

Summary
Study of disease-relevant immune cells, namely monocytes and macrophages, is limited based on availability of primary tissue, a limitation that can be remedied using human induced pluripotent stem cell (hiPSC) technology. Here, we present a protocol for differentiation of monocytes and macrophages from hiPSCs. We describe steps for hiPSC maintenance, mesoderm lineage induction, hematopoietic progenitor cells (HPCs) commitment and expansion, and myeloid lineage induction. We then detail procedures for monocyte formation and functional macrophage formation and polarization.
For complete details on the use and execution of this protocol, please refer to Chen et al.1
Subject areas: Cell Biology, Flow Cytometry, Immunology, Microscopy, Stem Cells, Cell Differentiation
Graphical abstract
Highlights
-
•
Simple monolayer-based differentiation of hiPSCs to monocytes and macrophages
-
•
Checkpoints at multiple stages to assess established cell-surface markers
-
•
Steps to assess phagocytic activity of monocytes and macrophages
-
•
Steps for polarization of macrophages to M0, M1, and M2 subtypes
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Study of disease-relevant immune cells, namely monocytes and macrophages, is limited based on availability of primary tissue, a limitation that can be remedied using human induced pluripotent stem cell (hiPSC) technology. Here, we present a protocol for differentiation of monocytes and macrophages from hiPSCs. We describe steps for hiPSC maintenance, mesoderm lineage induction, hematopoietic progenitor cells (HPCs) commitment and expansion, and myeloid lineage induction. We then detail procedures for monocyte formation and functional macrophage formation and polarization.
Before you begin
This protocol describes differentiation of myeloid lineage cells from human iPSCs obtained from multiple donors. Extensive characterization was performed on cell lines that consistently produced the largest number of floating hematopoietic progenitor cells (HPCs), monocytes and macrophages. In total, all seven lines that were tested were capable of producing myeloid lineage cells. We outline potential sources of variability to address difficultly in differentiating lines, primarily being flexible on the timelines for cells to reach each stage outlined in the protocol. All cells were cultured in standard incubators at 37°C (5% CO2, 20% O2) and all medium changes/non-terminal characterization steps were performed in sterile conditions.
Preparation of reagents for human iPSC (hiPSC) culture
Timing: ∼1 day
-
1.Prepare hiPSC culture StemFlex medium.Note: We have had success with this protocol using hiPSCs maintained with E8 medium as well but prefer StemFlex for its pluripotency maintaining ability.
-
a.Thaw 450 mL StemFlex basal medium at 4°C overnight.
-
b.Thaw 50 mL StemFlex supplement in 37°C water bath for 1 h or at 4°C overnight.
-
c.Add entire volume of StemFlex supplement to 450 mL basal medium to make complete medium.
-
d.Store completed StemFlex media at 4°C for up to 4 weeks.
-
e.Make separate 50 mL StemFlex medium aliquot supplemented with Rock inhibitor (Y-27632) by adding 50 μL of 10 mM Rock inhibitor stock solution (10 μM final concentration).
-
a.
-
2.Prepare Matrigel human Embryonic Stem Cell (hESC) qualified matrix solution-coated plates.Note: Alternative solutions such as Vitronectin may be used as well.
-
a.Thaw one 5 mL vial of Matrigel hESC-qualified matrix stock at 4°C overnight.
-
b.Thaw one 500 mL bottle of DMEM/F12 at 4°C overnight. Aliquot 50 mL into conical tube for use in the next step.
-
c.Dilute Matrigel stock 1:100 in DMEM/F12 medium in 50 mL conical tube.
-
d.To coat one 6-well plate, add 1 mL diluted Matrigel to each well.
-
e.Incubate coated plate at 37°C for at least 30 min before use to culture cells if immediate use is required. For longer term storage, coated plates can be kept at 37°C for up to ∼1 week prior to use.
-
a.
Preparation of growth factor stocks
All growth factor stocks are reconstituted in PBS containing + 0.1% low endotoxin bovine serum albumin (BSA) to generate stock solutions of 100 μg/mL. To avoid repeated freeze-thaw cycles, it is recommended to aliquot the growth factor stock solutions in small volumes (∼100 μL), aliquots are stable for up to 1 year at −20°C to −80°C.
Preparation of glass coverslips
-
3.Carefully clean glass coverslips before proceeding with cell seeding.
-
a.Transfer coverslips to glass container containing 1 M HCl.
-
b.Incubate 16–20 h at 20°C–22°C.
-
c.Transfer coverslips to glass container containing 70% EtOH.
-
d.Incubate 16–20 h at 20°C–22°C. Longer is optional.
-
e.Before use, dry coverslips in a sterile environment.
-
a.
Preparation of pHrodo Green Zymosan BioParticles conjugate for phagocytosis assessment
-
4.Reconstitute pHrodo bioparticles in DPBS +/+ with 20 mM HEPES (IMPORTANT: Check pH 7.4).
-
a.Warm pHrodo bioparticles and the buffer to 20°C–25°C.
-
b.Resuspend the pHrodo Bioparticles by adding 2.5 mL of PBS +/+ with 20 mM HEPES (pH 7.4) to a vial to generate a 0.4 mg/mL stock.
-
a.
-
5.Vortex or sonicate the vial to homogeneously disperse the particles. For best results, sonicate the vial of resuspended particles for 10 min.
-
a.If particle aggregates are still evident Vortex or sonication, repeat this step.
-
a.
Note: This passage is critical as particle aggregation immediately after resuspension is normal and particles monodispersion is required to improve the quality of the results obtained in the subsequent analysis.
-
6.
Once reconstituted, pHrodo Bioparticles Conjugates can be stored at 2°C–8°C for several weeks by adding sodium azide (2 mM). Do not freeze the resuspended pHrodo bioparticle conjugates.
Preparation of Zombie Aqua Fixable viability kit
-
7.
Prepare stock solution by adding 100 μL of DMSO to one vial of Zombie Aqua dye.
-
8.
Prepare 1:800 working solution by adding 1 μL Zombie Aqua dye stock to 39 μL H2O and 760 μL HBSS −/−.
Note: After reconstitution, use the reagent immediately, or store it up to one month at −20°C in a dry place and protected from light. Store reconstituted Zombie Aqua dye in aliquots to avoid freeze-thaw cycles.
