

Abstract
Initiation of reentry requires 2 factors: (1) a triggering event, most commonly focal excitations such as premature ventricular complexes (PVCs); and (2) a vulnerable substrate with regional dispersion of refractoriness and/or excitability, such as occurs during the T wave of the electrocardiogram when some areas of the ventricle have repolarized and recovered excitability but others have not. When the R wave of a PVC coincides in time with the T wave of the previous beat, this timing can lead to unidirectional block and initiation of reentry, known as the R-on-T phenomenon. Classically, the PVC triggering reentry has been viewed as arising focally from 1 region and propagating into another region whose recovery is delayed, resulting in unidirectional conduction block and reentry initiation. However, more recent evidence indicates that PVCs also can arise from the T wave itself. In the latter case, the PVC initiating reentry is not a separate event from the T wave but rather is causally generated from the repolarization gradient that manifests as the T wave. We call the former an “R-to-T” mechanism and the latter an “R-from-T” mechanism, which are initiation mechanisms distinct from each other. Both are important components of the R-on-T phenomenon and need to be taken into account when designing antiarrhythmic strategies. Strategies targeting suppression of triggers alone or vulnerable substrate alone may be appropriate in some instances but not in others. Preventing R-from-T arrhythmias requires suppressing the underlying dynamic tissue instabilities responsible for producing both triggers and substrate vulnerability simultaneously. The same principles are likely to apply to supraventricular arrhythmias.
Keywords: Initiation, R-on-T, Reentry, Ventricular arrhythmia
Ventricular arrhythmias including ventricular tachycardia (VT) and ventricular fibrillation (VF) are the leading cause of sudden cardiac death (SCD).1,2 Such arrhythmias can be caused by reentry, focal excitations, or a mixture of both.3,4 To develop effective therapies, understanding the mechanisms underlying the transition from normal sinus rhythm to these arrhythmic states is essential. In this article, we review the current understanding of this topic by revisiting one of the seminal concepts in this field, the R-on-T phenomenon. Figure 1A shows the electrocardiogram (ECG) of a patient undergoing a transition from sinus rhythm to VT and VF, in which premature ventricular complexes (PVCs), triplets, and nonsustained runs of PVCs precede VT and VF.5 It is known that in patients with ischemic heart disease, the mortality rate increases with the frequency and complexity of PVCs (Figure 1B).6–9 The observation that the risk of lethal ventricular arrhythmias is increased when the R wave of a PVC falls on a T wave (Figure 2A) was made as early as the late 1920s and subsequently documented clearly by Smirk10 in 1949. This so-called R-on-T phenomenon has been linked to a high risk of SCD in many subsequent studies,11 particularly in the setting of acute myocardial infarction.12–16 Although R-on-T is neither necessary nor sufficient for the occurrence of arrhythmias,13 it has been widely demonstrated to precede ventricular arrhythmias in a variety of diseased conditions17–20 as well as in the setting of pacemaker malfunction.21,22
Figure 1.
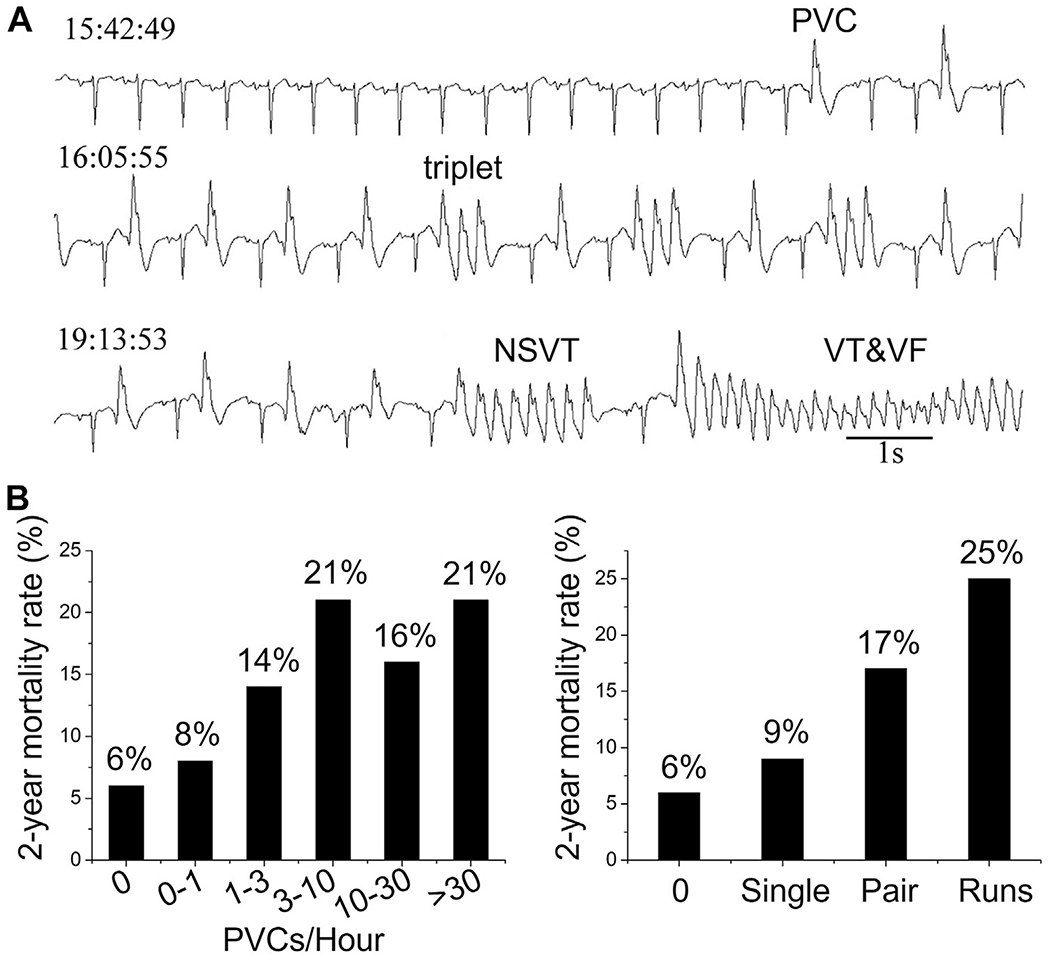
Premature ventricular complexes (PVCs) and ventricular arrhythmias. A: Electrocardiogram recorded from an 82-year-old woman with heart failure showing single PVCs, triplets, nonsustained ventricular tachycardia (NSVT), and ventricular tachycardia/ventricular fibrillation (VT/VF) leading to sudden cardiac death. (Modified from Glass.5) B: Mortality rate vs PVC frequency and complexity in patients 2 years after myocardial infarction. (Data from Bigger et al.8).
Figure 2.
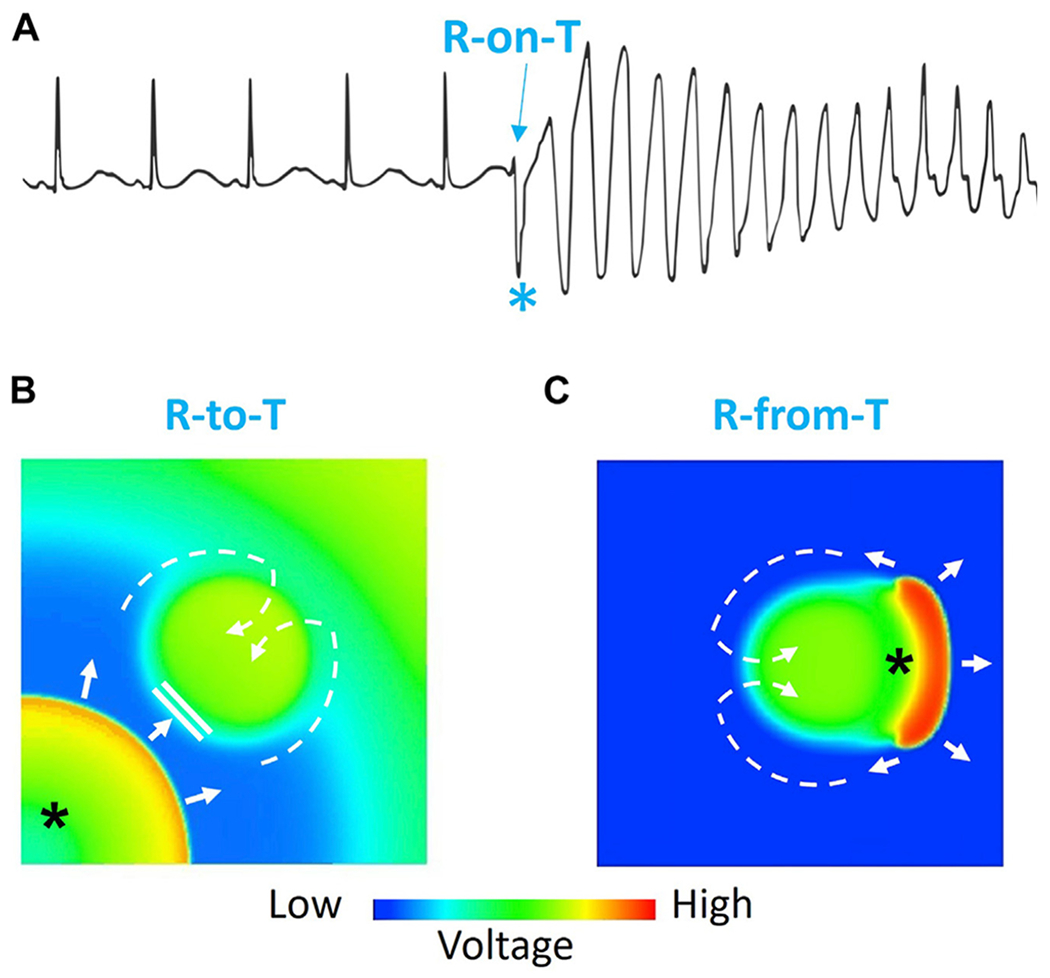
R-on-T phenomenon. A: Electrocardiogram showing an R-on-T event (*) initiating torsades de pointes. B: Voltage snapshot illustrating the “R-to-T” mechanism in which a PVC (*) propagates toward the delayed repolarization region, where it blocks locally and then proceeds around the heterogeneous region to reenter it from a distant site (dashed arrows), thereby initiating figure-of-8 reentry (see Figure 3B for a time sequence of snapshots). C: Voltage snapshot showing the “R-from-T” mechanism in which the PVC (*) is generated directly by the repolarization gradient and conducts around the heterogeneous region to initiate figure-of-8 reentry (dashed arrows). The central region is the long APD region, which repolarizes much later than the rest of the tissue (see Figure 5B or Figure 5C for a time sequence of snapshots).
The arrhythmogenic mechanism of the R-on-T phenomenon has been linked to experimental observations of a “vulnerable period” or “vulnerable window” of the heart, in which an extrastimulus occurring when ventricular repolarization is still incomplete has an increased probability of inducing reentrant arrhythmias.23–27 The vulnerable period in the cardiac cycle typically occurs during the repolarization phase when some regions have recovered excitability but others have not, that is, near the peak of the T wave on the ECG. Here, we first briefly review the classic mechanisms underlying the R-on-T phenomenon first described nearly a century ago, which we refer to as “R-to-T” mechanisms. In this scenario, the PVC triggering reentry arises focally from 1 region and propagates into an adjacent region with prolonged refractoriness where the unidirectional conduction block initiating reentry occurs (Figure 2B). We next proceed to more recently described mechanisms in which the PVC initiating reentry is not a separable event but instead is causally generated from the previous beat’s repolarization pattern during its T wave, which we refer to as “R-from-T” mechanisms (Figure 2C). We also describe other scenarios in which initiation of reentry does not involve the R-on-T phenomenon, which we group into the “R-after-T” category. Finally, we discuss how all of these scenarios must be taken into consideration to develop more effective treatments for ventricular (and supraventricular) arrhythmias in the future.
