

Abstract
Purpose:
YKT6 plays important roles in multiple intracellular vesicle trafficking events but has not been associated with Mendelian diseases.
Methods:
We report 3 unrelated individuals with rare homozygous missense variants in YKT6 who exhibited neurological disease with or without a progressive infantile liver disease. We modeled the variants in Drosophila. We generated wild-type and variant genomic rescue constructs of the fly ortholog dYkt6 and compared their ability in rescuing the loss-of-function phenotypes in mutant flies. We also generated a dYkt6KozakGAL4 allele to assess the expression pattern of dYkt6.
Results:
Two individuals are homozygous for YKT6 [NM_006555.3:c.554A>G p.(Tyr185Cys)] and exhibited normal prenatal course followed by failure to thrive, developmental delay, and progressive liver disease. Haplotype analysis identified a shared homozygous region flanking the variant, suggesting a common ancestry. The third individual is homozygous for YKT6 [NM_006555.3:c.191A>G p.(Tyr64Cys)] and exhibited neurodevelopmental disorders and optic atrophy. Fly dYkt6 is essential and is expressed in the fat body (analogous to liver) and central nervous system. Wild-type genomic rescue constructs can rescue the lethality and autophagic flux defects, whereas the variants are less efficient in rescuing the phenotypes.
Conclusion:
The YKT6 variants are partial loss-of-function alleles, and the p.(Tyr185Cys) is more severe than p.(Tyr64Cys).
Keywords: Autophagy, Drosophila, Failure to thrive, Fat body, Syrian Christians of India
Introduction
Soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs) are key proteins that function in mediating vesicle trafficking and membrane fusion by forming a 4-helix bundle with other SNAREs and bridging the vesicle and target membranes to initiate fusion.1,2 YKT6 (YKT6 v-SNARE HOMOLOG [MIM: 606209]) encodes a special SNARE protein. Unlike most of the other SNAREs, YKT6 lacks a C-terminal transmembrane domain and cycles between the cytosol and cellular membranes.3 YKT6 plays multiple functions in different membrane trafficking events, including endoplasmic reticulum to Golgi transport, early/recycling endosome to trans-Golgi network transport, constitutive secretory vesicles-plasma membrane fusion, and autophagosome-lysosome fusion.4–10
The roles of YKT6 and its orthologs in autophagy are conserved across species. In mammalian cells, YKT6 mainly localizes to autophagosomes upon autophagy induction where it forms a SNARE complex with SNAP29 and STX7 and functions in autophagosome-lysosome fusion.7,11 In Drosophila fat body cells, the dYkt6 protein mainly localizes to lysosome where it forms a SNARE complex with Syx17 and Snap29.8 Despite these differences, YKT6 is a critical SNARE that mediates autophagosome-lysosome fusion and is the only metazoan SNARE involved in this process that has a homolog in yeast.12 Additionally, YKT6 plays a role in axonal autophagosome retrograde transport.13 In human, YKT6 is ubiquitously expressed.14
Among the SNAREs that are involved in autophagosome-lysosome fusion, SNAP29 (MIM: 604202) causes autosomal recessive cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma syndrome (MIM: 609528).15–17 Other components of the autophagosome-lysosome fusion SNAREs, including YKT6, have not been previously associated with Mendelian disorders in OMIM.
Materials and Methods
Molecular genetics
For individual 1, the exome sequencing and analysis was performed at the clinical laboratory as previously described,18 and it was negative. Research reanalysis of the exome sequencing data for family 1 was performed as previously described.19,20 For individual 2, the exome sequencing was performed as part of clinical care in the Baylor Genetics lab. The analysis was performed as previously described.21–23 For individual 3, quad (both siblings and both parents) exome sequencing and analyses were performed based on previously described method.24 Sanger sequencing was performed for variant confirmation and segregation purposes.
For the haplotype analysis of individuals 1 and 2, variant calling was done with Deep-Variant (V.1.5.0), and variants from chromosome 7 (hg38;NC_000007.14) with a Genotype Quality score ≥25 were used. The Bcftools (V.1.17) RoH tool along with South Asian population allele frequency data from Genome Aggregation Database (gnomAD) v2.1.1 release (with exome sequencing analyses of 15,308 South Asian individuals) and the genetic map from the HapMap project (reformatted and hosted at https://storage.googleapis.com/broad-alkesgroup-public/Eagle/downloads/tables/genetic_map_hg38_withX.txt.gz) were used to identify the stretches of homozygosity on chromosome 7, including the region flanking the YKT6 variant of interest.25–29 The homozygous regions in the 2 affected individuals were compared and shared stretches were identified (Supplemental Table 2).
Drosophila husbandry and generation of the dYkt6KozakGAL4 allele and genomic rescue strains
The fly strains used in this study were either generated in house, obtained from the Bloomington Drosophila Stock Center, or requested from other labs. The genotypes and strain identifiers are provided in Supplemental Table 3 or described in the Materials and Methods section. All the flies were raised and maintained on standard fly food at the temperature indicated in each experiment.
The dYkt6KozakGAL4 CRISPR-Mediated Integration Cassette (CRIMIC) allele was designed and generated following the strategy described previously,30 using sgRNA1-GGCTGCCAAGACTGGGTCTGCGG, and sgRNA2-GAGAGGATTAGCGTGTGGTGGGG.
The dYkt6GR wild-type genomic rescue (GR) construct was generated by cloning a genomic region of ~5.2 kb that covers dYkt6 and parts of nearby genes into the pattB vector (Drosophila Genomics Resource Center, #1420). The DNA sequence of the construct is provided in the Supplemental Methods.
The variant GR constructs were generated by site-directed mutagenesis strategy using Q5 Hot Start High-Fidelity 2x Master Mix (NEB, #M0494S) and DpnI restriction enzyme (NEB, # R0176L). The following primers were used for mutagenesis:
For dYkt6GR-Y186C:
Forward: 5′-CAAGGCGTTCTGCAAGACGGCGAAAAAG-3′
Reverse: 5′-GCCGTCTTGCAGAACGCCTTGCTCTGC-3′
For dYkt6GR-Y65C:
Forward: 5′- CAGGATGCCTGCATGTGTCATGTCTATGTG-3′
Reverse: 5′- CATGACACATGCAGGCATCCTGTTTCACCG-3′
All the constructs were sanger verified and injected, and each was inserted into the VK33 (PBac{y[+]-attP} fVK00033) docking site using ϕC31-mediated transgenesis.31,32
Drosophila behavioral assays
The climbing assay to assess the negative geotaxis and locomotion ability of the flies was performed following previously described methods33,34 with some modifications. The flies were isolated in an empty plastic vial, allowed to rest for 20 minutes, tapped to the bottom of the vial, and allowed to climb for 15 seconds. The maximum distance from the bottom to the top of the vial was set at 18.5 cm.
For the lifespan assay, flies were raised at 25 °C, and newly eclosed male flies were collected and maintained at 29 °C (10 flies per vial). The flies were transferred to a new vial and the number of dead flies was counted every 2 days.
Generation of dYkt6 RNAi flippase-out clone
dYkt6 RNAi clones in fat body were generated by crossing UAS-dYkt6 RNAi lines to y w hsFlp, UAS-Dcr-2; pmCherry-Atg8a; Act>CD2>GAL4, UAS-nlsGFP/TM6B35 or hsFlp, UAS-Dcr-2; Act>CD2>GAL4, UAS-nlsGFP/TM6B.36,37 The CD2 cassette inserted between Actin promoter and GAL4 sequence is flanked by FRT sites. The flies were kept at 29 °C to induce spontaneous Flippase expression that removes the CD2 insertion to activate Act-GAL4 and drive the expression of dYkt6 RNAi in GFP-marked cells.
Immunofluorescence
Fly tissues were dissected in ×1 phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde. For the larval tissues, fixation was at room temperature for 20 minutes. For the adult brain, fixation was at 4 °C overnight. For antibody staining, samples were penetrated in Triton X-100 in PBS (PBST, 0.1% for larval tissues, 2% for adult brains), blocked in 5% bovine serum albumin or 5% normal goat serum, and incubated with primary antibody at 4 °C overnight (2 nights for anti-cleaved Caspase-3). Samples were washed with 0.1% PBST (3 × 10 minutes) and incubated with secondary antibody for 2 hours at room temperature and washed in 0.1% PBST (3 × 10 minutes). Larval central nervous system (CNS) and adult brain were mounted in Rapiclear (Cedarlane, #RC147001), and other larval tissues were mounted in Vectashield (Vector Labs #H1200 and #H1000). Primary antibodies are rat anti-Drosophila Elav (1:250, Developmental Studies Hybridoma Bank [DSHB], #7E8A10), mouse anti-Drosophila Repo (1:50, DSHB, #8D12), rabbit anti-Ref(2)P (1:500, Abcam, #ab178440), and rabbit anti-cleaved Caspase-3 (Asp175) (1:150, Cell Signaling Technology, #9661S). Secondary antibodies are goat anti-rat-647 (1:250, Jackson ImmunoResearch, #112–605-003), goat anti-mouse-Cy5 (1:250, Invitrogen, #A10524), and goat anti-Rabbit (1:250, conjugated to Alexa 488 or Cy3, Jackson ImmunoResearch #111–545-003 and #111–165-144, respectively). The images were obtained with confocal microscopes (Leica SP8X and Zeiss LSM 880 Airyscan) and processed using the ImageJ software.38
Immunoblotting
Western blots were performed following previously described methods39 with some modifications. The fly adult heads were homogenized on ice in radio-immunoprecipitation assay buffer (Thermo Fisher, # 89900) supplemented with ethylenediaminetetraacetic acid and protease inhibitor cocktail (GenDEPOT, # P3100–001) at 50 μl per 5 heads. Isolated lysates were heated in an appropriate volume of Laemmli buffer (Bio-RAD, #1610747), loaded onto gels (Bio-RAD, # 456–1094), separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and transferred onto polyvinylidene difluoride membranes (Millipore). Membranes were incubated with primary and secondary antibodies, and signal was revealed using Supersignal chemiluminescent substrate (Thermo Fisher, # 34580). Primary antibodies are rabbit anti-Ref(2)P (1:500, Abcam, #ab178440), rabbit anti-Drosophila Atg8 (1:1000, gift from Dr. Linda Partridge), and mouse anti-alpha-Tubulin (1:5000, DSHB, #AA4.3). Horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, # 111–035-144 and # 115–035-146) were used at 1:5000.
