

Abstract
Dual targeting by a single molecule has emerged as a promising strategy for fighting cancer. In this study, a new set of 2-thioquinazolin-4(3H)-ones as potential anti-cancer surrogates endowed with dual EGFR/VEGFR-2 kinases inhibitory activities were synthesized. The anti-tumor potency of the newly synthesized candidates 4–27 was evaluated against a panel of four cancer cell lines. The prepared candidates 4–27 showed comparable activity to that of the standard drug sorafenib. For instance, compound 4 (IC50 = 1.50–5.86 μM) and compound 20 (IC50 = 4.42–6.39 μM) displayed superior potencies against all cell lines compared to sorafenib (IC50 = 5.47–7.26 μM). Dual EGFR/VEGFR-2 inhibitory activities of the most active analogues (4, 11, and 20) were investigated. Compound 4 showed comparable EGFR/VEGFR-2 inhibitory activity to the used control drugs. Flow cytometric analysis indicates that the most potent analogue 4 stopped the cell cycle at the G1 phase and induced 46.53% total apoptosis in HCT-116 cells that was much more powerful than the untreated cells with 2.15% apoptosis. Molecular docking and dynamic simulations of 4, 11, and 20 with EGFR and VEGFR-2 were performed to examine the binding mode and interaction within the enzyme binding pockets.
Novel quinazolin-4(3H)-ones 4–27 were synthesized. Compounds 4, 11, and 20 showed potential anti-tumour activities. 4, 11, and 20 blocked EGFR/VEGFR-2 and triggered apoptosis. Compound 4 showed the most potent activity against EGFR/VEGFR-2.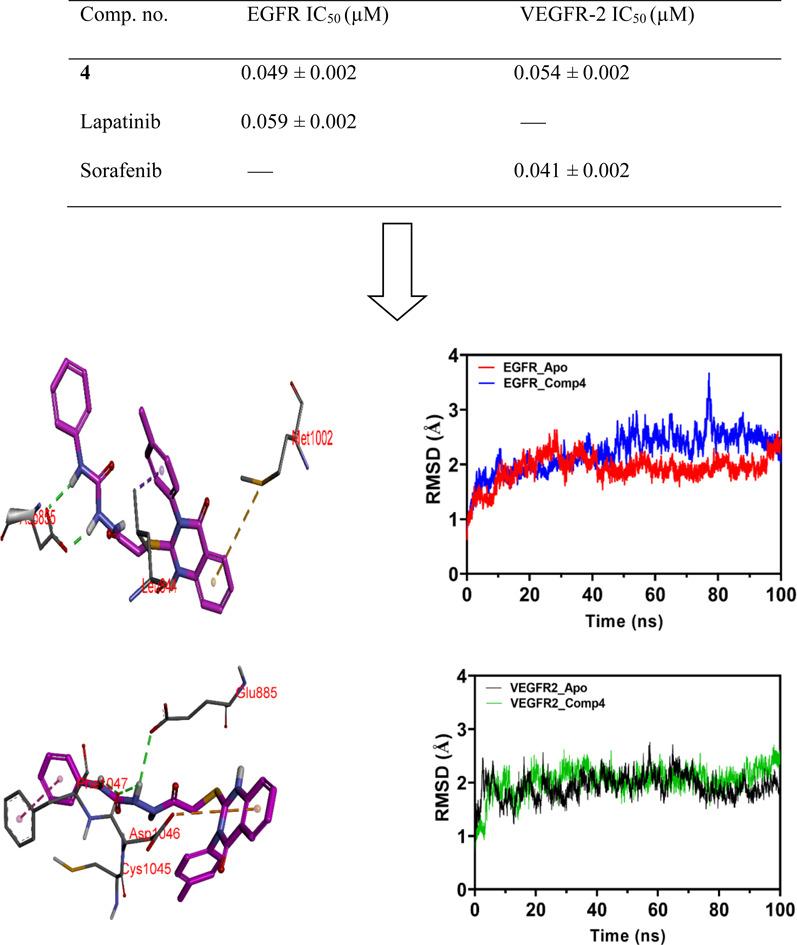
1. Introduction
Cancer is a fatal illness caused by unchecked cell proliferation. By 2050, it will be the leading cause of death worldwide, primarily in low- and middle-income countries (LMIC).1,2 Ten million cancer deaths and 19.3 million new cases were reported globally in the 2020 Global Cancer Statistics Report.3 Over the past few years, there has been a discernible increase in the acquired chemotherapeutic resistance of various cancer types.4 These considerations lead to a great demand for and development of novel anti-cancer medications that are more active and selective.5–8 The discipline of medicinal chemistry relies heavily on the nitrogen-containing heterocyclic scaffold exemplified by quinazoline.9–14 Quinazoline heterocyclic compounds exhibit a variety of biological effects, such as antidiabetic,15 anti-inflammatory,16,17 anti-cancer activity,18–24 carbonic anhydrase inhibition,25–28 and antimalarial29 properties.
The tyrosine kinase family member epidermal growth factor receptor (EGFR) is essential for cell signal transduction, influencing cell proliferation, survival, migration, and differentiation.30–34 EGFR is currently overexpressed in many human cancer cells.30–34 As a result, the EGFR protein has emerged as a critical therapeutic target for cancer treatment, and several EGFR inhibitors have been created.35–38 Representative EGFR inhibitors with considerable EGFR inhibitory action include Gefitinib (I), Erlotinib (II), Vandetanib (III), and Afatinib (IV) (Fig. 1).39–43
Fig. 1. The design strategy of our target compounds is guided by previously reported antitumour drugs/agents.

Angiogenesis, or constructing new blood vessels from pre-existing ones, is a normal physiological process for most blood vessel formation during growth and development, inflammation, and wound healing.44 Angiogenesis is a critical phase in transforming a benign tumor into a malignant tumor in which new blood vessels penetrate tumor masses, providing them with oxygen and nutrients to promote tumor development and metastasis.45,46 As a result, inhibiting angiogenesis could be a successful technique for slowing tumor growth.
Vascular endothelial growth factor (VEGF) is a crucial element in angiogenesis, increasing the number of blood vessels in a given network and driving both healthy and pathological angiogenesis.45–47 VEGF overexpression has been observed in a variety of cancers, including colorectal cancer,48 hepatocellular carcinoma,49 and breast cancer.50 Endothelial cells express VEGFR-2, a tyrosine kinase receptor, which has been shown to play an essential role in tumor angiogenesis control.51–53 As a result, VEGF/VEGFR-2 signaling represents an appealing therapeutic target in cancer treatment.36,54 Representative VEGFR-2 inhibitors with considerable EGFR inhibitory action include Sunitinib (V), Sorafenib (VI), Tivozanib (VII) and Pazopanib (VIII) (Fig. 1).55 As a result, inhibiting VEGFR-2 or down-regulating its signaling is a primary method for developing novel treatments for various human angiogenesis-dependent cancers.56
Based on our earlier findings54 and the significance of tyrosine kinases as attractive targets for the development of new anti-cancer medicines, we chose to introduce new quinazolin-4(3H)-one derivatives with dual inhibitory effects against EGFR/VEGFR-2-TK. Molecular hybridization is an appealing and efficient method for generating multi-targeted compounds that can synergize biological activity and bring significant value to cancer treatment. Furthermore, the synthesized compounds were tested for antiproliferative activity against colon, liver, breast, and lung cancer cell lines expressing EGFR/VEGFR-2. The most active compounds were evaluated for their ability to inhibit EGFR/VEGFR-2-TK and cause apoptosis in the HCT116 cell line. Finally, the examined compounds were docked with their molecular targets to investigate their binding patterns to the predicted targets.
2. The rationale of design strategy
In an extension of our previous research,54 which demonstrated the potential anti-cancer effects of novel quinazoline chemical agents, we designed and synthesised a new series of quinazoline-4(3H)-one derivative. The analysis of structure–activity correlations (SAR) and common pharmacophoric characteristics shared by diverse EGFR/VEGFR-2 antagonists indicated that the majority of them exhibit four key features, as illustrated in (Fig. 1).56,57 These characteristics are as follows: (i) a flat central heteroaromatic (CHA, grey region) ring system with at least one H-bond acceptor (the N-atom is favoured, or C O group). This H-bond acceptor binds hydrogen with the catalytic ATP-binding domain's essential amino acid residue (Cys917).55 (ii) A central hydrophobic aryl ring (hydrophobic group, HG, green region) that sits between the ATP-binding domain and the DFG domain.58 This hydrophobic group varies in size between EGFR and VEGFR-2 known inhibitors. In EGFR-TK, these groups comprise an aryl group with small lipophilic functionality at the distal end of this aryl ring. In VEGFR-TK, the hydrophobic group is composed of 3 parts. Part I is a central aryl spacer (S, cyan region) between the ATP-binding and DFG domains.55–59 Part II is a pharmacophore moiety composed of two groups of an H-bond acceptor/donor (HBA/HBD, red region) (e.g., urea or amide). This pharmacophore binds to two critical residues in the DFG (Asp-Phe-Gly) motif (Glu883 and Asp1044). The NH motifs of amide or urea moieties often establish two hydrogen bonds with Glu883, whilst the C O motif creates another hydrogen bond with Asp1044.55–59 Part III is a terminal hydrophobic tail (HT, blue region) that occupies the generated allosteric hydrophobic pocket via various hydrophobic contacts.59 Furthermore, the X-ray structural characterisation of numerous VEGFR-2 inhibitors indicated enough space for different substituents surrounding the terminal hetero aromatic ring.60,61
Surveying the literature, we found a series of quinazoline-4(3H)-one derivative that can develop into EGFR/VEGFR-2 inhibitors. Ghorab et al. prepared compound (IX) with N3-phenyl-quinazoline-4(3H)-one core bearing S-alkylated substituent in 2-position, taking into consideration the required features of Erlotinib (II). Compound IX was evaluated against the EGFR tyrosine kinase enzyme with an IC50 value of 58.26 μM. Superimposition of IX and erlotinib showed that the phenyl ring (hydrophobic tail) of the thioacetamide moiety in analogues is expected to bind to Lys721 in the back hydrophobic pocket similar to the free phenyl ring of Erlotinib (II).62 Wu et al. synthesised compound IX with a longer spacer on the S-alkylated branch at the 2-position of the same scaffold of compound (X). This structural change resulted in a 100-fold boost in EGFR IC50 value. This indicates tolerance of the increase of bulkiness in this side of these molecule series.63 On the other hand, using the VEGFR-2 as mentioned earlier pharmacophoric model, Ghorab et al. designed and synthesised compound (XI) with N3-phenyl-quinazoline-4(3H)-one core with bulkier S-alkylated substituent in 2-position. The S-substituent would comprise a spacer, HBA/HBD, and hydrophobic tail, essential to fill the back pocket for potent VEGFR-2 inhibition. They obtained XI with VEGFR-2 inhibition IC50 value of 0.46 μM.64 Compound (XII)65 and compound (XIII)66 were synthesised. They showed a dramatic decrease in the binding affinity to VEGFR-2, which could be attributed to the partial occupation of the back pocket in the VEGFR-2 active site due to smaller S-alkylated substituent in both compounds. Another compound (XIII) observation is the tolerance of introducing bulky bromo on the para-position of N3-phenyl of the quinazolinone core. This aligned with our previously synthesised compound (XIV)54 by introducing the 4-bromostyryl group at the 2-position of the 4-arylaminoquizoline scaffold. Compound XIV showed an IC50 value of 0.06 μM concerning VEGFR-2 inhibition.
