271–281. doi: 10.1111/j.1742-7843.2005.pto960401.x [DOI] [PubMed] [Google Scholar]
- 8. CDC . 2022. COVID-19: U.S. impact on antimicrobial resistance, special report 2022. GA US Dep Heal Hum Serv, Atlanta. [Google Scholar]
- 9. AHA annual survey database. 2022. Available from: http://www.ahadata.com
- 10. Saier MH Jr, Reddy VS, Tamang DG, Västermark Å. 2014. The transporter classification database. Nucl Acids Res 42:D251–D258. doi: 10.1093/nar/gkt1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saier MH, Yen MR, Noto K, Tamang DG, Elkan C. 2009. The transporter classification database: recent advances. Nucleic Acids Res 37:D274–D278. doi: 10.1093/nar/gkn862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saier MH, Tran CV, Barabote RD. 2006. TCDB: The transporter classification database for membrane transport protein analyses and information. Nucleic Acids Res 34:D181–6. doi: 10.1093/nar/gkj001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Davidson AL, Chen J. 2004. ATP-binding cassette transporters in bacteria. Annu Rev Biochem 73:241–268. doi: 10.1146/annurev.biochem.73.011303.073626 [DOI] [PubMed] [Google Scholar]
- 14. Saier Jr MH, Beatty JT, Goffeau A, Harley KT, Heijne WH, Huang S-C, Jack DL, Jahn PS, Lew K, Liu J. 1999. The major facilitator superfamily. J Mol Microbiol Biotechnol 1:257–279. [PubMed] [Google Scholar]
- 15. Brown MH, Paulsen IT, Skurray RA. 1999. The multidrug efflux protein NorM is a prototype of a new family of transporters. Mol Microbiol 31:394–395. doi: 10.1046/j.1365-2958.1999.01162.x [DOI] [PubMed] [Google Scholar]
- 16. Saier MH, Tam R, Reizer A, Reizer J. 1994. Two novel families of bacterial membrane proteins concerned with nodulation, cell division and transport. Mol Microbiol 11:841–847. doi: 10.1111/j.1365-2958.1994.tb00362.x [DOI] [PubMed] [Google Scholar]
- 17. Kobylka J, Kuth MS, Müller RT, Geertsma ER, Pos KM. 2020. AcrB: a mean, keen, drug efflux machine. Ann N Y Acad Sci 1459:38–68. doi: 10.1111/nyas.14239 [DOI] [PubMed] [Google Scholar]
- 18. Morrison EA, DeKoster GT, Dutta S, Vafabakhsh R, Clarkson MW, Bahl A, Kern D, Ha T, Henzler-Wildman KA. 2012. Antiparallel EmrE exports drugs by exchanging between asymmetric structures. Nature 481:45–50. doi: 10.1038/nature10703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hassan KA, Liu Q, Elbourne LDH, Ahmad I, Sharples D, Naidu V, Chan CL, Li L, Harborne SPD, Pokhrel A, Postis VLG, Goldman A, Henderson PJF, Paulsen IT. 2018. Pacing across the membrane: the novel PACE family of efflux pumps is widespread in Gram-negative pathogens. Res Microbiol 169:450–454. doi: 10.1016/j.resmic.2018.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klenotic PA, Moseng MA, Morgan CE, Yu EW. 2021. Structural and functional diversity of resistance-nodulation-cell division transporters. Chem Rev 121:5378–5416. doi: 10.1021/acs.chemrev.0c00621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tal N, Schuldiner S. 2009. A coordinated network of transporters with overlapping Specificities provides a robust survival strategy. Proc Natl Acad Sci U S A 106:9051–9056. doi: 10.1073/pnas.0902400106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nikaido H. 2001. Preventing drug access to targets: cell surface permeability barriers and active efflux in bacteria. Semin Cell Dev Biol 12:215–223. doi: 10.1006/scdb.2000.0247 [DOI] [PubMed] [Google Scholar]
- 23. Nikaido H. 1998. The role of outer membrane and efflux pumps in the resistance of Gram-negative bacteria. Can we improve drug access? Drug Resist Updat 1:93–98. doi: 10.1016/s1368-7646(98)80023-x [DOI] [PubMed] [Google Scholar]
- 24. Zgurskaya HI, Rybenkov VV. 2020. Permeability barriers of Gram-negative pathogens. Ann N Y Acad Sci 1459:5–18. doi: 10.1111/nyas.14134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murakami S, Nakashima R, Yamashita E, Yamaguchi A. 2002. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 419:587–593. doi: 10.1038/nature01050 [DOI] [PubMed] [Google Scholar]
- 26. Das D, Xu QS, Lee JY, Ankoudinova I, Huang C, Lou Y, DeGiovanni A, Kim R, Kim SH. 2007. Crystal structure of the multidrug efflux transporter AcrB at 3.1 Å resolution reveals the N-terminal region with conserved amino acids. J Struct Biol 158:494–502. doi: 10.1016/j.jsb.2006.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rosenberg EY, Ma D, Nikaido H. 2000. AcrD of Escherichia coli is an aminoglycoside efflux pump. J Bacteriol 182:1754–1756. doi: 10.1128/JB.182.6.1754-1756.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nishino K, Yamaguchi A. 2001. Analysis of a complete library of putative drug transporter genes in Escherichia coli. J Bacteriol 183:5803–5812. doi: 10.1128/JB.183.20.5803-5812.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aires JR, Nikaido H. 2005. Aminoglycosides are captured from both periplasm and cytoplasm by the AcrD multidrug efflux transporter of Escherichia coli . J Bacteriol 187:1923–1929. doi: 10.1128/JB.187.6.1923-1929.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang Z, Morgan CE, Cui M, Yu EW. 2023. Cryo-EM structures of AcrD illuminate a mechanism for capturing aminoglycosides from its central cavity. mBio 14. doi: 10.1128/mbio.03383-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lau SY, Zgurskaya HI. 2005. Cell division defects in Escherichia coli deficient in the multidrug efflux transporter AcrEF-TolC. J Bacteriol 187:7815–7825. doi: 10.1128/JB.187.22.7815-7825.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baranova N, Nikaido H. 2002. The BaeSR two-component regulatory system activates transcription of the yegMNOB (mdtABCD) transporter gene cluster in Escherichia coli and increases its resistance to novobiocin and deoxycholate. J Bacteriol 184:4168–4176. doi: 10.1128/JB.184.15.4168-4176.