

Abstract
Gene replacement using Adeno-associated viral (AAV) vectors is a promising therapeutic approach for many diseases. However, this therapeutic modality is challenged by the packaging capacity of AAVs (~4.7 kb), limiting its application for disorders involving large coding sequences, such as Duchenne muscular dystrophy, with a 14 kb mRNA. Here we developed a novel method for expressing large dystrophins by utilizing the protein trans-splicing mechanism mediated by split inteins. We identified several split intein pairs that efficiently join two or three fragments to generate a large midi-dystrophin or the full-length protein. We show that delivery of two or three AAVs into dystrophic mice results in robust expression of large dystrophins and significant physiological improvements compared with micro-dystrophins. Moreover, using the potent myotropic AAVMYO, we demonstrate that low total doses (2×1013 vg/kg) are sufficient to express large dystrophins in striated muscles bodywide with significant physiological corrections in dystrophic mice. Our data show a clear functional superiority of large dystrophins over micro-dystrophins that are being tested in clinical trials. This novel method could benefit many patients with Duchenne or Becker muscular dystrophy regardless of genotype and could be adapted to numerous other disorders caused by mutations in large genes exceeding the AAV capacity.
Duchenne muscular dystrophy (DMD) results from loss-of-function mutations in the 2.2 MB dystrophin (DMD) gene and is among the most common human genetic disorders1,2. In muscle, dystrophin is expressed as a 427 kDa protein that performs a critical role in protecting cells from mechanical stress and it also modulates several intracellular signaling pathways by providing a scaffold to a variety of proteins3,4,5,6. The absence of dystrophin leads to cycles of necrosis and regeneration, progressive muscle wasting, inflammatory infiltration, and eventual cardiac and respiratory failure7. To develop gene therapy for DMD, we and others have previously generated “micro-dystrophins” (μDys)8–10 small enough to be delivered by AAV vectors. Nonetheless, these micro-proteins (~33% of full-length dystrophin) lack critical functional domains and show incomplete phenotypic rescue in animal models and patients11–14. These results suggest that expression of larger dystrophins with improved function could lead to a more robust therapy.
Several approaches to generate larger dystrophins by co-delivery of two or three AAVs have been tested15–18. Using homologous recombination or DNA trans-splicing, AAV genomes harboring partial dystrophin sequences could be reassembled inside muscles to encode larger proteins. However, these DNA-based approaches showed reduced efficiency and depended on the joining of vector genomes in a specific orientation. In addition, unwanted products resulting from vector concatemerization are formed, which could complicate their use in the clinic. An alternative method relies instead on the use of split inteins to join two proteins. Split inteins are small polypeptides that undergo a unique post-translational auto-processing event termed protein trans-splicing (PTS), in which the flanking N- and C-terminal residues (exteins) are spontaneously ligated into one functional protein accompanied by the removal of the reconstituted intein19. These genetic elements have been successfully implemented in numerous biological applications, including protein purification and labeling steps and, more recently, gene replacement in retinal or liver diseases involving large genes20–24. One early study explored such a strategy combining dual AAV vector administration and PTS but failed to show significant dystrophin expression or functional benefit after intramuscular administration25.
Here, we identified several efficient intein pairs that can reconstitute a highly functional midi-dystrophin (midi-Dys ΔSR5–15) or full-length dystrophin (Dp427) following dual or triple AAV vector administration. Importantly, this method does not require an increase in total vector dose and leads to expression levels the same or higher than are obtained with μDys. Using recently published myotropic AAV vectors, we show that these larger dystrophins can be generated in muscles using doses significantly lower than are currently being tested in the clinic for DMD. We demonstrate therapeutic correction of numerous functional deficits in striated muscles of young and very old dystrophic mdx4cv mice that are superior to those obtained using current μDys vectors. The combined use of these split intein constructs, myotropic serotypes of AAV, and strong muscle-specific promoters has the potential to significantly increase the therapeutic impact of DMD gene therapy as well as other disorders resulting from mutations in large genes.
In vitro screening and optimization of split inteins
More than 30 split inteins have been characterized19. However, most studies were performed on purified proteins, making it difficult to extrapolate for in vivo applications. Another caveat is that all inteins highly depend on the adjacent residues found in the native extein (host protein) to catalyze the final ligation. These residues are consensually defined by the last and first tripeptide sequences of N- and C- terminal halves of the extein. To reconstitute any protein of interest using the PTS process, these six residues must be included in the extein/intein boundaries and are left behind as “footprints”, which could alter the function of the protein.
To compare their efficacy, we generated a library of 23 short but ultrafast split inteins26–28. The library was divided into two sub-groups: Group1 comprises 19 split inteins that share high sequence homology, including the 6-residue footprint, while Group2 encompasses four unique pairs (Extended data Table 1, Extended data Table 2). We developed a split green fluorescent protein (GFP) system to perform a multistep screening of the different split intein pairs. GFP has a barrel-like tri-dimensional conformation with several connecting loops that can accommodate insertions of up to 10 amino acids (AA) without seriously disturbing the protein folding29, and hence its fluorescence (Extended data Fig. 1a). We evaluated two sites on linkers between β-sheets for their tolerance to up to 8-residue insertions. Although the expression of these chimeric constructs in Human Embryonic Kidney 293 (HEK293) cells resulted in lower fluorescence intensity than Wild-type (WT) GFP, site#2 (Glu172-Asp173) was more permissive to 8-residue insertion than site#1 (Gln157-Lys158), with almost one log higher fluorescence (Extended data Fig. 1b–c). Therefore, we used site#2 to cleave the GFP into two fragments and insert split inteins to screen their PTS activity.
For each intein, two plasmids were cloned, where the N- or C-terminal half were inserted in-frame with a GFP moiety. In addition, corresponding GFPs with 6-residue footprint insertions were generated and used as controls. All plasmids were transfected into HEK293 cells to monitor the GFP fluorescence. Although the fluorescence emitted by the reconstituted GFP was lower than WT GFP, it was comparable to values recorded with the GFP-footprint controls (Extended data Fig. 1d). In Group1, six of the 19 split inteins tested showed a high ligation efficacy, while in Group2, three of the pairs joined the two GFP halves into functional GFPs.
A second screening was performed with these nine pairs to determine the specificity of an N-terminal split intein towards its C-terminal partner. Because of the limited packaging capacity of AAVs (<5 kb), expression of full-length dystrophin (Dp427) would require co-administration of three vectors encoding N-terminal, middle, and C-terminal fragments that must be precisely reassembled using two split intein pairs. We found that split inteins from Group1 showed poor specificity and cross-spliced with different inteins of the same group as reflected by the variable GFP fluorescence signals (Extended data Fig. 1e). In contrast, split inteins from Group2 demonstrated a high specificity towards the other half of the same intein and did not cross-splice with any other split intein. These data revealed the lack of specificity of pairs from Group1 and highlight the orthogonality of split gp41.1, IMPDH, and Nrdj1, suggesting the possibility of using them simultaneously to join multiple fragments without generating unwanted products. Based on these data, we selected split Aha from Group1 and gp41.1, IMPDH, and Nrdj1 from Group2 for further testing.
In a third screening, we sought to minimize the hexapeptide footprint that results from intein splicing. We tested several combinations with progressive deletion of one residue from the split intein/GFP linker on either the N- or C-terminal half (Extended data Fig. 1f). With split Aha, the same splicing efficiency was achievable with a three-residue linker (AEY at the N-terminal side) as the native AEY/CFN (AEY at the N-terminal side and CFN at the C-terminal end). Similarly, using gp41.1, efficient GFP ligation was obtained with GY/S instead of SGY/SSS. Consequently, the footprint left by these two split inteins is three residues instead of six. This footprint was even shorter for split IMPDH and Nrdj1 with only G/S and C/S, respectively. These results identified flexibility of the selected split inteins and reveal that similar PTS activity can be obtained while leaving a minimal footprint ‘scar’ in the ligated protein.
Validation of the selected split inteins for dystrophins
The small size of the pre-selected split inteins offers the possibility to deliver and express a large midi-dystrophin (midi-Dys) using two AAVs. Here, we used a 5’-clone, encoding sequences from the N-terminal domain to the end of Spectrin-like Repeat (SR) 19, but lacking SRs 5–15, with a 3’-clone encoding sequences from Hinge3 through the C-terminal domain (CT). Fusion of these two fragments via the PTS mediated by split inteins generates a large midi-Dys (ΔSR5–15) containing 68% of the dystrophin protein sequence, with four hinges, 13 SRs, the-N-terminal actin binding domain (ABD), the nNOS-localization domain, a putative cardioprotective domain, the cysteine-rich (CR) domain and the entire C-terminal domain, several of which are missing from μDys (Fig. 1a Extended data Fig. 2). Moreover, analysis of the peptide sequence showed the presence of four splitting sites (GpS1, GpS2, GpS3, and GpS4) located between SR19 and Hinge3 that partially match the gp41.1 minimal footprint determined above (Extended data Fig. 2). To test these options, HEK293 cells were transfected with either a control plasmid expressing the entire midi-Dys ΔSR5–15 or dual plasmids encoding N- or C-terminal split intein/midi-Dys. Western blot analysis revealed the production of a strong band at ~291 kDa corresponding to the reconstituted midi-Dys (Fig. 1b). Furthermore, the split intein approach resulted in 5 to 8-fold higher expression than was obtained with the ΔSR5–15 single plasmid, perhaps due to transfection efficiency differences between large and small plasmids. The highest expression was detected when using split sites GpS2 and GpS4 (Fig. 1b). This observation reflects the efficacy of the selected splitting sites and the optimized split gp41.1.
Figure 1: In vitro validation of selected split inteins using dystrophin sequence.

