

ABSTRACT
Japanese encephalitis virus (JEV) is an arthropod-borne, plus-strand flavivirus causing viral encephalitis in humans with a high case fatality rate. The JEV non-structural protein 5 (NS5) with the RNA-dependent RNA polymerase activity interacts with the viral and host proteins to constitute the replication complex. We have identified the multifunctional protein Nucleolin (NCL) as one of the several NS5-interacting host proteins. We demonstrate the interaction and colocalization of JEV NS5 with NCL in the virus-infected HeLa cells. The siRNA-mediated knockdown of NCL indicated that it was required for efficient viral replication. Importantly, JEV grew to higher titers in cells over-expressing exogenous NCL, demonstrating its pro-viral role. We demonstrated that NS5 interacted with the RRM and GAR domains of NCL. We show that the NCL-binding aptamer AS1411 containing the G-quadruplex (GQ) structure and the GQ ligand BRACO-19 caused significant inhibition of JEV replication. The antiviral effect of AS1411 and BRACO-19 could be overcome in HeLa cells by the overexpression of exogenous NCL. We demonstrated that the synthetic RNAs derived from the 3′-NCR of JEV genomic RNA containing the GQ sequence could bind NCL in vitro. The replication complex binding to the 3′-NCR is required for the viral RNA synthesis. It is likely that NCL present in the replication complex destabilizes the GQ structures in the genomic RNA, thus facilitating the movement of the replication complex resulting in efficient virus replication.
IMPORTANCE
Japanese encephalitis virus (JEV) is endemic in most parts of South-East Asia and the Western Pacific region, causing epidemics of encephalitis with a high case fatality rate. While a tissue culture-derived JEV vaccine is available, no antiviral therapy exists. The JEV NS5 protein has RNA-dependent RNA polymerase activity. Together with several host and viral proteins, it constitutes the replication complex necessary for virus replication. Understanding the interaction of NS5 with the host proteins could help design novel antivirals. We identified Nucleolin (NCL) as a crucial host protein interactor of JEV NS5 having a pro-viral role in virus replication. The NS5-interacting NCL binds to the G-quadruplex (GQ) structure sequence in the 3′-NCR of JEV RNA. This may smoothen the movement of the replication complex along the genomic RNA, thereby facilitating the virus replication. This study is the first report on how NCL, a host protein, helps in JEV replication through GQ-binding.
KEYWORDS: protein-protein interaction, flavivirus, RNA-dependent RNA polymerase, nuclear protein, cytoplasmic translocation
INTRODUCTION
Japanese encephalitis virus (JEV) is an enveloped, single-stranded, plus-sense RNA virus. The virus belongs to the family Flaviviridae containing over 70 viruses, many of which cause arboviral diseases in humans. These include medically important dengue (DEN), yellow fever (YF), and West Nile (WN) viruses. JEV is responsible for the disease Japanese encephalitis (JE) and is the main cause of viral encephalitis in humans. The disease is endemic in most parts of South-East Asia and Western Pacific regions, and frequent epidemics of JE have been reported from different parts of India (1). Although a variety of live attenuated, recombinant, and inactivated JE vaccines are licensed, no virus-specific treatment is available. Development of novel, efficacious, and affordable antivirals against JEV is, therefore, a high priority. Understanding the virus replication that involves the interaction of the viral and host proteins will greatly benefit in this direction.
The JEV genomic RNA is ~11 kb long, carrying a 5′ type 1 cap, 5′- and 3′-non-coding regions (NCRs), and a single open reading frame (ORF). The NCRs are highly conserved among flaviviruses and form secondary structures that have a role in viral replication (2). The ORF encodes a single polyprotein that is cleaved into three structural (Capsid, C; pre-Membrane, prM; and Envelope, E) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5) by viral and host proteases. The structural proteins play a role in viral entry into the host cell, assembly, and egress. The non-structural proteins, together with several host proteins, form the viral replication complex inside the host cell (3). The largest and the most conserved protein among the Flaviviridae is the non-structural protein 5 (NS5) (4). The 103 kDa protein consists of two domains—the N-terminal methyltransferase (MTase) domain, and the C-terminal RNA-dependent RNA polymerase (RdRp) domain. The MTase domain of NS5 is responsible for the 5′ cap formation (5). The RdRp domain replicates the viral genome (6). The MTase and RdRp domains are coupled via a short linker (7). The enzyme forms a multimeric replication complex with several viral and host proteins and plays a central role in the viral genome replication and other events in the virus life cycle (8–11).
The flavivirus NS5 interacts with genomic RNA and initiates RNA replication at the 3′-NCR (12, 13). NS3 is the major viral protein associated with NS5 in the replication complex (14). However, only little information is available on the host proteins that constitute the replication complex for most flaviviruses. For example, NS5 of the DEN virus interacts with GBF1 to regulate intracellular transport (15). Its interaction with STAT2 and UBR4 inhibits the type 1 interferon-mediated signaling pathway during DEN virus infection (16). In the case of JEV, HnRNP A2, and DDX3 bind to the viral RNA through interaction with NS5 (17, 18).
We have studied the JEV NS5 interactome using two different methods and have identified nucleolin (NCL) as an important host protein interactor. NCL is a multifunctional nucleolar protein that can be found in different locations within a cell depending upon the physiological conditions (19). Here, we demonstrate a pro-viral role of NCL in JEV replication that involves its binding to the G-quadruplex (GQ) structures in the 3′-NCR.
MATERIALS AND METHODS
Cells and virus
The mouse neuroblastoma Neuro-2a cells (N2a) and human epithelial HeLa cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (HiMedia) supplemented with 10% fetal bovine serum (FBS). The African green monkey kidney cells (Vero) were cultured in Minimum essential media (MEM) (HiMedia) with 10% FBS. The mosquito cells (C6/36) were grown in Leibovitz’s L-15 media (HiMedia) with 10% FBS. HAP1 cells (a near-haploid human cell line derived from the KBM-7 chronic myelogenous leukemia cell line) were grown in Iscove’s modified Dulbecco’s medium (IMDM) (HiMedia) supplemented with 10% FBS. The N2a, HeLa, and Vero cells were grown at 37°C in a humidified atmosphere with 5% CO2, while C6/36 were grown at 28°C without CO2. All cultures were supplemented with 1× penicillin streptomycin glutamine (PSG) (HiMedia). The P20778 strain of JEV was used for this study (GenBank accession number AF080251).
Reagents and antibodies
The rabbit polyclonal antibody against NS5 was produced in the lab using purified JEV NS5 protein as previously described (20). The following commercial primary antibodies were used in this study: rabbit anti-NCL (Bethyl Laboratories; A300-709A), mouse anti-NCL (Santa Cruz Biotechnology; C23-H6 sc-55486), rabbit anti-JEV NS5 (Genetex; GTX131359), mouse anti-Myc tag (Abcam; ab18185), rabbit anti-GAPDH (Genetex; GTX100118), mouse anti-GAPDH (Invitrogen; MA1-16757), mouse anti-Lamin A/C (Cell Signalling Technology; 4777S), mouse anti-JEV NS1 (The Native Antigen Company; MAB 12176). The secondary antibodies used were as follows: anti-rabbit HRP (Invitrogen; 65-6120), anti-mouse HRP (Abcam; ab205719), anti-mouse Alexa Fluor 488 (Abcam; ab150113), anti-mouse Alexa Fluor 568 (Invitrogen; A-11031), anti-rabbit Alexa Fluor 488 (Invitrogen; A-11008), anti-rabbit Alexa Fluor 568 (Invitrogen; A-11011).
Plasmid construction
The JEV NS5 cDNA was amplified from the expression plasmid described previously (21) and transferred to the BioID2 vector (Addgene; 92308) for the production of BioID-NS5 plasmid expressing Myc-BirA-13xLinker-NS5 fusion protein. The empty vector BioID-Empty expressing Myc-BirA-13xLinker was used as the control. For the overexpression studies, the full-length and truncated forms of NCL cDNA were amplified using total cellular RNA and cloned into the pCMV-Myc vector (Clontech; 631604). All constructs were sequenced to verify the nucleotide sequence. The primers used for the plasmid construction are listed in Table 1.
TABLE 1.
Primers used in the study
| Amplicon | Primer | Nucleotide sequence (5′−3′) | Restriction sitea |
|---|---|---|---|
| Myc-BirA-13XLinker-NS5 | Forward | ATGCGCGGCCGCTCAGGGGAAGGCCCGGG | NotI |
| Reverse | ATGCGGTACCCTAGATGACCCTGTCTTCCTGGG | KpnI | |
| Myc-NCL | Forward | ATGCGAATTCTTGTGAAGCTCGCGAAGGCAGG | EcoRI |
| Reverse | ATGCGGTACCCTATTCAAACTTCGTCTTCTTTCCTTGTGG | KpnI | |
| Myc-1234R (tNCL) | Forward | ATGCGAATTCTTTTCAATCTCTTTGTTGGAAAC | EcoRI |
| Reverse | ATGCGGTACCCTATTCAAACTTCGTCTTCTTTCCT | KpnI | |
| Myc-123 (tNCL) | Forward | ATGCGAATTCTTTTCAATCTCTTTGTTGGAAAC | EcoRI |
| Reverse | ATGCGGTACCCTACAACTCCAGCCTGATTGCTC | KpnI | |
| Myc-34R (tNCL) | Forward | ATGCGAATTCTTTCAAAAACTCTGGTTTTAAGCA | EcoRI |
| Reverse | ATGCGGTACCCTATTCAAACTTCGTCTTCTTTCCT | KpnI | |
| Myc-4R (tNCL) | Forward | ATGCGAATTCTTAAAACTCTGTTTGTCAAAGG | EcoRI |
| Reverse | ATGCGGTACCCTATTCAAACTTCGTCTTCTTTCCT | KpnI |
Restriction sites are underlined in the nucleotide sequence.
Generation of stable cell line
N2a cells were transfected with BglII (New England Biolabs; R0144S)-linearized 500 ng BioID-NS5 or BioID-Empty vector using Lipofectamine 2000 (Thermo Fisher Scientific, 11668019). The transfected cells were incubated at 37°C for 24 h before the Lipofectamine 2000-containing medium was replaced with the fresh medium containing increasing concentrations of G418 (Gibco, 10131035) (0.1 –-1.5 mg/mL) with media change every 3 days. After 10 days, cell death was checked by cytotoxicity assay. For maintaining the stably-transfected cells, 0.6 mg/mL G418 was used (22).
Generation of nucleolin knockout cell line
HAP1 cells were transfected with the CRISPR/Cas9 NCL-knockout plasmid synthesized by GeneScript. The knockout plasmid was based on GenScript plasmid eSpCas9-2A-Puro (PX459) V2.0. The sgRNA sequence (3′-GATGAAGAAGATGATAGCAG-5′) was selected from the GeCKO v2 Human CRISPR Knockout Pooled Library and reviewed by the CHOPCHOP web tool. The sgRNA/Cas9 plasmid was transfected using Lipofectamine 3000. The transfected cells were selected using puromycin (InvivoGen; ant-pr) at a concentration of 0.8 µg/mL. The cells were single-cell cloned and stored. The absence of the NCL expression in the knockout cells was established by western blotting.
Cell transfection
For the overexpression studies, HeLa cells were transfected with plasmid using Lipofectamine 3000 (Thermo Fisher Scientific, L3000015) according to the manufacturer’s protocol. The protein knock-down studies were performed by transfecting HeLa cells with siRNA (30 nM) using Lipofectamine RNAiMAX (Thermo Fisher Scientific, 13778075) according to the manufacturer’s protocol. Cell cytotoxicity/viability was checked by the MTT Assay (Sigma Aldrich; TOX1) as per the product manual.
Western blotting
The cells were harvested and lysed using the lysis buffer containing 25 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 5% glycerol, pH 7.4. The protease inhibitor cocktail (Sigma Aldrich; P8340) was added immediately before lysing the cells. The lysates were incubated on ice for 1 h with periodic sonication and spun down, and the supernatant was collected for protein estimation by the Bradford Assay kit (Thermo Scientific; 23200). The 5× SDS-PAGE loading dye was then added to the lysates and heated at 95°C for 10 min. Equal amounts of protein lysates were then resolved on 8%–12% SDS-PAGE and transferred onto a polyvinylidene difluoride (PVDF) membrane. The membrane was then blocked by 5% non-fat milk for 2 h at room temperature, followed by overnight incubation in the primary antibody at 4°C. The membrane was then washed with PBST (PBS with 0.1% Tween-20) and incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h. The protein bands were visualized using the SuperSignal West Pico PLUS Chemiluminescent substrate (Thermo Scientific; 34577).