Institutional permissions
This study was approved by the National Heart Lung and Blood Institute’s institutional review board. Primary monocytes were obtained from peripheral blood mononuclear cells (PBMCs) isolated from buffy coats of four healthy volunteers who signed consent forms in accordance with the Declaration of Helsinki and with clinical protocols approved by the Institutional Review Board of Humanitas Research Hospital (Ethical Committee of 30/01/2007, name of the protocol “Impiego di buffycoat nella ricerca corrente “processamento dei campioni di sangue”).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE-conjugated mouse monoclonal antibody anti-human CD34 (clone: 561, 1:20) | BioLegend | Cat# 343606; RRID: AB_1732033 |
| APC-conjugated mouse monoclonal antibody anti-human CD45 (clone: H130, 1:20) | BioLegend | Cat# 304012; RRID: AB_314399 |
| FITC-conjugated mouse monoclonal antibody anti-human CD14 (clone: GCD14, 1:20) | BioLegend | Cat# 325604; RRID: AB_830677 |
| PE Cy5-conjugated mouse monoclonal antibody anti-human CD11b (clone: IRCF44, 1:20) | BioLegend | Cat# 301308; RRID: AB_314160 |
| PE-conjugated mouse monoclonal antibody anti-human CD209 (clone: DCN46, 1:20) | BD | Cat# 551265; RRID: AB_394123 |
| FITC-conjugated mouse monoclonal antibody anti-human CD80 (clone: L307-4, 1:20) | BD | Cat# 557226; RRID: AB_396605 |
| Mouse monoclonal antibody anti-human CD68 (clone: KP1, 1:200) | Dako | Cat# M0814; RRID: AB_2314148 |
| AF568-conjugated goat anti-mouse antibody (1:1,000) | Invitrogen | Cat# A-11004; RRID: AB_2534072 |
| Chemicals, peptides, and recombinant proteins | ||
| Matrigel hESC-qualified matrix | Corning | 354277 |
| DMEM/F-12 | Thermo Fisher Scientific | 11320033 |
| StemFlex medium | Thermo Fisher Scientific | A3349401 |
| STEMdiff APEL 2 medium | STEMCELL Technologies | 05275 |
| Protein-free hybridoma mixture II (PFHMII) | Invitrogen | 12040-077 |
| Penicillin/streptomycin (50 U Pen G/50 mg streptomycin sulfate) | Invitrogen | 15140122 |
| ReLeSR (enzyme-free dissociation reagent) | STEMCELL Technologies | 100-0483 |
| Y-27632 dihydrochloride (Rock inhibitor) | Tocris | 1254 |
| Recombinant human (rh) vascular endothelial growth factor (rhVEGF) | PeproTech | 100-20 |
| rh bone morphogenetic protein 4 (rhBMP4) | R&D Systems | 314-BP-050 |
| rh basic fibroblast growth factor (rhbFGF) | PeproTech | 100-18B |
| rh stem cell factor (rhSCF) | STEMCELL Technologies | 02830 |
| rh Flt-3 ligand (rhFlt-3L) | STEMCELL Technologies | 02941 |
| rh thrombopoietin (rhTPO) | STEMCELL Technologies | 02720 |
| rh granulocyte-macrophage colony-stimulating factor (rhGM-CSF) | PeproTech | 300-03 |
| rh macrophage colony-stimulating factor (rhM-CSF) | PeproTech | 300-25 |
| Lipopolysaccharides (LPS) purified from Escherichia coli (E. coli) 055:B5 | Sigma-Aldrich | L2880-10MG |
| rh interferon-gamma (rhIFN-γ) | PeproTech | 300-02 |
| rh interleukin-4 (rhIL-4) | PeproTech | 200-04 |
| RPMI 1640 medium without L-glutamine with phenol red | Euroclone | ECB9006L |
| L-glutamine 200 mM | PAN-Biotech | P04-80100 |
| PENICILLIN-STREPTOMICIN 100X | Euroclone | ECB3001D |
| Fetal bovine serum (FBS) | Sigma-Aldrich | F0926 |
| Accutase | Merck-Millipore | SCR005 |
| Low endotoxin bovine serum albumin (BSA) | Sigma-Aldrich | A9418-5G |
| Dulbecco’s phosphate-buffered saline (PBS) without calcium (Ca++) and magnesium (Mg++) (DPBS −/−) | Sigma-Aldrich | D8537 |
| H2O | ||
| Glass coverslip (round, 14 mm) | Thermo Fisher Scientific | 10337423 |
| Deionized H2O | ||
| Hydrochloric acid (HCl) | Sigma-Aldrich | 320331-500ML |
| Ethyl alcohol (EtOH) | Sigma-Aldrich | 792802 |
| Dulbecco’s phosphate-buffered saline (DPBS) containing calcium (Ca++) and magnesium (Mg++) (DPBS +/+) | Sigma-Aldrich | D1283 |
| Paraformaldehyde solution (PFA) 4% in PBS | Santa Cruz | 30525-89-4 |
| Tween 20 | Sigma-Aldrich | P1379-100ML |
| Triton X-100 | Sigma-Aldrich | X100-100ML |
| Normal goat serum (NGS) | Abcam | ab7481 |
| DAPI (4′,6-diamidino-2-phenylindole, dihydrochloride) | Invitrogen | D1306 |
| ProLong Gold antifade reagent | Invitrogen | P36934 |
| EDTA solution pH 8.0 (0.5 M) | Euroclone | APA48920500 |
| Human TruStain FcX (Fc receptor blocking solution) | BioLegend | 422301 |
| Zombie Aqua fixable viability kit | BioLegend | 423102 |
| BD CompBeads anti-mouse Ig, κ/negative control compensation particles set | BD | 552843 |
| HEPES | Sigma-Aldrich | H4034-25G |
| Hanks’ balanced salt solution (HBSS) without calcium (Ca++) and magnesium (Mg++) (HBSS −/−) | Thermo Fisher Scientific | 14170120 |
| Critical commercial assays | ||
| pHrodo Green Zymosan BioParticles conjugate for phagocytosis | Invitrogen | P35365 |
| Experimental models: Cell lines | ||
| hiPSC NC6 (line 1 in figures, protocol 10-H-0126, control) | In-house generated | N/A |
| hiPSC NC8 (line 2 in figures, protocol 10-H-0126, control) | In-house generated | N/A |
| hiPSC ND7C (line 3 in figures, protocol 10-H-0126, disease) | In-house generated | N/A |
| hiPSC NC10 (protocol 10-H-0126, control) | In-house generated | N/A |
| hiPSC NC4 (protocol 10-H-0126, control) | In-house generated | N/A |
| hiPSC ND7B (protocol 10-H-0126, disease) | In-house generated | N/A |
| hiPSC ND7D (protocol 10-H-0126, disease) | In-house generated | N/A |
| Human primary monocytes | In-house generated | N/A |
| Software and algorithms | ||
| FlowJo X 10.0.7r2 software | BD | https://www.bdbiosciences.com/en-us/products/software/flowjo-v10-software; RRID: SCR_008520 |
| Other | ||
| Non-pyrogenic Costar clear TC-treated multiple well tissue culture plates | Corning | 3506 (6-well), 3512 (12-well), 3527 (24-well) |
| Corning 100 mm (10 cm) TC-treated culture dish | Corning | 430167 |
| 0.22 mm filter units-50 mL | Sigma | SCGP00525 |
| ZEISS Primovert inverted cell culture dissection microscope | ZEISS | N/A |
| TC20 automated cell counter | Bio-Rad | 1450102 |
| Cell counting slides for TC20 | Bio-Rad | 1450015 |
| Olympus Fluoview FV1000 (laser scanning confocal microscopy) | Olympus | N/A |
| MACSQuant Analyzer 10 flow cytometer | Miltenyi Biotec | N/A |
| LSRFortessa flow cytometer | Beckton Dickinson | N/A |
| FACSAria | Beckton Dickinson | N/A |
Materials and equipment
Matrigel hESC-qualified matrix solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Matrigel hESC-qualified matrix stock | N/A | 500 μL |
| DMEM/F12 | N/A | 49.5 mL |
| Total | N/A | 50 mL |
Note: Store at 4°C for maximum 4 weeks. Pre-warm the culture medium for 15 min at room temperature before use.
hiPSC culture StemFlex medium
| Reagent | Final concentration | Amount |
|---|---|---|
| StemFlex Basal Medium | N/A | 450 mL |
| StemFlex Supplement | 10% | 50 mL |
| Rock inhibitor, 10 mM (the day of passage only) | 10 μM | 10 μL in 10 mL medium |
| Total | N/A | 500 mL |
Note: Sterilize media using a 0.22 μm membrane filter and store at 4°C for maximum 14 days. Pre-warm the culture medium for 15 min at room temperature before use.