The goal of our research is to create novel anti-tumour agents that have the same essential pharmacophoric properties as previously reported and clinically used EGFR/VEGFR-2 inhibitors while also being molecularly hybridised with efficient anti-tumour moieties in an attempt to create more potent anti-tumour compounds. Our molecular design reasoning was built around bio-isosteric modification techniques of EGFR/VEGFR-2 inhibitors (IX–XIII) at two different sites (Fig. 1). Due to the presence of the conventional linker's significant features (HBA–HBD linker), our modification incorporates the usage of a quinazolinone core as a central heteroaromatic core, with S-alkylated moiety chosen for the spacer at the 2-position of the quinazolinone ring. The first position was the heterocyclic aromatic ring to explore the para-position of the N3-phenyl ring on the 2-S-alkylated-3-phenylquinazolin-4(3H)-one scaffold. The second position was the linker (spacer) region at the 2-mercapto group on the quinazolinone. The linker was modified to be N-acylthioacetohydrazide with the nitrogen atoms acting as HBA/HBD, resulting in high VEGFR-2 potency. The third position was the terminal hydrophobic tail on the far end of the hydrazide group. This various aryl group was directed to fill the back pocket of VEGFR-2 to achieve interaction with Lys721 and gain the noticed increase in binding affinity as in compound X. The numerous structural alterations allowed us to investigate the SAR of these quinazolinone compounds as efficient anti-cancer drugs with possible dual EGFR/VEGFR-2 inhibitory actions, which is a critical goal of our research. Fig. 1 depicts and summarises all modification paths and molecular design reasoning. Compounds 4–27, which varied like S-alkylated substituent and para-substituent on the N3-phenyl ring of the quinazolinone scaffold (Fig. 1), were developed and synthesised. The most effective compounds were tested as EGFR/VEGFR-2 inhibitors after in vitro cytotoxic activities against a panel of four cell lines. The influence of the most potent compound on the cell cycle, its ability to decrease cell migration, and its apoptotic effect were also investigated. A docking study was carried out to obtain insight into the molecular interaction and determine a potential mode of action.
3. Results and discussion
3.1. Chemistry
Synthesis of the target compounds 4–27 is depicted in Scheme 1. 2-Mercapto-3-arylquinazolin-4(3H)-ones 1a–c were synthesized by reaction of anthranilic acid with different aryl isothiocyanates. Alkylation on the free mercapto group on compounds 1a–c was obtained by reaction of quinazolinones 1a–c with ethyl bromoacetate using K2CO3 in acetone at room temperature for 8 hours. 2-S-Acetatequinazolin-4(3H)-ones 2a–c were further reacted with hydrazine hydrate in ethanol under reflux conditions to generate the acid hydrazide derivatives 3a–c. The previously mentioned 1a–c, 2a–c and 3a–c were synthesized following the detailed procedure in previous literature. The subsequent reaction of the obtained hydrazides 3a–c with aryl isocyanates in DMF at 80 °C overnight furnished the final target 4–27 in 61–76% yield.67–69 The chemical structures of all the final targeted 4–27 were confirmed based on the spectroscopic data (IR, 1H-NMR, 13C-NMR, and ASAP-MS). IR analyses showed the existence of urea functionality by distinct stretching bands around 1630 and 3300, which match the C O and NH groups, respectively. 1H NMR spectra for the targeted compounds showed downfield signals of δ 8.02–10.14 ppm corresponding to the urea and hydrazide three exchangeable NH protons. In addition, 13C NMR spectra of the targeted 4–24 showed distinct signals resonating at the range of δ 156.7–167.6 ppm, which corresponds to the C O carbon for the urea group.
Scheme 1. Synthesis of quinazoline analogues 4–27 with various S-linked fragments.

4. Biological activities
4.1. In vitro anti-tumor activity and structure–activity relationship (SAR)
The newly synthesized quinazolinone analogues 4–27 were tested for in vitro anti-cancer activity utilizing four human tumour cell lines: normal human lung fibroblast (WI38), hepatocellular carcinoma (HEPG-2), colorectal carcinoma (HCT-116), and breast cancer cell lines (MCF-7).70,71 As a reference control drug, sorafenib was employed. Table 1 summarizes the data given as IC50 values, denoting the concentration that produces a net 50% impairment of cell viability. The results showed that the synthesized compounds had varying inhibitory effects against the investigated four human tumor cell lines. The IC50 values against the HEPG-2 cell line demonstrated that 4, 11, and 20 showed remarkable anti-tumor activities with IC50 values of 3.92, 8.1, and 5.17 μM, respectively. In addition, 6, 12 and 21 displayed moderate activity with IC50 values of 14.15, 19.5 and 11.53 μM, respectively. The anti-tumor activity against the HCT-116 cell line revealed that 4, 6, 11, 20 and 21 showed potent binding affinity with IC50 values of 1.50, 7.83, 6.72, 9.52, 4.42 and 2.83 μM, respectively. Moreover, 9 and 17 displayed moderate anti-tumor activity with IC50 values of 18.66 and 13.5 μM, respectively. Regarding the HCT-116 cell line, 4 demonstrated IC50 of 6.59 μM, the best activity against HCT-116 cells exceeding sorafenib (positive control) (IC50 = 5.47 μM). With the comparison of IC50 values of our synthesized compounds against MCF-7 cells, 4, 11, and 20 showed significant inhibitory activity with IC50 of 5.86, 9.43, and 6.39 μM, respectively. Furthermore, 6, 12 and 21 showed moderate cytotoxic activities with IC50 values of 18.18, 12.49, and 10.23 μM. 4, 11, and 20 exhibited anti-tumor solid activities with an IC50 range of 1.5–8.1 μM.
In vitro anti-tumor activities (IC50) of the synthesized 4–27 against lung, liver, colon and breast cancer cell lines compared to sorafenib.
| ||||||
|---|---|---|---|---|---|---|
| Comp. no. | R | Ar | IC50a (μM) | |||
| WI-38 | HEPG-2 | HCT-116 | MCF-7 | |||
| 4 | –CH3 | Ph | 45.61 ± 2.4 | 3.92 ± 0.2 | 1.50 ± 0.1 | 5.86 ± 0.2 |
| 5 | 4-FPh | >100 | 58.92 ± 3.4 | 39.79 ± 2.3 | 52.49 ± 2.9 | |
| 6 | 4-ClPh | 66.23 ± 3.9 | 14.15 ± 1.2 | 7.83 ± 0.6 | 18.18 ± 1.3 | |
| 7 | 4-CH3Ph | >100 | 76.41 ± 4.3 | 69.43 ± 3.7 | 65.99 ± 3.7 | |
| 8 | 4-OCH3Ph | 31.83 ± 2.2 | >100 | 74.56 ± 4.1 | 76.07 ± 3.8 | |
| 9 | 2,6-(CH3)2Ph | 62.19 ± 3.5 | 53.10 ± 3.0 | 18.66 ± 1.4 | 25.20 ± 2.0 | |
| 10 | 2-Cl-4-CF3Ph | 86.85 ± 4.4 | 49.64 ± 2.7 | 44.95 ± 2.5 | 38.39 ± 2.3 | |
| 11 | –Bn | 54.06 ± 3.0 | 8.10 ± 0.6 | 6.72 ± 0.3 | 9.43 ± 0.7 | |
| 12 | –Cl | Ph | 75.51 ± 4.0 | 19.50 ± 1.3 | 9.52 ± 0.8 | 12.94 ± 1.0 |
| 13 | 4-FPh | 47.46 ± 2.6 | 63.48 ± 3.7 | 24.83 ± 1.9 | 32.65 ± 2.2 | |
| 14 | 4-ClPh | 16.74 ± 1.4 | 29.78 ± 1.8 | 35.01 ± 2.2 | 36.31 ± 2.3 | |
| 15 | 4-CH3Ph | 43.16 ± 2.5 | 68.60 ± 3.9 | 67.71 ± 3.8 | 59.49 ± 3.3 | |
| 16 | 4-OCH3Ph | 17.82 ± 1.3 | 46.60 ± 2.6 | 51.46 ± 2.7 | 43.75 ± 2.4 | |
| 17 | 2,6-(CH3)2Ph | 89.43 ± 4.7 | 22.38 ± 1.5 | 13.50 ± 1.1 | 21.41 ± 1.8 | |
| 18 | 2-Cl-4-CF3Ph | 84.39 ± 4.2 | 34.59 ± 2.0 | 28.79 ± 2.1 | 26.31 ± 1.9 | |
| 19 | –Bn | >100 | 73.49 ± 4.2 | 79.47 ± 4.3 | 68.07 ± 3.9 | |
| 20 | –OCH3 | Ph | 42.86 ± 2.6 | 5.17 ± 0.4 | 4.42 ± 0.2 | 6.39 ± 0.4 |
| 21 | 4-FPh | 27.63 ± 1.9 | 11.53 ± 0.9 | 2.83 ± 0.1 | 10.23 ± 0.9 | |
| 22 | 4-ClPh | 15.48 ± 1.2 | 37.75 ± 2.1 | 64.34 ± 3.5 | 55.53 ± 3.1 | |
| 23 | 4-CH3Ph | 63.88 ± 3.7 | 41.29 ± 2.4 | 57.12 ± 3.2 | 48.68 ± 2.6 | |
| 24 | 4-OCH3Ph | 71.38 ± 3.8 | >100 | >100 | 93.74 ± 4.7 | |
| 25 | 2,6-(CH3)2Ph | 56.35 ± 3.3 | 83.56 ± 4.5 | >100 | 78.32 ± 4.1 | |
| 26 | 2-Cl-4-CF3Ph | 30.17 ± 2.1 | 72.80 ± 4.0 | 85.24 ± 4.5 | 61.92 ± 3.5 | |
| 27 | –Bn | >100 | 87.27 ± 4.6 | 91.16 ± 4.9 | 82.75 ± 4.3 | |
| Sorafenib | — | — | 10.65 ± 0.8 | 9.18 ± 0.6 | 5.47 ± 0.3 | 7.26 ± 0.3 |
IC50 value defines the compound concentration required to achieve 50% inhibition of cancer cell proliferation. Data are shown as the mean of three independent experiments ± SD. IC50, (μM) values are classified as follows: very strong (1–10 μM), strong (11–20 μM), moderate (21–50 μM), weak (51–100 μM), very weak (100–200 μM), inactive (above 200 μM).
Anti-tumor activity against the HEPG-2, HCT-116 and MCF-7 cell lines revealed some essential SAR features (Fig. 2). 4, 12 and 20 equipped with unsubstituted phenyl group at the distal end of urea moiety (IC50 values of 1.5–19.5 μM) possessed anti-tumor activity more than the corresponding congers with different substituents at the same phenyl ring. 11 with the benzyl group at the urea group showed a superior inhibitory activity (IC50 values of 8.10 μM) compared to the substituted Ar analogues (5–10). The introduction of this methylene spacer between the urea functionality and the phenyl ring in 11 showed a 2–4-fold decrease in IC50 value compared to 4. 21 with 4-fluorophenyl on the urea group showed comparable activity with 20 as both shared a 4-methoxy group on the distal end of the N3-phenyl group.