Nuclear cytoplasmic fractionation
HeLa cells were infected with JEV at 1 multiplicity of infection (MOI) and harvested at 6, 12, 18, and 24 h post-infection (pi). The nuclear and cytoplasmic fractions were obtained using the NE-PER nuclear and cytoplasmic extraction reagent (Thermo Fisher, 78833) according to the manufacturer’s protocol. GAPDH and Lamin A/C were used as the indicators of the purity of cytoplasmic and nuclear fractions, respectively.
Pull-down assays and co-immunoprecipitation
The BirA-NS5-expressing stable cell line harbors Bir-A, the mutant biotin ligase R118G that can promiscuously add biotin to NS5-interacting proteins (23–25). For in situ biotinylation, 50 µM biotin (Sigma Aldrich, B4501) was added to BioID-NS5 stable cells or control BioID-Empty cells. The cells were incubated for 24 h at 37°C, after which they were washed twice with 1× PBS to remove any residual biotin. The cells were lysed using RIPA lysis buffer (50 mM Tris HCl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.1% SDS, 0.5% sodium deoxycholate, 1 mM DTT, protease inhibitor cocktail) and the protein concentration was determined by the Bradford assay. The biotinylated proteins were pulled down using Dynabeads MyOne Streptavidin T1 beads (Invitrogen; 65601) as per the product manual. The proteins were eluted by boiling at 95°C in 1× SDS-PAGE loading dye for 5 min and resolved by gel electrophoresis.
For the NCL co-immunoprecipitation (Co-IP), HeLa cells were infected with JEV at 5 MOI for 24 h. Cells were washed with 1× PBS and lysed using RIPA lysis buffer. NCL was co-immunoprecipitated from 1 mg cell lysate with the help of a Co-IP kit (Thermo Scientific; 26149) using 10 µg anti-NS5 antibody. The eluted proteins were western blotted using the anti-NCL antibody.
Immunofluorescence assay
HeLa cells were plated on glass coverslips and mock-infected or infected with JEV at 5 MOI. At indicated time points post-infection (pi), cells were washed with 1× PBS, fixed with 4% paraformaldehyde (PFA) for 15 min, and permeabilized in 0.1% Triton X-100 for 15 min or 20 µg/mL digitonin for 1–3 min followed by blocking in 5% BSA for 1 h. Primary antibody staining for NCL, NS5, or Myc was done at 4°C overnight in 1% BSA followed by staining with fluorescently labeled secondary antibody in 1% BSA for 1 h. The primary and secondary antibody staining was followed by 3 washes for 15 min each with 1% BSA. Coverslips were then mounted, and the nucleus was stained with DAPI (Invitrogen; P36935). Image acquisition was done on a Leica SP8 confocal microscope with a 63× oil immersion objective. The co-localization analysis was done using the Leica Application Suite X (LAS-X) software.
Proximity ligation assay
HeLa cells were mock-infected or infected with JEV at 5 MOI. Cells were washed, fixed, and permeabilized at 24 h pi as above. The cells were processed further as per the Duolink in situ PLA kit (Sigma Aldrich; DUO92101-1KT) protocol. Briefly, the cells were treated with the blocking solution and incubated with primary antibody at 4°C overnight. Plus (anti-rabbit) and minus (anti-mouse) probes from different species, matching the host species of the primary antibodies, were then added followed by ligation and rolling circle amplification. The coverslips were finally mounted, and the nucleus was stained with the Duolink in situ mounting medium with DAPI. The image acquisition was done as above. The number of PLA signals per cell was quantified using the ImageJ software.
RNA isolation and quantitative real-time PCR
The total RNA was isolated from the cells using the RNAiso Plus reagent (Takara; 9109) and purified using the phenol-chloroform method. The first strand of the cDNA was synthesized using 0.1–1 μg total RNA with the ImProm II Reverse Transcriptase kit (Promega; A3802). The quantitative real-time PCR (qRT-PCR) was set up using the TB Green Premix Ex Taq (Takara; RR420A). The reactions were performed using QuantStudio 6 (Applied Biosystems). The primers (5′−3′) used in the study were—JEV: F-AGAGCACCAAGGGAATGAAATAGT, R-AATAAGTTGTAGTTGGGCACTCTG; GAPDH: F-GGTGAAGGTCGGAGTCAACG, R-AGGGATCTCGCTCCTGGAAG; NCL: F-GGTGGTCGTTTCCCCAACAAA, R- GCCAGGTGTGGTAACTGCT.
Plaque assay
JEV titer was determined by plaque assay (26). Briefly, a 6-well plate with the monolayers of Vero cells with ~80% confluency was infected with serial dilutions of virus-containing supernatants. The dilutions were prepared in the incomplete MEM and added to cells. Post 1 h of incubation, cells were washed and incubated at 37°C with an overlay consisting of 2% agarose and 2× MEM (supplemented with 10% FBS and PSG) for 5 days. The cell monolayers were then fixed with 4% formaldehyde and stained with crystal violet to visualize the plaques.
Electrophoretic mobility shift assay
The GQ sequence RNAs tagged with TAMRA at the 3′-end and the control RNA with biotin at the 3′-end (Table 2) were commercially synthesized (Sigma, USA). The GQ formation was induced by heating the RNAs to 80°C on a thermal heating block and cooling to 4°C at a rate of 2°C/min in 100 mM KCl. HeLa cells were transfected with plasmids expressing NCL or tNCL-34GAR. The cells were harvested 24 h later, and the total cell lysates were prepared by solubilizing the cells in lysis buffer (150 mM NaCl, 1 mM EDTA, 50 mM Tris pH 8, 1% Triton X-100, 1 mM PMSF, 1× protease inhibitor cocktail). The RNA-protein binding reactions were performed in a final volume of 30 µL using 20 ng RNA and 50 µg cell lysate in the binding buffer (14 mM HEPES pH 7.5, 1 mM EDTA, 60 mM KCl, 6 mM Tris-Cl pH 8, 1 mM DTT, RNAse inhibitor). After incubation for 30 min at 25°C, the RNA-protein complex was UV-crosslinked for 30 min on ice. The reaction mix was loaded on a 12% native polyacrylamide gel. Both the gel and the electrophoresis buffer contained 0.5× TBE buffer (45 mM Tris base, 45 mM boric acid, and 0.5 mM EDTA). Electrophoresis was performed at 10 V/cm at 4°C. The fluorescent image acquisition was done by the GE Typhoon FLA 9000 Gel Scanner.
TABLE 2.
Nucleotide sequences
| Name | Sequence |
|---|---|
| AS1411 | 5′-GGTGGTGG TGGTTGTGGTGGTGGTGG-3′ |
| CRO | 5′-CCTCCTCCTCCTTCTCCTCCTCCTCC-3′ |
| GQ1 | 5′-GGUGUAAGGACUAGAGGUUAGAGGA-3′-TAMRA |
| GQ2 | 5′-GGAGGUGGAAGGACUAGAGGUUAGAGGA-3′-TAMRA |
| Control RNA | 5′-GUGAUUCAGAGGUUUUUGACUUUAGUAAAUCGCCCAUCCUUUGAUAGA-3′-Biotin |
Computational analysis
For the generation of the protein-protein interaction (PPI) networks, only proteins scored in the experimental samples but not in the control samples were considered. All protein interactors were subjected to Gene Ontology (GO) by performing a database search in DAVID Knowledgebase v2022q3 (27). GO terms with P < 0.05 were considered for analysis. Venny v2.1.0 was used to determine the interactors common between the BioID and Co-IP experimental samples. OrthoDB v11 was used to determine the orthologs of common interactors. A PPI network of selected orthologs was constructed and enriched by StringDB v11.5. The networks were then exported to Cytoscape v3.9.1. The protein orthologs were identified from M. musculus to H. sapiens. To find the hub proteins, the interactors identified in BioID and Co-IP methods were pooled together and subjected to StringDB enrichment with a minimum required interaction score of 0.4 for each protein. The network was visualized by Cytoscape. Twenty-five proteins with the highest node degree were selected and compared to the common interactors as found via Venny.
For the in-silico docking, the crystal structure of full-length JEV NS5 was used (PDB ID—4K6M). The structure of human NCL was taken from the AlphaFold protein structure prediction database (P19338). The truncated forms of NCL were generated using Chimera v1.16 by removing unstructured N- and C-terminal regions with a low per-residue confidence score (pLDDT < 50). For docking, full-length JEV NS5 and truncated forms of NCL were used in the Cluspro protein-protein docking tool (28). The top-ranked models with the lowest scores were visualized and analyzed using Chimera.
The prediction of the GQ structure forming RNA sequence was done by web-based server QGRS mapper (29). The multiple sequence alignment of GQ1 and GQ2 sequences was done using the Multiple Sequence Alignment Tool (BV-BRC v3.31.12).
Statistical analysis
The data are presented as mean ± SEM with statistical significance calculated by paired Student’s t-test using GraphPad Prism v8. The P-values where significant are presented in the figures as ***P < 0.001, **P < 0.002, and *P < 0.033.
RESULTS
Bi-modal approach to identify NS5-interacting proteins
To study the JEV NS5 interactome, the pull-down of NS5 and its interacting proteins were performed by two approaches. First, for the BioID pull-down assay, the 2,718 bp cDNA encoding JEV NS5 was PCR amplified and cloned into the BioID plasmid. N2a cells were transfected with linearized NS5-encoding plasmid (pNS5) or the empty vector (pEV) followed by G418 selection to generate stable cell lines which were then clonally expanded. The expression of the Myc-tagged BirA-NS5 fusion protein was confirmed by western blotting (Fig. 1A). When excess biotin was supplemented in the culture media, due to the presence of the biotin ligase (BirA), all the proteins in its vicinity, and, in turn, in the vicinity of NS5, were promiscuously biotinylated in Myc-tagged BirA-NS5 fusion protein-expressing stable cell line. The cells transfected with pEV containing only the Myc-tagged BirA gene were processed in parallel as a negative control. The biotinylated proteins were captured and pulled down using the streptavidin-coated beads. The pull-down was electrophoresed on an SDS-PAGE and the proteins were in-gel digested by trypsin for mass spectrometric analysis. In the second approach, the Co-IP was performed to pull down JEV NS5 and its interacting proteins from the lysate of JEV-infected N2a cells using the rabbit polyclonal NS5 antibody. The immunoprecipitated proteins were digested in-solution and analyzed by mass spectrometry. Following the liquid chromatography and tandem mass spectrometry (LC-MS/MS), the proteins were selected based on the presence of the peptide spectra in the experimental sample but not in the control (Table S1). The MS data were screened for the NCBI IDs and the identified proteins were then subjected to various gene ontology (GO) categories (Fig. 1B and C). The two methods produced different outputs for the biological process; the BioID method identified 34 proteins while Co-IP identified 74 proteins. There was significant enrichment in annotated genes for viral genome replication, translation, cytoplasmic localization, RNA binding, and protein-binding functions that are likely to have a direct role in viral replication.
Fig 1.

The JEV NS5 interactome. (A) The schematic shows the linearized BioID plasmid constructs encoding Myc-tagged BirA-NS5 (pNS5) or the empty vector (pEV). N2a cells were transfected with these vectors, and the stable transfected cells were selected using G418. The cell lysates were western blotted with anti-Myc antibody. The unique bands, as indicated by the arrows, of Myc-BirA fusion and Myc-BirA-NS5 fusion were seen in the pEV- or pNS5-transfected cells, respectively (right panel). The molecular weight markers in KDa are shown on the right side of the blot. (B) GO analysis of NS5 interactors from the BioID pull-down assay using DAVID is presented. (C) GO analysis of NS5 interactors from the Co-IP assay using DAVID is presented. The heat maps show significant P-values for each list. The gene count is represented by bars on the y-axis.
Identification of NCL as a potential JEV NS5-interacting host protein
For the identification of important NS5-interacting host proteins, the proteomics data produced by the BioID and Co-IP methods were screened for the common proteins identified by the two methods. Four such proteins identified were Vinculin (Vcl), Asap2, Eukaryotic Translation Elongation Factor 1 Alpha 1 (eEF1α−1), and NCL (Fig. 2A). Additionally, a PPI network was generated from the proteomics data to find the hub proteins (Fig. 2C). Thus, the interactome data from the BioID and Co-IP methods were merged and the interaction network was generated using StringDB with a medium cut-off score of 0.4. The network was enriched and the degree of each node was determined (Table S2). Twenty-five proteins from the proteomics data were chosen with the highest degree of interaction. It was found that two proteins (eEF1α-1 and NCL) out of the four common proteins identified above had a high degree of interaction and were hub proteins (Fig. 2C). These methods, thus, identified NCL as a potential JEV NS5-interacting protein.