R-to-T mechanisms: The classic component of the R-on-T phenomenon
The vast majority of PVCs are benign and in normal hearts do not confer an increased risk of SCD.28–30 Even in patients with severe heart disease, the risk of any given PVC initiating a lethal arrhythmia is extremely rare. For example, in patients with chronic heart disease, 2 PVCs per minute, which translates to 1 million PVCs per year, is a commonly observed level of ventricular ectopy, yet the incidence of SCD is measured in months or years, not minutes. This is because in order to initiate a lethal arrhythmia, a properly timed PVC must propagate into tissue that is momentarily vulnerable. In the classic explanation of R-on-T phenomenon, this rare event happens when the PVC propagates from its focal site of origin and encounters a region of prolonged refractoriness (the vulnerable substrate) into which its propagation is blocked. The PVC then propagates around the borders of that region until it has recovered excitability and then reenters the region from a distal site (Figure 2B). In this scenario, the sites of PVC origination and unidirectional conduction block occur at spatially distinct sites, even if they are generated by the same underlying process (eg, early afterdepolarizations [EADs] causing both triggered activity at 1 site and dispersion of refractoriness elsewhere). Alternatively, the processes generating the PVC and the vulnerable substrate can be altogether different (eg, a delayed afterdepolarization [DAD] in the His-Purkinje system generating the PVC and an anatomic obstacle such as a scar creating the vulnerable substrate).
Initiation of reentry
There are 3 common scenarios of initiation of reentry by the R-to-T mechanism (Figure 3). The first is anatomic reentry, that is, reentry around an anatomic obstacle, such as a scar embedding a channel of surviving excitable myocardial tissue that makes connections with adjacent normal tissue at 2 or more sites (Figure 3A).31–33 Here, a properly timed PVC during the T wave conducts successfully into the channel through the first site (the entrance) while blocking at a second site that has a longer refractory period (Figure 3A, left panels). The impulse then propagates from the entrance at the first site through the channel and, if the delay is sufficiently long for the second site to recover, reenters the normal tissue. Thus, the first site becomes the entrance site and the second site the exit site completing the reentrant circuit. In this scenario, the PVC does not generally originate from the exit site and so has a different morphology from the first beat of reentrant tachycardia. In another scenario, the PVC can originate from within the myocardial channel itself, where DADs or EADs arising from electrophysiological and structural remodeling within the channel have a higher probability of eliciting a PVC because of the more favorable source-sink relationship in a quasi–1-dimensional strand of tissue than in a 3-dimensional syncytium.34–36 In this case, unidirectional block can result when the scar-encased channel has a narrow opening at one of its connections with the larger mass of more normal tissue (Figure 3A, right panels). If the resulting source-sink mismatch at this connection is sufficient to prevent propagation in this direction,35,37 the PVC then exits the scar through a different, wider opening (the exit site) and reenters the narrow opening from the other direction (entrance site). The PVC initiating reentry therefore has the same QRS morphology as the subsequent tachycardia, as often observed clinically.38 Although refractoriness can potentiate the formation of reentry, this mechanism does not require the PVC to occur during the refractory period of the tissue and so may manifest itself as either R-on-T or R-after-T.
Figure 3.
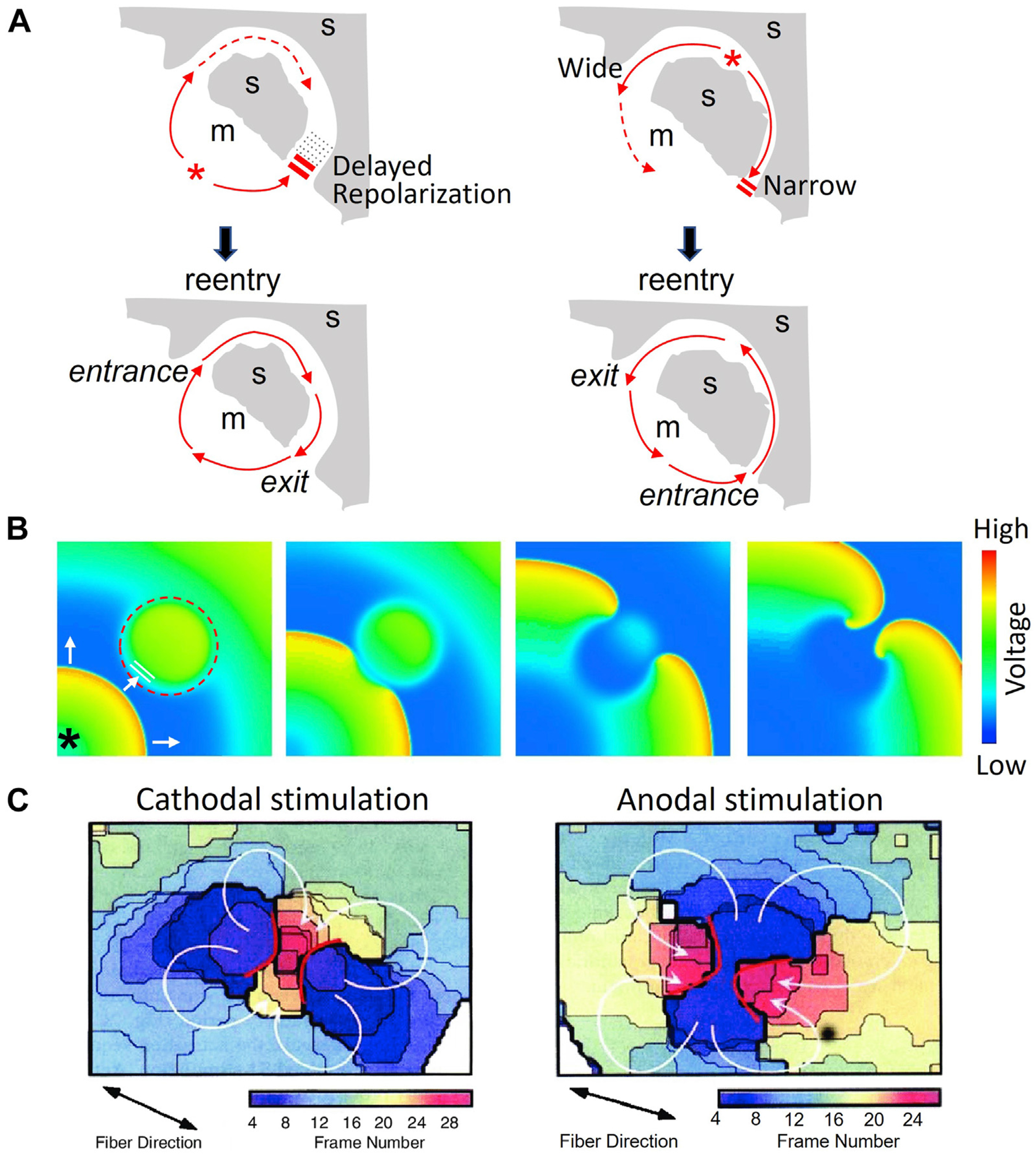
Reentry initiation via trigger (premature ventricular complex [PVC]) and substrate interactions. A: Schematic diagrams showing a PVC inducing anatomic reentry using a myocardial channel embedded in a scar (s) (gray shading). Left panels: Due to a local longer refractory period, the PVC (*) is blocked from entering the channel at its lower connection with normal myocardium (m) (no shading) but conducts successfully through the upper connection with a shorter refractory period. Meanwhile, the lower channel recovers excitability, allowing the PVC to exit to the normal myocardium and completing the reentrant circuit. Right panels: A PVC (*) arising from within the channel is blocked due to the source-sink mismatch at the narrow lower connection to normal myocardium but propagates successfully through the wide upper connection and then reenters the channel through its lower connection to complete the reentrant circuit. B: Voltage snapshots from a computer simulation showing a PVC (*) inducing spiral wave reentry in a 2-dimensional tissue model with a central region whose refractory period is prolonged (dashed circle). The PVC from the lower-left corner blocks in the central region, conducts around its borders, and reenters the central region from the other side, initiating figure-of-8 reentry. C: Shock-induced quatrefoil reentry (arrows) in the vulnerable period. Isochronal maps showing the activation wavefronts for a cathodal (left) and an anodal (right) stimulus in the vulnerable window. (Taken from Lin et al.47).
The second case is spiral/scroll wave or functional reentry, which can occur in both structurally normal and diseased tissue (Figure 3B).39–42 Here, a PVC propagates toward a region of still refractory tissue, conducts around its borders until the region recovers excitability, and then reenters the now-recovered region from the opposite side to form a figure-of-8 reentrant pattern.
The third case (Figure 3C) is reentry initiated by strong stimuli, such as a powerful electrical impulse (shock)26,27,43 or mechanical impact (commotio cordis)44,45 delivered during the vulnerable period of the T wave. Complex reentrant patterns can occur due to the virtual electrodes caused by the electrical impulse46,47 and propagated graded responses.27 Once initiated, reentry can self-terminate or degenerate to VF. Both the timing with respect to the T wave and the strength of the stimulus are important, and can be quantified as the VF threshold in normal and diseased hearts.48
Mechanisms of triggering events
PVCs and other focal excitations can originate from any of the following mechanisms. (1) Automaticity (phase 4 depolarization). Automaticity is a spontaneous oscillatory behavior of myocytes that can be induced by multiple mechanisms, including persistent opening of Na channels49 and other inward currents such as the Na/Ca exchange current,50,51 or decreases in outward currents that stabilize the resting membrane potential like IK1. Fundamentally, the mechanisms of automaticity are similar to that of sinoatrial nodal cells driven by the coupled interactions of a membrane current clock and an intracellular Ca cycling clock.52 (2) EADs. EADs are afterdepolarizations that occur during the repolarization phases (phase 2 or 3) of the action potential.53,54 They tend to occur when action potential duration (APD) is lengthened, such as in acquired and inherited long QT syndromes (LQTSs) and heart failure (HF). (3) DADs. DADs are afterdepolarizations due to activation of inward currents (primarily the Na/Ca exchange current) by spontaneous sarcoplasmic reticulum Ca release during diastole. They tend to occur in inherited diseases such as catecholaminergic polymorphic ventricular tachycardia (CPVT)55,56 or acquired HF when Ca cycling is dysfunctional.57,58 Once the DAD amplitude reaches the threshold for Na current activation,59 a full action potential is triggered. Similar to automaticity, DAD-triggered action potentials are potentiated by reduction of IK1 and increases in INCX. The number of DAD-triggered beats can range from single to multiple and even can become self-sustained.60,61
Mechanisms of substrate vulnerability
A tissue substrate becomes vulnerable to reentry when there is an increased spatial dispersion of refractoriness and/or excitability, such that an impulse can propagate in some directions but not in others. Increased dispersion can be caused by structural factors such as fibrosis and scars, disease-related remodeling (neural, vascular, gap junction, ion channel, Ca cycling), drugs, genetic channelopathies, and inflammation. Experimental data62,63 and theoretical analysis64,65 show that for unidirectional conduction block to occur, a minimum spatial gradient of refractoriness of the order of several milliseconds per millimeter is required in normal tissue. For example, Laurita and Rosenbaum62 showed that in normal guinea pig ventricular epicardium, a minimum APD gradient of 3.2 ms/mm was required for unidirectional conduction block. Theory predicts that the minimum gradient becomes larger with weaker gap junction coupling,64,65 which is in agreement with the finding that in wedge preparations of failing dog hearts,63 polymorphic VT could be induced only when the transmural APD gradient was >10 ms/mm. The minimum gradient required also decreased at faster heart rates.64,65
Dispersion of refractoriness also can be caused by dynamic instabilities66,67 arising from changes in heart rate, autonomic tone, electrolyte disturbances, and abnormal temperature. For example, spatially discordant repolarization alternans is a dynamic factor that creates dispersion of refractoriness.66,68,69 It can occur transiently after single extrasystoles, thus setting the stage for additional extrasystoles to initiate reentry, or become sustained during rapid pacing, resulting directly in wavebreak and initiation of reentry. Clinically, this is likely to be an important mechanism by which rapid triggered activity arising from the His-Purkinje system induces idiopathic VT/VF in susceptible patients with otherwise normal hearts.70,71 If the sites of triggered activity in the His-Purkinje system can be isolated by catheter ablation to prevent them from reaching ventricular muscle, these patients can be cured, similar to patients in whom triggers arising from the pulmonary veins can be isolated from the bulk of atrial tissue by catheter ablation to prevent AF.72
Evaluating substrate vulnerability using programmed electrical stimulation
The most common method used to assess substrate vulnerability to reentry is programmed electrical stimulation (PES), in which timed extrasystoles are induced artificially via electrical pacing to simulate 1 or more PVCs.73–75 The technique applies the paced extrasystoles at shorter and shorter coupling intervals during diastole until the pacing stimulus fails to elicit a response, which defines the tissue’s refractory period. If a single paced extrasystole fails to induce reentry, then a second paced extrastimulus can be introduced at progressively shorter coupling intervals during the T wave of first paced extrasystole, and so forth, typically up to 3–5 paced extrasystoles. Alternatively, rapid burst pacing at short cycle lengths (200–300 ms) serves a similar purpose by causing the R wave of the paced extrasystole to fall on the T wave of the previous paced extrasystole. Either method works reasonably well for inducing reentry when tissue vulnerability is related to an anatomic obstacle (eg, monomorphic VT around a scar, atrioventricular [AV] nodal reentry, or AV reentry due to a bypass tract).