a) Schematic representation of structural domains of full-length dystrophin (muscle isoform Dp427), midi-dystrophin (ΔSR5–15) expressed using split intein in dual vector approach, mini-dystrophin (ΔH2-SR19) reported in asymptomatic DMD patients carrying a large deletion (exons 17–48) of the DMD gene, and μDys5 currently evaluated in clinical trials. ABD: actin binding domain, CR: cysteine-rich, CT: C-terminal domain, H: Hinge, R: Spectrin-like Repeat, nNOS: neuronal nitric oxide synthase binding site, PCPD: putative cardioprotective domain, DgBD: dystroglycan binding domain, SBD: syntrophin binding domain, DbBD: dystrobrevin binding domain. b) Representative western blot of HEK293 lysates showing the 290 kDa midi-Dys (ΔSR5–15). In control midi-Dys, cells were transfected with a plasmid expressing the entire midi-Dys (ΔSR5–15). In split midi-Dys/intein, cells were co-transfected with both N- and C-terminal plasmids. Each lane represents a selected split site between SR19 and Hinge3. c) Western blot example of HEK293 lysates showing efficient full-length dystrophin expression following triple plasmid transfection and using two split intein combinations. In the control plasmid, cells were transfected with a plasmid expressing full-length human dystrophin. M.W: molecular weight. kDa: kiloDalton. Ab: Antibody.
We next tested the possibility of expressing full-length dystrophin by ligating three fragments using two split inteins (Fig. 1c; Extended data Fig. 3). Since split gp41.1 showed efficient ligation of SR19 to Hinge3, it was maintained at this position to join the middle to the C-terminal fragment. Split Aha, IMPDH, and Nrdj1 were compared for their ability to join the N-terminal to the middle fragment at different locations between SR7 and SR8 (Extended data Fig. 4a–d). The efficacy of each split intein was evaluated individually to reconstitute full-length dystrophin, initially using two constructs. HEK 293 cells were co-transfected with these two plasmids: one expressing a third of dystrophin fused to one side of the split intein, while the second plasmid encoding the remaining two-thirds of dystrophin fused to the other half of the split intein (Extended data Fig. 4). Analysis of protein expression revealed a strong band at the predicted molecular weight (~400 kDa) with IMPDH, Nrdj1, or gp41.1 (but not with Aha) inserted at different locations (Extended data Fig. 4e).
Finally, split IMPDH, Nrdj1, and gp41.1 were simultaneously evaluated in a triple vector approach using two combinations. In full-Dys1, IMPDH was used to ligate the N-terminal to the middle fragment, while in full-Dys2, Nrdj1 was used. In both cases, split gp41.1 was used to ligate the middle to the C-terminal portion (Extended data Fig. 4f). HEK293 cells were transfected with different conditions (single or dual plasmids as controls, or triple plasmids to test simultaneous PTS). Protein expression analysis showed that both combinations resulted in the expression of a strong band of ~427 kDa, 2- to 4-fold higher than the control full-length plasmid (Fig. 1c). These data demonstrate the feasibility of robust reconstitution of the entire full-length dystrophin from three plasmids via simultaneous PTS mediated by two efficient split inteins.
In vivo validation of split intein/Dys constructs
To validate these constructs in vivo, split dystrophin/intein clones for each combination were cloned downstream of the muscle-specific enhancer/promotor CK8e and packaged into pseudotyped AAV6 vectors10. 5×1010 vector genomes (vg) of different pairs were injected at 1:1 ratio into the tibialis anterior (TA) muscles of three-week-old dystrophin-null mdx4cv males. Five weeks post-injection, treated muscles were collected for analysis. Using C-terminal antisera, a robust midi-Dys band at 291 kDa was detected in treated muscles (Fig. 2a, 2b). A second band at ~150 kDa representing the unspliced C-terminal fragment accounted for 50–70% of the detected products (Fig. 2a, 2c). The expression of both bands was noticeably higher using midi-Dys2 with 4 times higher expression of the final product (Fig. 2b), indicating higher stability of protein fragments and efficient PTS with split gp41.1 when inserted in split site GpS4. Immunolabeling of muscle cross-section using antibodies against the N- or C-terminal fragments showed colocalization of these protein fragments at the sarcolemma (Fig. 2d). Of note, the injection of individual fragments led to their expression at the sarcolemma membrane without any noticeable adverse effects (Extended data Fig. 5a, 5b). Importantly, adjusting the ratios of the N- versus C-terminal vectors led to similar expression of midi-Dys2 and histology improvements while eliminating excess expression of the C-terminal fragment (Extended data Fig. 5c–e).
Figure 2: in vivo validation of AAV6 split intein/Dys constructs in mdx4cv mice.

a) Western blot of T.A muscle lysates from controls or injected with 5×1010 vg AAV6 N- and/or C-terminal split mini-Dys/intein. b) Protein expression normalized to GAPDH and c) ratio of spliced (top) or unspliced (bottom) bands determined by densitometry (WT n=4, n=5 for injected muscles). d) Cross-sections of TA muscles treated with 2 different dual vector combinations and stained with hematoxylin and eosin (H&E, top row. Scale bar: 50 μm) or immunolabeled with antibodies against the dystrophin N- or C-terminal domains (lower panels. Scale bar: 50 μm). e) Western blot showing expression of full-length dystrophin in TA muscles treated with the triple AAV vector strategy. f) Full-length dystrophin expression normalized to GAPDH and g) different product ratios were quantified using densitometry (n=4 muscles per group). h) Transverse muscle cryosections stained with H&E (top row. Scale bar: 50 μm) or immunolabeled with antibodies against the N-, middle or C-terminal dystrophin fragments (lower panels. Scale bar: 50 μm). Western blots showing protein expression of dystrophins in T.A (i) or heart (j) muscles of WT, saline-injected mdx4cv, or mdx4cv mice treated systemically with a total dose of 2×1014 vg/kg of the single, dual or triple AAV vectors. k) H&E (top row. Scale bar: 50 μm) and immunofluorescence staining (lower panel. Scale bar: 100 μm) for dystrophin or laminin of TA muscle cross-sections. μDys: micro-dystrophin. m-Dys: midi-dystrophin. f-Dys: full-length dystrophin. Midi-Dys1 and midi-Dys2 result from split GpS2 and split GpS4, respectively (See Extended data Fig. 2). Full-Dys1 is expressed following simultaneous trans-splicing of three fragments via IMPDH (split site: ImS4) and gp41.1 (split site: GpS4), while in Full-Dys2 Nrdj-1 (split site: NrS4) and gp41.1 gp41.1 (split site: GpS4) are used (See Extended data Fig. 4). Data represented as means±s.e.m Ab: antibody. kDa: kilodalton. MW: molecular weight.
Similarly, muscles injected intramuscularly with the triple vectors showed strong expression of full-length dystrophin when using the full-Dys2 combination at a level ~9-fold higher than endogenous dystrophin levels in WT muscles (Fig. 2e, 2f). Other bands were detected at ~150 kDa (which represents the unfused C-terminal fragment expressed at 67% with Full-Dys1 and 33% with Full-Dys2), and two medium-size bands at ~260 and ~280 kDa in conditions where both the middle and C- terminal were administrated (Fig. 2e, 2g). The latter most likely result from the fusion of the middle to the C-terminal fragments. The slight difference in the molecular size (~30 kDa) may correspond to the forms with or without reassembled gp41.1 intein during the different splicing steps. Importantly, no additional bands were detected in muscles injected with only N- and C-terminal fragments, which confirms the orthogonality of split gp41.1 towards IMPDH or Nrdj1. Triple immunostaining of muscle sections using antibodies against N-, middle, or C-terminal fragments showed that all three moieties are located at the myofiber periphery (Fig. 2h). Analysis of general muscle morphology revealed significant morphometric improvements with a dramatic reduction in centrally-nucleated myofibers using both the dual and triple vector strategies, with ~30% centrally-nucleated myofibers versus ~68% in controls (Fig. 2h, Extended data Fig. 5f).
These data demonstrate the in vivo efficacy of the optimized split inteins in expressing highly functional midi-Dys (ΔSR5–15) or full-length dystrophin by joining two or three fragments transportable by AAV.
Robust expression in muscles following systemic delivery
An important consideration for this new approach is to infuse therapeutic AAV doses without compromising safety. The maximal dose injected in ongoing clinical trials for musculoskeletal disorders is ~2–3×1014 vg/kg (with AAV serotype 9 or rh74)30. To test the feasibility of the split intein approach using systemic delivery, a total dose of 2×1014 vg/kg of dual or triple AAV6 vectors was administrated intravascularly into eight-week-old mdx4cv males. A control group was treated with a single AAV vector expressing μDys5 that is being evaluated in DMD patients (NCT03368742). Three months post-injection, analysis of protein expression revealed dystrophin expression at 75% of WT levels for midi-Dys, ~31% for full-Dys, and ~8% with μDys5 (Fig. 2i) in TA muscles. These proteins were more abundant in cardiac muscle, with ~62% full-length dystrophin following infusion of the triple vector, ~178% midi-Dys from the dual vector strategy, and ~66% with the single μDys5 vector (Fig. 2j). Expression of dystrophin was confirmed by immunolabelling of TA and heart sections (Fig. 2k, Extended data Fig. 6a). On average, ~40% of TA myofibers were positive for dystrophin staining in groups treated with the dual or triple vector strategies, while only ~5% were expressing μDys5 (Fig. 2k, Extended data Fig. 6b). In contrast, heart sections showed uniform and widespread dystrophin expression in almost every cardiomyocyte (Extended data Fig. 6a). Furthermore, dystrophins expression in TA muscles resulted in modest improvement in general muscle morphology with increase of fiber size and diameter (Extended data Fig. 6c, 6d).
These data support the feasibility of expressing large dystrophins using split intein-mediated PTS in striated muscles. Nonetheless, injecting these adult mdx4cv mice with AAV6 resulted in lower expression of dystrophin constructs in hindlimb skeletal muscles compared with cardiac muscle. These observations are most likely due to limited efficacy of AAV6 in skeletal muscles from young adult mice in the midst of a degeneration/regeneration crisis31,32, as we have previously obtained significantly higher expression levels when infusing neonatal and adolescent mice8,10,33.
Improvement of dystrophic phenotype in young mice
Given the mosaic expression of dystrophin found in adult mouse TA cross-sections, we concluded that systemic delivery of AAV6 at a dose of 2×1014 vg/kg might be insufficient to ensure therapeutic levels of dystrophins with uniform expression in adult mice. Consequently, we tested the recently described myotropic capsid AAVMYO34.
As an initial study, the single (μDys5) and dual (midi-Dys2) vector strategies were compared using a low dose of 2×1013 (total) vg/kg, and a high dose of 2×1014 (total) vg/kg of AAVMYO. As a control, AAV9 was used to deliver μDys5. AAV particles were infused into the tail vein of eight-week-old, young adult mdx4cv males. Three months later, analyses of in situ TA muscle force showed that AAV9 μDys5 did not improve muscle performance at the low dose (Fig. 3a), while the specific force of TA muscles treated with low dose AAVMYO μDys5 exhibited a significant increase (~41%) versus the saline group; however, neither μDys5 group displayed a significant protection from contraction-induced injury (Fig. 3b). Encouragingly, mdx4cv mice treated with a low dose of AAVMYO dual (midi-Dys2) vectors showed normalization of the TA specific force to WT values and significant protection from contraction-induced injury. Similar normalization of mechanical properties was found in groups treated with high doses of AAVMYO using either the single (μDys5) or dual (midi-Dys2) vector strategies. Furthermore, analysis of serum creatine kinase levels showed a slight reduction in the group treated with low-dose dual AAVMYO versus control mdx4cv treated with saline, but groups treated with high doses of single or dual AAVMYO exhibited normal values (Fig. 3c). Conversely, no improvements of serum creatine kinase levels were observed in mice treated with a low dose of AAV9 or AAVMYO μDys5.
Figure 3: Systemic delivery of AAVMYO split intein/Dys enhances muscle targeting at low doses.

