

Abstract
Neuropilin-1 acts as a coreceptor with vascular endothelial growth factor receptors to facilitate binding of its ligand, vascular endothelial growth factor. Neuropilin-1 also binds to heparan sulfate, but the functional significance of this interaction has not been established. A combinatorial library screening using heparin oligosaccharides followed by molecular dynamics simulations of a heparin tetradecasaccharide suggested a highly conserved binding site composed of amino acid residues extending across the b1 and b2 domains of murine neuropilin-1. Mutagenesis studies established the importance of arginine513 and lysine514 for binding of heparin to a recombinant form of Nrp1 composed of the a1, a2, b1, and b2 domains. Recombinant Nrp1 protein bearing R513A,K514A mutations showed a significant loss of heparin-binding, heparin-induced dimerization, and heparin-dependent thermal stabilization. Isothermal calorimetry experiments suggested a 1:2 complex of heparin tetradecasaccharide:Nrp1. To study the impact of altered heparin binding in vivo, a mutant allele of Nrp1 bearing the R513A,K514A mutations was created in mice (Nrp1D) and crossbred to Nrp1+/– mice to examine the impact of altered heparan sulfate binding. Analysis of tumor formation showed variable effects on tumor growth in Nrp1D/D mice, resulting in a frank reduction in tumor growth in Nrp1D/– mice. Expression of mutant Nrp1D protein was normal in tissues, suggesting that the reduction in tumor growth was due to the altered binding of heparin/heparan sulfate to neuropilin-1. These findings suggest that the interaction of neuropilin-1 with heparan sulfate modulates its stability and its role in tumor formation and growth.
Introduction
Members of the neuropilin (Nrp) family of proteins (Nrp1 and Nrp2) are plasma membrane surface receptors that participate in angiogenesis and axon guidance.1−5 In angiogenesis, Nrp1 acts as a coreceptor for vascular endothelial growth factor receptor-2 (VEGFR2) and facilitates VEGF binding.5,6 In the central nervous system, Nrp1 forms a coreceptor with members of the plexin family to facilitate Sema3a binding and axon guidance.7,8 Nrp1 has also been identified as a coreceptor for human T-cell lymphotropic virus type 1 entry and recently as a potential receptor for SARS-CoV-2 virus.9−12 Thus, there is great interest in understanding the structure and function of the Nrp proteins in angiogenesis, axon guidance, and infectious disease.
Nrp1 and Nrp2 are evolutionarily conserved, possessing almost identical domain structures and 44% amino acid identity.2,4 The domain structure of Nrp1 includes two complement binding CUB domains (a1 and a2), two coagulation factor domains (b1 and b2), a MAM (meprin, A5, mu) domain (c), a transmembrane domain, and a short C-terminal cytoplasmic tail.13−15 The a1 and a2 domains are responsible for binding to the Sema domain of Sema3 ligands, and they facilitate interaction with the VEGF.16,17 The b1 and b2 domains bind the C-terminal domains of both VEGF and Sema3 ligands.17,18 The c domain does not directly facilitate Sema3 or VEGF binding.18 Therefore, other roles have been suggested for the c domain including regulating signaling, promoting proper positioning of the ectodomain, and facilitating other protein–protein interactions relevant to the Nrp1 function.19,20 Evolutionary conservation of the neuropilin structure emphasizes its importance as a cell surface receptor and likely participation in angiogenesis and axon guidance across species.
An electropositive patch in the b1b2 domains has been implicated as a ligand binding site for heparin.17,21 An oligosaccharide composed of at least eight sugar residues (degree of polymerization [dp] 8 oligosaccharide) was needed to compete with the binding of native heparin (∼dp50) to Nrp1, and dp20 heparin oligosaccharides were able to completely inhibit binding.17 Heparin has also been shown to induce oligomerization of Nrp1, and the effect is size dependent.21 Heparin is a form of heparan sulfate generated by connective tissue mast cells and is derived from porcine or bovine intestinal mucosa.22 Extensive sulfation of heparin makes it one of the most electronegative polymers found in nature. In contrast, heparan sulfate is made by all animal cells and varies significantly in the overall structure from heparin, especially in sulfation. How these structural variations impact Nrp1 is unknown.
In this report, we elucidated the structure of the heparin/heparan sulfate binding site in Nrp1 using molecular modeling and biochemical binding assays. To explore the functional significance of the heparin/heparan sulfate interaction with Nrp1, key amino acid residues required for binding were identified and mutated. Inactivation of the capacity of Nrp1 to bind heparin/heparan sulfate resulted in destabilization of the protein to thermal inactivation and reduced dimerization of the protein in vitro. Replacement of the wild-type Nrp1 gene in mice with the mutant reduced tumor formation in vivo.
Results
To locate and understand the heparin/heparan sulfate binding site in Nrp1 at the residue level, we first resorted to computational simulations and analysis. The surface of mouse Nrp1 (PDB ID: 4GZ9) was divided into 13 potential binding sites (BS1–BS13, Figure S1B) in an unbiased manner followed by implementation of the combinatorial virtual library screening (CVLS) algorithm. The CVLS algorithm is a genetic algorithm that has now been implemented on dozens of heparin/heparan sulfate binding proteins to identify the preferred binding site and preferred structures of oligosaccharide chains.23,24 The algorithm, which has been parametrized primarily for highly sulfated structures, utilizes a dual-filter strategy consisting of GOLDScore as the first filter followed by an RMSD filter. In combination, the two filters segregate the library of oligosaccharide chains into two bins, those that engage the putative binding site with high GOLDScore and low RMSD versus the rest. The genetic-screening function within the CVLS algorithm studies thousands of configurational and conformational variations to extract the best set of 6 poses for each preferred oligosaccharide sequence, which are then used to calculate the GOLDScore, a measure of “in silico affinity”, and RMSD (see Figure S1A), a measure of “in silico selectivity”.
For Nrp1, we implemented the CVLS algorithm on each of the 13 putative binding sites, utilizing a library of 72 heparin/heparan sulfate disaccharide topologies. When the docked poses of disaccharides were overlaid onto the 13 binding sites (Figure S1B), the higher affinity/selectivity disaccharides preferentially aligned with the electropositive cleft formed by the b1 and b2 domains (Figure 1A). Following this, a library of 288 hexasaccharides was generated from all possible combinations of disaccharides reported earlier.25 This library, containing hexasaccharides with 8 to 12 sulfate groups, was then studied using the dual filter CVLS algorithm. Figure 1B compiles the results of these experiments, which led to the conclusion that BS1, BS4, and BS5 are the three primary putative sites for binding of heparan sulfate hexasaccharides. When the best-fit hexasaccharides (Figure 1C–E), having the sequences IdoA-GlcNS-GlcA2S-GlcNS3S6S-IdoA-GlcNS3S6S, GlcNAc6S-IdoA2S-GlcNS6S-GlcA2S-GlcNS3S6S-GlcA, and GlcA-GlcNS6S-IdoA2S-GlcNAc-GlcA2S-GlcNS3S6S, respectively, were simultaneously projected onto the three binding sites, considerable overlap was noted between the best ranking sequences, suggesting a strong possibility of recognition of an extended heparan sulfate chain within the electropositive cleft in the b1 and b2 domains.
Figure 1.
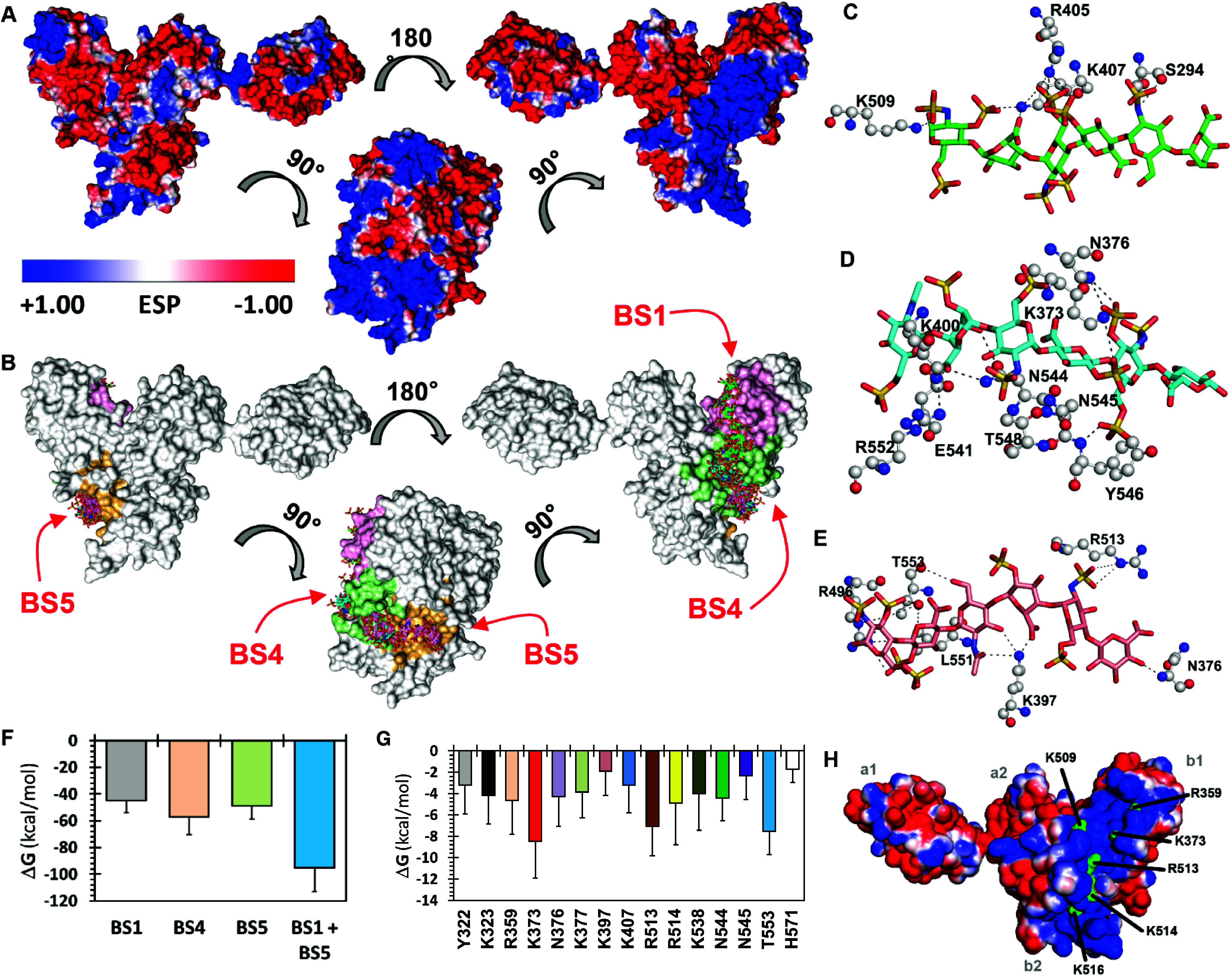
Combinatorial virtual library screening (CVLS) and molecular dynamics (MD) based identification of binding sites, key amino acid residues, and minimal chain length. A, Electrostatic potential surface of mouse Nrp1 (PDB: 4GZ9) using APBS tool in Pymol. Blue areas represent electropositive surfaces, and red areas represent electronegative surfaces. B, CVLS-predicted hexasaccharide sequences binding to three preferred sites (BS1, BS4 and BS5) on mNrp1 from a library of 288 sequences containing a range of sulfate groups (0–12) per sequence. Shown are overlays of the docked poses of hexasaccharide sequences that bind mNrp1 by satisfying the CVLS dual-filter strategy. Sequences that bound at the preferred binding sites: BS1 (green atom sticks), BS4 (cyan atom sticks), and BS5 (pink atom sticks) are shown. Note: sequences at BS4 and BS5 were found to partially overlap with each other. C–E, Interaction of heparin/heparan sulfate hexasaccharides at BS1 (C), BS4 (D) and BS5 (E). Hydrogen-bonding profile of hexasaccharide sequences with key residues of mNrp1 (shown as black dotted lines; hexasaccharides are shown as green (BS1), cyan (BS4) and pink (BS5) sticks). F, Total free energy value in kcal/mol, from 200 ns MD simulations of hexasaccharide bound to BS1, BS4, and BS5, respectively. Note: total free energy decreased (better binding) when BS1 and BS5 were simultaneously occupied. G, Free energy contributions for the 14-mer GAG sequence interacting amino acid residues in the binding region of the Nrp1 protein. H, Electrostatic potential plot of Nrp1 with selected residues colored green.