Fig 2.

Identification of the JEV NS5 interacting proteins. (A) The NS5 interactors that were common between the BioID and Co-IP assays were identified by a Venn diagram in Venny. (B) The StringDB PPI networks (gray edges) between the common interactors in M. Musculus (blue) were compared with their orthologs (dot edges) in H. sapiens (green). (C) The PPI network of 116 interactors of JEV NS5 was created from the combined proteomics data of BioID and CoIP methods using STRING. The network was enriched in StringDB and visualized in Cytoscape. The heat map shows proteins with higher degree of interactions.
JEV NS5 interacts with NCL in vivo
The Co-IP assay was performed to validate the interaction of JEV NS5 with NCL. The lysates of JEV- or mock-infected HeLa cells were employed to immunoprecipitate interacting proteins using JEV NS5 antibody (Fig. 3A). The western blotting of the immunoprecipitate with the NCL antibody showed a ~110 kDa NCL band only in JEV-infected cells confirming the in vivo interaction of JEV NS5 with NCL. NCL displays self-cleaving activity and usually exhibits multiple bands (30). The 110 kDa NCL was the major species in the actively dividing HeLa cells.
Fig 3.

Interaction of NCL with JEV-NS5. (A) HeLa cells were mock infected or infected with JEV at 5 MOI. The cells were harvested at 24 h pi, and the lysates were prepared. The immunoprecipitation was performed using the JEV NS5 antibody, and the western blots were stained using the NCL antibody. The molecular weight markers in kDa are shown at the right. (B) HeLa cells were mock infected or infected with JEV at 5 MOI. The cells were fixed and permeabilized with digitonin at 24 h pi and immunostained with the NS5 and NCL antibodies. The images were taken under a Leica SP8 confocal microscope. The zoom image shows the area under the inset. The line plot for the green and red fluorescence is shown at the right. (C) HeLa cells were mock infected or infected with JEV at 5 MOI. The cells were processed 24 h pi for the PLA. The left panel shows the representative images acquired using the Leica SP8 microscope. The red dots signify the interaction or very close proximity between the two proteins. The right panel presents the relative number of red dots observed per cell, where the number of dots in the mock-infected sample was taken as 1. The NS5-NS1 and NS5-GAPDH interactions were taken as the positive and negative controls, respectively. The data presented are from three independent experiments.
We then used confocal microscopy to see if JEV NS5 colocalizes with endogenous NCL in JEV-infected HeLa cells (Fig. 3B). Both the NCL and NS5 proteins could be nicely stained in the cytoplasm of the cells permeabilized with digitonin. Notably, the two proteins were co-localizing in the cytoplasm (Pearson correlation coefficient 0.42 ± 0.02). These results suggest that NCL and JEV NS5 may be interacting during the virus infection. This was further confirmed by studies in the JEV-infected HeLa cells expressing Myc-tagged NCL (Fig. 7B).
We also studied the interaction of JEV NS5 and endogenous NCL in virus-infected HeLa cells by in situ proximity ligation assay (PLA) (Fig. 3C). PLA is a sensitive method to detect PPI. The PLA probes bind to primary antibodies and undergo rolling circle amplification only when two proteins are in close proximity/interacting with each other. These amplified signals are visible as discrete red spots. The JEV-infected HeLa cells showed a significantly increased count of red dots compared to the mock-infected cells confirming the interaction between NCL and NS5. In flaviviruses, NS1 protein is known to interact with NS5 (31). The NS1-NS5 interaction, used here as a positive control, was clearly seen in the PLA. No interaction of NS5 with GAPDH was seen. This served as a negative control for the PLA (Fig. 3C).
JEV infection modulates NCL expression
We studied the NCL expression during the JEV infection of HeLa cells by determining the NCL mRNA levels by qRT-PCR and the protein levels by western blotting. As the virus infection proceeded, the JEV RNA levels peaked in the cells at 24 h pi. The NCL mRNA levels in the cells were significantly elevated at 18 h pi when these were ~2-fold higher in JEV-infected cells than seen in the mock-infected cells (Fig. 4A). The western blots of the total cell lysates showed that the NCL levels were ~1.2-fold higher in virus-infected cells at 12 and 18 h pi. While this increase was small and statistically not significant, it was seen reproducibly (Fig. 4B).
Fig 4.

JEV infection upregulates NCL expression and affects its cellular distribution. (A) HeLa cells were mock infected or infected with JEV at 1 MOI, and the total RNA was extracted at different time points. The level of JEV RNA and NCL mRNA was determined by qRT-PCR. The left panel shows the JEV RNA where GAPDH was used as the internal control. The right panel shows the NCL mRNA level as fold-change compared to the mock-infected sample at each time point which was assigned as 1. GAPDH was used as the internal control to normalize the data. The data from three independent experiments are shown. (B) JEV-infected (1 MOI) or mock-infected HeLa cells were harvested at different time points pi, and the level of NCL was analyzed by western blot. A representative western blot is shown in the left panel. The NCL expression was quantified relative to GAPDH, and the fold-change in the NCL expression level was calculated compared to that in the mock-infected sample for each time point. The fold change is shown at the top. The molecular weight marker in kDa is shown at the right. The data from three independent experiments are presented in the right panel. (C) The JEV-infected (1 MOI) or mock-infected HeLa cells were harvested at different time points pi, and the nuclear and cytoplasmic fractions were prepared. The levels of NCL and NS5 were analyzed by western blot in both fractions for each time point. GAPDH served as the cytoplasmic marker while Lamin A/C served as the nuclear marker. The NCL distribution in the nucleus and cytoplasm was quantified with respect to Lamin A/C and GAPDH, respectively. The ratio of the cytoplasmic to nuclear NCL in the mock-infected cells at each time point was taken as 1. A representative western blot is shown in the top panel. The lane M has the molecular weight marker in kDa shown at the bottom of the marker band. The data from three independent experiments are presented in the lower panel.
NCL is majorly a nuclear protein (Fig. S1) that is reported to redistribute from the nucleus to the cytoplasm during the Polio and Seneca Valley virus infection (32, 33). We studied the localization of NCL during the JEV infection. To this end, nuclear and cytoplasmic fractions from the JEV- or mock-infected HeLa cells were prepared at different time points during the course of virus infection. The western blot analysis showed that there was an increased accumulation of NCL in the cytoplasmic fractions of the virus-infected cells; the NCL levels in the cytoplasm were highest at 18 h pi (Fig. 4C).
Together, these data show that the JEV infection led to enhanced NCL expression and its accumulation in the cytoplasm in HeLa cells.
NCL has a pro-viral role in JEV replication
The siRNA-mediated knock-down of NCL was used to study its role in JEV replication. HeLa cells were treated with NCL siRNA or a non-targeting (NT) control siRNA for 48 h. This resulted in ~80% reduction in NCL mRNA expression in the NCL siRNA-treated cells, while no effect on cell viability was seen. The western blotting showed a very significant knock-down of the NCL protein in the siNCL-treated cells (Fig. 5A). At this point, cells were infected with JEV, and 24 h later, the viral RNA in the cells and the virus titers in the culture supernatant were determined. The NCL siRNA-treated cells showed ~40%–70% reduction in the viral RNA levels depending upon the MOI. This resulted in significantly reduced viral titers; these were ~30%–50% lower in the siNCL-treated cells than in the siNT-treated control cells. (Fig. 5A).
Fig 5.

NCL has a pro-viral role during JEV replication. (A) HeLa cells were transfected with non-targeting control siRNA (siNT) or NCL siRNA (siNCL) (30 nM) and incubated for 48 h. The cells were then infected with JEV at different MOIs. The NCL mRNA level at 48 h post-transfection was analyzed by qRT-PCR (upper left panel), and the cell cytotoxicity was measured by the MTT assay (upper middle panel). The upper right panel has the representative picture showing the level of NCL protein by western blotting at 48 h post-transfection. The molecular weight marker in kDa is shown at the left. The left lower panel shows the JEV RNA at 24 h pi as determined by qRT-PCR at different MOI, and the lower right panel shows the virus titers. The data from three independent experiments are presented. (B) HeLa cells were transfected with the plasmid expressing Myc-tagged NCL under the control of the CMV promoter (CMV-NCL) or the empty vector (CMV-EV) and incubated for 48 h. The cells were then infected with JEV (1 MOI). The top panels show the NCL mRNA level at 48 h post-transfection analyzed by qRT-PCR and the cell cytotoxicity measured by the MTT assay from three independent experiments. A representative picture showing the level of the exogenous NCL protein by western blotting at 48 h post-transfection is presented. The molecular weight marker in kDa is shown at the left. The bottom panels show the JEV RNA level at 24 h pi as determined by qRT-PCR and the virus titers. The data from three independent experiments are presented.
Since JEV is a neurotropic virus, we also studied the role of NCL in JEV replication in the human neuroblastoma cell line SH-SY5Y. The cells were treated with NCL siRNA or a non-targeting (NT) control siRNA for 48 h. The western blotting showed ~80% knock-down of the NCL protein in the siNCL-treated cells (Fig. S2B). At this point, cells were infected with JEV, and 24 h later, the viral RNA in the cells and the virus titers in the culture supernatant were determined (Fig. S2). The NCL siRNA-treated cells showed ~70% reduction in the viral RNA levels. This resulted in significantly reduced viral titers; these were ~80% lower in the siNCL-treated cells than in the siNT-treated control cells.
To validate these findings further and rule out any off-target effect of the NCL siRNA used above, we created the NCL knockout (KO) HAP1 cells. The KO cells were infected with JEV, and 24 h later, the viral RNA in the cells and the virus titers in the culture supernatant were determined. The NCL KO cells showed ~95% reduction in the viral RNA levels and ~85% reduction in the virus titers (Fig. S3).
We then studied the effect of the NCL overexpression on JEV replication. HeLa cells were transfected with the plasmid expressing Myc-tagged NCL (CMV-NCL) under the control of the CMV promoter or the empty vector (CMV-EV). At 48 h post-transfection, the expression of the NCL mRNA was found to be significantly higher in CMV-NCL-transfected cells than in CMV-EV-transfected cells. The cell viability was not affected due to the plasmid transfection or NCL overexpression. The expression of the exogenous NCL was clearly seen in the CMV-NCL-transfected cells by western blotting. At this point, cells were infected with JEV, and 24 h pi, the viral RNA in the cells and the virus titer in the culture supernatant were determined. There was a small increase of ~20% seen in the levels of viral RNA. The viral titers were ~50% enhanced in the NCL-overexpressing cells compared to that in the control (Fig. 5B). We also studied the effect of NCL over-expression on JEV replication in SH-SY5Y cells. The JEV titers were 4–8 times enhanced in the cells where NCL or its truncated form (described below) was exogenously expressed (Fig. S2C). Taken together, these data demonstrate a pro-viral role of NCL during JEV replication.
JEV NS5 interacts with RRM3, RRM4, and GAR domains of NCL
The JEV NS5 consists of two domains—the N-terminal MTase and the C-terminal RdRp, while the human NCL consists of three domains—the N-terminal domain rich in the acidic stretches, the central RNA-binding domain (RBD) consisting of four RNA Recognition Motifs (RRM), and the C-terminal domain rich in Gly/Arg residues (GAR). The in silico docking of JEV NS5 with human NCL generated different NS5-NCL-binding conformations (Fig. 6). The top two best-predicted models indicated the binding of the RRM4 and RRM3 of NCL with NS5. The GAR domain may also be involved in the binding since RRM4 showed relatively more interactions than RRM3 with the NS5 in both the top two best-predicted models.
Fig 6.

In silico interaction of NCL and JEV NS5. (A) The left panel shows the crystal structure of full-length JEV NS5 (PDB ID 4K6M). The structure of human NCL containing central RRMs taken from the AlphaFold protein structure prediction database (P19338) is shown in the right panel. Individual domains of NS5 and NCL are labeled. (B) The top two best-predicted models of NS5-NCL-binding conformations were generated by the Cluspro protein-protein docking tool. The best (left panel) and second best (right panel) conformations show the binding of RRM4 and RRM3 from NCL to NS5. RRM4 shows more interactions than RRM3 with the NS5. (C) The location of the flexible GAR region around the Mtase domain suggests a possible involvement of the GAR region during the NCL-NS5 complex formation. The figure was generated by the superposition of RRM34-GAR from the AlphaFold model on the top-ranked NCL(RRM1234)-NS5 binding conformations.