One of the limitations of PES in assessing substrate vulnerability is that as the number of premature extrasystoles or the rate and duration of rapid pacing increases, diseased tissue becomes susceptible to forms of reentry that are not observed clinically, and even completely normal hearts become susceptible to reentry that quickly degenerates from polymorphic VT to VF. For example, if paced too rapidly, unidirectional conduction block initiating reentry can occur in localized regions with longer refractory periods if dispersion of refractoriness is present, either pre-existing or dynamically amplified by spatially discordant repolarization alternations. We consider this to fall into the R-to-T category because the rapid heart beats, whether arising from pacing or an endogenous mechanism, create R waves that fall on the T waves of previous beat(s). As noted earlier, a high-amplitude paced extrasystole delivered during the T wave in normal cardiac tissue can also induce spiral/scroll wave reentry manifested as polymorphic VT/VF (the VF threshold).76–78
For these reasons, PES, which can be complemented by electroanatomic mapping and personalized heart modeling,74,75 is most useful for predicting risk and managing treatment of anatomic reentry manifesting itself as clinically documented monomorphic tachycardia, such as monomorphic VT in the ventricles or supraventricular tachycardias due to accessory AV pathways, dual AV nodal pathways, or atrial flutter. PES is less useful for managing polymorphic VT and VF because of the difficulty in distinguishing whether the arrhythmias induced are clinically significant or nonspecific responses to an aggressive PES protocol.79
R-from-T mechanisms: The nonclassic component of the R-on-T phenomenon
In addition to the classic R-to-T component of the R-on-T phenomenon, reentry can arise in the wake of a single beat that self-generates both a vulnerable substrate and the crucial triggering event(s) simultaneously from the same region of tissue. In this case, both the triggering event(s) and vulnerable substrate arise simultaneously as emergent properties of a dynamic instability of the tissue state.80,81 To distinguish this nonclassic component of the R-on-T phenomenon from the R-to-T mechanism described earlier, we refer to it as the R-from-T mechanism, to indicate that the R wave of the extrasystole initiating reentry is not an independent event but instead is causally generated from the previous beat’s repolarization pattern during its T wave (Figure 2C). The R-from-T mechanism has been described primarily in the settings of excess repolarization reserve,81–84 as in early repolarization syndromes, and reduced repolarization reserve, as in acquired and congenital LQTSs.80,81,85–89
Excess repolarization reserve and phase 2 reentry
The first direct experimental evidence for the R-from-T mechanism came from studies by Di Diego and Antzelevitch82 and Lukas and Antzelevitch.83 They showed in canine hearts subjected to simulated ischemia that the spike-and-dome action potential morphology in ventricular epicardium, where the transient outward potassium current (Ito) is large, could lead to spontaneous initiation of reentry. In this case, the simulated ischemic conditions led to a sudden loss of the action potential dome and early repolarization in some regions of the epicardium but not in others, creating marked dispersion of refractoriness (ie, a vulnerable substrate). At the same time, the resulting electrotonic current flow from the remaining spike-and-dome regions into the spike-only regions was sufficient to re-excite the spike-only tissue, creating a PVC (the triggering event), which then reinvaded the spike-and-dome region after it had repolarized, initiating reentry (Figure 4A). They called this mechanism phase 2 reentry (P2R). We view P2R as the first unequivocal experimental evidence supporting the R-from-T scenario. Extensive studies by the same group subsequently implicated P2R as an important mechanism initiating reentrant VT/VF in Brugada syndrome, short QT syndromes, and other early repolarization (J-wave) syndromes characterized by excess repolarization reserve.90,91
Figure 4.
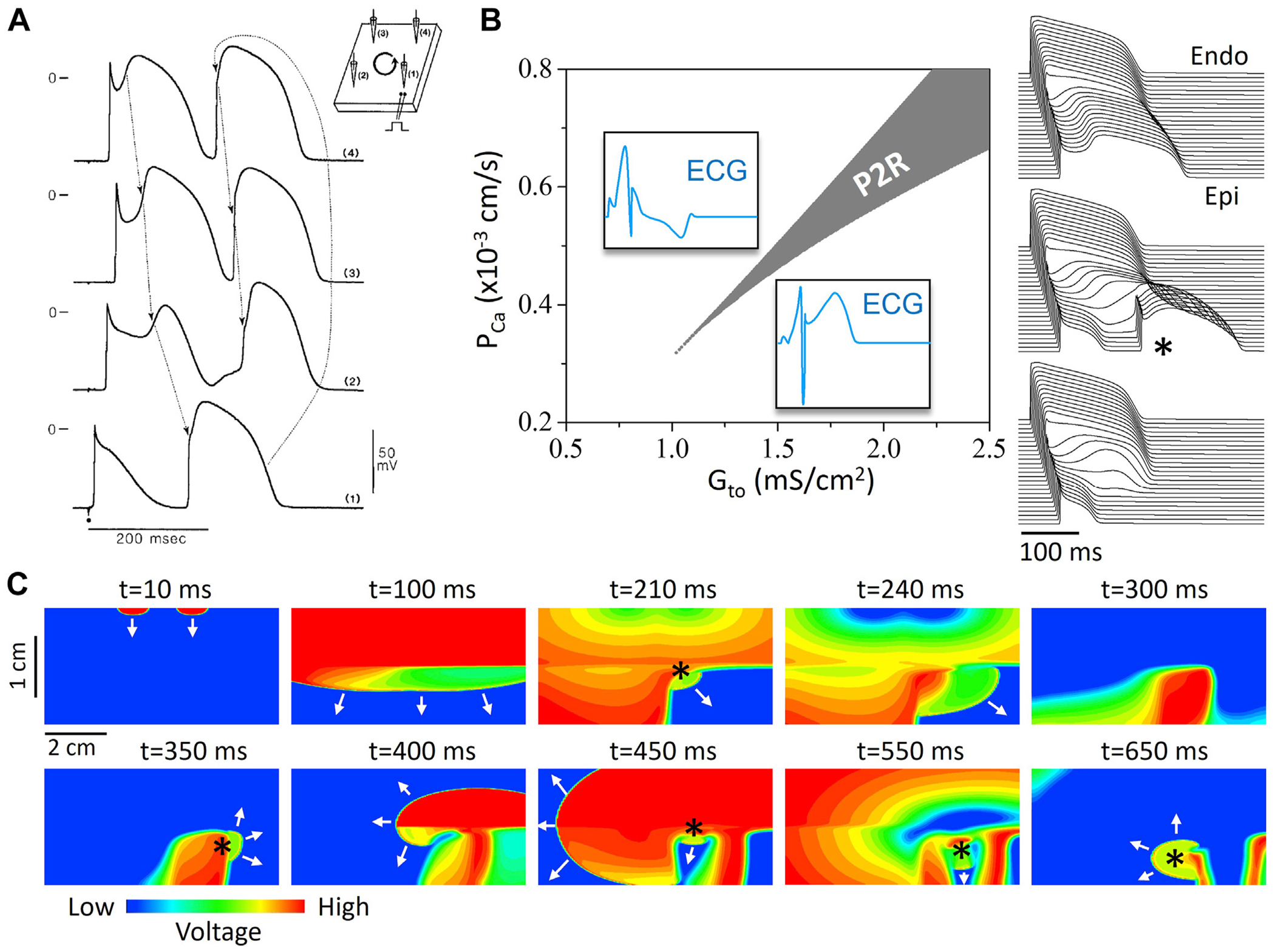
R-from-T. Phase 2 reentry (P2R) resulting from the spike-and-dome action potential (AP) morphology. A: Microelectrode recording of P2R in canine ventricular tissue.83 Arrows show how the AP dome propagates from top to bottom to re-excite the region of early repolarization (bottom) and initiate P2R. B: Computer simulation of a 1-dimensional cable showing the P2R region and its dependence on the conductances of Ito (Gto) and ICa,L (PCa). Left: One-dimensional cable simulation showing P2R region vs Gto and PCa. Insets show the characteristic pseudo-electrocardiograms (ECGs) above and below the P2R region. Right: Time-space plot of voltage from above, within, and below the P2R region. (From Zhang et al.84) C: Computer simulation showing P2R and wave dynamics in 2-dimensional tissue, showing voltage snapshots at different time points. Arrows indicate propagating wavefronts. A wave elicited from 2 endocardial stimulus sites at top (t = 10 ms) propagates to the epicardium (t = 100 ms) at bottom. Due to the early repolarization in the lower-right quadrant, P2R occurs (t = 210 ms, marked by *), propagates toward the tissue boundary (t = 240 ms), and disappears. The repolarization pattern remains heterogeneous (t = 300 ms), and a new P2R occurs (t = 350 ms) that propagates as a spiral wave reentry (t = 400 ms) before wandering off the tissue boundary (t = 450 ms). New P2Rs (*) also occur in snapshots at t = 450, 550, and 650 ms. (From Zhang et al.84).
More recently, computer simulations have provided detailed mechanistic insights into the conditions required to generate P2R.81,84,92,93 In general, P2R is potentiated by reduction of inward currents or increase of outward currents that, in conjunction with Ito, facilitate sudden early repolarization. The occurrence of P2R may depend sensitively on parameter settings in computer models (Figure 4B) and can manifest as a single PVC or cause nonsustained or sustained reentry as long as the size and geometry of the heterogeneity are appropriate (Figure 4C). Moreover, because P2R is due to an ongoing dynamic tissue instability, once the first episode of P2R occurs, the repolarization heterogeneities become very dynamic, resulting in additional P2R events perpetuating the arrhythmia.
It is important to note that reentrant arrhythmias in the setting of excess repolarization reserve are not exclusively due to the R-from-T mechanism but also can involve the classic R-to-T mechanism. For example, a PVC arising from a region that fails to induce reentry locally at the same site may then propagate to a distant site in which repolarization has been delayed, resulting in conduction block and initiation of reentry. Although the PVC at the first site and vulnerable substrate at the distant site both result from the same underlying dynamic process, we classify the latter scenario as R-to-T because the PVC origination and the unidirectional conduction block initiating reentry occur at different sites. Moreover, in Brugada syndrome, an entirely different mechanism has been proposed, called the depolarization hypothesis, hypothesizing that structural changes due to fibrosis in regions of the ventricular epicardium are the main cause of unidirectional block initiating reentry rather than P2R (called the repolarization hypothesis) (for a discussion of this controversy, see Weiss,94 Wilde et al,95 and Nademanee and Wilde96). Thus, both R-from-T and R-to-T mechanisms may contribute to reentry initiation in these settings.
Reduced repolarization reserve and EAD- and repolarization gradient-induced reentrant arrhythmias
The P2R studies described indicate that when all-or-none early repolarization occurs regionally in ventricular tissue, tissue APD gradients (reflecting tissue vulnerability) can become large enough such that electrotonic current flow from depolarized spike-and-dome regions can trigger re-excitation of recovered spike-only regions, generating an extrasystole (triggering event) that initiates reentry (P2R) by the R-from-T mechanism. Experimental and computer modeling evidence indicates that an analogous situation can occur when decreased repolarization reserve makes the heart susceptible to EADs, such as in congenital and acquired LQTSs and HF. In these cases, a large tissue APD gradient can arise between regions exhibiting EADs or markedly prolonged APD and adjacent regions without EADs and less APD prolongation. Electrotonic current flow from the still depolarized regions then can re-excite the repolarized shorter APD regions to cause an extrasystole that initiates re-entry. Computer simulation studies have revealed 2 variations of this scenario in simulated LQTSs (Figure 5A)80,81,85–89: (1) EAD-induced PVCs and (2) APD gradient-induced PVCs. In the EAD-induced PVC mechanism, a region with cells exhibiting phase 2 EADs (ie, where dV/dt becomes transiently positive during phase 2) can trigger a PVC in adjacent repolarized tissue without EADs (Figure 5A, left panel) when the APD gradient falls in a critical range (neither too small nor too large; see recent studies by Zhang and Qu81 and Zhang et al89). In the APD gradient-induced PVC mechanism, a region with prolonged APD but no EADs per se (ie, dV/dt remains negative throughout phase 2) still can generate PVCs (Figure 5A, middle and right panels ). This is known as “prolonged repolarization-dependent reexcitation” caused by the repolarization gradient.97 However, as demonstrated in computer simulations, a large repolarization gradient alone is not sufficient, and a minimum strength of the L-type of Ca current (ICa,L) also is required.80,81,89 The computer simulation studies have shown that for both the EAD-induced and APD gradient-induced mechanisms, generation of PVCs is the result of dynamic instabilities in the tissue.80,81,89 The EAD-induced PVC mechanism occurs in a narrow, finely tuned parameter range (similar to the narrow, finely tuned parameter region for P2R shown in Figure 4B) due to the requirement for dynamic instabilities, and only generates a single PVC no matter how many EADs occur in the long APD region. In contrast, the APD gradient-induced PVC mechanism can occur in a much wider range of parameters and generate single or multiple PVCs (Figure 5A, middle and right panels) depending on the APD gradient.