To identify the preferred chain length, we used molecular dynamics (MD) simulations on 1:1 docked complexes of hexasaccharides from CVLS onto Nrp1. The MD technology was now been successfully implemented on dozens of GAG–protein complexes to understand thermodynamic and mechanistic features of interactions.26,27 We implemented MD simulations in explicit water molecule box (TIP3P) using a constant temperature, constant pressure (NPT) ensemble of each high affinity/high selectivity hexasaccharide sequence bound in its preferred site, i.e., BS1, BS4, or BS5. Each hexasaccharide was found to bind well in its preferred binding site in the presence of water. MMGBSA-based free energy calculations revealed the order of preference with regard to sites of binding (BS4 > BS5 > BS1; see Figure 1F). It is important to note that the MMGBSA approach, which is a well-established method for calculating free energies from MD trajectories, is an excellent tool for qualitative comparison of closely related structures and, therefore, particularly suited for revealing preferred sites of binding. We then studied the effect of simultaneous, dual-site occupancy and found that the combined occupancy of BS1 and BS5 with their preferred hexasaccharide sequences was 2-fold better than single occupancy at any of three BSs (Figure 1F). Furthermore, simultaneous triple-site occupancy was found to be detrimental to binding (not shown). Rather, it was computationally possible to identify a disaccharide sequence that binds in BS4 and optimally connects on either side with the best hexasaccharide sequences of BS1 and BS5. This led to the hypothesis that a distinct tetradecasaccharide (dp14) would be predicted to bind in the long electropositive cleft of Nrp1.
To support this hypothesis, we studied the stability of the dp14 oligosaccharide in complex with Nrp1 using a 1 μs MD simulation (see Figures 1, S1C and S1D, see also Supporting Information Video 1). The average fluctuations of the dp14 oligosaccharide over a long simulation time (>100,000 structures) was 4.36 Å, while for Nrp1, it was 2.20 Å. Visualization of the full MD trajectory (Supporting Information Video 1) showed that the ligand remained anchored to the contiguous site formed by BS1, BS4, and BS5.
A better insight into the nature of interactions within the site of binding can be derived from the hydrogen-bond occupancy and enthalpy contributions of each amino acid present in the site of binding. Such an analysis has provided keen predictive insights into a range of proteins with minimal information on their recognition of heparin/heparan sulfate.26,28 Thus, we evaluated hydrogen-bond occupancies (Figure S1E) and single-residue free energy contributions to binding (Figure 1G). Not surprisingly, the long dp14 chain length implicated an extensive set of amino acid residues in binding including Glu271, Ser321, Tyr322, Lys323, Arg359, Lys373, Asn376, Lys377, Ala378, Lys397, Lys407, Lys509, Arg513, Lys514, Lys538, Glu541, Asn544, Asn545, Thr553, and His571 (Figures 1G and S1E). Of the 20 residues, nine are evolutionarily identical across multiple animal species, whereas the other 11 positions have conservative substitutions (Figure S1F). Comparison of Nrp1 to Nrp2 in humans, mice, and rats reveals that six of these residues are identical, five have conservative substitutions, and nine are nonconserved residues between the two isoforms. Of note, the positively charged lysine and arginine residues are highly conserved, as would be expected for a site involved in binding negatively charged polysaccharides such as heparin or heparan sulfate.22 In addition, the b2 domain contains a conserved XBBXBX amino acid motif across amino acid residues 512–517 (MRKFKI), which corresponds to a Cardin-Weintraub glycosaminoglycan binding consensus sequence.21,29
Structural studies of the Nrp1 indicate that subdomains b1b2 are tightly packed and form a rigid scaffold, with a deep cleft or groove that runs across the interdomain junction, measuring ∼40 Å in length.21 This cleft has dimensions that roughly correspond to the length of a heparin dp12.21 Mutation of specific arginine and lysine residues to glutamate (R359E, K373E, R513E, K514E, and K516E) along the cleft abolished the heparin-induced dimerization.21 Examination of the interaction of recombinant dimeric Fc fusion of rat Nrp1 with heparin suggested amino acid residues involved in heparin binding were also present in the a1 domains,30 although molecular docking studies with the murine a1a2b1b2 protein did not support this hypothesis.
Recombinant Protein Production
In the following biochemical studies, we expressed wild-type and mutant forms of the a1a2b1b2 domain of mouse Nrp1 using a construct previously utilized in the crystallization of mouse Nrp1.8 Nrp1 a1a2b1b2 domain has been shown to have a 3-to-8-fold greater affinity for VEGF165 over the b1b2 domain alone.17 The mouse construct was selected in anticipation of in vivo studies to examine the functional significance of the heparin binding domain. Based on the modeling results, the location of conserved amino acid residues, and previous mutational studies, the amino acid residues R359, K373, K509, R513, K514, and K516 were converted to alanine residues (Figure 1H). Robust expression was achieved using ExpiCHO cells to allow glycosylation of the two Asn-linked glycosylation sites in the a2 domain,31 which could affect binding to heparin/heparan sulfate. Previous studies employed recombinant protein expressed in E. coli which lacks the glycosylation enzymes found in mammalian systems.21 The resulting recombinant proteins, wild-type and seven alanine substitution mutants, were purified by Ni2+-affinity chromatography to take advantage of the His6Tag added to the N-terminus. All the recombinant proteins expressed at comparable levels (0.8–2.4 mg/L). SDS-PAGE analysis showed that all the proteins had the expected molecular weight and appeared to be stable based on the retention of the His6Tag and recognition by anti-Nrp1 antibody in western blots (Figure S2A). Each recombinant protein was also N-glycosylated as demonstrated by a comparable decrease in molecular weight following treatment of the purified proteins with PNGase to release the Asn-linked glycans (Figure S2B). The overall conformation of the recombinant proteins appeared similar based on their comparable migration on native PAGE (Figure S2C).
Binding of Recombinant Nrp1 Mutants to Heparin
To elucidate the contribution of these candidate amino acids to the interaction of Nrp1 a1a2b1b2 with heparan sulfate, recombinant proteins were subjected to a series of heparin and heparan sulfate binding assays. Heparin, a highly sulfated fractionated form of heparan sulfate, is commercially available in large quantities and is used as a surrogate for more typical heparan sulfate found on cells.22 To assess the interaction of the recombinant proteins with heparin, samples were bound to heparin sepharose and eluted with a salt concentration gradient from 0.15 to 0.6 M NaCl. Several of the recombinant mutant proteins showed small reductions in the salt concentration needed for elution compared to the wild-type a1a2b1b2 protein (0.43–0.5 M NaCl versus 0.52 M for wild-type Nrp1, Figure S2D, Table 1). R513A, K514A, and K373A mutants eluted at much lower salt concentrations (0.35, 0.43, and 0.43 M NaCl, respectively), suggesting these amino acids contributed significantly to the interaction of Nrp1 a1a2b1b2 with heparin (Figures 2A, S2D, Table 1). Recombinant Nrp1 a1a2b1b2 protein containing two mutations (R513A,K514A) showed an additive effect, eluting at 0.28 M NaCl (Δ = −0.24 M NaCl for the double mutant vs −0.17 and −0.09 M NaCl for mutants R513A and K514A, respectively).
Table 1. Salt Concentrations for Heparin Sepharose Elutions and Kd and Bmax Values for BSA-Heparin ELISAs.
| heparin
sepharose |
heparin-BSA
ELISA |
||||
|---|---|---|---|---|---|
| conductivity (mS/cm) | salt (M) | Δsalt (mM) | Kd (μM, 95% CI) | Bmax (95% CI) | |
| Nrp1 | 48 | 0.52 | 0 | 0.10–0.14 | 0.69–0.76 |
| R359A | 43 | 0.47 | –50 | 0.39–0.52 | 0.76–0.85 |
| K373A | 40 | 0.43 | –90 | 0.50–0.73 | 0.67–0.80 |
| K509A | 44 | 0.48 | –40 | 0.34–0.38 | 0.58–0.61 |
| R513A | 33 | 0.35 | –170 | 0.43–0.61 | 0.64–0.75 |
| K514A | 39 | 0.43 | –90 | 0.45–0.66 | 0.77–0.91 |
| K516A | 46 | 0.50 | –20 | 0.34–0.45 | 0.68–0.75 |
| R513A,K514A | 28 | 0.28 | –240 | 1.64–4.61 | 0.33–0.69 |
Figure 2.

Heparin binding of Nrp1 a1a2b1b2 mutants. A, Elution profiles of Nrp1 mutants on heparin sepharose. Wild-type Nrp1a1a2b1b2 and mutants were passed over heparin sepharose, and the concentration of NaCl required for elution from the column was determined. Dashed vertical line at 10 mL is the beginning of the elution phase. B, Binding of wild-type Nrp1 a1a2b1b2 and mutants to immobilized heparin-BSA. C, Increases in melting temperatures of the Nrp1 a1a2b1b2 proteins in the presence of heparin due to heparin-induced stabilization. Statistics denote a one-way ANOVA with Tukey’s multiple comparison test. **** p < 0.0001.
These results were validated in a different assay format in which binding of recombinant proteins was evaluated by ELISA using plates coated with heparin-BSA (Figure 2B, Figure S2E). Incubation with the wild-type protein showed saturable binding with an apparent Kd value of 0.10–0.14 μM (95% Cl). All of the single amino acid mutants showed reduced affinity, but the extent of binding based on Bmax values was comparable (Table 1). The double mutant R513A,K514A bound with much reduced affinity (Kd = 1.64–4.61 μM, 95% CI), and the extent of binding was reduced as well.
Heparin Stabilizes Neuropilin-1 against Thermal Denaturation
Binding of glycosaminoglycans to proteins often stabilizes them against denaturation compared to their unbound state.32,33 To determine the effect of heparin binding on Nrp1 stability, we utilized differential scanning fluorimetry to monitor the thermal denaturation of Nrp1 in the presence and absence of heparin. Nrp1 was mixed with 5X SYPRO Orange Protein Gel Stain, a hydrophobic dye that binds to the hydrophobic domains of the protein exposed by denaturation. Heparin-enhanced thermal stability appeared as an increase in the melting temperature. Wild-type Nrp1 a1a2b1b2 demonstrated a characteristic melting curve (Figure S2F), which yielded a melting temperature of 49.4 °C based on the maximum of the first derivative of the dye binding curves (Figure S2G). Increasing the molar ratio of heparin:protein from 1:1 to 10:1 resulted in further stabilization reflected as higher melting temperatures (50.6–51.6 °C) (Figure S2H). Analysis of the mutant proteins with a 10:1 molar ratio of heparin:protein showed that mutants K509A and K516A were similarly stabilized by heparin compared to the wildtype, whereas R359A, K373A, R513A, and K514A mutants showed less heparin-induced stabilization (Figure 2C). Heparin had no effect on the R513A,K514A double mutant as reflected by the lack of increase at the melting temperature (Figure 2C). As a control, we also examined melting of each protein construct in the absence of heparin. Some mutants exhibited slightly altered melting temperature in the absence of heparin indicating some differences in stability (K509A, R513A, R516A; Figure S2I). Nevertheless, the stabilization by heparin suggests that the heparin-binding sites can still engage heparin. The lack of stabilization by heparin of the double R513/K514A mutant is consistent with diminished levels of binding.