To validate these predicted interactions in the cell culture, Myc-tagged truncated mutants of NCL (tNCL) were constructed by cloning the relevant cDNAs under the CMV promoter (Fig. 7A). The different tNCLs had the N-terminal domain completely deleted while containing some of the above-described protein domains. HeLa cells were transfected with these tNCL plasmids and 48 h later infected with JEV. The cells were immunostained 24 h pi with anti-NS5 and anti-Myc antibodies and examined under a confocal microscope to check for the co-localization of JEV NS5 with tNCL (Fig. 7B). Based on the Pearson’s correlation coefficient, tNCL harboring RRM3 in combination with RRM4 and GAR domain showed significant co-localization with JEV NS5 indicating their close interaction with NS5, thus validating the in silico prediction.
Fig 7.

JEV NS5 interacts with RRM3, RRM4, and GAR domains of NCL. (A) Schematic of the full-length NCL and the tNCL forms is shown in the left panel. The right panel shows the expression of each of the Myc-tagged tNCL forms by transfecting HeLa cells with the respective plasmid and western blotting of the lysates 24 h after transfection using anti-Myc antibody. (B) HeLa cells were transfected with plasmids expressing different tNCL forms and infected 48 h later with JEV at 5 MOI. The cells were fixed a day later and immunostained with NS5 and Myc antibodies. The left panel has the confocal images of the stained cells. The right panel has the bar graph showing Pearson’s Correlation Coefficient depicting the colocalization of NS5 and the NCL forms. The data are from three independent experiments. (C) HeLa cells were transfected with plasmids expressing different tNCL forms and infected 48 h later with JEV at 1 MOI. The culture supernatants were collected at 24 h pi, and the virus titer was determined by plaque assay. Fold change in the virus titer is presented with respect to the control where cells were transfected with EV.
We also checked what effect the NCL deletion mutants had on JEV replication in HeLa cells. As seen before, the full-length NCL enhanced the virus titers by ~65%. Interestingly, JEV titers were ~600% higher in cells expressing the tNCL containing RRM3 in combination with RRM4 and GAR domain (Fig. 7C).
These data show that the RRM3, in combination with RRM4 and the GAR domain of NCL, interacted with JEV NS5, and this interaction had a strong pro-viral role in enhancing the JEV replication significantly.
JEV NS5 interacts with tNCL in cells transfected with the expression plasmids
In the above experiments, the truncated NCL (tNCL-34GAR) was found to interact with the NS5 protein during the JEV infection. To examine if NS5 alone was sufficient for this interaction, we transfected HeLa cells with the plasmids expressing tNCL-34GAR and JEV NS5 and studied the colocalization of these two proteins in the cell by confocal microscopy (Fig. 8A). The PCC in the cells transfected with the plasmids expressing tNCL-34GAR and JEV NS5 was significantly higher than in the control cells transfected with the plasmid expressing tNCL-34GAR and the empty vector. However, the degree of colocalization, as defined by the PCC, was lower in plasmid-transfected cells than in JEV-infected cells. This may be because of the possibility that the NS5-NCL interaction in the JEV-infected cells involves other viral proteins.
Fig 8.

JEV NS5 interacts with NCL and causes its cytoplasmic accumulation. (A) HeLa cells were transfected with the plasmids expressing tNCL-34GAR and JEV NS5 or with the plasmid expressing tNCL-34GAR and the empty vector (pEV) as the negative control. The cells were fixed 48 h later and immunostained with the NS5 and NCL antibodies. The nucleus was stained with DAPI. The images were taken under a Leica SP8 confocal microscope (upper panel). The Pearson’s correlation coefficients determined for the overlapping green and red fluorophores are presented (lower panel). (B) HeLa cells were transfected with the plasmid expressing JEV NS5 or with the empty vector (pEV) as the negative control. The cells were harvested at different time points post-transfection, and the nuclear and cytoplasmic fractions were prepared. The levels of NCL and NS5 were analyzed by western blotting. GAPDH served as the cytoplasmic marker. The NCL distribution in the cytoplasm was quantified with respect to GAPDH; the numbers at the top show the NCL/GAPDH ratio. The lane M has the molecular weight markers shown in kDa at the left.
We also studied the localization of NCL in HeLa cells transfected with the plasmid expressing JEV NS5, or the empty vector (pEV) as the negative control (Fig. 8B). The proteins from the nuclear and cytoplasmic fractions of the transfected cells were western blotted at different times pi. Interestingly, NCL was found to increasingly localize to the cytoplasm in the pNS5-transfected cells at 6 and 18 h pi when compared with the cells transfected with pEV.
These data show that JEV NS5 alone could interact with NCL in HeLa cells, and the expression of JEV NS5 led to an increased NCL accumulation in the cytoplasm.
NCL-binding aptamer AS1411 reduces JEV replication
NCL has been shown to exhibit a remarkable binding preference for the GQ structures (34–36). The AS1411 aptamer is a GQ-forming oligonucleotide that is internalized into cells and hijacks NCL away from other processes (37). We studied the effect of AS1411 binding to NCL on JEV replication. JEV-infected HeLa cells were incubated with increasing concentrations of AS1411 or the negative control aptamer CRO. The treatment of cells with AS1411 resulted in a dose-dependent decrease in the JEV RNA and virus titers (Fig. 9A). The JEV RNA levels were 40%–70% lower, and the virus titers were 60%–80% lower in the AS1411-treated cells when compared to those in the control-treated cells. The cytotoxicity assay showed that AS1411 or CRO treatment did not affect cell viability (Fig. 9B).
Fig 9.

NCL-binding aptamer AS1411 retards JEV replication in a dose-dependent manner. (A) HeLa cells were infected with JEV at 1 MOI. The cells were treated at 2 h pi with AS1411 or a non-specific random control aptamer CRO at increasing concentrations. The cells were harvested 24 h pi to analyze the level of intracellular JEV RNA by qRT-PCR, and the culture supernatants were used to determine the virus titers. Fold-change in viral RNA levels and titers were calculated with respect to the control for each concentration. (B) To check the cytotoxic effect of oligodeoxynucleotide aptamers on cell viability, HeLa cells were treated with AS1411 or CRO at 7 µM concentration and incubated for 24 h. The cell cytotoxicity was measured by the MTT assay. (C) HeLa cells were infected with JEV at 1 MOI. The cells were treated at 2 h pi with AS1411 or the control aptamer CRO at 3 µM concentration. The co-localization of JEV NS5 and NCL was studied by the PLA at 24 h pi.
We reasoned that the reduced viral titers were caused by a reduced interaction of NCL with JEV NS5. To this end, we conducted PLA-based studies to demonstrate that the aptamer AS1411 inhibited the JEV NS5 interaction with NCL (Fig. 9C). While the control aptamer CRO did not affect NS5-NCL interaction, the interaction was significantly reduced by AS1411.
We then studied if AS1411-mediated retardation of JEV replication could be overcome by exogenously-produced NCL. HeLa cells were transfected with the plasmid expressing NCL or tNCL-34GAR, or empty vector (EV), and 48 h later infected with JEV in the presence or absence of AS1411. The EV-transfected cells treated with AS1411 showed around 60% reduction in JEV titer compared to the control (Fig. 10). The reduction in JEV titer was overcome in cells transfected with the plasmid expressing NCL or tNCL-34GAR. As seen before, tNCT-34GAR was able to overcome the virus titer reduction more efficiently than the full-length NCL.
Fig 10.
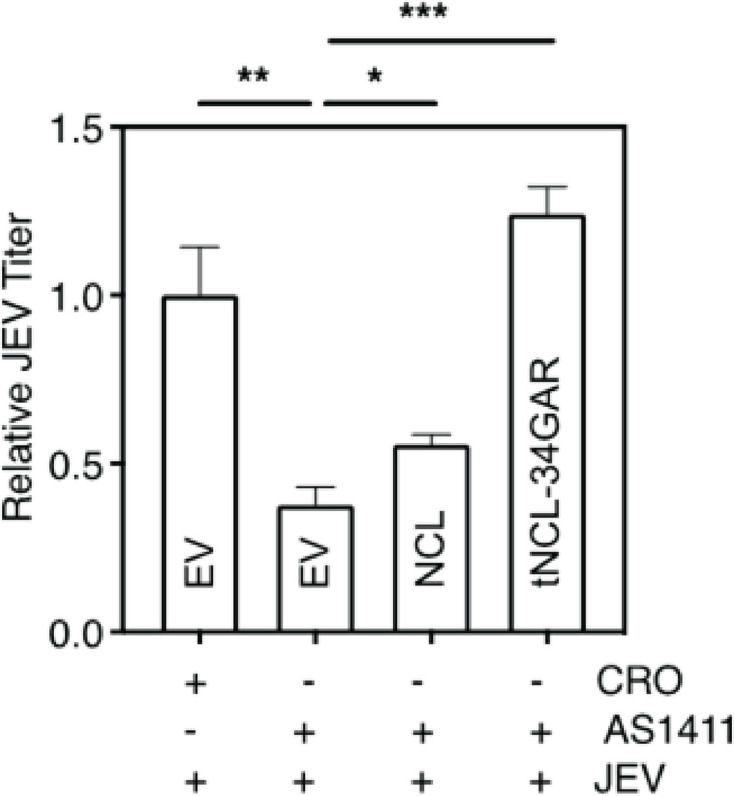
AS1411-mediated retardation of JEV replication can be overcome by exogenous NCL. HeLa cells were transfected with the empty vector (EV) or plasmid expressing NCL or tNCL-34GAR. The cells were infected with JEV (MOI 1) 48 h post-transfection and incubated in the presence of 3 µM AS1411 or CRO. The culture supernatants were harvested at 24 h pi, and JEV titers were determined by plaque assay.
These results further confirm that NCL is an important pro-viral host factor, and its interaction with NS5 and the GQ-structures supports an enhanced JEV replication.
NCL binds the GQ sequences in the 3′-NCR of JEV genomic RNA
The 5′- and 3′- NCRs of the flavivirus genome are involved in the binding of the replication complex, of which NS5 protein is a key component shown above to be interacting with NCL. NCL, in turn, has been shown to readily interact with GQ sequences and is involved in its unfolding (35, 38, 39). We, therefore, searched for the GQ sequences in the JEV NCRs. No such sequence was found in the 5′-NCR of JEV genomic RNA. However, two GQ sequences (GQ1 and GQ2) were identified in the 3′-NCR (Fig. 11A). These sequences showed a range of conservation across various flaviviruses suggesting an important functional role. Importantly, the GQ2 nucleotide sequence was better conserved than GQ1 across different flaviviruses. Expectedly, these sequences showed a higher degree of conservation among different JEV strains (Fig. S4).
Fig 11.

GQ sequences in the JEV 3′-NCR bind with NCL. (A) The nucleotide sequence alignment of the two predicted GQ sequences in the 3′-NCR of different flaviviruses, and their strains is shown. The top of the panel has the WebLogo representation of the two predicted GQ sequences in the 3′-NCR of JEV genomic RNA showing the conservation of G-bases among different flaviviruses. The height of the letters represents the frequency of their occurrence at that particular position. (B) Synthetic RNA containing the GQ sequences or a random control RNA was incubated with the cell lysate containing the exogenously expressed Myc-tagged NCL or tNCL-34GAR. The complexes were UV cross-linked and separated by native gel electrophoresis. The NCL-RNA and tNCL-RNA complexes were resolved on an 8% or 10% native PAGE, respectively. The GQ RNA was detected by fluorescence (upper panel). The gel was western blotted with the anti-Myc antibody to detect the exogenous NCL or tNCL-34GAR (lower panel).