Results
Individuals with homozygous variants in YKT6 exhibit neurological disorders with or without hepatic issues
Here, we report 3 unrelated individuals with homozygous rare missense variants in YKT6 who exhibited overlapping neurological symptoms and, in 2 infants, progressive cholestatic liver disease evolving to micronodular cirrhosis with development of hepatocellular carcinoma in 1. The pedigrees of the 3 families are shown in Figure 1A.
Figure 1. Three affected individuals have biallelic variants in YKT6.
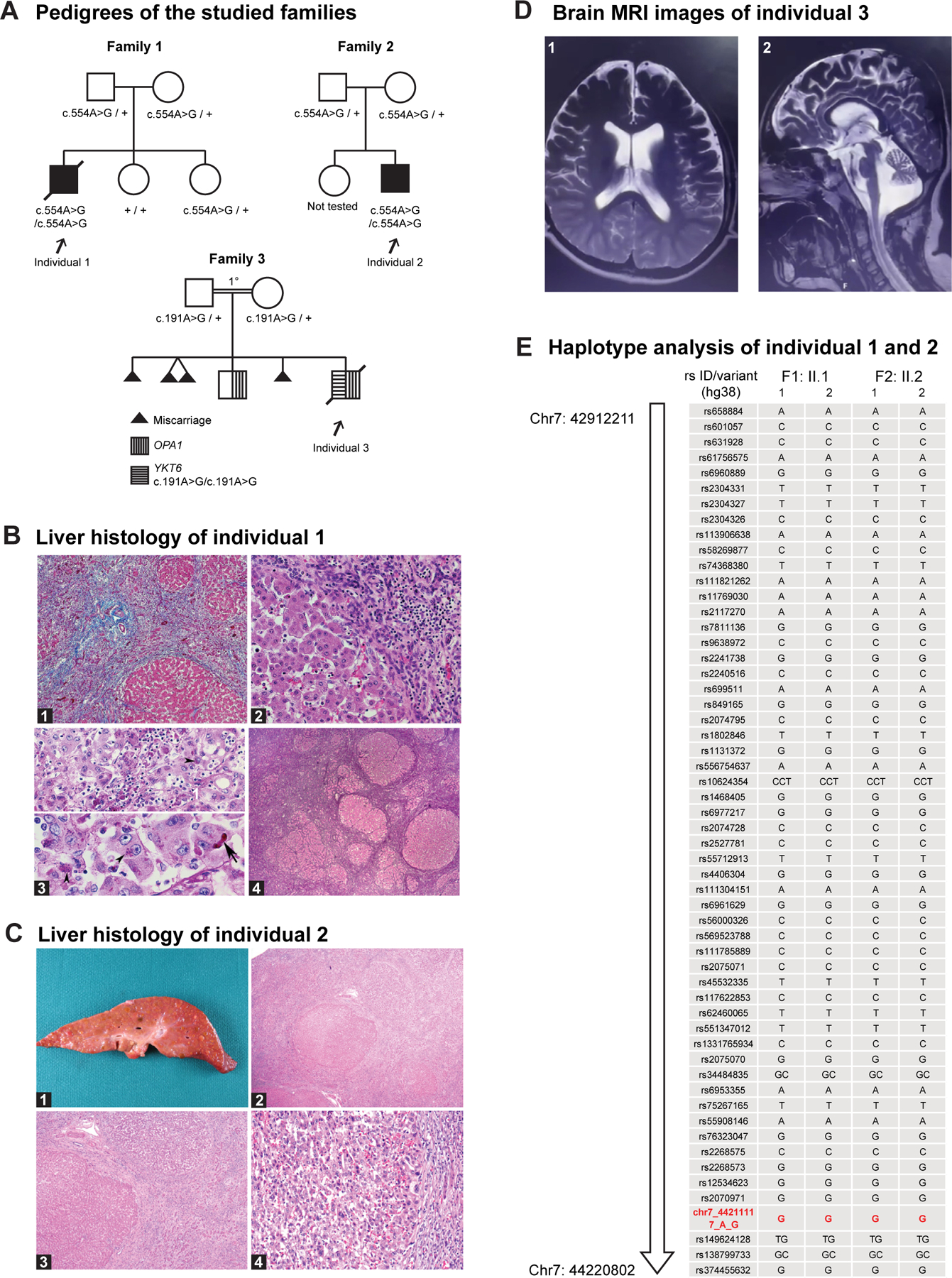
A. Pedigrees of the studied families. The individuals with homozygous YKT6 variants are affected. Individual 1 and 2 have the c.554A>G p.(Tyr185Cys) variant; individual 3 has the c.191A>G p.(Tyr64Cys) variant. The parents in family 3 are first cousins. B. Liver histology of individual 1. B1. Liver parenchyma shows massive swaths of collapsed parenchyma in which regenerative tubules are intermixed with a mononuclear cell infiltrate and nodules of surviving hepatocytes. H&E stain ×50. B2. Margin of large residual nodule along the zone of collapse shows preserved hepatocytes in which reactive or regressive features are rare. Interface inflammatory activity is lacking. H&E stain ×200. B3. Upper panel: Progressive local loss of hepatocytes within parenchymal nodules is associated with mononuclear inflammatory infiltrate, prominent pigment granules in the cytoplasm of regressing hepatocytes (arrowhead), and aggregates of pigment-laden Kupffer cells. The pigment is interpreted as ceroid. PAS diastase stain ×400. Lower panel: reactive enlarged hepatocytes have prominent nucleoli, occasional canalicular bile plugs (arrow), and focal aggregates of ceroid pigment (arrowheads). PAS diastase stain ×400. B4. Generalized zones of parenchymal collapse and nodular regeneration of residual parenchyma, features of early cirrhosis, coexist with signs of ongoing hepatocyte degeneration and loss. Reticulin stain ×25. C. Liver histology of individual 2. C1. Liver explant 203 g (expected for age: 288 g) cut surface with diffuse, variable sized nodularity ranging in size from less than 0.1 to 1.5 cm. C2. Liver parenchyma with nodules of hepatocytes surrounded by thick fibrous bands without central veins. C3. Focus of hepatocellular carcinoma showing perinodular sclerotic rim H&E stain ×200. C4. Numerous diffuse regenerative and dysplastic nodules. H&E stain ×40. D. Brain MRI of individual 3. D1. Axial T2 MRI image showing enlarged lateral ventricles and extraaxial space increase in the frontotemporal regions. D2. Midsagittal T2 MRI image showing cerebellar atrophy and extraaxial space increase in the frontal regions. E. Analysis of the SNPs on chromosome 7 revealed a shared contiguous stretch of homozygosity (Chr7:42912211–44220802, hg38, 1.3Mb; SNPs n = 100) including YKT6, in the 2 affected individuals from the unrelated families 1 and 2. For each individual, F represents the family, and the 2 alleles are represented as 1 and 2. The YKT6 variant seen in the 2 families is shown in red (Chr7:44211117A>G, hg38).
Individual 1 was born at term with a birth weight of 3401 g (40th percentile) and was without medical concerns until 4 months of age when failure to thrive became apparent. Growth failure persisted despite oral feeding supplementation, with weight z-score −3.3 at 7 months of age. At 6 months, laboratory studies demonstrated elevated alkaline phosphatase >2330 U/L (normal 45–117 U/L), total bilirubin 42.76 μmol/L (2.5 mg/dL, normal <1.0 mg/dL), Aspartate aminotransferase (AST) 285 U/L (normal 15–37 U/L), and alanine aminotransferase (ALT) 61 U/L (normal 16–61 U/L). Conjugated bilirubin was elevated since birth DOL (day of life) 3: 11.97 μmol/L (0.7 mg/dL), DOL 4: 23.95 μmol/L (1.4 mg/dL, newborn range: 0–0.6 mg/dL), 2 months of age: 10.26 μmol/L (0.6 mg/dL), 8 months of age: 11.97 μmol/L (0.7 mg/dL, range 0.1–0.5 mg/dL). Gamma-glutamyltransferase (GGT) was normal at 8 months of age: 53 U/L (normal 15–85 U/L). At 7 months, a liver ultrasound demonstrated coarse echotexture of the liver parenchyma and high resistance flow in the hepatic artery, suggestive of parenchymal liver disease. He was delayed in achieving his gross motor milestones, first sitting independently at 8.5 months. He stopped breathing and died at 9 months of age while enroute to an urgent care facility for symptoms including fussiness and dark urine. An autopsy showed micronodular cirrhosis, ascites, mild splenomegaly, and an atrophic thymus. As shown in Figure 1B, the paraffin sections of formalin fixed liver biopsy sample showed massive swaths of collapsed parenchyma in which regenerative tubules were intermixed with a mononuclear cell infiltrate and nodules of surviving hepatocytes. Progressive local loss of hepatocytes within parenchymal nodules was associated with mononuclear inflammatory infiltrate, prominent ceroid pigment granules in the cytoplasm of regressing hepatocytes and aggregates of pigment-laden Kupffer cells. Generalized zones of parenchymal collapse and nodular regeneration of residual parenchyma, which are features of early cirrhosis, coexisted with signs of ongoing hepatocyte degeneration and loss. A small subcapsular, galeal tissue hemorrhage over the right parietal bone was seen, likely due to a clotting factor deficiency secondary to liver dysfunction. There was no family history of liver disease, autoimmune disease, or other genetic disorders. Trio exome sequencing identified a rare homozygous missense variant c.554A>G p.(Tyr185Cys) in YKT6 in the proband. This variant is absent in gnomAD (v2.1.1) and ExAC,25,40,41 TOPMED,42 greater Middle East variome,43 and IndiGenomes population databases44 and seen at a very low allele frequency in heterozygous individuals in gnomAD (v4.0.0; allele frequency 0.000004105).41
Individual 2 was born at 36.5 weeks of gestation with a birth weight 2500 g (4th percentile). Development was unremarkable at 2 and 4 months, but weight gain was poor, with a weight z-score of −2.2 at 4 months of age. Following a mild acute respiratory illness (later diagnosed as cytomegalovirus) at 5 months of age, he was admitted to the hospital for new-onset seizures and was found to have a severe coagulopathy (international normalized ratio of 6) with right parietal lobe intraparenchymal hemorrhage. Vitamin K infusion significantly improved the coagulopathy, indicating that vitamin K malabsorption associated with chronic cholestasis likely played a major role in the severity of the coagulopathy. AST, ALT, and conjugated bilirubin were elevated, but GGT was mostly normal (<100 U/L) on multiple rechecks after hospital admission. Abdominal ultrasound revealed multiple, small lesions throughout the liver, and serum alpha-fetoprotein level was 22.8 μg/L (22,800 ng/mL, normal up to 12 ng/mL). Targeted liver biopsy revealed micronodular cirrhosis, and lesional tissue was consistent with a regenerative nodule. He was subsequently listed for liver transplant because of decompensated cirrhosis. Serum alpha-fetoprotein levels remained elevated, and a magnetic resonance imaging (MRI) performed at 10 months of age revealed multiple regenerative nodules and a single sub-centimeter suspicious lesion with arterial phase hyperenhancement in the right hepatic lobe worrisome for hepatocellular carcinoma. He underwent liver transplant at 1 year of age, and the explanted liver demonstrated numerous, diffuse regenerative and dysplastic nodules and numerous foci of hepatocellular carcinoma (Figure 1C), consistent with cirrhotomimetic hepatocellular carcinoma, which has not been previously described in a pediatric patient.45 The child has had an unremarkable post-transplant course and has not had cancer recurrence or additional seizures at 5.5 years of age but has significant verbal and motor developmental delays. Neurology evaluation at 4.5 years of age noted mild spastic left hemiparesis, excessive gag reflex, photophobia, probable phonophobia, and stereotypies with gratification phenomenon. He needed assistance in eating with utensils, could walk fast but not run, and was able to speak a few phrases but otherwise mostly single words and gibberish speech. He is socially interactive and makes good eye contact and attends a school for children with special needs. Trio exome sequencing identified a homozygous c.554A>G p.(Tyr185Cys) variant in YKT6. There is no known parental consanguinity or family history of liver or neurodevelopmental disease.