Basal early-stage differentiation medium
| Reagent | Final concentration | Amount |
|---|---|---|
| STEMdiff APEL 2 medium | N/A | 47.5 mL |
| Protein free hybridoma medium (PFHM-II) | 5% | 2.5 mL |
| Total | N/A | 50 mL |
Note: Store at 4°C for maximum 14 days. Pre-warm the culture medium for 15 min at room temperature before use.
Mesoderm induction medium (days 0–3)
| Reagent | Final concentration | Amount |
|---|---|---|
| BMP4 | 10 ng/mL | 5 μL |
| VEGF | 10 ng/mL | 5 μL |
| bFGF | 10 ng/mL | 5 μL |
| Basal early-stage differentiation medium | N/A | 50 mL |
| Total | N/A | 50 mL |
Note: Sterilize media using a 0.22 μm membrane filter and store at 4°C for maximum 7 days. Pre-warm the culture medium for 15 min at room temperature before use.
HPC commitment medium (days 3–7)
| Reagent | Final concentration | Amount |
|---|---|---|
| BMP4 | 10 ng/mL | 5 μL |
| VEGF | 10 ng/mL | 5 μL |
| bFGF | 10 ng/mL | 5 μL |
| SCF | 50 ng/mL | 25 μL |
| TPO | 50 ng/mL | 25 μL |
| FLT-3L | 50 ng/mL | 25 μL |
| Basal early-stage differentiation medium | N/A | 50 mL |
| Total | N/A | 50 mL |
Note: Sterilize media using a 0.22 μm membrane filter and store at 4°C for maximum 7 days . Pre-warm the culture medium for 15 min at room temperature before use.
HPC expansion medium (days 7–10)
| Reagent | Final concentration | Amount |
|---|---|---|
| BMP4 | 10 ng/mL | 5 μL |
| VEGF | 10 ng/mL | 5 μL |
| bFGF | 10 ng/mL | 5 μL |
| SCF | 50 ng/mL | 25 μL |
| TPO | 50 ng/mL | 25 μL |
| FLT-3L | 50 ng/mL | 25 μL |
| GM-CSF | 100 ng/mL | 50 μL |
| Basal early-stage differentiation medium | N/A | 50 mL |
| Total | N/A | 50 mL |
Note: Sterilize media using a 0.22 μm membrane filter and store at 4°C for maximum 7 days. Pre-warm the culture medium for 15 min at room temperature before use.
Basal late-stage differentiation medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Fetal bovine serum (FBS) | 10% | 5 mL |
| RPMI 1640 medium | N/A | 45 mL |
| Total | N/A | 50 mL |
Note: Sterilize media using a 0.22 μm membrane filter and store at 4°C for maximum 7 days. Pre-warm the culture medium for 15 min at room temperature before use.
Myeloid lineage induction medium (days 10–14)
| Reagent | Final concentration | Amount |
|---|---|---|
| SCF | 50 ng/mL | 25 μL |
| TPO | 50 ng/mL | 25 μL |
| FLT-3L | 50 ng/mL | 25 μL |
| GM-CSF | 100 ng/mL | 50 μL |
| Basal late-stage differentiation medium | N/A | 50 mL |
| Total | N/A | 50 mL |
Note: Sterilize media using a 0.22 μm membrane filter and store at 4°C for maximum 7 days. Pre-warm the culture medium for 15 min at room temperature before use.
Monocyte formation medium (days 14–21)
| Reagent | Final concentration | Amount |
|---|---|---|
| GM-CSF | 100 ng/mL | 50 μL |
| M-CSF | 100 ng/mL | 50 μL |
| Basal late stage differentiation medium | N/A | 50 mL |
| Total | N/A | 50 mL |
Note: Sterilize media using a 0.22 μm membrane filter and store at 4°C for maximum 7 days. Pre-warm the culture medium for 15 min at room temperature before use.
Basal macrophage M0 medium (days 21–28)
| Reagent | Final concentration | Amount |
|---|---|---|
| L-Glutamine | N/A | 500 μL |
| Penicillin-streptomycin | N/A | 500 μL |
| M-CSF | 100 ng/mL | 50 μL |
| Basal late stage differentiation medium | N/A | 50 mL |
| Total | N/A | 51 mL |
Note: Sterilize media using a 0.22 μm membrane filter and store at 4°C for maximum 7 days. Pre-warm the culture medium for 15 min at room temperature before use.
M1 differentiation medium (days 28–29)
| Reagent | Final concentration | Amount |
|---|---|---|
| Lipopolysaccharides | 100 ng/mL | 50 μL |
| IFN-γ | 20 ng/mL | 10 μL |
| Basal macrophage M0 medium | N/A | 50 mL |
| Total | N/A | 50 mL |
Note: Sterilize media using a 0.22 μm membrane filter and store at 4°C for maximum 7 days. Pre-warm the culture medium for 15 min at room temperature before use.
M2 polarization media (days 28–29)
| Reagent | Final concentration | Amount |
|---|---|---|
| IL-4 | 20 ng/mL | 10 μL |
| Basal macrophage M0 medium | N/A | 50 mL |
| Total | N/A | 50 mL |
Note: Sterilize media using a 0.22 μm membrane filter and store at 4°C for maximum 7 days. Pre-warm the culture medium for 15 min at room temperature before use.
Step-by-step method details
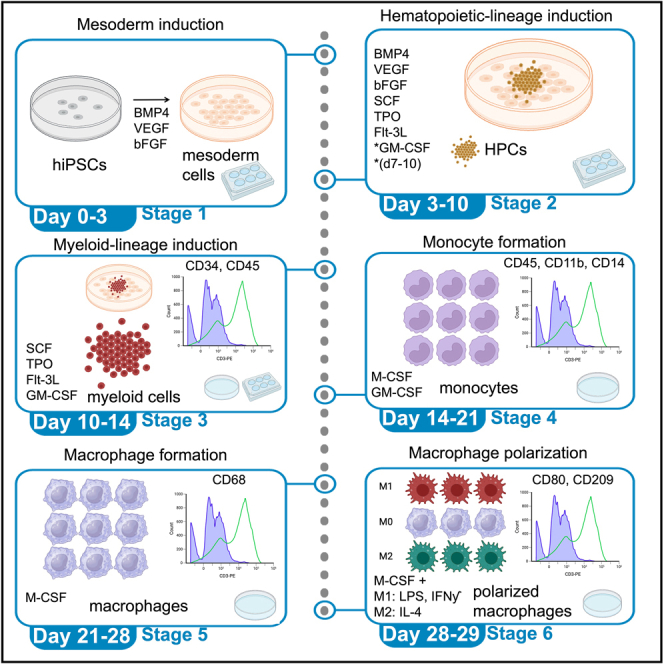
This protocol describes a method to induce several specific stages of hiPSC differentiation to myeloid lineage cells akin to the myeloid cell maturation process in human embryos in vivo that sequentially express markers such as CD34, CD45, CD11b, and CD14.2 These steps include: (1) formation of mesoderm progenitor cells, (2) formation of floating HPCs, (3) formation of myeloid progenitor cells, (4) formation of mature monocytes, and finally (5) formation of macrophages that can be polarized to either M1 or M2 macrophages (Figure 1).