To examine if NCL could bind these RNA sequences, electrophoretic mobility shift assay (EMSA) was done using synthetic RNAs and lysates prepared from cells overexpressing exogenous Myc-tagged NCL or Myc-tagged tNCL-34GAR. A retarded RNA band could be seen for the GQ2 sequence when lysate containing exogenous NCL was used (Fig. 11B). However, we failed to detect NCL in this band on western blotting with antibody to Myc-tag or NCL. Similarly, a retarded RNA band could be seen for the GQ2 sequence when lysate containing exogenous tNCL-34GAR was used. The western blot with Myc-tag antibody showed the presence of tNCL-34GAR in the retarded band. This band showed a supershift in the presence of the Myc-tag antibody (Fig. S5). We did not see a similar supershift for the NCL retarded band. However, the intensity of the GQ2-NCL complex was reduced in the presence of the specific antibody. If the monoclonal NCL antibody and GQ2 RNA bind to the same, overlapping, or nearby location on NCL, we may see a diminishing band of the RNA-protein complex and not see the complex supershift.
We did not see the binding of NCL or tNCL-34GAR with GQ1 RNA in the EMSA although a faint band showing the binding of tNCL-34GAR was seen in the western blot. This can happen if the protein amount in the band is too small or below the western blot detection limit.
These findings suggest that the 34GAR region of NCL binds to a specific GQ-forming stretch (nts. 10,816–10,842) located in the 3′-NCR of JEV genomic RNA.
GQ-binding ligand BRACO-19 inhibits JEV replication
BRACO-19 is a small molecule extensively used as a GQ structure-binding and stabilizing ligand (40–43). It should, therefore, compete with NCL for binding to GQ structures in the JEV genomic RNA and affect JEV replication. We found that BRACO-19-treated cells showed a dose-dependent decrease in the levels of JEV RNA and virus titer when compared with control cells (Fig. 12A). BRACO-19 used at a concentration of 25 µM showed no significant cytotoxicity (Fig. 12B). We also observed that the exogenous expression of NCL or tNCL-34GAR rescued JEV replication from the antiviral effect of BRACO-19 (Fig. 12C). Importantly, we see that tNCL-34GAR showed an enhanced rescuing effect as compared to NCL in this experiment. EGFP used as a negative control was unable to rescue the virus replication.
Fig 12.

GQ-binding ligand BRACO-19 reduces JEV replication in a dose-dependent manner. (A) HeLa cells were infected with JEV at 1 MOI. The cells were treated at 2 h pi with BRACO-19 at increasing concentrations. The cells were harvested 24 h pi to analyze the level of intracellular JEV RNA by qRT-PCR, and the culture supernatants were used to determine the virus titers. Fold-change in viral RNA levels and titers were calculated with respect to the DMSO-only control. (B) HeLa cells were treated with BRACO-19 at 25 µM concentration and incubated for 24 h. The cell cytotoxicity was measured by the MTT assay. (C) HeLa cells were transfected with empty vector (EV) or the plasmid expressing NCL, tNCL-34GAR, or EGFP. The cells were infected 48 h later with JEV (MOI 1) and treated 2 h pi with BRACO-19 at 25 µM concentration. The culture supernatants were harvested at 24 h pi, and the virus titer was determined. The titer shown is relative to that obtained in cells transfected with EV and not treated with BRACO-19.
Taken together, these data show that the interaction of the 34GAR region of NCL with the GQ structures in JEV genomic RNA is a plausible mode of pro-viral action of NCL.
AS1411 and BRACO-19 target the early stage of JEV replication
To understand the time of action of the inhibitory compounds AS1411 and BRACO-19 during the virus replication, these were added to the JEV-infected cells at different times pi. The compounds were added at the minimum inhibitory concentrations from 2 to 12 h pi at every 2 h interval, and the culture supernatants were collected at 24 h pi. The viral titer analysis showed that both AS1411 and BRACO-19 inhibited the virus replication when present within the first 4 h of infection (Fig. 13). These compounds were unable to inhibit the viral replication when added at 6 h pi or later, suggesting that they acted at the early stage of JEV replication. It may be noted that the synthesis of the negative-strand RNA intermediate, which takes place during the early phase of virus infection, would require the destabilization of the GQ structures in the 3′-NCR. Thus, the antiviral action of AS1411 and BRACO-19 is probably due to the maintenance of the stable GQ structures at the 3′-NCR of JEV RNA in the presence of these compounds, resulting in the inhibition of virus replication.
Fig 13.

The time of action of AS1411 and BRACO-19. HeLa cells were infected with JEV at 0.5 MOI, and AS1411 (3 µM) or BRACO-19 (25 µM) was added to the virus-infected cells at different times pi indicated on the x-axis. CRO (3 µM) was added to the cells used as the control for the AS1411-treated cells. DMSO was added to the cells used as control for the BRACO-19-treated cells. The culture supernatants were collected at 24 h pi, and the virus titers were determined. The fold change in the virus titer was calculated in relation to the respective control at each time point. The upper panel depicts the experimental plan. Each experiment had biological triplicates, and data from two independent experiments are shown in the lower panel.
DISCUSSION
The flavivirus NS5 protein having the RdRp activity, together with the host and viral proteins, constitutes the replication complex required for the viral genome replication. NS5 is, therefore, a key protein for virus replication (44). Novel antivirals could be designed if we identify and understand the functioning of these host proteins and their interaction with NS5. Accordingly, we set out to identify the host proteins that interact with the JEV NS5 protein. We used the BioID proximity labeling method, which has high sensitivity and specificity and, hence, provides extended coverage of the interactome (24). We acknowledge that some interactions may only take place in the context of virus infections and would not be captured here. So, we also used the co-immunoprecipitation (CoIP) method using JEV-infected cells. Our study identified a small number of common cellular proteins that were revealed by both methods. With a series of investigations that utilize proteomics data, network analyses, and bioinformatics tools, we were able to identify the JEV NS5-interacting host proteins that have orthologs in humans. One such protein was NCL which appeared both in the BioID and CoIP interactomes and was mapped to be highly interconnected.
We observed minimal overlap between the two interactomes identified by the BioID and CoIP methods. An obvious explanation would lie in the significant difference in the experimental conditions between the two methods. While CoIP was performed on JEV-infected N2a cells, the BioID was performed on uninfected N2a cells overexpressing exogenous JEV NS5 protein. The composition of our JEV NS5 interactomes also differed from that reported by Kovanich et al. (8) who used HEK293T cells overexpressing the exogenous EGFP-tagged JEV NS5. Importantly, we did find HSPs and MYO proteins in our interactomes. These are some of the host proteins described in the literature as interacting with JEV NS5 (8).
We validated the interaction of JEV NS5 with NCL by co-immunoprecipitation, and the colocalization of the two proteins by the immunofluorescence method using confocal microscopy. The close interaction of the two proteins in JEV-infected cells was convincingly demonstrated by the PLA method, which is a more sensitive method for PPI detection and quantification as compared to traditional immunofluorescence methods (45). Altogether, these data provide evidence of NS5-NCL interaction during the JEV infection. Similarly, NCL has been shown to interact with the NS5B protein of the hepatitis C virus (HCV), a flavivirus (46), to form the virus replication complex. The HCV NS5B protein has the RdRp activity as is the case with JEV NS5 protein. Besides, NCL was also shown to interact with the RdRp of caliciviruses such as rabbit hemorrhagic disease virus (47) and Feline calicivirus (48).
The mammalian NCL is a 707-amino acid long evolutionarily conserved protein that has three structural and multifunctional domains. The N-terminal domain contains several acidic amino acids and is involved in the cell cycle (49–52). The central domain is made up of four RNA-recognition motifs (RRMs) and is involved in RNA binding (53–57). The C-terminal domain (GAR or RGG domain) is rich in arginine and glycine residues and has several Arg-Gly-Gly (RGG) repeats (58–60). The GAR domain, also known as the protein interaction domain, has been shown to interact with several proteins, such as urokinase-type plasminogen activator (uPA) and its receptor uPAR, midkine (MK), and lactoferrin (61–65). We studied the interaction of NCL structural domains with JEV NS5 and found that the deletion of the N-terminal domain enhanced the interaction of the truncated NCL protein with NS5. We studied the interaction of different combinations of RRMs and GAR domains with NS5 and concluded that the GAR domain contributed significantly to NCL interaction with JEV NS5 and virus replication.
NCL primarily accumulates in the nucleolus although it has been reported to be present in the cell membrane (66–69) as well as in the cytoplasm (32, 33, 58, 70–72) depending on the physiological state of the cell. It is a multifunctional protein involved in several important cellular functions, such as the transcription and maturation of ribosomal RNA and ribosome assembly and transport (58, 73). The protein is also involved in the transcription, splicing, stability, transport, and translation of different mRNAs (74–76). On the cell surface, NCL acts as a co-receptor of cytokines, growth factors, and matrix proteins (77). It also has a role as the virus receptor/co-receptor or in the uptake process in the case of respiratory syncytial virus (RSV), enterovirus A71 (EVA71), human immunodeficiency virus (HIV), influenza A virus (IAV), human parainfluenza virus 3 (HPIV-3), rabbit hemorrhagic disease virus (RHDV), Crimean-Congo hemorrhagic fever virus (CCHFV), and coxsackie virus B (CVB) (19). Besides, NCL has been reported to play a multifunctional role during the proliferation of different viruses. For example, it binds to the capsid protein of the DEN virus and Adeno-associated virus-2 (AAV-2) and has a role in virus egress (78, 79). NCL is also reported to interact with the US11 protein of HSV-1 and has a role in egress (71). A more recent study shows that NCL interacts with the internal ribosome entry site (IRES), promoting IRES-driven translation of Foot-and-mouth disease virus RNA (70). NCL has also been shown to bind to viral RNA and serves different functions, such as promoting the translation of poliovirus RNA (33), suppressing HCV replication (80) and HIV-1 viral transcription (81).
In light of the above findings, we studied the role of NCL interaction with NS5 protein during the JEV infection. We found that the expression of NCL progressively increased early during the JEV infection, suggesting a role for NCL in the JEV replication. Our western blot data show that NCL accumulates in the cytoplasm as the JEV infection progresses. Flavivirus replication takes place in the virus-induced membrane-bound replication complexes localized to the perinuclear site in the host cell cytoplasm. Accordingly, the majority of NS5 protein is located in the cytoplasm. Since JEV replication takes place in the cytoplasm, NCL’s presence in the cytoplasm would be required for it to interact with NS5 and form the replication complex. NCL is known as a shuttling protein that migrates constantly back and forth between the nucleus and cytoplasm (82). Our studies showed an increased accumulation of NCL in the cytoplasm during the JEV infection. Interestingly, JEV NS5 alone caused an increased accumulation of NCL in the cytoplasm, as seen in HeLa cells transfected with JEV NS5-expressing plasmid. Similar translocation of NCL to cytoplasm has been reported upon infection with other viruses such as HSV-1, Poliovirus, Seneca Valley virus, and more recently SARS-CoV-2 (32, 33, 83, 84). NCL has also been studied extensively for its RNA-binding properties in the cytoplasm, which, in turn, influences the stability and translation of the RNAs (74). The causes and mechanism of this redistribution are not well understood although it has been reported that, under stress conditions, NCL is mobilized from the nucleolus to the nucleoplasm in a p53-dependent manner (85).
The multiple functions and subcellular localizations of NCL reflect its complex structure. A series of deletion mutants of NCL were constructed in the present study to identify the protein domain/s interacting with JEV NS5 and their role in JEV replication. We observed that the truncated mutants devoid of the N-terminus were localized in the cytoplasm, and they showed an increased co-localization with NS5 as well as enhanced JEV replication. As predicted by the in silico docking, the NCL mutant harboring the RRM3, RRM4, and GAR domains showed the maximum enhancement of virus replication. This shows that cytoplasmic localization signal was exposed by the deletion of the N-terminal domain of NCL, suggesting that the signals involved in cytoplasmic targeting were masked or that cytoplasmic localization is restricted to some cleaved forms of NCL. The mechanism of NCL shuttling to various cellular locations under different conditions needs to be explored further.
NCL was found to have a pro-viral role during the JEV replication as siRNA-mediated knockdown of NCL resulted in a significant reduction of JEV RNA and titer, whereas, transient overexpression of NCL led to an enhanced viral replication in HeLa as well as SH-SY5Y cells. Additionally, in the NCL knockout HAP1 cells, there was a significant reduction in the JEV RNA levels and virus titers. A similar observation was made in the case of flaviviruses DEN and HCV where siRNA-mediated knockdown of NCL resulted in a significant reduction in viral replication (46, 78). The siRNA-based studies indicated a pro-viral role of NCL in viruses from other families such as rabies virus (86), herpes simplex virus type 1 (83), and rabbit hemorrhagic disease virus (47). Interestingly, NCL was shown to have an antiviral role in the case of H5N1 avian influenza virus (87), Peste des pestis ruminants virus (88), and human influenza A virus (89). In all of these cases, NCL interacts with different viral or host proteins, and this could explain the different roles that NCL plays in each of these different viruses.