Heparin-Induced Dimerization of Neuropilin-1
Heparin binding by Nrp1 has been shown to induce dimerization of human b1b2 domains based on size exclusion chromatography.21 To explore the impact of heparin-binding on the oligomerization of murine a1a2b1b2 protein, we examined heparin oligosaccharides of different lengths. Wild-type protein was incubated with size-specific heparin oligosaccharides (dp8–dp20), and the resulting mixtures were analyzed by size-exclusion chromatography with multiangle light scattering (SEC-MALS). Murine Nrp1 a1a2b1b2 protein ran as a monomer with an apparent molecular mass of 74 ± 2% kDa by gel filtration (Table 2), consistent with the expected mass based on the amino acid sequence as well as previously determined molecular mass for Nrp1 a1a2b1b2.8 The consistent molecular mass across the peak suggested homogeneity (Figure 3). The extended heparin binding cleft accommodated dp6 to dp18 oligosaccharides, resulting in a shift in apparent molecular masses approximately equal to the summation of the masses of the individual components (Table 2). Incubation with dp20 heparin oligosaccharides resulted in a complex with an apparent molecular mass consistent with a dimer (∼160 kDa; Figure 3H, Table 2). These results suggest that heparin oligosaccharides shorter than dp20 can bind; the intermediate apparent molecular weights of these complexes most likely reflect the dynamic process of complex assembly and disassembly during the gel filtration run. Smaller trailing peaks in the OD280 traces may be due to the presence of a double bond at the nonreducing end of the heparin oligosaccharides that arose from enzymatic processing of heparin during production of the oligosaccharides (Figure 3O).
Table 2. Determined Molar Masses (MMs) for Nrp1 a1a2b1b2:Heparin Oligosaccharide Complexes.
| sample | oligo size (kDa) | MM (kDa) | error (%) | ratio to monomer | expected MM 1:1 complex (kDa) | expected MM 2:1 complex (kDa) |
|---|---|---|---|---|---|---|
| Nrp1 | 74 | 2 | 1.0 | 74 | ||
| Nrp1 + dp6 | 1.8 | 73 | 4 | 1.0 | 76 | 149 |
| Nrp1 + dp8 | 2.4 | 73 | 4 | 1.0 | 76 | 150 |
| Nrp1 + dp10 | 3.0 | 77 | 3 | 1.0 | 77 | 150 |
| Nrp1 + dp12 | 3.6 | 94 | 5 | 1.3 | 78 | 151 |
| Nrp1 + dp14 | 4.1 | 106 | 3 | 1.4 | 78 | 152 |
| Nrp1 + dp16 | 4.7 | 130 | 6 | 1.8 | 79 | 152 |
| Nrp1 + dp18 | 5.2 | 134 | 3 | 1.8 | 79 | 153 |
| Nrp1 + dp20 | 5.8 | 162 | 2 | 2.2 | 80 | 153 |
Figure 3.

Size-dependent heparin-induced dimerization of Nrp1. SEC-MALS chromatogram of Nrp1 a1a2b1b2 and A, heparin hexasaccharide (dp6); B, heparin octasaccharide (dp8); C, heparin decasaccharide (dp10); D, heparin dodecasaccharide (dp12); E, heparin tetradecasaccharide (dp14); F, heparin hexadecasaccharide (dp16); G, heparin octadecasaccharide (dp18); and H, heparin icosasaccharide (dp20). SEC-MALS chromatogram of I, R513A + heparin octadecasaccharide (dp18); J, K514A + heparin octadecasaccharide (dp18); K, R513A,K514A + heparin octadecasaccharide (dp18); L, R513A + heparin icosasaccharide (dp20); M, K514A + heparin icosasaccharide (dp20); N, R513A,K514A + heparin icosasaccharide (dp20); and O, a mix of heparin oligosaccharides (dp20, dp14, dp8). Gray lines are protein only, and black lines are protein incubated with oligosaccharide.
Alanine substitution of positively charged amino acid residues in the electropositive cleft in mouse Nrp1 a1a2b1b2 decreased its ability to bind heparin (Figure 2A), suggesting that heparin-mediated dimerization might be altered, as well. Incubation of dp20 heparin oligosaccharides with the R513A and K514A mutant proteins resulted in complexes of intermediate apparent molecular weights compared to wild-type protein (83 and 97 kDa versus 180 kDa, respectively; Figure 3L,M). A bound complex consisting of one heparin dp20 chain and one a1a2b1b2 monomer can account for the apparent molecular mass of the complexes formed by the single R513A and K514A mutants with dp20 heparin oligosaccharides (Table 3). The R513A,K514A double mutant showed no shift in molecular mass by gel filtration, supporting its weak interaction with heparin (Figure 3N; Table 3). Experiments with dp18 heparin oligosaccharides yielded similar results (Figure 3I–K; Table 3).
Table 3. Determined Molar Masses (MMs) for Nrp1 a1a2b1b2:dp18 and Nrp1 a1a2b1b2:dp20 complexes.
| solo |
+dp18 (5.2 kDa) |
+dp20 (5.8 kDa) |
expected
size + dp18 |
expected
size + dp20 |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| MM (kDa) | error (%) | MM (kDa) | error (%) | MM (kDa) | error (%) | 1:1 complex | 2:1 complex | 1:1 complex | 2:1 complex | |
| Nrp1 | 80 | 5 | 140 | 7 | 183 | 3 | 85 | 165 | 86 | 166 |
| R513A | 79 | 1 | 80 | 3 | 83 | 3 | 84 | 163 | 85 | 163 |
| K514A | 79 | 2 | 92 | 2 | 97 | 4 | 84 | 163 | 85 | 163 |
| R513A, K514A | 78 | 2 | 79 | 1 | 77 | 1 | 83 | 161 | 84 | 161 |
Neuropilin-1 Binds Heparin Oligosaccharides in a 1:2 Heparin:Protein Complex
The accuracy of SEC-MALS was not sufficient to accurately determine the stoichiometry of the heparin binding. Thus, we analyzed the complexes with isothermal calorimetry (ITC). Titration of wild-type a1a2b1b2 protein with a heparin dp14 oligosaccharide produced a KD value of 0.69 μM and a stoichiometry of 0.419 ± 0.004, indicating that the complex most likely consists of one heparin oligosaccharide and two a1a2b1b2 proteins (Figure 4A). Analysis of complexes generated by admixture of wild-type Nrp1 a1a2b1b2 with dp20 yielded similar results (KD of 0.49 μM and a stoichiometry of 0.404 ± 0.005, Figure S4A). The R513A,K514A double mutant engaged poorly with the heparin tetradecasaccharide, and the shape of the binding curve did not allow accurate determination of a KD value or stoichiometry (Figure 4B). Control titration of heparin oligosaccharides (dp14) into buffer alone showed appropriate peak magnitudes (Figure S4B,C). The stoichiometry of the 1:2 dp14:protein complex suggests that a single heparin oligosaccharide is adequate to dimerize the protein, even in the absence of other domains and other ligands.
Figure 4.

Nrp1 and heparin oligosaccharide form a 1:2 heparin:Nrp1 a1a2b1b2 complex. Isothermal calorimetry was used to assess complex formation of Nrp1 a1a2b1b2 with heparin tetrasaccharides. Nrp1 a1a2b1b2 (A) and the R513A,K514A mutant (B).
Interaction of Neuropilin-1 with Heparan Sulfate
While heparin is commonly used as a surrogate for heparan sulfate, Nrp1 on the plasma membrane of cells more likely engages heparan sulfate in the form of proteoglycans present in the glycocalyx. Prior studies showed that human recombinant human Nrp1 b1b2 domain can bind cell surface heparan sulfate based on flow cytometry.34 Wild-type mouse Nrp1 a1a2b1b2 protein bound to wild-type CHO-K1 cells, and binding was proportional to concentration up to 3 μM, the highest concentration that was tested (Figure 5A). The recombinant mutant proteins showed various levels of binding to the cells when tested at 1 μM; R359A and K516A mutants retained similar levels of binding as the wild-type protein, whereas K373A, K509A, R513A, and K514A bound to a lesser extent (Figure 5B). Engagement of the recombinant R513A,K514A mutant protein with the cell surface was especially attenuated. Prior treatment of the cells with a combination of heparin lyases I, II, and III (HSase), which removes cell surface heparan sulfate, diminished binding of wildtype and mutants to a similar baseline level (Figure 5B). Staining of untreated and HSase-treated CHO-K1 cells with an antiheparan sulfate antibody (10E4) demonstrated efficient removal of heparan sulfate from the cell surface (Figure S5). Binding was also measured in human umbilical cord endothelial cells (HUVECs). Binding showed more typical signs of saturability, but HSase treatment only partially diminished the level of binding (Figure 5C). Furthermore, binding of the R513A,K514A mutant was reduced to a level like that observed for wild-type protein after treatment of the endothelial cells with HSase (Figure 5D), whereas binding of the R513A,K514A mutant was minimally affected by HSase treatment. Together, these results show that the reduced binding of the a1a2b1b2 mutants to heparin was recapitulated in studies of recombinant protein with native heparan sulfate present on mammalian cells.
Figure 5.
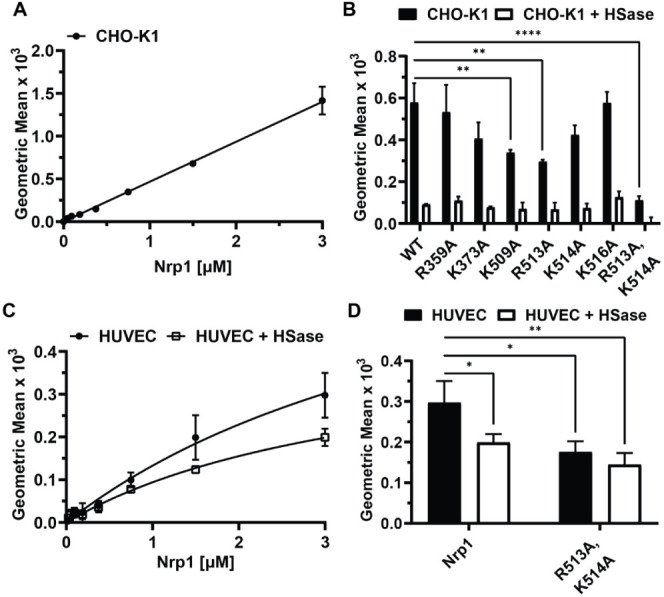
Neuropilin-1 binding to cell surface heparan sulfate. A, CHO-K1 cells titrated with recombinant wild-type Nrp1 a1a2b1b2 protein. B, Binding of wild-type Nrp1 a1a2b1b2 and mutants (1 μM) to CHO-K1 cells with and without treatment with heparin lyases I, II, and III (HSase). Statistics denote a one-way ANOVA with Tukey’s multiple comparison test. ** p < 0.002 and **** p < 0.0001. C, HUVECs were titrated with recombinant wild-type Nrp1 a1a2b1b2 protein. D, Binding of wild-type Nrp1 a1a2b1b2 and mutant (3 μM) to HUVECs with and without HSase treatment. Specific binding (total binding–nonspecific binding) is shown for each condition in this figure. Statistics denote a two-way ANOVA with Tukey’s multiple comparison test. * p < 0.033 and ** p < 0.002.
Generation of Heparan Sulfate Binding Deficient Nrp1 (Nrp1D) Mouse Model
Nrp1 plays important roles in developmental processes including axon guidance and angiogenesis based on the phenotype of mice bearing null alleles of Nrp1 or mutant alleles in which binding to VEGFA or Sema3A were altered.1,35−38 To assess how Nrp-1 interaction with heparan sulfate could influence its functions in vivo, we created a mouse model expressing full length Nrp1 with the double mutation R513A;K514A in order to systemically reduce its interaction with heparan sulfate. CRISPR/Cas9 gene editing was employed with an sgRNA designed to target Nrp1 near amino acid residues 513 and 514 along with a donor DNA oligonucleotide to allow for the alanine substitution of these target residues. Sequencing of the resulting heparan sulfate binding-deficient mutant confirmed the desired change in genomic sequence (Figure S6A). This sequence alteration was further validated by the loss of a BspHI restriction enzyme site in the Nrp1D sequence, rendering PCR-generated amplicons of the site resistant to digestion (Figure S6B).
