

Summary
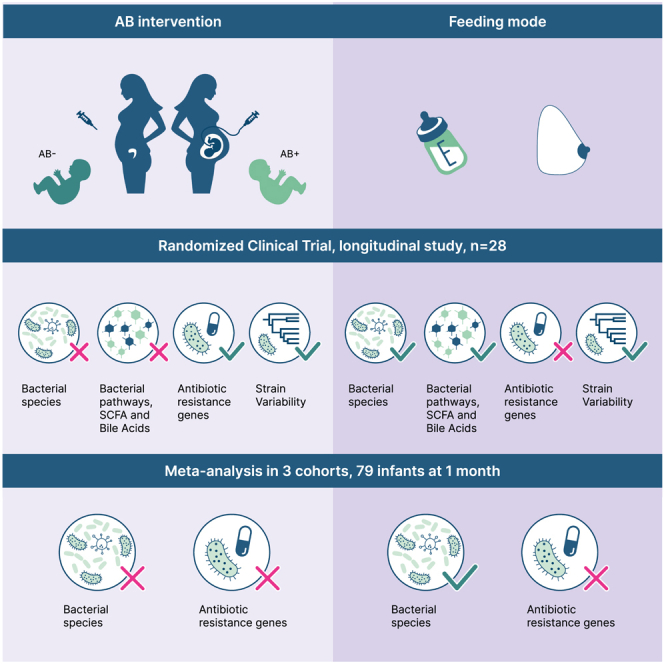
Pregnant women undergoing a cesarean section (CS) typically receive antibiotics prior to skin incision to prevent infections. To investigate if the timing of antibiotics influences the infant gut microbiome, we conducted a randomized controlled trial (NCT06030713) in women delivering via a scheduled CS who received antibiotics either before skin incision or after umbilical cord clamping. We performed a longitudinal analysis on 172 samples from 28 infants at 8 post-birth time points and a cross-sectional analysis at 1 month in 79 infants from 3 cohorts. Although no significant associations with bacterial composition, metabolic pathways, short-chain fatty acids, and bile acids were found, we observed subtle differences between the groups at the bacterial strain level and in the load of antibiotic resistance genes. Rather, feeding mode was a predominant and defining factor impacting infant microbial composition. In conclusion, antibiotic administration during CS has only limited effects on the early-life gut microbiome.
Keywords: maternal prophylactic antibiotics, cesarean section, infant gut microbiome, feeding mode, resistome, antibiotic resistance genes, strain variability
Graphical abstract
Highlights
-
•
Feeding mode profoundly influences infant gut microbiome and bile acid composition
-
•
Prophylactic antibiotics given to mother during CS transfer to infant cord blood
-
•
Antibiotics during CS exert minor effects on infant gut microbiome composition
-
•
Antibiotics during CS subtly impact strain variability and antibiotic resistance genes
Sinha et al. conducted a randomized controlled trial and meta-analysis to assess the effects of maternal antibiotics during cesarean sections on the infant gut microbiome and metabolites. They found that these antibiotics have a minor impact compared with feeding mode but noted minor alterations in bacterial strains and antibiotic resistance genes.
Introduction
There is increasing evidence that the gut microbiome plays a functional role in driving immune development in the newly born and in the development of chronic conditions later in life.1 Antibiotic exposure in childhood has been associated with increased risk of diabetes, obesity, inflammatory bowel disease, asthma, and allergies later in life.2,3,4 Consequently, there have been efforts to avoid unnecessary antibiotic exposure in infants, including cautiousness in prescribing prophylactic antibiotics given to the mother before and during birth. Over the last few years, obstetric guidelines related to cesarean delivery (cesarean section [CS]) have been revised to reduce the incidence of maternal infections and neonatal sepsis.5 Most guidelines recommend administering prophylactic antibiotics to the mother about 20 min before a CS skin incision, rather than after clamping the umbilical cord.5,6,7 This policy has been shown to reduce maternal risk of infectious morbidities, particularly of endometritis and wound infections.5 Consequently, in line with these guidelines, all infants born by CS are currently exposed to antibiotics antepartum via the umbilical cord, as these antibiotics rapidly cross the placenta. However, while there are studies that look at overall neonatal outcome,8,9,10 the effect of these antenatal antibiotics on the infant gut microbiome has not been extensively studied. The latest World Health Organization guidelines explicitly state that there is an urgent need to study the effects of prophylactic antibiotics on vital neonatal health outcomes and the neonatal microbiome.7 It is thus extremely important that high-quality evidence for immediate benefits to the mother should be weighed against equally good evidence about any potential risks of short- and long-term harm to the infant. We hypothesized that antenatal exposure to antibiotics in infants delivered by CS influences gut microbiome seeding and colonization and thereby impacts health outcomes in later life. In this randomized controlled trial (RCT), CS Baby Biome, we assessed the impact of intrauterine antibiotic exposure on microbiome composition and functionality, using deep metagenomic sequencing in a longitudinal study in the first year of life of infants born via CS. To enhance the robustness of our findings, we supplemented our data with data from the MAMI trial, a separate RCT with similar objectives.11,12 Additionally, we incorporated data from the Lifelines NEXT population-based cohort study to augment our sample size and strengthen the overall analysis.13