a) Plots representing TA specific force development of adult WT or mdx4cv mice treated intravenously with saline, AAV serotype 9, or AAVMYO at low (2×1013 vg/kg) or high (2×1014 vg/kg) dose for 12 weeks. b) Muscle forces measured following a 15%-lengthening contraction beyond the optimal L0. c) Circulating Creatine Kinase levels measured in serum samples after 12 weeks of AAV treatment. d) Protein expression of μDys, midi-Dys (ΔSR5–15), or full-length dystrophin in T.A muscle lysates of control (WT or mdx4cv) or AAV-treated mice. For a and b n=5 mice for low dose AAV9 or AAVMYO μDys5, n= 6 mice per group other groups, while in c, n=4 per group was used. Data represent means±s.e.m. *p<0.05, **p<0.01, ***p<0.001 versus mdx4cv mice treated with saline, and $p<0.05, $ $p<0.01, $ $ $p<0.001 versus WT mice (ANOVA test followed by Tukey’s post hoc). μDys: micro-dystrophin. m-Dys: midi-dystrophin. Ab: antibody. kDa: kilodalton. MW: molecular weight. vg: viral genome. Kg: Kilogram.
These phenotypic observations were correlated with the μDys5 and midi-Dys2 expression levels. No dystrophin expression was detected in mdx4cv mice treated with a low dose of AAV9 (Fig. 3d), while groups treated with AAVMYO showed strong expression of μDys5 and midi-Dys2. The expression levels of both μDys and midi-Dys2 were at least twice as high when high dose AAVMYO vectors were administered. Similar observations were found by immunofluorescence staining, where ~80% and ~98% of myofibers were dystrophin-positive in TA cross-sections of mice treated with, respectively, low or high doses of AAVMYO μDys5 or midi-Dys2, while ~10% of myofibers were positive for dystrophin with AAV9 (Fig. 4a, b). Expression of μDys5 or midi-Dys also restored α-sarcoglycan and β-dystroglycan localization (Extended data Fig. 7a). The general muscle morphology was clearly improved with both doses using either μDys5 or midi-Dys2. However, only TA muscles treated with midi-Dys exhibited a reduced percentage of centrally-nucleated myofibers and normal myofiber area and diameter (Fig. 4c, Extended data Fig. 7b, 7c). Other striated muscles, such as diaphragm and heart, exhibited uniform dystrophin staining in groups treated with AAVMYO at either low or high doses (Fig. 4a, 4d), resulting in a substantial increase of fiber size and diameter (Fig. 4e, Extended data Fig. 7d). Similar to hindlimb muscles, dystrophin expression levels were doubled in groups treated with the higher dose in both diaphragms and hearts (Extended data Fig. 7e, 7f).
Figure 4: Muscle histology improvement following systemic delivery of AAVMYO.

a) T.A and diaphragm cross-sections stained with H&E (top rows. Scale bar: 50 μm) or immunolabeled for dystrophin (N-terminal antibody) or laminin (lower panels. Scale bar: 100 μm).
b) Percentage of dystrophin-positive myofibers determined from T.A cross-sections (n=4, more than 1000 fiber per sample). c) T.A myofiber minimal diameter measured from cross-sections stained with laminin (n=4, ~1000 fibers per sample). d) Percentage of dystrophin myofibers in diaphragm samples (n=4, more than 300 fiber per sample). e) Minimal diameter of diaphragm myofibers determined from sections stained with laminin (n=4, more than 300 fibers per sample). For b and d, Data represent means±s.e.m For c and e, the average values are shown on top of the violin bars. The solid line represents the median, while the dashed lines show the quartiles. μDys: micro-dystrophin. m-Dys: midi-dystrophin. Dys+: Dystrophin positive. vg: viral genome. kg: Kilogram
Overall, these data demonstrate the superiority of AAVMYO for delivery of μDys5 or the split midi-Dys2 components. Successful assembly of a large midi-dys (ΔSR5–15) results in significant phenotypic improvements compared with μDys following low dose infusion of AAVMYO vectors.
Restoration of DMD defect in old mdx4cv mice
Despite dystrophin deficiency, young mdx mice develop only a mild dystrophic phenotype35,36. However, the phenotype progressively deteriorates as they age, leading to severe diaphragm dysfunction, cardiomyopathy, and greater skeletal muscle impairment with pronounced fibrosis37,38. To explore whether the split intein vector approach can protect and/or reverse these defects, 17-month-old mdx4cv males were treated for seven months with a total dose of 2×1013 vg/kg AAV9 or AAVMYO to express μDys5 or midi-Dys (ΔSR5–15). Robust expression of dystrophins was found in TA, gastrocnemius, and soleus muscles from groups treated with AAVMYO, where we observed ~60% dystrophin-positive myofibers; in contrast, AAV9 led to ~5% transduction of μDys5 (Fig. 5a, 5b, Extended data Fig. 8a–d, 9a–c). Analysis of muscle histology showed highly fibrotic and infiltrated muscles in untreated 17-month-old and saline-injected 24-month-old mdx4cv mice, as well as in the low dose of AAV9-μDys5 group. A significant improvement in muscle morphology was found in the AAVMYO μDys5 mdx4cv group, but a far greater amelioration was noticed in mice treated with dual AAVMYO (Fig. 5a, 5c, Extended data Fig. 8a, 8e, 9a, 9d). Further histological analyses revealed a high percentage of centrally nucleated myofibers in all AAV-treated groups (~40%), but a slight increase in the myofiber area and diameter was found in muscle expressing μDys5 or midi-Dys2 following AAVMYO treatment (Extended data Fig. 8f–j, 9e, 9f), suggesting intermediate muscle remodeling. Nonetheless, TA muscles exhibited significant protection against injury (Figure 5d). The group treated with dual AAVMYO showed normalization of TA specific force to WT level (Fig. 5d, 5e), whereas μDys5 groups failed to show clear amelioration when delivered at low doses with either AAV9 or AAVMYO.
Figure 5: Long-term expression of midi-Dys2 in very old mdx4cv mice reverses skeletal muscle defect to wild-type levels.

a) T.A muscle cross-section from 17- (as baseline) or 24-month-old WT or mdx4cv stained with H&E, Trichrome (Scale bar: 50 μm, whole sections with large fields are shown in Extended data Fig. 8), or immunolabeled with antibodies against dystrophin or laminin (Scale bar: 200 μm). b) Western blot analysis of dystrophin expression with GAPDH as loading control from T.A muscle lysates. c) Quantification of fibrosis percentage measured from T.A cross-section stained with Trichrome (n=5 per group). d) Muscle forces drop measured following a 15%-stretching beyond the optimal L0. e) Maximal developed specific forces of T.A measured in situ at L0. For c and e, AAV9-μDys: n=4, control groups: n=5, AAVMYO group: n=6. Data represent means±s.e.m. N.S: no statistical significance. *p<0.05, **p<0.01 and ***p<0.001. (ANOVA test followed by Tukey’s post hoc). μDys: micro-dystrophin. m-Dys: midi-dystrophin.
Assessment of isolated and perfused hearts showed superior protective effects of midi-Dys2 versus μDys5. Hearts from mdx4cv mice treated with dual AAVMYO presented responses to high workload on left ventricular function induced by high calcium concentration that were not different from WT hearts (Figure 6a, 6b, Extended data Fig. 10a, 10b). In contrast, both μDys5 groups showed defective protection. Analysis of heart cross-sections revealed uniform expression of dystrophins with both AAV serotypes and amelioration of the general morphology but significant reduction of fibrosis only with midi-Dys2 (Extended data Fig. 10c, 10d). Surprisingly, no difference in μDys5 protein expression was found between AAV9- or AAVMYO-treated mdx hearts, but enrichment of midi-Dys2 was observed (Fig. 6c). This may indicate a longer half-life or enhanced stability of the large midi-Dys2. These observations were consistent with protein expression assessed by western blot using diaphragm samples, which showed strong expression of midi-Dys2 and slightly lower amounts of μDys5 (Fig. 6d). This led to a moderate increase in diaphragm specific force with μDys5, but significant improvements with dual AAVMYO (midi-Dys2) compared with both the age-matched saline controls and the 17-month-old group (Fig. 6e). Thus, expression of the large midi-Dys resulted in clear protective effects and therapeutic amelioration over time in old dystrophic hearts and noticeable protection of the diaphragm. Indeed, mdx4cv mice exhibited a dramatic loss of muscle mass between the 17- and 24-month-old groups with significant inter-myofiber infiltration and collagen deposition and smaller fibers (Fig. 6f, Extended data Fig. 9g–j), as has previously been noted37,39. In contrast, AAVMYO delivery prevented muscle wasting and led to uniform dystrophin expression, fiber size and diameter, which increased strength in 2-year-old dystrophic mice.
Figure 6: Protection of heart and diaphragm muscles of old mdx4cv mice with dual AAVMYO midi-Dys approach.