Fig. 2. Summary of structure–activity correlation of target s-alkylated quinazoline-4(3H)-ones 4–27.

4.2. In vitro cytotoxicity against human normal cell
The safety margin of the newly synthesized quinazolinone analogues was evaluated by assessing their cytotoxicity on the WI-38 standard fibroblast cell line to determine if the synthesized derivatives showed preferential cytotoxicity towards tumor over normal cells (Table 1). Notably, the tested compounds had decreased cytotoxicity against normal fibroblast cells WI-38, as indicated by their IC50 values. When compared to sorafenib (IC50 = 10.65 μM), the most active 4, 11, and 20 have a reduced damaging effect on WI-38, with IC50 values of 45.61, 54.06, and 42.86 μM, respectively.
4.3. EGFR/VEGFR-2 enzyme inhibition assay
Candidates with the highest anti-cancer activity against the tested cell lines were chosen to investigate their dose-related enzymatic inhibition of EGFR/VEGFR-2 further at five different concentrations to determine their IC50 values.36,54,72 The investigated 4, 11, and 20 had significant EGFR/VEGFR-2 inhibitory activity with IC50 values in the nanomolar range, as shown in Table 2. 4 was the most potent analogue for strongly inhibiting EGFR, with an IC50 of 0.049 μM compared to the conventional medication lapatinib (IC50 of 0.059 μM). Also, 4 showed the lowest IC50 value (0.054 μM) amongst the tested analogues regarding VEGFR-2 inhibition (sorafenib has an IC50 of 0.041 μM). From the obtained results, we could deduce that the best scenario for this series of a compound is to have 4-methylphenyl groups at the N3-position of the quinazolinone core and unsubstituted phenyl on the far end of the urea group at the 2-position of the quinazolinone scaffold, as in 4. The introduction of a methylene spacer between the phenyl ring and the urea group, as in 11, resulted in a 2–3-fold reduction of IC50 value against EGFR/VEGFR-2, compared to 4. 20 showed the highest IC50 value against EGFR and VEGFR-2 between the tested analogues. It showed a 9–10-fold decrease in binding affinity, reflecting the detrimental effect of the 4-methoxy group at the para-position on the N3-phenyl moiety.
In vitro EGFR/VEGFR-2 inhibitory effects of the synthesized 4, 11 and 20 compared to lapatinib and sorafenib.
| Comp. no. | EGFR IC50 (μM) | VEGFR-2 IC50 (μM) |
|---|---|---|
| 4 | 0.049 ± 0.002 | 0.054 ± 0.002 |
| 11 | 0.117 ± 0.005 | 0.156 ± 0.007 |
| 20 | 0.362 ± 0.014 | 0.474 ± 0.02 |
| Lapatinib | 0.059 ± 0.002 | — |
| Sorafenib | — | 0.041 ± 0.002 |
4.4. Cell cycle analysis
The cell cycle involves a series of events in development and growth that culminate in DNA replication and cell division.73 Flow cytometry is used to evaluate cell proliferation at several cell cycle phases (subG1, G1, S, and G2/M).35,74–76 Cell cycle distribution and apoptosis induction on HCT-116 cells were investigated using propidium iodide staining, compared to DMSO as control, to gather additional data about the mechanism of 4 in cancer cell growth inhibition.77 HCT-116 cells were treated with 4 (using a concentration at the IC50 value for inhibition of this cell line) for 24 hours before being stained with propidium iodide (PI) and analyzed using a flow cytometer (FCM). The results (Table 3 and Fig. 3) demonstrated that 4 treatment resulted in a significant increase in the ratio of HCT-116 cells in the G0/G1 phase to 55.17% compared to 43.93% (untreated cells).
Effect of 4 on the cell cycle progression in HCT-116 cells compared to DMSO control.
| Comp. no | Cell cycle distribution (%) | ||
|---|---|---|---|
| G0–G1 | S | G2-M | |
| 4 | 55.17 | 36.18 | 8.65 |
| Control | 43.93 | 36.53 | 19.54 |
Fig. 3. Effect of 4 (left panel) and DMSO as a control (right panel) on DNA-ploidy flow cytometric analysis of HCT-116 cells after treatment for 24 h.

4.5. Detection of apoptosis
One of the promising strategies for cancer treatment is apoptosis targeting and the creation of apoptosis inducers.74–77 To measure the proportion of apoptosis produced by 4 in HCT-116 cells, a double staining flow cytometry test utilising Annexin V-FITC/propidium iodide was performed. 4 causes early apoptosis in HCT-116 cells by (26.34%) over control DMSO-treated cells (0.62%), as depicted in Fig. 4. Furthermore, it induces late apoptotic by 16.5% compared to untreated control (0.21%). 4 triggered total apoptosis with 46.53%, higher than the apoptotic effect in control untreated cells (2.15%). In addition, 4 showed necrosis by only 3.69% compared to untreated cells (1.32%) (Fig. 4 and 5).
Fig. 4. Compound 4 affects the percentage of annexin V-FITC-positive staining in HCT-116 cells. The cells were treated with DMSO as control and 4 for 24 hours. Dead (necrotic) cells exist in the Q1 quadrant; late apoptosis is represented in the Q2 quadrant; live cells are shown in the Q3 quadrant; early apoptosis is depicted in the Q4 quadrant.

Fig. 5. Total, late and early apoptosis induced by 4 compared to DMSO control.

5. Molecular modeling study
The molecular modeling method explores all 3D structures of ligands and molecular targets to predict plausible binding interactions within the putative active site of the protein of interest and study the molecular properties of the target compounds.11,14,78,79 This methodology bridges experimental and theoretical models, thus enabling the design of more potent compounds.80,81
5.1. Molecular docking
5.1.1. Molecular docking studies with the EGFR using 4, 11, and 20
The docking study of 4, 11, 20, and co-crystallized ligand (lapatinib) with the EGFR enzyme provides insightful data for the comparative analysis of these potential inhibitors. Lapatinib demonstrates a compelling binding profile with a notably high affinity docking score (−10.69 kcal mol−1). Lapatinib, the co-crystallized inhibitor, showed several interactions, such as regular hydrogen bonds, extensive hydrophobic contacts, and Pi interactions, indicating a stable interaction with EGFR (Fig. 6 and Table S1†). In the context of interactions of the target compounds with EGFR, each compound demonstrates unique binding characteristics that could potentially inform their efficacy as inhibitors, as evidenced by their IC50 values. 4 exhibits the highest affinity for EGFR among the tested compounds (IC50 = 0.049 μM). The potent inhibition could be attributed to its favorable binding energy (−9.58 kcal mol−1) and two crucial hydrogen bonds formed with amino acid residue ASP855, a residue within the DFG motif essential for ATP binding and kinase activity (Fig. 6 and Table S1†). The extensive hydrophobic interactions and the unique Pi–sulfur interaction with the amino acid residue MET1002 likely contribute to the compound's high affinity and specificity for EGFR. These observations suggest that 4 might block the ATP binding site, function as a competitive inhibitor, and be a promising candidate for further drug development.82
Fig. 6. Visual representation of molecular interactions within the EGFR active site. This 3D model showcases the comparative binding patterns of investigated 4 (in pink), 11 (in yellow), 20 (in cyan), and reference compound LPB (in metallic hue). Each compound exhibits distinct conformational orientations and interactions within the kinase domain, highlighting their potential differential impacts on EGFR's (PDB: 1xkk) enzymatic activity and signaling pathways.

11 exhibits slightly less inhibitory activity than 4 (IC50 = 0.049 μM) with an IC50 of 0.117 μM and demonstrates significant binding affinity, as evidenced by its docking score of −8.80 kcal mol−1. The interaction of 11 with CYS797 and the DFG motif's ASP855, primarily through hydrogen bonds, is noteworthy (Fig. 6 and Table S1†). These interactions and its extensive Pi and hydrophobic interactions could contribute to its potency as an EGFR inhibitor. Interestingly, the interaction of 11 with residues in both the N-lobe and C-lobe suggests a potential influence on the kinase's overall conformation and activation state, indicating a sophisticated mechanism of inhibition beyond mere competitive ATP blockage.
The least inhibitory 20 (IC50 of 0.362 μM), despite its favorable docking score (−8.67 kcal mol−1), might be the specificity of interactions within the ATP binding site or the molecular conformation that accounts for this relative decrease in activity. The hydrogen bond with ASP855 and multiple hydrophobic interactions, including those with critical residues like LEU844, CYS797, and LYS745, suggest a strong affinity for the binding site (Fig. 6 and Table S1†). Still, the overall orientation and interactions might not induce the same level of inhibition as observed with 4 or lapatinib.82
5.1.2. Molecular docking studies with the VEGFR2 using compounds 4,11, and 20
The docking studies of 4,11, 20, and sorafenib with the VEGFR2 kinase domain present a profound understanding of the molecular interaction governing these compounds' binding affinity and inhibitory potential. Each compound exhibits a distinct interaction profile with the VEGFR2 active site, correlating somewhat with their biological inhibitory effect (IC50s) against VEGFR2. Sorafenib, the reference drug, showcases the most potent inhibition with an IC50 of 0.041 μM, supported by its extensive interaction network within the VEGFR2 active site. Essential hydrogen bonds with critical residues like ASP1046 and GLU885 and its halogen bonds due to the fluorine constituent underpin its high affinity and potent inhibitory action (Fig. 7 and Table S2†). The hydrophobic interactions, especially within the HYD II region, and its strategic occupation of the ATP site underscore sorafenib's mechanism of locking the kinase in an inactive DFG-out conformation, a commonality it shares with the test compounds.
Fig. 7. Visual representation of molecular interactions within the VEGFR-2 active site. The 3D model showcases the binding patterns of the investigated 4 (in pink), 11 (in yellow), 20 (in cyan), and the reference compound SRB (in green). Each compound exhibits unique conformational orientations and interactions within the kinase domain, highlighting their potential differential impacts on VEGFR-2's enzymatic activity and signaling pathways (PDB: 3wze). This visual representation offers valuable insights into the compounds' molecular mechanisms and potential effects on VEGFR-2 and its associated signaling pathways.

The following, 4, has an IC50 of 0.054 μM, which strongly inhibits VEGFR-2. Its hydrogen bonding with ASP1046 and GLU885 is reminiscent of Sorafenib's interaction profile, highlighting the significance of these residues in high-affinity binding. An exciting feature is the sulfur–X interaction, a unique aspect suggesting an additional avenue for binding that could be explored for novel inhibitory mechanisms (Fig. 7 and Table S2†). Like sorafenib, the hydrophobic interactions of 4 play a crucial role in its stability within the catalytic cleft, albeit with a different set of residues. This compound's affinity, although slightly lower than sorafenib, coupled with its unique binding features, makes it a promising scaffold for further optimisation.83
11 exhibits moderate VEGFR2 inhibition with an IC50 of 0.156 μM. Its hydrogen bonds with GLU885 and ASP1046 emphasize the universal importance of these residues in inhibitor binding. The presence of Pi-alkyl interactions points towards a reliance on hydrophobic forces for stability within the active site, a strategy employed by all discussed compounds (Fig. 7 and Table S2†). However, the reduced number of these interactions, especially in the HYD regions, may account for its lower affinity relative to sorafenib and 4.