Individual 3 was previously included in a cohort of individuals with neurological manifestations (Index BAB5177).24 His parents are first cousins from Turkey, and he has a sibling (BAB5176) who also has neurological issues. BAB5177 was mildly delayed at 8 months based on the Denver Developmental Screening Test.46 He showed developmental regression and died at the age of 15 years because of lung infection. He had severe neurodevelopmental delay and was nonverbal and non-ambulatory. He had cerebellar and brainstem atrophy (Figure 1D). There was no record of liver disease. Exome sequencing previously identified a likely pathogenic homozygous variant in OPA1 (NM_130832.3:c.814C>T p.(Arg272Trp)) in this child and his similarly affected sibling. They both exhibited optic atrophy and neurodevelopmental delay. BAB5177 is also homozygous for a missense variant in YKT6 (NM_006555.3:c.191A>G p.(Tyr64Cys)) and had more severe developmental delay and hypotonia than his sibling, who is heterozygous for the YKT6 variant. This YKT6 variant is observed in gnomAD (v4.0.0)25,41 in heterozygous individuals at a frequency of 6.08e–4. The genetic and clinical features are summarized in Table 1 and detailed clinical reports of the affected individuals are provided in the Case reports in Supplemental Materials.
Table 1.
Genetic and clinical features of the individuals with homozygous variants of YKT6
| Variant Details | Individual 1 | Individual 2 | Individual 3 |
|---|---|---|---|
|
YKT6 variant (NM_006555.3) |
c.554A>G p.(Tyr185Cys) | c.191A>G p.(Tyr64Cys) | |
| CADD | 33 | 31 | |
| M-CAP | 0.8133, damaging | 0.7912, damaging | |
| REVEL | 0.643 | 0.749 | |
| PolyPhen2 hDiv (rare allele) |
0.912, probably damaging | 0.912, probably damaging | |
| PolyPhen2 hVar (Mendelian Disease) |
0.9296, probably damaging | 0.9296, probably damaging | |
| MutationTaster | 0.8100, disease causing | 0.8100, Disease causing | |
| Count in gnomAD (v4.0.0) | 6/1,461,480 alleles, 0 homozygous | 982/1,613,908 alleles, 0 homozygous | |
| Clinical Features | Individual 1 | Individual 2 | Individual 3 |
|
| |||
| Sex | male | male | male |
| Onset Age | failure to thrive onset 4 months old | liver disease diagnosed 5 months old preceded by failure to thrive | onset 6 months old |
| Current Age | 9 months old passed away | 5.5 years old | 15 years 4 month old passed away |
| Growth and Development | unremarkable neonatal period, normal development to 4 months of age followed by failure to thrive | speech delay, gross and fine motor delay, mild spastic diplegia following intracranial hemorrhage | severe neurodevelopmental delay (no speech, no walking) |
| Seizure | N/A | new-onset seizure at 5 months old associated with intracranial hemorrhage and coagulopathy, no subsequent seizures (off antiepileptic medications) | staring spells with normal EEG |
| Structural brain abnormality/brain pathology | autopsy: minimal thin subcapsular, galeal tissue hemorrhage over the right parietal bone | acute right frontoparietal hemorrhage | cerebellar and brainstem atrophy |
| Liver disorder | elevated liver enzymes. At 6 months: total bilirubin 42.76 μmol/L (2.5 mg/dL), alkaline phosphatase >2330 U/L, AST/ALT 285/61). At 8 months (total bilirubin 30.79 μmol/L (1.8 mg/dL), alkaline phosphatase >2330 U/L, AST/ALT 171/50) | normal GGT (<100 U/L), cholestasis with severe fat soluble vitamin deficiencies and coagulopathy, micronodular cirrhosis, diffuse cirrhotomimetic, hepatocellular carcinoma | no record of liver symptoms, liver enzymes normal |
| Others | hypothyroidism, normocytic anemia | liver transplantation, explanted liver with numerous regenerative, dysplastic, and hepatocellular carcinoma nodules | hypotonia, optic atrophy, sensorineural hearing loss, high palate, claw hand, syndactyly, motor neuropathy |
| Ancestry | Indian, Syrian Christian from Kerala | Turkish | |
ALT, alanine transaminase; AST, Aspartate transaminase; CADD, Combined Annotation Dependent Depletion; EEG, electroencephalogram; GGT, gamma-glutamyltransferase; N/A, not available.
The pLI score of YKT6 based on gnomAD (v4.0.0)25,41 is 0.46 (o/e = 0.39), and there are several splice, stop-gain, or frameshift alleles present in the reference population, but none of these individuals are homozygous. The YKT6 missense variants identified from the affected individuals have high Combined Annotation Dependent Depletion scores47 and are predicted to be deleterious based on multiple pathogenicity predictions48–52 (Table 1).
Haplotype analysis indicates that the YKT6 p.(Tyr185Cys) variant seen in individuals 1 and 2 likely originated from a common ancestor
Both family 1 (Jacobite sub-sect) and family 2 (Marthoma sub-sect) are of Syrian Christian ancestry originating from the state of Kerala in Southern India. The parents, although unrelated, carry the same heterozygous YKT6 variant c.554A>G p.(Tyr185Cys). Haplotype analysis using single-nucleotide variants from the exome sequencing data, in a region on chromosome 7 flanking YKT6, revealed a stretch of homozygous shared variants spanning 1.3 Mb from Chr7:42912211–44220802 (hg38) in the 2 affected unrelated individuals, indicating a common ancestor (Figure 1E, Supplemental Table 2). The data imply a founder variant within the Syrian Christians who later split into multiple sub-sects, including the Marthoma Syrian Christian Church and the Jacobite Syrian Orthodox Church53 (for more information about the history of the Syrian Christians, see Web Resources).
The fly ortholog dYkt6 is essential
To functionally assess the 2 YKT6 missense variant alleles, we performed experiments in Drosophila. The sole fly ortholog of human YKT6 is Drosophila Ykt6 (dYkt6) with a high Drosophila RNAi Screening Center Integrative Ortholog Prediction Tool (DIOPT) score of 16 out of 18 (DIOPT version 9.0).54 The encoded proteins are composed of conserved domains, sharing 61% identity and 78% similarity. The Tyr64 and Tyr185 residues localize to the Longin domain and Synaptobrevin domain (also known as SNARE core domain) of human YKT6, respectively (Figure 2A), and both amino acid residues are conserved in dYKT6 (Figure 2B). Multiple fly strains of dYKT6 are available including several loss-of-function alleles (Figure 2C). dYkt6[G808] carries a P-element insertion in the 5’UTR region of dYkt6 and is lethal at the L1 stage.55 dYkt6L162Q carries a missense allele affecting a residue in the SNARE core domain and causes pupal lethality. It was isolated in a forward genetic screen aimed to identify genes required in the nervous system.56 We also generated a CRIMIC allele dYkt6KozakGAL4 by replacing the entire coding sequence of dYkt6 with a Kozak-GAL4 cassette and a dominant marker,30 creating a null allele that allows assessment of complete loss-of-function phenotypes, as well as the gene expression pattern in combination with the GAL4/UAS system (Figure 2C). dYkt6KozakGAL4 causes lethality at the L1 stage. Hence, the allelic series from most to least severe are: dYkt6KozakGAL4 ≥ dYkt6[G808] > dYkt6L162Q. Note that dYkt6 is on the X chromosome and lethality of the hemizygotes/homozygotes/transheterozygotes of the 3 alleles can be fully rescued by introducing a ~73 kb P(acman) genomic rescue construct (Dp(1;3)DC495,PBac{DC495}VK00033, abbreviated as Dp)57 that covers dYkt6 and several nearby genes or partially rescued by introducing a smaller 5 kb genomic rescue construct (dYkt6GR, abbreviated as GR) that only covers dYkt6 (Figure 2D). These data show that dYkt6 is an essential gene required for normal development and that the lethality caused by the dYkt6 alleles are, indeed, due to the specific loss of dYkt6.
Figure 2. YKT6 is evolutionally conserved and the fly ortholog is dYkt6.