Cardiac function was assessed ex vivo using isolated and perfused hearts in the Langendorf chamber. a) Left ventricle developed pressure and b) negative rate of pressure change measured 5min following high workload induction by high calcium concentration. For a and b, Saline and AAV9-μDys: n=4, WTs, untreated, and AAVMYO mDys2: n=5, AAVMYO μDys: n=6. Western blot analysis of c) heart or d) diaphragm lysates showing a successful assembly and enrichment of midi-Dys2 in heart and diaphragm muscles following a long-term treatment AAV low dose (total dose: 2×1013 vg/kg). e) Plots representing the maximal force measured in vitro using diaphragm strips from WT or mdx4cv mice (AAV9-μDys: n=4, WTs, Saline, and AAVMYO μDys: n=5, AAVMYO midiDys2: n=6). f) H&E, Trichrome staining (Scale bar: 50 μm), or double-immunolabeling (Scale bar: 100 μm) of dystrophin and laminin of diaphragm cross-sections. Data represent means±s.e.m. *p<0.05, **p<0.01 and ***p<0.001. (ANOVA test followed by Tukey’s post hoc).
Discussion:
Gene replacement methods using AAV have been challenging for DMD and some other disorders due in part to a modest carrying capacity and the need for very high doses. In this study, we demonstrated the feasibility of expressing large genes by splitting the coding sequence into two or three parts transportable by AAV, which are then efficiently reconstituted into a large functional protein through the highly specific PTS mechanism mediated by split inteins.
Our results showed superior therapeutic effects of large dystrophins in comparison to micro-dystrophin in skeletal and cardiac muscles of both young and old dystrophic mice. Of note, the midi-Dys2 tested in this study represents the largest construct that can be expressed by administrating two AAV vectors and is not necessarily the most efficient or optimal mid-sized dystrophin. This construct carries 13 properly-phased SRs and all 4 hinges, together with the entire N-terminal actin-binding, cysteine-rich, and C-terminal domains. England et al. reported that a patient carrying a deletion of 46% (exons 17–48) of the DMD gene was largely asymptomatic and remained ambulatory until the 7th decade while expressing a highly functional mini-dystrophin40. We previously showed that a slightly smaller mini-dystrophin generated by properly phasing the SRs (ΔH2-SR19) was even more functional than the Δexons17–48 dystrophin in mdx mice12. Although this study did not directly compare the larger midi-Dys (ΔSR5–15) to the smaller patient mini-dystrophin (ΔH2-SR19), the data with midi-Dys (ΔSR5–15) imply that expressing a large dystrophin with additional functional domains (Fig. 1) results in better phenotypic outcomes and higher protein expression in comparison to far smaller micro-dystrophins. This could be due to differences in AAV biodistribution, protein expression, PTS efficiency, biomechanical function and/or protein stability.
Our data also demonstrate the feasibility of fusing three different dystrophin sub-fragments to generate full-length dystrophin in striated muscles using two very specific split inteins. The thorough multi-step screening of a split intein library using the split GFP system allowed identification of several pairs that can be used simultaneously without cross-splicing. The orthogonality of various candidates was also validated using dystrophin.
In addition to efficient split intein pairs, optimal gene reconstitution in skeletal muscles requires a potent AAV vector with high muscle tropism as well as strong muscle-restricted expression cassettes. Several new myotropic capsids have been engineered using directed evolution or DNA shuffling34,41,42. In this study, we confirmed the superiority and the high muscle tropism of AAVMYO, allowing the administration of 10-fold lower doses than are being used in clinical trials for DMD. Use of the potent CK8e tissue-specific expression cassette restricts expression to muscle, avoiding off-target expression in other organs, such as the liver, and further reduces potential immune reactions (the use of muscle-restricted promoters has been shown to minimize transgene immune responses43,44).
In this proof-of-principle study, all vectors carrying split intein/dystrophin were delivered at equimolar ratios, but the expression levels of each component were assessed using qualitative or semi-quantitative assays. The development of accurate assays to precisely quantify the protein levels is crucial to determine the expression efficacy and stability of each fragment before PTS occurs and to optimize their stoichiometry in the administrated AAV cocktail (Extended data Fig. 5). Nevertheless, the relative expression levels of each sub-fragment can also be adjusted by using regulatory cassettes with different transcriptional activities for different moieties.
Finally, while our study clearly demonstrates greater functional improvements in both young and old mdx4cv mice, and in both skeletal and cardiac muscle, that are attributable to both the split intein and myotropic AAV technologies, additional investigations are required to address the safety of this approach. First, the split inteins used in the current study contain ~125–150 total amino acid residues, all of whose sequences are of bacterial origins with unknown intracellular half-life. Second, the expression of each protein half, as well as reconstituted proteins containing residual intein “footprint” in patient cells, could contribute to an immune reaction. Third, the expression of exogenous dystrophins in dystrophin-deficient cells might be immunogenic, as reported with a few DMD patients treated with AAV-μDys45. The region involved in this immune reaction was mapped to the region centered around Hinge1. Whether this reaction is due intrinsically to Hinge1 or non-natural junctions of H1/SR1 to other adjacent sequences of μDys remains unclear. Although the intein-generated midi-Dys (ΔSR5–15) contains Hinge1, they are juxtaposed to continuous sequences as in normal dystrophin, which may lower the risk of exposing immunodominant epitopes.
Overall, this work addresses important limitations of AAV-based gene replacement and presents a novel method to efficiently express large and highly functional extra-large proteins at vector doses lower than currently being used in the clinic for DMD. This new method could be implemented for many other diseases involving large genes exceeding the packaging capacity of AAV vectors.
Methods
Intein selection, design, and cloning
23 split inteins were selected from “The Intein Database and Registry” (InBase) for their short sequences and previously described fast and efficient protein trans-splicing (full sequences listed in Extended Data Table 1 and Extended Data Table 2). To maximize their protein expression in mammalian cells, split inteins’ DNA sequences were codon-optimized, then DNA fragments were synthesized (Twist Biosciences, San Francisco, CA). Using the Golden Gate assembly, each half of a split intein was cloned in a plasmid containing either the N- (from methionine 1 to glutamic acid 172) or C-terminal half (from aspartic acid 173 to arginine 240) of the green fluorescent protein (GFP) driven by Cytomegalovirus (CMV) promoter. All plasmids were sequenced following bacterial transformation and miniprep plasmid purification.
In vitro pre-screening of split inteins:
Human Embryonic Kidney 293 (HEK293) cells were seeded in 96-well plates with clear bottom and black wall (Greiner Bio-One) and incubated overnight at 37°C in phenol-free DMEM (Gibco) containing 10% FBS (Sigma) and 1% PenStrep (Gibco). Using Lipofectamine 3000, 80% confluent cultures were transfected with a control plasmid expressing the wild-type GFP, GFP containing a six-residue footprint that would remain after splicing of each split intein, or dual plasmids expressing N- or C-terminal halves of split intein/GFP constructs. 24 hours later, GFP fluorescence (excitation filter: 485 nm; emission filter: 535 nm) was measured in living cells using SpectraMax® iD5 microplate reader. All conditions were tested in the same 96-well plate. Three to six biological and independent replicates were performed as described in the figure legends.
In vitro validation of split intein/dystrophin constructs:
All dystrophin constructs were subcloned from a plasmid containing the entire coding sequence of the human muscle isoform Dp427 driven by the CMV promoter, which was used in this study to express the full-length dystrophin. Following the multi-step GFP screening, the selected split intein sequences were PCR-amplified, then inserted in-frame next to dystrophin fragments cDNA using Golden Gate assembly. In addition, a midi-dystrophin version (lacking sequences of Spectrin-like repeat 5 to 15) was engineered using standard cloning techniques. The μDys5 plasmid was previously cloned and validated1. Upon cloning and successful sequencing, HEK293 cells were transfected with control plasmids that express either the full-length dystrophin or midi-Dys (ΔSR5–15), or plasmids encoding split intein/dystrophin fragments. 48 hours later, transfected cells were collected for protein extraction and analysis.
AAV vector cloning and production:
Split intein/dystrophin constructs were subcloned into pAAV plasmid containing the muscle-specific creatine kinase 8 (CK8e) regulatory cassette and small synthetic polyA flanked by two AAV serotype 2 inverted terminal repeats (ITRs). Using calcium phosphate solution, the final pAAV plasmids were co-transfected with the pDG6, pDG9, or pDG9MYO packaging plasmids (containing the AAV2 rep, and cap genes of serotype 6, 9 or MYO) into HEK293 cells in 850-cm2 roller bottle to generate recombinant AAV6, AAV9 or AAVMYO vectors as previously described2. AAV6 capsids were purified via heparin-affinity chromatography, while AAV9 and AAVMYO were precipitated using PEG8000, then full capsids were separated using CsCl gradient. All AAV preps were concentrated using sucrose gradient ultracentrifugation. Finally, the viral titers were determined by Southern blot analyses and qPCR before in vivo assessment.
Animals:
All animal experiments were performed in accordance with the University of Washington’s Institutional Animal Care and Use Committee (IACUC). In this study, male wild-type (C57BL/6) and dystrophic mdx4cv mice were used.
Intramuscular injection:
For initial screening, 3-week-old mdx4cv mice were anesthetized using isoflurane (Piramal Critical Care). 5×1010 viral genome (vg) of AAV6 was administrated into one tibialis anterior (TA) muscle, while the contralateral leg was injected with a sterile saline solution as a sham manipulation. 5 weeks later, mice were euthanized, and TA muscles were collected for analysis.
Systemic delivery:
In this study, two doses were evaluated by systemic delivery using an intravenous route. A low dose (2×1013 vg/kg) of AAVMYO or AAV9; or a high dose (2×1014 vg/kg) of AAV6 or AAVMYO were administrated into tail veins of 8-week-old or 17-month-old mdx4cv mice for a period of 3 or 6 months, respectively. Before the injection, mice were anesthetized using isoflurane. Once the AAV solution was successfully administrated, injected mice were kept in a warm cage and monitored for one hour. Also, mice were assigned a serial identification number on the injection day to conduct unbiased and blinded analyses. These numbers were used throughout the study, and the treatment history of each mouse was determined after completing the data collection.
Skeletal muscle contractile properties:
TA muscle functional analysis:
In situ specific force generation and susceptibility to contraction-induced injury of TA muscles were assessed after sciatic nerve stimulations (Aurora Scientific, model 701C). Briefly, mice were deeply anesthetized with isoflurane. Once an appropriate depth of anesthesia that prevents any response to tactile stimuli was achieved, the distal tendon of the TA muscle was detached and tied to a lever of a force transducer (Aurora Scientific, model 305B-LR) using a silk suture. The optimal muscle fiber length (L0) that produces the maximal isometric twitch was determined from the micromanipulation of muscle length. While held at L0, maximum isometric tetanic force (P0) was determined by stimulating the TA muscle at 200 Hz. The specific muscle force values were obtained by normalizing the maximum isometric force at L0 by the muscle cross-sectional area (CSA)1,3. Eccentric contractions were performed by subjecting TA muscles to a series of progressively increasing lengths (from 0% to 15% of the optimal length) under maximum stimulation. The peak isometric force generated just prior to the subsequent lengthening contraction was recorded and represented as a percentage versus the initial measurement.
In vitro diaphragm muscle performances:
Once the TA muscle force analyses were completed, the anesthetized mice were euthanized, and the entire diaphragm muscle with the surrounding ribcage was isolated and quickly transferred into a dish containing carbogenated Tyrode’s solution1. Using a light microscope, diaphragm strips composed of longitudinal and intact muscle fibers with a portion of the central tendon and rib bones were dissected. Both ends were firmly tied with surgical silk, then sutures were immediately secured to a temperature-controlled in vitro horizontal bath (Aurora Scientific, model 809A) filled with carbogen-bubbling Tyrode’s solution. L0 that led to the production of maximal isometric twitch was determined from micro-adjustments of muscle length (Aurora Scientific, model 300). Then, specific muscle force was calculated by normalizing the maximal isometric tetanic force (P0) by the muscle CSA4.
Ex vivo cardiac function assessment:
Ex vivo cardiac function was analyzed on isolated and perfused hearts using the Langendorff chamber as previously described5,6. Briefly, hearts were isolated from deeply anesthetized WT or mdx4cv mice and quickly perfused at a constant pressure of 80 mmHg with a modified Krebs-Henseleit buffer supplemented with glucose and pyruvate (118 mM NaCl, 25 mM NaHCO3, 5.3mM KCl, 2.0 mM CaCl2, 1.2 mM MgSO4, 0.5 mM ethylenediaminetetraacetic acid, 10.0 mM glucose, and 0.5 mM pyruvate) and equilibrated with 95% O2 and 5% CO2 at pH 7.4. Temperature was maintained at 37.5°C throughout the protocol. The left ventricle developed pressure, and the minimum and maximum rate of pressure change in the ventricle (±dP/dt) were measured by inserting a water-filled balloon into the LV, which was connected to a pressure transducer (PowerLab, ADInstruments, Colorado Springs, Colorado). After 5 min of stabilization, the perfusate was changed to an identical buffer as above, except for the addition of 4.0 mM CaCl2 to simulate a high workload challenge for 20 min.
Protein extraction and western blot:
Total proteins were extracted from HEK293 cell pellets or serial muscle cryosections using radioimmunoprecipitation analysis buffer (RIPA) supplemented with 1 mM PMSF and 5% protease inhibitor cocktail (P8340, Sigma). Total protein concentration was determined using the Pierce BCA assay kit (ThermoFisher). Samples were denatured at 100 °C for 10 min, then 30 μg of protein lysates were separated in NuPage 4–12% Bis-Tris polyacrylamide gels (Invitrogen). Protein transfer to 0.45 μm PVDF membranes (Amersham hybond) was performed at 120 volts at 4 °C for 2 h. Membranes were blocked for 2 h in Tris-Buffered Saline containing 5% non-fat dry milk and 0.005% Tween20 before overnight incubation with antibodies against dystrophin N-terminal (mouse Manex1011b, DSHB) or C-terminal (mouse NCL-DYS2, Leica); or Gapdh (rabbit, Sigma G9545) as a loading control. Secondary antibodies coupled to horseradish peroxidase were goat anti-rabbit (Jackson 111–035-144), anti-murine IgG2a (Jackson 115–035-206), or anti-murine IgG1 (Jackson 115–035-205). The full list of antibodies, sources, and concentrations is described in Extended Data Table 3. Blots were incubated for 2 h at room temperature, then they were visualized in Chemidoc MP imaging system (BioRad) using Clarity Western ECL substrate (BioRad). Band densitometry measurements were performed on unsaturated images using Fiji image analysis software.
Muscle histology analysis:
Following mice euthanasia, skeletal and cardiac muscles were quickly dissected, and weighed, while diaphragm muscles (a half of the diaphragm segment) were embedded in OTC. Samples were snap-frozen in liquid nitrogen-cooled isopentane and stored at −80°C for subsequent analysis.
General muscle morphology:
10 μm transversal cryo-sections of TA, gastrocnemius, soleus, heart, or diaphragm samples were stained with Hematoxylin and Eosin (H&E) or Trichrome. Whole sections were imaged with the Hamamatsu NanoZoomer slide scanner. The percentage of myofibers with centralized nuclei, the myofiber size, minimal fiber diameter (miniFeret), and the fibrosis area were measured using Fiji image analysis software.
Immunofluorescence:
Cross-sections of TA, gastrocnemius, soleus, heart, or diaphragm were stained with antibodies against dystrophin N-terminal (homemade rabbit 246)7, middle rod-domain (mouse IgG2a NCL-DYS1, Leica) or C-terminal (mouse IgG1 NCL-DYS2, Leica); Laminin2 (L0663 Rat, Sigma), alpha-sarcoglycan (mouse IgG1 NCL-a-SARC Leica), beta-dystroglycan (mouse IgG2a NCL-b-DG Leica). Secondary antibodies were goat anti-mouse IgG1 Alexa488 (A-21121, Invitrogen), IgG2a Alexa594 (A-21135, Invitrogen), anti-rat Alexa488 (Jackson 112–545-167), anti-rat Alexa594 (A-11007, Invitrogen), anti-rabbit Alexa700 (A-21038, Invitrogen), anti-rabbit Alexa594 (A-11012, Invitrogen), anti-rabbit Alexa488 (A11034, Invitrogen), anti-rabbit Alexa350 (A-21068, Invitrogen). The full list of antibodies, sources, and concentrations is provided in Extended Data Table 4. Slides were mounted using Dapi-Fluoromount-G (SouthernBiotech), and images were captured on Nikon Eclipse 90i Microscope.
Serum CK level analysis:
Blood samples were collected by cardiac puncture from anesthetized mice. Clots were allowed to form while samples rested at room temperature for 15 min. Samples were then centrifuged at 3,500 g for 10 min at 4 °C, and serum was immediately collected and stored at −80 °C. Circulating CK levels were measured using the Creatine Kinase Activity Assay Kit (MAK116, Sigma) following the manufacturer’s instructions.
Data collection and statistical analysis:
Once all the data collection was completed, blinding was lifted, and data were organized by groups using Microsoft Excel. Graphs and curves were made using Prism software (GraphPad). Values were expressed as mean±s.e.m or in violin graphs to show sample distribution. Significant differences were determined using analysis of variances (ANOVA) followed by Dunnet’s or Tukey’s test to identify differences between means. Significance was accepted at p ≤ 0.05. Differences between means are indicated by different symbols as defined in the legends.
Extended Data
Extended Data Figure 1: Validation of GFP as a platform for split intein screening.