To address if the Nrp1D protein was expressed at levels comparable to wild-type Nrp1, homogenates of the mouse lungs were separated by SDS-PAGE and immunoblotted with antibodies to Nrp1 and β-actin (Figure S6C). Animals with either a single copy of the mutant allele (Nrp1D/+) or two copies of the mutant allele (Nrp1D/D) expressed similar levels of Nrp1 to that of wild-type mice (Figure S6C,D). Nrp1D/– mice express comparable levels of protein to Nrp1+/–, which was roughly half of the wild-type level of expression (Figure S6C,D). Additionally, Nrp1 mRNA levels were normal, as expected based on the protein expression data (Figure S6E).
While homozygous Nrp1 knockout animals (Nrp1–/–) are embryonic lethal,1,35Nrp1+/–, Nrp1D/D, and Nrp1D/– animals survived to adulthood and bred normally. Nrp1 is expressed in bone marrow stromal cells and endothelial cells that support the hematopoietic stem cell niche.39,40 Tibias of wildtype and Nrp1 mutant mice (Nrp1+/–, Nrp1D/+, and Nrp1D/–) were analyzed for total bone marrow cellularity (Figure S7A), c-Kit+ Sca-1+ Lin– (KSL) hematopoietic stem/progenitor cells (HSPCs) (Figure S7B), CD48– CD150+ KSL long-term hematopoietic stem cells (LT-HSCs) (Figure S7C), and c-Kit+ Sca-1– Lin– hematopoietic progenitor cells (Figure S7D). Although there was reduced total bone marrow cellularity concomitant with decreased numbers of HSPCs in the Nrp1+/– mice, HSPC frequency and mature blood cells were unchanged (Figure S7E), indicating a modest impact to the hematopoietic system in Nrp1 mutant mice.
Continual growth of solid tumors requires tumor angiogenesis to support their metabolic needs;41 specifically, in a subcutaneous B16 melanoma model, tumor growth has been shown to be dependent on VEGF-mediated angiogenesis.42,43 This model has also been used more recently to demonstrate that Nrp1 plays an important part in the VEGF-stimulated angiogenesis and tumor growth.36,44 To assess if heparan sulfate binding by Nrp1 affects tumor growth in this model, 5 × 105 B16–F10 cells were injected subcutaneously on the flank of the animals and yielded tumors over the next 15 days that were easily measured with calipers (Figure 6A). Tumors in Nrp1D/D animals showed reduced growth in one set of experiments (Figure 6B) but did not exhibit a diminution in growth when the experiment was repeated (Figure 6C), possibly reflecting differences in the overall injected dose and bilateral versus unilateral tumor burden. To sensitize tumor growth and obtain more consistent results, we reduced the amount of Nrp1 protein in half by using Nrp1D/– (Figure S6D) and comparing the growth of the unilateral tumors in Nrp1D/– to Nrp1+/– and Nrp1+/+ animals. Tumors in Nrp1D/– mice grew much slower and remained smaller than the other groups (Figure 6B,C6). This difference in growth is unlikely to be from the reduced expression of Nrp1 in these animals as normal tumor growth is seen in the Nrp1+/– mice, which express the same amount of protein and mRNA as the Nrp1D/– animals (Figure S6C,D).
Figure 6.
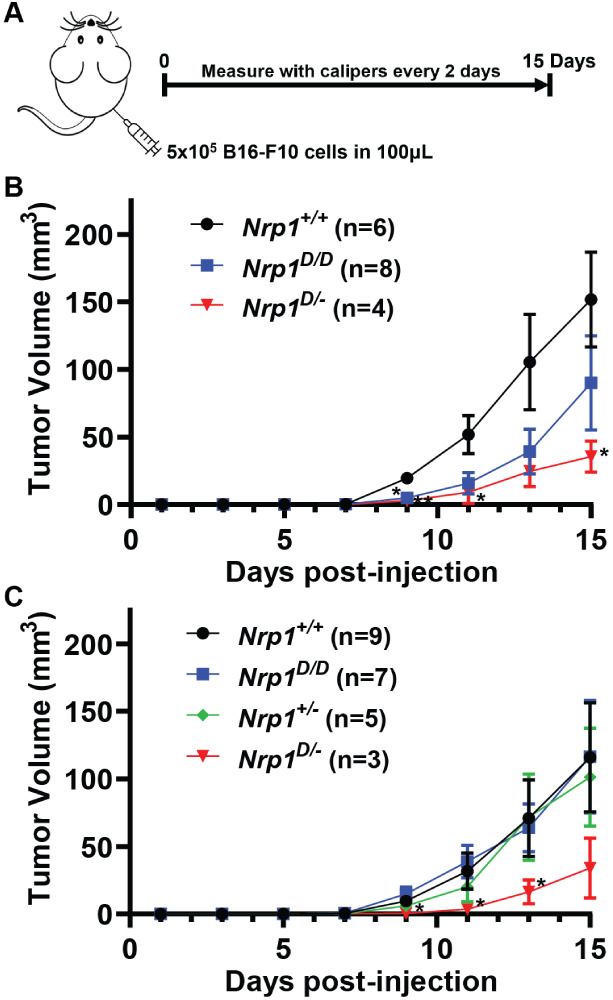
Alteration of neuropilin-1 and heparan sulfate binding impairs subcutaneous B16 tumor growth. A, Schematic of subcutaneous B16–F10 tumor model. B, Tumor growth curves of bilateral subcutaneous B16–F10 tumors in wild-type and mutant Nrp1 mice. Significance between Nrp1+/+ and Nrp1D/–. C, Tumor growth curves of unilateral subcutaneous B16–F10 tumors in wild-type and mutant Nrp1 mice. Significance between Nrp1D/D and Nrp1D/–. There is some variability between the bilateral and unilateral models, as observed from differences in the growth curves of the Nrp1D/D mice. Statistics denote a two-way ANOVA with the Geisser–Greenhouse correction followed by a Fisher’s LSD test for each set of data. * p < 0.05 and ** p < 0.01.
In summary, we mapped the heparin/heparan sulfate binding site in mouse neuropilin-1 and defined, through molecular modeling and biochemical evaluation of alanine substitution mutants, some of the amino acids crucial for Nrp1 interaction with heparin and heparan sulfate. Binding stabilizes the protein to thermal denaturation and induces dimerization in the absence of other ligands and domains of the protein. Dimerization of Nrp1 was found to be dependent on the length of the heparin chain, with dp14 oligosaccharides being able to induce a 1:2 heparin:Nrp1 complex. The mutations also affected the ability of Nrp1 to bind heparan sulfate on endothelial cells. Finally, reduction of binding to heparan sulfate by Nrp1 affects subcutaneous tumor growth.
Discussion
Nrp1 plays critical roles in the development of cardiovascular and peripheral nervous systems. In the nervous system, Nrp1 modulates axon guidance by acting as a coreceptor with plexin receptors and different semaphorins.8 In the vascular system, Nrp1 regulates vasculogenesis and angiogenesis by forming complexes with vascular endothelial growth factors (VEGFs) and VEGF receptors.1,45 These developmental processes are also modulated by heparin and heparan sulfate, which can bind both Nrp1, VEGF receptors, and certain VEGF isoforms, such as VEGF165. Previous studies have focused on heparin and heparan sulfate interactions with Nrp1 in vitro using rat and human forms of Nrp1 subdomains, in particular, b1b2 and a1a2b1b2 subdomains. Here we characterized the binding of heparin and heparan sulfate to murine a1a2b1b2 domains and examined protein stability, oligomerization, and the stoichiometry of binding and as a segue to in vivo studies of tumor growth in the mouse.
It is well established that both Nrp1 and VEGF165 can bind heparin, and in fact binding can enhance the formation of VEGF165/Nrp1 complexes,5 specifically through the b1b2 domain.17 Nrp1 can also bind heparin directly, and chains of 8 monosaccharides (dp8) are sufficient.17 In contrast, a dp20-dp24 heparin chain was necessary for forming a complex with VEGF165 and Nrp1, consistent with the idea that the heparin chain acts as a scaffold that binds both VEGF and Nrp1. This complex facilitates VEGF-mediated endothelial cell proliferation in vitro, suggesting that complexes that form in vivo with endothelial heparan sulfate can play a role in vasculogenesis and angiogenesis.
Heparin binding patches and clefts on proteins tend to contain many electropositive amino acids that form salt bridges with the uronic acids and sulfate groups in heparin/heparan sulfate.22 These ionic interactions account for a substantial fraction of the free binding energy of the total interaction.22 The structure of human Nrp1 b1b2 determined by X-ray crystallography and the modeling studies of murine Nrp1 a1a2b1b2 presented here are in general agreement regarding the location and general size of the heparin binding site.21 Residues identified in previous and current studies through biochemical means were also identified in the CVLS simulation and cover a ∼40 Å cleft spanning both b1 and b2 domains, which would correspond to a heparin oligomer of ∼12 saccharides21 (Figure 1). Other studies have suggested heparin binding sites in other domains of the protein, including a1, c (MAM), and the region between the c domain and the transmembrane domain.30 The b2 domain contains a conserved XBBXBX amino acid motif (512–517; MRKFKI) which corresponds to a Cardin-Weintraub glycosaminoglycan binding consensus sequence.29 We found through mutagenesis that R513 and K514, which lie in the middle of the electropositive groove, contributed highly to the interaction of Nrp1 with heparin and heparan sulfate. These two residues may act as “gatekeepers” to allow the heparin/heparan sulfate chain further access to the protein surface. In contrast, the K516 residue, despite being part of the conserved Cardin-Weintraub motif, does not seem to contribute significantly to the interaction. K516 may be positioned past a small bend in the tertiary structure and, therefore, is displaced from the primary binding cleft that contains R513 and K514. In addition, based on our modeling studies, the positioning of the heparin/heparan sulfate chain suggests that the chain wraps around to the back of the protein to engage other amino acids (K397, K538, E541, N544, N545, T553, H571) instead of continuing down to the bottom of the b1 domain (Supporting Information Video 1). Mutational manipulation of these residues should be explored singularly or in combination in order to further elucidate their contribution to heparin/heparan sulfate binding.
Structural information from X-ray crystallography and size exclusion chromatography suggests that in solution recombinant Nrp1 b1b2, a2b1b2, and a1a2b1b2 behave as monomers,8,21,31 The absence of crystal structures of Nrp1 in complex with either VEGFR1/VEGF or plexin/Sema3a makes it difficult to predict the orientation of the b1b2 domains in these higher order functional signaling complexes. Superposition of Nrp1 dimers in the plexin/Sema3a complex positions the Nrp1 dimer as a bridge locking the components of opposing semaphorin-plexin pairs within the overall structure,8 but this does not provide any additional insight into the orientation of the heparin ligand. Inclusion of heparin induces dimerization of human b1b2 domain and the need for a stoichiometric excess of heparin to drive dimer formation in vitro suggested a 2:2 stoichiometry of human recombinant Nrp1 b1b2 with heparin.21 A model was proposed in which the two heparin oligosaccharides were oriented toward each other, but charge repulsion of two heparin chains makes this orientation unlikely. Here, we provide direct evidence based on isothermal calorimetry that the complex of murine Nrp1 a1a2b1b2 is best characterized as a 1:2 complex of heparin oligosaccharide to Nrp1. We imagine that a single heparin oligosaccharide chain acts as a scaffold, serving as the interface between the b1b2 monomers in the Nrp1 dimer. A similar arrangement has been proposed for FGF1/FGFR2 complexes,46 RAGE dimer,47 and the dimeric E2 domain of APLP122.