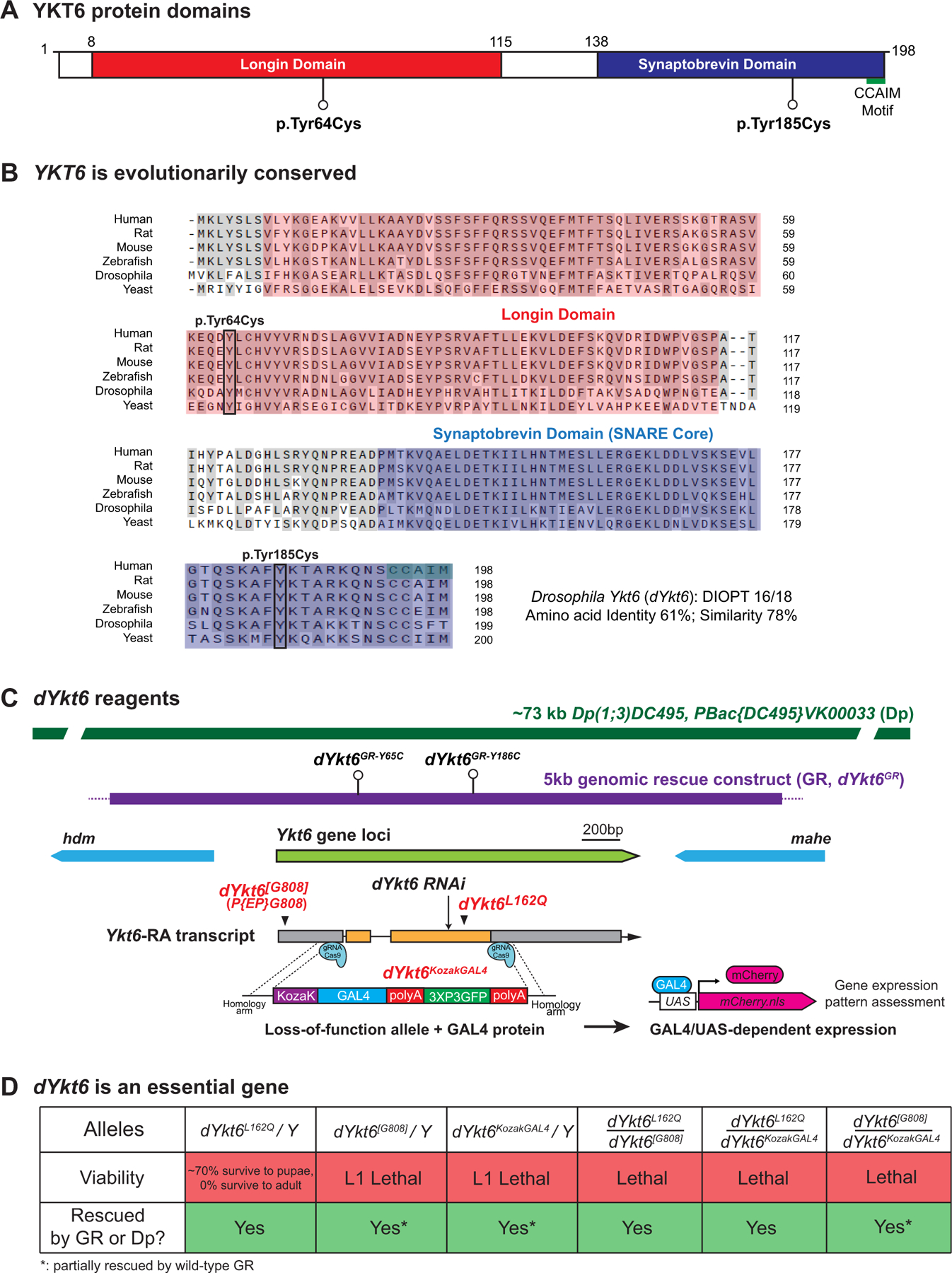
A. Schematic of human YKT6 protein domains and the position of the variants identified from the affected individuals. Domain annotation is based on the PROSITE database. B. Alignment of YKT6 and the homologous proteins. The Longin domain is in red, the Synaptobrevin domain is in blue, and the CCAIM motif is marked in green. The 2 variants are marked with boxes. The variants affect the residues with conserved amino acid across species. The following isoforms were used for alignment: NP_006546.1 (Human), NP_113880.2 (Rat), NP_062635.2 (Mouse), NP_957386.1 (Zebrafish), NP_572423.1 (Drosophila), and NP_012725.1 (Baker’s yeast). C. Schematic of fly dYkt6 genomic span, transcript, and the reagents, including a P-element insertion allele dYkt6[G808], a point mutation allele dYkt6L162Q, a CRIMIC allele dYkt6KozakGAL4, a 73 kb duplication (Dp) construct, a 5 kb genomic rescue (GR) construct (dYkt6GR), and 2 GR constructs that carry the corresponding variants (dYkt6GR-Y65C and dYkt6GR-Y186C). D. The flies with hemizygous/trans-heterozygous dYkt6 alleles are lethal. The lethality can be rescued by either the Dp or the wild-type GR constructs.
dYkt6 is expressed in the fat body and CNS, analogous to the vertebrate liver and CNS
Because the affected individuals present with hepatic and neurologic defects, we explored the expression pattern of dYkt6 in the fat body and central nervous system. The fat body in flies is analogous to the human liver and has been used to study hepatic diseases.58–60 We crossed dYkt6KozakGAL4 flies with UAS-mCD8::RFP (membrane-bound RFP) flies to label the dYkt6-expressing cells in heterozygous female progeny (dYkt6KozakGAL4/+;; UAS-mCD8::RFP/+) because we have previously shown that this approach reflects the expression pattern of genes.61–63 As shown in Figure 3A, dYkt6 is expressed in all the fat body cells of 3rd instar larvae (left panel). In the larval CNS, we observe expression in the mushroom bodies, the optic lobes, and the neuropils of the central brain and ventral nerve cord (Figure 3A, middle panel). In the adult brain, the mCD8::RFP signal is detected in the mushroom bodies, optic lobes, and antennal lobes. We also observed punctate RFP-positive structures, which may correspond to glia cells (Figure 3A, right panel). To identify the cell types that express dYkt6 in the CNS, we crossed dYkt6KozakGAL4 flies with UAS-mCherry.nls (nuclear-localized mCherry) flies and stained the brain tissues of heterozygous female progeny (dYkt6KozakGAL4/+;; UAS-mCherry.nls/+) with the nuclear pan-neuronal marker Elav or glial marker Repo. The mCherry signals partially overlap with Elav, as well as Repo (Figure 3B), showing that dYkt6 is expressed in the neurons and glia in both the larval CNS and adult brain.
Figure 3. dYkt6 is expressed in fly fat body and CNS, analogs of human liver and CNS.
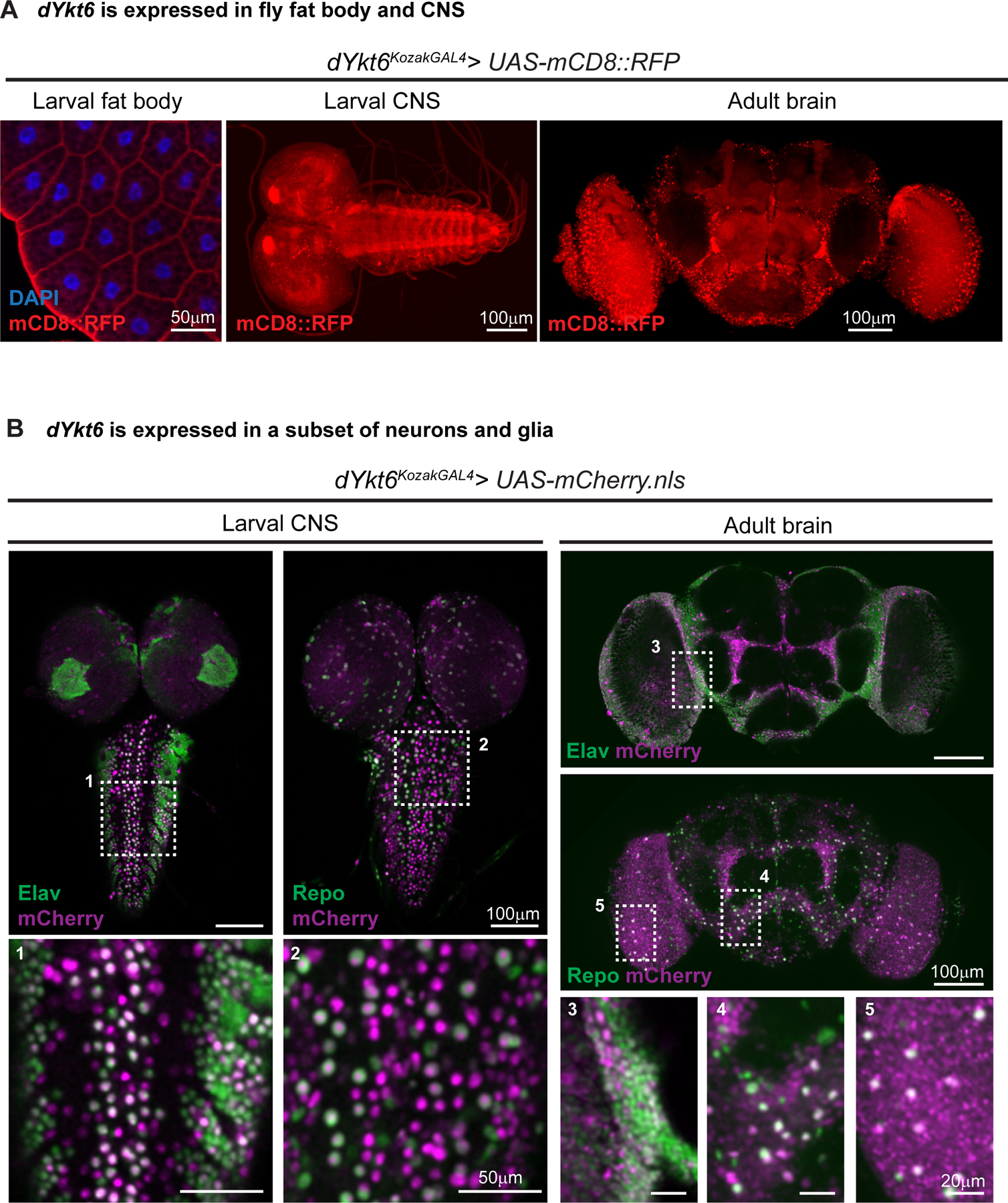
A. The L3 fat body, larval CNS and adult brain of the dYkt6KozakGAL4 > UAS-CD8::RFP (membrane-targeted RFP labeling the dYkt6 expressing cells in red) flies showing that dYkt6 is expressed in both tissues. Nuclei are labeled by DAPI (blue). Scale bars, 50 μm in the fat body image, 100 μm in the CNS images. B. The larval CNS and adult brain of the dYkt6KozakGAL4 > UAS-mCherry.nls (nuclear-localized mCherry labeling the dYkt6 expressing cells in magenta) flies documenting the expression pattern of dYkt6 in the CNS. The tissues are costained with the pan-neuronal marker Elav or panglia marker Repo (green). Higher magnification images of the regions indicated by dashed rectangles (1–5) are shown. Several z-stack sections were processed. Scale bars, 50 μm in the magnified larval CNS images (1 and 2), 20 μm in the magnified adult brain images (3, 4, and 5), and 100 μm in other images.