NCL shows a remarkable binding preference for GQ structures in DNA as well as RNA. The aptamer AS1411 forms a stable GQ structure and has extensively been used as an NCL ligand. Notably, AS1411 has been shown to bind the GAR domain of NCL (35). Our PLA studies showed that AS1411 interfered with the NCL-NS5 interaction, and this resulted in a significantly reduced JEV replication. The exogenously expressed NCL in the AS1411-treated virus-infected cells was found to rescue the retarded JEV replication. This suggested that the role of NCL in JEV replication might involve its GQ binding property.
GQs are nucleic acid secondary structures that may form in the G-rich sequences. They are based on the formation of G-quartets, which are stabilized by Hoogsteen H-bonds between guanines. G-quartets stack on top of each other to give rise to GQs that are further stabilized by the interaction with monovalent cations (90). GQs occur in functionally important regions of the genome (91) suggesting the possibility that they behave as structural switches of cellular processes, therefore, providing a basis for therapeutic intervention (92). The functions of GQ structures are regulated by GQ-binding proteins (38). Some of these binding proteins, including hnRNPs, CIRBP, TLS/FUS, and EWS have common nucleic acid-binding domains, such as the RRM and GAR domains as found in NCL. Previous bioinformatic analysis has revealed that the GAR domain has an evolutionarily conserved sequence and at least 31 different protein isoforms contain this domain (93). This domain is known as both a GQ folding and unfolding domain (94–97). It is reported that the GAR domain in NCL is important for GQ binding although the domain alone does not show obvious binding to the GQ (35). It has also been previously shown that NCL can naturally produce cleaved forms that lack the N-terminal but retain full binding capabilities. Thus, the N-terminal of NCL is dispensable for its GQ-binding activity (81).
One of the most potent GQ ligands is BRACO-19, a 3,6,9-trisubstituted acridine derivative designed to stabilize the quadruplex DNA structures formed in human telomeres (98). The mode of interaction of these ligands may be through end-stacking, groove binding, or interaction with the backbone or loop sequences. The molecules fit themselves within these GQ structures and bind through various non-covalent bonds, thus stabilizing the structure (99). In viruses, evidence of significant GQ regions is increasingly being recognized where BRACO-19 is able to exert antiviral activity by stabilizing the GQ conformations. In HSV-1, GQs are likely to form in the viral DNA when the duplex is stimulated to unwind, i.e., during replication and transcription events. BRACO-19 stabilized all tested GQ sequences, and it was able to arrest viral DNA processing in vitro. In infected cells, this activity resulted in overall inhibition of viral production (40). We have shown that the exogenous NCL or tNCL-34GAR was able to overcome the antiviral effect of BRACO-19, indicating the role of GQs in virus replication. We have also shown that the overexpression of exogenous NCL or tNCL-34GAR counteracts the effect of AS1411 and rescues the JEV replication, possibly by destabilizing and unwinding the GQs.
Our EMSA data confirmed the binding of NCL-34GAR to one of the two GQ-forming stretches present at the 3′-NCR of the JEV genome. Notably, the specificity of this interaction could be further validated by the supershift assay. However, we did not see the supershift in the case of the NCL interaction with the GQ sequence. We speculate that this might not be seen if the RNA and antibody binding sites on the NCL protein are same, overlapping, or in the close vicinity.
The 3′-NCR is pivotal to the replication of the negative-sense genomic RNA which may be governed by the secondary structures adopted by it. JEV is an RNA virus with a high mutation rate, and thus, the conservation of these GQs suggests their functional importance in the viral life cycle. We see that the G-bases in the GQ2 structure are more conserved and its binding to NCL-34GAR is more prominent than to GQ1. This suggests that the interaction is both conformation- and sequence-dependent.
Our data show that NCL binds to JEV NS5 as well as the GQ sequence in the 3′-NCR of the (+)-strand RNA during the JEV replication. The NCL protein in the virus replication complex may facilitate the destabilization and unfolding of the GQ structures, thus promoting genomic RNA synthesis and virus replication. BRACO-19 and AS1411 exhibit anti-JEV activity through the NCL-NS5-3′-NCR axis during the JEV replication. BRACO-19 may stabilize the GQ structures in the 3′-NCR. These stable RNA structures may impede the smooth movement of NS5 protein, resulting in reduced RNA synthesis and lower virus titer. On the other hand, AS1411 may inhibit the replication complex formation by sequestering the endogenous NCL. Depletion of NCL may result in maintaining the stable GQ structures in the genomic RNA, stalling the progress of the replication complex and, thus, retarded genomic RNA synthesis and reduced viral titers. The NCL interaction with the GQ sequence in the 3′-NCR of JEV RNA, therefore, presents a novel target for antiviral development.
Flavivirus replication takes place in the virus-induced membrane-bound replication complexes localized to the perinuclear site in the host cell cytoplasm (3). Accordingly, the majority of NS5 protein is located in the cytoplasm. However, a significant proportion (20%) of the total RdRp activity in cells infected with WNV, JEV, and dengue viruses has been reported within the nucleus (100). Subsequent studies have shown the presence of NS5 in the nucleus of the host cells infected with dengue (101, 102), Zika (103), and WNV (104). Since NCL is primarily a nuclear protein, it would be interesting to study its interaction with the nuclear NS5 in the JEV-infected cells and what role it might play in the replication of JEV.
ACKNOWLEDGMENTS
We acknowledge the CSIR research fellowship to A.D. The work benefitted from the SERB grant no. JCB/2021/000015 to S.V.
We would like to thank Manjula Kalia, Arup Banerjee, Ambadas Rode, Payal Gupta, Shivani Balyan, and Brohmomoy Basu for helpful advice on experimental methods.
Contributor Information
Sudhanshu Vrati, Email: vrati@rcb.res.in.
Mark T. Heise, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jvi.00858-24.
NCL is primarily a nuclear protein.
Role of NCL in JEV replication in SH-SY5Y cells.
JEV replication in NCL knockout cells.
GQ sequences in the JEV 3'-NCR.
Supershift of GQ2 RNA complex with NCL and tNCL-34GAR.
Proteins identified by BioID and Co-IP methods.
Proteins from StringDB.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Tiwari S, Singh RK, Tiwari R, Dhole TN. 2012. Japanese encephalitis: a review of the Indian perspective. Braz J Infect Dis 16:564–573. doi: 10.1016/j.bjid.2012.10.004 [DOI] [PubMed] [Google Scholar]
- 2. Markoff L. 2003. 5'- and 3'-noncoding regions in flavivirus RNA. Adv Virus Res 59:177–228. doi: 10.1016/s0065-3527(03)59006-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van den Elsen K, Quek JP, Luo D. 2021. Molecular insights into the flavivirus replication complex. Viruses 13:956. doi: 10.3390/v13060956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brand C, Bisaillon M, Geiss BJ. 2017. Organization of the flavivirus RNA replicase complex. Wiley Interdiscip Rev RNA 8:1–14. doi: 10.1002/wrna.1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sampath A, Padmanabhan R. 2009. Molecular targets for flavivirus drug discovery. Antiviral Res 81:6–15. doi: 10.1016/j.antiviral.2008.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davidson AD. 2009. Chapter 2. new insights into flavivirus nonstructural protein 5. Adv Virus Res 74:41–101. doi: 10.1016/S0065-3527(09)74002-3 [DOI] [PubMed] [Google Scholar]
- 7. Lu G, Gong P. 2017. A structural view of the RNA-dependent RNA polymerases from the flavivirus genus. Virus Res 234:34–43. doi: 10.1016/j.virusres.2017.01.020 [DOI] [PubMed] [Google Scholar]
- 8. Kovanich D, Saisawang C, Sittipaisankul P, Ramphan S, Kalpongnukul N, Somparn P, Pisitkun T, Smith DR. 2019. Analysis of the zika and Japanese encephalitis virus NS5 interactomes. J Proteome Res 18:3203–3218. doi: 10.1021/acs.jproteome.9b00318 [DOI] [PubMed] [Google Scholar]
- 9. Le Breton M, Meyniel-Schicklin L, Deloire A, Coutard B, Canard B, de Lamballerie X, Andre P, Rabourdin-Combe C, Lotteau V, Davoust N. 2011. Flavivirus NS3 and NS5 proteins interaction network: a high-throughput yeast two-hybrid screen. BMC Microbiol 11:234. doi: 10.1186/1471-2180-11-234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krishnan MN, Ng A, Sukumaran B, Gilfoy FD, Uchil PD, Sultana H, Brass AL, Adametz R, Tsui M, Qian F, Montgomery RR, Lev S, Mason PW, Koski RA, Elledge SJ, Xavier RJ, Agaisse H, Fikrig E. 2008. RNA interference screen for human genes associated with West Nile virus infection. Nature 455:242–245. doi: 10.1038/nature07207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hirano M, Kaneko S, Yamashita T, Luo H, Qin W, Shirota Y, Nomura T, Kobayashi K, Murakami S. 2003. Direct interaction between nucleolin and hepatitis C virus NS5B. J Biol Chem 278:5109–5115. doi: 10.1074/jbc.M207629200 [DOI] [PubMed] [Google Scholar]
- 12. Alvarez DE, Lodeiro MF, Ludueña SJ, Pietrasanta LI, Gamarnik AV. 2005. Long-range RNA-RNA interactions circularize the Dengue virus genome. J Virol 79:6631–6643. doi: 10.1128/JVI.79.11.6631-6643.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gebhard LG, Filomatori CV, Gamarnik AV. 2011. Functional RNA elements in the Dengue virus genome. Viruses 3:1739–1756. doi: 10.3390/v3091739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kapoor M, Zhang L, Ramachandra M, Kusukawa J, Ebner KE, Padmanabhan R. 1995. Association between NS3 and NS5 proteins of Dengue virus type 2 in the putative RNA replicase is linked to differential phosphorylation of NS5. J Biol Chem 270:19100–19106. doi: 10.1074/jbc.270.32.19100 [DOI] [PubMed] [Google Scholar]