a) Diagram illustrating the topology of the GFP folding pattern with the chromophore, alpha helices, beta sheets, and the connecting loops with the two tested splitting sites (site#1: Glutamine 157-Lysine158, site#2: Glutamic acid 172-Aspartic acid 173). The numbers represent the delimiting residues at the beginning and the end of the secondary structures. b) Brightfield and fluorescent microscopy pictures of living HEK293 cells transfected with the WT or mutated GFP. c) Mean fluorescence intensities of transfected HEK293 cells with either WT GFP or two mutated GFP harboring 8-amino acid insertions within the tested sites. Site#2 was more permissive to the insertion of an octapeptide and, thus, was selected as a splitting site where different split inteins were inserted for initial screening. d) GFP fluorescence intensity measured on living HEK293 cells 24h post-transfection with a single plasmid expressing either WT GFP, control (ctrl) GFP with a six-residue footprint (checkered bars), or a dual plasmid expressing split intein/GFP. The protein ligation efficiency of each split intein (ratio of GFP fluorescence of a given intein to its internal control) is labeled on each bar (n=6). e) Characterization of the orthogonality of the preselected split inteins. The matrix shows cross-reactivity between split inteins from Group 1 but high specificity with pairs from Group 2. f) Determination of minimal extein AA required for efficient PTS of the top four split inteins using a combination of intein halves with variable linkers (n=3). Values are represented as an average of independent transfections ± s.e.m, that are normalized to values from cells transfected with WT GFP.
Extended Data Fig. 2: Illustration of splitting sites tested to generate the midi-Dys (ΔSR5–15) using split gp41.1.

Split gp41.1 with an optimized footprint (Gly-Tyr/Ser) was tested in four locations. The resulting midi-Dys (SR5–15) harbor a footprint between one to three residues. These additional residues were inserted in loops between Helix B and C of Spectrin-like repeat 19, the linker between Helix C and Hinge 3, or within Hinge 3 to minimize their impact on protein folding.
Extended Data Fig. 3: Illustration of splitting sites tested with triple AAV vector strategy to express the full-length dystrophin.

Optimized split Aha, IMPDH, and Nrdj1 with minimal footprints were inserted in loops located between Helix A & B or Helix B & C of Spectrin-like repeat 7 as well as Helix A & B of Spectrin-like repeat 8. These combinations join the N-terminal (from ABD to Spectrin-like repeat 7/8) to the middle fragment (from Spectrin-like repeat 7/8 to repeat 19/Hinge3). Split gp41.1 was evaluated to fuse the middle fragment to the C-terminal fragment at the same four splitting sites validated above with the dual strategy.
Extended Data Fig. 4: Evaluation of PTS to express full-length dystrophin using the optimized split inteins.

a) Optimized split Aha with a minimal footprint (Ala-Glu-Tyr) was tested in four sites located in loops between Helix B & C of Spectrin-like repeat 7, or Helix A & B of Spectrin repeat 8. By using the native amino acids of dystrophin as part of the catalyzing reaction, the PTS mediated by the optimized Aha results in a minimal footprint with the insertion of only one residue (Tyr in AhS1 or AhS2, or Ala in AhS3 or AhS4). b) Optimized split IMPDH with a minimal footprint (Gly/Ser) was inserted in four sites located in loops between Helix A & B of Spectrin-like repeat 7 or Spectrin-like repeat 8. The PTS mediated by the optimized IMPDH results in a minimal footprint with the insertion of one (Gly in ImS1) or two residues (Gly-Ser in Ims3 & ImS4), while in ImS2 native Gly and Ser were used as catalyzing amino acids. c) The same splitting sites tested with IMPDH were evaluated with Nrdj-1. The PTS mediated by the optimized Nrdj-1 results in a minimal footprint with either substitution of the native Asp (D) by Cys (in NrS1), or insertion of a new residue (Cys in NrS2). Similarly, two residues (Cys-Ser) were inserted in NrS3 in the loop between Helix A and B of Spectrin repeat 8, while in NrS4 substitution of native Tyr-lys (K) by the footprint (Cys-Ser). d) Optimized split gp41.1 was tested to join an N-terminal dystrophin fragment carrying two-thirds of the protein (from ABD to Spectrin-like repeat 19/Hinge 3) to the C-terminal one-third of dystrophin (from Spectrin-like repeat 19/Hinge 3 to C-terminus) using the same split sites used to reconstitute the midi-Dys (SR5–15). e) The efficacy of each split intein to reconstitute full-length dystrophin from two fragments (one-third + two-thirds of dystrophin) was evaluated in vitro at four splitting sites. f) Two split inteins, IMPDH (split site: ImS4) and Nrdj-1 (split site: NrS4), were selected from previous screenings and tested for their ability to join the N-terminal to the middle fragment in a triple vector strategy. Split gp41.1 was inserted at the GpS4 split site to join the middle fragment to the C-terminal fragment.
Extended Data Fig. 5: Histology analysis of WT or mdx4cv T.A cross-sections injected saline, dual AAV midi-Dys2 at different ratios, or individual fragments:

a) Cross-sections stained with H&E (top) or immunolabeled with antibodies against the midi-Dys2 N- or C-terminal fragments (bottom, scale bars: 50μm). b) Quantification of myofibers with central nuclei was determined from sections stained with H&E (WT and N-ter alone: n=4; C-ter alone and dual vector: n=5; Saline: n=6). c) Western blot of protein lysate from T.A muscle injected with various ratios of AAV6 midi-Dys moieties shows a similar expression of the final product (midi-Dys ΔSR5–15). However, using 1:1 ratio showed accumulation of the C-terminal fragment. d) The percentage of myofibers with central nuclei was determined using (WT: n=4; [1:1]: n=5; Saline, [3:1], and [6:1]: n=6) e) T.A cross-sections stained with H&E (scale bars: 50μm). f) Quantification of myofibers with central nuclei following 5 weeks post AAV6 intramuscular injection (WT and dual vector: n=5; Triple vector: n=6; Saline: n=12) Data represent mean ± s.e.m. *p<0.05, **p<0.01, ***p<0.001 for muscles treated AAV vs saline (ANOVA test followed by Dunnett’s post hoc). mDys: midi-dystrophin. fDys: Full-length dystrophin
Extended Data Fig. 6: General morphology analysis of T.A and hearts from AAV6-treated cohort.