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nagakubo S, Nishino K, Hirata T, Yamaguchi A. 2002. The putative response regulator BaeR stimulates multidrug resistance of Escherichia coli via a novel multidrug exporter system. J Bacteriol 184:4161–4167. doi: 10.1128/JB.184.15.4161-4167.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim HS, Nikaido H. 2012. Different functions of MdtB and MdtC subunits in the heterotrimeric efflux transporter MdtB2C complex of Escherichia coli. Biochemistry 51:4188–4197. doi: 10.1021/bi300379y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bohnert JA, Schuster S, Fähnrich E, Trittler R, Kern WV. 2007. Altered spectrum of multidrug resistance associated with a single point mutation in the Escherichia coli RND-type MDR efflux pump YhiV (MdtF). J Antimicrob Chemother 59:1216–1222. doi: 10.1093/jac/dkl426 [DOI] [PubMed] [Google Scholar]
- 36. Kobayashi N, Nishino K, Yamaguchi A. 2001. Novel macrolide-specific ABC-type efflux transporter inEscherichia coli. J Bacteriol 183:5639–5644. doi: 10.1128/JB.183.19.5639-5644.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Long F, Su C-C, Zimmermann MT, Boyken SE, Rajashankar KR, Jernigan RL, Yu EW, Edward WY. 2010. Crystal structures of the CusA efflux pump suggest methionine-mediated metal transport. Nature 467:484–488. doi: 10.1038/nature09395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Su C-C, Long F, Lei H-T, Bolla JR, Do SV, Rajashankar KR, Yu EW. 2012. Charged amino acids (R83, E567, D617, E625, R669, and K678) of CusA are required for metal ion transport in the Cus efflux system. J Mol Biol 422:429–441. doi: 10.1016/j.jmb.2012.05.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moseng MA, Lyu M, Pipatpolkai T, Glaza P, Emerson CC, Stewart PL, Stansfeld PJ, Yu EW. 2021. Cryo-EM structures of cusa reveal a mechanism of metal-ion export. mBio 12:e00452-21. doi: 10.1128/mBio.00452-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yu E. 2011. Crystal structures of the Cusa efflux pump suggest methionine mediated metal transport. Biophysical Journal 100:382a. doi: 10.1016/j.bpj.2010.12.2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nies DH. 2003. Efflux-mediated heavy metal resistance in prokaryotes. FEMS Microbiol Rev 27:313–339. doi: 10.1016/S0168-6445(03)00048-2 [DOI] [PubMed] [Google Scholar]
- 42. Tseng TT, Gratwick KS, Kollman J, Park D, Nies DH, Goffeau A, Saier MH. 1999. The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J Mol Microbiol Biotechnol 1:107–125. [PubMed] [Google Scholar]
- 43. Eicher T, Seeger MA, Anselmi C, Zhou W, Brandstätter L, Verrey F, Diederichs K, Faraldo-Gómez JD, Pos KM. 2014. Coupling of remote alternating-access transport mechanisms for protons and substrates in the multidrug efflux pump AcrB. Elife 3:e03145. doi: 10.7554/eLife.03145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Z, Fan G, Hryc CF, Blaza JN, Serysheva II, Schmid MF, Chiu W, Luisi BF, Du D. 2017. An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pump. Elife 6. doi: 10.7554/eLife.24905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Murakami S, Nakashima R, Yamashita E, Matsumoto T, Yamaguchi A. 2006. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 443:173–179. doi: 10.1038/nature05076 [DOI] [PubMed] [Google Scholar]
- 46. Seeger MA, Schiefner A, Eicher T, Verrey F, Diederichs K, Pos KM. 2006. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 313:1295–1298. doi: 10.1126/science.1131542 [DOI] [PubMed] [Google Scholar]
- 47. Sennhauser G, Amstutz P, Briand C, Storchenegger O, Grütter MG. 2006. Drug export pathway of multidrug exporter AcrB revealed by DARPin inhibitors. PLoS Biol 5:e7. doi: 10.1371/journal.pbio.0050007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nakashima R, Sakurai K, Yamasaki S, Nishino K, Yamaguchi A. 2011. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 480:565–569. doi: 10.1038/nature10641 [DOI] [PubMed] [Google Scholar]
- 49. Nikaido H, Takatsuka Y. 2009. Mechanisms of RND multidrug efflux pumps. Biochim Biophys Acta 1794:769–781. doi: 10.1016/j.bbapap.2008.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eicher T, Cha H, Seeger MA, Brandstätter L, El-Delik J, Bohnert JA, Kern WV, Verrey F, Grütter MG, Diederichs K, Pos KM. 2012. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc Natl Acad Sci U S A 109:5687–5692. doi: 10.1073/pnas.1114944109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nakashima R, Sakurai K, Yamasaki S, Hayashi K, Nagata C, Hoshino K, Onodera Y, Nishino K, Yamaguchi A. 2013. Structural basis for the inhibition of bacterial multidrug exporters. Nature 500:102–106. doi: 10.1038/nature12300 [DOI] [PubMed] [Google Scholar]
- 52. Ruggerone P, Murakami S, Pos KM, Vargiu AV. 2013. RND efflux pumps: structural information translated into function and inhibition mechanisms. Curr Top Med Chem 13:3079–3100. doi: 10.2174/15680266113136660220 [DOI] [PubMed] [Google Scholar]
