

Abstract
Aerocyanidin and amycomicin are two antibiotics derived from long-chain acids with a rare epoxy isonitrile moiety, the complexity of which renders the total synthesis of these two natural products rather challenging. How this functionality is biosynthesized has also remained obscure. While the biosynthetic gene clusters for these compounds have been identified, both appear to be deficient in genes encoding enzymes seemingly necessary for the oxidative modifications observed in these antibiotics. Herein, the biosynthetic pathways of aerocyanidin and amycomicin are fully elucidated. They share a conserved pathway to isonitrile intermediates that involves a bifunctional thioesterase and a nonheme iron α-ketoglutarate-dependent enzyme. In both cases, the isonitrile intermediates are then loaded onto an acyl carrier protein (ACP) catalyzed by a ligase. The isonitrile-tethered ACP is subsequently processed by polyketide synthase(s) to undergo chain extension, thereby assembling a long-chain γ-hydroxy isonitrile acid skeleton. The epoxide is installed by the cupin domain-containing protein AecF to conclude the biosynthesis of aerocyanidin. In contrast, three P450 enzymes AmcB, AmcC, and AmcQ are involved in epoxidation and keto formation to finalize the biosynthesis of amycomicin. These results thus explain the sequence of oxidation events that result in the final structures of aerocyanidin and amycomicin as well as the biosynthesis of the key γ-hydroxy epoxy isonitrile functional group.
Graphical Abstract
INTRODUCTION
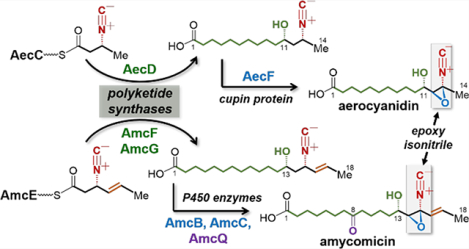
Aerocyanidin (1),1 YM-47515 (2)2 and amycomicin (3)3 isolated from Chromobacterium violaceum, Micromonospora echinospora and Amycolatopsis sp. AA4, respectively, are the only three long-chain fatty acid derivatives featuring a γ-hydroxyl-α,β-epoxy isonitrile functionality (Figure 1A). These natural products are antibiotics showing activity against Grampositive bacteria including several methicillin-resistant Staphylococcus aureus (MRSA) strains.1–3 Both YM-47515 (2) and amycomicin (3) are homologues of aerocyanidin (1) with their chains extended by two and four more carbons, respectively. Amycomicin also contains an extra carbonyl group at C8 and an olefin moiety between C16 and C17 compared to aerocyanidin and YM-47515. The epoxy isonitrile group is known to be essential for the antimicrobial activity of these compounds,4 and amycomicin has recently been shown to target FabH in fatty acid biosynthesis such that it holds promise to treat infections with S. aureus.3 Chemical preparation of this class of compounds has been challenging due to instability of the γ-hydroxyl-α,β-epoxy isonitrile moiety, which can readily undergo Payne rearrangement (5 → 6) under both alkaline and acidic conditions, resulting in an epoxy ketone product with concomitant release of cyanide (Figure 1C).4 Nevertheless, trichoviridin (4),5,6 a highly functionalized epoxy isonitrile-containing cyclopentane natural product useful for the control of crop pathogens,7 has been successfully synthesized by Baldwin and co-workers.8 Despite this progress, how the rare and highly reactive epoxy isonitrile moiety in these secondary metabolites is biosynthesized remains largely unexplored.
Figure 1.
(A) Epoxy isonitrile-containing antibiotics. Arrows indicate the oxidative modifications on the long-chain acid backbone in 1 and 3. (B) Isonitrile-containing lipopeptides. (C) Payne rearrangement as a part of decomposition of the epoxy isonitrile functionality.
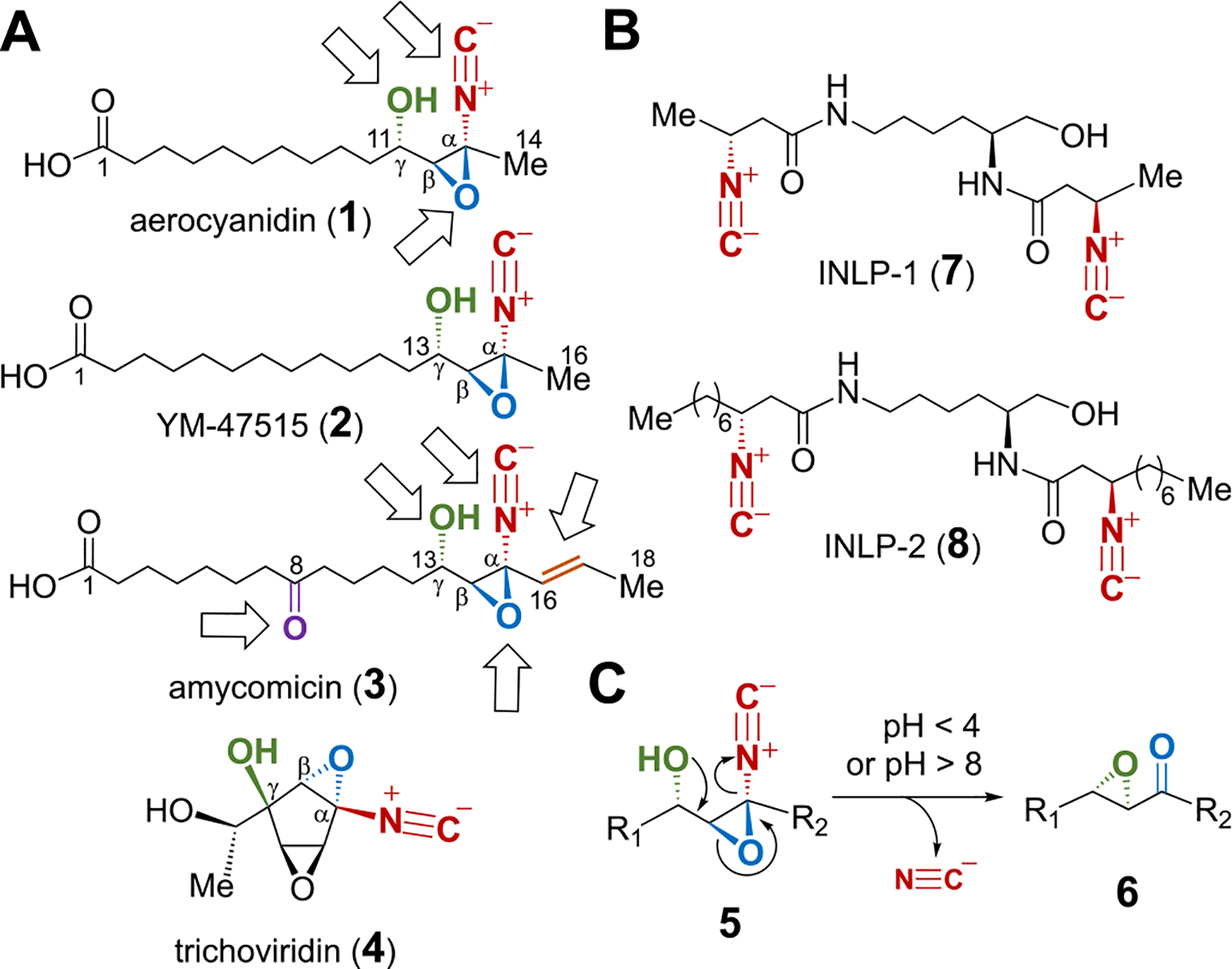
The gene clusters, aec and amc, for the production of aerocyanidin (1) and amycomicin (3), respectively, have recently been identified by Clardy and co-workers (Figure 2A).3 The presence of polyketide synthase (PKS) genes (aecD and amcF/G) in these clusters suggests that the backbone in 1 and 3 does not originate entirely from fatty acid biosynthesis as previously surmised but also polyketide biosynthesis. Both clusters are in part homologous to the sco and mma gene clusters from Streptomyces coeruleorubidus and Mycobacterium marinum, respectively. Among them, the sco cluster is involved in the biosynthesis of the isonitrile lipopeptide (INLP) 7, while the mma cluster is proposed to produce 8 (Figures 1B and 2A).9 Importantly, the functions of the gene products encoded in the sco and mma clusters have been fully established by Zhang and co-workers.9,10 As illustrated in Figure 2B, biosynthesis of INLP-1 (7) begins with the ScoC-mediated loading of crotonic acid (9) onto the acyl carrier protein (ACP) ScoB. The bifunctional thioesterase ScoD then catalyzes a Michael addition of glycine (11) to the crotonyl-ScoB (10) followed by hydrolysis of the thioester bond of 12 to give 13. Subsequent oxidation catalyzed by the nonheme iron α-ketoglutarate-dependent (Fe/α-KG) enzyme ScoE converts 13 to the isonitrile product 14. Compound 14 will be reloaded onto ScoB and processed by the nonribosomal peptide synthetase (NRPS) ScoA to yield the final product 7 (Figure S2). The four genes scoB/C/D/E in the sco cluster are conserved in the aec and amc gene clusters, corresponding to aecC/E/B/A and amcE/H/D/A, respectively. This gene conservation implies that biosynthesis of the isonitrile moieties in 1 and 3 may involve a similar series of reactions (Figure 2B).