Results
Study design and inclusion in the CS Baby Biome trial
During the inclusion period of CS Baby Biome, 348 women were scheduled for a CS in the University Medical Center Groningen (UMCG) (excluding the period of research inclusion stop due to the COVID-19 outbreak). The most common indication for scheduled CS was previous CS (85%), with a few other indications like previous uterus surgery, previous shoulder dystocia in infant, and placenta previa. Of these women scheduled for a CS, we counseled 161 who fulfilled the inclusion criteria, and 33 (20.5%) agreed to participate in the study and signed the informed consent. After signing of informed consent and after randomization, there were 5 dropouts (4 cases of no fecal sample collection and 1 emergency CS) (for flowchart of study inclusion see Figure S1). Of the 28 mother-infant pairs ultimately included in the analysis, 12 women received antibiotic prophylaxis prior to skin incision (AB+ group) and 16 after clamping of the umbilical cord (AB− group). Mother and infant phenotypic data are shown in Table 1. Only pre-pregnancy BMI differed significantly between the two groups (Table 1), and this difference was considered in all analyses (see STAR Methods). Fecal microbiome samples were collected longitudinally from infants at weeks 1–6 and at months 6 and 12 and were compared between the two groups. In terms of our secondary outcomes, the rates of maternal endometritis and wound infections, there were no differences between the two groups. None of the women were diagnosed with early wound infections. One woman in the AB− group developed a late wound infection. This analysis was however limited by the low power to study these outcomes.
Table 1.
Clinical characteristics of gravidae and infant
| Variable | AB+ group (n = 12) | AB− group (n = 16) | p value |
|---|---|---|---|
| Cefazolin, median (range), mg/L | 25.23 (9.06–39.57) | 0 (0–0) | 1.29 × 10−6∗ |
| Pre-pregnancy BMI, median (range), kg/m2 | 21.6 (16.61–28.1) | 25.6 (19.6–41.2) | 0.01113∗ |
| Weight gain in pregnancy, median (range), kg | 13.7 (8–24) | 17 (5.5–24.5) | 0.8484 |
| Gestational age, median (range), weeks | 39.35 (38.0–39.6) | 39.30 (38.1–40.0) | 0.9624 |
| Gravidity, median (range) | 3.5 (1–5) | 3 (1–10) | 0.981 |
| Parity, median (range) | 2 (0–4) | 2 (0–3) | 0.9807 |
| Sex, n (%) | – | – | 0.1212 |
| Female | 5 (41.7) | 12 (75.0) | – |
| Male | 7 (58.3) | 4 (25.0) | – |
| Birthweight, median (range), g | 3,740 (3,165–4,480) | 3,598 (2,770–4,580) | 0.6952 |
| Maternal age | 33.5 (27–42) | 34.5 (29–41) | 0.9288 |
| Apgar score, median (range) | |||
| 1 min | 9 (8–9) | 9 (6–9) | 0.5805 |
| 5 min | 10 (8–10) | 10 (9–10) | 0.9735 |
| p value birthweight, n (%) | – | – | 0.9233 |
| p < 10 | 0 (0.0) | 1 (6.3) | – |
| p 10–50 | 2 (16.7) | 2 (12.5) | – |
| p 51–90 | 8 (66.7) | 9 (56.3) | – |
| p > 90 | 2 (16.7) | 4 (25.0) | – |
| Feeding mode, n (%) | – | – | 0.151 |
| Breastfeeding | 7 (58.3) | 4 (25.0) | – |
| Formula feeding | 2 (16.7) | 8 (50.0) | – |
| Mixed feeding | 3 (25.0) | 4 (25.0) | – |
| Living environment, n (%) | – | – | 0.430 |
| City | 7 (58.3) | 5 (31.3) | – |
| Village | 5 (41.7) | 9 (56.3) | – |
| Farm | 0 (0.0) | 2 (12.5) | – |
| Pets, n (%) | – | – | 0.6424 |
| Yes | 2 (22.2) | 5 (41.7) | – |
| No | 7 (77.8) | 7 (58.3) | – |
| Technical variable: read depth, median (range) | 14,188,948 (2,030,988–23,119,622) | 13,526,312 (4,015,235–23,688,302) | 0.4545 |
| Technical variable: DNA concentration, median (range), ng/μL | 2.09 (0.23–41.61) | 2.08 (0.01–68.60) | 0.7685 |
AB+ group consists of infants of gravidae who received antibiotics before skin incision. AB− group consists of infants of gravidae who received antibiotics after skin incision. Comparisons between the groups were done with the Fisher’s exact test for dichotomous and categorical variables, Wilcoxon rank-sum test for non-parametric continuous data, and t test for parametric continuous data. N/As (not available) are not included in the calculation of %.