- 53. Blair JMA, Smith HE, Ricci V, Lawler AJ, Thompson LJ, Piddock LJV. 2015. Expression of homologous RND efflux pump genes is dependent upon AcrB expression: implications for efflux and virulence inhibitor design. J Antimicrob Chemother 70:424–431. doi: 10.1093/jac/dku380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Piddock LJV. 2006. Clinically relevant chromosomally encoded multidrug resistance efflux pumps in bacteria. Clin Microbiol Rev 19:382–402. doi: 10.1128/CMR.19.2.382-402.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sulavik MC, Houseweart C, Cramer C, Jiwani N, Murgolo N, Greene J, DiDomenico B, Shaw KJ, Miller GH, Hare R, Shimer G. 2001. Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes. Antimicrob Agents Chemother 45:1126–1136. doi: 10.1128/AAC.45.4.1126-1136.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nichols RJ, Sen S, Choo YJ, Beltrao P, Zietek M, Chaba R, Lee S, Kazmierczak KM, Lee KJ, Wong A, Shales M, Lovett S, Winkler ME, Krogan NJ, Typas A, Gross CA. 2011. Phenotypic landscape of a bacterial cell. Cell 144:143–156. doi: 10.1016/j.cell.2010.11.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Su C-C, Long F, Zimmermann MT, Rajashankar KR, Jernigan RL, Yu EW. 2011. Crystal structure of the CusBA heavy-metal efflux complex of Escherichia coli. Nature 470:558–562. doi: 10.1038/nature09743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Du D, Wang Z, James NR, Voss JE, Klimont E, Ohene-Agyei T, Venter H, Chiu W, Luisi BF. 2014. Structure of the AcrAB-TolC multidrug efflux pump. Nature 509:512–515. doi: 10.1038/nature13205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kumar N, Su C-C, Chou T-H, Radhakrishnan A, Delmar JA, Rajashankar KR, Yu EW. 2017. Crystal structures of the Burkholderia multivorans hopanoid transporter HpnN. Proc Natl Acad Sci U S A 114:6557–6562. doi: 10.1073/pnas.1619660114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nikaido H. 2018. RND transporters in the living world. Res Microbiol 169:363–371. doi: 10.1016/j.resmic.2018.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Su C-C, Klenotic PA, Bolla JR, Purdy GE, Robinson CV, Yu EW. 2019. MmpL3 is a lipid transporter that binds trehalose monomycolate and phosphatidylethanolamine. Proc Natl Acad Sci U S A 116:11241–11246. doi: 10.1073/pnas.1901346116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang B, Li J, Yang X, Wu L, Zhang J, Yang Y, Zhao Y, Zhang L, Yang X, Yang X, Cheng X, Liu Z, Jiang B, Jiang H, Guddat LW, Yang H, Rao Z. 2019. Crystal structures of membrane transporter MmpL3, an anti-TB drug target. Cell 176:636–648. doi: 10.1016/j.cell.2019.01.003 [DOI] [PubMed] [Google Scholar]
- 63. Su CC, Klenotic PA, Cui M, Lyu M, Morgan CE, Yu EW. 2021. Structures of the mycobacterial membrane protein MmpL3 reveal its mechanism of lipid transport. PLoS Biol 19:e3001370. doi: 10.1371/journal.pbio.3001370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Adams O, Deme JC, Parker JL, Fowler PW, Lea SM, Newstead S. 2021. Cryo-EM structure and resistance landscape of M. tuberculosis MmpL3: an emergent therapeutic target. Structure 29:1182–1191. doi: 10.1016/j.str.2021.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zwama M, Yamaguchi A. 2018. Molecular mechanisms of AcrB-mediated multidrug export. Res Microbiol 169:372–383. doi: 10.1016/j.resmic.2018.05.005 [DOI] [PubMed] [Google Scholar]
- 66. Su CC, Li M, Gu R, Takatsuka Y, McDermott G, Nikaido H, Yu EW. 2006. Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J Bacteriol 188:7290–7296. doi: 10.1128/JB.00684-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Goldberg M, Pribyl T, Juhnke S, Nies DH. 1999. Energetics and topology of CzcA, a cation/proton antiporter of the resistance-nodulation-cell division protein family. J Biol Chem 274:26065–26070. doi: 10.1074/jbc.274.37.26065 [DOI] [PubMed] [Google Scholar]
- 68. Pos KM. 2009. Drug transport mechanism of the AcrB efflux pump. Biochim Biophys Acta 1794:782–793. doi: 10.1016/j.bbapap.2008.12.015 [DOI] [PubMed] [Google Scholar]
- 69. Takatsuka Y, Nikaido H. 2007. Site-directed disulfide cross-linking shows that cleft flexibility in the periplasmic domain is needed for the multidrug efflux pump AcrB of Escherichia coli. J Bacteriol 189:8677–8684. doi: 10.1128/JB.01127-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Seeger MA, von Ballmoos C, Eicher T, Brandstätter L, Verrey F, Diederichs K, Pos KM. 2008. Engineered disulfide bonds support the functional rotation mechanism of multidrug efflux pump AcrB. Nat Struct Mol Biol 15:199–205. doi: 10.1038/nsmb.1379 [DOI] [PubMed] [Google Scholar]
- 71. Vargiu AV, Ramaswamy VK, Malvacio I, Malloci G, Kleinekathöfer U, Ruggerone P. 2018. Water-mediated interactions enable smooth substrate transport in a bacterial efflux pump. Biochim Biophys Acta Gen Subj 1862:836–845. doi: 10.1016/j.bbagen.2018.01.010 [DOI] [PubMed] [Google Scholar]
- 72. Takatsuka Y, Nikaido H. 2009. Covalently linked trimer of the AcrB multidrug efflux pump provides support for the functional rotating mechanism. J Bacteriol 191:1729–1737. doi: 10.1128/JB.01441-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mishima H, Oshima H, Yasuda S, Kinoshita M. 2015. Statistical thermodynamics for functionally rotating mechanism of the multidrug efflux transporter AcrB. J Phys Chem B 119:3423–3433. doi: 10.1021/jp5120724 [DOI] [PubMed] [Google Scholar]
- 74. Lu W, Chai Q, Zhong M, Yu L, Fang J, Wang T, Li H, Zhu H, Wei Y. 2012. Assembling of AcrB trimer in cell membrane. J Mol Biol 423:123–134. doi: 10.1016/j.jmb.2012.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Yu L, Lu W, Wei Y. 2011. AcrB trimer stability and efflux activity, insight from mutagenesis studies. PLoS One 6:e28390. doi: 10.1371/journal.pone.0028390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Raimondi V, Grinzato A. 2021. A basic introduction to single particles cryo-electron microscopy. AIMS Biophys. doi: 10.3934/BIOPHY.2022002 [DOI] [Google Scholar]
- 77. Murali A. 2021. Chapter 7, Electron microscopy and single particle analysis for solving three-dimensional structures of macromolecules, p 141–154. In Molecular docking for computer-aided drug design: fundamentals, techniques, resources and applications. Academic Press. [Google Scholar]