Figure 2.
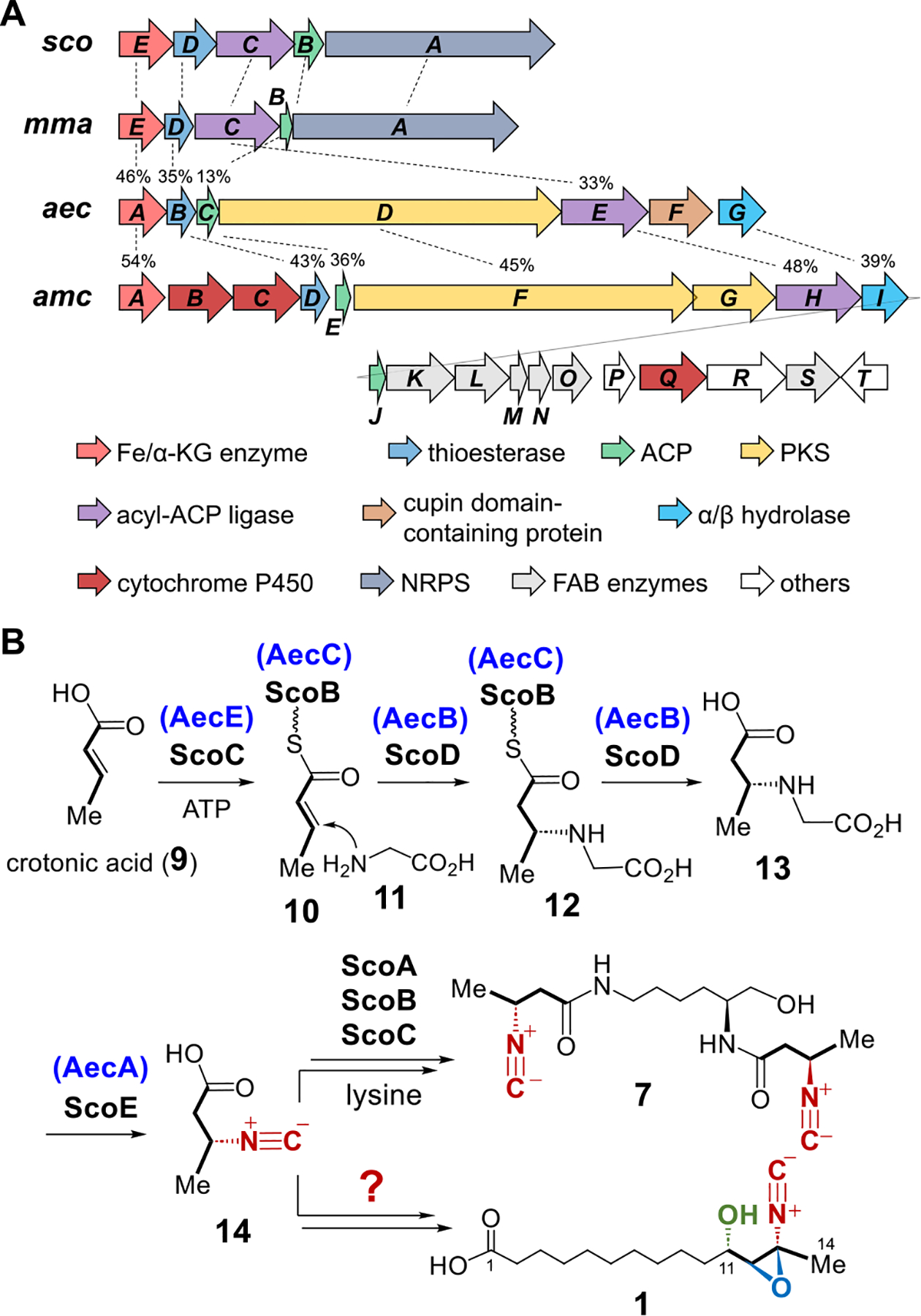
(A) Gene clusters for INLP-1 (7) (sco), INLP-2 (8) (mma), aerocyanidin (1) (aec), and amycomicin (3) (amc). Percentages indicate the sequence identity between the two homologous genes. (B) Biosynthesis of 7 and 1 may share the same intermediate 14.
While previous study of INLP biosynthesis has shed light on isonitrile formation in aerocyanidin and amycomicin, how the hydroxyl group as well as the epoxide are introduced in 1 and 3 remains obscure. In particular, aerocyanidin biosynthesis involves three oxidation reactions including isonitrile formation, C12−C13 epoxidation and C11 hydroxylation; yet, there are only two putative oxidase genes (i.e., aecA and aecF) in the cluster and the aecA-encoded gene product has already been assigned to isonitrile formation.11 Interestingly, compared with the aec cluster, the amc cluster has an aecA homologue (amcA), but no aecF gene homologue, which suggests a distinct difference between amycomicin biosynthesis and that of aerocyanidin, especially since the former is more oxidized than the latter. In addition to isonitrile formation which likely involves amcA, six two-electron oxidations of the backbone are expected to install the C8 ketone, C13 hydroxyl, C14−C15 epoxide, and C16−C17 double bond in 3 (see arrows in Figure 1A). However, only three oxidase genes (i.e., amcB, amcC and amcQ), which all encode P450 enzymes, can be located in the amc cluster. Moreover, it is not apparent why the amc cluster also includes six genes normally associated with fatty acid biosynthesis (FAB) (i.e., amcK, amcL, amcM, amcN, amcO and amcS) as the long-chain acid backbone of amycomicin (3) is likely derived from a polyketide. The present work addresses these questions investigating the activity of the putative oxidases in vitro along with several other enzymes encoded in the gene clusters and thereby establishing the aerocyanidin and amycomicin biosynthetic pathways.
RESULTS AND DISCUSSION
To study the proposed functions of AecA/B/C/E, these four proteins were heterologously overexpressed and purified from Escherichia coli as N-His6-tagged constructs and assayed in vitro. Co-incubation of the putative ligase AecE with crotonic acid (9), the ACP AecC (apo form), coenzyme A (CoA), phosphopantetheinyl transferase (Sfp), ATP, and MgCl2 did not yield the anticipated crotonyl-AecC adduct. Inclusion of the reductant tris(2-carboxyethyl)phosphine (TCEP) in the preceding assay did not produce crotonyl-AecC, either. It is thus possible that formation of crotonyl-AecC may not be required in the pathway. Considering that crotonyl-CoA (15) is a common intermediate in fatty acid metabolism,12 crotonyl-CoA instead of crotonyl-AecC may instead be the substrate of AecB. To test this hypothesis, N-acetylcysteamine activated crotonic acid (16, the SNAc analog of crotonyl-CoA) was synthesized and incubated with AecB as well as glycine (11). In this case, the glycine adduct 13 was indeed produced as shown in Figure S3A. The fact that 16 can be processed by AecB indicated that crotonyl-CoA is likely the true substrate for AecB in vivo. Marfey’s analysis13 revealed that the stereochemistry at C3 in 13 is R (Figure S4A). Moreover, coincubation of chemically synthesized 13 with AecA, α-KG, Fe(NH4)2(SO4)2, and ascorbic acid resulted in isonitrile formation to yield 14 (Figure S3B). The stereochemistry of C3 was the same in both 13 and 14 (Figure S4B). These results are consistent with a pathway in which the isonitrile 14 serves as a common intermediate in the biosynthesis of both INLP-1 (7) and aerocyanidin (1). However, formation of crotonyl-AecC appears to be unnecessary in the latter pathway.
During the biosynthesis of 7, ScoC catalyzes reloading of 14 onto the ACP ScoB. The resulting 14-ScoB is then processed by the NRPS ScoA followed by reductive release from ScoA catalyzed by its own reductase domain to generate the final product 7 (Figures 2B and S2).9,10 It was thus hypothesized that the ligase AecE may catalyze ligation of 14 with the ACP AecC in the aerocyanidin pathway. Indeed, 14-tethered AecC (17) was observed when 14 was incubated with AecC and AecE (Figure 3A,C). The presence of the type I PKS gene aecD in the cluster suggested that 17 may be the starter unit for polyketide chain extension catalyzed by AecD to generate the hydrocarbon chain in 1. Based on this assumption, the C11 hydroxyl group in aerocyanidin may thus result from β-keto reduction catalyzed by the ketoreductase (KR) domain of AecD in the first round of chain elongation (18 → 19, Figure 3C). The following four rounds of extension would furnish the 14-carbon skeleton of 1 (19 → 24, Figure 3C). In order to test this hypothesis, aecD was heterologously coexpressed with aecABCE in E. coli K207. LCMS analysis of the culture medium identified a species with the same molecular weight as 24 (calcd for C15H26NO3− [M−H]−: 268.1918, found: 268.1916, Figure 3B). Furthermore, it also has an identical retention time with the chemically synthesized standard of 24. In the absence of aecD, this species was absent, indicating that production of this species is aecD-dependent (Figure 3B).