Figure 5.
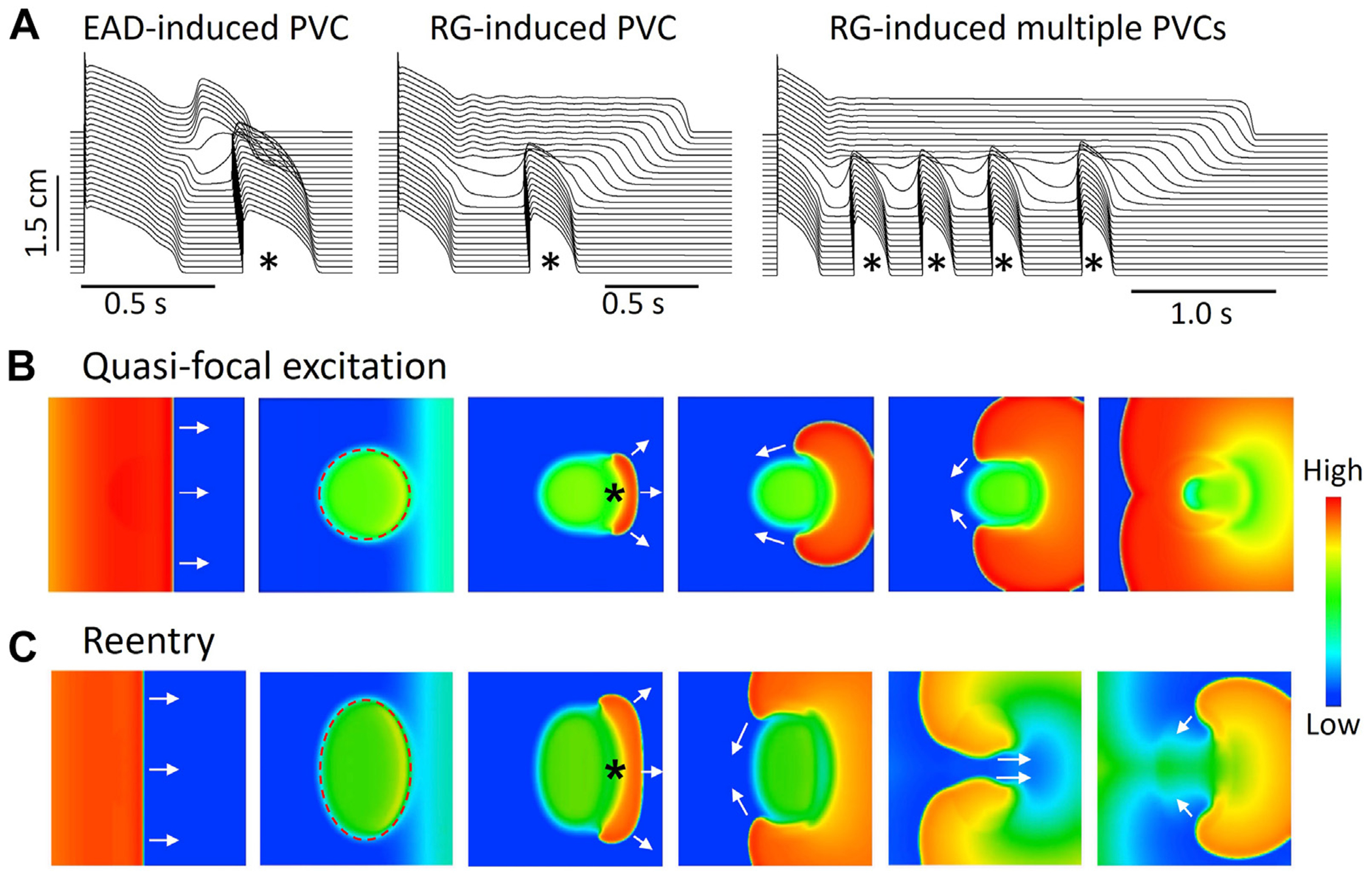
R-from-T. QT prolongation-induced premature ventricular complex (PVC) and reentry in computer models. A: Computer simulation showing an early afterdepolarization (EAD)-induced PVC (left), a repolarization gradient (RG)-induced PVC (middle), and RG-induced multiple PVCs in a 1-dimensional cable model after a single pacing beat.89 PVCs are marked by asterisks. B: Voltage snapshots at different time points from a computer simulation showing an R-from-T quasi-focal excitation induced by a RG gradient in a 2-dimensional tissue model. Potassium current conductance is reduced in the dashed circular region, resulting in a longer APD. A pacing stimulus was applied at the left side of the tissue and propagated toward the right side (first and second panels). A unidirectionally propagating PVC (asterisk) occurs spontaneously in the gradient region (third panel), forming 2 tips that conduct around the long APD region (fourth and fifth panels). The 2 wave tips then collide in the other side of the long APD region (sixth panel), forming a targetlike pattern or a quasi-focal excitation. Note that although the heterogeneous region is centrally located and symmetric, propagation of the pacing beat causes the PVC to originate on the right side of the long APD region. In real tissue, the origin of the PVC will depend on the spatial geometry of the heterogeneity and the conduction direction of the sinus beat, and can even fire simultaneously for the entire border, resulting in a true target pattern. C: Same as B but with a larger heterogeneous region (dashed oval). Because the long APD region is large enough to allow recovery of the central region, the 2 wave tips do not collide but reenter the central region to initiate figure-of-8 reentry. (Simulation details were shown in Huang et al.80).
In both mechanisms, the breakthrough site of the PVC occurs in the APD gradient region, such that the PVC propagates in only 1 direction, from the gradient region toward the shorter APD region. Therefore, in heterogeneous tissue, a PVC arising from the APD gradient region circles around the region of heterogeneity. Depending on the length of the refractory period, the size, and the geometry of the region of heterogeneity, it may manifest as a quasi-focal excitation (Figure 5B), or, if conditions are appropriate, it may initiate reentry (Figure 5C). This mechanism could underlie the short-long-short sequence that often induces torsades de pointes (TdP), nonsustained polymorphic VT, and the degeneration to VF in LQTS.88 In this setting, the long pause promotes bradycardia-dependent EADs or APD prolongation that generate the heterogeneous repolarization pattern from which the PVC emerges by the R-from-T mechanism to initiate reentry.
Experimental microelectrode and optical mapping studies investigating the initiation of TdP due to QT prolongation80,98–104 directly support the modeling predictions (Figure 5).80,89 Yan et al98,99 used microelectrode recordings in rabbit and canine left ventricular wedge preparations to demonstrate how phase 2 EADs and APD prolongation were linked to the spontaneous initiation of TdP. Figure 6A shows a representative recording of spontaneous TdP in their experiments. In the first paced beat, a long APD with phase 2 EADs occurred in the endocardial layer and a PVC originated in the epicardial layer. In the second beat, APD became even longer in the endocardium with multiple EADs, and TdP occurred spontaneously. Although it is unclear whether the PVCs and TdP were induced by the EADs or the endo-to-epi APD gradient due to only 2 recording sites, Yan et al98,99 observed that increased endo-to-epi APD gradient played an important role. In an optical mapping study using a canine left ventricular wedge preparation (Figure 6B), Liu and Laurita101 observed that the breakthrough site of triggered activity always occurred where the local repolarization gradients were largest, that is, the triggered beat did not originate from the region of longest APD (the M cell or the endocardial region) where EADs were more likely to occur. Other optical mapping studies have similarly found that the triggered beats tend to originate from the steep APD gradient region (Figure 6C), not from where APD is the longest.80,102–104 These experimental studies demonstrate that the genesis of PVCs and initiation of reentry causing TdP are closely associated with and emerge from the APD gradient region as predicted by the R-from-T mechanism in P2R. It also is possible that a PVC originating from the APD gradient in 1 region can fail to induce reentry locally and propagate as a target wave to a distant APD gradient location where unidirectional conduction block occurs and initiates reentry by the R-to-T mechanism, although this scenario was not reported in these particular studies.
Figure 6.
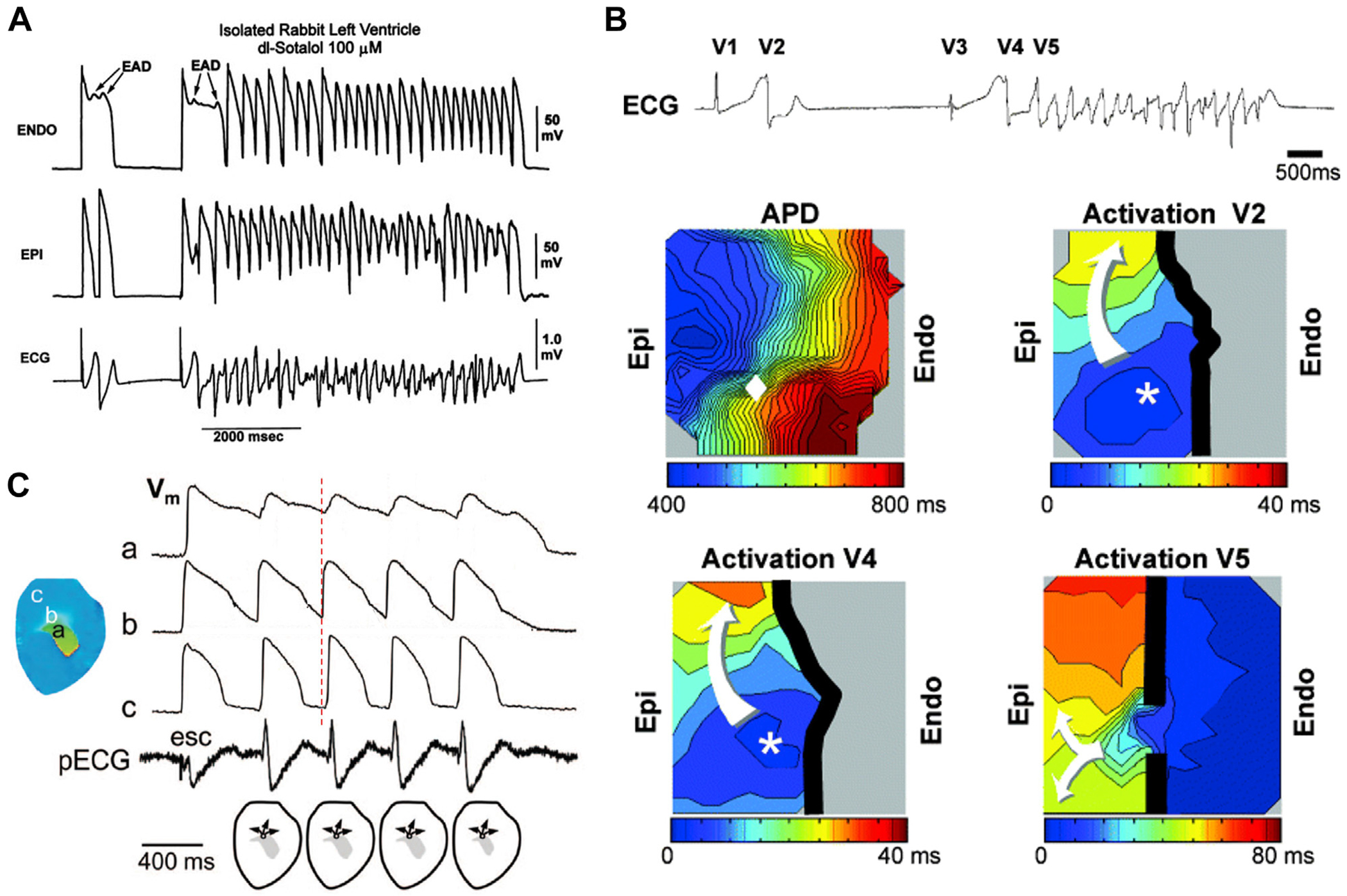
R-from-T. Spontaneous initiation of torsades de pointes (TdP) caused by QT prolongation in experimental settings. A: Spontaneous TdP in arterially perfused rabbit left ventricle pretreated with d,l-sotalol. Shown are transmembrane potentials from the endocardial (top) and epicardial (middle) sites and a pseudo-electrocardiogram (ECG) (bottom). (Reproduced from Yan et al.98) B: Spontaneous TdP induced by a pause (or the short-long-short sequence) in a canine ventricular wedge preparation. Top: ECG shows a spontaneous beat (V1) followed by a spontaneous PVC (V2). After a pause, a second spontaneous beat (V3) and a PVC (V4) occurred preceding TdP. Middle left: Contour map showing action potential duration (APD) after a pause (1700 ms) from a similar recording in the same wedge preparation corresponding to V3 but without an ectopic beat. The maximum APD gradient in the mapping field is marked by the diamond. Middle right: Contour map showing activation of V2. The breakthrough site (*) occurred exactly at the site where the local APD gradient was largest, and conduction block (thick black line) occurred in the direction of longest APD. Bottom left: Contour map showing breakthrough activation of V4. Bottom right: Contour map showing activation of the first beat of TdP entering from outside the mapping field (V5). (Reproduced from Liu and Laurita.101) C: Multiple PVCs or nonsustained ventricular tachycardia recorded in an optical mapping experiment of rabbit heart. Vertical dashed line is a reference for timing, which shows that depolarization at site b is earlier than at sites a and c, indicating the breakthrough site for PVCs is in the gradient region rather than the site with the longest APD, agreeing with the recording in B. (Reproduced from Maruyama et al.102).