The studies reported here demonstrate the participation of heparin/heparan sulfate–protein interactions in dimerization, but we did not explore possible protein–protein interactions that might further stabilize the complex. Previous studies of the contributions of various domains of Nrp1 to its ability to dimerize collectively suggest that dimerization necessitates multiple domains and their interactions. Since heparin is able to dimerize the extracellular domains (a1a2b1b2) without the c or transmembrane domains, these interactions may be superfluous in vivo given that the cell surface is covered with a glycocalyx rich in heparan sulfate. In addition, heparan sulfate likely stabilizes the dimerization of soluble forms of Nrp1, which usually contain only a1a2b1b2 domains. Nevertheless, there may be protein–protein contacts between the b1b2 monomers that further stabilize the complex and are worthy of consideration. If the dimer involves weak protein–protein interactions, this might explain why longer oligosaccharides provide greater stabilization to the complex during gel filtration. Lastly, it is possible that binding of heparin/heparan sulfate to Nrp1 may induce a conformational change in the protein, exposing an interface that facilitates dimerization.
Earlier studies examining the glycosaminoglycan-binding properties of Nrp1 utilized heparin as a mimetic for heparan sulfate. However, heparin is a fractionated form of heparan sulfate, derived from connective mast cells resident in the mucosa of porcine and bovine intestines.22 Like heparan sulfate, heparin is a linear polysaccharide comprised of alternating glucosamine and uronic acid units.22,48 A family of sulfotransferases add sulfate groups at C2 of uronic acid and at N-, C3, and C6 of the glucosamine residues.49 In heparan sulfate, sulfation occurs substoichiometrically and within short segments of the chain, creating a variety of partially sulfated sequences interspersed by nonsulfated or poorly sulfated domains of the chain. In contrast, in heparin, the chains are more fully sulfated and exhibit a higher extent of uronic acid epimerization. Thus, one must be careful about extrapolating the behavior of systems analyzed by the addition of heparin compared with native heparan sulfate. Indeed, studies of Nrp1 indicate that affinity for heparan sulfate is much reduced compared to heparin.50
The modification of the glucosamine units with a 3-O-sulfate group is of interest with respect to Nrp1. Sulfation at the C3 position of glucosamine residues occurs at low frequency compared to sulfation at other positions, but can induce high affinity interaction with proteins, such as antithrombin.51 The 3-O-sulfate modification has also been assessed for its specific interactions with tau52−54 and apoE.55 In recent years, revised methods for detection of the 3-O-sulfate group56,57 as well as disaccharide and tetrasaccharide standards have been developed.58,59 Potential explanations for how 3-O-sulfation promotes specific protein binding include alteration of the topology of the heparan sulfate chain60 as well as reduction of the chain size needed for protein engagement.61 Thacker et al. showed that 3-O-sulfation of glucosamine residues in heparan sulfate enhanced binding of Nrp1 to heparan sulfate immobilized on plastic plates and to heparan sulfate present on cultured cells.34 Chemoenzymatically synthesized 3-O-sulfated heparan sulfate dp12 oligosaccharides protected Nrp1 from thermal denaturation and inhibited Nrp1-dependent, Sema3A-induced growth cone collapse of neurons derived from murine dorsal root ganglia.34 3-O-sulfated heparan sulfate also enhanced the inhibition of endothelial cell sprouting by exogenous heparan sulfate.34 Moreover, alteration of the enzyme Hs3st2, which positions 3-O-sulfate groups in a unique way, specifically affected growth cone collapse.34 Attempts to determine the amino acid residues in Nrp1 that interact specifically with the 3-O-sulfate group, for example by comparing binding of the mutant recombinant Nrp1 proteins to cells expressing specific 3-O-sulfotransferases, were unsuccessful possibly due to the complex network of electrostatic and nonelectrostatic interactions that facilitate ligand binding. The CVLS and MD studies can be used to study this problem further, since it can account virtually for all possible sulfation configurations. Further exploration of the effect of 3-O-sulfation could be achieved by utilizing cell lines that lack specific 3-O-sulfotransferases.
Previous studies have demonstrated that alterations in Nrp1 function reduce tumor growth, likely due to changes in tumor angiogenesis. Blocking Nrp1 function through antibodies reduced subcutaneous H1299 lung tumor growth and demonstrated additive effects with an antibody targeting VEGF.62 Tumor growth was also heavily impacted in subcutaneous B16 melanoma models in mice with VEGFA-binding-deficient Nrp1 (Y297A/Y297A) or endothelial specific loss of Nrp136,.44 Thus, we predicted that a change in Nrp1 function due to its reduced ability to bind heparan sulfate would likely alter tumor growth as well. To understand the participation of heparan sulfate binding in Nrp1 activities, we created a knock-in mouse expressing Nrp1 containing the R513A,K514A allele (Figure S6). Variable penetrance of the mutation in the context of tumor growth was noted, which became penetrant when the level of Nrp1 was reduced by heterozygous mutation (Nrp1D/–). The R513A,K514A mutation only partially reduces heparan sulfate binding of Nrp1 in endothelial cells (Figure 5D) and therefore, the effect of the Nrp1/HS interaction on tumor growth is likely underestimated. In addition, our mutation does not prevent Nrp1 interaction with its protein binding partners, independently of heparan sulfate. Further studies are needed to examine how compounding the Nrp1D/D mutation with a loss of function alleles in heparan sulfate might exacerbate the phenotype. Additional targeted genetic studies are also needed to pinpoint defects in angiogenesis or vascular permeability in the tumor environment.
Methods and Materials
Computational Studies to Elucidate the GAG-Binding Site in Nrp1
The coordinates of mouse Nrp1 were extracted from the Protein Data Bank (ID:4GZ9).8 Protein preparation for modeling was carried out in a sequential manner by first removing water molecules from the crystal structure, adjusting the protonation states of amino acids to pH 7.2, and finally adding hydrogen atoms. The structure was then minimized with fixed heavy-atom coordinates using the Tripos force field for a maximum of 10 000 iterations subject to a termination gradient of 0.05 kcal/(mol-Å), followed by calculation of the electrostatic surface potential (ESP) of the protein (a1a2b1b2 subdomains, see Figure 1A) using the Adaptive Poisson–Boltzmann Solver (APBS) tool in PyMOL. Based on the ESP, we divided the Nrp1 surface into 13 different plausible GAG-binding sites for implementation of the CVLS algorithm (see Figure S1B). The radii of the binding sites for CVLS studies were assigned as 12 and 18 Å for di- and hexa-saccharides, respectively. The coordinates of structures in the heparin/heparan sulfate libraries were obtained from our in-house database, which was constructed earlier.25,63,64 For this study, we utilized 72 disaccharides and 288 hexasaccharide sequences. CVLS screening of the disaccharides was performed on each of the 13 sites using the GOLD version 5.6,65 which uses a genetic algorithm (GA) to optimize the docking of ligands into the binding site. Each sequence was docked using 100 GA runs, each consisting of 100 000 iterations. The GA runs were allowed to terminate early if the top three solutions displayed an RMSD of 2.5 Å or lower. The two best poses from each GA run were stored and analyzed at the end of the docking experiment. Experiments were carried out in triplicate, which yielded at least six solutions for each sequence. GOLDScore and RMSD were used to assess the fitness of the docked poses, as described earlier.23
Molecular Dynamics (MD)
The initial structures for MD runs were taken from the CVLS output files, which provided the docked complexes for the best sequence(s) in the preferred sites of binding. The residue and atom labeling of the bound hexasaccharide (HS06) were altered to match the GLYCAM library (see http://glycam.org/docs/force field/glycam-naming-2/). Both, protein (PDB ID: 4GZ9) and ligand (HS06), were loaded in XLEAP of the AMBER18 suite,66 and glycosidic linkages and the formal charge of HS06 were rechecked to ensure their appropriateness. Residues 1 to 143 of the protein were deleted for MD simulations because they do not play any role in heparan sulfate binding. The overall charge of the protein–ligand system was neutralized by adding appropriate number of counterions (Na+). Amber-ff14SB and GLYCAM06 force field parameters were used for protein and ligand preparation, respectively.67,68 This charge-neutralized complex was solvated using a three-point water (TIP3P) molecule box with a minimum distance of 12 Å between the walls of any atom of the complex. Finally, the initial parameters and coordinate files were saved for each heparan sulfate-protein complex, respectively. Each solvated heparan sulfate-protein complex was minimized in two steps with 10 Å nonbonded cutoff. In the first step, the solute atoms were restrained with a force constant of 100 kcal/(mol Å2), while the water molecules were relaxed using 500 cycles of steepest descent and 2000 cycles of conjugate gradient method. In the second step, the whole system was relaxed using conjugate gradient minimization of 2500 cycles without any restraints.
Each solvated heparan sulfate-protein complex was equilibrated in three phases to achieve the desired temperature and pressure with an integration step of 2 fs. In the first phase, the temperature was brought to 300 K using the temperature coupling with a time constant of 2 ps. In the second phase, the system was brought to a constant pressure using isotropic position scaling. Equilibration was carried out for 1 ns with initial strong restraints on the solute, which were systematically reduced. The production run was performed in an NPT ensemble with the integration time step of 2 fs. Bonds involving hydrogen atoms were constrained by using the SHAKE algorithm. Maxwell distribution was used to assign the initial velocities. Each MD trajectory was computed for one microsecond. Equilibration and simulation processes were validated by monitoring physical observables of the system including the energy (total, potential, and kinetic), temperature, and pressure as a function of the simulation time, which confirmed NPT ensemble settings (not shown). During the entire process, the ring puckers of IdoA/IdoA2S were maintained in their respective 1C4 or 2SO conformers using a weak torsional restraint.23
The hydrogen bond interaction between the donor–acceptor atoms of the protein and HS06 oligosaccharide were calculated from the trajectory using the CPPTRAJ tool of AMBER using a donor–acceptor distance cutoff of 3.5 Å and angle cutoff of 135°.69 Free energy calculations on Nrp1-HS06 complexes were computed using postprocessing MM-PB(GB)SA method70 from the MD trajectories. MM-GBSA employed single residue energy decomposition (SRED) to estimate the energy contributions of each receptor residue in the bound state. Energy calculations were performed using the default parameter settings by employing the python version of the MM-PB(GB)SA module from Amber Tools (refer to http://ambermd.org/tutorials/advanced/tutorial3/). Typically, these calculations were performed using the entire simulation trajectory by taking strictures at equal intervals of time of a total of 12 500 frames.
Expression and Purification of Nrp1
The Nrp1 plasmid containing the cDNA for murine a1a2b1b2 domains (residues 22–586) and a C-terminal His tag was obtained from Jones’ lab.8 Nrp1 mutant plasmids were developed through Gibson Assembly using NEB HiFi Assembly Master Mix (NEB) and fragments generated from synthesized geneblocks (IDT) or from Q5 PCR amplification of the template wild-type plasmid. Primers for the Q5 PCR were designed by using the NEBuilder Assembly Tool (NEB) to generate fragments containing the desired mutation. Mutant plasmids were confirmed by sequencing (Genewiz and Eton Bioscience).
Recombinant protein was produced by transfecting ExpiCHO cells (6 × 106 cells/mL) with 0.8 μg/mL plasmid DNA using ExpiFectamine and OptiPRO SFM following the ExpiCHO Expression (ThermoFisher) protocol with Max Titer feeding schedule. Cells were cultured post-transfection at 37 °C in the ExpiCHO expression medium. On day 1 after transfection, ExpiFectamine CHO Enhancer and ExpiCHO Feed were added to the culture. On day 5 after transfection, ExpiCHO Feed was added to the culture. On day 12 post-transfection, conditioned media was collected, treated with a EDTA-free protease inhibitor (Roche Applied Science) and adjusted to 30 mM imidazole before filtration through a 0.2 μm filter. Recombinant protein was initially purified on a 1 mL Ni2+-Sepharose 6 Fast Flow column (GE Healthcare) on an AKTA pure protein purification system (GE Healthcare). Recombinant protein was loaded onto the column followed by washing with 30 mM imidazole and 0.5 M NaCl in 25 mM HEPES buffer (pH 7.5) and eluting with 0.3 M imidazole and 0.5 M NaCl in 25 mM HEPES buffer (pH 7.5). Recombinant protein was concentrated by Amicon Centrifugal Filter Units (Millipore Sigma) or Pierce Protein Concentrator PES (ThermoFisher Scientific). Concentrated protein was further purified by size-exclusion chromatography (HiLoad 16/60 Superdex 200, preparation grade, GE Healthcare) with 0.2 M NaCl and 5% glycerol in a 25 mM HEPES buffer (pH 7.5). Wild-type and mutant forms of Nrp1 were purified by this method.