Figure 3.
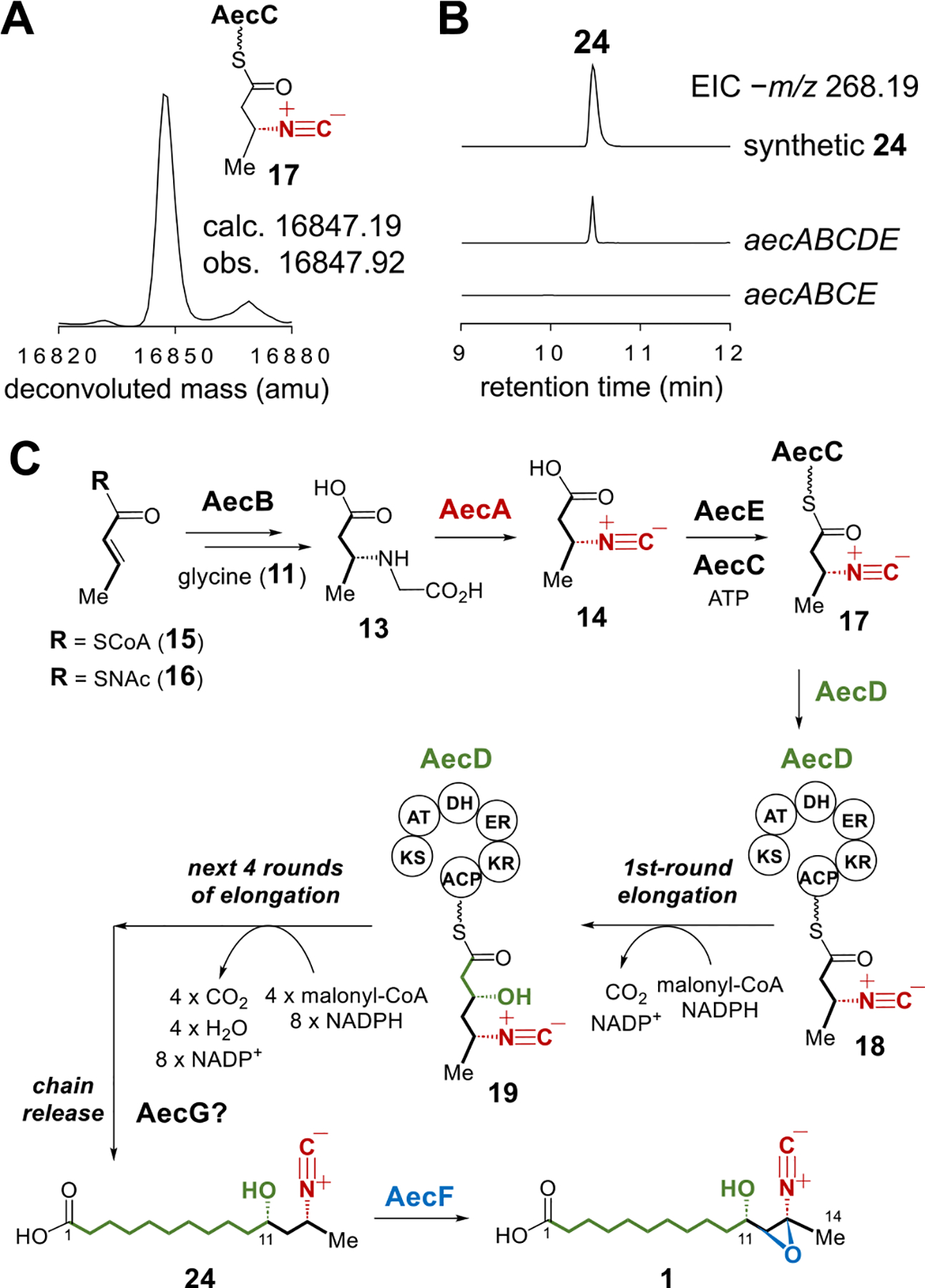
(A) MS analysis of 17. (B) LCMS analysis of the heterologous expression experiments of aecABCDE. (C) Proposed biosynthetic pathway of aerocyanidin (1).
In order to better characterize the chain elongation process, AecD was isolated from E. coli as a recombinant N-His6-tagged construct and incubated with chemically prepared 14, CoA, apo-AecC, MgCl2, TCEP, Sfp, ATP, AecE, malonic acid, the malonyl CoA synthetase MatB, and NADPH. The reaction mixture was analyzed by LCMS after hydrolysis of the thioester bonds to release the products from AecD at pH 9.5. As shown in Figure 4A, 24 was observed consistent with the preceding heterologous expression experiments. Moreover, the C11 stereochemistry of 24 is S as its C11-epimer (11-epi-24) has a different retention time (Figure S5). The S-configuration at C11 of 24 is also consistent with the prediction based on sequence analysis of the reductase domain of AecD, which harbors the conserved Asp residue that dictates the stereoselectivity to generate D-hydroxyl groups (Figure S6).14,15 Besides, intermediates with various carbon chain lengths were also identified by comparison with the reaction using [2-13C]-malonic acid (Figures 4A and S7). Collectively, these findings imply that AecE mediates linkage of 14 to AecC to form 17. The acyl group of 17 is then transferred to the PKS AecD to initiate carbon chain elongation whereupon five rounds of chain extension lead to the production of 24 (Figure 3C). It is also noted that AecD lacks a thioesterase (TE) domain. Thus, the offloading of 24 from the ACP domain may be catalyzed by AecG, which is annotated as an α/β hydrolase and thus may serve as a trans-acting thioesterase. However, this activity could not be verified since AecG forms inclusion bodies upon overexpression.
Figure 4.
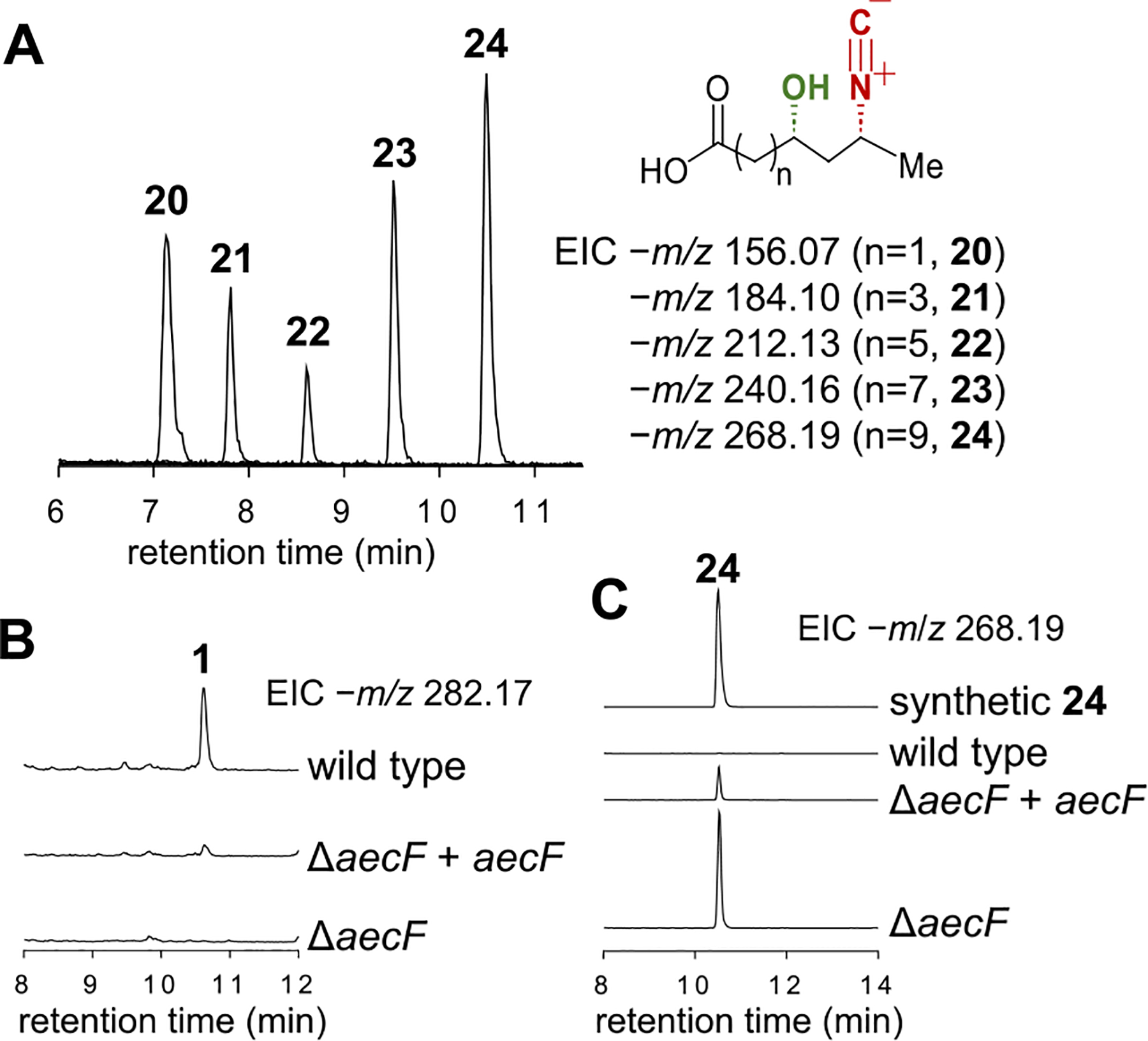
(A) LCMS analysis of the in vitro AecD reaction. (B, C) LCMS analysis of the gene deletion and complementation experiments of aecF.