Figure 1.

Summary of differentiation protocol
hiPSCs are passaged to form small colonies, the next day at day 0, treatment with the first growth factor combination is induced from days 0–3 to form mesoderm progenitor cells. From days 3–7, new growth factors are introduced to form hematopoietic progenitor cells (HPCs). From days 7–10 new growth factors are introduced to enhance HPC proliferation. From days 10–14 new growth factors are introduced to promote development of myeloid progenitor cells that float on top of the monolayer culture. Floating myeloid cells are replated on low-attached plates and treated with growth factors from day 14–21 to enhance production of monocytes and other mature myeloid lineage cells. Monocytes can be further cultured to resting M0 macrophages from days 21–28 and these cells can be polarized to classical M1 and M2 states by additional growth factor treatment for 24 h. Created with license from BioRender.
hiPSC passage prior to differentiation (stage 0, day −1 to 0)
Undifferentiated hiPSCs should be regularly passaged once every ∼3–4 days when reaching ∼80% confluency at dilution rations of between 1:5-1:10 in order to maintain pluripotency. It is also critical to regularly replace hiPSC culture medium between passages (at least every 2 days) to avoid possible auto-differentiation prior to cell passaging (Figure 2). To begin a differentiation, passage hiPSCs for maintenance as normal and additionally split cells into a new plate for differentiation using the below protocol.
Note: If obtaining hiPSCs through purchase of a frozen stock, follow manufacturer’s instructions to set up initial hiPSC culture and passage at least 2 times before differentiation.
-
1.
Prepare a Matrigel coated plate prior to passaging hiPSCs as described in the before you begin section.
-
2.Split hiPSCs into each well of a 6-well plate when cells reach ∼80% confluency.
-
a.Aspirate StemFlex medium from plates.
-
b.Wash cells with 1 mL DPBS by adding to the well and aspirating.
-
c.Add 0.5 mL of ReLeSR cell passaging reagent to and swirl for ∼30 s.
-
d.Aspirate ReLeSR and place cells at 37°C for ∼5 min to allow cells to detach.
-
e.Add 1 mL hiPSC culture StemFlex medium and pipette gently to detach cells.
-
f.Remove Matrigel from previously coated well and add 1.5 mL StemFlex medium with Rock Inhibitor as described in the materials and equipment section.
-
g.Add cells from prior well to new well at dilution ratio of ∼1:100 (10 μL from 1 mL) and swirl the plate to mix and ensure even distribution throughout the well.
-
h.Place cells at 37°C overnight until starting mesodermal induction the next day (the next day is defined at day 0 when treatment with differentiation media begins).
-
a.
CRITICAL: In this step, the size of colonies and density prior to start of differentiation is critical. In our experience, splitting at 1:100 from 80% confluency yields an ideal density.
Note: It is recommended to view cells under a dissection microscope to ensure small (10–30 cell) colony sizes that we have found to be optimal for promoting differentiation efficiency (Figure 3A,1 see troubleshootingproblem 1 for more details).
Figure 2.

Healthy and unhealthy hiPSC colony examples
(A) Example of healthy hiPSCs at typical confluence for cell passaging prior to differentiation.
(B) Example of unhealthy hiPSCs colonies with evidence of auto-differentiation.
Scale bar = 200 μm and is applicable to all images.
Figure 3.

Examples of cell morphology through monocyte formation
(A) Day 0 hiPSCs following passaging one day before.
(B) Day 2: cells during mesoderm induction.
(C) Day 5 cells during HPC induction, monolayer culture often grows rapidly and may reach near full confluency at this stage.
(D) Day 9: floating HPCs on top of monolayer culture.
(E) Day 14: myeloid progenitor cells at day 14 derived from replated floating HPCs.
(F) Day 21: mature myeloid cells with noteworthy cell size increase from prior stages.
Scale bar = 200 μm and is applicable to all images.
Mesoderm induction (stage 1, day 0–3)
The first step in this differentiation process is designed to induce mesoderm lineage cells that can be further directed to hematopoietic progenitor cells. From the small starting colonies derived from hiPSC passaging, cell clusters should grow steadily in size through this stage (see Figure 3B for example of day 2 cell colonies).
Note: This is defined as day 0 of the differentiation process.
-
3.
Prepare basal early-stage differentiation medium as described in the materials and equipment section, this will be used as the basis for medium for days 0–10 (3 stages).
-
4.
Prepare mesoderm induction medium for days 0–3 as described in the materials and equipment section.
-
5.
Gently aspirate StemFlex medium from the prior day.
-
6.
Add 1.5 mL mesoderm induction medium and place the cells back at 37°C from day 0–2.
-
7.
At day 2, aspirate mesoderm induction medium and replace with 1.5 mL fresh mesoderm induction medium.
HPC commitment and expansion (stage 2 and 3, day 3–10)
The second stage in this process is designed to commit cells to the hematopoietic lineage. During the early days (∼day 3–7), the monolayer culture should grow significantly and may reach near confluence (Figure 3C). During the later phases (∼day 7–10), characteristic hematopoietic progenitor cells (HPCs) start appear to be “floating” above monolayer culture cells (Figure 3D). We have defined two media necessary for these stages noted above, the first is designed to enhance commitment of HPCs and the second to promote HPC expansion.
Note: At this stage, variability between cell lines in their timeframe and capability to produce HPCs becomes readily apparent. In robustly differentiating lines, floating HPCs are apparent typically by day 8–10 but some lines may lack these cells by day 10 (See troubleshootingproblem 2 for more details).
-
8.
Prepare HPC commitment medium as described in the materials and equipment section.
-
9.
At day 3, aspirate mesoderm induction medium and replace with 2 mL HPC commitment medium. Place cells at 37°C from day 3–5.
-
10.
At day 5, aspirate medium and replace with 2 mL HPC commitment medium. Place cells at 37°C from day 5–7.
-
11.
At day 7, check cells under dissection microscope and determine if HPCs are visible on top of monolayer culture.
-
12.
Prepare HPC expansion medium as described in the materials and equipment section.
-
13.
At day 7, aspirate medium and replace with 2 mL HPC expansion medium. Place cells at 37°C from day 7–9.
-
14.
At day 9, aspirate and replace with 2 mL HPC expansion medium.
-
15.At day 10, determine if floating HPCs are present on top of monolayer culture and collect into a separate plate (size determined by density via cell count (based on Table 1).