20, despite its lowest affinity (IC50 of 0.474 μM), demonstrates a binding profile that combines features observed in sorafenib and the other compounds. While less extensive, its hydrogen bonding involves critical residues like GLU885 and ASP1046 (Fig. 7 and Table S2†). Furthermore, its Pi-anion and hydrophobic interactions suggest an attempt to stabilize within the VEGFR2 active site similarly to the higher affinity compounds. The comparative lack of these interactions, possibly in the ATP or DFG site, could partially explain its reduced inhibitory potency.
5.2. The Molecular Dynamics Simulation (MDS)
The Molecular Dynamics Simulation (MDS) results and discussion reveal insightful information about the conformational dynamics and interactions of EGFR and VEGFR2 with 4. Analyzing the root mean square deviation (RMSD) for both the apo (unbound) and 4-bound states of EGFR, a pronounced rise in RMSD was observed in the initial phase, with the apo form experiencing a more significant increase than the bound form and suggesting that 4 may induce structural dynamics or rearrangements in EGFR. Towards the end of the simulation, both states exhibit analogous RMSD values, but fluctuations are more evident in the apo form. A similar trend is observed in VEGFR2, indicating that 4 introduces variability in the structure, though the protein–ligand complex remains relatively stable (Fig. 8).
Fig. 8. Root Mean Square Deviation (RMSD) trends for EGFR and VEGFR2 in its apo form (depicted in red and black, respectively) and when bound to 4 with EGFR and VEGFR2 (illustrated in blue and green, respectively) over the simulation time. The plot highlights the dynamic conformational changes of EGFR and VEGFR2 in both states.

The Root Mean Square Fluctuation (RMSF) analysis provides detailed insights into local flexibility. In EGFR, 4 stabilizes regions near the N-lobe, increasing flexibility in the α-helix C segment. In VEGFR2, 4 influences hydrophobic regions, the linker, and the DFG motif, suggesting its potential role as a type II inhibitor (Fig. S1†). The Solvent Accessible Surface Area (SASA) analysis indicates that 4 binding results in a more compact conformation for both EGFR and VEGFR2, with reduced variability in the complex (Fig. S2†). The radius of gyration (Rg) trends also supports the idea of 4 inducing a stable and compact structure in both receptors (Fig. S3†).
The hydrogen bond analysis of 4 with EGFR and VEGFR2 reveals a dynamic interaction with multiple residues, including those crucial for ATP binding (Fig. S4†). 4 exhibits a hybrid mechanism, interacting with residues associated with type I and type II inhibitors. The diverse hydrogen bond network, stability, and relatively high number of hydrogen bonds (H-bonds) suggest that 4 may have enhanced solubility and could be biologically relevant. Overall, these findings shed light on the structural dynamics, flexibility, and interaction mechanisms of EGFR and VEGFR2 in the presence of 4, providing valuable insights for understanding the compound's potential therapeutic applications.
6. Conclusions
4–27 are designed and synthesized as novel N3-phenyl-2-S-alkylated quinazolinones to explore the fragmented nature at the 2,3-positions on the quinazolinone core. The biological efficacy of our synthesized compounds was confirmed through in vitro anti-tumor efficacy, EGFR/VEGFR-2 inhibition, and apoptotic induction. 4, 11, and 20 were the most active against HepG-2, HCT-116, and MCF-7 cancer cell lines with IC50 values of 1.5–9.43 μM, which was comparable to sorafenib activity (IC50 = 5.4–9.18 μM). 4 showed potent inhibition for EGFR and VEGFR-2, equivalent to the control drug, with an IC50 value of 0.049 μM and 0.054 μM, respectively. The most potent analog 4 induced apoptosis in HCT-116 cancer cells to 46.53% (DMSO-treated cells showed only 2.15%), and the cell cycle ceased at the G1 phase. The anti-tumor and EGFR/VEGFR-2 inhibitory properties of 4, 11, and 20 were further explained using molecular docking and dynamic simulation. Molecular Dynamics Simulations revealed that 4 induces structural dynamics, stabilizes specific regions, and influences the conformational flexibility of EGFR and VEGFR2, suggesting its potential as a versatile inhibitor with implications for therapeutic applications. The prompt information gathered from docking and dynamic simulation can be utilized for further lead optimization to design and rationalize novel and more effective inhibitors.
7. Experimental
7.1. General chemistry
Melting values (°C) are uncorrected and calculated with the Stuart apparatus (SMP 30). The FT-IR 200 spectrophotometer ( cm−1) at the Faculty of Pharmacy, Mansoura University, Egypt, was used to record the IR spectra (KBr). Using TMS as an internal standard, the 1H-NMR (400 MHz) and 13C-NMR (100 MHz) spectra were measured in (DMSO-d6) at the NMR Unit, Faculty of Pharmacy, Mansoura University, Egypt. At the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University in Egypt, mass spectra were obtained using a Thermo Scientific Gas Chromatography-Mass Spectrometry (GCMS) model I.S.Q. on the direct inlet portion of the mass analyzer. For a GC/MS analysis, Electron Ionization (EI) was employed in full scan mode. The mass spectrometer operates over a mass-to-charge (m/z) range of 40–1000, with an electron energy set at 70 electron volts (eV). The chemicals and reagents were bought from commercial sources, including Aldrich Chemicals Co. in the United States. The essential precursors, thiols (1a–c), ethyl esters (2a–c), and hydrazides (3a–c), could be easily prepared according to the previously described literature procedures.67–69
cm−1) at the Faculty of Pharmacy, Mansoura University, Egypt, was used to record the IR spectra (KBr). Using TMS as an internal standard, the 1H-NMR (400 MHz) and 13C-NMR (100 MHz) spectra were measured in (DMSO-d6) at the NMR Unit, Faculty of Pharmacy, Mansoura University, Egypt. At the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University in Egypt, mass spectra were obtained using a Thermo Scientific Gas Chromatography-Mass Spectrometry (GCMS) model I.S.Q. on the direct inlet portion of the mass analyzer. For a GC/MS analysis, Electron Ionization (EI) was employed in full scan mode. The mass spectrometer operates over a mass-to-charge (m/z) range of 40–1000, with an electron energy set at 70 electron volts (eV). The chemicals and reagents were bought from commercial sources, including Aldrich Chemicals Co. in the United States. The essential precursors, thiols (1a–c), ethyl esters (2a–c), and hydrazides (3a–c), could be easily prepared according to the previously described literature procedures.67–69
7.1.1. Synthetic procedures
General procedure for the synthesis of 3,4-dihydroquinazolin-(2-yl)thioacetylhydrazinecarboxamides (4–27)
A mixture of hydrazide derivatives 3a–c (0.2 mmol) and appropriate substituted phenyl isocyanate (1.1 eq.) in DMF (2 mL) was stirred at 80 °C overnight. Then, the reaction mixture was poured into ice to afford precipitates filtered off, washed with water, and recrystallized with ethyl acetate to give target products of 4–27.
2-(2-((4-Oxo-3-(p-tolyl)-3,4-dihydroquinazolin-2-yl)thio)acetyl)-N-phenylhydrazinecarboxamide (4)
White solid (0.065 g, 71%). M.p. 235–237 °C. IR (νmax cm−1): 3330, 3280, 2985, 2925, 1700, 1620, 1545, 1260. 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.67 (s, 1H), 8.18 (s, 1H), 8.10 (d, J = 7.5 Hz, 1H), 7.85 (d, J = 7.2 Hz, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.50 (t, J = 7.5 Hz, 1H), 7.44–7.38 (m, 4H), 7.35 (d, J = 8.3 Hz, 2H), 7.25 (t, J = 7.8 Hz, 2H), 6.96 (t, J = 7.3 Hz, 1H), 3.99 (s, 2H), 2.43 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 167.4, 161.2, 157.5, 156.4, 147.6, 140.3, 139.4, 135.3, 133.6, 130.6, 129.6, 128.6, 127.0, 126.7, 126.6, 126.0, 125.7, 120.0, 35.2, 21.1. GC-MS EI m/z (%): 459.28 (M+, 20.69).
N-(4-Fluorophenyl)-2-(2-((4-oxo-3-(p-tolyl)-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (5)
White solid (0.06 g, 63%). M.p. 235–237 °C. IR (νmax cm−1): 3335, 3280, 2985, 2925, 1700, 1625, 1545, 1260. 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H), 8.72 (s, 1H), 8.21 (s, 1H), 8.09 (d, J = 7.3 Hz, 1H), 7.87–7.83 (m, 1H), 7.67 (d, J = 7.6 Hz, 1H), 7.51–7.34 (m, 7H), 7.10 (d, J = 8.2 Hz, 2H), 3.99 (s, 2H), 2.43 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.5, 161.2, 159.1 (d, JC–F = 240.0 Hz), 155.8, 147.6, 140.2, 136.2, 135.3, 133.6, 130.6, 129.6, 127.0, 126.7 (d, JC–F = 12.0 Hz), 126.5, 120.9, 120.0, 115.7 (d, JC–F = 20.0 Hz), 115.5, 35.1, 21.3. GC-MS EI m/z (%): 477.23 (M+, 22.19).
N-(4-Chlorophenyl)-2-(2-((4-oxo-3-(p-tolyl)-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (6)
White solid (0.069 g, 70%). M. p. 239–241 °C. IR (νmax cm−1): 3340, 3200, 3025, 2920, 1695, 1615, 1550, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.82 (s, 1H), 8.27 (s, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.86 (t, J = 7.2 Hz, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.56 (d, J = 8.4 Hz, 2H), 7.48 (s, 1H), 7.40 (d, J = 8.4 Hz, 2H), 7.39–7.30 (m, 4H), 3.99 (s, 2H), 2.43 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.5, 161.2, 157.4, 156.4, 147.6, 140.2, 139.2, 139.0, 135.3, 133.6, 130.5, 129.6, 128.9, 127.0, 126.7, 126.5, 125.9, 120.6, 35.1, 21.4. GC-MS EI m/z (%): 493.83 (M+, 18.68).
2-(2-((4-Oxo-3-(p-tolyl)-3,4-dihydroquinazolin-2-yl)thio)acetyl)-N-(p-tolyl)hydrazinecarboxamide (7)
White solid (0.068 g, 72%). M.p. 240–242 °C. IR (νmax cm−1): 3335, 3200, 3020, 2920, 1690, 1615, 1550, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.06 (s, 1H), 8.55 (s, 1H), 8.12 (s, 1H), 8.10 (d, J = 7.9 Hz, 1H), 7.85 (t, J = 7.5 Hz, 1H), 7.69 (d, J = 7.9 Hz, 1H), 7.50 (t, J = 7.2 Hz, 1H), 7.38–7.21 (m, 6H), 7.06 (d, J = 7.9 Hz, 2H), 3.98 (s, 2H), 2.43 (s, 3H), 2.23 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.5, 161.2, 157.4, 155.7, 147.6, 140.2, 137.3, 135.4, 133.6, 131.3, 130.6, 129.6, 129.5, 127.0, 126.7, 126.6, 120.0, 119.2, 35.0, 21.3, 20.8. GC-MS EI m/z (%): 473.73 (M+, 20.61).