PKS-mediated formation of 24 renders subsequent oxidation at C11 unnecessary leaving epoxide installation as the remaining step in aerocyanidin biosynthesis. As mentioned above, the only uncharacterized oxidase gene in the aec cluster is aecF, which has been annotated as a cupin-domain-containing protein. This class of proteins is known to bind transition metals in their conserved β-barrel structures16 and catalyze oxygenation reactions.17,18 In addition, a BLAST search suggests that the cupin domain of AecF resembles the JmjC domain that is found in Fe/α-KG enzymes.19 HHpred analysis20 consistently revealed the Fe/α-KG halogenase OocP (PDB ID: 8A82) as a top hit for AecF. Therefore, AecF was hypothesized to be responsible for catalyzing the epoxidation of 24 to form 1. However, recombinant AecF could not be expressed in E. coli as a soluble protein. Therefore, in vivo experiments were performed in which aecF was deleted inframe from the producing strain C. violaceum. Upon deletion of aecF, the production of 1 was abolished in the mutant strain (Figure 4B), whereas complementation of aecF into the ΔaecF mutant strain restored aerocyanidin production albeit not to the extent of the wild type (Figure 4B). This partial restoration could be due to a decrease in the expression level of the aecF gene in the complementation strain or another effect related to the trans-expression system. In addition, compound 24 was also observed to accumulate in the culture broth of the ΔaecF mutant strain as well as the aecF complementation strain but at a reduced level (ca. 20% that from the ΔaecF mutant strain); however, 24 was not observed with the wildtype strain (Figure 4C). This is consistent with the reduced production level of 1 in the aecF complementation strain as opposed to the wild type and the conclusion that AecF catalyzes the epoxidation of 24 to yield the final product 1. However, the possibility that AecF acts prior to offloading of the carbon chain of 24 from AecD cannot yet be excluded.
The fact that aerocyanidin (1) and amycomicin (3) share similar structures and partially conserved gene clusters implies that biosynthesis of the core structure of 3 may be similar to that of 1. As proposed in Figure 5, 2-hexenoyl-ACP (fatty acyl carrier protein or its CoA derivative) (25) could be sequentially processed by the bifunctional thioesterase AmcD and the Fe/α-KG enzyme AmcA to form the isonitrile intermediate 29 (25 → 27 → 29, Figure 5). The ligase AmcH may then catalyze loading of 29 onto the ACP AmcE (29 → 31, Figure 5). The PKSs AmcF and AmcG would accept and process 31 to afford the 18-carbon chain product 37 (31 → 33 → 35 → 37, Figure 5). While a homologue of aecF is absent in the amc cluster, the cluster instead encodes three putative P450 enzymes (i.e., amcB, amcC, and amcQ), which may be responsible for oxidative installation of the epoxide, the C8 keto and the C16 alkene functionalities (37 → 3, Figure 5).
Figure 5.
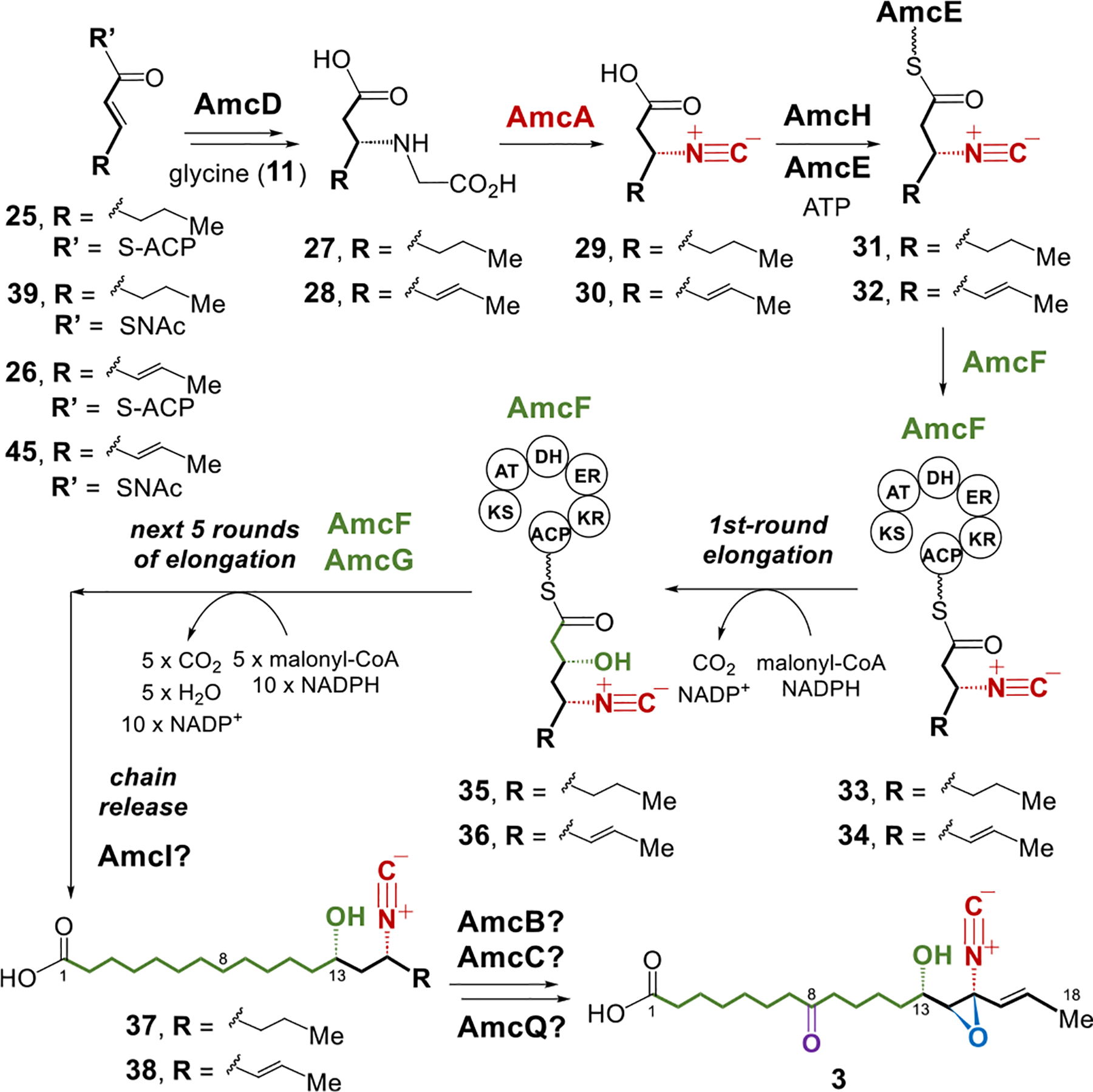
Proposed biosynthetic pathways of amycomicin (3).
Consistent with this proposal, the glycine adduct 27 was found to be generated when the SNAc analog (39) of 2-hexenoyl-ACP was synthesized and incubated with glycine and AmcD (Figure S8A). Moreover, 27 was converted in the presence of AmcA to the isonitrile product 29 (Figure S8A), which could be loaded onto AmcE in the presence of AmcH to give 31 (Figure S8B). However, one-pot reaction of chemically synthesized 29 with AmcE, AmcH, and the two PKS gene products AmcF and AmcG did not give rise to the expected chain-elongated product 37 (Figure 6B). Instead, 44 (calcd for C17H30NO3 − [M−H]−: 296.2231, found: 296.2232) was found to be the major product, the carbon chain of which is two carbons shorter than 37 and 3 (see Figures 6B and S9 for the structure of 44). This observation indicated that the final round of elongation to extend a 16-carbon chain to an 18-carbon chain (44 → 37) did not happen, probably due to the inability of the ketosynthase (KS) domain of AmcF or AmcG to accommodate the 16-carbon chain substrate for another round of elongation (Figure S9A). Accordingly, 31 may not be the true starter unit for PKS elongation, and 2-hexenoyl-ACP (25) is not the precursor for amycomicin biosynthesis. These observations prompted us to consider an alternative starter unit of chain elongation catalyzed by AmcF.
Figure 6.