- 15. Carpp LN, Rogers RS, Moritz RL, Aitchison JD. 2014. Quantitative proteomic analysis of host-virus interactions reveals a role for golgi brefeldin a resistance factor 1 (GBF1) in Dengue infection. Mol Cell Proteomics 13:2836–2854. doi: 10.1074/mcp.M114.038984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morrison J, Laurent-Rolle M, Maestre AM, Rajsbaum R, Pisanelli G, Simon V, Mulder LCF, Fernandez-Sesma A, García-Sastre A. 2013. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog 9:e1003265. doi: 10.1371/journal.ppat.1003265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li C, Ge L, Li P, Wang Y, Dai J, Sun M, Huang L, Shen Z, Hu X, Ishag H, Mao X. 2014. Cellular DDX3 regulates Japanese encephalitis virus replication by interacting with viral un-translated regions. Virology 449:70–81. doi: 10.1016/j.virol.2013.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Katoh H, Mori Y, Kambara H, Abe T, Fukuhara T, Morita E, Moriishi K, Kamitani W, Matsuura Y. 2011. Heterogeneous nuclear ribonucleoprotein A2 participates in the replication of Japanese encephalitis virus through an interaction with viral proteins and RNA. J Virol 85:10976–10988. doi: 10.1128/JVI.00846-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tonello F, Massimino ML, Peggion C. 2022. Nucleolin: a cell portal for viruses, bacteria, and toxins. Cell Mol Life Sci 79:271. doi: 10.1007/s00018-022-04300-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sehrawat S, Khasa R, Deb A, Prajapat SK, Mallick S, Basu A, Surjit M, Kalia M, Vrati S. 2021. Valosin-containing protein/p97 plays critical roles in the Japanese encephalitis virus life cycle. J Virol 95:e02336-20. doi: 10.1128/JVI.02336-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Surana P, Satchidanandam V, Nair DT. 2014. RNA-dependent RNA polymerase of Japanese encephalitis virus binds the initiator nucleotide GTP to form a mechanistically important pre-initiation state. Nucleic Acids Res 42:2758–2773. doi: 10.1093/nar/gkt1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stuchbury G, Münch G. 2010. Optimizing the generation of stable neuronal cell lines via pre-transfection restriction enzyme digestion of plasmid DNA. Cytotechnology 62:189–194. doi: 10.1007/s10616-010-9273-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Roux KJ, Kim DI, Raida M, Burke B. 2012. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196:801–810. doi: 10.1083/jcb.201112098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, Roux KJ. 2016. An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell 27:1188–1196. doi: 10.1091/mbc.E15-12-0844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheah JS, Yamada S. 2017. A simple elution strategy for biotinylated proteins bound to streptavidin conjugated beads using excess biotin and heat. Biochem Biophys Res Commun 493:1522–1527. doi: 10.1016/j.bbrc.2017.09.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vrati S, Agarwal V, Malik P, Wani SA, Saini M. 1999. Molecular characterization of an Indian isolate of Japanese encephalitis virus that shows an extended lag phase during growth. J Gen Virol 80 (Pt 7):1665–1671. doi: 10.1099/0022-1317-80-7-1665 [DOI] [PubMed] [Google Scholar]
- 27. Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. doi: 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 28. Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S. 2017. The ClusPro web server for protein–protein docking. Nat Protoc 12:255–278. doi: 10.1038/nprot.2016.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kikin O, D’Antonio L, Bagga PS. 2006. QGRS Mapper: a web-based server for predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res 34:W676–82. doi: 10.1093/nar/gkl253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen CM, Chiang SY, Yeh NH. 1991. Increased stability of nucleolin in proliferating cells by inhibition of its self-cleaving activity. J Biol Chem 266:7754–7758. [PubMed] [Google Scholar]
- 31. Nestorowicz A, Chambers TJ, Rice CM. 1994. Mutagenesis of the yellow fever virus NS2A/2B cleavage site: effects on proteolytic processing, viral replication, and evidence for alternative processing of the NS2A protein. Virology 199:114–123. doi: 10.1006/viro.1994.1103 [DOI] [PubMed] [Google Scholar]
- 32. Song J, Quan R, Wang D, Liu J. 2022. Seneca valley virus 3C pro mediates cleavage and redistribution of nucleolin to facilitate viral replication. Microbiol Spectr 10:e0030422. doi: 10.1128/spectrum.00304-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Waggoner S, Sarnow P. 1998. Viral ribonucleoprotein complex formation and nucleolar-cytoplasmic relocalization of nucleolin in poliovirus-infected cells. J Virol 72:6699–6709. doi: 10.1128/JVI.72.8.6699-6709.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lago S, Tosoni E, Nadai M, Palumbo M, Richter SN. 2017. The cellular protein nucleolin preferentially binds long-looped G-quadruplex nucleic acids. Biochim Biophys Acta Gen Subj 1861:1371–1381. doi: 10.1016/j.bbagen.2016.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Masuzawa T, Oyoshi T. 2020. Roles of the RGG domain and RNA recognition motif of nucleolin in G-quadruplex stabilization. ACS Omega 5:5202–5208. doi: 10.1021/acsomega.9b04221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Saha A, Duchambon P, Masson V, Loew D, Bombard S, Teulade-Fichou MP. 2020. Nucleolin discriminates drastically between long-loop and short-loop quadruplexes. Biochemistry 59:1261–1272. doi: 10.1021/acs.biochem.9b01094 [DOI] [PubMed] [Google Scholar]
- 37. Doron-Mandel E, Koppel I, Abraham O, Rishal I, Smith TP, Buchanan CN, Sahoo PK, Kadlec J, Oses-Prieto JA, Kawaguchi R, Alber S, Zahavi EE, Di Matteo P, Di Pizio A, Song D-A, Okladnikov N, Gordon D, Ben-Dor S, Haffner-Krausz R, Coppola G, Burlingame AL, Jungwirth P, Twiss JL, Fainzilber M. 2021. The glycine arginine-rich domain of the RNA-binding protein nucleolin regulates its subcellular localization. EMBO J. 40:e107158. doi: 10.15252/embj.2020107158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brázda V, Hároníková L, Liao JCC, Fojta M. 2014. DNA and RNA quadruplex-binding proteins. Int J Mol Sci 15:17493–17517. doi: 10.3390/ijms151017493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. James SH, Prichard MN. 2014. Current and future therapies for herpes simplex virus infections: mechanism of action and drug resistance. Curr Opin Virol 8:54–61. doi: 10.1016/j.coviro.2014.06.003 [DOI] [PubMed] [Google Scholar]
- 40. Artusi S, Nadai M, Perrone R, Biasolo MA, Palù G, Flamand L, Calistri A, Richter SN. 2015. The herpes simplex virus-1 genome contains multiple clusters of repeated G-quadruplex: implications for the antiviral activity of a G-quadruplex ligand. Antiviral Res 118:123–131. doi: 10.1016/j.antiviral.2015.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Majee P, Shankar U, Pasadi S, Muniyappa K, Nayak D, Kumar A. 2020. Genome-wide analysis reveals a regulatory role for G-quadruplexes during adenovirus multiplication. Virus Res 283:197960. doi: 10.1016/j.virusres.2020.197960 [DOI] [PubMed] [Google Scholar]
- 42. Ruggiero E, Richter SN. 2018. G-quadruplexes and G-quadruplex ligands: targets and tools in antiviral therapy. Nucleic Acids Res 46:3270–3283. doi: 10.1093/nar/gky187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Burger AM, Dai F, Schultes CM, Reszka AP, Moore MJ, Double JA, Neidle S. 2005. The G-quadruplex-interactive molecule BRACO-19 inhibits tumor growth, consistent with telomere targeting and interference with telomerase function. Cancer Res 65:1489–1496. doi: 10.1158/0008-5472.CAN-04-2910 [DOI] [PubMed] [Google Scholar]
- 44. Kumar S, Verma A, Yadav P, Dubey SK, Azhar EI, Maitra SS, Dwivedi VD. 2022. Molecular pathogenesis of Japanese encephalitis and possible therapeutic strategies. Arch Virol 167:1739–1762. doi: 10.1007/s00705-022-05481-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hegazy M, Cohen‐Barak E, Koetsier JL, Najor NA, Arvanitis C, Sprecher E, Green KJ, Godsel LM. 2020. Proximity ligation assay for detecting protein-protein interactions and protein modifications in cells and tissues in situ. Curr Protoc Cell Bio 89:e115. doi: 10.1002/cpcb.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shimakami T, Honda M, Kusakawa T, Murata T, Shimotohno K, Kaneko S, Murakami S. 2006. Effect of hepatitis C virus (HCV) NS5B-nucleolin interaction on HCV replication with HCV subgenomic replicon. J Virol 80:3332–3340. doi: 10.1128/JVI.80.7.3332-3340.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhu J, Miao Q, Guo H, Tang A, Dong D, Tang J, Wang F, Tong G, Liu G. 2022. Nucleolin interacts with the rabbit hemorrhagic disease virus replicase RdRp, nonstructural proteins p16 and p23, playing a role in virus replication. Virol Sin 37:48–59. doi: 10.1016/j.virs.2022.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cancio-Lonches C, Yocupicio-Monroy M, Sandoval-Jaime C, Galvan-Mendoza I, Ureña L, Vashist S, Goodfellow I, Salas-Benito J, Gutiérrez-Escolano AL. 2011. Nucleolin interacts with the feline calicivirus 3′ untranslated region and the protease-polymerase NS6 and NS7 proteins, playing a role in virus replication. J Virol 85:8056–8068. doi: 10.1128/JVI.01878-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jia W, Yao Z, Zhao J, Guan Q, Gao L. 2017. New perspectives of physiological and pathological functions of nucleolin (NCL). Life Sciences 186:1–10. doi: 10.1016/j.lfs.2017.07.025 [DOI] [PubMed] [Google Scholar]
- 50. Peter M, Nakagawa J, Dorée M, Labbé JC, Nigg EA. 1990. Identification of major nucleolar proteins as candidate mitotic substrates of cdc2 kinase. Cell 60:791–801. doi: 10.1016/0092-8674(90)90093-T [DOI] [PubMed] [Google Scholar]
- 51. Caizergues-Ferrer M, Belenguer P, Lapeyre B, Amalric F, Wallace MO, Olson MOJ. 1987. Phosphorylation of nucleolin by a nucleolar type NII protein kinase. Biochemistry 26:7876–7883. doi: 10.1021/bi00398a051 [DOI] [PubMed] [Google Scholar]
- 52. Belenguer P, Caizergues-Ferrer M, Labbé JC, Dorée M, Amalric F. 1990. Mitosis-specific phosphorylation of nucleolin by p34cdc2 protein kinase. Mol Cell Biol 10:3607–3618. doi: 10.1128/mcb.10.7.3607-3618.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bouvet P, Allain FHT, Finger LD, Dieckmann T, Feigon J. 2001. Recognition of pre-formed and flexible elements of an RNA stem-loop by nucleolin. J Mol Biol 309:763–775. doi: 10.1006/jmbi.2001.4691 [DOI] [PubMed] [Google Scholar]
- 54. Serin G, Joseph G, Ghisolfi L, Bauzan M, Erard M, Amalric F, Bouvet P. 1997. Two RNA-binding domains determine the RNA-binding specificity of nucleolin. J Biol Chem 272:13109–13116. doi: 10.1074/jbc.272.20.13109 [DOI] [PubMed] [Google Scholar]
- 55. Ghisolfi L, Kharrat A, Joseph G, Amalric F, Erard M. 1992. Concerted activities of the RNA recognition and the glycine-rich C-terminal domains of nucleolin are required for efficient complex formation with pre-ribosomal RNA. Eur J Biochem 209:541–548. doi: 10.1111/j.1432-1033.1992.tb17318.x [DOI] [PubMed] [Google Scholar]
- 56. Bugler B, Bourbon H, Lapeyre B, Wallace MO, Chang JH, Amalric F, Olson MO. 1987. RNA binding fragments from nucleolin contain the ribonucleoprotein consensus sequence. J Biol Chem 262:10922–10925. [PubMed] [Google Scholar]
- 57. Allain FHT, Bouvet P, Dieckmann T, Feigon J. 2000. Molecular basis of sequence-specific recognition of pre-ribosomal RNA by nucleolin. EMBO J 19:6870–6881. doi: 10.1093/emboj/19.24.6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ginisty H, Sicard H, Roger B, Bouvet P. 1999. Structure and functions of nucleolin. J Cell Sci 112 (Pt 6):761–772. doi: 10.1242/jcs.112.6.761 [DOI] [PubMed] [Google Scholar]
- 59. Lapeyre B, Amalric F, Ghaffari SH, Rao SV, Dumbar TS, Olson MO. 1986. Protein and cDNA sequence of a glycine-rich, dimethylarginine-containing region located near the carboxyl-terminal end of nucleolin (C23 and 100 kDa). J Biol Chem 261:9167–9173. [PubMed] [Google Scholar]
- 60. Lischwe MA, Cook RG, Ahn YS, Yeoman LC, Busch H. 1985. Clustering of glycine and NG,NG-dimethylarginine in nucleolar protein C23. Biochemistry 24:6025–6028. doi: 10.1021/bi00343a001 [DOI] [PubMed] [Google Scholar]
- 61. Legrand D, Vigié K, Said EA, Elass E, Masson M, Slomianny M-C, Carpentier M, Briand J-P, Mazurier J, Hovanessian AG. 2004. Surface nucleolin participates in both the binding and endocytosis of lactoferrin in target cells. Eur J Biochem 271:303–317. doi: 10.1046/j.1432-1033.2003.03929.x [DOI] [PubMed] [Google Scholar]
- 62. Hovanessian AG. 2006. Midkine, a cytokine that inhibits HIV infection by binding to the cell surface expressed nucleolin. Cell Res 16:174–181. doi: 10.1038/sj.cr.7310024 [DOI] [PubMed] [Google Scholar]