- 78. Nagano K, Nikaido H. 2009. Kinetic behavior of the major multidrug efflux pump AcrB of Escherichia coli. Proc Natl Acad Sci U S A 106:5854–5858. doi: 10.1073/pnas.0901695106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Takatsuka Y, Chen C, Nikaido H. 2010. Mechanism of recognition of compounds of diverse structures by the multidrug efflux pump AcrB of Escherichia coli. Proc Natl Acad Sci U S A 107:6559–6565. doi: 10.1073/pnas.1001460107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ramaswamy VK, Vargiu AV, Malloci G, Dreier J, Ruggerone P. 2017. Molecular rationale behind the differential substrate specificity of bacterial RND multi-drug transporters. Sci Rep 7:8075. doi: 10.1038/s41598-017-08747-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kinana AD, Vargiu AV, Nikaido H. 2016. Effect of site-directed mutations in multidrug efflux pump AcrB examined by quantitative efflux assays. Biochem Biophys Res Commun 480:552–557. doi: 10.1016/j.bbrc.2016.10.083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Matsumoto Y, Hayama K, Sakakihara S, Nishino K, Noji H, Iino R, Yamaguchi A. 2011. Evaluation of multidrug efflux pump inhibitors by a new method using microfluidic channels. PLoS One 6:e18547. doi: 10.1371/journal.pone.0018547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Reading E, Ahdash Z, Fais C, Ricci V, Wang-Kan X, Grimsey E, Stone J, Malloci G, Lau AM, Findlay H, Konijnenberg A, Booth PJ, Ruggerone P, Vargiu AV, Piddock LJV, Politis A. 2020. Perturbed structural dynamics underlie inhibition and altered efflux of the multidrug resistance pump AcrB. Nat Commun 11. doi: 10.1038/s41467-020-19397-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lin Jun, Michel LO, Zhang Q. 2002. CmeABC functions as a multidrug efflux system in Campylobacter jejuni. Antimicrob Agents Chemother 46:2124–2131. doi: 10.1128/AAC.46.7.2124-2131.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lin J, Sahin O, Michel LO, Zhang Q. 2003. Critical role of multidrug efflux pump CmeABC in bile resistance and in vivo colonization of Campylobacter jejuni. Infect Immun 71:4250–4259. doi: 10.1128/IAI.71.8.4250-4259.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Pumbwe L, Piddock LJV. 2002. Identification and molecular characterisation of CmeB, a Campylobacter jejuni multidrug efflux pump. FEMS Microbiol Lett 206:185–189. doi: 10.1111/j.1574-6968.2002.tb11007.x [DOI] [PubMed] [Google Scholar]
- 87. Su C-C, Yin L, Kumar N, Dai L, Radhakrishnan A, Bolla JR, Lei H-T, Chou T-H, Delmar JA, Rajashankar KR, Zhang Q, Shin Y-K, Yu EW. 2017. Structures and transport dynamics of a Campylobacter jejuni multidrug efflux pump. Nat Commun 8:1–11. doi: 10.1038/s41467-017-00217-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Tam HK, Foong WE, Oswald C, Herrmann A, Zeng H, Pos KM. 2021. Allosteric drug transport mechanism of multidrug transporter AcrB. Nat Commun 12. doi: 10.1038/s41467-021-24151-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Blair JMA, Bavro VN, Ricci V, Modi N, Cacciotto P, Kleinekathӧfer U, Ruggerone P, Vargiu AV, Baylay AJ, Smith HE, Brandon Y, Galloway D, Piddock LJV. 2015. AcrB drug-binding pocket substitution confers clinically relevant resistance and altered substrate specificity. Proc Natl Acad Sci U S A 112:3511–3516. doi: 10.1073/pnas.1419939112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Yao H, Shen Z, Wang Y, Deng F, Liu D, Naren G, Dai L, Su CC, Wang B, Wang S, Wu C, Yu EW, Zhang Q, Shen J. 2016. Emergence of a potent multidrug efflux pump variant that enhances Campylobacter resistance to multiple antibiotics. mBio 7:e01543-16. doi: 10.1128/mBio.01543-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Smolarchuk C, Wensley A, Padfield S, Fifer H, Lee A, Hughes G. 2018. Persistence of an outbreak of gonorrhoea with high-level resistance to azithromycin in England, November 2014‒May 2018. Euro Surveill 23:1800287. doi: 10.2807/1560-7917.ES.2018.23.23.1800287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Wadsworth CB, Arnold BJ, Sater MRA, Grad YH. 2018. Azithromycin resistance through interspecific acquisition of an epistasis-dependent efflux pump component and transcriptional regulator in Neisseria gonorrhoeae. mBio 9:e01419-18. doi: 10.1128/mBio.01419-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zhang Z, Lizer N, Wu Z, Morgan CE, Yan Y, Zhang Q, Yu EW. 2023. Cryo-electron microscopy structures of a Campylobacter multidrug efflux pump reveal a novel mechanism of drug recognition and resistance. Microbiol Spectr 11. doi: 10.1128/spectrum.01197-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Sennhauser G, Bukowska MA, Briand C, Grütter MG. 2009. Crystal structure of the multidrug exporter MexB from Pseudomonas aeruginosa. J Mol Biol 389:134–145. doi: 10.1016/j.jmb.2009.04.001 [DOI] [PubMed] [Google Scholar]
- 95. Bolla JR, Su C-C, Do SV, Radhakrishnan A, Kumar N, Long F, Chou T-H, Delmar JA, Lei H-T, Rajashankar KR, Shafer WM, Yu EW. 2014. Crystal structure of the Neisseria gonorrhoeae MtrD inner membrane multidrug efflux pump. PLoS One 9:e97903. doi: 10.1371/journal.pone.0097903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Su C-C, Morgan CE, Kambakam S, Rajavel M, Scott H, Huang W, Emerson CC, Taylor DJ, Stewart PL, Bonomo RA, Yu EW. 2019. Cryo-electron microscopy structure of an Acinetobacter baumannii multidrug efflux pump. mBio 10:e01295-19. doi: 10.1128/mBio.01295-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Yu EW, McDermott G, Zgurskaya HI, Nikaido H, Koshland DE. 2003. Structural basis of multiple drug-binding capacity of the AcrB multidrug efflux pump. Science 300:976–980. doi: 10.1126/science.1083137 [DOI] [PubMed] [Google Scholar]