The dYkt6 variants are less efficient in rescuing loss-of-function phenotypes in mutant flies, and the rescue ability suggests that the p.(Tyr185Cys) variant is a more severe hypomorph than the p.(Tyr64Cys) variant
Given the conservation of the affected amino acids, we induced the missense variant p.(Tyr186Cys) in the 5kb dYkt6GR construct and generated the dYkt6GR-Y186C transgenic flies (Figure 2C) to model the human YKT6 p.(Tyr185Cys) variant. As shown in Figure 4A, heterozygous female flies carrying the dYkt6 mutant alleles were crossed with male transgenic flies that carry wild-type or variant GR constructs. The wild-type dYkt6GR rescues the lethality associated with the dYkt6L162Q allele at ~90% of the expected frequency. In contrast, dYkt6GR-Y186C rescues the dYkt6L162Q allele at ~40% (Figure 4B, left panel). Moreover, the dYkt6L162Q flies rescued by dYkt6GR-Y186C are not healthy and exhibit locomotor defects (Figure 4C), as well as severely reduced lifespan (Supplemental Figure 1A), compared with the flies rescued by the wild-type dYkt6GR. Hence, these data indicate that the corresponding human YKT6 p.(Tyr185Cys) is a partial loss-of-function variant.
Figure 4. dYKT6GR-Y186C is a more severe hypomorphic allele than dYKT6GR-Y65C.
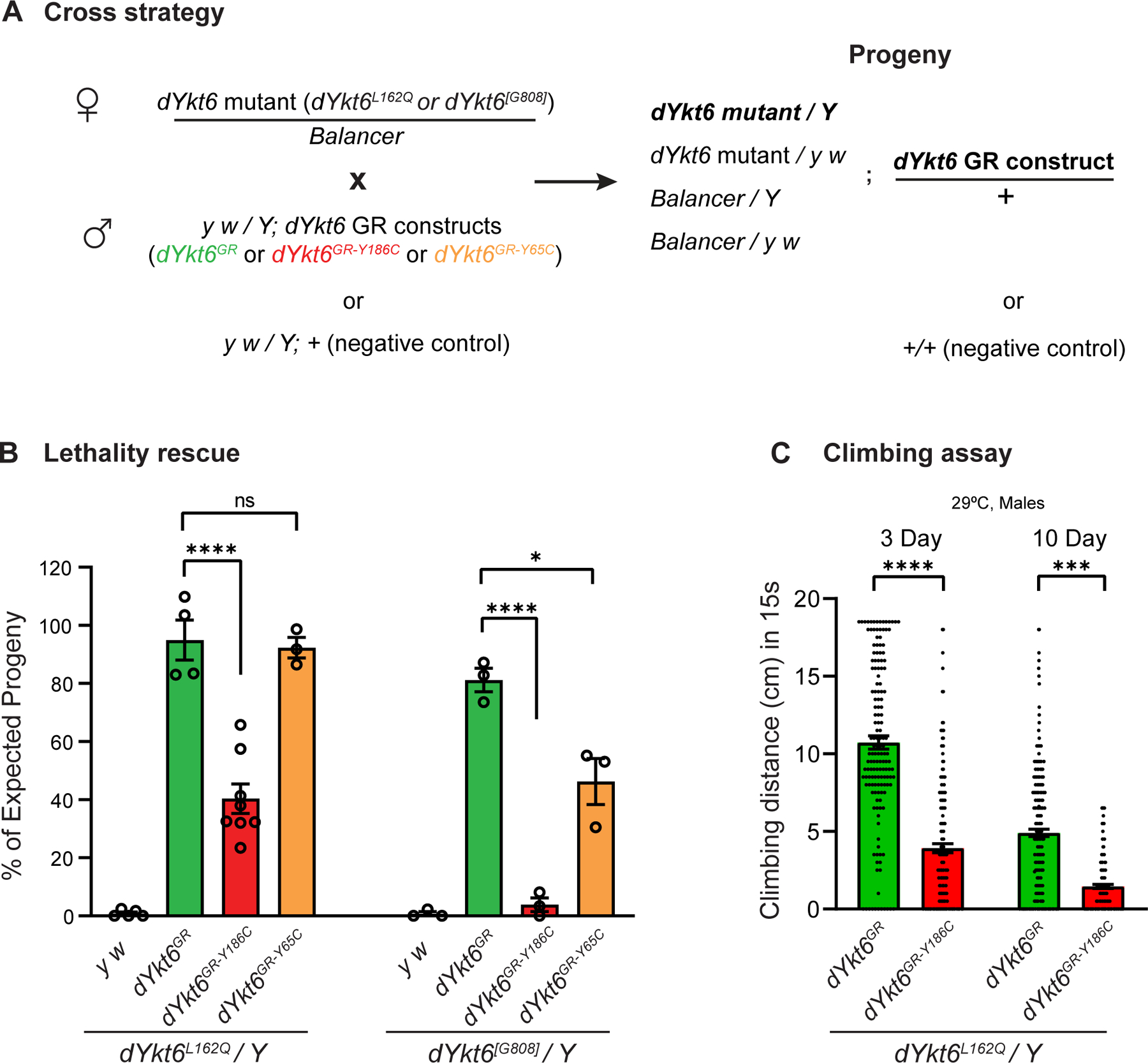
A. The cross strategy of the lethality rescue experiments. Heterozygous female flies carrying dYkt6 mutant alleles were crossed with male transgenic flies with wild-type or variant GR constructs, or with the y w/Y flies (as a negative control). Based on Mendelian ratio, the number of the dYkt6 mutant hemizygotes with GR constructs should be one-quarter of the total number of progeny flies. B. Graph showing the observed/expected percentage of the progeny flies. The dYkt6GR-Y186C construct is significantly less efficient than the wilt-type dYkt6GR in rescuing the lethality caused by the dYkt6L162Q (left panel) or the dYkt6[G808] (right panel) allele. The dYkt6GR-Y65C construct can rescue the lethality caused by the dYkt6L162Q allele (left panel) but only partially rescue the lethality caused by the dYkt6[G808] allele (right panel). The flies were raised at 25 °C. Each dot in the graph represents 1 independent cross. One-way ANOVA with Tukey’s multiple comparisons test, *P < .05, ****P < .0001, mean ± SEM. C. Climbing assay of dYkt6L162Q mutant flies rescued by dYkt6GR or dYkt6GR-Y186C. The climbing ability of the mutant flies rescued by dYkt6GR-Y186C is significantly poorer than the ones rescued by wild-type dYkt6GR. The flies were kept at 29 °C and were tested 3 and 10 days after eclosion. Each dot represents 1 tested fly. Unpaired t test, ***P < .001, ****P < .0001, mean ± SEM.
To assess the function of the p.(Tyr64Cys) variant, we created dYkt6GR-Y65C transgenic flies and tested the variant in the dYkt6L162Q mutant background. In contrast to dYkt6GR-Y186C, the dYkt6GR-Y65C rescued the lethality, locomotion, and lifespan of the dYkt6L162Q mutants and exhibited no discernible differences from the wild-type dYkt6GR in these assays (Figure 4B left panel, Supplemental Figure 1A and B). We therefore tested the GR constructs in more severe dYkt6 mutants. The dYkt6GR-Y186C barely rescues the lethal phenotype (<5%) associated with dYkt6[G808] whereas the dYkt6GR-Y65C partially rescues the lethality of dYkt6[G808] (~40%) when compared with the wild-type dYkt6GR (~80%) (Figure 4B, right panel). These data indicate that both variants affect the function of dYkt6 and that the corresponding human YKT6 p.(Tyr185Cys) variant is a more severe hypomorph than the p.(Tyr64Cys) variant.
Previous studies have shown that YKT6 and its orthologs play important roles in autophagic flux.7,64 In fly fat body cells, loss of dYkt6 leads to accumulation of Atg8a and Ref(2)P.8,65 Atg8a is the fly homolog of mammalian ATG8/LC3, which is a marker for autophagosome formation. Ref(2)P is the fly homolog of mammalian SQSTM1/p62, an autophagic cargo adaptor that binds other target cargos for autophagosomal degradation, and it is typically accumulated when the autophagic flux is impaired.65,66 RNAi-mediated dYkt6 knockdown in the fat body cell clones (see Methods) causes accumulation of mCherry-Atg8a+ vesicular structures and Ref(2)P+ puncta in the cells expressing the dYkt6 RNAi (Supplemental Figure 2). These results confirm that loss of dYkt6 affects autophagic flux in the fat body cells and provides an assay to test the variants.
To assess the functional impact of the variants, we compared the ability of the wild-type 5 kb GR construct versus the variant constructs in rescuing the Ref(2)P accumulation phenotype of the dYkt6 mutant flies. High levels of Ref(2)P are observed in the fat body cells of dYkt6L162Q mutant flies upon starvation-induced autophagy (Supplemental Figure 2B). The dYkt6L162Q mutant flies rescued by the wild-type GR construct do not cause Ref(2)P accumulation. However, in the presence of dYkt6GR-Y186C, there is an obvious accumulation of Ref(2)P. Moreover, elevated levels of Ref(2)P are also detected in adult head samples of dYkt6GR-Y186C based on immunoblotting analysis (approximately 1.25-fold change compared with wild-type dYkt6GR, Figure 5B), suggesting that dYkt6GR-Y186C is a loss-of-function variant that causes phenotypes both in fat body and brain.
Figure 5. The YKT6 variants identified from the affected individuals are associated with autophagic flux defects.