-
a.Swirl the plate to detach floating HPCs.Note: Not all cells with HPC morphology will detach, but later collection time points should collect these cells as they mature.
-
b.Collect medium including detached HPCs into 15 mL conical tube.
-
c.Add 1 mL DPBS to monolayer and collect that into the same 15 mL conical tube.
-
d.Centrifuge 15 mL conical tube of HPCs at 600 g for 3 min at room temperature.Note: All centrifugation steps should be performed at room temperature.
- e.
-
f.Keep the monolayer culture and continually collect floating HPCs.CRITICAL: Starting ∼ day 10, floating HPCs will be moved to new plate(s) while the monolayer culture is still cultured separately. Plating the floating HPCs at the recommended densities below are seen to enhanced proliferation of HPCs while they are further committed to the myeloid lineage.
-
a.
Table 1.
Recommended density for floating HPC seeding per plate type
| Plates | Surface area (cm2) | Number of seeded cells | Volume of medium (mL) |
|---|---|---|---|
| Standard 10 cm plate | 58.1 | 4.7 × 106 | 12 |
| 6-well | 9.6 | 7.7 × 105 | 2 |
| 12-well | 3.5 | 2.8 × 105 | 1 |
| 24-well | 1.9 | 1.5 × 105 | 0.5 |
Myeloid lineage induction (stage 3, day 10–14)
This stage in the differentiation process is designed to induce floating HPCs to become myeloid lineage committed cells. Following collection of floating HPCs and plating at the recommended densities above, HPCs proliferate but appear to stay in small clusters or as isolated cells (Figure 3E).
Note: At this stage, ideally HPCs, that were attached to the monolayer culture, have been collected and are now re-plated at the appropriate density. We recommend maintaining the monolayer culture using the same medium for the resuspended HPCs in order to collect additional batches of HPCs that may arise.
-
16.
Prepare basal late-stage differentiation medium as in the materials and equipment section, this will be used as the basis for medium for day 10 to the end of differentiation (3 stages).
-
17.
Prepare myeloid lineage induction medium for days 10–14 as described in the materials and equipment section.
-
18.
[Monolayer] At day 10, aspirate media and add 2 mL myeloid lineage induction medium to cells. Place cells back in 37°C incubator until day 12.
-
19.
[Isolated HPCs] At day 10, plate HPCs in appropriate volume myeloid lineage induction medium (based on Table 1). Place cells back in 37°C incubator until day 12.
-
20.
At day 12, analyze monolayer culture under dissection microscope and determine if there are more floating HPCs to collect. If so, perform same steps as noted above in stage 2 and combine with already collected HPCs.
-
21.
[Monolayer] If not collecting additional HSCPs, aspirate medium and replace with the same volume of myeloid lineage induction medium.
Note: In most cells lines we observed floating HPCs being produced through day 14 and thus would continue culture past day 12 even if there are not HPCs to collect.
-
22.[Isolated HPCs] Refresh medium for HPCs.
-
a.Collect medium/resuspended cells in 15 mL conical tubes.
-
b.Add ½ the appropriate volume (based on Table 1) myeloid lineage induction medium to the wells while performing centrifugation to maintain resuspended cells.
-
c.Centrifuge collected cells at 600 g for 3 min.
-
d.Aspirate supernatant and add the same volume of myeloid lineage induction medium as (b).
-
e.Pipette to mix cells and add back to original plate.
-
f.Place cells back in 37°C incubator until day 14.
-
a.
-
23.
At day 14, analyze monolayer culture under dissection microscope and determine if there are more floating HPCs to collect. If so, perform same steps as noted above in stage 2 and combine with already collected HPCs.
Note: This is typically the last collection point for HPCs in the cell lines tested.
Optional: Hematopoietic/early myeloid-cell phenotypic characterization
Floating HPCs collected at any time point (day 10, 12, 14, etc) may be analyzed for expression of hematopoietic and myeloid-cell markers CD34 and CD45. We recommend using at least 20,000 cells for analysis and adding additional controls for non-stained cells as well as isogenic antibody staining if enough cells are present. In robustly differentiating lines, we typically see ∼85% CD34+ and 90% CD45+ cells at this stage (Figure 4).
-
24.Label cells for flow cytometry to assess expression of markers CD34 and CD45.
-
a.Collect HPCs from unattached plate following separation from monolayer into 15 mL conical tube.
-
b.Centrifuge cells at 600 g for 3 min.
-
c.Aspirate medium and resuspend cells in 1 mL DPBS.Optional: Count cells to estimate volume needed for 20,000 cells.
-
d.Resuspend to obtain a single-cell suspension and transfer appropriate volume of cells to 1.5 mL Eppendorf tube.
-
e.Centrifuge cells at 600 g for 3 min.
-
f.Aspirate medium and replace with 50 μL DPBS.
-
g.Incubate cells with appropriate CD34 and CD45 antibodies, at the concentration defined by manufacturer’s instructions, for 30 min at 4°C.Note: As negative controls to define positivity threshold, incubate cells with isogenic antibodies or prepare a Fluorescence minus one (FMO) control for each analyzed marker.
-
h.Add 1 mL DPBS and centrifuge at 600 g for 3 min.
-
i.Aspirate medium and add 200 μL of MACS buffer (1000 mL PBS, 5 g BSA, 4 mL 0.5 M EDTA).
-
a.
-
25.
Run flow cytometry according to manufacturer’s instructions.
Figure 4.

Phenotypic assessment of hiPSC-derived HPCs
(A) Representative overlay showing expression of hematopoietic progenitor cell marker CD34 across 3 genetically independent hiPSC-derived cells lines at day 14 in floating HPCs (Average = 86.5%, N = 3).
(B) Representative overlay showing expression of leukocyte marker CD45 across 3 genetically independent hiPSC-derived cells lines at day 14 in floating HPCs (Average = 93.7%, N = 3). Expression from staining with the appropriate isotype antibodies is shown as negative control for all groups (gray).
Monocyte formation (stage 4, day 14–21)
Following production of HPCs, the next stage aims to develop mature myeloid cells and in particular monocyte cells. During this stage, differentiating cells become larger in size and often form cell clusters toward the bottom of the low-attached plates (Figure 3F).
Note: We observe variability in the maturation time required for different cells lines once switched into monocyte formation medium. Phenotypic measurement of the pan-leukocyte marker CD45, the myeloid marker CD11b and the monocyte marker CD144 can be used to evaluate when cells may be ready for downstream stages (see troubleshootingproblem 3 for more details).
Note: The total amount of hiPSC-derived monocytes at this stage varies by line in a correlated manner to differentiation efficiency. Robustly differentiating lines produced ∼12–18 million cells starting from 2 wells of a 6-well plate whereas poorly differentiating lines varied from ∼0.5–5 million cells.
-
26.
Prepare monocyte formation medium as described in the materials and equipment section.
-
27.
Add ½ appropriate medium volume (based on Table 1) to new plate (same size from prior stage).
-
28.
Collect cells from myeloid lineage induction medium and centrifuge at 600 g for 3 min.
-
29.
Aspirate medium following centrifugation and add ½ appropriate volume (based on Table 1) of monocyte formation medium.
-
30.
Transfer cells to the new plate prepared above, place cells back in 37°C incubator until day 16.