a) Heart sections of control mice or mdx4cv males treated with single, dual, or triple AAV6 vectors stained with H&E or double-immunolabeled for dystrophin and laminin (Scale bars: 100μm). Dashed rectangles represent a higher magnification view. b) Percentage of dystrophin-positive myofibers, c) fiber area, d) minimal fiber diameter measured using T.A cross sections (n=4, more than 1000 fiber per each sample). For b, data represent mean ± s.e.m. In c and d, the average values are shown on top of the violin bars. The solid line represents the median, while the dashed lines show the quartiles. μDys: micro-dystrophin. mDys: midi-dystrophin. fDys: Full-length dystrophin. Dys+: Dystrophin positive. vg: viral genome. kg: Kilogram
Extended Data Fig. 7: Histology and protein expression analyses from mice treated with low or high doses of AAVs.

a) Immunostaining of T.A cross-sections showing that expression of dystrophins restores the localization of α-sarcoglycan and β-dystroglycan. Cross-sections were triple immunolabeled with antibodies against dystrophin, α-sarcoglycan or β-dystroglycan (Scale bars: 100μm). Bottom rows are heart transverse cryosections stained with H&E or immunolabelled for dystrophin and laminin (Scale bars: 100μm). b) Percentage of centrally nucleated myofibers measured using H&E-stained T.A sections (~1000 fiber quantified per section). Data represent mean ± s.e.m. Fiber area determined from c) T.A muscles (n=1000 fibers) or d) diaphragms (n=400 fiber) using cross-sections stained for laminin. The average values are shown on top of the violin bars. The solid line represents the median, while the dashed lines show the quartiles. Western blot example of heart (e) and diaphragm (f) samples collected from mice treated with AAV9 or AAVMYO at a low or high dose and probed with antibodies against the N-terminal portion of dystrophin or GAPDH as a loading control. μDys: micro-dystrophin. mDys: midi-dystrophin. Ab: antibody. kDa: kilodalton. MW: molecular weight.
Extended Data Fig. 8: Histology analysis of T.A and gastrocnemius muscles from the old cohort.

a) Whole T.A cross-sections stained with Trichrome (top, scale bars: 500μm) with a large view of the selected field in a white rectangle represented underneath (scale bars: 100μm). The yellow rectangles (zoomed-in view) are represented in Figure 5a. The bottom rows represent gastrocnemius cross-sections stained with H&E, Trichrome (scale bars: 100μm), or immunolabeled with antibodies against dystrophin or laminin (scale bars: 250μm). b) western blot analysis of dystrophin expression with GAPDH as loading control from gastrocnemius muscle lysates. Percentage of dystrophin-myofibers determined from c) T.A or d) gastrocnemius muscles (n=4 samples, 1000 fibers per sample). e) Fibrosis area measured using gastrocnemius sections stained with Trichrome (n=5). f) Percentage of myofibers with central nuclei in T.A muscle sections (n=4 samples, 1000 fibers per sample). For c-f, data represent mean ± s.e.m. g) myofiber area and h) minimal diameter measured in T.A muscle sections. i) Area and j) minimal diameter of gastrocnemius myofibers. (n=4 samples, 1000 fibers per sample). The average values are shown on top of the violin bars. The solid line represents the median, while the dashed lines show the quartiles. *p<0.05, ***p<0.001. (ANOVA test followed by Tukey’s post hoc).
Extended Data Fig. 9: Histology analysis and protein expression in soleus muscles from the old cohort.

a) Soleus cross-sections stained with H&E or Trichrome (scale bars large view: 250μm, zoomed-in view: 50μm), or immunostained with antibodies against dystrophin or laminin (scale bars: 250μm). b) Western blot analysis of dystrophin expression with GAPDH as loading control from soleus muscle lysates. c) Percentage of dystrophin-myofibers determined from soleus muscles (n=4 samples, ~400 fibers per sample). d) Fibrosis area measured using soleus sections stained with Trichrome (n=5). e) Myofiber area and f) minimal diameter determined from soleus cross-sections (n=4 samples, ~400 fibers per sample). g) Fibrosis area (n=5) and h) percentage of dystrophin-positive myofibers determined from diaphragm samples (n=4). i) Myofiber area and j) minimal diameter determined from diaphragm cross-sections (n=4 samples, ~400 fibers per sample). In c, d, g, and h, data represent mean ± s.e.m. ***p<0.001 (ANOVA test followed by Tukey’s post hoc). For e, f, i, and j, the average values are shown on top of the violin bars. The solid line represents the median, while the dashed lines show the quartiles.
Extended Data Fig. 10: Functional and histology analyses of hearts from the old cohort.