N-(4-Methoxyphenyl)-2-(2-((4-oxo-3-(p-tolyl)-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (8)
White solid (0.071 g, 72%). M.p. 237–239 °C. IR (νmax cm−1): 3335, 3290, 2985, 2925, 1695, 1620, 1545, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.04 (s, 1H), 8.49 (s, 1H), 8.12 (s, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.85 (t, J = 7.5 Hz, 1H), 7.67 (d, J = 8.1 Hz, 1H), 7.50 (t, J = 7.5 Hz, 1H), 7.40 (d, J = 7.9 Hz, 2H), 7.35 (d, J = 7.9 Hz, 2H), 7.31 (d, J = 8.8 Hz, 2H), 6.84 (d, J = 8.8 Hz, 2H), 3.98 (s, 2H), 3.71 (s, 3H), 2.43 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.5, 161.2, 157.4, 155.9, 155.0, 147.6, 140.2, 135.3, 133.5, 132.9, 130.5, 129.6, 127.0, 126.7, 126.5, 121.0, 120.0, 114.3, 55.6, 35.1, 21.3. GC-MS EI m/z (%): 489.40 (M+, 25.69).
N-(2,6-Dimethylphenyl)-2-(2-((4-oxo-3-(p-tolyl)-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (9)
White solid (0.063 g, 65%). M.p. 241–243 °C. IR (νmax cm−1): 3320, 3280, 2990, 2925, 1700, 1630, 1545, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H), 8.45 (s, 1H), 8.15 (s, 1H), 8.10 (d, J = 7.8 Hz, 1H), 7.86 (t, J = 7.5 Hz, 1H), 7.20–7.13 (m, 6H), 7.04–7.00 (m, 2H), 6.63–6.59 (m, 1H), 4.01 (s, 2H), 2.43 (s, 3H), 2.20 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 161.2, 157.5, 155.6, 147.6, 140.3, 139.7, 138.0, 135.4, 133.6, 130.6, 129.6, 127.0, 126.7, 126.6, 124.1, 120.0, 116.8, 35.1, 21.5, 21.3. GC-MS EI m/z (%): 487.65 (M+, 28.60).
N-(2-Chloro-4-(trifluoromethyl)phenyl)-2-(2-((4-oxo-3-(p-tolyl)-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (10)
White solid (0.079 g, 71%). M.p. 230–232 °C. IR (νmax cm−1): 3340, 3220, 2980, 2920, 1700, 1620, 1550, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.11 (s, 1H), 9.16 (s, 1H), 8.52 (s, 1H), 8.10 (d, J = 7.6 Hz, 1H), 8.05–8.02 (m, 1H), 7.85 (t, J = 7.3 Hz, 1H), 7.77–7.73 (m, 1H), 7.68 (d, J = 8.0 Hz, 1H), 7.60 (d, J = 8.6 Hz, 1H), 7.50 (t, J = 7.4 Hz, 1H), 7.41 (d, J = 7.8 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 4.00 (s, 2H), 2.43 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 161.2, 157.4, 155.6, 147.6, 140.3, 139.7, 135.3, 133.6, 132.4, 130.5, 129.6, 127.0, 126.9, 126.7, 126.5, 124.6, 123.8, 122.9, 121.9, 120.0, 35.0, 21.3. GC-MS EI m/z (%): 561.93 (M+, 15.55).
N-Benzyl-2-(2-((4-oxo-3-(p-tolyl)-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (11)
White solid (0.072 g, 76%). M.p. 242–244 °C. IR (νmax cm−1): 3360, 3220, 2980, 2920, 1695, 1615, 1550, 1260; 1H NMR (400 MHz, DMSO-d6) δ 9.99 (s, 1H), 8.06 (d, J = 7.9 Hz, 1H), 8.02 (s, 1H), 7.83 (t, J = 7.3 Hz, 1H), 7.63 (d, J = 7.9 Hz, 1H), 7.48 (t, J = 7.3 Hz, 1H), 7.40 (d, J = 7.6 Hz, 2H), 7.36–7.25 (m, 4H), 7.25–7.10 (m, 3H), 6.86–6.82 (m, 1H), 4.16 (d, J = 5.1 Hz, 2H), 3.92 (s, 2H), 2.43 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 161.1, 158.5, 157.6, 147.5, 140.7, 140.2, 135.3, 133.5, 130.5, 129.6, 128.5, 127.2, 127.1, 127.1, 127.0, 126.5, 120.0, 42.9, 35.1, 21.3. GC-MS EI m/z (%): 473.23 (M+, 25.75).
2-(2-((3-(4-Chlorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)-N-phenylhydrazinecarboxamide (12)
White solid (0.07 g, 75%). M.p. 239–241 °C. IR (νmax cm−1): 3340, 3200, 2985, 2915, 1695, 1625, 1545, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.66 (s, 1H), 8.13 (d, J = 8.1 Hz, 2H), 7.86 (s, 1H), 7.92 (d, J = 7.6 Hz, 2H), 7.61 (d, J = 7.6 Hz, 2H), 7.51–7.2 (m, 6H), 6.96 (s, 1H), 4.01 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 167.4, 161.1, 156.7, 155.6, 147.6, 139.9, 135.5, 135.3, 135.1, 131.9, 130.2, 129.1, 127.0, 126.7, 126.6, 122.5, 120.0, 119.1, 35.3. GC-MS EI m/z (%): 479.63 (M+, 22.69).
2-(2-((3-(4-Chlorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)-N-(4-fluorophenyl)hydrazinecarboxamide (13)
White solid (0.072 g, 72%). M.p. 238–240 °C. I.R. (νmax cm−1): 3320, 3200, 3020, 2920, 1690, 1615, 1560, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.72 (s, 1H), 8.15 (s, 1H), 8.10 (d, J = 7.8 Hz, 1H), 7.85 (t, J = 7.9 Hz, 1H), 7.76–7.38 (m, 8H), 7.10 (d, J = 8.0 Hz, 2H), 4.02 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 167.4, 161.1, 156.8 (d, JC–F = 242.0 Hz), 155.7, 147.6, 136.2, 135.4, 135.3, 135.1, 131.9, 130.2, 127.1, 126.7 (d, JC–F = 8.0 Hz), 126.6, 121.3, 120.2, 116.3, 115.5 (d, JC–F = 22.0 Hz), 35.1. GC-MS EI m/z (%): 497.63 (M+, 24.09).
N-(4-Chlorophenyl)-2-(2-((3-(4-chlorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (14)
White solid (0.067 g, 65%). M.p. 244–246 °C. IR (νmax cm−1): 3330, 3220, 2985, 2920, 1700, 1615, 1550, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 8.83 (s, 1H), 8.29 (s, 1H), 8.10 (d, J = 7.8 Hz, 1H), 7.87 (t, J = 7.9 Hz, 1H), 7.72–7.66 (m, 3H), 7.61–7.45 (m, 5H), 7.34–7.30 (m, 2H), 4.02 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 167.4, 161.1, 156.7, 155.5, 147.6, 139.0, 135.5, 135.3, 135.1, 131.9, 130.2, 128.9, 127.0, 126.7, 126.6, 126.0, 120.6, 120.0, 35.1. GC-MS EI m/z (%): 514.30 (M+, 21.61).
2-(2-((3-(4-Chlorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)-N-(p-tolyl)hydrazinecarboxamide (15)
White solid (0.069 g, 70%). M.p. 242–244 °C. IR (νmax cm−1): 3340, 3220, 2985, 2925, 1695, 1615, 1550, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.08 (s, 1H), 8.56 (s, 1H), 8.12 (s, 1H), 8.10 (d, J = 7.8 Hz, 1H), 7.66–7.60 (m, 7H), 7.24–7.16 (m, 4H), 4.02 (s, 2H), 2.24 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.4, 161.1, 157.3, 155.7, 147.6, 137.3, 135.5, 135.3, 135.1, 132.0, 131.3, 130.3, 129.5, 127.0, 126.7, 126.6, 120.0, 119.1, 35.1, 20.8. GC-MS EI m/z (%): 493.92 (M+, 15.22).
2-(2-((3-(4-Chlorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)-N-(4-methoxyphenyl)hydrazinecarboxamide (16)
White solid (0.075 g, 74%). M.p. 239–241 °C. IR (νmax cm−1): 3335, 3200, 3020, 2920, 1700, 1615, 1550, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.05 (s, 1H), 8.48 (s, 1H), 8.15 (s, 1H), 8.10 (d, J = 7.8 Hz, 1H), 7.86 (t, J = 7.9 Hz, 1H), 7.69–7.65 (m, 3H), 7.56–7.50 (m, 3H), 7.30 (d, J = 7.4 Hz, 2H), 6.84 (d, J = 7.7 Hz, 2H), 4.01 (s, 2H), 3.71 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.4, 161.1, 157.7, 155.9, 155.1, 147.6, 135.5, 135.3, 135.1, 133.7, 132.1, 130.2, 127.0, 126.7, 126.6, 121.0, 120.0, 114.3, 55.6, 35.1. GC-MS EI m/z (%): 509.75 (M+, 18.69).
2-(2-((3-(4-Chlorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)-N-(2,6-dimethylphenyl)hydrazinecarboxamide (17)
White solid (0.069 g, 68%). M.p. 250–252 °C. IR (νmax cm−1): 3360, 3230, 2985, 2915, 1695, 1620, 1550, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H), 8.45 (s, 1H), 8.15 (s, 1H), 8.10 (d, J = 7.8 Hz, 1H), 7.86 (t, J = 7.5 Hz, 1H), 7.69 (d, J = 8.4 Hz, 3H), 7.58–7.52 (m, 3H), 7.02 (s, 2H), 6.60 (s, 1H), 4.01 (s, 2H), 2.20 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 167.4, 161.1, 156.8, 155.6, 147.6, 139.7, 138.0, 135.5, 135.3, 135.1, 131.9, 130.2, 127.1, 127.0, 124.1, 120.0, 116.9, 35.1, 21.5. GC-MS EI m/z (%): 507.89 (M+, 19.55).
N-(2-Chloro-4-(trifluoromethyl)phenyl)-2-(2-((3-(4-chlorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (18)
White solid (0.08 g, 69%). M.p. 220–222 °C. IR (νmax cm−1): 3350, 3230, 2985, 2920, 1695, 1620, 1550, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.12 (s, 1H), 9.17 (s, 1H), 8.53 (s, 1H), 8.10 (d, J = 7.4 Hz, 1H), 8.06–8.03 (m, 1H), 7.85 (d, J = 7.0 Hz, 1H), 7.76–7.72 (m, 1H), 7.73–7.66 (m, 3H), 7.63–7.49 (m, 4H), 4.03 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 167.4, 161.1, 156.7, 155.7, 147.6, 139.7, 135.4, 135.3, 135.1, 132.4, 131.9, 130.2, 127.2, 127.0, 126.9, 126.8, 126.6, 124.7, 123.9.9, 123.0, 120.0, 35.1. GC-MS EI m/z (%): 582.25 (M+, 25.69).