Analytical Heparin-Sepharose Chromatography
Recombinant proteins were loaded in Buffer A (Gibco DPBS adjusted to 150 mM total NaCl, pH 7.2) onto a 1 mL HiTrap heparin sepharose column (GE Healthcare). The column was washed with 10 column volumes of Buffer A followed by gradient elution from Buffer A to Buffer B (DPBS adjusted to 0.6 M NaCl, pH 7.2) over 10 column volumes. The salt concentration of the elution peak was determined for each protein.
Nrp1 Binding to Immobilized Heparin/HS-BSA
The preparation of heparin-BSA conjugates was performed as previously described.71 High binding microplates were coated with heparin-BSA (50 μL of 2 ng/μL solution) overnight while rotating at 4 °C. The plates were blocked for 1 h at 32 °C with 1% BSA in PBST (Gibco DPBS with 0.05% Tween). Nrp1 proteins were incubated in the wells in triplicate, and bound protein was detected with THE His Tag antibody [HRP] (Genscript) diluted 1:4000 in 1% BSA in PBST. The wells were developed with 100 μL of TMB turbo substrate for 6 m followed by quenching with 100 μL of 1 M sulfuric acid. The absorbance of the wells was measured at 450 and 540 nm for background correction. Prism 8 (Graphpad) was used for data analysis and statistical analysis of the curves. Non-specific binding was assessed by incubation with antibody in the absence of Nrp1 proteins.
Size Exclusion Chromatography and Multiangle Light Scattering (SEC-MALS)
Nrp1 protein was concentrated and exchanged into gel filtration buffer (75 mM NaCl in 10 mM Phosphate Buffer, pH 7.4) in a Pierce Protein Concentrator PES (10 KDa MWCO, ThermoFisher Scientific). Concentrated protein (∼2.5–3 mg mL–1) was then incubated with a 5-fold molar excess of heparin oligosaccharides (Iduron). For analysis by SEC-MALS, 100 μL of the protein-heparin mixtures in gel filtration buffer was injected onto a Superdex 200 Increase 10/300 GL Column (GE Life Sciences). miniDAWN TREOS and Optilab T-rex detectors (Wyatt Technology) were used to collect light scattering and refractive index data, respectively. ASTRA v.6 software (Wyatt Technology) was used to calculate the molar masses of the complexes.
Differential Scanning Fluorimetry (DSF)
Recombinant Nrp1 (2 μM), heparin oligosaccharides (2–20 uM, Iduron), and 5X SYPRO Orange Protein Gel Stain (Thermo Fisher Scientific) were added to 150 mM NaCl in 10 mM HEPES (pH 7.5) buffer. Thermal denaturation was achieved using a temperature gradient from 25 to 95 °C at a rate of (0.5 °C/30 s) on a CFX96 real-time PCR system (Biorad). Melting temperatures were determined from the first derivatives of the melting curve data, assuming a Gaussian distribution (Prism 8).
Cell Surface Binding
Cells were grown to 70–80% confluency and lifted with 10 mM EDTA in DPBS (Gibco). Cells were centrifuged and then washed with 0.5% BSA in DPBS (Gibco; HUVEC wash buffer contained 5 mM EDTA). For some experiments, some of the cells were partitioned and treated with heparinases (HSase; 2.5 mU/mL Heparinase II and 5 mU/mL Heparinase III; IBEX) for 30 min at 37 °C in 0.5% BSA in DPBS. The cells were then incubated with his-tagged Nrp1 protein (recombinant a1a2b1b2) in 0.5% BSA in DPBS for 60 min at 4 °C. After washing the cells twice in 0.5% BSA in DPBS, the cells were incubated with an antiHis6 antibody (1:500 THE His Tag Antibody iFluor 488, Genscript) in 0.5% BSA in DPBS for 30 min at 4 °C. The cells were washed twice with 0.5% BSA in DPBS and then analyzed on a FACSCanto flow cytometer (BD Bioscience). Data were analyzed using FlowJo software (BD Bioscience) and Prism 8. Non-specific binding was assessed by incubation with antibody in the absence of Nrp1 proteins.
Generation and Genotyping of Mouse cell Lines
Nrp1 floxed mice (Nrp1f/f) B6.129(SJL)-Nrp1tm2Ddg/J) were purchased from the Jackson Laboratory.38 Nrp1f/f mice were crossed to EIIa-Cre+ mice on C57Bl/6 background to generate a Nrp1 knockout allele.72 This line was then backcrossed into the C57Bl/6 background over 5 generations. To generate a mouse bearing an allele for a heparan sulfate deficient Nrp1 protein (Nrp1D), a CRISPR RNA (crRNA) sequence and homology directed repair (HDR) donor oligonucleotide were designed using the Deskgen platform to target and mutate the R513,K514 amino acids into alanine residues. crRNA: GAG AAA ACA AGG UGU UCA UG; HDR ssoligo donor: CTTGCTGTCATCCATGATAGTTTTCCAGTCAGAGCCATTGTTACTATAGGCGATCTTGAACgcCgcCATGAACACCTTGTTTTCTCGGTGCTTCCCACCCTGAATGATGACACCTCTTACTATCTT.
Alt-R S.p. Cas9 protein, a transacting CRISPR RNA (TracrRNA), crRNA, and HDR oligonucleotides were from IDT. An in vitro cleavage assay was used to determine whether the sgRNA (TracrRNA + crRNA) would induce cleavage in a DNA fragment containing the target sequence, by combining 0.6 μg of Cas9 protein, 0.25 μg of sgRNA (tracrRNA+crRNA), 0.15 μg targeting substrate (PCR product), 1 μL of 10X NEB Buffer, and 1 μL of 10X BSA buffer, and nuclease free buffer up to 10 μL. After incubation for 1 h at 37 °C, RNase was added to the reaction mixture, which was then incubated for 15 min at 37 °C. Stop solution (1 μL; 30% glycerol, 1.2% SDS, 250 mM EDTA, pH 8.0) was added, and after 15 min at 37 °C, the samples were analyzed by agarose gel electrophoresis. The following mixture was used for blastocyst injection: Alt-R S.p. Cas9 protein: 0.6 uM; TracrRNA: 0.6 uM; crRNA: 0.6 uM; HDR Oligo: 20 ng/μL and diluted with IDTE buffer to 30 μL.
Resulting offspring were screened for transgenesis by amplifying the genomic DNA surrounding the mutation site by PCR using a Q5 High-Fidelity polymerase (New England Biolabs) and ligated into a PUC19 vector. The ligated plasmids were transformed into 10-beta competent E. coli (New England Biolabs) and spread on plates treated with X-gal (800ug; Apex) and IPTG (800ug; Sigma). White colonies were selected for colony PCR using M13Forward and M13Reverse primers. Resulting amplicons were purified by using the QIAquick PCR purification kit (QIAGEN). Sequencing of these amplicons confirmed the editing of the Nrp1 gene (Figure S7A) and validating the genotyping method involving BspHI digestion.
For genotyping of the Nrp1 alleles, the following primers were used. Nrp1 Forward: AAG GAG TGG CAC AGC ATC TT; Nrp1 Reverse: TCA CAC CCA AAC TTC CTT CC; Nrp1 Exon2–3B: GGG TGA ACT CAG CCA CTT GT; Nrp1-HSBD Forward: GCA TCC AAT CAA GCC GAC AG; Nrp1-HSBD Reverse: GCT TGG GAG GTA GAG ATG CA. The Nrp1 Forward and Reverse primers were used to identify the floxed gene, and the Nrp1 Forward and Nrp1 Exon2-3B primers were used to determine knockout alleles. The Nrp1-HSBD forward and reverse primers were used to generate a PCR-amplified region around the mutation site. The resulting amplicons are then digested with BspHI and analyzed by agarose gel electrophoresis. All animal husbandry and experiments were performed in accordance with relevant guidelines and regulations following standards and procedures approved by the UC San Diego Institutional Animal Care and Use Committee (protocol #S99127).
Tumor Experiments
B16–F10 melanoma cells were grown to 80% confluency in DMEM (Gibco) with 10% (v/v) FBS, 1% (v/v) penicillin–streptomycin (Gibco) in an environment of 37 °C, 5% CO2, and 95% air. Cells were lifted with 0.05% trypsin (Corning), which was inactivated by adding in DMEM with 10% FBS. Cells were sedimented by centrifugation, washed with DPBS (Gibco) twice, resuspended in DPBS and counted twice before being diluted to 5 × 106 cells/mL in DPBS. Cell suspensions (100 μL) were injected subcutaneously into the shaved flanks of anesthetized mice of various genotypes. The tumors were initially done bilaterally, but concerns about the overall tumor burden led us to continue with only unilateral injections. Tumors were measured with calipers every 2 days. From these measurements, tumor volume was calculated using the formula volume = length × width2 × 0.5236.73,74 Tumors were grown until they reached the terminal end point (>10 mm in one dimension). Animals were sacrificed at the end point or when ulcers were found, in accordance with our animal protocol. Size comparison was done at 15 days, when the first tumors reached terminal end point and all animals were alive. Tumors that ulcerated before day 15 were removed from the data set. Those tumors deemed to be growing intramuscularly by palpation and later dissection were also removed from the data set. Tumor volumes were plotted, and statistical analysis was performed in Prism 8.
Acknowledgments
We appreciate the assistance by the UCSD Microscopy Core (M. Erb and J. Santini), the La Jolla Institute of Immunology Microscopy and Histology Core (K. Dobaczewska), the Sanford Burnham Prebys Medical Discovery Institute Protein Analysis Facility (A. Bobkov), the UCSD Transgenic Mouse Core (J. Zhao and E. Kothari), the UCSD Murine Hematopathology Core (Q. Chen and C. Yu). We also gratefully thank S. Dowdy at the University of California, San Diego for providing B16–F10 melanoma cells and E. Jones at the University of Oxford for the mNrp1 plasmids. This work was originally conducted for and included in a doctoral dissertation for C. Painter at the University of California, San Diego. Funding sources include NINDS P30 NS047101 to UCSD Microscopy, NIH grants P42 ES010377, P30 CA023100, & P30 DK063491 to UCSD Transgenic Mouse Core, NIGMS 5R01GM145913-02 to K.G., and NIH grants R35 GM144121 to K.D.C., 1K01DK126989 to C.M.T., P01 HL151333, U01 CA241951, & K12 HL141954 to U.R.D., and P01 HL131474 & AR073031 to J.D.E.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.4c00389.