a) The left ventricle developed pressure (the difference between systolic and diastolic pressures), and b) the negative rate of pressure change calculated by the first derivative of the descending LV pressure wave (−dP/dt) of isolated and perfused hearts were measured ex vivo at a baseline (BL), then for 20min under high workload condition (HWL) using a glucose-pyruvate buffer containing high calcium (4.0 mmol/l). Saline, AAV9-μDys: n=4, WTs; untreated, and AAV-MYO-μDys and midi-Dys2: n=6. c) Heart transverse cryosections stained with H&E, Trichrome or immunolabelled (Scale bars: 100μm) for dystrophin and laminin (Scale bars: 200μm). d) Fibrosis area measured using heart sections stained with Trichrome (n=5). data represent mean ± s.e.m. **p<0.01. (ANOVA test followed by Tukey’s post hoc).
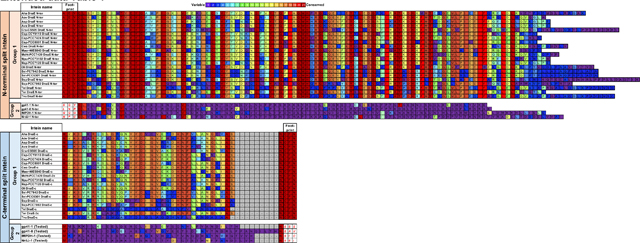
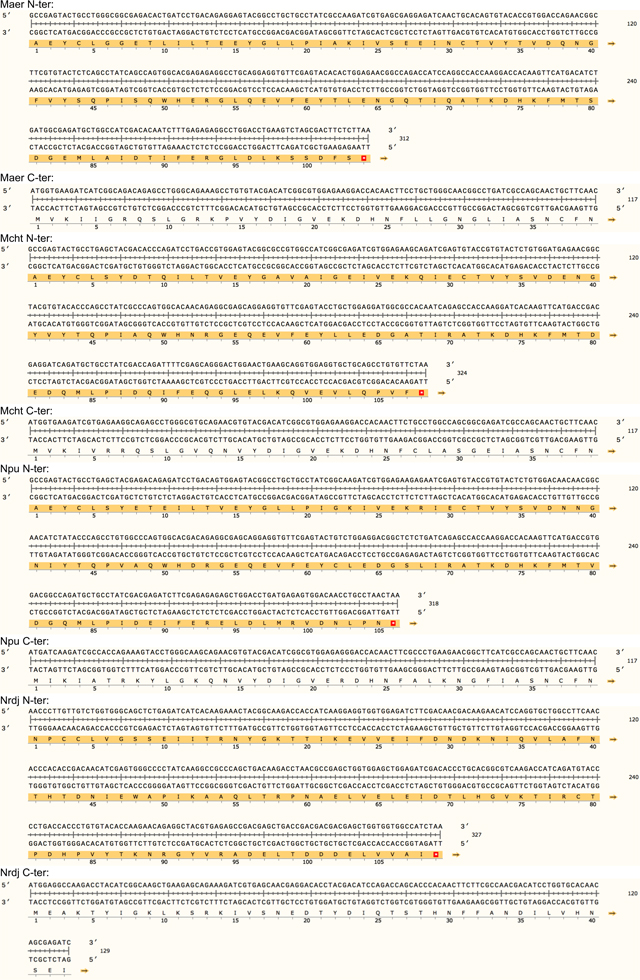
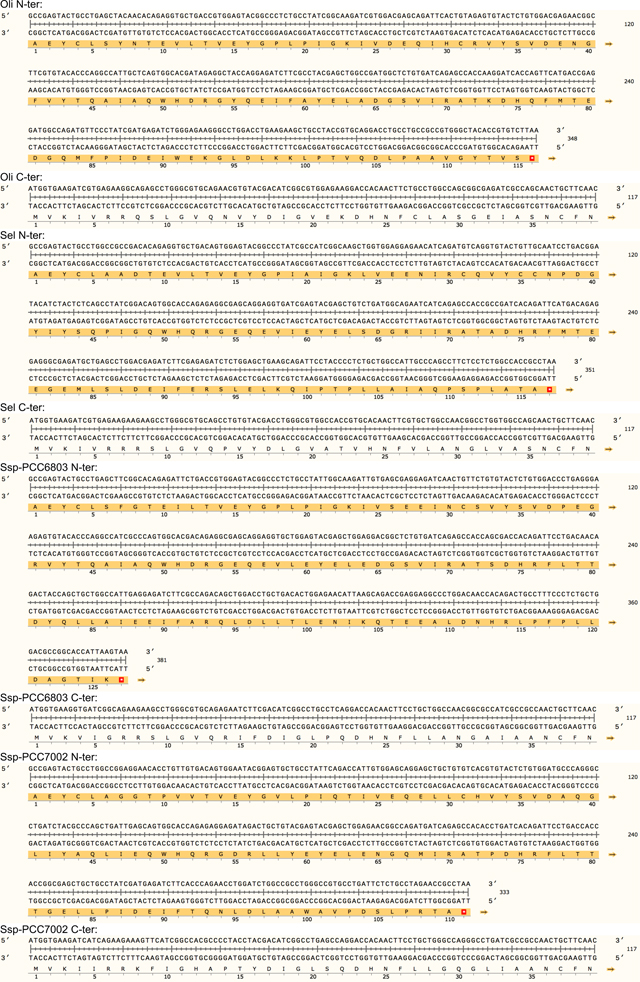
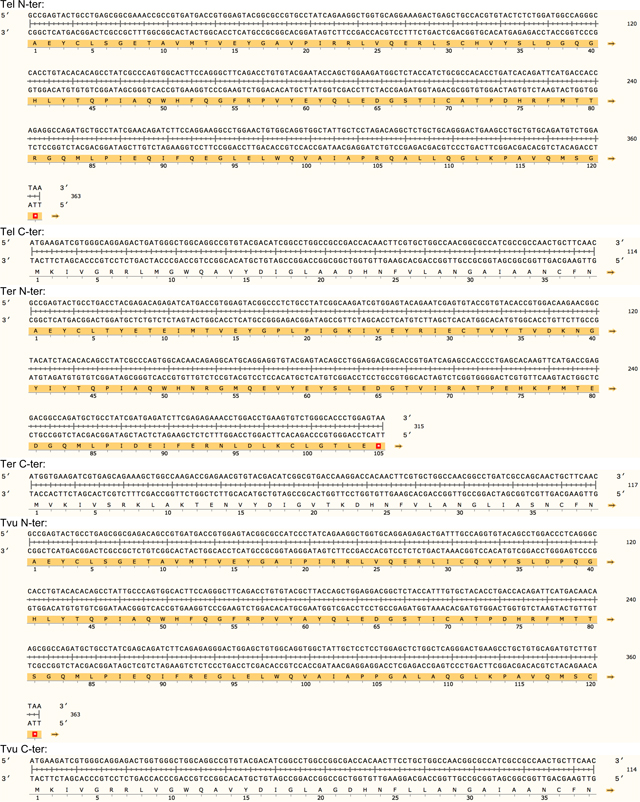
Extended data Table 1. Protein sequence of the 23 split inteins with amino acid conservation.
The intein library was divided into two subgroups. Group 1 contains 19 split intein pairs that share high sequence homology including the 6-residue footprint (i.e AEY and CFN at respectively the N- and the C-terminal end). Group 2 was composed of four candidates that are unique with 6-residue footprints for each pair (i.e. SGY/SSS for gp41.1, LNR/SAV for gp41.8, GGG/SIC for IMPDH and NPC/SEI for Nrdj1).
|
Extended data Table 2.
Codon-optimized DNA sequences of the tested split inteins.
|
|
|
|
|
|
Extended data Table 3.
List of antibodies used for western blot.
| Figure | Protein | Primary Antibody | Secondary antibody | ||
|---|---|---|---|---|---|
| ID, source | Concentration | ID, source | Concentration | ||
| Figure 1b, Figure 1c, Figure 2i, Figure 2j, Figure 3d, Figure 5b, Figure 6c, Figure 6d, Extended data Fig. 4e, Extended data Fig. 7e, Extended data Fig. 7f, Extended data Fig. 8b, Extended data Fig. 9b | Dystrophin (N-ter) | Manex1011B (Developmental Studies Hybridoma Bank) | 1:500 | Peroxidase-conjugated Goat Anti-Mouse IgG, Fcγ subclass 2a specific, Jackson 111-035-206 | 1:5,000 |
| Figure 2a, Figure 2e Extended data Fig. 5c | Dystrophin (C-ter) | Leica NCL-DYS2 | 1:500 | Peroxidase-conjugated Goat Anti-Mouse IgG, Fcγ subclass 1 specific, Jackson 111-035-205 | 1:5,000 |
| Figure 1b, Figure 1c, Figure 2a, Figure 2e, Figure 2i, Figure 2j, Figure 3d, Figure 5b, Figure 6c, Figure 6d, Extended data Fig. 4e, Extended data Fig. 5c, Extended data Fig. 7e, Extended data Fig. 7f, Extended data Fig. 8b, Extended data Fig. 9b | Gapdh | Sigma G9545 | 1:20,000 | Peroxidase-conjugated Goat Anti-Rabbit IgG (H+L), Jackson 111-035-144 | 1:10,000 |
Extended data Table 4.
List of antibodies used for immunofluorescence.
| Figure | Protein | Primary Antibody | Secondary antibody | ||
|---|---|---|---|---|---|
| ID, source | Concentration | ID, source | Concentration | ||
| Figure 2d, Figure 2k, Figure 4a, Figure 5a, Figure 6f, Extended data Fig. 5a, Extended data Fig. 6a, Extended data Fig. 7a (heart samples), Extended data Fig. 8a, Extended data Fig. 9a | Dystrophin (N-ter) | Rabbit 246 (homemade) | 1:400 | Goat anti-Rabbit IgG (H+L) Alexa Fluor 594, Invitrogen A-11012 | 1:400 |
| Figure 2d, Extended data Fig. 5a | Dystrophin (C-ter) | Leica NCL-DYS2 | 1:400 | Goat anti-mouse IgG1 Alexa Fluor 488, Invitrogen A-21121 | 1:400 |
| Figure 2h | Dystrophin (N-ter) | Rabbit 246 (homemade) | 1:400 | Goat anti-Rabbit IgG (H+L) Alexa Fluor 700, Invitrogen A-21038 | 1:200 |
| Dystrophin (middle) | Leica NCL-DYS1, | 1:400 | Goat anti-Mouse IgG2a Alexa 594, Invitrogen A- 21135 | 1:400 | |
| Dystrophin (C-ter) | Leica NCL-DYS2 | 1:400 | Goat anti-mouse IgG1 Alexa Fluor 488, Invitrogen A-21121 | 1:400 | |
| Extended data Fig.7a (Tibialis anterior samples) | Dystrophin (N-ter) | Rabbit 246 (homemade) | 1:400 | Goat anti-Rabbit IgG (H+L) Alexa 350, Invitrogen A-21068 | 1:400 |
| Alpha-sarcoglycan | Leica, NCL-a-SARC | 1:400 | Goat anti-mouse IgG1 Alexa Fluor 488, Invitrogen A-21121 | 1:400 | |
| Beta-dystroglycan | Leica NCL-b-DG | 1:400 | Goat anti-Mouse IgG2a Alexa 594, Invitrogen A- 21135 | 1:400 | |
| Figure 2k, Figure 4a, Figure 5a, Figure 6f, Extended data Fig. 6a, Extended data Fig. 7a (heart samples), Extended data Fig. 8a, Extended data Fig. 9a | Laminin-2 | L0663 Rat, Sigma | 1:400 | Goat Anti-Rat IgG (H+L) Alexa 488, Jackson 112–545-167 | 1:400 |
| Extended data Fig. 10c | Dystrophin (N-ter) | Rabbit 246 (homemade) | 1:400 | Goat anti-Rabbit IgG (H+L) Alexa 488, Invitrogen A-11034 | 1:400 |
| Laminin-2 | L0663 Rat, Sigma | 1:400 | Goat Anti-Rat IgG (H+L) Alexa 594, Invitrogen A-11007 | 1:400 | |
Supplementary Material
Acknowledgments:
We thank the Histology and Imaging Core of the University of Washington for the excellent technical assistance. This work was supported by research grants from Muscular Dystrophy Association (MDA, USA). H.T was supported by fellowships from Bettencourt-Schueller Foundation, Philippe Foundation, and Association Française Contre Les Myopathies (AFM-Telethon).
Footnotes
Competing interests:
The University of Washington has intellectual property based on the findings of this study. J.S.C and S.D.H are inventors of patents covering ΔH2-R19/ΔCT μDys, μDys5 and CK8e expression cassettes. D.G is an inventor of a patent describing the development of the AAVMYO capsid. The other authors declared no competing interests.
Data availability:
All relevant data that support the findings of this study are available in supplementary information files and from the corresponding authors upon reasonable request.
References Methods:
- 1.Ramos JN et al. Development of Novel Micro-dystrophins with Enhanced Functionality. Mol Ther 27, 623–635 (2019). 10.1016/j.ymthe.2019.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halbert CL, Allen JM & Chamberlain JS AAV6 Vector Production and Purification for Muscle Gene Therapy. Methods Mol Biol 1687, 257–266 (2018). 10.1007/978-1-4939-7374-3_18 [DOI] [PubMed] [Google Scholar]
- 3.Dellorusso C, Crawford RW, Chamberlain JS & Brooks SV Tibialis anterior muscles in mdx mice are highly susceptible to contraction-induced injury. J Muscle Res Cell M 22, 467–475 (2001). https://doi.org:Doi 10.1023/A:1014587918367 [DOI] [PubMed] [Google Scholar]
- 4.Gregorevic P, Plant DR, Leeding KS, Bach LA & Lynch GS Improved contractile function of the mdx dystrophic mouse diaphragm muscle after insulin-like growth factor-I administration. Am J Pathol 161, 2263–2272 (2002). https://doi.org:Doi 10.1016/S0002-9440(10)64502-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kolwicz SC Jr. & Tian R Assessment of cardiac function and energetics in isolated mouse hearts using 31P NMR spectroscopy. J Vis Exp (2010). 10.3791/2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kolwicz SC Jr. et al. Gene Therapy Rescues Cardiac Dysfunction in Duchenne Muscular Dystrophy Mice by Elevating Cardiomyocyte Deoxy-Adenosine Triphosphate. JACC Basic Transl Sci 4, 778–791 (2019). 10.1016/j.jacbts.2019.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rafael JA et al. Forced expression of dystrophin deletion constructs reveals structure-function correlations. J Cell Biol 134, 93–102 (1996). https://doi.org:Doi 10.1083/jcb.134.1.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 1.Monaco AP et al. Detection of Deletions Spanning the Duchenne Muscular-Dystrophy Locus Using a Tightly Linked DNA Segment. Nature 316, 842–845 (1985). https://doi.org:DOI 10.1038/316842a0 [DOI] [PubMed] [Google Scholar]
- 2.Kunkel LM et al. Analysis of Deletions in DNA from Patients with Becker and Duchenne Muscular-Dystrophy. Nature 322, 73–77 (1986). https://doi.org:DOI 10.1038/322073a0 [DOI] [PubMed] [Google Scholar]
- 3.Danialou G et al. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. Faseb J 15, 1655-+ (2001). 10.1096/fj.01-0030fje [DOI] [PubMed] [Google Scholar]
- 4.Petrof BJ, Shrager JB, Stedman HH, Kelly AM & Sweeney HL Dystrophin Protects the Sarcolemma from Stresses Developed during Muscle-Contraction. P Natl Acad Sci USA 90, 3710–3714 (1993). https://doi.org:DOI 10.1073/pnas.90.8.3710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ervasti JM & Campbell KP Membrane Organization of the Dystrophin-Glycoprotein Complex. Cell 66, 1121–1131 (1991). https://doi.org:Doi 10.1016/0092-8674(91)90035-W [DOI] [PubMed] [Google Scholar]
- 6.Ervasti JM & Campbell KP A Role for the Dystrophin-Glycoprotein Complex as a Transmembrane Linker between Laminin and Actin. J Cell Biol 122, 809–823 (1993). https://doi.org:DOI 10.1083/jcb.122.4.809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Emery AEH The muscular dystrophies. Lancet 359, 687–695 (2002). https://doi.org:Doi 10.1016/S0140-6736(02)07815-7 [DOI] [PubMed] [Google Scholar]