N-Benzyl-2-(2-((3-(4-chlorophenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (19)
White solid (0.073 g, 74%). M.p. 247–249 °C. IR (νmax cm−1): 3355, 3230, 2985, 2915, 1695, 1620, 1550, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.00 (s, 1H), 8.12–7.99 (m, 2H), 7.84 (s, 1H), 7.69–7.62 (m, 3H), 7.58–7.48 (m, 3H), 7.36–7.16 (m, 5H), 6.83 (s, 1H), 4.17 (s, 2H), 3.95 (s, 2H); 13C NMR (100 MHz, DMSO-d6) δ 167.5, 161.0, 158.5, 156.9, 147.5, 140.7, 135.5, 135.3, 135.1, 131.9, 130.2, 128.5, 127.2, 127.1, 127.0, 126.6, 126.5, 119.9, 42.9, 35.1. GC-MS EI m/z (%): 493.76 (M+, 26.07).
2-(2-((3-(4-Methoxyphenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)-N-phenylhydrazinecarboxamide (20)
White solid (0.065 g, 68%). M.p. 220–222 °C. IR (νmax cm−1): 3350, 3215, 2990, 2925, 1690, 1620, 1550, 1255; 1H NMR (400 MHz, DMSO-d6) δ 10.06 (s, 1H), 8.65 (s, 1H), 8.12 (s, 1H), 8.10 (d, J = 7.6 Hz, 1H), 7.85 (s, 1H), 7.68 (s, 1H), 7.49 (s, 1H), 7.43–7.66 (m, 4H), 7.26–7.24 (m, 2H), 7.16–7.12 (m, 2H), 6.98–6.64 (m, 1H), 3.99 (s, 2H), 3.86 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 161.4, 160.7, 155.6, 147.6, 139.9, 135.3, 131.1, 129.1, 128.6, 127.0, 126.7, 126.5, 122.5, 120.0, 119.1, 115.2, 115.1, 55.9, 35.1. GC-MS EI m/z (%): 475.29 (M+, 19.77).
N-(4-Fluorophenyl)-2-(2-((3-(4-methoxyphenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (21)
White solid (0.074 g, 75%). M.p. 217–219 °C. IR (νmax cm−1): 3365, 3220, 2984, 2900, 1690, 1615, 1550, 1258; 1H NMR (400 MHz, DMSO-d6) δ 10.05 (s, 1H), 8.70 (s, 1H), 8.19 (s, 1H), 8.09 (d, J = 6.8 Hz, 1H), 7.84 (s, 1H), 7.66 (d, J = 7.0 Hz, 1H), 7.44–7.38 (m, 4H), 7.18–7.04 (m, 5H), 3.98 (s, 2H), 3.86 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 161.4, 160.6, 157.8 (d, JC–F = 237.0 Hz), 155.8, 147.6, 136.2, 135.3, 131.1, 128.6, 127.0, 126.7 (d, JC–F = 12.0 Hz), 126.5, 121.0, 120.0, 115.7 (d, JC–F = 22.0 Hz), 115.5, 115.2, 559, 35.1. GC-MS EI m/z (%): 493.33 (M+, 26.09).
N-(4-Chlorophenyl)-2-(2-((3-(4-methoxyphenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (22)
White solid (0.065 g, 64%). M.p. 230–232 °C. IR (νmax cm−1): 3350, 3220, 2984, 2910, 1700, 1615, 1550, 1260; 1H NMR (400 MHz, DMSO-d6) δ 10.07 (s, 1H), 8.82 (s, 1H), 8.26 (s, 1H), 8.09 (d, J = 7.6 Hz, 1H), 7.85 (t, J = 7.1 Hz, 1H), 7.67 (d, J = 7.9 Hz, 1H), 7.53–7.43 (m, 3H), 7.39 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 8.4 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H), 3.98 (s, 2H), 3.86 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 161.4, 160.7, 157.8, 155.6, 147.6, 138.9, 135.3, 131.1, 128.9, 128.6, 127.0, 126.7, 126.5, 126.1, 120.6, 120.0, 115.2, 55.9, 35.1. GC-MS EI m/z (%): 509.92 (M+, 25.88).
2-(2-((3-(4-Methoxyphenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)-N-(p-tolyl)hydrazinecarboxamide (23)
White solid (0.071 g, 72%). M.p. 225–227 °C. IR (νmax cm−1): 3355, 3220, 2984, 2900, 1690, 1615, 1550, 1258; 1H NMR (400 MHz, DMSO-d6) δ 10.05 (s, 1H), 8.54 (s, 1H), 8.10 (d, J = 8.7 Hz, 2H), 7.85 (t, J = 7.4 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.49 (t, J = 7.4 Hz, 1H), 7.39 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 7.9 Hz, 2H), 7.13 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 7.9 Hz, 2H), 3.98 (s, 2H), 3.86 (s, 3H), 2.23 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 161.4, 160.7, 157.8, 155.7, 147.6, 137.3, 135.3, 131.1, 129.5, 129.4, 128.7, 128.6, 127.0, 126.7, 126.5, 119.2, 115.2, 55.9, 35.1, 20.8. GC-MS EI m/z (%): 489.36 (M+, 28.62).
N-(4-Methoxyphenyl)-2-(2-((3-(4-methoxyphenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (24)
White solid (0.075 g, 74%). M.p. 220–222 °C. IR (νmax cm−1): 3340, 3215, 2990, 2925, 1695, 1620, 1550, 1250; 1H NMR (400 MHz, DMSO-d6) δ 10.02 (s, 1H), 8.47 (s, 1H), 8.12 (s, 1H), 8.09 (d, J = 7.9 Hz, 1H), 7.84 (t, J = 7.1 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.49 (t, J = 7.2 Hz, 1H), 7.39 (d, J = 8.3 Hz, 2H), 7.30 (d, J = 8.5 Hz, 2H), 7.13 (d, J = 8.3 Hz, 2H), 6.84 (d, J = 8.5 Hz, 2H), 3.98 (s, 2H), 3.86 (s, 3H), 3.71 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.5, 161.6, 160.7, 157.9, 155.9, 155.1, 147.6, 135.3, 132.9, 131.1, 128.6, 127.1, 126.7, 126.5, 121.0, 120.0, 115.2, 114.3, 55.9, 55.6, 35.1. GC-MS EI m/z (%): 505.50 (M+, 24.65).
N-(2,6-Dimethylphenyl)-2-(2-((3-(4-methoxyphenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (25)
White solid (0.074 g, 73%). M.p. 219–221 °C. IR (νmax cm−1): 3350, 3215, 2990, 2925, 1690, 1620, 1550, 1255; 1H NMR (400 MHz, DMSO-d6) δ 10.05 (s, 1H), 8.42 (s, 1H), 8.19–8.05 (m, 2H), 7.84 (t, J = 7.3 Hz, 1H), 7.68 (d, J = 7.9 Hz, 1H), 7.49 (t, J = 7.3 Hz, 1H), 7.39 (d, J = 8.5 Hz, 2H), 7.13 (d, J = 8.5 Hz, 2H), 7.00 (s, 2H), 6.60 (s, 1H), 3.97 (s, 2H), 3.86 (s, 3H), 2.21 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 161.4, 160.7, 157.9, 155.6, 147.6, 139.6, 138.0, 135.3, 131.1, 128.6, 127.0, 126.7, 126.6, 124.1, 120.0, 116.9, 115.4, 55.9, 35.1, 21.5. GC-MS EI m/z (%): 503.44 (M+, 23.55).
N-(2-Chloro-4-(trifluoromethyl)phenyl)-2-(2-((3-(4-methoxyphenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (26)
White solid (0.08 g, 69%). M.p. 228–230 °C. IR (νmax cm−1): 3360, 3220, 2984, 2910, 1700, 1615, 1550, 1258; 1H NMR (400 MHz, DMSO-d6) δ 10.09 (s, 1H), 9.15 (s, 1H), 8.49 (s, 1H), 8.09 (d, J = 7.7 Hz, 1H), 8.03 (s, 1H), 7.84 (t, J = 7.3 Hz, 1H), 7.75–7.66 (m, 2H), 7.59 (d, J = 8.5 Hz, 1H), 7.49 (t, J = 7.0 Hz, 1H), 7.38 (d, J = 8.3 Hz, 2H), 7.13 (d, J = 8.3 Hz, 2H), 3.99 (s, 2H), 3.86 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.7, 161.4, 160.7, 157.8, 155.6, 147.6, 139.6, 135.3, 132.4, 131.1, 128.6, 127.1, 126.7, 126.5, 124.6, 123.9, 123.0, 121.9, 120.0, 117.8, 115.2, 55.9, 35.1. GC-MS EI m/z (%): 577.85 (M+, 24.29).
N-Benzyl-2-(2-((3-(4-methoxyphenyl)-4-oxo-3,4-dihydroquinazolin-2-yl)thio)acetyl)hydrazinecarboxamide (27)
White solid (0.06 g, 61%). M.p. 225–227 °C. IR (νmax cm−1): 1H NMR (400 MHz, DMSO-d6) δ 9.99 (s, 1H), 8.06 (d, J = 7.9 Hz, 1H), 8.02 (s, 1H), 7.83 (t, J = 7.3 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.48 (t, J = 7.2 Hz, 1H), 7.36–7.25 (m, 2H), 7.25–7.10 (m, 3H), 7.18 (d, J = 8.3 Hz, 2H), 6.97 (d, J = 8.3 Hz, 2H), 6.83 (s, 1H), 4.16 (d, J = 5.1 Hz, 2H), 3.92 (s, 2H), 3.86 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 167.6, 161.1, 158.5, 157.6, 155.8, 147.5, 140.2, 135.3, 133.5, 130.5, 128.8, 127.2, 127.1, 127.0, 126.5, 125.0, 120.0, 114.5, 42.9, 55.9, 35.1. GC-MS EI m/z (%): 489.53 (M+, 28.68).
7.2. Biological assays
7.2.1. Anti-tumor screening
The in vitro evaluation for the anti-tumour activity of the synthesized quinazolinone analogues was carried out using MTT assay according to the previously reported method.70,71
7.2.2. In vitro EGFR/VEGFR-2 kinase inhibition assay
The EGFR/VEGFR-2 enzyme assay was done following the steps described in the previous report.71,72
7.2.3. Flow cytometry analysis of the cell cycle distribution
According to our previous report, cell cycle analysis was done using a FACSCalibur flow cytometer on the HCT-116 cell lines stained with propidium iodide (PI).71,74
7.2.4. Analysis of cellular apoptosis
Apoptosis induction was carried out using the HCT-116 cell lines, and the Annexin 5-FITC/PI detection kit was used similarly to the previously reported procedure.71,73
7.3. Molecular modeling
7.3.1. Molecular docking procedure
Crystallographic data of the protein structures were sourced from the RCSB Protein Databank for VEGFR2, which complexed with Sorafenib, and (PDB code 3wze) and EGFR, which was complexed with Lapatinib (PDB code 1XKK). The molecular docking details of 4, 11, and 20 are presented in the ESI.†
7.3.2. Molecular Dynamics (MD) methodology
The binding configurations of 4, showcasing strong affinity towards both EGFR and VEGFR2, underwent MD analysis. Molecular Dynamic simulation methods are shown in detail in the ESI.†
7.4. Statistical analysis
The obtained data were plotted and analyzed to determine the IC50 values of the tested compounds using nonlinear regression with a variable slope. IC50 values were calculated based on the mean and standard deviation (SD) for each concentration. All analyses were performed using GraphPad Prism version 9.0 (GraphPad Software, San Diego, CA, USA).