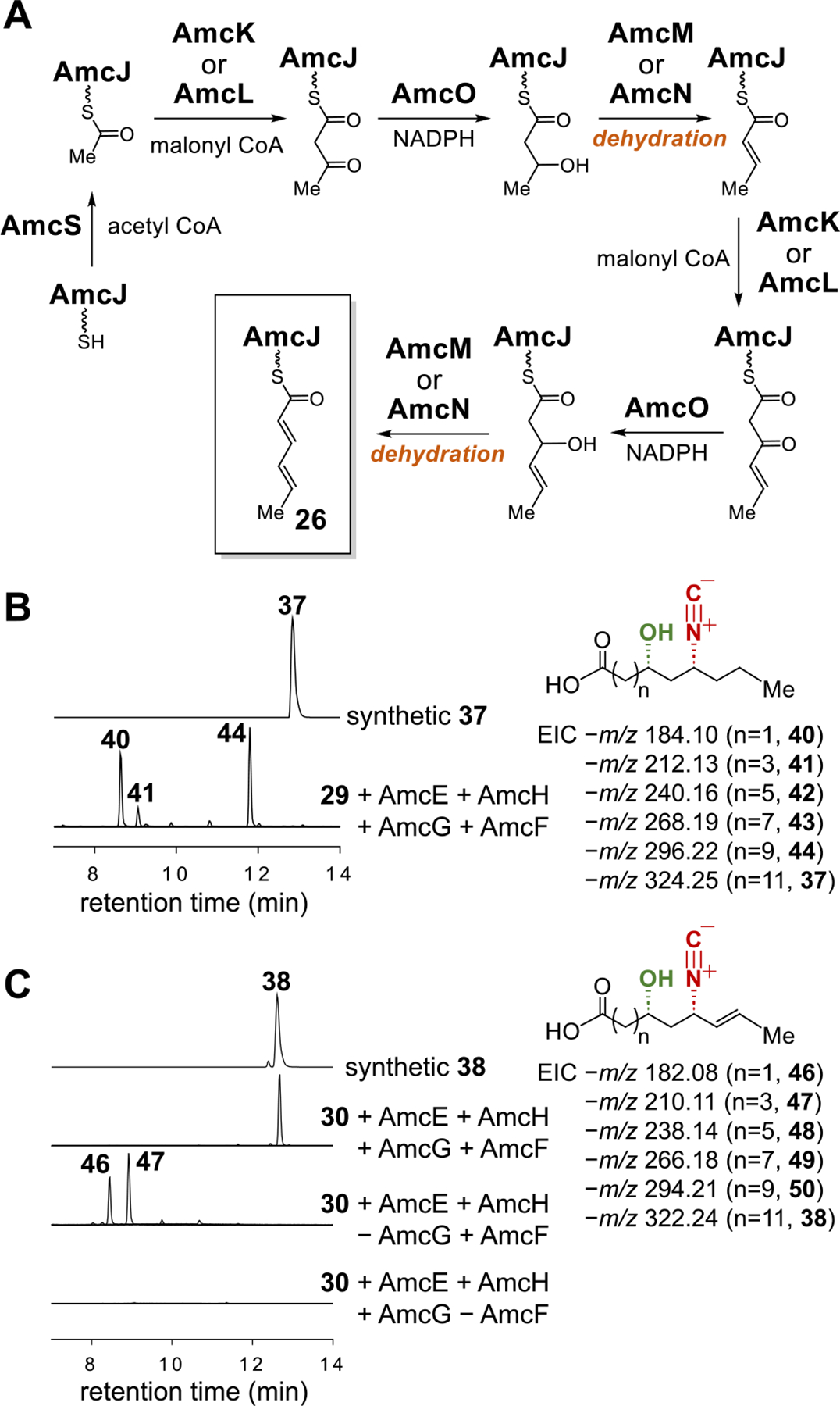
(A) Proposed biosynthetic pathway for sorbic-ACP (26 with AmcJ serving as ACP). (B) LCMS analysis of the in vitro PKS reaction with 29 and (C) with 30.
As mentioned earlier, the amc cluster also contains an additional set of genes that are related to FAB (see Figure 2A). These include the ACP amcJ, the two fabF (ketosynthase for chain elongation) homologues amcK and amcL, the fabA (dehydratase) homologue amcM, the fabZ (dehydratase) homologue amcN, the fabG (ketoreductase) homologue amcO, and the fabH (ketosynthase for initiation of FAB) homologue amcS. Therefore, the enzymes encoded by these genes may play an important role in the construction of the productive six-carbon starter unit used in the chain initiation of amycomicin biosynthesis. Based on their annotations, these FAB enzymes may produce 2,4-hexadienoyl-ACP, also known as sorbic-ACP (26) (Figure 6A), which could potentially serve as the six-carbon precursor for the biosynthesis of amycomicin (Figure 5).
To investigate this biosynthetic model, the SNAc analog (45) of sorbic-ACP (26), was synthesized and tested. As shown in Figure S10, 45 can be processed by AmcD, AmcA, and AmcE/AmcH in sequence to form 30 and then 32. More importantly, incubation of a synthetic racemate of 30 with AmcE, AmcH, AmcF and AmcG successfully produced the chain-elongated product 38 (calcd for C19H32NO3 − [M−H]−: 322.2388, found: 322.2396) with the correct chain length of 18 carbons (Figures 6C and S11D). This result showed 32 to be the starter unit for PKS elongation catalyzed by AmcF, thereby indicating that sorbic-ACP (26) is the true entry point to amycomicin biosynthesis. Omitting AmcG from the above reaction led to no production of 38 but accumulation of 46 (calcd for C9H12NO3− [M−H]−: 182.0823, found: 182.0835) and 47 (calcd for C11H16NO3− [M−H]−: 210.1136, found: 210.1150) (Figures 6C and S11B,C). These findings suggested that the ten-carbon acyl chain of 47 cannot be accepted by the KS domain of AmcF but requires the standalone KS AmcG to catalyze further elongation (Figure S11A).
Having established the assembly of the 18-carbon chain intermediate 38, the subsequent installations of the epoxide and the keto group are believed to be catalyzed by the three P450 enzymes, AmcB, AmcC and AmcQ. To interrogate the roles of these P450 enzymes, a synthetic racemate of 38 was incubated with AmcB, AmcC or AmcQ individually along with other necessary reaction components (see Supporting Information). LCMS analysis of the reaction mixture of AmcQ with 38 showed that 38 was depleted with concomitant formation of four oxygenated species, P1, P2, P3 and P4 (Figure 7B). According to the m/z values, P1 (calcd for C19H32NO4− [M−H]−: 338.2337, found: 338.2345) can be assigned as the monohydroxylated product (+16 Da) of 38, while P2 (calcd for C19H30NO4− [M−H]−: 336.2180, found: 336.2185) is the dehydrogenated product (−2 Da) of P1. Furthermore, P3 (calcd for C19H30NO5− [M−H]−: 352.2129, found: 352.2120) may result from hydroxylation of P2, and P4 (calcd for C19H28NO5− [M−H]−: 350.1973, found: 350.1967) is the dehydrogenated product of P3. Time course analysis of the AmcQ assay revealed that 38 undergoes two consecutive rounds of hydroxylation and dehydrogenation to form P4 (38 → P1 → P2 → P3 → P4, Figure S12). However, attempts to isolate and characterize these products were unsuccessful perhaps due to their instability and scarcity.
Figure 7.