- 98. Lyu M, Moseng MA, Reimche JL, Holley CL, Dhulipala V, Su C-C, Shafer WM, Yu EW. 2020. Cryo-EM structures of a gonococcal multidrug efflux pump illuminate a mechanism of drug recognition and resistance. mBio 11. doi: 10.1128/mBio.00996-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lyu M, Ayala JC, Chirakos I, Su C-C, Shafer WM, Yu EW. 2022. Structural basis of peptide-based antimicrobial inhibition of a resistance-nodulation-cell division multidrug efflux pump. Microbiol Spectr 10:e0299022. doi: 10.1128/spectrum.02990-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zhang Z, Morgan CE, Bonomo RA, Yu EW. 2021. Cryo-EM determination of eravacycline-bound structures of the ribosome and the multidrug efflux pump AdeJ of Acinetobacter baumannii. mBio 12. doi: 10.1128/mBio.01031-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Morgan CE, Zhang Z, Bonomo RA, Yu EW. 2022. An analysis of the novel fluorocycline TP-6076 bound to both the ribosome and multidrug efflux pump AdeJ from Acinetobacter baumannii. mBio 13. doi: 10.1128/mbio.03732-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Elkins CA, Nikaido H. 2002. Substrate specificity of the RND-type multidrug efflux pumps AcrB and AcrD of Escherichia coli is determined predominately by two large periplasmic loops. J Bacteriol 184:6490–6498. doi: 10.1128/JB.184.23.6490-6499.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Yu EW, Aires JR, McDermott G, Nikaido H. 2005. A periplasmic drug-binding site of the AcrB multidrug efflux pump: a crystallographic and site-directed mutagenesis study. J Bacteriol 187:6804–6815. doi: 10.1128/JB.187.19.6804-6815.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Kobayashi N, Tamura N, van Veen HW, Yamaguchi A, Murakami S. 2014. βLactam selectivity of multidrug transporters AcrB and AcrD resides in the proximal binding pocket. J Biol Chem 289:10680–10690. doi: 10.1074/jbc.M114.547794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Husain F, Bikhchandani M, Nikaido H. 2011. Vestibules are part of the substrate path in the multidrug efflux transporter AcrB of Escherichia coli. J Bacteriol 193:5847–5849. doi: 10.1128/JB.05759-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Alebachew Woldu M. 2016. Klebsiella pneumoniae and its growing concern in healthcare settings. Clin Exp Pharmacol 06. doi: 10.4172/2161-1459.1000199 [DOI] [Google Scholar]
- 107. Pourajam S, Kalantari E, Talebzadeh H, Mellali H, Sami R, Soltaninejad F, Amra B, Sajadi M, Alenaseri M, Kalantari F, Solgi H. 2022. Secondary bacterial infection and clinical characteristics in patients with COVID-19 admitted to two intensive care units of an academic hospital in Iran during the first wave of the pandemic. Front Cell Infect Microbiol 12:784130. doi: 10.3389/fcimb.2022.784130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Santajit S, Indrawattana N. 2016. Mechanisms of antimicrobial resistance in ESKAPE pathogens. Biomed Res Int 2016:2475067. doi: 10.1155/2016/2475067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Micozzi A, Gentile G, Santilli S, Minotti C, Capria S, Moleti ML, Barberi W, Cartoni C, Trisolini SM, Testi AM, Iori AP, Bucaneve G, Foà R. 2021. Reduced mortality from KPC-K.pneumoniae bloodstream infection in high-risk patients with hematological malignancies colonized by KPC-K.pneumoniae. BMC Infect Dis 21:1079. doi: 10.1186/s12879-021-06747-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Tacconelli E, Carrara E, Savoldi A, Harbarth S, Mendelson M, Monnet DL, Pulcini C, Kahlmeter G, Kluytmans J, Carmeli Y, et al. 2018. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis 18:318–327. doi: 10.1016/S1473-3099(17)30753-3 [DOI] [PubMed] [Google Scholar]
- 111. Fang CT, Chen HC, Chuang YP, Chang SC, Wang JT. 2002. Cloning of a cation efflux pump gene associated with chlorhexidine resistance in Klebsiella pneumoniae. Antimicrob Agents Chemother 46:2024–2028. doi: 10.1128/AAC.46.6.2024-2028.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Li DW, Onishi M, Kishino T, Matsuo T, Ogawa W, Kuroda T, Tsuchiya T. 2008. Properties and expression of a multidrug efflux pump AcrAB-KocC from Klebsiella pneumoniae. Biol Pharm Bull 31:577–582. doi: 10.1248/bpb.31.577 [DOI] [PubMed] [Google Scholar]
- 113. Ni RT, Onishi M, Mizusawa M, Kitagawa R, Kishino T, Matsubara F, Tsuchiya T, Kuroda T, Ogawa W. 2020. The role of RND-type efflux pumps in multidrug-resistant mutants of Klebsiella pneumoniae. Sci Rep 10. doi: 10.1038/s41598-020-67820-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Padilla E, Llobet E, Doménech-Sánchez A, Martínez-Martínez L, Bengoechea JA, Albertí S. 2010. Klebsiella pneumoniae AcrAB efflux pump contributes to antimicrobial resistance and virulence. Antimicrob Agents Chemother 54:177–183. doi: 10.1128/AAC.00715-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Zhang Z, Morgan CE, Bonomo RA, Yu EW. 2023. Cryo-EM structures of the Klebsiella pneumoniae AcrB multidrug efflux pump. mBio 14:e0065923. doi: 10.1128/mbio.00659-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ornik-Cha A, Wilhelm J, Kobylka J, Sjuts H, Vargiu AV, Malloci G, Reitz J, Seybert A, Frangakis AS, Pos KM. 2021. Structural and functional analysis of the promiscuous AcrB and AdeB efflux pumps suggests different drug binding mechanisms. Nat Commun 12:6919. doi: 10.1038/s41467-021-27146-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Falagas ME, Skalidis T, Vardakas KZ, Voulgaris GL, Papanikolaou G, Legakis N, Hellenic TP-6076 Study Group . 2018. Activity of TP-6076 against carbapenem-resistant Acinetobacter baumannii isolates collected from inpatients in Greek hospitals. Int J Antimicrob Agents 52:269–271. doi: 10.1016/j.ijantimicag.2018.03.009 [DOI] [PubMed] [Google Scholar]