R-after-T: Reentry initiation without the R-on-T phenomenon
Although the magnitude of the ECG T wave or Tpeak-Tend interval is a measure of the repolarization gradient in the heart,105,106 conduction block is not due to repolarization per se but is caused by refractoriness. In normal tissue, repolarization is a reliable measure of refractoriness because Na channel recovery conferring excitability is rapid. However, this is not always the case in the presence of disease or drugs that slow Na channel recovery, and dispersion of refractoriness conferring susceptibility to unidirectional conduction block may also be present at a later time during the cardiac cycle. For instance, an R wave may induce reentry by encountering still refractory tissue after the T wave. Four typical settings for this R-after-T scenario are described here.
Postrepolarization refractoriness
In either acute or chronic ischemia, Na channel recovery is slowed due to either elevation of the resting potential107 or remodeling,108 prolonging the refractory period beyond repolarization, known as postrepolarization refractoriness.108,109 Na channel-blocking drugs also commonly slow the recovery of Na channels from inactivation. Under these conditions, an R wave that falls after the T wave may cause unidirectional conduction block when the PVC encounters a region with postrepolarization refractoriness.
Source-sink mismatch
In fibrotic tissue, source-sink mismatches can induce unidirectional conduction block,35,37 also allowing a late R wave to induce reentry in repolarized tissue after the T wave (Figure 3A, bottom panels).
Acute myocardial ischemia
Acute regional ischemia following coronary artery occlusion leads to extracellular K accumulation that elevates the resting membrane potential in the ischemic region, causing injury currents to flow electrotonically across the border zone into adjacent nonischemic tissue during diastole. This diastolic electrotonic current flow, if large enough, can depolarize the recovered nonischemic tissue and elicit a PVC, which then propagates and reenters the ischemic zone at a distal location to induce reentry.32,110,111 Although this scenario remains controversial, its timing can allow an R wave after the T wave to induce reentry and VT/VF following coronary occlusion.112,113 The observation that infusing elevated K selectively into a coronary artery branch in dogs reproduces these arrhythmias supports the key arrhythmogenic role of ischemic extracellular K accumulation in the genesis of lethal arrhythmias following coronary occlusion.114 This is not the only arrhythmogenic mechanism causing VT/VF during acute ischemia, however. As noted earlier, simulated acute ischemia also promotes P2R initiating VT/VF by the R-from-T mechanism.
Ca cycling instabilities and DAD-related arrhythmias
DADs and DAD-induced triggered activity result from intracellular Ca cycling instabilities.61,115–118 Classically, DADs are considered triggers generating PVCs in the form of triggered activity when the DAD amplitude reaches the Na current activation threshold.119,120 Single DADs occur in diastole and may serve as triggers to initiate R-after-T arrhythmias if a vulnerable substrate is present following the T wave (eg, from postrepolarization refractoriness). DAD-mediated runs of triggered activity may mimic fast pacing initiating reentry by either the R-to-T or R-after-T mechanism, depending on whether the rate of triggered activity is rapid enough to encroach upon the preceding T wave.
In addition to these roles of suprathreshold DADs in generating triggers, recent computer simulations have also demonstrated that subthreshold DADs that do not trigger overt PVCs can create a substrate vulnerable to unidirectional conduction block (Figure 7A).121 In this case, elevation of the resting membrane potential during the subthreshold DAD causes inactivation of Na channels, which lowers excitability sufficiently to cause conduction block of a PVC propagating into that region. This possibility is supported experimentally in engineered cardiac tissue.122 Thus, in principle, DADs can generate both triggers and a substrate vulnerable to unidirectional conduction block and initiation of reentry. Due to heterogeneities in excitability, DAD-induced PVCs may propagate in all directions as target waves or in only 1 direction (Figure 7B). Thus, as in the LQTS and P2R cases, a PVC propagating in 1 direction may spontaneously initiate reentry depending on the size and geometry of the subthreshold DAD region.121 Because DADs and triggered activity are potentiated by fast heart rates, once DAD-induced PVCs occur, they can potentiate more DAD-induced PVCs due to a positive feedback loop between synchronization of Ca release and triggered action potentials.60,61 This positive feedback loop results in a bistable phenomenon,61 which may provide a dynamic mechanism explaining DAD-triggered arrhythmias.
Figure 7.
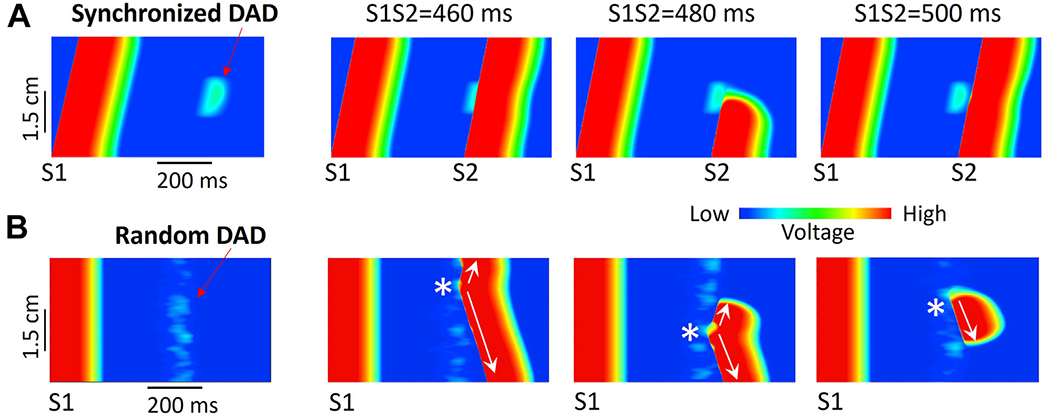
R-after-T. Delayed afterdepolarizations (DADs) can generate both triggers and a vulnerable substrate simultaneously. A: Line scans from computer simulations of a 1-dimensional (1D) cable illustrating a subthreshold DAD causing conduction block. S1 and S2 stimuli were given from the bottom end of the cable, and only the central region developed a DAD. First panel: A subthreshold DAD in the center region of the cable following the S1 stimulus. Second panel: An S2 delivered at 460 ms propagates successfully through the DAD region. Third panel: However, an S2 delivered at 480 ms beat is blocked. Fourth panel: An S2 delivered at 500 ms again propagates successfully. B: Line scans from computer simulations of a 1D cable in which DADs developed randomly. The S1 stimulus was given simultaneously to all cells. First panel: Random DADs following an S1 stimulation. Second panel: A suprathreshold DAD-induced premature ventricular contraction (PVC) (*) propagates successfully in both directions. Third panel: A suprathreshold DAD-induced PVC (*) initially propagates in both directions, but upward propagation is then blocked by subthreshold DADs in the upper portion of the cable. Fourth panel: A suprathreshold DAD-induced PVC (*) initially propagates only in the downward direction (unidirectional conduction block) and later is blocked by a subthreshold DAD a distance away. (Simulation details were shown in Liu et al.121).
Clinical implications of the R-to-T, R-from-T, and R-after-T concepts
In Table 1, we summarize how the R-to-T, R-from-T, and R-after-T mechanisms are involved in reentrant ventricular arrhythmias causing SCD in various clinical settings. We have not included nonreentrant arrhythmias, such as pure triggered activity from EADs, DADs, or automaticity that also can cause VT, but we do include arrhythmias that can exhibit mixed focal and reentrant components such as polymorphic VT/VF and TdP.
Table 1.
Relevance of R-to-T, R-from-T, and R-after-T mechanisms to clinical reentrant ventricular arrhythmias
| Clinical setting | Reentrant arrhythmia | R-to-T | R-from T | R-after-T |
|---|---|---|---|---|
| Acute ischemia | Polymorphic VT/VF | ✓ | ✓ | |
| J-wave syndromes (Brugada, short QT, early repolarization syndromes) | Polymorphic VT/VF | ✓ | ✓ | |
| Chronic ischemic HD | Scar-related monomorphic VT | ✓ | ||
| Bundle branch macroreentry | ✓ | |||
| Polymorphic VT/VF | ✓ | ✓ | ✓ | |
| Chronic nonischemic HD | Scar-related monomorphic VT | ✓ | ||
| Bundle branch macroreentry | ✓ | |||
| Polymorphic VT/VF | ✓ | ✓ | ✓ | |
| Long QT syndromes | TdP, polymorphic VT/VF | ✓ | ✓ | |
| CPVT and digitalis toxicity | Polymorphic VT/VF | ✓ | ✓ | |
| Idiopathic VF | HPS triggers initiate VF | ✓ |
CPVT = catecholaminergic polymorphic ventricular tachycardia; HD = heart disease; HPS = His-Purkinje system; TdP = torsades de pointes; VF = ventricular fibrillation; VT = ventricular tachycardia.
In all of the clinical settings in which ventricular reentry takes the form of reentrant polymorphic VT/VF or TdP, individual episodes may involve different operative mechanisms. Only monomorphic VT in chronic ischemic or nonischemic heart disease is likely to involve a single mechanism, namely, anatomic reentry initiated by the R-on-T mechanism. Most commonly, the anatomic reentrant circuit in these cases involves surviving myocardial channels in the border zone of an infarct in ischemic heart disease or a densely scarred area in nonischemic heart disease, although bundle branch macroreentry can also be a cause.123 Reentrant monomorphic VT is more common in ischemic than in nonischemic heart disease, but the risk of polymorphic VT/VF is increased in both settings because HF remodeling associated with these conditions both reduces repolarization reserve,124 predisposing the heart to EAD-mediated arrhythmias similar to LQTSs, and alters intracellular Ca cycling,125 predisposing the heart to DAD-mediated arrhythmias similar to CPVT. Both EADs and DADs can generate triggers to induce monomorphic VT by the R-to-T mechanism but also can create vulnerable substrates that facilitate initiation of reentry and polymorphic VT/VF by the R-to-T, R-from-T, and R-after-T mechanisms. In ischemic heart disease, the possibility of acute ischemia from coronary artery occlusion inducing P2R-induced or injury current-induced polymorphic VT/VF represents an additional arrhythmogenic threat.