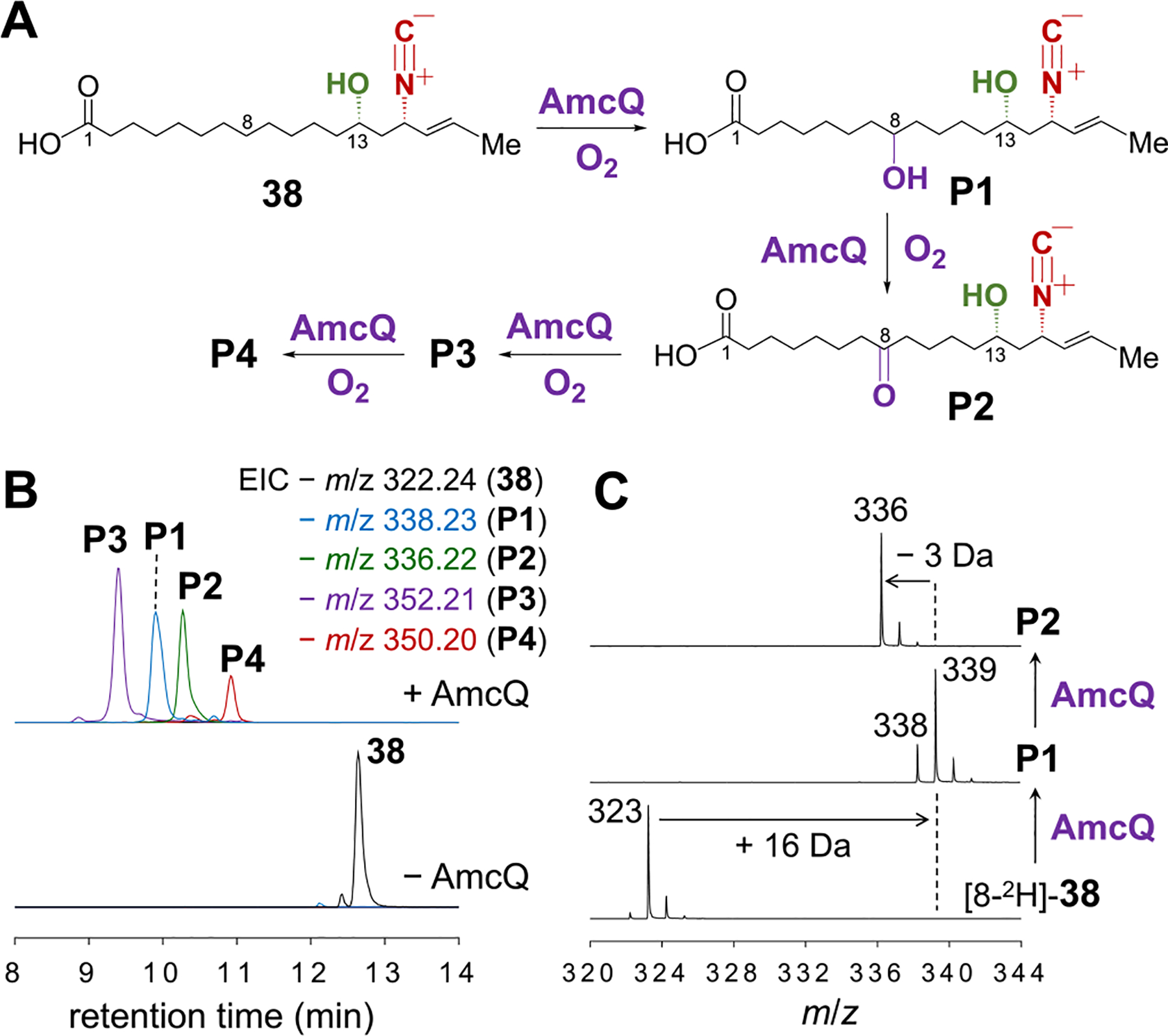
(A) Scheme and (B) LCMS analysis of the AmcQ reaction with 38. (C) Mass analysis of P1 and P2 generated in the AmcQ reaction with [8-2H]-38.
Three isotopologs of 38 including [8-2H]-38, [14-2H2]-38, and [15-2H]-38, all in their racemic forms, were synthesized in order to probe the loci where oxidation takes place in AmcQ catalyzed reactions. Retention of deuterium labels were observed in all four products (P1 to P4) when [14-2H2]-38 or [15-2H]-38 was used as the substrate (Figure S13C,D) indicating that C14 and C15 are not affected by the AmcQ catalyzed reaction. In contrast, reaction of AmcQ with [8-2H]-38 led to partial retention of 8-2H in P1 and disappearance from P2, P3 and P4 (Figures 7C and S13B). These results suggested that AmcQ first catalyzes hydroxylation at C8 to form P1 followed by dehydrogenation to generate the C8 keto product P2. While the site of subsequent hydroxylation and dehydrogenation to yield P3 and P4 was not determined, it must be at a carbon other than C14 or C15. To uncover the source of the oxygen atoms, 18O-isotope tracer experiments with 18O2 and H218O were performed. The results shown from Figure S14A to C demonstrated that molecular oxygen is incorporated once into P1 and P2 as well as twice into P3 consistent with the mechanism of P450-catalyzed oxygenation.21 In contrast, H2O was incorporated at both sites in P4 (Figure S14D). As P4 is the dehydrogenated product of P3, it is likely that P4 bears two keto groups including the one at C8, which are prone to solvent exchange.
Despite the fact that AmcQ catalyzes C8 hydroxylation and dehydrogenation of 38 to yield the ketone P2, the overoxidation of P2 to P3 and P4 suggested that 38 may not be the true substrate for AmcQ. Instead, epoxidation of 38 between C14 and C15 may occur prior to C8 oxygenation. The remaining two P450 enzymes AmcB and AmcC are thus hypothesized to catalyze the epoxidation step. To test this hypothesis, AmcB-AmcC coupled reaction with 38 was conducted. However, the anticipated epoxidation product (51) was not detected by LCMS analysis. Rather, a new peak was observed eluting at 10.8 min (P5) (traces i and iv, Figure 8B). By comparison with the authentic standard, P5 was established to be 13-hydroxytridecanoic acid (52, traces iv and v, Figure 8B). When [8-2H]-38 was used as the substrate, the mass of P5 increased by 1 Da (Figure S15), suggesting that P5 is indeed derived from 38. It is noteworthy that incubation of 38 with either AmcB or AmcC alone also generated P5 but with much lower efficiency (traces ii to iv, Figure 8B). Collectively, these results suggest that 38 is transformed to an unstable product in the presence of AmcB and AmcC, which rapidly decomposes to P5 (52) presumably via C−C bond breakage between C13 and C14. The details regarding this process are still under investigation.
Figure 8.
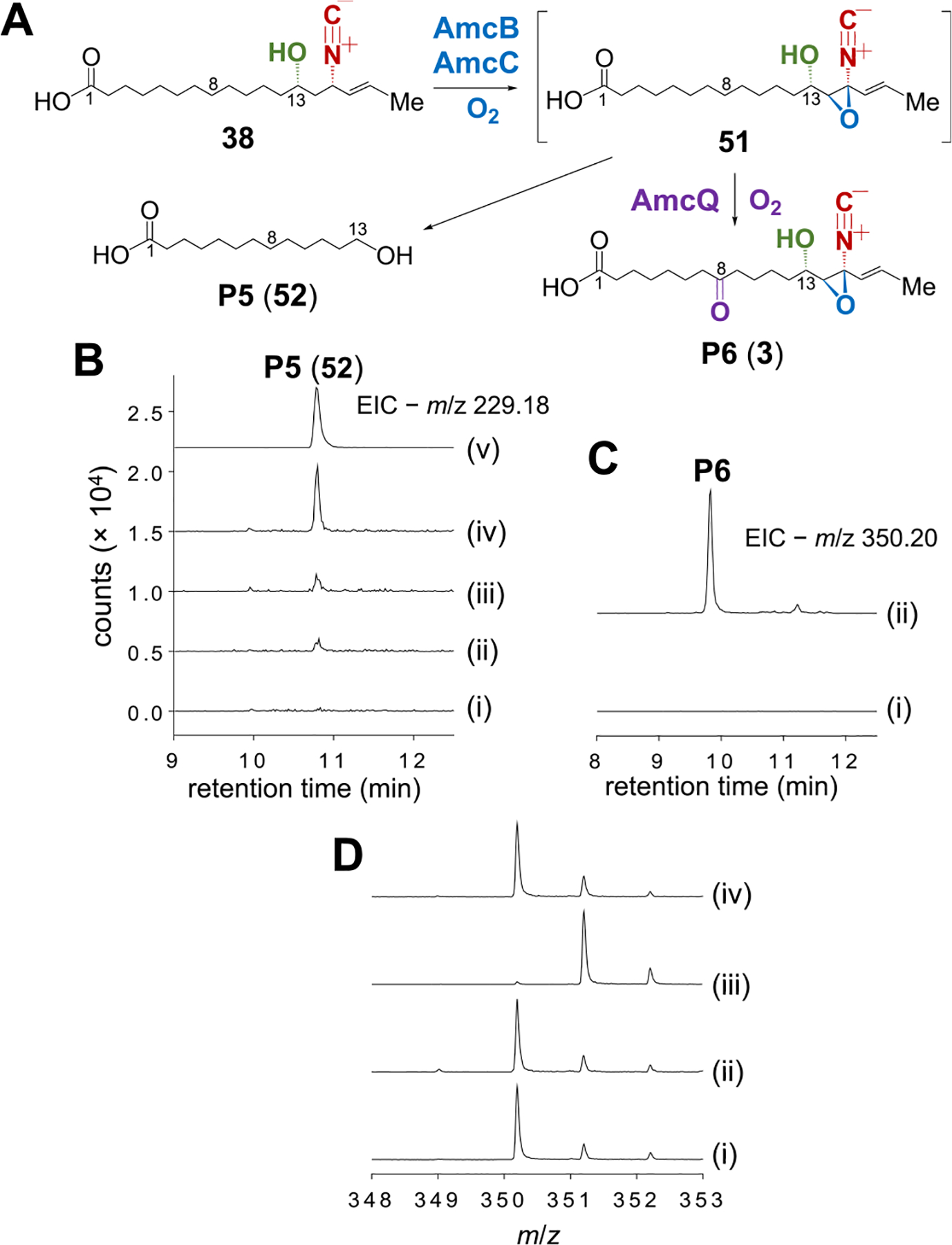
(A) Scheme of the final steps in amycomicin (3) biosynthesis involving AmcB, AmcC and AmcQ. (B) LCMS analysis of the AmcB-AmcC reaction with 38: (i) without AmcB or AmcC; (ii) with AmcB only; (iii) with AmcC only; (iv) with both AmcB and AmcC; (v) standard of 52. (C) LCMS analysis of the AmcB-AmcC-AmcQ coupled reaction with 38: (i) incubation of 38 with AmcB and AmcC for 30 min; (ii) incubation of 38 with AmcB and AmcC for 10 min followed by addition of AmcQ and incubation for another 20 min. (D) Mass analysis of P6 produced in the AmcB-AmcC-AmcQ coupled reaction with (i) unlabeled 38, (ii) [8-2H]-38, (iii) [14-2H2]-38, and (iv) [15-2H]-38.