Evolutionary conservation, SDS-PAGE/western blotting methods, RNA isolation and qPCR analysis, immunoblotting of purified Nrp1, gel electrophoresis, hematopoietic analysis mouse husbandry, bone marrow processing, bone marrow cellularity, hematopoietic stem cell and progenitor flow cytometric analyses, and supporting figures (PDF) (MPG)
Author Contributions
C.D.P., N.V.S., B.N., T.M.C., C.M.T., U.R.D, and J.D.E. contributed to conceptualization; C.D.P., N.V.S., B.N., T.M.C., A.M.V.W., R.N.P., D.R.S., K.G., C.W.V.K., C.M.T., K.D.C, U.R.D., and J.D.E. contributed to methodology; C.D.P., N.V.S., B.N., T.M.C., A.M.V.W., R.N.P., J.P.C., K.G., C.W.V.K., K.D.C, U.R.D., and J.D.E. contributed to resources; N.V.S., B.N., and U.R.D. contributed to software; C.D.P., N.V.S., B.N., T.M.C., A.M.V.W., N.J.S., J.P., P.L.B., R.N.P., and G.J.V. contributed to investigation; C.D.P., N.V.S., B.N., T.M.C., A.M.V.W., N.J.S., J.P., P.L.S.M.G, K.DC. C.M.T., U.R.D., and J.D.E. contributed to formal analysis; C.D.P., N.V.S., B.N., A.M.V.W., N.J.S., K.DC. C.M.T., U.R.D., and J.D.E. contributed to data curation; C.D.P., N.V.S., B.N., T.M.C., A.M.V.W., N.J.S., K.D.C, C.M.T., U.R.D., and J.D.E. contributed to validation; C.D.P, N.V.S., B.N., N.J.S, C.M.T., U.R.D., and J.D.E contributed to visualization; C.D.P. and J.D.E. contributed to writing – original draft; C.D.P, C.M.T., U.R.D., and J.D.E. contributed to writing – review and editing; C.D.P., C.M.T., U.R.D., and J.D.E. contributed to project administration; C.W.V.K., P.L.S.M.G, C.M.T., U.R.D., and J.D.E. contributed to supervision; K.G., K.DC. C.M.T., U.R.D., and J.D.E. contributed to funding acquisition.
The authors declare no competing financial interest.
Supplementary Material
References
- Kawasaki T.; Kitsukawa T.; Bekku Y.; Matsuda Y.; Sanbo M.; Yagi T.; Fujisawa H. A requirement for neuropilin-1 in embryonic vessel formation. Development 1999, 126, 4895–4902. 10.1242/dev.126.21.4895. [DOI] [PubMed] [Google Scholar]
- Kolodkin A. L.; Levengood D. V.; Rowe E. G.; Tai Y. T.; Giger R. J.; Ginty D. D. Neuropilin is a semaphorin III receptor. Cell 1997, 90, 753–762. 10.1016/S0092-8674(00)80535-8. [DOI] [PubMed] [Google Scholar]
- He Z.; Tessier-Lavigne M. Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell 1997, 90, 739–751. 10.1016/S0092-8674(00)80534-6. [DOI] [PubMed] [Google Scholar]
- Chen H.; Chedotal A.; He Z.; Goodman C. S.; Tessier-Lavigne M. Neuropilin-2, a novel member of the neuropilin family, is a high affinity receptor for the semaphorins Sema E and Sema IV but not Sema III. Neuron 1997, 19, 547–559. 10.1016/S0896-6273(00)80371-2. [DOI] [PubMed] [Google Scholar]
- Soker S.; Takashima S.; Miao H. Q.; Neufeld G.; Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell 1998, 92, 735–745. 10.1016/S0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- Klagsbrun M.; Takashima S.; Mamluk R. The role of neuropilin in vascular and tumor biology. Adv. Exp. Med. Biol. 2002, 515, 33–48. 10.1007/978-1-4615-0119-0_3. [DOI] [PubMed] [Google Scholar]
- Nakamura F.; Tanaka M.; Takahashi T.; Kalb R. G.; Strittmatter S. M. Neuropilin-1 extracellular domains mediate semaphorin D/III-induced growth cone collapse. Neuron 1998, 21, 1093–1100. 10.1016/S0896-6273(00)80626-1. [DOI] [PubMed] [Google Scholar]
- Janssen B. J.; Malinauskas T.; Weir G. A.; Cader M. Z.; Siebold C.; Jones E. Y. Neuropilins lock secreted semaphorins onto plexins in a ternary signaling complex. Nat. Struct. Mol. Biol. 2012, 19, 1293–1299. 10.1038/nsmb.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly J. L.; Simonetti B.; Klein K.; Chen K. E.; Williamson M. K.; Anton-Plagaro C.; Shoemark D. K.; Simon-Gracia L.; Bauer M.; Hollandi R.; et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science 2020, 370, 861–865. 10.1126/science.abd3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti-Castelvetri L.; Ojha R.; Pedro L. D.; Djannatian M.; Franz J.; Kuivanen S.; Kallio K.; Kaya T.; Anastasina M.; Smura T.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. 10.1126/science.abd2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghez D.; Lepelletier Y.; Lambert S.; Fourneau J. M.; Blot V.; Janvier S.; Arnulf B.; van Endert P. M.; Heveker N.; Pique C.; et al. Neuropilin-1 is involved in human T-cell lymphotropic virus type 1 entry. J. Virol. 2006, 80, 6844–6854. 10.1128/JVI.02719-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert S.; Bouttier M.; Vassy R.; Seigneuret M.; Petrow-Sadowski C.; Janvier S.; Heveker N.; Ruscetti F. W.; Perret G.; Jones K. S.; et al. HTLV-1 uses HSPG and neuropilin-1 for entry by molecular mimicry of VEGF165. Blood 2009, 113, 5176–5185. 10.1182/blood-2008-04-150342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi S.; Hirata T.; Agata K.; Mochii M.; Eguchi G.; Fujisawa H. The A5 antigen, a candidate for the neuronal recognition molecule, has homologies to complement components and coagulation factors. Neuron 1991, 7, 295–307. 10.1016/0896-6273(91)90268-5. [DOI] [PubMed] [Google Scholar]
- Lee C. C.; Kreusch A.; McMullan D.; Ng K.; Spraggon G. Crystal structure of the human neuropilin-1 b1 domain. Structure 2003, 11, 99–108. 10.1016/S0969-2126(02)00941-3. [DOI] [PubMed] [Google Scholar]
- Beckmann G.; Bork P. An adhesive domain detected in functionally diverse receptors. Trends Biochem. Sci. 1993, 18, 40–41. 10.1016/0968-0004(93)90049-S. [DOI] [PubMed] [Google Scholar]
- Gu C.; Limberg B. J.; Whitaker G. B.; Perman B.; Leahy D. J.; Rosenbaum J. S.; Ginty D. D.; Kolodkin A. L. Characterization of neuropilin-1 structural features that confer binding to semaphorin 3A and vascular endothelial growth factor 165. J. Biol. Chem. 2002, 277, 18069–18076. 10.1074/jbc.M201681200. [DOI] [PubMed] [Google Scholar]
- Mamluk R.; Gechtman Z.; Kutcher M. E.; Gasiunas N.; Gallagher J.; Klagsbrun M. Neuropilin-1 binds vascular endothelial growth factor 165, placenta growth factor-2, and heparin via its b1b2 domain. J. Biol. Chem. 2002, 277, 24818–24825. 10.1074/jbc.M200730200. [DOI] [PubMed] [Google Scholar]
- Giger R. J.; Urquhart E. R.; Gillespie S. K.; Levengood D. V.; Ginty D. D.; Kolodkin A. L. Neuropilin-2 is a receptor for semaphorin IV: insight into the structural basis of receptor function and specificity. Neuron 1998, 21, 1079–1092. 10.1016/S0896-6273(00)80625-X. [DOI] [PubMed] [Google Scholar]
- Yelland T.; Djordjevic S. Crystal Structure of the Neuropilin-1 MAM Domain: Completing the Neuropilin-1 Ectodomain Picture. Structure 2016, 24, 2008–2015. 10.1016/j.str.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Windwarder M.; Yelland T.; Djordjevic S.; Altmann F. Detailed characterization of the O-linked glycosylation of the neuropilin-1 c/MAM-domain. Glycoconj. J. 2016, 33, 387–397. 10.1007/s10719-015-9602-x. [DOI] [PubMed] [Google Scholar]
- Vander Kooi C. W.; Jusino M. A.; Perman B.; Neau D. B.; Bellamy H. D.; Leahy D. J. Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 6152–6157. 10.1073/pnas.0700043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D.; Esko J. D. Demystifying heparan sulfate-protein interactions. Annu. Rev. Biochem. 2014, 83, 129–157. 10.1146/annurev-biochem-060713-035314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan N. V.; Nagarajan B.; Desai U. R. So you think computational approaches to understanding glycosaminoglycan-protein interactions are too dry and too rigid? Think again!. Curr. Opin. Struct. Biol. 2018, 50, 91–100. 10.1016/j.sbi.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan N. V.; Sarkar A.; Desai U. R.; Mosier P. D. Designing ″high-affinity, high-specificity″ glycosaminoglycan sequences through computerized modeling. Methods Mol. Biol. 2015, 1229, 289–314. 10.1007/978-1-4939-1714-3_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan N. V.; Desai U. R. Toward a robust computational screening strategy for identifying glycosaminoglycan sequences that display high specificity for target proteins. Glycobiology 2014, 24, 1323–1333. 10.1093/glycob/cwu077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan B.; Holmes S. G.; Sankaranarayanan N. V.; Desai U. R. Molecular dynamics simulations to understand glycosaminoglycan interactions in the free- and protein-bound states. Curr. Opin. Struct. Biol. 2022, 74, 102356. 10.1016/j.sbi.2022.102356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan B.; Sankaranarayanan N. V.; Desai U. R. Perspective on computational simulations of glycosaminoglycans. Wiley Interdiscip. Rev.: comput. Mol. Sci. 2019, 9 (2), e1388 10.1002/wcms.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepuru K. M.; Nagarajan B.; Desai U. R.; Rajarathnam K. Structural basis, stoichiometry, and thermodynamics of binding of the chemokines KC and MIP2 to the glycosaminoglycan heparin. J. Biol. Chem. 2018, 293, 17817–17828. 10.1074/jbc.RA118.004866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin A. D.; Weintraub H. J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. 10.1161/01.ATV.9.1.21. [DOI] [PubMed] [Google Scholar]
- Uniewicz K. A.; Ori A.; Ahmed Y. A.; Yates E. A.; Fernig D. G. Characterisation of the interaction of neuropilin-1 with heparin and a heparan sulfate mimetic library of heparin-derived sugars. PeerJ. 2014, 2, e461 10.7717/peerj.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton B. A.; Wu P.; Maloney J.; Yin J.; Liang W. C.; Stawicki S.; Mortara K.; Bowman K. K.; Elliott J. M.; Desmarais W.; et al. Structural studies of neuropilin/antibody complexes provide insights into semaphorin and VEGF binding. Embo J. 2007, 26, 4902–4912. 10.1038/sj.emboj.7601906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uniewicz K. A.; Ori A.; Xu R.; Ahmed Y.; Wilkinson M. C.; Fernig D. G.; Yates E. A. Differential scanning fluorimetry measurement of protein stability changes upon binding to glycosaminoglycans: a screening test for binding specificity. Anal. Chem. 2010, 82, 3796–3802. 10.1021/ac100188x. [DOI] [PubMed] [Google Scholar]
- Sandoval D. R.; Gomez Toledo A.; Painter C. D.; Tota E. M.; Sheikh M. O.; West A. M. V.; Frank M. M.; Wells L.; Xu D.; Bicknell R.; et al. Proteomics-based screening of the endothelial heparan sulfate interactome reveals that C-type lectin 14a (CLEC14A) is a heparin-binding protein. J. Biol. Chem. 2020, 295, 2804–2821. 10.1074/jbc.RA119.011639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker B. E.; Seamen E.; Lawrence R.; Parker M. W.; Xu Y.; Liu J.; Vander Kooi C. W.; Esko J. D. Expanding the 3-O-Sulfate Proteome--Enhanced Binding of Neuropilin-1 to 3-O-Sulfated Heparan Sulfate Modulates Its Activity. ACS Chem. Biol. 2016, 11, 971–980. 10.1021/acschembio.5b00897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitsukawa T.; Shimizu M.; Sanbo M.; Hirata T.; Taniguchi M.; Bekku Y.; Yagi T.; Fujisawa H. Neuropilin-semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron 1997, 19, 995–1005. 10.1016/S0896-6273(00)80392-X. [DOI] [PubMed] [Google Scholar]