- 118. Wienken CJ, Baaske P, Rothbauer U, Braun D, Duhr S. 2010. Protein-binding assays in biological liquids using microscale thermophoresis. Nat Commun 1:100. doi: 10.1038/ncomms1093 [DOI] [PubMed] [Google Scholar]
- 119. Verma P, Tiwari M, Tiwari V. 2021. Efflux pumps in multidrug-resistant Acinetobacter baumannii: current status and challenges in the discovery of efflux pumps inhibitors. Microb Pathogen 152:104766. doi: 10.1016/j.micpath.2021.104766 [DOI] [PubMed] [Google Scholar]
- 120. Gordon NC, Wareham DW. 2010. Multidrug-resistant Acinetobacter baumannii: mechanisms of virulence and resistance. Int J Antimicrob Agents 35:219–226. doi: 10.1016/j.ijantimicag.2009.10.024 [DOI] [PubMed] [Google Scholar]
- 121. Coyne S, Rosenfeld N, Lambert T, Courvalin P, Périchon B. 2010. Overexpression of resistance-nodulation-cell division pump AdeFGH confers multidrug resistance in Acinetobacter baumannii. Antimicrob Agents Chemother 54:4389–4393. doi: 10.1128/AAC.00155-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Magnet S, Courvalin P, Lambert T. 2001. Resistance-nodulation-cell division-type efflux pump involved in aminoglycoside resistance in Acinetobacter baumannii strain BM4454. Antimicrob Agents Chemother 45:3375–3380. doi: 10.1128/AAC.45.12.3375-3380.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Higgins PG, Wisplinghoff H, Stefanik D, Seifert H. 2004. Selection of topoisomerase mutations and overexpression of adeB mRNA transcripts during an outbreak of Acinetobacter baumannii. J Antimicrob Chemother 54:821–823. doi: 10.1093/jac/dkh427 [DOI] [PubMed] [Google Scholar]
- 124. Ruzin A, Keeney D, Bradford PA. 2007. AdeABC multidrug efflux pump is associated with decreased susceptibility to tigecycline in Acinetobacter calcoaceticus-Acinetobacter baumannii complex. J Antimicrob Chemother 59:1001–1004. doi: 10.1093/jac/dkm058 [DOI] [PubMed] [Google Scholar]
- 125. Gallagher LA, Ramage E, Weiss EJ, Radey M, Hayden HS, Held KG, Huse HK, Zurawski DV, Brittnacher MJ, Manoil C. 2015. Resources for genetic and genomic analysis of emerging pathogen Acinetobacter baumannii. J Bacteriol 197:2027–2035. doi: 10.1128/JB.00131-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Peleg AY, Adams J, Paterson DL. 2007. Tigecycline efflux as a mechanism for nonsusceptibility in Acinetobacter baumannii. Antimicrob Agents Chemother 51:2065–2069. doi: 10.1128/AAC.01198-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sun J-R, Chan M-C, Chang T-Y, Wang W-Y, Chiueh T-S. 2010. Overexpression of the adeB gene in clinical isolates of tigecycline-nonsusceptible Acinetobacter baumannii without insertion mutations in adeRS. Antimicrob Agents Chemother 54:4934–4938. doi: 10.1128/AAC.00414-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lari AR, Ardebili A, Hashemi A. 2018. Ader-ades mutations & overexpression of the AdeABC efflux system in ciprofloxacin-resistant Acinetobacter baumannii clinical isolates. Indian J Med Res 147:413. doi: 10.4103/ijmr.IJMR_644_16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kim YJ, Kim SI, Kim YR, Hong KW, Wie SH, Park YJ, Jeong H, Kang MW. 2012. Carbapenem-resistant Acinetobacter baumannii: diversity of resistant mechanisms and risk factors for infection. Epidemiol Infect 140:137–145. doi: 10.1017/S0950268811000744 [DOI] [PubMed] [Google Scholar]
- 130. Wieczorek P, Sacha P, Czaban S, Hauschild T, Ojdana D, Kowalczuk O, Milewski R, Poniatowski B, Nikliński J, Tryniszewska E. 2013. Distribution of AdeABC efflux system genes in genotypically diverse strains of clinical Acinetobacter baumannii. Diagn Microbiol Infect Dis 77:106–109. doi: 10.1016/j.diagmicrobio.2013.06.017 [DOI] [PubMed] [Google Scholar]
- 131. Naidu V, Bartczak A, Brzoska AJ, Lewis P, Eijkelkamp BA, Paulsen IT, Elbourne LDH, Hassan KA. 2023. Evolution of RND efflux pumps in the development of a successful pathogen. Drug Resist Updat 66:100911. doi: 10.1016/j.drup.2022.100911 [DOI] [PubMed] [Google Scholar]
- 132. Sugawara E, Nikaido H. 2014. Properties of AdeABC and AdeIJK efflux systems of Acinetobacter baumannii compared with those of the AcrAB-TolC system of Escherichia coli. Antimicrob Agents Chemother 58:7250–7257. doi: 10.1128/AAC.03728-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Leus IV, Weeks JW, Bonifay V, Smith L, Richardson S, Zgurskaya HI. 2018. Substrate specificities and efflux efficiencies of RND efflux pumps of Acinetobacter baumannii. J Bacteriol 200:e00049-18. doi: 10.1128/JB.00049-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Rosenfeld N, Bouchier C, Courvalin P, Périchon B. 2012. Expression of the resistance-nodulation-cell division pump AdeIJK in Acinetobacter baumannii is regulated by AdeN, a TetR-type regulator. Antimicrob Agents Chemother 56:2504–2510. doi: 10.1128/AAC.06422-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Fernando DM, Xu W, Loewen PC, Zhanel GG, Kumar A. 2014. Triclosan can select for an AdeIJK-overexpressing mutant of Acinetobacter baumannii ATCC 17978 that displays reduced susceptibility to multiple antibiotics. Antimicrob Agents Chemother 58:6424–6431. doi: 10.1128/AAC.03074-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Li H, Wang X, Zhang Y, Zhao C, Chen H, Jiang S, Zhang F, Wang H. 2015. The role of RND efflux pump and global regulators in tigecycline resistance in clinical Acinetobacter baumannii isolates. Future Microbiol 10:337–346. doi: 10.2217/fmb.15.7 [DOI] [PubMed] [Google Scholar]