Implications for antiarrhythmic therapy: Lessons from the past
The most straightforward form of ventricular reentry to treat and potentially cure is monomorphic VT due to anatomic reentry around a scar or the bundle branches in the setting of ischemic or nonischemic heart disease. If the anatomic reentrant circuit can be identified by electroanatomic mapping and then ablated, the success rate for curing monomorphic VT is substantial.79 Likewise, patients with idiopathic VF in which rapid triggered activity arising from the His-Purkinje system initiates polymorphic VT/VF can be cured using catheter ablation to prevent the triggers from reaching the ventricular myocardium,70 analogous to pulmonary vein isolation in AF.72 However, patients with chronic heart disease remain at risk for EAD- and DAD-associated polymorphic VT/VF, for which catheter and surgical ablation have limited direct applicability due to the nonlocalized nature of shifting reentrant circuits. With the exception of beta-blockade to counter the proarrhythmogenic effects of sympathetic stimulation, pharmacological approaches targeting PVC triggers or substrate vulnerability, such as those used in the CAST (Cardiac Arrhythmia Suppression Trial)126,127 and SWORD (Survival With ORal D-Sotalol)128,129 trials, have been disappointing because of the multiplicity of the mechanisms listed in Table 1.
The premise of the CAST study, which was designed to test the PVC hypothesis in patients with a previous myocardial infarction, was eminently reasonable. If PVCs encountering a vulnerable substrate are necessary to trigger ventricular reentry, then reducing the number of PVCs should translate to a reduction in mortality from ventricular arrhythmias. However, this is true only if the treatment to reduce PVCs does not exacerbate substrate vulnerability such that the probability of a PVC inducing reentry increases. In the CAST study, the frequency of PVCs was reduced 5-fold by the class I antiarrhythmic Na channel-blocking drugs tested but mortality was doubled, implying that the probability of a PVC inducing lethal ventricular reentry had increased 10-fold. We now appreciate that Na channel-blocking drugs enhance substrate vulnerability by slowing Na channel recovery and inducing postrepolarization refractoriness (R-after-T), as well as by potentiating spatially discordant repolarization alternans (R-to-T).66,130
Similarly, the SWORD trial was designed to reduce substrate vulnerability by prolonging refractoriness with the class III antiarrhythmic K channel-blocking drug D-sotalol. We now know that D-sotalol further potentiated the reduced repolarization reserve associated with chronic heart disease, increasing the risk of EAD-mediated TdP and polymorphic VT/VF (both R-to-T and R-from-T) so that these drugs are now generally contraindicated in patients with chronic heart disease.131 Thus, at the present time, we have no reliable strategy to prevent the initiation of polymorphic VT/VF in any of the clinical conditions listed in Table 1 and must rely on implantable cardioverter-defibrillator insertion to reduce SCD risk.
Implications for antiarrhythmic therapy: Lessons for the future
What can we learn about designing future antiarrhythmic strategies from the data in Table 1? For the R-to-T and R-after-T mechanisms, the relationship between the PVC trigger and the vulnerable substrate is incidental. PVCs arise independently and propagate toward a vulnerable region that dispersion of refractoriness makes susceptible to unidirectional conduction block (Figure 2A). In these cases, an intervention that suppresses triggers without enhancing the vulnerability of the substrate (eg, an ideal antiarrhythmic drug) or conversely reduces the vulnerability of the substrate without increasing triggers (eg, ablation of an anatomic pathway) should be effective.
For the R-from-T mechanism, however, the relationship between the PVC triggers and the vulnerable substrate is not incidental but causal. Instead of the PVC propagating into a vulnerable region and blocking unidirectionally (Figure 2B), the PVC is generated by a repolarization gradient in the vulnerable region and then reenters the same region to initiate reentry (Figure 2C). Although R-to-T and R-from-T appear identical electrocardiographically, they are intrinsically different. For the R-from-T mechanism, the PVC trigger and vulnerable substrate are not separable and therefore cannot be targeted individually. Instead, the appropriate targets are the dynamic tissue instabilities that generate both together. For example, computer modeling80,88,132 and experimental studies133,134 have identified the L-type Ca window current is a critical parameter underlying the tissue instabilities that promote EAD-mediated arrhythmias when repolarization reserve is reduced. In simulations and experiments, interventions selectively inhibiting the L-type Ca window current were found to be highly effective at suppressing both EADs and EAD-mediated arrhythmias. Recent computer modeling has also shown that suppressing the L-type Ca window current may be effective at preventing P2R in the setting of excess repolarization reserve such as Brugada syndrome.84
Another approach to targeting dynamic instabilities is to take advantage of tissue scale parameters, such as source-sink relationships. For example, in a genetically altered mouse model of CPVT due to a mutation in the protein calsequestrin, gene therapy with a vector to overexpress wild-type calsequestrin prevented DAD-mediated arrhythmias despite infecting only about 50% of the myocytes in the tissue.135 This was due to electronic load of the infected myocytes without DADs attenuating the DADs of the uninfected cells, thereby preventing them from reaching the threshold for triggered activity.136 A similar strategy was developed in silico to prevent both DAD- and EAD-mediated arrhythmias by overexpressing inward rectifier K channels to stabilize a minority of myocytes (around 20%) interspersed among the arrhythmogenic myocytes. The stabilized cells were incapable of generating EAD- or DAD-mediated triggered activity on their own, and acted as a distributed electrotonic sink that prevented the bulk of unmodified arrhythmogenic myocytes in the tissue from generating triggered activity.136 This strategy has not yet been tested experimentally, however.
Relevance to supraventricular arrhythmias
The results in Table 1 are generalizable to supraventricular arrhythmias, although the equivalent ECG signatures are more hidden. As in the ventricles, supraventricular arrhythmias due to anatomic reentry, including AV nodal reentrant tachycardia, AV reentrant tachycardia using an accessory pathway, and atrial flutter, are readily treated by ablation procedures targeting the vulnerable components of the reentrant circuit (ie, the vulnerable substrate). Paroxysmal AF can also be successfully treated by isolating the anatomic structures, such as the pulmonary veins, from which the triggers inducing AF emerge. However, in persistent and permanent AF, extensive structural and electrical remodeling throughout the body of atrial myocardium markedly expand the mass of tissue contributing to generation of triggers and increased substrate vulnerability so that the success of catheter ablation decreases, although the surgical maze procedure remains effective.137 Pharmacological approaches suffer from the same limitations as in ventricular arrhythmias because the same corresponding mechanisms of R-to-T, R-from-T, and R-after-T mechanisms in ventricular myocardium are likely to apply to atrial myocardium (substituting the P and atrial repolarization waves for R and T waves, respectively).
Summary and conclusion
The main message from the data in Table 1 is that multiple mechanisms contribute to the initiation of lethal ventricular reentry in all of the clinical settings listed and have their correlates in supraventricular reentry as well. To be effective, an antiarrhythmic strategy targeting prevention of reentry by 1 mechanism must not exacerbate any of the other relevant mechanisms. That was the hard lesson learned from the CAST, SWORD, and many other antiarrhythmic trials. Only through a detailed understanding of the spectrum of potential mechanisms putting a patient at risk will it be possible to make progress in preventing lethal and nonlethal arrhythmias. Progress is being made, but the challenge expressed in the statement from the Sicilian Gambit Investigators more than 2 decades ago that “…, it is the investigators’ belief that until we are ready to contend with the diverse mechanisms for arrhythmias and the ever more diverse targets for drug action we will not be able to prevent and treat arrhythmias pharmacologically in consistently satisfactory fashion”138 still holds true today.
Funding Sources:
This study was supported by National Institutes of Health Grants R01 HL134709, R01 HL139829, R01 HL133294, R01 HL134346, P01 HL078931, and OT2OD028190; and the Burn & Allen Chair in Cardiology Research of the Cedars-Sinai Medical Center to Dr Chen. Disclosures: The authors have no conflicts of interest to disclose. This manuscript was processed by a Guest Editor.
References
- 1.Zipes DP, Wellens HJ. Sudden cardiac death. Circulation 1998;98:2334–2351. [DOI] [PubMed] [Google Scholar]
- 2.Myerburg RJ, Interian A, Mitrani RM, Kessler KM, Castellanos A. Frequency of sudden cardiac death and profiles of risk. Am J Cardiol 1997;80:10F–19F. [DOI] [PubMed] [Google Scholar]
- 3.Qu Z, Weiss JN. Mechanisms of ventricular arrhythmias: from molecular fluctuations to electrical turbulence. Annu Rev Physiol 2015;77:29–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aras KK, Kay MW, Efimov IR. Ventricular fibrillation: rotors or foci? Both! Circ Arrhythm Electrophysiol 2017;10:e006011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glass L. Multistable spatiotemporal patterns of cardiac activity. Proc Natl Acad Sci U S A 2005;102:10409–10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruberman W, Weinblatt E, Goldberg JD, Frank CW, Chaudhary BS, Shapiro S. Ventricular premature complexes and sudden death after myocardial infarction. Circulation 1981;64:297–305. [DOI] [PubMed] [Google Scholar]
- 7.Ruberman W, Weinblatt E, Goldberg JD, Frank CW, Shapiro S. Ventricular premature beats and mortality after myocardial infarction. N Engl J Med 1977;297:750–757. [DOI] [PubMed] [Google Scholar]
- 8.Bigger JT, Fleiss JL, Kleiger R, Miller JP, Rolnitzky LM. The relationships among ventricular arrhythmias, left ventricular dysfunction, and mortality in the 2 years after myocardial infarction. Circulation 1984;69:250–258. [DOI] [PubMed] [Google Scholar]
- 9.Lerma C, Gorelick A, Ghanem RN, Glass L, Huikuri HV. Patterns of ectopy leading to increased risk of fatal or near-fatal cardiac arrhythmia in patients with depressed left ventricular function after an acute myocardial infarction. Europace 2013. [DOI] [PubMed] [Google Scholar]
- 10.Smirk FH. R waves interrupting T waves. Br Heart J 1949;11:23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smirk FH, Palmer DG. A myocardial syndrome: with particular reference to the occurrence of sudden death and of premature systoles interrupting antecedent T waves. Am J Cardiol 1960;6:620–629. [DOI] [PubMed] [Google Scholar]
- 12.El-Sherif N, Myerburg RJ, Scherlag BJ, et al. Electrocardiographic antecedents of primary ventricular fibrillation. Value of the R-on-T phenomenon in myocardial infarction. Br Heart J 1976;38:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engel TR, Meister SG, Frankl WS. The “R-on-T” phenomenon: an update and critical review. Ann Intern Med 1978;88:221–225. [DOI] [PubMed] [Google Scholar]
- 14.Chou T-C, Wenzke F. The importance of R on T phenomenon. Am Heart J 1978;96:191–194. [DOI] [PubMed] [Google Scholar]
- 15.Campbell RW, Murray A, Julian DG. Ventricular arrhythmias in first 12 hours of acute myocardial infarction. Natural history study. Br Heart J 1981;46:351–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiladakis JA, Karapanos G, Davlouros P, Aggelopoulos G, Alexopoulos D, Manolis AS. Significance of R-on-T phenomenon in early ventricular tachyarrhythmia susceptibility after acute myocardial infarction in the thrombolytic era. Am J Cardiol 2000;85:289–293. [DOI] [PubMed] [Google Scholar]