- Fantin A.; Herzog B.; Mahmoud M.; Yamaji M.; Plein A.; Denti L.; Ruhrberg C.; Zachary I. Neuropilin 1 (NRP1) hypomorphism combined with defective VEGF-A binding reveals novel roles for NRP1 in developmental and pathological angiogenesis. Development 2014, 141, 556–562. 10.1242/dev.103028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelfand M. V.; Hagan N.; Tata A.; Oh W. J.; Lacoste B.; Kang K.-T.; Kopycinska J.; Bischoff J.; Wang J.-H.; Gu C. Neuropilin-1 functions as a VEGFR2 co-receptor to guide developmental angiogenesis independent of ligand binding. Elife 2014, 3, e03720 10.7554/eLife.03720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C.; Rodriguez E. R.; Reimert D. V.; Shu T.; Fritzsch B.; Richards L. J.; Kolodkin A. L.; Ginty D. D. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev. Cell 2003, 5, 45–57. 10.1016/S1534-5807(03)00169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tordjman R.; Ortega N.; Coulombel L.; Plouet J.; Romeo P. H.; Lemarchandel V. Neuropilin-1 is expressed on bone marrow stromal cells: a novel interaction with hematopoietic cells?. Blood 1999, 94, 2301–2309. 10.1182/blood.V94.7.2301.419k24_2301_2309. [DOI] [PubMed] [Google Scholar]
- Termini C. M.; Pang A.; Fang T.; Roos M.; Chang V. Y.; Zhang Y.; Setiawan N. J.; Signaevskaia L.; Li M.; Kim M. M.; Tabibi O.; Lin P. K.; Sasine J. P.; Chatterjee A.; Murali R.; et al. Neuropilin 1 regulates bone marrow vascular regeneration and hematopoietic reconstitution. Nat. Commun. 2021, 12, 6990. 10.1038/s41467-021-27263-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaaf M. B.; Garg A. D.; Agostinis P. Defining the role of the tumor vasculature in antitumor immunity and immunotherapy. Cell Death Dis. 2018, 9, 115. 10.1038/s41419-017-0061-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prewett M.; Huber J.; Li Y.; Santiago A.; O’Connor W.; King K.; Overholser J.; Hooper A.; Pytowski B.; Witte L.; Bohlen P.; Hicklin D. J. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res. 1999, 59 (20), 5209–5218. [PubMed] [Google Scholar]
- Franco S.; Segura I.; Riese H. H.; Blasco M. A. Decreased B16F10 melanoma growth and impaired vascularization in telomerase-deficient mice with critically short telomeres. Cancer Res. 2002, 62, 552–559. [PubMed] [Google Scholar]
- Benwell C. J.; Johnson R. T.; Taylor J.; Price C. A.; Robinson S. D. Endothelial VEGFR Coreceptors Neuropilin-1 and Neuropilin-2 Are Essential for Tumor Angiogenesis. Cancer Res. Commun. 2022, 2, 1626–1640. 10.1158/2767-9764.CRC-22-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y.; Takakura N.; Yasue H.; Ogawa H.; Fujisawa H.; Suda T. Exogenous clustered neuropilin 1 enhances vasculogenesis and angiogenesis. Blood 2001, 97, 1671–1678. 10.1182/blood.V97.6.1671. [DOI] [PubMed] [Google Scholar]
- Pellegrini L.; Burke D. F.; von Delft F.; Mulloy B.; Blundell T. L. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature 2000, 407, 1029–1034. 10.1038/35039551. [DOI] [PubMed] [Google Scholar]
- Xu D.; Young J. H.; Krahn J. M.; Song D.; Corbett K. D.; Chazin W. J.; Pedersen L. C.; Esko J. D. Stable RAGE-heparan sulfate complexes are essential for signal transduction. ACS Chem. Biol. 2013, 8, 1611–1620. 10.1021/cb4001553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko J. D.; Selleck S. B. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem. 2002, 71, 435–471. 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- Thacker B. E.; Xu D.; Lawrence R.; Esko J. D. Heparan sulfate 3-O-sulfation: a rare modification in search of a function. Matrix Biol. 2014, 35, 60–72. 10.1016/j.matbio.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teran M.; Nugent M. A. Synergistic Binding of Vascular Endothelial Growth Factor-A and Its Receptors to Heparin Selectively Modulates Complex Affinity. J. Biol. Chem. 2015, 290, 16451–16462. 10.1074/jbc.M114.627372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pejler G.; Backstrom G.; Lindahl U.; Paulsson M.; Dziadek M.; Fujiwara S.; Timpl R. Structure and affinity for antithrombin of heparan sulfate chains derived from basement membrane proteoglycans. J. Biol. Chem. 1987, 262, 5036–5043. 10.1016/S0021-9258(18)61150-0. [DOI] [PubMed] [Google Scholar]
- Mah D.; Zhao J.; Liu X.; Zhang F.; Liu J.; Wang L.; Linhardt R.; Wang C. The Sulfation Code of Tauopathies: Heparan Sulfate Proteoglycans in the Prion Like Spread of Tau Pathology. Front. Mol. Biosci. 2021, 8, 671458. 10.3389/fmolb.2021.671458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira A.; Royaux I.; Liu J.; Wang Z.; Su G.; Moechars D.; Callewaert N.; De Muynck L. The 3-O sulfation of heparan sulfate proteoglycans contributes to the cellular internalization of tau aggregates. BMC Mol. Cell Biol. 2022, 23 (1), 61. 10.1186/s12860-022-00462-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J.; Zhu Y.; Song X.; Xiao Y.; Su G.; Liu X.; Wang Z.; Xu Y.; Liu J.; Eliezer D.; et al. 3-O-Sulfation of Heparan Sulfate Enhances Tau Interaction and Cellular Uptake. Angew. Chem., Int. Ed. 2020, 59, 1818–1827. 10.1002/anie.201913029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah D.; Zhu Y.; Su G.; Zhao J.; Canning A.; Gibson J.; Song X.; Stancanelli E.; Xu Y.; Zhang F.; Linhardt R. J.; Liu J.; Wang L.; Wang C. Apolipoprotein E Recognizes Alzheimer’s Disease Associated 3-O Sulfation of Heparan Sulfate. Angew. Chem., Int. Ed. 2023, 62, e202212636. 10.1002/anie.202212636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Wei J.; Hogan J. D.; Chopra P.; Joshi A.; Lu W.; Klein J.; Boons G. J.; Lin C.; Zaia J. Negative Electron Transfer Dissociation Sequencing of 3-O-Sulfation-Containing Heparan Sulfate Oligosaccharides. J. Am. Soc. Mass Spectrom. 2018, 29, 1262–1272. 10.1007/s13361-018-1907-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Arnold K.; Dhurandahare V. M.; Xu Y.; Pagadala V.; Labra E.; Jeske W.; Fareed J.; Gearing M.; Liu J. Analysis of 3-O-Sulfated Heparan Sulfate Using Isotopically Labeled Oligosaccharide Calibrants. Anal. Chem. 2022, 94, 2950–2957. 10.1021/acs.analchem.1c04965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhurandhare V. M.; Pagadala V.; Ferreira A.; Muynck L.; Liu J. Synthesis of 3-O-Sulfated Disaccharide and Tetrasaccharide Standards for Compositional Analysis of Heparan Sulfate. Biochemistry 2020, 59, 3186–3192. 10.1021/acs.biochem.9b00838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson R.; Chopra P.; Joshi A.; Yang Z.; Vakhrushev S. Y.; Clausen T. M.; Painter C. D.; Szekeres G. P.; Chen Y. H.; Sandoval D. R.; Hansen L.; Esko J. D.; Pagel K.; Dyer D. P.; Turnbull J. E.; et al. Dissecting structure-function of 3-O-sulfated heparin and engineered heparan sulfates. Sci. Adv. 2021, 7, eabl6026. 10.1126/sciadv.abl6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes S. G.; Nagarajan B.; Desai U. R. 3-O-Sulfation induces sequence-specific compact topologies in heparan sulfate that encode a dynamic sulfation code. Comput. Struct. Biotechnol. J 2022, 20, 3884–3898. 10.1016/j.csbj.2022.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandy L. A.; Canning A. J.; Lou H.; Xia K.; He P.; Su G.; Cairns T.; Liu J.; Zhang F.; Linhardt R. J.; Cohen G.; Wang C. Molecular determinants of the interaction between HSV-1 glycoprotein D and heparan sulfate. Front. Mol. Biosci. 2022, 9, 1043713. 10.3389/fmolb.2022.1043713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q.; Chanthery Y.; Liang W. C.; Stawicki S.; Mak J.; Rathore N.; Tong R. K.; Kowalski J.; Yee S. F.; Pacheco G.; et al. Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell 2007, 11, 53–67. 10.1016/j.ccr.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Sankarayanarayanan N. V.; Strebel T. R.; Boothello R. S.; Sheerin K.; Raghuraman A.; Sallas F.; Mosier P. D.; Watermeyer N. D.; Oscarson S.; Desai U. R. A Hexasaccharide Containing Rare 2-O-Sulfate-Glucuronic Acid Residues Selectively Activates Heparin Cofactor II. Angew. Chem., Int. Ed. 2017, 56 (9), 2312–2317. 10.1002/anie.201609541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan N. V.; Bi Y.; Kuberan B.; Desai U. R. Combinatorial virtual library screening analysis of antithrombin binding oligosaccharide motif generation by heparan sulfate 3-O-Sulfotransferase 1. Comput. Struct. Biotechnol. J. 2020, 18, 933–941. 10.1016/j.csbj.2020.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Case D. A.; Ben-Shalom I. Y.; Brozell S. R.; Cerutti D. S.; Cheatham T. E.; Cruzeiro V. W. D.; Darden T. A.; Duke R. E.; Ghoreishi D.; Gilson M. K.; Gohlke H.; Goetz A. W.; Greene D.; Harris R.; Homeyer N., et al. AMBER 2018, University of California; 2018. [Google Scholar]
- Tian C.; Kasavajhala K.; Belfon K. A. A.; Raguette L.; Huang H.; Migues A. N.; Bickel J.; Wang Y.; Pincay J.; Wu Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. 10.1021/acs.jctc.9b00591. [DOI] [PubMed] [Google Scholar]
- Kirschner K. N.; Yongye A. B.; Tschampel S. M.; Gonzalez-Outeirino J.; Daniels C. R.; Foley B. L.; Woods R. J. GLYCAM06: a generalizable biomolecular force field. Carbohydrates. J. Comput. Chem. 2008, 29, 622–655. 10.1002/jcc.20820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe D. R.; Cheatham T. E. 3rd. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Miller B. R. 3rd; McGee T. J.; Swails J. M.; Homeyer N.; Gohlke H.; Roitberg A. E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- Porell R. N.; Follmar J. L.; Purcell S. C.; Timm B.; Laubach L. K.; Kozirovskiy D.; Thacker B. E.; Glass C. A.; Gordts P.; Godula K. Biologically Derived Neoproteoglycans for Profiling Protein-Glycosaminoglycan Interactions. ACS Chem. Biol. 2022, 17, 1534–1542. 10.1021/acschembio.2c00205. [DOI] [PubMed] [Google Scholar]
- Lakso M.; Pichel J. G.; Gorman J. R.; Sauer B.; Okamoto Y.; Lee E.; Alt F. W.; Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 5860–5865. 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janik P.; Briand P.; Hartmann N. R. The effect of estrone-progesterone treatment on cell proliferation kinetics of hormone-dependent GR mouse mammary tumors. Cancer Res. 1975, 35 (12), 3698–3704. [PubMed] [Google Scholar]
- Neukirch L.; Nielsen T. K.; Laursen H.; Daradoumis J.; Thirion C.; Holst P. J. Adenovirus based virus-like-vaccines targeting endogenous retroviruses can eliminate growing colorectal cancers in mice. Oncotarget 2019, 10, 1458–1472. 10.18632/oncotarget.26680. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.