- 137. Yoon EJ, Chabane YN, Goussard S, Snesrud E, Courvalin P, Dé E, Grillot-Courvalin C. 2015. Contribution of resistance-nodulation-cell division efflux systems to antibiotic resistance and biofilm formation in Acinetobacter baumannii. mBio 6:e00309-15. doi: 10.1128/mBio.00309-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Damier-Piolle L, Magnet S, Brémont S, Lambert T, Courvalin P. 2008. AdeIJK, a resistance-nodulation-cell division pump effluxing multiple antibiotics in Acinetobacter baumannii. Antimicrob Agents Chemother 52:557–562. doi: 10.1128/AAC.00732-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Yoon EJ, Balloy V, Fiette L, Chignard M, Courvalin P, Grillot-Courvalin C. 2016. Contribution of the ade resistance-nodulation-cell division-type efflux pumps to fitness and pathogenesis of Acinetobacter baumannii. mBio 7:e00697-16. doi: 10.1128/mBio.00697-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Morgan CE, Glaza P, Leus IV, Trinh A, Su C-C, Cui M, Zgurskaya HI, Yu EW. 2021. Cryoelectron microscopy structures of adeb illuminate mechanisms of simultaneous binding and exporting of substrates. mBio 12. doi: 10.1128/mBio.03690-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Vargiu AV, Nikaido H. 2012. Multidrug binding properties of the AcrB efflux pump characterized by molecular dynamics simulations. Proc Natl Acad Sci U S A 109:20637–20642. doi: 10.1073/pnas.1218348109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Nguyen F, Starosta AL, Arenz S, Sohmen D, Dönhöfer A, Wilson DN. 2014. Tetracycline antibiotics and resistance mechanisms. Biol Chem 395:559–575. doi: 10.1515/hsz-2013-0292 [DOI] [PubMed] [Google Scholar]
- 143. Steitz TA. 2008. A structural understanding of the dynamic ribosome machine. Nat Rev Mol Cell Biol 9:242–253. doi: 10.1038/nrm2352 [DOI] [PubMed] [Google Scholar]
- 144. Seifert H, Stefanik D, Olesky M, Higgins PG. 2020. In vitro activity of the novel fluorocycline TP-6076 against carbapenem-resistant Acinetobacter baumannii. Int J Antimicrob Agents 55:105829. doi: 10.1016/j.ijantimicag.2019.10.010 [DOI] [PubMed] [Google Scholar]
- 145. Quillin SJ, Seifert HS. 2018. Neisseria gonorrhoeae host adaptation and pathogenesis. Nat Rev Microbiol 16:226–240. doi: 10.1038/nrmicro.2017.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Unemo M, Shafer WM. 2014. Antimicrobial resistance in Neisseria gonorrhoeae in the 21st century: past, evolution, and future. Clin Microbiol Rev 27:587–613. doi: 10.1128/CMR.00010-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Newman L, Rowley J, Vander Hoorn S, Wijesooriya NS, Unemo M, Low N, Stevens G, Gottlieb S, Kiarie J, Temmerman M. 2015. Global estimates of the prevalence and incidence of four curable sexually transmitted infections in 2012 based on systematic review and global reporting. PLoS One 10:e0143304. doi: 10.1371/journal.pone.0143304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hagman KE, Shafer WM. 1995. Transcriptional control of the mtr efflux system of Neisseria gonorrhoeae. J Bacteriol 177:4162–4165. doi: 10.1128/jb.177.14.4162-4165.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Warner DM, Shafer WM, Jerse AE. 2008. Clinically relevant mutations that cause derepression of the Neisseria gonorrhoeae MtrC-MtrD-MtrE Efflux pump system confer different levels of antimicrobial resistance and in vivo fitness. Mol Microbiol 70:462–478. doi: 10.1111/j.1365-2958.2008.06424.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Veal WL, Shafer WM. 2003. Identification of a cell envelope protein (MtrF) involved in hydrophobic antimicrobial resistance in Neisseria gonorrhoeae. J Antimicrob Chemother 51:27–37. doi: 10.1093/jac/dkg031 [DOI] [PubMed] [Google Scholar]
- 151. Delahay RM, Robertson BD, Balthazar JT, Shafer WM, Ison CA. 1997. Involvement of the gonococcal MtrE protein in the resistance of Neisseria gonorrhoeae to toxic hydrophobic agents. Microbiology (Reading) 143 (Pt 7):2127–2133. doi: 10.1099/00221287-143-7-2127 [DOI] [PubMed] [Google Scholar]
- 152. Lucas CE, Hagman KE, Levin JC, Stein DC, Shafer WM. 1995. Importance of lipooligosaccharide structure in determining gonococcal resistance to hydrophobic antimicrobial agents resulting from the mtr efflux system. Mol Microbiol 16:1001–1009. doi: 10.1111/j.1365-2958.1995.tb02325.x [DOI] [PubMed] [Google Scholar]
- 153. Lei H-T, Chou T-H, Su C-C, Bolla JR, Kumar N, Radhakrishnan A, Long F, Delmar JA, Do SV, Rajashankar KR, Shafer WM, Yu EW. 2014. Crystal structure of the open state of the Neisseria gonorrhoeae MtrE outer membrane channel. PLoS One 9:e97475. doi: 10.1371/journal.pone.0097475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Hagman KE, Lucas CE, Balthazar JT, Snyder L, Nilles M, Judd RC, Shafer WM. 1997. The MtrD protein of Neisseria gonorrhaeae is a member of the resistance/nodulation/division protein family constituting part of an efflux system. Microbiology (Reading) 143:2117–2125. doi: 10.1099/00221287-143-7-2117 [DOI] [PubMed] [Google Scholar]