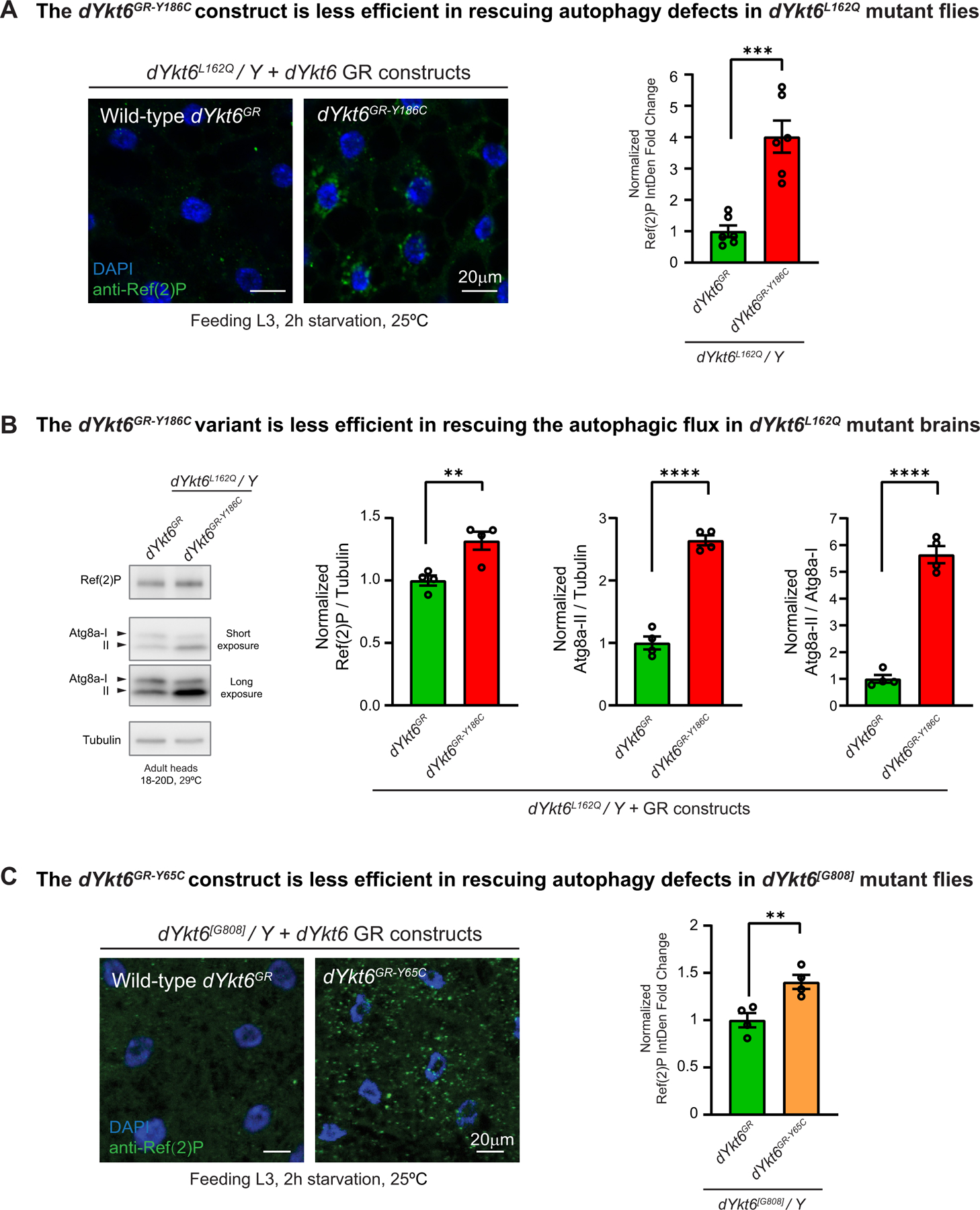
A. Fat body cells of the starved L3 larvae (feeding stage) were stained with the autophagic cargo adaptor Ref(2)P (green). The dYkt6L162Q mutant hemizygotes with wild-type dYkt6GR reduce the Ref(2)P level, whereas the ones with dYkt6GR-Y186C still show Ref(2)P accumulation. Nuclei are labeled by DAPI (blue). Scale bars, 20 μm. For the quantification, each dot represents 1 image as shown on the left. The average integrated density was calculated for 5–7 cells randomly selected in each image. At least 3 animals were dissected for imaging for each genotype. Unpaired t test, ***P < .001, mean ± SEM. B. Immunoblots of anti-Ref(2)P and anti-Atg8a of protein lysates from adult heads with quantification of Ref(2)P/Tubulin ratio, Atg8a-II/Tubulin ratio, and Atg8a-II/Atg8a-I ratio. Different parts of the blotting membranes were incubated with Ref(2)P or Atg8a antibodies. Subsequently, the membranes were stripped and incubated with Tubulin antibody. Low-exposure images were used for quantification. The eclosed flies were kept at 29 °C, and the heads were collected 18 to 20 days after eclosion. Each dot represents 1 biologically independent samples. Unpaired t test, **P < .01, ****P < .0001, mean ± SEM. C. Fat body cells of the starved L3 larvae (feeding stage) were stained with Ref(2)P (green). The dYkt6[G808] mutant hemizygotes with wild-type dYkt6GR reduce the Ref(2)P level, whereas the ones with dYkt6GR-Y65C cannot fully rescue the Ref(2)P accumulation phenotype. Nuclei are labeled by DAPI (blue). Scale bars, 20 μm. For the quantification, each dot represents 1 image. The average integrated density was calculated for 5 to 7 cells randomly selected in each image. At least 3 animals were dissected for imaging for each genotype. Unpaired t test, **P < .01, mean ± SEM.
The accumulation of Ref(2)P may result from impairment of any step of the autophagic flux, including the formation of autophagosome, the fusion of autophagosome and lysosome, and the degradation of autolysosome contents.67 We hence analyzed the immunoblotting of Atg8a and its conversion (Atg8a-I to Atg8a-II), which is a reliable indicator of autophagy initiation.68 Upon autophagy induction, the cytosolic Atg8a-I undergoes lipidation and is converted to the membrane-tethered Atg8a-II, which is presented on the inner and outer autophagosome membranes. The Atg8a-II on the inner membrane is degraded after fusion of autophagosomes with lysosomes. As shown in Figure 5B, the dYkt6L162Q mutant flies rescued by dYkt6GR-Y186C exhibit approximately a 2.5-fold increase in Atg8a-II levels and a 5.5-fold increase in the Atg8a-II/Atg8a-I ratio when compared with the mutant flies rescued by wild-type dYkt6GR. Hence, the dYkt6GR-Y186C variant leads to an abnormal accumulation of Ref(2)P, elevated Atg8a-II levels, and an increased Atg8a-I to Atg8a-II ratio. These data argue that the autophagic phenotypes are not due to the autophagy initiation steps but rather result from impairment in fusion of autophagosome with the lysosomes or the degradation of the content.
The impact of the dYkt6GR-Y65C variant on autophagy was assessed in the dYkt6[G808] mutant flies. As shown in Figure 5C, dYkt6GR-Y65C is less efficient in reducing the Ref(2)P accumulation in the fat body cells when compared with wild-type dYkt6GR (Figure 5C), suggesting that dYkt6GR-Y65C is a mild loss-of-function allele.
Given that impaired autophagy can trigger apoptosis69 and increased cell death is observed in the brains of flies with autophagy defects,70,71 we assessed the levels of apoptosis in the adult brain of dYkt6 mutants. As shown in Supplemental Figure 2C, the 20-day-old dYkt6L162Q mutant flies rescued by dYkt6GR-Y186C exhibit a high level of apoptosis in brains when compared with those rescued by wild-type dYkt6GR. In contrast, dYkt6GR-Y65C causes only a mild increase in apoptosis (Supplemental Figure 2C). In summary, these data corroborate the previous findings that the 2 YKT6 variants are hypomorphic and the p.(Tyr185Cys) variant is a more severe hypomorph than the p.(Tyr64Cys) variant.
Discussion
YKT6 is a special SNARE that has no transmembrane domains but instead contains a “CCAIM” motif at its C terminus. Previous studies demonstrated that the lipidation state of the cysteine motif is important to the localization and function of YKT6. One model is that YKT6 cycles between the cytosolic and membrane-bound states (Supplemental Figure 3A), which is facilitated by the irreversible farnesylation at the second cysteine and the further reversible palmitoylation at the first cysteine.72,73 Cytosolic YKT6 is autoinhibited in a closed conformation with the N-terminal Longin domain folding back onto the C-terminal SNARE core domain and forming a hydrophobic groove that masks the farnesyl moiety.3,74,75 Once activated, YKT6 switches to an open conformation and is recruited to the membrane, where it associates with the membrane through the exposed farnesyl moiety and is further stabilized at the membrane by the palmitoyl moiety.73 The membrane-anchored YKT6 can interact with other SNARE components to mediate membrane fusion, after which it is released from membranes and regains the inactivated closed conformation in the cytosol.10 Another model revealed by a recent study suggested that YKT6 is doubly prenylated with a farnesyl group attached to the second cysteine and a geranylgeranyl group subsequently attached to the first cysteine. The double prenylation is essential for the function of YKT6 as a Golgi SNARE.76 Wen et al77 crystalized the rat YKT6 protein and found that the Tyr64 and Tyr185 residues are at the interface of the Longin/SNARE core, which is critical for the conformational switch that regulates the subcellular localization and activation of YKT6. The authors generated Tyr to Glu substitution at the variant residues (Tyr64Glu and Tyr185Glu, respectively) and found that the mutant proteins have a more open conformation compared with wild-type YKT6. The mutant proteins are more membrane-bound in HeLa cells and show enhanced binding affinity with the cognate SNAREs,77 suggesting that these 2 residues play important roles in the regulating the function of YKT6. It is interesting that the 2 variants identified from the probands are both Tyr to Cys substitution. The Cys residues could lead to aberrant disulfide formation and abnormal intramolecular interaction. We used AlphaFold78,79 to predict the 3D protein structures and label the p.(Tyr64Cys) and p.(Tyr185Cys) variants. The models indicate that these variants cause local differences in protein structures (Supplemental Figure 3B). Tyr185Cys mutation can potentially disrupt the intracellular interactions between Tyr185 and nearby residues in the SNARE core (Leu167, Asp168, and Arg169) (Supplemental Figure 3C), which may affect the stability of the SNARE core. The impact of Tyr64Cys mutation is not obvious in this context (Supplemental Figure 3D).
We modeled the YKT6 variants in Drosophila by assessing the function of the corresponding missense variants in fly dYkt6. Compared with the wild-type genomic rescue construct dYkt6GR, the dYkt6GR-Y186C exhibited very limited rescue ability in all of the tested rescue assays. However, dYkt6GR-Y65C can be distinguished from the wild-type dYkt6GR when the function of the endogenous dYkt6 is strongly lost. The results of our functional assays argue that the 2 missense YKT6 variants are hypomophic alleles with different severity. The clinical manifestations of the probands align with these observations. The 2 individuals with the p.(Tyr185Cys) variant exhibited neurological, as well as hepatic issues, whereas the individual with the p.(Tyr64Cys) variant exhibited mainly neurological disorders. It is possible that some symptoms observed in the affected individuals are due to a secondary effect. Nonetheless, our work indicates that different tissues exhibit different requirement and sensitivity of YKT6 function.