Data availability
The data supporting this study's findings are available from the corresponding author, S. S. T., upon reasonable request.
Conflicts of interest
The authors declare that they have no conflict of interest.
Supplementary Material
Acknowledgments
The authors extend their appreciation to the Researchers Supporting Project number (RSPD2024R1049), King Saud University, Riyadh, Saudi Arabia for funding this research.
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04828h
References
- Marklew R. E. Jackson A. A. Wiseman M. J. Wootton S. A. Trends Food Sci. Technol. 2022;130:3–10. [Google Scholar]
- Hamdi A. Said E. A Farahat A. AA El-Bialy S. AM Massoud M. Lett. Drug Des. Discovery. 2016;13:912–920. [Google Scholar]
- Sung H. Ferlay J. Siegel R. L. Laversanne M. Soerjomataram I. Jemal A. Bray F. Ca-Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- Christiea E. L. Bowtell D. D. L. Ann. Oncol. 2017;28:viii13–viii15. doi: 10.1093/annonc/mdx446. [DOI] [PubMed] [Google Scholar]
- Al-Sanea M. M. Chilingaryan G. Abelyan N. Mamikonyan M. Gasparyan H. Hovhannisyan S. Hamdi A. Ali A. R. Selim S. Mohamed A. A. PLoS One. 2022;17:e0272065. doi: 10.1371/journal.pone.0272065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanazi A. M. Alaa A.-M. Al-Suwaidan I. A. Abdel-Hamide S. G. Shawer T. Z. El-Azab A. S. Eur. J. Med. Chem. 2014;79:446–454. doi: 10.1016/j.ejmech.2014.04.029. [DOI] [PubMed] [Google Scholar]
- El-Ayaan U. Abdel-Aziz A. A.-M. Al-Shihry S. Eur. J. Med. Chem. 2007;42:1325–1333. doi: 10.1016/j.ejmech.2007.02.014. [DOI] [PubMed] [Google Scholar]
- Al-Suwaidan I. A. Abdel-Aziz N. I. El-Azab A. S. El-Sayed M. A.-A. Alanazi A. M. El-Ashmawy M. B. A-M Abdel-Aziz A. J. Enzyme Inhib. Med. Chem. 2015;30:679–687. doi: 10.3109/14756366.2014.960863. [DOI] [PubMed] [Google Scholar]
- Gomaa H. A. M. Chem. Biol. Drug Des. 2022;100:639–655. doi: 10.1111/cbdd.14129. [DOI] [PubMed] [Google Scholar]
- Asif M. Int. J. Med. Chem. 2014;2014:395637. doi: 10.1155/2014/395637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Azab A. S. Al-Dhfyan A. Abdel-Aziz A. A.-M. Abou-Zeid L. A. Alkahtani H. M. Al-Obaid A. M. Al-Gendy M. A. J. Enzyme Inhib. Med. Chem. 2017;32:935–944. doi: 10.1080/14756366.2017.1344981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanazi A. M. Al-Suwaidan I. A. Alaa A.-M. Mohamed M. A. El-Morsy A. M. El-Azab A. S. Med. Chem. Res. 2013;22:5566–5577. [Google Scholar]
- Mohamed M. A. Ayyad R. R. Shawer T. Z. Alaa A.-M. El-Azab A. S. Eur. J. Med. Chem. 2016;112:106–113. doi: 10.1016/j.ejmech.2016.02.002. [DOI] [PubMed] [Google Scholar]
- Al-Suwaidan I. A. Abdel-Aziz A. A.-M. Shawer T. Z. Ayyad R. R. Alanazi A. M. El-Morsy A. M. Mohamed M. A. Abdel-Aziz N. I. El-Sayed M. A.-A. El-Azab A. S. J. Enzyme Inhib. Med. Chem. 2016;31:78–89. doi: 10.3109/14756366.2015.1004059. [DOI] [PubMed] [Google Scholar]
- Haider K. Pathak A. Rohilla A. Haider M. R. Ahmad K. Yar M. S. Eur. J. Med. Chem. 2019;184:111773. doi: 10.1016/j.ejmech.2019.111773. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziz A. A.-M. Abou-Zeid L. A. ElTahir K. E. H. Mohamed M. A. El-Enin M. A. A. El-Azab A. S. Bioorg. Med. Chem. 2016;24:3818–3828. doi: 10.1016/j.bmc.2016.06.026. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziz A. A.-M. Abou-Zeid L. A. ElTahir K. E. H. Ayyad R. R. Magda A.-A. El-Azab A. S. Eur. J. Med. Chem. 2016;121:410–421. doi: 10.1016/j.ejmech.2016.05.066. [DOI] [PubMed] [Google Scholar]
- Farouk A. Abdelrasheed Allam H. Rashwan E. George R. F. Abbas S. E. Bioorg. Chem. 2022;128:106099. doi: 10.1016/j.bioorg.2022.106099. [DOI] [PubMed] [Google Scholar]
- Kharb R. Haider K. Neha K. Yar M. S. Arch. Pharm. 2020;353:e2000081. doi: 10.1002/ardp.202000081. [DOI] [PubMed] [Google Scholar]
- Al-Suwaidan I. A. Alanazi A. M. Abdel-Aziz A. A.-M. Mohamed M. A. El-Azab A. S. Bioorg. Med. Chem. Lett. 2013;23:3935–3941. doi: 10.1016/j.bmcl.2013.04.056. [DOI] [PubMed] [Google Scholar]
- Alanazi A. M. Abdel-Aziz A. A.-M. Shawer T. Z. Ayyad R. R. Al-Obaid A. M. Al-Agamy M. H. Maarouf A. R. El-Azab A. S. J. Enzyme Inhib. Med. Chem. 2016;31:721–735. doi: 10.3109/14756366.2015.1060482. [DOI] [PubMed] [Google Scholar]
- El-Azab A. S. Abdel-Aziz A. A.-M. Ghabbour H. A. Al-Gendy M. A. J. Enzyme Inhib. Med. Chem. 2017;32:1229–1239. doi: 10.1080/14756366.2017.1368504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Azab A. S. Alkahtani H. M. AlSaif N. A. Al-Suwaidan I. A. Obaidullah A. J. Alanazi M. M. Al-Obaid A. M. Al-Agamy M. H. Abdel-Aziz A. A.-M. J. Mol. Struct. 2023;1278:134928. [Google Scholar]
- Alkahtani H. M. Zen A. A. Obaidullah A. J. Alanazi M. M. Almehizia A. A. Ansari S. A. Aleanizy F. S. Alqahtani F. Y. Aldossari R. M. Algamdi R. A. Al-Rasheed L. S. Abdel-Hamided S. G. Abdel-Aziz A. A.-M. El-Azab A. S. Molecules. 2022;28:120. doi: 10.3390/molecules28010120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Azab A. S. Abdel-Aziz A. A.-M. Bua S. Nocentini A. El-Gendy M. A. Mohamed M. A. Shawer T. Z. AlSaif N. A. Supuran C. T. Bioorg. Chem. 2019;87:78. doi: 10.1016/j.bioorg.2019.03.007. [DOI] [PubMed] [Google Scholar]
- El-Azab A. S. Abdel-Aziz A. A.-M. Bua S. Nocentini A. AlSaif N. A. Alanazi M. M. El-Gendy M. A. Ahmed H. E. A. Supuran C. T. J. Enzyme Inhib. Med. Chem. 2020;35:733–743. doi: 10.1080/14756366.2020.1742117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Azab A. S. Abdel-Aziz A. A.-M. Bua S. Nocentini A. Bakheit A. H. Alkahtani H. M. Hefnawy M. M. Supuran C. T. Saudi Pharm. J. 2023;31:101866. doi: 10.1016/j.jsps.2023.101866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Azab A. S. Abdel-Aziz A. A.-M. Ahmed H. E. A. Bua S. Nocentini A. AlSaif N. A. Obaidullah A. J. Hefnawy M. M. Supuran C. T. J. Enzyme Inhib. Med. Chem. 2020;35:598–609. doi: 10.1080/14756366.2020.1722121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson P. R. Tan C. Jarman K. E. Lowes K. N. Curtis J. M. Nguyen W. Di Rago A. E. Bullen H. E. Prinz B. Duffy S. Baell J. B. Hutton C. A. Jousset Subroux H. Crabb B. S. Avery V. M. Cowman A. F. Sleebs B. E. J. Med. Chem. 2017;60:1171–1188. doi: 10.1021/acs.jmedchem.6b01673. [DOI] [PubMed] [Google Scholar]
- Gschwind A. Fischer O. M. Ullrich A. Nat. Rev. Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- Popper H. H. Ryska A. Timar J. Olszewski W. Transl. Lung Cancer Res. 2014;3:291–300. doi: 10.3978/j.issn.2218-6751.2014.10.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R. Weihua Z. Front. Oncol. 2019;9:800. doi: 10.3389/fonc.2019.00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigismund S. Avanzato D. Lanzetti L. Mol. Oncol. 2018;12:3–20. doi: 10.1002/1878-0261.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry D. W. Kraker A. J. McMichael A. Ambroso L. A. Nelson J. M. Leopold W. R. Connors R. W. Bridges A. J. Science. 1994;265:1093–1095. doi: 10.1126/science.8066447. [DOI] [PubMed] [Google Scholar]
- El-Azab A. S. Abdel-Aziz A. A.-M. AlSaif N. A. Alkahtani H. M. Alanazi M. M. Obaidullah A. J. Eskandrani R. O. Alharbi A. Bioorg. Chem. 2020;104:104345. doi: 10.1016/j.bioorg.2020.104345. [DOI] [PubMed] [Google Scholar]
- Alkahtani H. M. Alanazi M. M. Aleanizy F. S. Alqahtani F. Y. Alhoshani A. Alanazi F. E. Almehizia A. A. Abdalla A. N. Alanazi M. G. El-Azab A. S. Abdel-Aziz A. A.-M. Saudi Pharm. J. 2019;27:682–693. doi: 10.1016/j.jsps.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkahtani H. M. Abdalla A. N. Obaidullah A. J. Alanazi M. M. Almehizia A. A. Alanazi M. G. Ahmed A. Y. Alwassil O. I. Darwish H. W. Abdel-Aziz A. A.-M. El-Azab A. S. Bioorg. Chem. 2020;95:103461. doi: 10.1016/j.bioorg.2019.103461. [DOI] [PubMed] [Google Scholar]