- 155. Rouquette-Loughlin CE, Reimche JL, Balthazar JT, Dhulipala V, Gernert KM, Kersh EN, Pham CD, Pettus K, Abrams AJ, Trees DL, St Cyr S, Shafer WM. 2018. Mechanistic basis for decreased antimicrobial susceptibility in a clinical isolate of Neisseria gonorrhoeae possessing a mosaic-like mtr efflux pump locus . mBio 9:1–15. doi: 10.1128/mBio.02281-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Shafer WM. 2018. Mosaic drug efflux gene sequences from commensal neisseria can lead to low-level azithromycin resistance expressed by Neisseria gonorrhoeae clinical isolates . mBio 9. doi: 10.1128/mBio.01747-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Tzeng YL, Ambrose KD, Zughaier S, Zhou X, Miller YK, Shafer WM, Stephens DS. 2005. Cationic antimicrobial peptide resistance in Neisseria meningitidis . J Bacteriol 187:5387–5396. doi: 10.1128/JB.187.15.5387-5396.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Tzeng YL, Berman Z, Toh E, Bazan JA, Turner AN, Retchless AC, Wang X, Nelson DE, Stephens DS. 2019. Heteroresistance to the model antimicrobial peptide polymyxin B in the emerging Neisseria meningitidis lineage 11.2 urethritis clade: mutations in the pilMNOPQ operon. Mol Microbiol 111:254–268. doi: 10.1111/mmi.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Cox DL, Sun Y, Liu H, Lehrer RI, Shafer WM. 2003. Susceptibility of Treponema pallidum to host-derived antimicrobial peptides. Peptides 24:1741–1746. doi: 10.1016/j.peptides.2003.07.026 [DOI] [PubMed] [Google Scholar]
- 160. Chitsaz M, Booth L, Blyth MT, O’Mara ML, Brown MH. 2019. Multidrug resistance in Neisseria gonorrhoeae: identification of functionally important residues in the MtrD efflux protein. mBio 10:e02277-19. doi: 10.1128/mBio.02277-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Shafer WM, Qu XD, Waring AJ, Lehrer RI. 1998. Modulation of Neisseria gonorrhoeae susceptibility to vertebrate antibacterial peptides due to a member of the resistance/nodulation/division efflux pump family. Proc Natl Acad Sci U S A 95:1829–1833. doi: 10.1073/pnas.95.4.1829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Parmley SF, Smith GP. 1988. Antibody-selectable filamentous fd phage vectors: affinity purification of target genes. Gene 73:305–318. doi: 10.1016/0378-1119(88)90495-7 [DOI] [PubMed] [Google Scholar]
- 163. Smith GP, Scott JK. 1993. Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol 217:228–257. doi: 10.1016/0076-6879(93)17065-d [DOI] [PubMed] [Google Scholar]
- 164. Cortese R, Monaci P, Nicosia A, Luzzago A, Felici F, Galfré G, Pessi A, Tramontano A, Sollazzo M. 1995. Identification of biologically active peptides using random libraries displayed on phage. Curr Opin Biotechnol 6:73–80. doi: 10.1016/0958-1669(95)80012-3 [DOI] [PubMed] [Google Scholar]
- 165. Scott JK, Smith GP. 1990. Searching for peptide ligands with an epitope library. Science 249:386–390. doi: 10.1126/science.1696028 [DOI] [PubMed] [Google Scholar]
- 166. Cwirla SE, Peters EA, Barrett RW, Dower WJ. 1990. Peptides on phage: a vast library of peptides for identifying ligands. Proc Natl Acad Sci U S A 87:6378–6382. doi: 10.1073/pnas.87.16.6378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Devlin JJ, Panganiban LC, Devlin PE. 1990. Random peptide libraries: a source of specific protein binding molecules. Science 249:404–406. doi: 10.1126/science.2143033 [DOI] [PubMed] [Google Scholar]
- 168. Lyu M, Su C-C, Kazura JW, Yu EW. 2021. Structural basis of transport and inhibition of the Plasmodium falciparum transporter PfFNT. EMBO Rep 22:e51628. doi: 10.15252/embr.202051628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Zughaier SM, Svoboda P, Pohl J, Stephens DS, Shafer WM. 2010. The human host defense peptide LL-37 interacts with Neisseria meningitidis capsular polysaccharides and inhibits inflammatory mediators release. PLoS One 5:e13627. doi: 10.1371/journal.pone.0013627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Kato T, Okada U, Hung LW, Yamashita E, Kim HB, Kim CY, Terwilliger TC, Schweizer HP, Murakami S. 2023. Crystal structures of multidrug efflux transporters from Burkholderia pseudomallei suggest details of transport mechanism. Proc Natl Acad Sci U S A 120:e2215072120. doi: 10.1073/pnas.2215072120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. De Angelis F, Lee JK, O’Connell JD 3rd, Miercke LJW, Verschueren KH, Srinivasan V, Bauvois C, Govaerts C, Robbins RA, Ruysschaert J-M, Stroud RM, Vandenbussche G. 2010. Metal-induced conformational changes in ZneB suggest an active role of membrane fusion proteins in efflux resistance systems. Proc Natl Acad Sci U S A 107:11038–11043. doi: 10.1073/pnas.1003908107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Moseng MA, Lyu M, Pipatpolkai T, Glaza P, Emerson CC, Stewart PL, Stansfeld PJ, Yu EW. 2021. Cryo-EM structures of CusA reveal a mechanism of metal-ion export. mBio 12:e00452-21. doi: 10.1128/mBio.00452-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Franke S, Grass G, Nies DH. 2001. The product of the ybdE gene of the Escherichia coli chromosome is involved in detoxification of silver ions. Microbiology (Reading) 147:965–972. doi: 10.1099/00221287-147-4-965 [DOI] [PubMed] [Google Scholar]
- 174. Franke S, Grass G, Rensing C, Nies DH. 2003. Molecular analysis of the copper-transporting efflux system CusCFBA of Escherichia coli.. J Bacteriol 185:3804–3812. doi: 10.1128/JB.185.13.3804-3812.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Banci L, Bertini I, Cantini F, Felli IC, Gonnelli L, Hadjiliadis N, Pierattelli R, Rosato A, Voulgaris P. 2006. The Atx1-Ccc2 complex is a metal-mediated protein-protein interaction. Nat Chem Biol 2:367–368. doi: 10.1038/nchembio797 [DOI] [PubMed] [Google Scholar]
- 176. Stevens CM, Babii SO, Pandya AN, Li W, Li Y, Mehla J, Scott R, Hegde P, Prathipati PK, Acharya A, Liu J, Gumbart JC, North J, Jackson M, Zgurskaya HI. 2022. Proton transfer activity of the reconstituted Mycobacterium tuberculosis MmpL3 is modulated by substrate mimics and inhibitors. Proc Natl Acad Sci U S A 119:e2113963119. doi: 10.1073/pnas.2113963119 [DOI] [PMC free article] [PubMed] [Google Scholar]