- 8.Banks GB, Judge LM, Allen JM & Chamberlain JS The Polyproline Site in Hinge 2 Influences the Functional Capacity of Truncated Dystrophins. Plos Genet 6 (2010). https://doi.org:ARTN e1000958 10.1371/journal.pgen.1000958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gregorevic P et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med 12, 787–789 (2006). 10.1038/nm1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramos JN et al. Development of Novel Micro-dystrophins with Enhanced Functionality. Mol Ther 27, 623–635 (2019). 10.1016/j.ymthe.2019.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bostick B et al. AAV micro-dystrophin gene therapy alleviates stress-induced cardiac death but not myocardial fibrosis in > 21-m-old mdx mice, an end-stage model of Duchenne muscular dystrophy cardiomyopathy. J Mol Cell Cardiol 53, 217–222 (2012). 10.1016/j.yjmcc.2012.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harper SQ et al. Modular flexibility of dystrophin: Implications for gene therapy of Duchenne muscular dystrophy. Nat Med 8, 253–261 (2002). https://doi.org:DOI 10.1038/nm0302-253 [DOI] [PubMed] [Google Scholar]
- 13.Wasala LP et al. The implication of hinge 1 and hinge 4 in micro-dystrophin gene therapy for Duchenne muscular dystrophy. Hum Gene Ther (2022). 10.1089/hum.2022.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birch SM et al. Assessment of systemic AAV-microdystrophin gene therapy in the GRMD model of Duchenne muscular dystrophy. Sci Transl Med 15 (2023). https://doi.org:ARTN abo1815 10.1126/scitranslmed.abo1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koo T, Popplewell L, Athanasopoulos T & Dickson G Triple Trans-Splicing Adeno-Associated Virus Vectors Capable of Transferring the Coding Sequence for Full-Length Dystrophin Protein into Dystrophic Mice. Human Gene Therapy 25, 98–108 (2014). 10.1089/hum.2013.164 [DOI] [PubMed] [Google Scholar]
- 16.Lai Y et al. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat Biotechnol 23, 1435–1439 (2005). 10.1038/nbt1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lostal W, Kodippili K, Yue YP & Duan DS Full-Length Dystrophin Reconstitution with Adeno-Associated Viral Vectors. Human Gene Therapy 25, 552–562 (2014). 10.1089/hum.2013.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Odom GL, Gregorevic P, Allen JM & Chamberlain JS Gene therapy of mdx mice with large truncated dystrophins generated by recombination using rAAV6. Mol Ther 19, 36–45 (2011). 10.1038/mt.2010.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shah NH & Muir TW Inteins: nature’s gift to protein chemists. Chem Sci 5, 446–461 (2014). 10.1039/c3sc52951g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Esposito F et al. Liver gene therapy with intein-mediated F8 trans-splicing corrects mouse haemophilia A. Embo Mol Med 14 (2022). https://doi.org:ARTN e15199 10.15252/emmm.202115199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li YF Split-inteins and their bioapplications. Biotechnol Lett 37, 2121–2137 (2015). 10.1007/s10529-015-1905-2 [DOI] [PubMed] [Google Scholar]
- 22.Padula A et al. Full-length ATP7B reconstituted through protein trans-splicing corrects Wilson disease in mice. Mol Ther Methods Clin Dev 26, 495–504 (2022). 10.1016/j.omtm.2022.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tornabene P et al. Inclusion of a degron reduces levels of undesired inteins after AAV-mediated protein trans-splicing in the retina. Mol Ther-Meth Clin D 23, 448–459 (2021). 10.1016/j.omtm.2021.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tornabene P et al. Intein-mediated protein trans-splicing expands adeno-associated virus transfer capacity in the retina. Sci Transl Med 11 (2019). https://doi.org:ARTN eaav4523 10.1126/scitranslmed.aav4523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J, Sun WC, Wang B, Xiao X & Liu XQ Protein trans-splicing as a means for viral vector-mediated in vivo gene therapy. Human Gene Therapy 19, 958–964 (2008). 10.1089/hum.2008.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carvajal-Vallejos P, Pallissé R, Mootz HD & Schmidt SR Unprecedented Rates and Efficiencies Revealed for New Natural Split Inteins from Metagenomic Sources. J Biol Chem 287, 28686–28696 (2012). 10.1074/jbc.M112.372680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caspi J, Amitai G, Belenkiy O & Pietrokovski S Distribution of split DnaE inteins in cyanobacteria. Mol Microbiol 50, 1569–1577 (2003). 10.1046/j.1365-2958.2003.03825.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shah NH, Dann GP, Vila-Perelló M, Liu ZH & Muir TW Ultrafast Protein Splicing is Common among Cyanobacterial Split Inteins: Implications for Protein Engineering. J Am Chem Soc 134, 11338–11341 (2012). 10.1021/ja303226x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abedi MR, Caponigro G & Kamb A Green fluorescent protein as a scaffold for intracellular presentation of peptides. Nucleic Acids Res 26, 623–630 (1998). https://doi.org:DOI 10.1093/nar/26.2.623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crudele JM & Chamberlain JS AAV-based gene therapies for the muscular dystrophies. Hum Mol Genet 28, R102–R107 (2019). 10.1093/hmg/ddz128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boer JM, de Meijer EJ, Mank EM, van Ommen GB & den Dunnen JT Expression profiling in stably regenerating skeletal muscle of dystrophin-deficient mice. Neuromuscular Disord 12, S118–S124 (2002). https://doi.org:Pii S0960-8966(02)00092-5Doi 10.1016/S0960-8966(02)00092-5 [DOI] [PubMed] [Google Scholar]
- 32.Torres LFB & Duchen LW The Mutant Mdx - Inherited Myopathy in the Mouse - Morphological-Studies of Nerves, Muscles and End-Plates. Brain 110, 269–299 (1987). https://doi.org:DOI 10.1093/brain/110.2.269 [DOI] [PubMed] [Google Scholar]
- 33.Bengtsson NE, Tasfaout H, Hauschka SD & Chamberlain JS Dystrophin Gene-Editing Stability Is Dependent on Dystrophin Levels in Skeletal but Not Cardiac Muscles. Mol Ther 29, 1070–1085 (2021). 10.1016/j.ymthe.2020.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weinmann J et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat Commun 11 (2020). https://doi.org:ARTN 5432 10.1038/s41467-020-19230-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lynch GS, Hinkle RT, Chamberlain JS, Brooks SV & Faulkner JA Force and power output of fast and slow skeletal muscles from mdx mice 6–28 months old. J Physiol-London 535, 591–600 (2001). https://doi.org:DOI 10.1111/j.1469-7793.2001.00591.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pastoret C & Sebille A Mdx Mice Show Progressive Weakness and Muscle Deterioration with Age. J Neurol Sci 129, 97–105 (1995). https://doi.org:Doi 10.1016/0022-510x(94)00276-T [DOI] [PubMed] [Google Scholar]
- 37.Stedman HH et al. The Mdx Mouse Diaphragm Reproduces the Degenerative Changes of Duchenne Muscular-Dystrophy. Nature 352, 536–539 (1991). https://doi.org:DOI 10.1038/352536a0 [DOI] [PubMed] [Google Scholar]
- 38.Lefaucheur JP, Pastoret C & Sebille A Phenotype of Dystrophinopathy in Old Mdx Mice. Anat Rec 242, 70–76 (1995). https://doi.org:DOI 10.1002/ar.1092420109 [DOI] [PubMed] [Google Scholar]
- 39.Chamberlain JS, Metzger J, Reyes M, Townsend DW & Faulkner JA Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. Faseb J 21, 2195–2204 (2007). 10.1096/fj.06-7353com [DOI] [PubMed] [Google Scholar]
- 40.England SB et al. Very Mild Muscular-Dystrophy Associated with the Deletion of 46-Percent of Dystrophin. Nature 343, 180–182 (1990). https://doi.org:DOI 10.1038/343180a0 [DOI] [PubMed] [Google Scholar]
- 41.El Andari J et al. Semirational bioengineering of AAV vectors with increased potency and specificity for systemic gene therapy of muscle disorders. Sci Adv 8 (2022). https://doi.org:ARTN eabn4704 10.1126/sciadv.abn4704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tabebordbar M et al. Directed evolution of a family of AAV capsid variants enabling potent muscle-directed gene delivery across species. Cell 184, 4919-+ (2021). 10.1016/j.cell.2021.08.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hartigan-O’Connor D, Kirk CJ, Crawford R, Mule JJ & Chamberlain JS Immune evasion by muscle-specific gene expression in dystrophic muscle. Mol Ther 4, 525–533 (2001). https://doi.org:DOI 10.1006/mthe.2001.0496 [DOI] [PubMed] [Google Scholar]
- 44.Cordier L et al. Muscle-specific promoters may be necessary for adeno-associated virus-mediated gene transfer in the treatment of muscular dystrophies. Human Gene Therapy 12, 205–215 (2001). https://doi.org:Doi 10.1089/104303401750061267 [DOI] [PubMed] [Google Scholar]
- 45.Boennemann CG et al. Dystrophin Immunity after Gene Therapy for Duchenne’s Muscular Dystrophy. New Engl J Med 388, 2294–2296 (2023). 10.1056/NEJMc2212912 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data that support the findings of this study are available in supplementary information files and from the corresponding authors upon reasonable request.