In summary, we report 3 individuals with homozygous missense variants in YKT6 who exhibit neurological disorders with or without severe infantile liver disease, and the affected individuals with hepatic dysfunction carry a risk for development of hepatocellular carcinoma (which occurred in 1 infant). The YKT6 p.(Tyr185Cys) variant seen in the 2 unrelated families originated from the largely endogamous Syrian Christian community of Kerala, India, a group currently estimated to be comprising about 5 million individuals worldwide, is a candidate for carrier screen in this population. In addition, our work suggests that children diagnosed with YKT6 liver disease will need to be screened for hepatocellular carcinoma. Our functional data strongly argue that the p.(Tyr185Cys) allele behaves as a severe loss-of-function variant, whereas the p.(Tyr64Cys) behaves as a milder hypomorphic variant allele that could also contribute to the symptoms. Additional patients along with further studies will be required to precisely understand the pathogenesis and to identify potential therapeutic targets.
Supplementary Material
Acknowledgments
The authors thank the individuals and families for their participation in this study. The authors thank Hongling Pan for transgenic fly lines. The authors thank Liwen Ma for helping with fly tissue dissection and preparation. The authors thank Dr. Meisheng Ma for suggestions about protein structure interpretation. The authors thank Dr. Sheela Nampoothiri and Dr. Vaidehi Jobanputra for their help. The authors thank the Bloomington Drosophila Stock Center for stocks and the Developmental Studies Hybridoma Bank for antibodies.
Funding
This work was supported by the Huffington Foundation, the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, and the Office of Research Infrastructure Programs of the National Institutes of Health (R24OD022005 and R24OD031447) to H.J.B.; the US National Human Genome Research Institute (NHGRI) and National Heart Lung and Blood Institute (NHBLI) to the Baylor-Hopkins Center for Mendelian Genomics (BHCMG, UM1 HG006542) and Neurological Disorders and Stroke (NINDS, R35NS105078) to J.R.L. D.P. is supported by Rett Syndrome Research Trust (RSRT), International Rett Syndrome Foundation (IRSF grant #3701-1), Doris Duke Charitable Foundation with grant #2023-0235, and Neurological Disorders and Stroke (NINDS, K23 NS125126-01A1). Confocal microscopy was performed in the BCM IDDRC Neurovisualization Core, supported by the National Institute of Child Health and Human Development (NICHD, U54HD083092).
Footnotes
Conflict of Interest
BCM and Miraca Holdings have formed a joint venture with shared ownership and governance of BG, which performs clinical microarray analysis, clinical ES (cES), and clinical biochemical studies. James R. Lupski serves on the Scientific Advisory Board of the BG. James R. Lupski has stock ownership in 23andMe, is a paid consultant for Genomics International, and is a coinventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting. Nhu Thao Nguyen Galvàn serves as a consultant for 3DSystems. Davut Pehlivan provides consulting service for Ionis Pharmaceuticals. Wendy K. Chung is on the Board of Directors of Prime Medicine and Rallybio. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics Laboratories. All other authors declare no conflicts of interest.
Ethics Declaration
Written informed consent was obtained for all the individuals. For individual 1, the study was approved by the Institutional Review Board of Columbia University. For individual 2, the study was under the Baylor College of Medicine IRB protocol number H-44779. Individual 3 was referred to Medical Genetics Department because of neurodevelopmental disorder and visual abnormalities. Because clinical molecular tests were unremarkable, the family was enrolled into Baylor-Hopkins Center for Mendelian Genomics research endeavor to identify the molecular etiology of underlying under the IRB protocol number H-29697.
Additional Information
The online version of this article (https://doi.org/10.1016/j.gim.2024.101125) contains supplemental material, which is available to authorized users.
Data Availability
The data supporting the findings of this study are available in the article and the Supplemental Material. All reagents generated in this study are available upon request from the corresponding authors. Requests for additional information will be considered subject to the data use agreement based on the participants’ consent. This study did not generate data sets.
References
- 1.Jahn R, Scheller RH. SNAREs–engines for membrane fusion. Nat Rev Mol Cell Biol 2006;7(9):631–643. 10.1038/nrm2002 [DOI] [PubMed] [Google Scholar]
- 2.Söllner T, Whiteheart SW, Brunner M, et al. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362(6418):318–324. 10.1038/362318a0 [DOI] [PubMed] [Google Scholar]
- 3.Tochio H, Tsui MM, Banfield DK, Zhang M. An autoinhibitory mechanism for nonsyntaxin SNARE proteins revealed by the structure of Ykt6p. Science. 2001;293(5530):698–702. 10.1126/science.1062950 [DOI] [PubMed] [Google Scholar]
- 4.Bas L, Papinski D, Licheva M, et al. Reconstitution reveals Ykt6 as the autophagosomal SNARE in autophagosome-vacuole fusion. J Cell Biol 2018;217(10):3656–3669. 10.1083/jcb.201804028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao J, Reggiori F, Ungermann C. A novel in vitro assay reveals SNARE topology and the role of Ykt6 in autophagosome fusion with vacuoles. J Cell Biol 2018;217(10):3670–3682. 10.1083/jcb.201804039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schoen TJ, Beebe DC, Clemmons DR, Chader GJ, Waldbillig RJ. Local synthesis and developmental regulation of avian vitreal insulin-like growth factor-binding proteins: a model for independent regulation in extravascular and vascular compartments. Endocrinology. 1992;131(6):2846–2854. 10.1210/endo.131.6.1280206 [DOI] [PubMed] [Google Scholar]
- 7.Matsui T, Jiang P, Nakano S, Sakamaki Y, Yamamoto H, Mizushima N. Autophagosomal YKT6 is required for fusion with lysosomes independently of syntaxin 17. J Cell Biol 2018;217(8):2633–2645. 10.1083/jcb.201712058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takáts S, Glatz G, Szenci G, et al. Non-canonical role of the SNARE protein Ykt6 in autophagosome-lysosome fusion. PLoS Genet 2018;14(4):e1007359. 10.1371/journal.pgen.1007359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gordon DE, Chia J, Jayawardena K, Antrobus R, Bard F, Peden AA. VAMP3/Syb and YKT6 are required for the fusion of constitutive secretory carriers with the plasma membrane. PLoS Genet 2017;13(4): e1006698. 10.1371/journal.pgen.1006698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McNew JA, Sogaard M, Lampen NM, et al. Ykt6p, a prenylated SNARE essential for endoplasmic reticulum-Golgi transport. J Biol Chem 1997;272(28):17776–17783. 10.1074/jbc.272.28.17776 [DOI] [PubMed] [Google Scholar]
- 11.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151(6):1256–1269. 10.1016/j.cell.2012.11.001 [DOI] [PubMed] [Google Scholar]
- 12.Kriegenburg F, Bas L, Gao J, Ungermann C, Kraft C. The multi-functional SNARE protein Ykt6 in autophagosomal fusion processes. Cell Cycle. 2019;18(6–7):639–651. 10.1080/15384101.2019.1580488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Li X, Xu Z, et al. Unbalanced regulation of Sec22b and Ykt6 blocks autophagosome axonal retrograde flux in neuronal ischemia-reperfusion injury. J Neurosci 2022;42(28):5641–5654. 10.1523/JNEUROSCI.2030-21.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348(6235):648–660. 10.1126/science.1262110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sprecher E, Ishida-Yamamoto A, Mizrahi-Koren M, et al. A mutation in SNAP29, coding for a SNARE protein involved in intracellular trafficking, causes a novel neurocutaneous syndrome characterized by cerebral dysgenesis, neuropathy, ichthyosis, and palmoplantar keratoderma. Am J Hum Genet 2005;77(2):242–251. 10.1086/432556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keser V, Lachance JB, Alam SS, et al. Snap29 mutant mice recapitulate neurological and ophthalmological abnormalities associated with 22q11 and CEDNIK syndrome. Commun Biol 2019;2:375. 10.1038/s42003-019-0601-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuchs-Telem D, Stewart H, Rapaport D, et al. CEDNIK syndrome results from loss-of-function mutations in SNAP29. Br J Dermatol 2011;164(3):610–616. 10.1111/j.1365-2133.2010.10133.x [DOI] [PubMed] [Google Scholar]
- 18.Retterer K, Juusola J, Cho MT, et al. Clinical application of whole-exome sequencing across clinical indications. Genet Med 2016;18(7):696–704. 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- 19.Zhu N, Gonzaga-Jauregui C, Welch CL, et al. Exome sequencing in children with pulmonary arterial hypertension demonstrates differences compared with adults. Circ Genom Precis Med 2018;11(4):e001887. 10.1161/CIRCGEN.117.001887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganapathi M, Padgett LR, Yamada K, et al. Recessive rare variants in deoxyhypusine synthase, an enzyme involved in the synthesis of hypusine, are associated with a neurodevelopmental disorder. Am J Hum Genet 2019;104(2):287–298. 10.1016/j.ajhg.2018.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng L, Pammi M, Saronwala A, et al. Use of exome sequencing for infants in intensive care units: ascertainment of severe single-gene disorders and effect on medical management. JAMA Pediatr 2017;171(12): e173438. 10.1001/jamapediatrics.2017.3438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med 2013;369(16):1502–1511. 10.1056/NEJMoa1306555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA 2014;312(18):1870–1879. 10.1001/jama.2014.14601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karaca E, Harel T, Pehlivan D, et al. Genes that affect brain structure and function identified by rare variant analyses of Mendelian neurologic disease. Neuron 2015;88(3):499–513. 10.1016/j.neuron.2015.09.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.International HapMap Consortium. The International HapMap project. Nature. 2003;426(6968):789–796. 10.1038/nature02168 [DOI] [PubMed] [Google Scholar]
- 27.Loh PR, Danecek P, Palamara PF, et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet 2016;48(11):1443–1448. 10.1038/ng.3679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poplin R, Chang PC, Alexander D, et al. A universal SNP and small-indel variant caller using deep neural networks. Nat Biotechnol 2018;36(10):983–987. 10.1038/nbt.4235 [DOI] [PubMed] [Google Scholar]