-
31.
At day 16, carefully aspirate ½ medium on top of cells.
Note: If handled gently, cells should stay toward bottom of the plate and thus this aspiration step should not remove cells. If cells do not appear to be attached at all then collect all cells for centrifugation as above and replate in new monocyte formation medium.
-
32.
Add ½ volume monocyte formation medium (based on Table 1) on top of cells, place cells back in 37°C incubator until day 18.
-
33.
At day 18, repeat medium refreshment steps 31–32.
-
34.
At day 20 repeat medium refreshment steps 31–32.
Optional: Hematopoietic/early myeloid-cell phenotypic characterization
At this stage, we strongly recommend phenotypic characterization of pan-leukocyte marker CD45, mature myeloid marker CD11b and monocyte marker CD14. This may be done as early as day 20–22 where robustly differentiating lines achieves ∼90% expression of CD45 and CD11b and ∼80% of CD14. These expression levels were on par with primary PBMC-derived monocytes as well (Figure 5).
Note: By this stage the vast majority of cells should be attached or partially attached to the bottom of the culture plate and will require chemical dissociation for cell collection.
-
35.Label cells for flow cytometry to assess expression of markers CD45, CD11b and CD14.
-
a.Carefully aspirate as much medium as possible from the plate, this should leave fully or partially attached cells behind.
-
b.Add appropriate volume of Accutase to the well (i.e., 1 mL for one well of a 6-well plate, 4 mL for 10 cm plate). Place cells at 37°C for 5 min.
-
c.Add double the Accutase volume of DMEM with 10% FBS.
-
d.Pipette cells to mix and then collect in 15 mL conical tube.
-
e.Centrifuge cells at 600 g for 3 min.
-
f.Aspirate medium and resuspend cells in 1 mL PBS.Optional: Count cells to estimate volume needed for 20,000 cells.
-
g.Resuspend to obtain a single-cell suspension and transfer appropriate volume of cells to 1.5 mL Eppendorf tube.
-
h.Centrifuge cells at 600 g for 3 min.
-
i.Aspirate medium and replace with 50 μL PBS.
-
j.Incubate cells with appropriate CD45, CD11b and CD14 antibodies, at the concentration defined by manufacturer’s instructions, for 30 min at 4°C.Note: As negative controls to define positivity threshold, incubate cells with isogenic antibodies or prepare a Fluorescence minus one (FMO) control for each analyzed marker.
-
k.Add 1 mL PBS and centrifuge at 600 g for 3 min.
-
l.Aspirate medium and add 200 μL of MACS buffer (1000 mL PBS, 5g BSA, 4 mL 0.5 M EDTA).
-
a.
-
36.
Run flow cytometry according to manufacturer’s instructions.
Optional: Freeze monocytes, recommended 1–3 million per vial, in 200 μL of CryoStor CS10 medium.
Figure 5.

Phenotypic assessment of hiPSC-derived and primary monocytes
(A) Representative overlay showing expression of leukocyte marker CD45 across 3 genetically independent hiPSC-derived cells lines at day 14 in monocytes (Average = 92.4%, N = 3).
(B) Representative overlay showing expression of mature myeloid cell marker CD11b across 3 genetically independent hiPSC-derived cells lines at day 14 in monocytes (Average = 92.0%, N = 3).
(C) Representative overlay showing expression of monocyte cell marker CD14 across 3 genetically independent hiPSC-derived cells lines at day 14 in monocytes (Average = 82.4%, N = 3).
(D–F) Expression of same markers from (A-C) in primary PBMC-derived monocytes. Expression from staining with the appropriate isotype antibodies is shown as negative control for all groups (gray).
Macrophage production and polarization (stage 5, day 21–28)
Following production of monocytes as characterized above, this stage describes how to further develop monocytes into macrophages. Macrophages can then be polarized from typical M0 states in either M1 or M2 states to study varying facets of inflammatory function.
-
37.
Prepare basal macrophage M0 medium as described in the materials and equipment section.
-
38.
Add ½ appropriate medium volume of basal macrophage M0 medium to new plate (based on Table 1) .
-
39.
Collect cells from prior stage medium and centrifuge at 600 g for 3 min.
-
40.
Aspirate medium following centrifugation and add ½ appropriate volume of basal macrophage M0 medium (based on Table 1).
-
41.
Transfer cells to a new plate prepared above, place cells back in 37°C incubator until day 23.
-
42.
At days 23 and 25, carefully aspirate ½ medium on top of cells and refresh with basal macrophage M0 medium.
-
43.At day 27, change cells to M1 or M2 polarization medium.
-
a.Prepare M1 and M2 polarization media as described in the materials and equipment section.
-
b.Collect medium with cells into 15 mL conical tube.Note: If cells are attached to plate, collect using the Accutase method noted above (detailed in step 35).
-
c.Add ½ appropriate medium volume of M1 or M2 polarization medium (based on Table 1) to the plate.
-
d.In the meanwhile, centrifuge collected medium with cells at 600 g for 3 min.
-
e.Aspirate medium following centrifugation, re-suspend the pellet with ½ appropriate volume of M1 or M2 polarization medium (based on Table 1) and add it to the same plate.
-
f.Place cells back at 37°C incubator for 24 h.Optional: Freeze macrophages, up to 3 million per vial, in 200 μL of CryoStor CS10 medium.
-
a.
Optional: Macrophage characterization by assessing CD68 expression by confocal microscopy
At this stage, macrophages may be characterized by immunofluorescence based on pan-monocyte/macrophage marker CD68. We have performed this staining on M0, M1 and M2 macrophages and included a comparison to primary human macrophages as well for comparison. (Figure 6A).
-
44.
Prepare glass coverslips as described in the before you begin section.
-
45.
Seed 0.25×106 cells/glass coverslip contained in 24 well plate (0.5 mL/well) with appropriate medium (M0, M1, or M2).
-
46.
Place cells back at 37°C incubator for 24 h.
-
47.Stain cells for imaging.
-
a.Aspirate basal macrophage medium.
-
b.Fix cells with 0.5 mL/well of 4% paraformaldehyde (PFA) in PBS for 15 min at room temperature.
-
c.Wash with 0.5 mL/well of PBS for 5 min at room temperature.
-
d.Blocking: incubate cells with 0.5 mL/well of Blocking Solution (0.1% Triton X-100 + 10% Normal Goat Serum (NGS) in DPBS+/+) for 30 min at room temperature.
-
e.Incubate cells with primary antibody anti-human CD68: incubate cells with 0.5 mL/well of mouse monoclonal antibody anti-human CD68 diluted 1:200 in Blocking Solution for 1 h at room temperature.
-
f.Wash cells with 0.5 mL/well of Wash Buffer (0.05% Tween 20 in DPBS +/+) for 5 min at room temperature. Repeat this step 3 times.
-
g.Incubate with secondary antibody: incubate cells with 0.5 mL/well of AF568-conjugated goat anti-mouse antibody diluted 1:1000 in Blocking Solution for 1 h at room temperature, in the dark.Note: starting from this step minimize the sample light exposure to avoid photo bleaching.
-
h.Wash cells with 0.5 mL/well of Wash Buffer (0.05% Tween 20 in DPBS+/+) for 5 min at room temperature. Repeat this step 3 times.