- 17.Viskin S, Lesh MD, Eldar M, et al. Mode of onset of malignant ventricular arrhythmias in idiopathic ventricular fibrillation. J Cardiovasc Electrophysiol 1997;8:1115–1120. [DOI] [PubMed] [Google Scholar]
- 18.Kakishita M, Kurita T, Matsuo K, et al. Mode of onset of ventricular fibrillation in patients with Brugada syndrome detected by implantable cardioverter defibrillator therapy. J Am Coll Cardiol 2000;36:1646–1653. [DOI] [PubMed] [Google Scholar]
- 19.Drew BJ, Ackerman MJ, Funk M, et al. Prevention of torsade de pointes in hospital settings: a scientific statement from the American Heart Association and the American College of Cardiology Foundation. Circulation 2010;121:1047–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Badri M, Patel A, Patel C,et al. Mexiletine prevents recurrent torsades de pointes in acquired long QT syndrome refractory to conventional measures. JACC Clin Electrophysiol 2015;1:315–322. [DOI] [PubMed] [Google Scholar]
- 21.Ren X, Hongo RH. Polymorphic ventricular tachycardia from R-on-T pacing. J Am Coll Cardiol 2009;53. 218–218. [DOI] [PubMed] [Google Scholar]
- 22.Oupadia P, Ramaswamy K. “R-on-T” phenomenon. N Engl J Med 1998;338. 1812–1812. [DOI] [PubMed] [Google Scholar]
- 23.Andrus EC, Carter EP. with an appendix by Harold A. Wheeler PD. The refractory period of the normally-beating dog’s auricle; with a note on the occurrence of auricular fibrillation following a single stimulus. J Exp Med 1930;51:357–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wiggers CJ, Wegria R. Ventricular fibrillation due to single, localized induction and condesor shocks applied during the vulnerable phase of ventricular systole. Am J Physiol 1940;128:500. [Google Scholar]
- 25.Lown B. Electrical reversion of cardiac arrhythmias. Br Heart J 1967;29:469–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen P-S, Wolf PD, Dixon EG, et al. Mechanism of ventricular vulnerability to single premature stimuli in open chest dogs. Circ Res 1988;62:1191–1209. [DOI] [PubMed] [Google Scholar]
- 27.Gotoh M, Uchida T, Mandel WJ, Fishbein MC, Chen PS, Karagueuzian HS. Cellular graded responses and ventricular vulnerability to reentry by a premature stimulus in isolated canine ventricle. Circulation 1997;95:2141–2154. [DOI] [PubMed] [Google Scholar]
- 28.Kennedy HL, Whitlock JA, Sprague MK, Kennedy LJ, Buckingham TA, Goldberg RJ. Long-term follow-up of asymptomatic healthy subjects with frequent and complex ventricular ectopy. N Engl J Med 1985;312:193–197. [DOI] [PubMed] [Google Scholar]
- 29.Conti CR. Ventricular arrhythmias: a general cardiologist’s assessment of therapies in 2005. Clin Cardiol 2005;28:314–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marcus GM. Evaluation and management of premature ventricular complexes. Circulation 2020;141:1404–1418. [DOI] [PubMed] [Google Scholar]
- 31.Mines GR. On dynamic equilibrium in the heart. J Physiol 1913;46:349–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cranefield PF. The Conduction of the Cardiac Impulse: The Slow Response and Cardiac Arrhythmias. Mt. Kisco, NY: Futura Publishing Company; 1975. [Google Scholar]
- 33.Moe GK. Evidence for reentry as a mechanism of cardiac arrhythmias. Rev Physiol Biochem Pharmacol 1975;72:55–81. [DOI] [PubMed] [Google Scholar]
- 34.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J 2010;99:1408–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nguyen TP, Qu Z, Weiss JN. Cardiac fibrosis and arrhythmogenesis: the road to repair is paved with perils. J Mol Cell Cardiol 2014;70C:83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campos FO, Shiferaw Y, Weber dos Santos R, Plank G, Bishop MJ. Microscopic isthmuses and fibrosis within the border zone of infarcted hearts promote calcium-mediated ectopy and conduction block. Front Phys 2018;6 10.3389/fphy.2018.00057. [DOI] [Google Scholar]
- 37.Rohr S, Kucera JP, Fast VG, Kleber AG. Paradoxical improvement of impulse conduction in cardiac tissue by partial cellular uncoupling. Science 1997;275:841–844. [DOI] [PubMed] [Google Scholar]
- 38.Roelke M, Garan H, McGovern BA, Ruskin JN. Analysis of the initiation of spontaneous monomorphic ventricular tachycardia by stored intracardiac electrograms. J Am Coll Cardiol 1994;23:117–122. [DOI] [PubMed] [Google Scholar]
- 39.Krinsky VI. Spread of excitation in an inhomogeneous medium. Biofizika 1966;11:676–683.6000627 [Google Scholar]
- 40.Allessie MA, Bonke FIM, Schopman FJC. Circus movement in rabbit atrial muscle as a mechanism of tachycardia. III. The “leading circle” concept: a new model of circus movement in cardiac tissue without the involvement of an anatomical obstacle. Circ Res 1977;41:9–41. [DOI] [PubMed] [Google Scholar]
- 41.Davidenko JM, Pertsov AM, Salomonsz R, Baxter W, Jalife J. Stationary and drifting spiral waves of excitation in isolated cardiac muscle. Nature 1992;355:349–351. [DOI] [PubMed] [Google Scholar]
- 42.Gray RA, Pertsov AM, Jalife J. Spatial and temporal organization during cardiac fibrillation. Nature (Lond) 1998;392:75–78. [DOI] [PubMed] [Google Scholar]
- 43.Frazier DW, Wolf PD, Wharton JM, Tang AS, Smith WM, Ideker RE. Stimulus-induced critical point. Mechanism for electrical initiation of reentry in normal canine myocardium. J Clin Invest 1989;83:1039–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kohl P, Nesbitt AD, Cooper PJ, Lei M. Sudden cardiac death by commotio cordis: role of mechano-electric feedback. Cardiovasc Res 2001;50:280–289. [DOI] [PubMed] [Google Scholar]
- 45.Kohl P. Recognition and prevention of commotio cordis. Heart Rhythm 2005;2:902–903. 902; author reply. [DOI] [PubMed] [Google Scholar]
- 46.Efimov IR, Cheng Y, Van Wagoner DR, Mazgalev T, Tchou PJ. Virtual electrode-induced phase singularity: a basic mechanism of defibrillation failure. Circ Res 1998;82:918–925. [DOI] [PubMed] [Google Scholar]
- 47.Lin SF, Roth BJ, Wikswo JP Jr. Quatrefoil reentry in myocardium: an optical imaging study of the induction mechanism. J Cardiovasc Electrophysiol 1999;10:574–586. [DOI] [PubMed] [Google Scholar]
- 48.Rodriguez B, Li L, Eason JC, Efimov IR, Trayanova NA. Differences between left and right ventricular chamber geometry affect cardiac vulnerability to electric shocks. Circ Res 2005;97:168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scherf D. Studies on auricular tachycardia caused by aconitine administration. Proc Soc Exp Biol Med 1947;64:839–844. [DOI] [PubMed] [Google Scholar]
- 50.Miake J, Marban E, Nuss HB. Biological pacemaker created by gene transfer. Nature 2002;419:132–133. [DOI] [PubMed] [Google Scholar]
- 51.Silva J, Rudy Y. Mechanism of pacemaking in IK1-downregulated myocytes. Circ Res 2003;92:261–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lakatta EG, Maltsev VA, Vinogradova TM. A coupled system of intracellular Ca2+ clocks and surface membrane voltage clocks controls the timekeeping mechanism of the heart’s pacemaker. Circ Res 2010;106:659–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clancy CE, Rudy Y. Linking a genetic defect to its cellular phenotype in a cardiac arrhythmia. Nature 1999;400:566–569. [DOI] [PubMed] [Google Scholar]
- 54.Qu Z, Xie L-H, Olcese R, et al. Early afterdepolarizations in cardiac myocytes: beyond reduced repolarization reserve. Cardiovasc Res 2013;99:6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knollmann B, Chopra N, Hlaing T, et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 2006;116:2510–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu N, Colombi B, Memmi M, et al. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res 2006;99:292–298. [DOI] [PubMed] [Google Scholar]
- 57.Hobai IA, O’Rourke B. Enhanced Ca2+-activated Na+-Ca2+ exchange activity in canine pacing-induced heart failure. Circ Res 2000;87:690–698. [DOI] [PubMed] [Google Scholar]
- 58.Yeh YH, Wakili R, Qi XY, et al. Calcium-handling abnormalities underlying atrial arrhythmogenesis and contractile dysfunction in dogs with congestive heart failure. Circ Arrhythm Electrophysiol 2008;1:93–102. [DOI] [PubMed] [Google Scholar]
- 59.Liu MB, Ko CY, Song Z, Garfinkel A, Weiss JN, Qu Z. A dynamical threshold for cardiac delayed afterdepolarization-mediated triggered activity. Biophys J 2016;111:2523–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lou Q, Belevych AE, Radwanski PB, et al. Alternating membrane potential/calcium interplay underlies repetitive focal activity in a genetic model of calcium-dependent atrial arrhythmias. J Physiol 2015;593:1443–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Song Z, Qu Z, Karma A. Stochastic initiation and termination of calcium-mediated triggered activity in cardiac myocytes. Proc Natl Acad Sci U S A 2017;114:E270–E279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Laurita KR, Rosenbaum DS. Interdependence of modulated dispersion and tissue structure in the mechanism of unidirectional block. Circ Res 2000;87:922–928. [DOI] [PubMed] [Google Scholar]
- 63.Akar FG, Rosenbaum DS. Transmural electrophysiological heterogeneities underlying arrhythmogenesis in heart failure. Circ Res 2003;93:638–645. [DOI] [PubMed] [Google Scholar]
- 64.Qu Z, Garfinkel A, Weiss JN. Vulnerable window for conduction block in a one-dimensional cable of cardiac cells, 1: single extrasystoles. Biophys J 2006;91:793–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Qu Z, Garfinkel A, Weiss JN. Vulnerable window for conduction block in a one-dimensional cable of cardiac cells, 2: multiple extrasystoles. Biophys J 2006;91:805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qu Z, Garfinkel A, Chen PS, Weiss JN. Mechanisms of discordant alternans and induction of reentry in simulated cardiac tissue. Circulation 2000;102:1664–1670. [DOI] [PubMed] [Google Scholar]
- 67.Sato D, Xie LH, Sovari AA, et al. Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc Natl Acad Sci U S A 2009;106:2983–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pastore JM, Girouard SD, Laurita KR, Akar FG, Rosenbaum DS. Mechanism linking T-wave alternans to the genesis of cardiac fibrillation. Circulation 1999;99:1385–1394. [DOI] [PubMed] [Google Scholar]
- 69.Ziv O, Morales E, Song YK, et al. Origin of complex behaviour of spatially discordant alternans in a transgenic rabbit model of type 2 long QT syndrome. J Physiol 2009;587:466–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Haïssaguerre M, Shoda M,Jaïs P, et al. Mapping and ablation of idiopathic ventricular fibrillation. Circulation 2002;106:962–967. [DOI] [PubMed] [Google Scholar]
- 71.Haissaguerre M, Vigmond E, Stuyvers B, Hocini M, Bernus O. Ventricular arrhythmias and the His–Purkinje system. Nat Rev Cardiol 2016;13:155–166. [DOI] [PubMed] [Google Scholar]
- 72.Parameswaran R, Al-Kaisey AM, Kalman JM. Catheter ablation for atrial fibrillation: current indications and evolving technologies. Nat Rev Cardiol 2021;18:210–225. [DOI] [PubMed] [Google Scholar]
- 73.Gelzer ARM, Koller ML, Otani NF, et al. Dynamic mechanism for initiation of ventricular fibrillation in vivo. Circulation 2008;118:1123–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arevalo HJ, Vadakkumpadan F, Guallar E, et al. Arrhythmia risk stratification of patients after myocardial infarction using personalized heart models. Nat Commun 2016;7:11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zahid S, Cochet H, Boyle PM, et al. Patient-derived models link re-entrant driver localization in atrial fibrillation to fibrosis spatial pattern. Cardiovasc Res 2016;110:443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Souza JJ, Malkin RA, Ideker RE. Comparison of upper limit of vulnerability and defibrillation probability of success curves using a nonthoracotomy lead system. Circulation 1995;91:1247–1252. [DOI] [PubMed] [Google Scholar]
- 77.Chen PS, Feld GK, Kriett JM, et al. Relation between upper limit of vulnerability and defibrillation threshold in humans. Circulation 1993;88:186–192. [DOI] [PubMed] [Google Scholar]
- 78.Chen PS, Swerdlow CD, Hwang C, Karagueuzian HS. Current concepts of ventricular defibrillation. J Cardiovasc Electrophysiol 1998;9:553–562. [DOI] [PubMed] [Google Scholar]
- 79.Stevenson WG, Soejima K. Catheter ablation for ventricular tachycardia. Circulation 2007;115:2750–2760. [DOI] [PubMed] [Google Scholar]