Given the transient nature of the AmcB-AmcC product, one-pot reaction of 38 with AmcB, AmcC, and AmcQ was carried out. However, the major products from this one-pot assay were P3 and P4 without any previously unnoticed products (Figure S16), indicating that a majority of the substrate had been consumed by AmcQ without the involvement of AmcB or AmcC. To overcome this, 38 was first incubated with AmcB and AmcC for 10 min before adding AmcQ. After another 20 min, enzymes were removed by centrifugal filtration. LCMS analysis of the reaction filtrate revealed a new EIC peak (P6, calcd for C19H28NO5− [M−H]−: 350.1973, found: 350.1967) (Figure 8C), the molecular weight of which is 28 Da greater than 38 consistent with the addition of two oxygen atoms, loss of four hydrogen atoms and thus the structure of amycomicin (3). While significant amounts of P1, P2, and P3 were also generated, P5 was not detected (Figure S17). This suggests that the product of the AmcB-AmcC reaction was not degraded to P5 but very likely converted to P6 by AmcQ. Consistent with this hypothesis, incubation of 38 with AmcQ followed by ultrafiltration to remove AmcQ prior to addition of AmcB and AmcC did not generate P6 (Figure S18), indicating that 38 must first be processed by AmcB and AmcC before AmcQ to produce P6.
To determine the source of the two added oxygen atoms, the reaction was run with 18O2 (97 atom %)/H216O and 16O2/H218O (65 atom %). Incorporation of two 18O isotopes into P6 from 18O2 were observed while no 18O incorporated from H218O was detected (Figure S14E). Moreover, these two oxygens are proposed to include one at C8 as a keto group and the other between C14 and C15 as an epoxide. Due to its extreme instability, preparation of an amycomicin standard for direct comparison with P6 proved challenging. Thus, site-specifically deuterated 38, including [8-2H]-38, [14-2H2]-38, and [15-2H]-38 were respectively assayed to establish the position of hydrogen abstraction from 38 to form P6. As a result, the deuterium in [8-2H]-38 was completely lost in P6 (spectrum ii, Figure 8D) consistent with formation of a ketone at C8. Furthermore, only one deuterium was lost from the [14-2H2]-38 while the deuterium in [15-2H]-38 was found to be completely depleted (spectra iii and iv, Figure 8D). This is consistent with addition of the second oxygen as an epoxide bridging C14 and C15 and thus the assignment of P6 as 3. Therefore, the final steps in amycomicin biosynthesis are established to involve the AmcB-AmcC catalyzed epoxidation of 38 between C14 and C15 followed by AmcQ catalyzed hydroxylation and dehydrogenation of C8 (38 → 51 → 3, Figure 8A).
Several putative mechanisms regarding the epoxidation catalyzed by the P450 enzymes AmcB/C can be envisioned as shown in Figures 9 and S19. The reactive Compound (Cpd) I may abstract the hydrogen atom at C14 followed by hydroxyl rebound to give 54 (38 → 53 → 54, Figure 9). The second hydrogen atom abstraction could be performed at C15 in 54 by a second Cpd I to result in the C15 radical 55. Oxidation of the C15 radical by Cpd II followed by cyclization could form the epoxide 51 (55 → 51, Figure 9). Alternatively, the initial C14 radical 53 could undergo one-electron oxidation then deprotonation of C15 to yield a double bond between C14 and C15 (53 → 56, Figure 9). Oxygen atom transfer from the second Cpd I to the C14=C15 double bond would generate 51 (56 → 51, Figure 9). Analogous reaction pathways beginning with hydrogen atom abstraction from C15 of 38 are also possible (see Figure S19).
Figure 9.
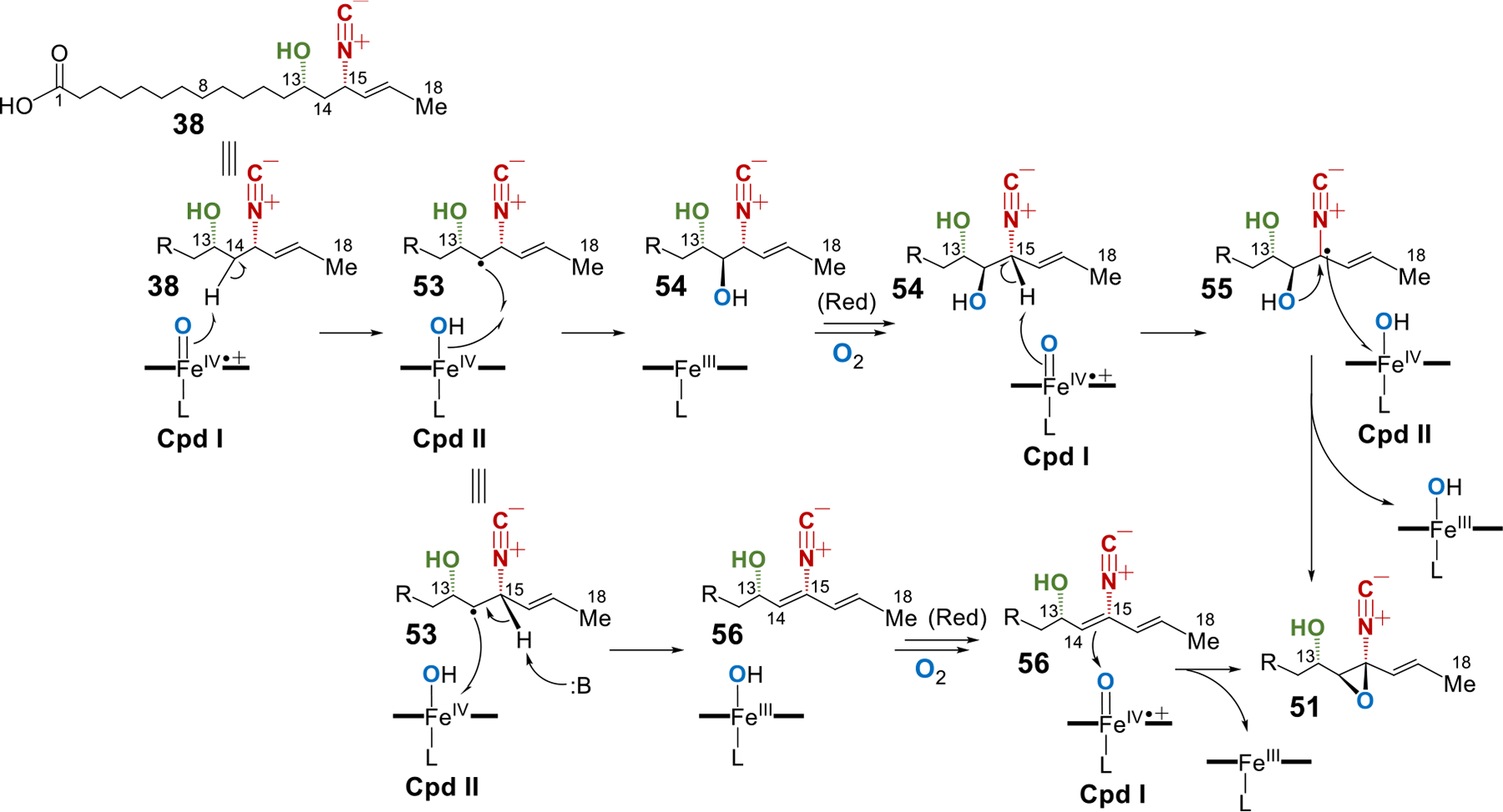
Proposed mechanisms for the AmcB/C catalyzed epoxidation involving the first hydrogen atom abstraction at C14 of 38. [Red] stands for the reduction that is required for the resting Fe(III) complex to react with O2 to form Cpd I.