-
i.Nuclear staining: incubate cells with 0.5 mL/well of DAPI (300 nM) diluted in PBS for 10 min at room temperature.
-
j.Wash cells with 0.5 mL/well of Wash Buffer (0.05% Tween 20 in DPBS+/+) for 5 min at room temperature. Repeat this step for 3 times
-
k.Mount coverslip with cells on glass slide using mounting reagent.
-
i.Add 1 drop of the mounting reagent (ProLong Antifade Gold Reagent) onto glass slide; try to use an amount that will just fill the space under the coverslip.
-
ii.Remove the coverslip with stained cells from the buffer.
-
iii.Blot excess buffer from the non-sample surface of the coverslip.
-
iv.Slowly tip the coverslip sample-side down onto the mounting medium on the glass slide, avoiding bubble creation.
-
i.
-
a.
-
48.Imaging of stained slides.
-
a.Use Olympus Fluoview FV1000 (Laser scanning confocal microscopy) or similar confocal microscope and image as per manufacturer’s instructions.
-
a.
Figure 6.

Phenotypic and functional assessment of hiPSC-derived and primary macrophages
(A) Representative images of immunofluorescence CD68 staining observed in primary macrophages (pMac, n = 5; first column) and iPSC-derived macrophages (iMac, n = 3; second column) polarized in M0, M1, and M2 macrophages (First, second and third row, respectively). Cells were stained for CD68 (red) and with DAPI for nuclear staining (blue).
(B and C) Representative overlay showing the expression of the M1 marker CD80 (B) and the M2 marker CD209 (C) on primary macrophages (pMac, n = 4; empty histograms) and iPSC-derived macrophages (iMac, n = 3; filled histograms) polarized in M0 (orange), M1 (red), and M2 (green) macrophages. The fluorescence minus one (FMO) used as negative control is shown (gray, filled histogram). (D-E) Phagocytic ability of M0, M1 and M2 primary macrophages (pMac) and iPSC-derived macrophages (iMac) was evaluated by using pHrodo Green Zymosan A Bioparticles and was assessed by flow cytometry and confocal microscopy.
(D) Representative images showing phagocytized Zymosan particles (in green) in primary macrophages (pMac, n = 3; first column) and iMac (iMac-Con, n = 3; second column) polarized in M0, M1, and M2 macrophages (First, second and third row, respectively). Nuclei were stained with DAPI (blue).
(E) Representative overlay showing the Zymosan phagocytosis in primary macrophages (pMac, n = 4; empty histograms) and iPSC-derived macrophages (iMac, n = 3; filled histograms) polarized in M0 (in orange, first graph), M1 (in red, second graph), and M2 (in green, third graph) macrophages. Samples incubated without Zymosan (gray histograms) and with Zymosan incubated at 4°C (light blue histograms), which were used as negative controls, are shown.
Scale bars = 25 μm and are applicable to all images.
Optional: Macrophage polarization characterization by flow cytometry
At this stage, polarized macrophages may be characterized by flow cytometry based on M1 marker CD80 and M2 marker CD209 expression. We have performed this experiment on M0, M1 and M2 macrophages and included a comparison to primary human macrophages as well for comparison (Figures 6B and 6C).
-
49.Label cells for flow cytometry to assess expression of markers CD80 and CD209.
-
a.Carefully aspirate as much medium as possible from the plate, this should leave fully or partially attached cells behind.
-
b.Add appropriate volume of Accutase to the well (i.e., 1 mL for one well of a 6-well plate, 4 mL for 10 cm plate). Place cells at 37°C for 5 min.
-
c.Add double the Accutase volume of DMEM with 10% FBS.
-
d.Pipette cells to mix and then collect in 15 mL conical tube.
-
e.Centrifuge cells at 600 g for 3 min.
-
f.Aspirate medium and resuspend cells in 1 mL PBS.Optional: Count cells to estimate volume needed for 20,000 cells.
-
g.Resuspend to obtain a single-cell suspension and transfer appropriate volume of cells to 1.5 mL Eppendorf tube.Optional: Stain cells with Aqua Zombie viability dye.
-
i.Centrifuge cells at 600 g for 3 min.
-
ii.Resuspend cells in 100 μL Aqua Zombie viability dye working solution prepared as described in the before you begin section.
-
iii.Incubate at room temperature for 15 min.
-
i.
-
h.Wash with 1 mL HBSS−/−.
-
i.Centrifuge cells at 600 g for 3 min.
-
j.Aspirate medium and replace with 50 μL PBS.
-
k.Incubate cells with appropriate CD80 and CD209 antibodies, at the concentration defined by manufacturer’s instructions, for 30 min at 4°C.Note: As negative controls to define positivity threshold, incubate cells with isogenic antibodies or prepare a Fluorescence minus one (FMO) control for each analyzed marker.
-
l.Add 1 mL PBS and centrifuge at 600 g for 3 min.
-
m.Aspirate medium and add 200 μL of MACS buffer (1000 mL PBS, 5g BSA, 4 mL 0.5 M EDTA).
-
a.
-
50.
Run flow cytometry according to manufacturer’s instructions.
Optional: Macrophage characterization by phagocytosis assay
At this stage, phagocytic activity of macrophages can be assessed by either imaging or flow cytometry using pHrodo Green Zymosan A Bioparticles (Invitrogen), a no-wash fluorogenic reagent. We have performed this experiment on M0, M1 and M2 macrophages and included a comparison to primary human macrophages as well for comparison (Figures 6D and 6E). In accordance with previous work that reported that LPS treatment inhibits phagocytosis in human macrophages,4 both hiPSC-derived and primary macrophages demonstrated reduced phagocytic ability of M1 macrophages compared to M0 and M2 macrophages.
-
51.
Prepare glass coverslips and Zymosan Bioparticles as described in the before you begin section.
-
52.
Seed 0.25×106 cells/glass coverslip contained in 24 well plate (0.5 mL/well) with appropriate medium (M0, M1, or M2).
-
53.
Place cells back at 37°C incubator for 24 h.
-
54.Aspirate culture medium and add 200 μL of pHrodo Green Zymosan A Bioparticles.
-
a.Incubate cells for 1h at 37°C.
-
a.
Note: As a negative control we recommend preparing additional wells with cells (1) untreated with Zymosan and kept at 37°C, and (2) treated with Zymosan but kept on ice at 4°C.
Note: Following incubation cells may be analyzed by microscopy and/or flow cytometry methods, both of which are detailed below.
-
55.Assess phagocytosis by microscopy.
-
a.Aspirate basal macrophage M0 medium.
-
b.Fix cells with 0.5 mL/well of 4% paraformaldehyde (PFA) in PBS for 15 min at room temperature.
-
c.Wash with 0.5 mL/well of PBS for 5 min at room temperature.
-
d.Nuclear staining: incubate cells with 0.5 mL/well of DAPI (300 nM) diluted in PBS for 10 min at room temperature.
-
e.Wash cells with 0.5 mL/well of Wash Buffer (0.05% Tween 20 in DPBS+/+) for 5 min at room temperature. Repeat this step for 3 times
-
f.Mount coverslip with cells on glass slide using mounting reagent.