Overall microbiome composition
Looking at the alpha diversity, as measured by Shannon diversity index and species richness, we did not observe statistically significant differences between the AB+ and AB− groups (Figures 1B and 1C; Tables S1A and S1B), although AB+ infants consistently had lower alpha diversity and richness in the first 6 weeks of life. At months 6 and 12, the AB+ infants continued to have lower diversity and lower species richness, but this was again not statistically significant, although this analysis was limited by low sample sizes (Figure S2).
Figure 1.

Study design and microbiome composition differences between the AB+ and AB− groups of infants
(A) Study design.
(B) Alpha diversity of the microbiome at the species level represented by Shannon diversity index.
(C) Species richness defined as the total number of non-zero species calculated at the species level. In (B) and (C), x axis depicts the time point after birth in weeks (weeks 1–6). p values are derived from a linear mixed-effect model without correction for other phenotypes. Boxplots visualize the median, hinges (25th and 75th percentiles), and whiskers extending up to 1.5 times the interquartile range from the hinges.
(D) Principal coordinates analysis (PCoA) plot using Aitchison distances. Dot color indicates feeding mode of the infant at each time point.
(E) Leading TCAM factors. Colors indicate the major feeding mode of the infant over 6 weeks of life. Each dot represents one infant. p value derived from a PERMANOVA test with 10,000 permutations without correction for other phenotypes.
(F) PCoA plot using Aitchison distances. Dot color indicates AB+ or AB− group.
(G) Leading TCAM factors. p value derived from a PERMANOVA test with 10,000 permutations without correction for other phenotypes. When correcting for infant feeding mode and maternal BMI, p value is 0.10 (Table S2C). Dot color indicates AB+ or AB− group. Each dot represents one infant. In (B)–(D) and (F), the sample sizes at each of the time points are as follows: group AB+: W01 (n = 12), W02 (n = 11), W03 (n = 12), W04 (n = 12), W05 (n = 12), W06 (n = 12); group AB−: W01 (n = 14), W02 (n = 14), W03 (n = 15), W04 (n = 15), W05 (n = 15), W06 (n = 15). In (E) and (G), sizes for each of the groups are as follows: AB+ (n = 12), AB− (n = 16).
To test the effect of timing of antibiotic administration on the overall microbiome on all samples in the first 6 weeks of life, we first used TCAM,14 a dimensionality reduction method for longitudinal multi-omics data analysis. This allowed us to compare the trajectories of the microbiome changes of each infant (Figures 1E and 1G). The AB+ group had a significant effect on the gut microbiome without correction for other factors influencing the overall gut microbiome composition (permutational multivariate analysis of variance [PERMANOVA] with 10,000 permutations R2 = 0.05, p = 0.02; Table S2A). However, after correcting for feeding mode and maternal pre-pregnancy BMI, which both had a significant effect on overall microbiome composition (R2 = 0.12, p = 0.0001 and R2 = 0.06, p = 0.02, respectively), the association of beta diversity trajectories and AB group was no longer significant (p = 0.10; Tables S2B and S2C). The levels of cephazolin measured in cord blood, however, did show significant association with beta diversity, even after correction for feeding mode and pre-pregnancy BMI (p = 0.03) (Table S2C). All these findings together suggest that the timing of antibiotic administration has a small effect on the overall microbiome composition (alpha and beta diversity), which is much less pronounced than the effect of feeding mode.
We then looked at the microbiome composition at species level, correcting for feeding mode, and observed that the abundances of 14 species were associated with antibiotic use, with nominal significance. Compared with AB infants, AB+ infants showed a lower abundance in Veillonella atypica, Lactobacillus gasseri, Bifidobacterium bifidum, Lancefieldella parvula, GGB4964 SGB6927, and Limosilactobacillus fermentum and a higher abundance of Klebsiella quasipneumoniae, Intestinibacter bartlettii, Veillonella sp. 3310, Varibaculum cambriense, Flavonifractor plautii, Staphylococcus hominis, Terrisporobacter othiniensis, and Lacticaseibacillus rhamnosus (Figure 2A; Table S3B, results not corrected for feeding mode shown in Table S3A). However, none of these associations remained significant after adjusting for multiple testing. Similarly, when assessing bacterial metabolic pathways, we found no associations with AB use after correction for feeding mode and adjusting for multiple testing (Tables S3C and S3D)
Figure 2.