- 29.Narasimhan V, Danecek P, Scally A, Xue Y, Tyler-Smith C, Durbin R. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics. 2016;32(11):1749–1751. 10.1093/bioinformatics/btw044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanca O, Zirin J, Hu Y, et al. An expanded toolkit for Drosophila gene tagging using synthesized homology donor constructs for CRISPR-mediated homologous recombination. Elife. 2022;11:e76077. 10.7554/eLife.76077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc Natl Acad Sci U S A 2007;104(9):3312–3317. 10.1073/pnas.0611511104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Venken KJ, He Y, Hoskins RA, Bellen HJ. P[acman]: a BAC transgenic platform for targeted insertion of large DNA fragments in D. melanogaster. Science. 2006;314(5806):1747–1751. 10.1126/science.1134426 [DOI] [PubMed] [Google Scholar]
- 33.Madabattula ST, Strautman JC, Bysice AM, et al. Quantitative analysis of climbing defects in a Drosophila model of neurodegenerative disorders. J Vis Exp 2015;100:e52741. 10.3791/52741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu S, Hernan R, Marcogliese PC, et al. Loss-of-function variants in TIAM1 are associated with developmental delay, intellectual disability, and seizures. Am J Hum Genet 2022;109(4):571–586. 10.1016/j.ajhg.2022.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Denton D, Chang TK, Nicolson S, et al. Relationship between growth arrest and autophagy in midgut programmed cell death in Drosophila. Cell Death Differ 2012;19(8):1299–1307. 10.1038/cdd.2012.43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma M, Zhang X, Zheng Y, et al. The fly homolog of SUPT16H, a gene associated with neurodevelopmental disorders, is required in a cell-autonomous fashion for cell survival. Hum Mol Genet 2023;32(6):984–997. 10.1093/hmg/ddac259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng Y, Buchwalter RA, Zheng C, Wight EM, Chen JV, Megraw TL. A perinuclear microtubule-organizing centre controls nuclear positioning and basement membrane secretion. Nat Cell Biol 2020;22(3):297–309. 10.1038/s41556-020-0470-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guichard A, Lu S, Kanca O, et al. A comprehensive Drosophila resource to identify key functional interactions between SARS-CoV-2 factors and host proteins. Cell Rep 2023;42(8):112842. 10.1016/j.celrep.2023.112842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen S, Francioli LC, Goodrich JK, et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature. 2024;625(7993):92–100. 10.1038/s41586-023-06045-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stilp AM, Emery LS, Broome JG, et al. A system for phenotype harmonization in the National Heart, Lung, and Blood Institute transomics for precision medicine (TOPMed) program. Am J Epidemiol 2021;190(10):1977–1992. 10.1093/aje/kwab115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scott EM, Halees A, Itan Y, et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat Genet 2016;48(9):1071–1076. 10.1038/ng.3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jain A, Bhoyar RC, Pandhare K, et al. IndiGenomes: a comprehensive resource of genetic variants from over 1000 Indian genomes. Nucleic Acids Res 2021;49(D1):D1225–D1232. 10.1093/nar/gkaa923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jakate S, Yabes A, Giusto D, et al. Diffuse cirrhosis-like hepatocellular carcinoma: a clinically and radiographically undetected variant mimicking cirrhosis. Am J Surg Pathol 2010;34(7):935–941. 10.1097/PAS.0b013e3181ddf52f [DOI] [PubMed] [Google Scholar]
- 46.Frankenburg WK, Dodds JB. The Denver developmental screening test. J Pediatr 1967;71(2):181–191. 10.1016/s0022-3476(67)80070-2 [DOI] [PubMed] [Google Scholar]
- 47.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46(3):310–315. 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J, Al-Ouran R, Hu Y, et al. MARRVEL: integration of human and model organism genetic resources to facilitate functional annotation of the human genome. Am J Hum Genet 2017;100(6):843–853. 10.1016/j.ajhg.2017.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- 50.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jagadeesh KA, Wenger AM, Berger MJ, et al. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 2016;48(12):1581–1586. 10.1038/ng.3703 [DOI] [PubMed] [Google Scholar]
- 52.Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet 2016;99(4):877–885. 10.1016/j.ajhg.2016.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zachariah KC. The Syrian christians of Kerala: Demographic and Socioeconomic Transition in the Twentieth Century. 2001. Accessed May 31, 2023. https://ideas.repec.org/p/ind/cdswpp/322.html
- 54.Hu Y, Comjean A, Rodiger J, et al. FlyRNAi.org-the database of the Drosophila RNAi screening center and transgenic RNAi project: 2021 update. Nucleic Acids Res 2021;49(D1):D908–D915. 10.1093/nar/gkaa936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rørth P A modular misexpression screen in Drosophila detecting tissue-specific phenotypes. Proc Natl Acad Sci U S A 1996;93(22):12418–12422. 10.1073/pnas.93.22.12418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto S, Jaiswal M, Charng WL, et al. A Drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell. 2014;159(1):200–214. 10.1016/j.cell.2014.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Venken KJ, Carlson JW, Schulze KL, et al. Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nat Methods. 2009;6(6):431–434. 10.1038/nmeth.1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Colombani J, Raisin S, Pantalacci S, Radimerski T, Montagne J, Léopold P. A nutrient sensor mechanism controls Drosophila growth. Cell. 2003;114(6):739–749. 10.1016/s0092-8674(03)00713-x [DOI] [PubMed] [Google Scholar]
- 59.Li S, Yu X, Feng Q. Fat body biology in the last decade. Annu Rev Entomol 2019;64:315–333. 10.1146/annurev-ento-011118-112007 [DOI] [PubMed] [Google Scholar]
- 60.Moraes KCM, Montagne J. Drosophila melanogaster: a powerful tiny animal model for the study of metabolic hepatic diseases. Front Physiol 2021;12:728407. 10.3389/fphys.2021.728407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagarkar-Jaiswal S, Lee PT, Campbell ME, et al. A library of MiMICs allows tagging of genes and reversible, spatial and temporal knockdown of proteins in Drosophila. Elife. 2015;4:e05338. 10.7554/eLife.05338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee PT, Zirin J, Kanca O, et al. A gene-specific T2A-GAL4 library for Drosophila. Elife. 2018;7:e35574. 10.7554/eLife.35574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lu S, Ma M, Mao X, et al. De novo variants in FRMD5 are associated with developmental delay, intellectual disability, ataxia, and abnormalities of eye movement. Am J Hum Genet 2022;109(10):1932–1943. 10.1016/j.ajhg.2022.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mizushima N, Matsui T, Yamamoto H. YKT6 as a second SNARE protein of mammalian autophagosomes. Autophagy. 2019;15(1):176–177. 10.1080/15548627.2018.1532262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lőrincz P, Mauvezin C, Juhász G. Exploring autophagy in Drosophila. Cells. 2017;6(3):22. 10.3390/cells6030022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.DeVorkin L, Gorski SM. Monitoring autophagic flux using Ref(2)P, the Drosophila p62 ortholog. Cold Spring Harb Protoc 2014;2014(9):959–966. 10.1101/pdb.prot080333 [DOI] [PubMed] [Google Scholar]
- 67.Nezis IP, Simonsen A, Sagona AP, et al. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol 2008;180(6):1065–1071. 10.1083/jcb.200711108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3(6):542–545. 10.4161/auto.4600 [DOI] [PubMed] [Google Scholar]
- 69.Boya P, González-Polo RA, Casares N, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005;25(3):1025–1040. 10.1128/MCB.25.3.1025-1040.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takáts S, Nagy P, Varga Á, et al. Autophagosomal Syntaxin17-dependent lysosomal degradation maintains neuronal function in Drosophila. J Cell Biol 2013;201(4):531–539. 10.1083/jcb.201211160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Juhász G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev 2007;21(23):3061–3066. 10.1101/gad.1600707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dietrich LE, Peplowska K, LaGrassa TJ, Hou H, Rohde J, Ungermann C. The SNARE Ykt6 is released from yeast vacuoles during an early stage of fusion. EMBO Rep 2005;6(3):245–250. 10.1038/sj.embor.7400350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fukasawa M, Varlamov O, Eng WS, Söllner TH, Rothman JE. Localization and activity of the SNARE Ykt6 determined by its regulatory domain and palmitoylation. Proc Natl Acad Sci U S A 2004;101(14):4815–4820. 10.1073/pnas.0401183101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pylypenko O, Schönichen A, Ludwig D, et al. Farnesylation of the SNARE protein Ykt6 increases its stability and helical folding. J Mol Biol 2008;377(5):1334–1345. 10.1016/j.jmb.2008.01.099 [DOI] [PubMed] [Google Scholar]
- 75.Hasegawa H, Yang Z, Oltedal L, Davanger S, Hay JC. Intramolecular protein-protein and protein-lipid interactions control the conformation and subcellular targeting of neuronal Ykt6. J Cell Sci 2004;117(Pt19):4495–4508. 10.1242/jcs.01314 [DOI] [PubMed] [Google Scholar]
- 76.Shirakawa R, Goto-Ito S, Goto K, et al. A SNARE geranylgeranyl-transferase essential for the organization of the Golgi apparatus. EMBO J. 2020;39(8):e104120. 10.15252/embj.2019104120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wen W, Yu J, Pan L, et al. Lipid-induced conformational switch controls fusion activity of longin domain SNARE Ykt6. Mol Cell. 2010;37(3):383–395. 10.1016/j.molcel.2010.01.024 [DOI] [PubMed] [Google Scholar]
- 78.Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–589. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Varadi M, Anyango S, Deshpande M, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res 2022;50(D1):D439–D444. 10.1093/nar/gkab1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
Web resources
- AlphaFold: https://alphafold.ebi.ac.uk/
- CADD: https://cadd.gs.washington.edu/
- DIOPT: https://www.flyrnai.org/diopt
- gnomAD: https://gnomad.broadinstitute.org/
- MARRVEL: http://marrvel.org/
- OMIM: https://www.omim.org/
- TOPMed: https://topmed.nhlbi.nih.gov/
- USCF Chimera: https://www.rbvi.ucsf.edu/chimera/
- History about Thomas Christians (Syrian Christians): https://gedsh.bethmardutho.org/Thomas-Christians and https://www.britannica.com/topic/Thomas-Christians
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available in the article and the Supplemental Material. All reagents generated in this study are available upon request from the corresponding authors. Requests for additional information will be considered subject to the data use agreement based on the participants’ consent. This study did not generate data sets.