-
g.Add 1 drop of the mounting reagent (ProLong Antifade Gold Reagent) onto glass slide; try to use an amount that will just fill the space under the coverslip.
-
h.Remove the coverslip with stained cells from the buffer.
-
i.Blot excess buffer from the non-sample surface of the coverslip.
-
j.Slowly tip the coverslip sample-side down onto the mounting medium on the glass slide, avoiding bubble creation.
-
a.
-
56.Imaging of stained slides.
-
a.Use Olympus Fluoview FV1000 (Laser scanning confocal microscopy) or similar confocal microscope and image as per manufacturer’s instructions.
-
a.
-
57.Assess phagocytosis by flow cytometry.
-
a.Carefully aspirate as much medium as possible from the plate, this should leave fully or partially attached cells behind.
-
b.Add appropriate volume of Accutase to the well (i.e., 1 mL for one well of a 6-well plate, 4 mL for 10 cm plate). Place cells at 37°C for 5 min.
-
c.Add double the Accutase volume of DMEM with 10% FBS.
-
d.Pipette cells to mix and then collect in 15 mL conical tube.
-
e.Centrifuge cells at 600 g for 3 min.
-
f.Aspirate medium and resuspend cells in 1 mL PBS.Optional: Count cells to estimate volume needed for 20,000 cells.
-
g.Transfer Pipette to mix to single-cell mixture and transfer appropriate volume of cells to 1.5 mL eppendorf tube.
-
h.Centrifuge cells at 600 g for 3 min.
-
i.Aspirate medium and replace with 200 μL MACS buffer (1000 mL PBS, 5 g BSA, 4 mL 0.5 M EDTA).
-
a.
-
58.
Run flow cytometry according to manufacturer’s instructions.
Expected outcomes
This protocol enables to rapid, simple production of mature myeloid lineage derived monocytes and functional macrophages from hiPSCs. Importantly, this protocol has been thoroughly tested across seven distinct hiPSC lines, each of which showed the capacity for generating monocytes and macrophages. In lines that consistently show robust differentiation capacity, we generated ∼12–18 × 106 monocytes from just two wells of a 6-well culture plate. We describe critical stages for phenotypic characterization based on established cell markers CD34, CD45, CD11b and CD142 using simple flow cytometry experiments. Comparisons to human primary monocytes with these markers showed similarities in phenotype and function to the hiPSC-derived myeloid cells produced with this protocol. Furthermore, hiPSC-derived macrophages highly expressed the macrophage marker CD68, similar to primary monocyte-derived macrophages, and the ability of macrophages to polarize to classically interrogated M1 and M2 states, and function in phagocytic assays was confirmed as well. Lastly, we have previously identified significant research results from downstream experiments using cells produced from this protocol.1
Limitations
The primary limitation involved in this protocol is that different cell lines vary in their capacity to form floating HPCs (∼day 10) and in capacity of those HPCs to differentiate into mature myeloid cells (∼day 21). The next section outlines how to troubleshoot these issues should they arise. Additionally due to the longer term nature of these cell culture conditions, 3–4 weeks depending on the end goal of monocytes or polarized macrophages, the introduction of non-sterile conditions and bacterial contamination must be vigilantly monitored.
Troubleshooting
Problem 1
Following hiPSC passaging at day −1, colony sizes between 10-30 cells are not formed.
Potential solution
Varying the amount of time cells are incubated following 30 s treatment with ReLeSR can help control resulting colony size. Extending the incubation time from ∼5 to ∼7 min leads to smaller colonies and decreasing time leads to larger colonies.
Problem 2
Initial floating HPCs are not produced in expected time range by of day ∼10.
Potential solution
Extending the cell culture time for the HPC commitment and/or expansion phases may be necessary for some lines. For example, we have observed extending the day 7–10 treatment window by 48 h until day 12 to aid in HPC production. Additionally, we recommend tracking HPC production across separate differentiation attempts, at least 3, within each line to determine if low HPC production can be attributed to the cell line being used or alternative technical issues such as starting cell density following hiPSC passaging.
Problem 3
Floating HPCs do not progress to mature myeloid cells/monocytes by ∼ day 21 based on examined flow cytometry markers CD45, CD11b, CD14.
Potential solution
Line to line and batch to batch variability often leads to varying levels of myeloid maturation at this stage. For lower efficiency cell lines, extending this treatment stage with the same medium (refreshing as above every 2 days) can strongly enhance myeloid maturation/monocyte formation. We have observed cell lines progress from ∼10-30% expression of CD14 to ∼40–60% following just 2 more days of treatment. Another option is to separate HPC collection batches as they are collected instead of pooling them prior to the monocyte induction stage starting at day 14.
Resource availability
Lead contact
Contact the lead contact Manfred Boehm (boehmm@nhlbi.nih.gov) for technical questions regarding this study.
Technical contact
Contact the technical contact Kevin Emmerich (emmerichkb@nih.gov) for technical questions regarding this study.
Materials availability
This study did not generate unique reagents.
Data and code availability
This study did not generate datasets or codes.
Acknowledgments
This research was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute with the National Institutes of Health (NIH) grant 1ZIAHL006079-10. We thank the NHLBI iPSC Core facility. This research was also funded by Ministero della Salute (PE-2016-02363915 to D.M.) and by Humanitas Research Hospital grants (Intramural Research and Clinical Funding Programs 5x1000) to D.M. R.C. was the recipient of a fellowship from the PhD program in Experimental Medicine of the University of Milan, Milan. Figure 1 was created using a licensed account through BioRender.com.
Author contributions
Conceptualization and investigation, K.E., F.C., X.T., G.C., E.P., S.D.B., D.M., and M.B. Methodology, validation, visualization, and analysis, K.E., F.C., X.T., E.P., R.C., G.C., D.Y., B.J., S.D.B., and I.V. Writing and editing, all authors. Acquisition of funding, D.M. and M.B.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Kevin Emmerich, Email: emmerichkb@nih.gov.
Manfred Boehm, Email: boehmm@nih.gov.
References
- 1.Chen G., Calcaterra F., Ma Y., Ping X., Pontarini E., Yang D., Oriolo F., Yu Z., Cancellara A., Mikulak J., et al. Derived myeloid lineage induced pluripotent stem as a platform to study human C-C chemokine receptor type 5Δ32 homozygotes. iScience. 2023;26 doi: 10.1016/j.isci.2023.108331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lambert C., Preijers F., Yanikkaya Demirel G., Sack U. Monocytes and macrophages in flow: an ESCCA initiative on advanced analyses of monocyte lineage using flow cytometry. Cytometry B Clin. Cytom. 2017;92:180–188. doi: 10.1002/cyto.b.21280. [DOI] [PubMed] [Google Scholar]
- 3.Jeong S., Chang H., Hong S.H. Protocol for differentiation of functional macrophages from human induced pluripotent stem cells. STAR Protoc. 2024;5 doi: 10.1016/j.xpro.2024.102925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Michlewska S., Dransfield I., Megson I.L., Rossi A.G. Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: key role for TNF-alpha. FASEB J. 2009;23:844–854. doi: 10.1096/fj.08-121228. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate datasets or codes.