- El-Husseiny W. M. El-Sayed M. A.-A. Abdel-Aziz N. I. El-Azab A. S. Ahmed E. R. Abdel-Aziz A. A.-M. J. Enzyme Inhib. Med. Chem. 2018;33:507–518. doi: 10.1080/14756366.2018.1434519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y. Yun C. H. Park E. Ercan D. Manuia M. Juarez J. Xu C. Rhee K. Chen T. Zhang H. Palakurthi S. Jang J. Lelais G. DiDonato M. Bursulaya B. Michellys P. Y. Epple R. Marsilje T. H. McNeill M. Lu W. Harris J. Bender S. Wong K. K. Janne P. A. Eck M. J. Nature. 2016;534:129–132. doi: 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Steen N. Caparello C. Rolfo C. Pauwels P. Peters G. J. Giovannetti E. OncoTargets Ther. 2016;9:6065–6074. doi: 10.2147/OTT.S97644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solca F. Dahl G. Zoephel A. Bader G. Sanderson M. Klein C. Kraemer O. Himmelsbach F. Haaksma E. Adolf G. R. J. Pharmacol. Exp. Ther. 2012;343:342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- Lau S. C. Chooback N. Ho C. Melosky B. Clin. Lung Cancer. 2019;20:e576–e583. doi: 10.1016/j.cllc.2019.05.003. [DOI] [PubMed] [Google Scholar]
- Maemondo M. Inoue A. Kobayashi K. Sugawara S. Oizumi S. Isobe H. Gemma A. Harada M. Yoshizawa H. Kinoshita I. Fujita Y. Okinaga S. Hirano H. Yoshimori K. Harada T. Ogura T. Ando M. Miyazawa H. Tanaka T. Saijo Y. Hagiwara K. Morita S. Nukiwa T. N. Engl. J. Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- Ribatti D., Recent Advances in Angiogenesis and Antiangiogenesis, Bentham Science Publishers, Sharjah, United Arab Emirates, 2009, pp. 1–133 [Google Scholar]
- Karamysheva A. F. Biochemistry. 2008;73:751–762. doi: 10.1134/s0006297908070031. [DOI] [PubMed] [Google Scholar]
- Kerbel R. S. Carcinogenesis. 2000;21:505–515. doi: 10.1093/carcin/21.3.505. [DOI] [PubMed] [Google Scholar]
- Stacker S. A. Caesar C. Baldwin M. E. Thornton G. E. Williams R. A. Prevo R. Jackson D. G. Nishikawa S. Kubo H. Achen M. G. Nat. Med. 2001;7:186–191. doi: 10.1038/84635. [DOI] [PubMed] [Google Scholar]
- Lee J. C. Chow N. H. Wang S. T. Huang S. M. Eur. J. Cancer. 2000;36:748–753. doi: 10.1016/s0959-8049(00)00003-4. [DOI] [PubMed] [Google Scholar]
- Poon R. T. Lau C. P. Cheung S. T. Yu W. C. Fan S. T. Cancer Res. 2003;63:3121–3126. [PubMed] [Google Scholar]
- Hlatky L. Tsionou C. Hahnfeldt P. Coleman C. N. Cancer Res. 1994;54:6083–6086. [PubMed] [Google Scholar]
- Holmes K. Roberts O. L. Thomas A. M. Cross M. J. Cell. Signal. 2007;19:2003–2012. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Strawn L. M. Shawver L. K. Expert Opin. Invest. Drugs. 1998;7:553–573. doi: 10.1517/13543784.7.4.553. [DOI] [PubMed] [Google Scholar]
- Shibuya M. Claesson-Welsh L. Exp. Cell Res. 2006;312:549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Hamdi A. El-Shafey H. W. Othman D. I. A. El-Azab A. S. AlSaif N. A. Abdel-Aziz A. A.-M. Bioorg. Chem. 2022;122:105710. doi: 10.1016/j.bioorg.2022.105710. [DOI] [PubMed] [Google Scholar]
- Lee K. Jeong K.-W. Lee Y. Song J. Y. Kim M. S. Lee G. S. Kim Y. Eur. J. Med. Chem. 2010;45:5420–5427. doi: 10.1016/j.ejmech.2010.09.002. [DOI] [PubMed] [Google Scholar]
- Abdullaziz M. A. Abdel-Mohsen H. T. El Kerdawy A. M. Ragab F. A. F. Ali M. M. Abu-Bakr S. M. Girgis A. S. El Diwani H. I. Eur. J. Med. Chem. 2017;136:315–329. doi: 10.1016/j.ejmech.2017.04.068. [DOI] [PubMed] [Google Scholar]
- Xie Q. Q. Xie H. Z. Ren J. X. Li L. L. Yang S. Y. J. Mol. Graphics Modell. 2009;27:751–758. doi: 10.1016/j.jmgm.2008.11.008. [DOI] [PubMed] [Google Scholar]
- Machado V. A. Peixoto D. Costa R. Froufe H. J. Calhelha R. C. Abreu R. M. Ferreira I. C. Soares R. Queiroz M. J. Bioorg. Med. Chem. 2015;23:6497–6509. doi: 10.1016/j.bmc.2015.08.010. [DOI] [PubMed] [Google Scholar]
- Dietrich J. Hulme C. Hurley L. H. Bioorg. Med. Chem. 2010;18:5738–5748. doi: 10.1016/j.bmc.2010.05.063. [DOI] [PubMed] [Google Scholar]
- Aziz M. A. Serya R. A. Lasheen D. S. Abdel-Aziz A. K. Esmat A. Mansour A. M. Singab A. N. Abouzid K. A. Sci. Rep. 2016;6:24460. doi: 10.1038/srep24460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garofalo A. Goossens L. Six P. Lemoine A. Ravez S. Farce A. Depreux P. Bioorg. Med. Chem. Lett. 2011;21:2106–2112. doi: 10.1016/j.bmcl.2011.01.137. [DOI] [PubMed] [Google Scholar]
- Ghorab W. M. El-Sebaey S. A. Ghorab M. M. Bioorg. Chem. 2023;131:106310. doi: 10.1016/j.bioorg.2022.106310. [DOI] [PubMed] [Google Scholar]
- Fang Z. Y. Zhang Y. H. Chen C. H. Zheng Q. Lv P. C. Ni L. Q. Sun J. Wu Y. F. Chem. Biodiversity. 2022;19:e202200189. doi: 10.1002/cbdv.202200189. [DOI] [PubMed] [Google Scholar]
- Zahran S. S. Ragab F. A. El-Gazzar M. G. Soliman A. M. Mahmoud W. R. Ghorab M. M. Eur. J. Med. Chem. 2023;245:114912. doi: 10.1016/j.ejmech.2022.114912. [DOI] [PubMed] [Google Scholar]
- Eissa I. H. Ibrahim M. K. Metwaly A. M. Belal A. Mehany A. B. M. Abdelhady A. A. Elhendawy M. A. Radwan M. M. ElSohly M. A. Mahdy H. A. Bioorg. Chem. 2021;107:104532. doi: 10.1016/j.bioorg.2020.104532. [DOI] [PubMed] [Google Scholar]
- Mahdy H. A. Ibrahim M. K. Metwaly A. M. Belal A. Mehany A. B. M. El-Gamal K. M. A. El-Sharkawy A. Elhendawy M. A. Radwan M. M. Elsohly M. A. Eissa I. H. Bioorg. Chem. 2020;94:103422. doi: 10.1016/j.bioorg.2019.103422. [DOI] [PubMed] [Google Scholar]
- Bello A. M. Wei L. Majchrzak-Kita B. Salum N. Purohit M. K. Fish E. N. Kotra L. P. Bioorg. Med. Chem. 2014;22:978–985. doi: 10.1016/j.bmc.2013.12.049. [DOI] [PubMed] [Google Scholar]
- Sonousi A. Hassan R. A. Osman E. O. Abdou A. M. Emam S. H. J. Enzyme Inhib. Med. Chem. 2022;37:2644–2659. doi: 10.1080/14756366.2022.2118735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed A. Mahmood S.-u. Flörke U. Turk. J. Chem. 2014;38:275–287. [Google Scholar]
- Mosmann T. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- El Hamaky N. F. Hamdi A. Bayoumi W. A. Elgazar A. A. Nasr M. N. Bioorg. Chem. 2024;148:107437. doi: 10.1016/j.bioorg.2024.107437. [DOI] [PubMed] [Google Scholar]
- Othman D. I. A. Hamdi A. Elhusseiny W. M. El-Azab A. S. Bakheit A. H. Hefnawy M. Abdel-Aziz A. A.-M. Saudi Pharm. J. 2023;31:101803. doi: 10.1016/j.jsps.2023.101803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormerod M. G. J. Immunol. Methods. 2002;265:73–80. doi: 10.1016/s0022-1759(02)00071-6. [DOI] [PubMed] [Google Scholar]
- Turky A. Bayoumi A. H. Ghiaty A. El-Azab A. S. Abdel-Aziz A. A.-M. Abulkhair H. S. Bioorg. Chem. 2020;101:104019. doi: 10.1016/j.bioorg.2020.104019. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziz A. A.-M. El-Azab A. S. AlSaif N. A. Obaidullah A. J. Al-Obaid A. M. Al-Suwaidan I. A. J. Enzyme Inhib. Med. Chem. 2021;36:1520–1538. doi: 10.1080/14756366.2021.1924698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdi A. Elhusseiny W. M. Othman D. I. Haikal A. Bakheit A. H. El-Azab A. S. Al-Agamy M. H. Abdel-Aziz A. A.-M. Eur. J. Med. Chem. 2022;244:114827. doi: 10.1016/j.ejmech.2022.114827. [DOI] [PubMed] [Google Scholar]
- Abass S. A. Elgazar A. A. El-kholy S. S. El-Refaiy A. I. Nawaya R. A. Bhat M. A. Farrag F. A. Hamdi A. Balaha M. El-Magd M. A. Molecules. 2024;29:1927. doi: 10.3390/molecules29091927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sanea M. M. Hamdi A. Brogi S. Tawfik S. S. Othman D. I. A. Elshal M. Rahman H. U. Parambi D. G. T. Elbargisy R. M. Selim S. Mostafa E. M. Mohamed A. A. B. J. Enzyme Inhib. Med. Chem. 2023;38:2162511. doi: 10.1080/14756366.2022.2162511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanazi A. M. El-Azab A. S. Al-Swaidan I. A. Maarouf A. R. El-Bendary E. R. Abu El-Enin M. A. Abdel-Aziz A. A.-M. Med. Chem. Res. 2013;22:6129–6142. [Google Scholar]
- Hamdi A. Yaseen M. Ewes W. A. Bhat M. A. Ziedan N. I. El-Shafey H. W. Mohamed A. A. B. Elnagar M. R. Haikal A. Othman D. I. A. Elgazar A. A. Abusabaa A. H. A. Abdelrahman K. S. Soltan O. M. Elbadawi M. M. J. Enzyme Inhib. Med. Chem. 2023;38:2231170. doi: 10.1080/14756366.2023.2231170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sanea M. M. Abdel-Maksoud M. S. El-Behairy M. F. Hamdi A. Rahman H. U. Parambi D. G. M Elbargisy R. Mohamed A. A. Bioorg. Chem. 2023;139:106716. doi: 10.1016/j.bioorg.2023.106716. [DOI] [PubMed] [Google Scholar]
- Roskoski Jr R. Pharmacol. Res. 2019;139:395–411. doi: 10.1016/j.phrs.2018.11.014. [DOI] [PubMed] [Google Scholar]
- McTigue M. Murray B. W. Chen J. H. Deng Y. L. Solowiej J. Kania R. S. Proc. Natl. Acad. Sci. U. S. A. 2012;109:18281–18289. doi: 10.1073/pnas.1207759109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this study's findings are available from the corresponding author, S. S. T., upon reasonable request.