- 63. Said EA, Krust B, Nisole S, Svab J, Briand JP, Hovanessian AG. 2002. The anti-HIV cytokine midkine binds the cell surface-expressed nucleolin as a low affinity receptor. J Biol Chem 277:37492–37502. doi: 10.1074/jbc.M201194200 [DOI] [PubMed] [Google Scholar]
- 64. Dumler I, Stepanova V, Jerke U, Mayboroda OA, Vogel F, Bouvet P, Tkachuk V, Haller H, Gulba DC. 1999. Urokinase-induced mitogenesis is mediated by casein kinase 2 and nucleolin. Curr Biol 9:1468–1476. doi: 10.1016/s0960-9822(00)80116-5 [DOI] [PubMed] [Google Scholar]
- 65. Stepanova V, Lebedeva T, Kuo A, Yarovoi S, Tkachuk S, Zaitsev S, Bdeir K, Dumler I, Marks MS, Parfyonova Y, Tkachuk VA, Higazi AAR, Cines DB. 2008. Nuclear translocation of urokinase-type plasminogen activator. Blood 112:100–110. doi: 10.1182/blood-2007-07-104455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mastrangelo P, Norris MJ, Duan W, Barrett EG, Moraes TJ, Hegele RG. 2017. Targeting host cell surface nucleolin for RSV therapy: challenges and opportunities. Vaccines (Basel) 5:27. doi: 10.3390/vaccines5030027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Larrucea S, González-Rubio C, Cambronero R, Ballou B, Bonay P, López-Granados E, Bouvet P, Fontán G, Fresno M, López-Trascasa M. 1999. Cellular adhesion mediated by factor J, a complement inhibitor: evidence for nucleolin involvement. J Biol Chem 273:31718–31725. doi: 10.1074/jbc.273.48.31718 [DOI] [PubMed] [Google Scholar]
- 68. Callebaut C, Blanco J, Benkirane N, Krust B, Jacotot E, Guichard G, Seddiki N, Svab J, Dam E, Muller S, Briand JP, Hovanessian AG. 1998. Identification of V3 loop-binding proteins as potential receptors implicated in the binding of HIV particles to CD4+ cells. J Biol Chem 273:21988–21997. doi: 10.1074/jbc.273.34.21988 [DOI] [PubMed] [Google Scholar]
- 69. Zhou G, Seibenhener ML, Wooten MW. 1997. Nucleolin is a protein kinase C-ζ substrate: connection between cell surface signaling and nucleus in PC12 cells. J Biol Chem 272:31130–31137. doi: 10.1074/jbc.272.49.31130 [DOI] [PubMed] [Google Scholar]
- 70. Han S, Wang X, Guan J, Wu J, Zhang Y, Li P, Liu Z, Abdullah SW, Zhang Z, Jin Y, Sun S, Guo H. 2021. Nucleolin promotes IRES-driven translation of foot-and-mouth disease virus by supporting the assembly of translation initiation complexes. J Virol 95:e0023821. doi: 10.1128/JVI.00238-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Greco A, Arata L, Soler E, Gaume X, Couté Y, Hacot S, Callé A, Monier K, Epstein AL, Sanchez J-C, Bouvet P, Diaz J-J. 2012. Nucleolin interacts with US11 protein of herpes simplex virus 1 and is involved in its trafficking. J Virol 86:1449–1457. doi: 10.1128/JVI.06194-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Westmark CJ, Malter JS. 2012. The regulation of AβPP expression by RNA-binding proteins. Ageing Res Rev 11:450–459. doi: 10.1016/j.arr.2012.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tajrishi MM, Tuteja R, Tuteja N. 2011. Nucleolin: the most abundant multifunctional phosphoprotein of nucleolus. Commun Integr Biol 4:267–275. doi: 10.4161/cib.4.3.14884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Abdelmohsen K, Gorospe M. 2012. RNA-binding protein nucleolin in disease. RNA Biol 9:799–808. doi: 10.4161/rna.19718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Abdelmohsen K, Tominaga K, Lee EK, Srikantan S, Kang MJ, Kim MM, Selimyan R, Martindale JL, Yang X, Carrier F, Zhan M, Becker KG, Gorospe M. 2011. Enhanced translation by nucleolin via G-rich elements in coding and non-coding regions of target mRNAs. Nucleic Acids Res 39:8513–8530. doi: 10.1093/nar/gkr488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sengupta TK, Bandyopadhyay S, Fernandes DJ, Spicer EK. 2004. Identification of nucleolin as an AU-rich element binding protein involved in bcl-2 mRNA stabilization. Journal of Biological Chemistry 279:10855–10863. doi: 10.1074/jbc.M309111200 [DOI] [PubMed] [Google Scholar]
- 77. Fujiki H, Watanabe T, Suganuma M. 2014. Cell-surface nucleolin acts as a central mediator for carcinogenic, anti-carcinogenic, and disease-related ligands. J Cancer Res Clin Oncol 140:689–699. doi: 10.1007/s00432-014-1587-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Balinsky CA, Schmeisser H, Ganesan S, Singh K, Pierson TC, Zoon KC. 2013. Nucleolin interacts with the Dengue virus capsid protein and plays a role in formation of infectious virus particles. J Virol 87:13094–13106. doi: 10.1128/JVI.00704-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Qiu J, Brown KE. 1999. A 110-kDa nuclear shuttle protein, nucleolin, specifically binds to adeno-associated virus type 2 (AAV-2) capsid. Virology 257:373–382. doi: 10.1006/viro.1999.9664 [DOI] [PubMed] [Google Scholar]
- 80. Bian WX, Xie Y, Wang XN, Xu GH, Fu BS, Li S, Long G, Zhou X, Zhang XL. 2019. Binding of cellular nucleolin with the viral core RNA G-quadruplex structure suppresses HCV replication. Nucleic Acids Res 47:56–68. doi: 10.1093/nar/gky1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Tosoni E, Frasson I, Scalabrin M, Perrone R, Butovskaya E, Nadai M, Palù G, Fabris D, Richter SN. 2015. Nucleolin stabilizes G-quadruplex structures folded by the LTR promoter and silences HIV-1 viral transcription. Nucleic Acids Res 43:8884–8897. doi: 10.1093/nar/gkv897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Borer RA, Lehner CF, Eppenberger HM, Nigg EA. 1989. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 56:379–390. doi: 10.1016/0092-8674(89)90241-9 [DOI] [PubMed] [Google Scholar]
- 83. Callé A, Ugrinova I, Epstein AL, Bouvet P, Diaz J-J, Greco A. 2008. Nucleolin is required for an efficient herpes simplex virus type 1 infection. J Virol 82:4762–4773. doi: 10.1128/JVI.00077-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Merino VF, Yan Y, Ordonez AA, Bullen CK, Lee A, Saeki H, Ray K, Huang T, Jain SK, Pomper MG. 2023. Nucleolin mediates SARS-CoV-2 replication and viral-induced apoptosis of host cells. Antiviral Res 211:105550. doi: 10.1016/j.antiviral.2023.105550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Daniely Y, Dimitrova DD, Borowiec JA. 2002. Stress-dependent nucleolin mobilization mediated by p53-nucleolin complex formation. Mol Cell Biol 22:6014–6022. doi: 10.1128/MCB.22.16.6014-6022.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Oksayan S, Nikolic J, David CT, Blondel D, Jans DA, Moseley GW. 2015. Identification of a role for nucleolin in rabies virus infection. J Virol 89:1939–1943. doi: 10.1128/JVI.03320-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Gao Z, Hu J, Wang X, Yang Q, Liang Y, Ma C, Liu D, Liu K, Hao X, Gu M, Liu X, Jiao X-A, Liu X. 2018. The PA-interacting host protein nucleolin acts as an antiviral factor during highly pathogenic H5N1 avian influenza virus infection. Arch Virol 163:2775–2786. doi: 10.1007/s00705-018-3926-3 [DOI] [PubMed] [Google Scholar]
- 88. Dong D, Zhu S, Miao Q, Zhu J, Tang A, Qi R, Liu T, Yin D, Liu G. 2020. Nucleolin (NCL) inhibits the growth of peste des petits ruminants virus. J Gen Virol 101:33–43. doi: 10.1099/jgv.0.001358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kumar D, Broor S, Rajala MS. 2016. Interaction of host nucleolin with influenza A virus nucleoprotein in the early phase of infection limits the late viral gene expression. PLoS One 11:e0164146. doi: 10.1371/journal.pone.0164146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sen D, Gilbert W. 1988. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 334:364–366. doi: 10.1038/334364a0 [DOI] [PubMed] [Google Scholar]
- 91. Huppert JL. 2008. Four-stranded nucleic acids: structure, function and targeting of G-quadruplexes. Chem Soc Rev 37:1375–1384. doi: 10.1039/b702491f [DOI] [PubMed] [Google Scholar]
- 92. Neidle S. 2010. Human telomeric G-quadruplex: the current status of telomeric G-quadruplexes as therapeutic targets in human cancer. FEBS J 277:1118–1125. doi: 10.1111/j.1742-4658.2009.07463.x [DOI] [PubMed] [Google Scholar]
- 93. Thandapani P, O’Connor TR, Bailey TL, Richard S. 2013. Defining the RGG/RG motif. Mol Cell 50:613–623. doi: 10.1016/j.molcel.2013.05.021 [DOI] [PubMed] [Google Scholar]
- 94. Takahama K, Sugimoto C, Arai S, Kurokawa R, Oyoshi T. 2011. Loop lengths of G-quadruplex structures affect the G-quadruplex DNA binding selectivity of the RGG motif in Ewing’s sarcoma. Biochemistry 50:5369–5378. doi: 10.1021/bi2003857 [DOI] [PubMed] [Google Scholar]
- 95. Takahama K, Takada A, Tada S, Shimizu M, Sayama K, Kurokawa R, Oyoshi T. 2013. Regulation of telomere length by G-quadruplex telomere DNA- and TERRA-binding protein TLS/FUS. Chem Biol 20:341–350. doi: 10.1016/j.chembiol.2013.02.013 [DOI] [PubMed] [Google Scholar]
- 96. González V, Hurley LH. 2010. The C-terminus of nucleolin promotes the formation of the C-MYC G-quadruplex and inhibits C-MYC promoter activity. Biochemistry 49:9706–9714. doi: 10.1021/bi100509s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Ghosh M, Singh M. 2018. RGG-box in hnRNPA1 specifically recognizes the telomere G-quadruplex DNA and enhances the G-quadruplex unfolding ability of UP1 domain. Nucleic Acids Res 46:10246–10261. doi: 10.1093/nar/gky854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Read M, Harrison RJ, Romagnoli B, Tanious FA, Gowan SH, Reszka AP, Wilson WD, Kelland LR, Neidle S. 2001. Structure-based design of selective and potent G quadruplex-mediated telomerase inhibitors. Proc Natl Acad Sci USA 98:4844–4849. doi: 10.1073/pnas.081560598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Majee P, Pattnaik A, Sahoo BR, Shankar U, Pattnaik AK, Kumar A, Nayak D. 2021. Inhibition of Zika virus replication by G-quadruplex-binding ligands. Mol Ther Nucleic Acids 23:691–701. doi: 10.1016/j.omtn.2020.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Uchil PD, Kumar AVA, Satchidanandam V. 2006. Nuclear localization of flavivirus RNA synthesis in infected cells. J Virol 80:5451–5464. doi: 10.1128/JVI.01982-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rawlinson SM, Pryor MJ, Wright PJ, Jans DA. 2009. CRM1-mediated nuclear export of Dengue virus RNA polymerase NS5 modulates interleukin-8 induction and virus production. J Biol Chem 284:15589–15597. doi: 10.1074/jbc.M808271200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Pryor MJ, Rawlinson SM, Butcher RE, Barton CL, Waterhouse TA, Vasudevan SG, Bardin PG, Wright PJ, Jans DA, Davidson AD. 2007. Nuclear localization of Dengue virus nonstructural protein 5 through its importin alpha/beta-recognized nuclear localization sequences is integral to viral infection. Traffic 8:795–807. doi: 10.1111/j.1600-0854.2007.00579.x [DOI] [PubMed] [Google Scholar]
- 103. Ng IHW, Chan KWK, Tan MJA, Gwee CP, Smith KM, Jeffress SJ, Saw WG, Swarbrick CMD, Watanabe S, Jans DA, Grüber G, Forwood JK, Vasudevan SG. 2019. Zika virus NS5 forms supramolecular nuclear bodies that sequester importin-α and modulate the host immune and pro-inflammatory response in neuronal cells. ACS Infect Dis 5:932–948. doi: 10.1021/acsinfecdis.8b00373 [DOI] [PubMed] [Google Scholar]
- 104. López-Denman AJ, Tuipulotu DE, Ross JB, Trenerry AM, White PA, Mackenzie JM. 2021. Nuclear localisation of West Nile virus NS5 protein modulates host gene expression. Virology 559:131–144. doi: 10.1016/j.virol.2021.03.018 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
NCL is primarily a nuclear protein.
Role of NCL in JEV replication in SH-SY5Y cells.
JEV replication in NCL knockout cells.
GQ sequences in the JEV 3'-NCR.
Supershift of GQ2 RNA complex with NCL and tNCL-34GAR.
Proteins identified by BioID and Co-IP methods.
Proteins from StringDB.