- 80.Huang X, Kim TY, Koren G, Choi B-R, Qu Z. Spontaneous initiation of premature ventricular complexes and arrhythmias in type 2 long QT syndrome. Am J Physiol Heart Circ Physiol 2016;311:H1470–H1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang Z, Qu Z. Life and death saddles in the heart. Phys Rev E 2021;103:062406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Di Diego JM, Antzelevitch C. Pinacidil-induced electrical heterogeneity and extrasystolic activity in canine ventricular tissues. Does activation of ATP-regulated potassium current promote phase 2 reentry? Circulation 1993;88:1177–1189. [DOI] [PubMed] [Google Scholar]
- 83.Lukas A, Antzelevitch C. Phase 2 reentry as a mechanism of initiation of circus movement reentry in canine epicardium exposed to simulated ischemia. Cardiovasc Res 1996;32:593–603. [PubMed] [Google Scholar]
- 84.Zhang Z, Chen P-S,Weiss JN, Qu Z. Why is only type 1 electrocardiogram diagnostic of Brugada syndrome? Mechanistic insights from computer modeling. Circ Arrhythm Electrophysiol 2022;15:e010365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dutta S, Mincholé A, Zacur E, Quinn TA, Taggart P, Rodriguez B. Early afterdepolarizations promote transmural reentry in ischemic human ventricles with reduced repolarization reserve. Prog Biophys Mol Biol 2016;120:236–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lu W, Kim TY, Huang X, et al. Mechanisms linking T-wave alternans to spontaneous initiation of ventricular arrhythmias in rabbit models of long QT syndrome. J Physiol 2018;596:1341–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vandersickel N, de Boer TP, Vos MA, Panfilov AV. Perpetuation of torsade de pointes in heterogeneous hearts: competing foci or re-entry? J Physiol 2016;594:6865–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu MB, Vandersickel N, Panfilov AV, Qu Z. R-From-T as a common mechanism of arrhythmia initiation in long QT syndromes. Circ Arrhythm Electrophysiol 2019;12:e007571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Z, Liu MB, Huang X, Song Z, Qu Z. Mechanisms of premature ventricular complexes caused by QT prolongation. Biophys J2021;120:352–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Antzelevitch C, Brugada P, Brugada J, et al. Brugada syndrome. Circ Res 2002;91:1114–1118. [DOI] [PubMed] [Google Scholar]
- 91.Antzelevitch C, Yan G-X, Ackerman MJ, et al. J-Wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. Heart Rhythm 2016;13:e295–e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miyoshi S, Mitamura H, Fujikura K, et al. A mathematical model of phase 2 reentry: role of L-type Ca current. Am J Physiol Heart Circ Physiol 2003;284:H1285–H1294. [DOI] [PubMed] [Google Scholar]
- 93.Maoz A, Christini DJ, Krogh-Madsen T. Dependence of phase-2 reentry and repolarization dispersion on epicardial and transmural ionic heterogeneity: a simulation study. Europace 2014;16:458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Weiss JN. Arrhythmias in Brugada syndrome: defective depolarization, repolarization or both? JACC Clin Electrophysiol 2021;7:271–272. [DOI] [PubMed] [Google Scholar]
- 95.Wilde AAM, Postema PG, Di Diego JM, et al. The pathophysiological mechanism underlying Brugada syndrome: depolarization versus repolarization. J Mol Cell Cardiol 2010;49:543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nademanee K, Wilde AAM. Repolarization versus depolarization defects in Brugada syndrome. JACC Clin Electrophysiol 2017;3:364–366. [DOI] [PubMed] [Google Scholar]
- 97.Brugada P, Wellens HJJ. Early afterdepolarizations: role in conduction block, “prolonged repolarization-dependent reexcitation,” and tachyarrhythmias in the human heart. Pacing Clin Electrophysiol 1985;8:889–896. [DOI] [PubMed] [Google Scholar]
- 98.Yan G-X, Wu Y, Liu T, Wang J, Marinchak RA, Kowey PR. Phase 2 early afterdepolarization as a trigger of polymorphic ventricular tachycardia in acquired long-QT syndrome: direct evidence from intracellular recordings in the intact left ventricular wall. Circulation 2001;103:2851–2856. [DOI] [PubMed] [Google Scholar]
- 99.Yan GX, Rials SJ, Wu Y, et al. Ventricular hypertrophy amplifies transmural repolarization dispersion and induces early afterdepolarization. Am J Physiol Heart Circ Physiol 2001;281:H1968–H1975. [DOI] [PubMed] [Google Scholar]
- 100.Choi BR, Burton F, Salama G. Cytosolic Ca2+ triggers early afterdepolarizations and torsade de pointes in rabbit hearts with type 2 long QT syndrome. J Physiol 2002;543:615–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liu J, Laurita KR. The mechanism of pause-induced torsade de pointes in long QT syndrome. J Cardiovasc Electrophysiol 2005;16:981–987. [DOI] [PubMed] [Google Scholar]
- 102.Maruyama M, Lin SF, Xie Y, et al. Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long-QT syndrome. Circ Arrhythm Electrophysiol 2011;4:103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim JJ, Němec J, Li Q, Salama G. Synchronous systolic subcellular Ca2+-elevations underlie ventricular arrhythmia in drug-induced long QT type 2. Circ Arrhythm Electrophysiol 2015;8:703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kim TY, Kunitomo Y, Pfeiffer Z, et al. Complex excitation dynamics underlie polymorphic ventricular tachycardia in a transgenic rabbit model of long QT syndrome type 1. Heart Rhythm 2015;12:220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Antzelevitch C, Antzelevitch C. Tpeak-Tend interval as an index of transmural dispersion of repolarization. Eur J Clin Invest 2001;31:555–557. [DOI] [PubMed] [Google Scholar]
- 106.Gupta P, Patel C, Patel H, et al. T(p-e)/QT ratio as an index of arrhythmogenesis. J Electrocardiol 2008;41:567–574. [DOI] [PubMed] [Google Scholar]
- 107.Joyner RW, Ramza BM, Osaka T, Tan RC. Cellular mechanisms of delayed recovery of excitability in ventricular tissue. AmJPhysiol 1991;260:H225–H233. [DOI] [PubMed] [Google Scholar]
- 108.Pu J, Boyden PA. Alterations of Na1 currents in myocytes from epicardial border zone of the infarct heart: as possible ionic mechanism for reduced excitability and postrepolarization refractoriness. Circ Res 1997;81:110–119. [DOI] [PubMed] [Google Scholar]
- 109.Pandit SV, Noujaim SF, Jalife J. Postrepolarization refractoriness in acute ischemia and after antiarrhythmic drug administration. Heart Rhythm 2012;9:e13–e14. author reply e14. [DOI] [PubMed] [Google Scholar]
- 110.Coronel R, Wilms-Schopman FJ, Opthof T, van Capelle FJ, Janse MJ. Injury current and gradients of diastolic stimulation threshold, TQ potential, and extracellular potassium concentration during acute regional ischemia in the isolated perfused pig heart. Circ Res 1991;68:1241–1249. [DOI] [PubMed] [Google Scholar]
- 111.Janse MJ, van Capelle FJ, Morsink H, et al. Flow of “injury” current and patterns of excitation during early ventricular arrhythmias in acute regional myocardial ischemia in isolated porcine and canine hearts. Evidence for two different arrhythmogenic mechanisms. Circ Res 1980;47:151–165. [DOI] [PubMed] [Google Scholar]
- 112.Imanishi S, Surawicz B. Automatic activity in depolarized guinea pig ventricular myocardium. Characteristics and mechanisms. Circ Res 1976;39:751–759. [DOI] [PubMed] [Google Scholar]
- 113.Tan RC, Osaka T, Joyner RW. Experimental model of effects on normal tissue of injury current from ischemic region. Circ Res 1991;69:965–974. [DOI] [PubMed] [Google Scholar]
- 114.Harris AS, Bistem A, Russell RA, Brigham JC, Firestone JE. Excitatory factors in ventricular tachycardia resulting from myocardial ischemia. Potassium a major excitant. Science 1954;119:200–203. [DOI] [PubMed] [Google Scholar]
- 115.Campos FO, Shiferaw Y, Prassl AJ, Boyle PM, Vigmond EJ, Plank G. Stochastic spontaneous calcium release events trigger premature ventricular complexes by overcoming electrotonic load. Cardiovasc Res 2015;107:175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Colman MA. Arrhythmia mechanisms and spontaneous calcium release: bidirectional coupling between re-entrant and focal excitation. PLOS Comput Biol 2019;15:e1007260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Song Z, Ko CY, Nivala M, Weiss JN, Qu Z. Calcium-voltage coupling in the genesis of early and delayed afterdepolarizations in cardiac myocytes. Biophys J 2015;108:1908–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen W, Aistrup G, Wasserstrom JA, Shiferaw Y. A mathematical model of spontaneous calcium release in cardiac myocytes. Am J Physiol Heart Circ Physiol 2011;300:H1794–H1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Katra RP, Laurita KR. Cellular mechanism of calcium-mediated triggered activity in the heart. Circ Res 2005;96:535–542. [DOI] [PubMed] [Google Scholar]
- 120.Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res 2012;110:1454–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Liu MB, de Lange E, Garfinkel A, Weiss JN, Qu Z. Delayed afterdepolarizations generate both triggers and a vulnerable substrate promoting reentry in cardiac tissue. Heart Rhythm 2015;12:2115–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Park S-J, Zhang D, Qi Y, et al. Insights into the pathogenesis of catecholaminergic polymorphic ventricular tachycardia from engineered human heart tissue. Circulation 2019;140:390–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Caceres J, Jazayeri M, McKinnie J, et al. Sustained bundle branch reentry as a mechanism of clinical tachycardia. Circulation 1989;79:256–270. [DOI] [PubMed] [Google Scholar]
- 124.Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res 2004;95:754–763. [DOI] [PubMed] [Google Scholar]
- 125.Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology 2006;21:380–387. [DOI] [PubMed] [Google Scholar]
- 126.Echt DS, Liebson PR, Mitchell LB, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med 1991;324:781–788. [DOI] [PubMed] [Google Scholar]
- 127.Epstein AE, Hallstrom AP, Rogers WJ, et al. Mortality following ventricular arrhythmia suppression by encainide, flecainide, and moricizine after myocardial infarction: the original design concept of the Cardiac Arrhythmia Suppression Trial (CAST). JAMA 1993;270:2451–2455. [PubMed] [Google Scholar]
- 128.Pratt CM, Camm AJ, Cooper W, et al. Mortality in the Survival With ORal D-Sotalol (SWORD) trial: why did patients die? Am J Cardiol 1998;81:869–876. [DOI] [PubMed] [Google Scholar]
- 129.Waldo AL, Camm AJ, deRuyter H, et al. Survival with oral d-Sotalol in patients with left ventricular dysfunction after myocardial infarction: rationale, design, and methods (the SWORD trial). Am J Cardiol 1995;75:1023–1027. [DOI] [PubMed] [Google Scholar]
- 130.Qu Z, Karagueuzian HS, Garfinkel A, Weiss JN. Effects of Na(+) channel and cell coupling abnormalities on vulnerability to reentry: a simulation study. Am J Physiol Heart Circ Physiol 2004;286:H1310–H1321. [DOI] [PubMed] [Google Scholar]
- 131.Yousuf O, Chrispin J, Tomaselli GF, Berger RD. Clinical management and prevention of sudden cardiac death. Circ Res 2015;116:2020–2040. [DOI] [PubMed] [Google Scholar]
- 132.Qu Z, Chung D. Mechanisms and determinants of ultralong action potential duration and slow rate-dependence in cardiac myocytes. PLoS One 2012;7:e43587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Madhvani RV, Angelini M, Xie Y, et al. Targeting the late component of the cardiac L-type Ca2+ current to suppress early afterdepolarizations. J Gen Physiol 2015;145:395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Angelini M, Pezhouman A, Savalli N, et al. Suppression of ventricular arrhythmias by targeting late L-type Ca2+ current. J Gen Physiol 2021;153:e202012584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Denegri M, Bongianino R, Lodola F, et al. Single delivery of an adeno-associated viral construct to transfer the CASQ2 gene to knock-in mice affected by catecholaminergic polymorphic ventricular tachycardia is able to cure the disease from birth to advanced age. Circulation 2014;129:2673–2681. [DOI] [PubMed] [Google Scholar]
- 136.Liu MB, Priori SG, Qu Z, Weiss JN. Stabilizer cell gene therapy. Circ Arrhythm Electrophysiol 2020;13:e008420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cox JL, Ad N, Palazzo T, et al. Current status of the maze procedure for the treatment of atrial fibrillation. Semin Thorac Cardiovasc Surg 2000;12:15–19. [DOI] [PubMed] [Google Scholar]
- 138.Rosen MR. Consequences of the Sicilian Gambit. Eur Heart J 1995;16:32–36. [DOI] [PubMed] [Google Scholar]