CONCLUSIONS
In summary, the biosynthetic pathways of aerocyanidin (1) and amycomicin (3) have been fully elucidated in this work. Both share the conserved INLP mechanism of isonitrile functionalization in 14 and 30. Unlike the NRPS machinery at work in the production of INLPs, the acyl group of the AecC-bound isonitrile intermediate 17 is transferred to the PKS enzyme AecD (17 → 18, Figure 3C) to undergo carbon chain extension in the assembly of aerocyanidin (1). The first round of elongation generates the β-hydroxy product 19, which accounts for the formation of the C11 hydroxyl group in 1. The following four rounds of fully reducing elongation and hydrolysis yield 24. Finally, the cupin domain-containing protein AecF catalyzes epoxidation to conclude the biosynthesis of 1. Construction of the isonitrile PKS starter unit during the biosynthesis of amycomicin is initiated with sorbic-ACP (26) which is a product of fatty acid biosynthesis. The following steps encompass reactions sequentially catalyzed by AmcD, AmcA, and AmcE/AmcH such that the starting material 26 is transformed to 32. Distinct from that in aerocyanidin biosynthesis where a single PKS is sufficient for the chain extension, the standalone KS AmcG is required in addition to the PKS AmcF to accomplish the conversion from 32 to 38. Specifically, AmcG is indispensable for the third round of elongation. Furthermore, unlike aerocyanidin biosynthesis, the cytochrome P450 enzymes AmcB and AmcC catalyze epoxidation of 38 to 51. Finally, the third P450 enzyme, AmcQ, catalyzes the keto formation at C8 to conclude the biosynthesis of 3 (51 → 3).
Fatty acid chains serve as the template for isonitrile formation during the biosynthesis of INLPs such as INLP-2 (8). This is followed by introduction of the peptide linkages to complete the production of these isonitrile-containing lipopeptides. Likewise, construction of the isonitrile moiety during the biosynthesis of aerocyanidin (1) and amycomicin (3) also occurs on a fatty acyl precursor. However, the subsequent chain elongation to assemble the hydrocarbon chain in the final structures are catalyzed by polyketide synthases in both cases. Importantly, these studies reveal that formation of the γ-hydroxy group in 1 and 3 are generated during PKS catalyzed chain elongation, and the olefinic moiety in 3 is from construction of the fatty acyl starter unit by FAB enzymes. All of them are not tailoring oxidation reactions as previously proposed. These findings thus help explain the apparent deficiency of oxidase genes in the aec and amc gene clusters. This work also demonstrates that biosynthesis of the epoxide group in 1 and 3 involves two different pathways. While accessing the epoxy isonitrile functionality synthetically has been challenging,22 this study unravels two distinctive enzymatic routes to introduce an epoxide at the aliphatic position α,β to an isonitrile group. These findings may be of use in developing a biocatalytic approach to the epoxy isonitrile moiety, which is a potentially useful pharmacophore.
Supplementary Material
ACKNOWLEDGMENTS
The heterologous expression host E. coli K207 was generously provided by Prof. Adrian Keatinge-Clay at the University of Texas at Austin. The plasmids camA/pET28b(+) and camB/pET28b(+) for expressing CamA and CamB proteins for P450 enzyme assays were kindly provided by Prof. Ikuro Abe at the University of Tokyo. We are grateful to Dr. Kazuo Shin-ya at the National Institute of Advanced Industrial Science and Technology for searching for the producing strain of YM-47515. We thank Dr. Daan Ren for providing the Sfp protein, and Dr. Yu-Hsuan Lee and Joseph Livy Franklin for providing the MatB protein. We thank Dr. Mark Ruszczycky for his valuable comments on this manuscript. This work was supported by the National Institutes of Health (GM040541 to H.-w.L. and R01AT009874 to J.C.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.4c06411.
Additional experimental details, materials, and methods, including chemical synthesis, gene deletion and complementation, heterologous expression experiments, protein purification, in vitro enzymatic assays, supplementary tables and figures, and NMR spectra for synthetic compounds (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.4c06411
The authors declare no competing financial interest.
Contributor Information
Ziyang Zheng, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States.
Jon Clardy, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School and Blavatnik Institute, Boston, Massachusetts 02115, United States.
Hung-wen Liu, Department of Chemistry, University of Texas at Austin, Austin, Texas 78712, United States; Division of Chemical Biology and Medicinal Chemistry, College of Pharmacy, University of Texas at Austin, Austin, Texas 78712, United States.
REFERENCES
- (1).Parker WL; Rathnum ML; Johnson JH; Wells JS; Principe PA; Sykes RB Aerocyanidin, a new antibiotic produced by Chromobacterium violaceum. J. Antibiot 1988, 41 (4), 454–460. [DOI] [PubMed] [Google Scholar]
- (2).Sugawara T; Tanaka A; Imai H; Nagai K; Suzuki K YM-47515, a novel isonitrile antibiotic from Micromonospora echinospora subsp. echinospora. J. Antibiot 1997, 50 (11), 944–948. [DOI] [PubMed] [Google Scholar]
- (3).Pishchany G; Mevers E; Ndousse-Fetter S; Horvath DJ; Paludo CR; Silva EA; Koren S; Skaar EP; Clardy J; Kolter R Amycomicin is a potent and specific antibiotic discovered with a targeted interaction screen. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (40), 10124–10129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ernouf G; Wilt IK; Zahim S; Wuest WM Epoxy isonitriles, a unique class of antibiotics: synthesis of their metabolites and biological investigations. ChemBioChem. 2018, 19 (23), 2448–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tamura A; Kotani H; Naruto S Trichoviridin and dermadin from Trichoderma sp. TK-1. J. Antibiot 1975, 28 (2), 161–162. [DOI] [PubMed] [Google Scholar]
- (6).Ollis WD; Rey M; Godtfredsen WO; Rastrupandersen N; Vangedal S; King TJ The constitution of the antibiotic trichoviridin. Tetrahedron 1980, 36 (4), 515–520. [Google Scholar]
- (7).Sood M; Kapoor D; Kumar V; Sheteiwy MS; Ramakrishnan M; Landi M; Araniti F; Sharma A Trichoderma: the ″secrets″ of a multitalented biocontrol agent. Plants 2020, 9 (6), 762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Baldwin JE; Adlington RM; Oneil IA; Russell AT; Smith ML The total synthesis of (±)-trichoviridin. Chem. Commun 1996, 41–42. [Google Scholar]
- (9).Harris NC; Sato M; Herman NA; Twigg F; Cai W; Liu J; Zhu X; Downey J; Khalaf R; Martin J; Koshino H; Zhang W Biosynthesis of isonitrile lipopeptides by conserved nonribosomal peptide synthetase gene clusters in Actinobacteria. Proc. Natl. Acad. Sci. U.S.A 2017, 114 (27), 7025–7030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Harris NC; Born DA; Cai W; Huang Y; Martin J; Khalaf R; Drennan CL; Zhang W Isonitrile formation by a nonheme iron(II)-dependent oxidase/decarboxylase. Angew. Chem., Int. Ed 2018, 57 (31), 9707–9710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chen T-Y; Zheng Z; Zhang X; Chen J; Cha L; Tang Y; Guo Y; Zhou J; Wang B; Liu H.-w.; Chang W-C. Deciphering the reaction pathway of mononuclear iron enzyme-catalyzed N≡C triple bond formation in isocyanide lipopeptide and polyketide biosynthesis. ACS Catal. 2022, 12 (4), 2270–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wilson MC; Moore BS Beyond ethylmalonyl-CoA: the functional role of crotonyl-CoA carboxylase/reductase homologs in expanding polyketide diversity. Nat. Prod. Rep 2012, 29 (1), 72–86. [DOI] [PubMed] [Google Scholar]
- (13).Bhushan R; Bruckner H Marfey’s reagent for chiral amino acid analysis: a review. Amino Acids 2004, 27 (3−4), 231–247. [DOI] [PubMed] [Google Scholar]
- (14).Keatinge-Clay AT; Stroud RM The structure of a ketoreductase determines the organization of the β-carbon processing enzymes of modular polyketide synthases. Structure 2006, 14 (4), 737–748. [DOI] [PubMed] [Google Scholar]
- (15).Khosla C; Tang Y; Chen AY; Schnarr NA; Cane DE Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu. Rev. Biochem 2007, 76, 195–221. [DOI] [PubMed] [Google Scholar]
- (16).Dunwell JM; Culham A; Carter CE; Sosa-Aguirre CR; Goodenough PW Evolution of functional diversity in the cupin superfamily. Trends Biochem. Sci 2001, 26 (12), 740–746. [DOI] [PubMed] [Google Scholar]
- (17).Ng TL; Rohac R; Mitchell AJ; Boal AK; Balskus EP An N-nitrosating metalloenzyme constructs the pharmacophore of streptozotocin. Nature 2019, 566 (7742), 94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).He H-Y; Henderson AC; Du Y-L; Ryan KS Twoenzyme pathway links L-arginine to nitric oxide in N-nitroso biosynthesis. J. Am. Chem. Soc 2019, 141 (9), 4026–4033. [DOI] [PubMed] [Google Scholar]
- (19).Klose RJ; Kallin EM; Zhang Y JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet 2006, 7 (9), 715–727. [DOI] [PubMed] [Google Scholar]
- (20).Alva V; Nam SZ; Söding J; Lupas AN The MPI bioinformatics toolkit as an integrative platform for advanced protein sequence and structure analysis. Nucleic Acids Res. 2016, 44 (W1), W410–W415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Meunier B; de Visser SP; Shaik S Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev 2004, 104 (9), 3947–3980. [DOI] [PubMed] [Google Scholar]
- (22).Baldwin JE; Chen DQ; Russell AT On the synthesis of vinyl isonitriles. Chem. Commun 1997, 2389–2390. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.