Microbial species, strain, and antibiotic resistance (AR) gene load differences between the AB+ and AB− groups of infants
(A) Heatmap of species nominally significant (p < 0.05) between the AB+ and AB− groups of infants as derived from a linear mixed model (see STAR Methods). The sample sizes at each of the time points are as follows: group AB+: W01 (n = 12), W02 (n = 11), W03 (n = 12), W04 (n = 12), W05 (n = 12), W06 (n = 12); group AB−: W01 (n = 14), W02 (n = 14), W03 (n = 15), W04 (n = 15), W05 (n = 15), W06 (n = 15).
(B) Phylogenetic trees of three species associated with AB group. Colors on the outer circle represent each individual infant. AB+ and AB− groups are depicted in the inner circles. Each tip represents a sample; samples sizes for each of the species-level genomic bins (SGBs) are as follows: Veillonella dispar (n = 67 samples from 22 infants), Bifidobacterium dentium (n = 49 samples from 13 infants), and Enterococcus faecalis (n = 54 samples from 15 infants).
(C) Total bacterial AR gene load between the two groups. x axis depicts the time point after birth in weeks (weeks 1–6). p value derived from a linear mixed-effect model without correction for other phenotypes. The sample sizes at each of the time points are as follows: group AB+: W01 (n = 12), W02 (n = 11), W03 (n = 12), W04 (n = 12), W05 (n = 12), W06 (n = 12); group AB−: W01 (n = 14), W02 (n = 14), W03 (n = 15), W04 (n = 15), W05 (n = 15), W06 (n = 15).
(D) Total AR gene load grouped by class cephalosporins. x axis depicts the time point after birth in weeks (weeks 1–6). p value derived from a linear mixed-effect model without correction for other phenotypes. The sample sizes at each of the time points are as follows: group AB+: W01 (n = 12), W02 (n = 11), W03 (n = 12), W04 (n = 12), W05 (n = 12), W06 (n = 12); group AB−: W01 (n = 14), W02 (n = 14), W03 (n = 15), W04 (n = 15), W05 (n = 15), W06 (n = 15). Boxplots visualize the median, hinges (25th and 75th percentiles), and whiskers extending up to 1.5 times the interquartile range from the hinges.
The within-species phylogenetic analysis revealed that microbial strains, once seeded in the infant gut, remained stable over the first 6 weeks of life for the majority of species. With regard to antibiotic use, three species—Veilonella dispar, Bifidobacterium dentium and Enterococcus faecalis—showed the presence of distinct clades associated with AB group (Figure 2B), whereas feeding mode was significantly associated to clades in two species—Clostridium innocuum and Clostridium perfringens.
We also looked at the abundance of antibiotic resistance (AR) genes and found that the total load of AR genes remained stable in the first 6 weeks of life. Using the marker gene approach, although the total load of AR genes was higher in the AB+ group in all time points, this difference was not significant (p = 0.057, Table S4A). We then specifically looked at genes that confer resistance to cefazolin and cefuroxim (CTX-M-3, CTX-M-27, CTX-M-15, CMY-2, CMY-136, porin OmpC, NmcA, TEM-1, PDC-5, PDC-3, PRC-1, LAQ-1, SCO-1, LAP-1, LAP-2, MAB, YRC, SED, EBR-5) and found that their abundance was not significantly increased in the AB+ group (Table S4B). AR gene profiles of bacterial genomes were further explored by identifying AR genes and quantifying their abundances in the recovered bacterial metagenome-assembled bacterial genomes (MAGs). In accordance with the trend in the previous analysis, we found a higher AR gene load in bacteria from AB+ infants, with nominally significant results (p = 0.017; Figure 2C; Table S4C). The bacterial AR gene load was also positively associated with cefazoline concentration in cord blood (p = 0.041; Table S4C), while no significant association was observed with other maternal or infant factors (Table S4C). Furthermore, we analyzed the abundances of AR genes categorized by antimicrobial class. We observed that, at nominal significance, genes associated with resistance to several antimicrobial classes displayed an increased abundance in AB+ infants (Table S4D). Importantly, we found that the abundance of genes conferring cephalosporin resistance was increased in infants from the AB+ group (p = 0.017) (Figure 2D; Table S4D). However, after applying multiple testing correction, none of these associations retained statistical significance.
SCFAs and BAs in infant stool
We further tested the influence of feeding mode and AB use on the functional capacity of the microbiome and measured the levels of three short-chain fatty acids (SCFAs) (acetate, propionate, and butyrate) at weeks 1 and 4 in 20 infants. While the levels of all three SCFAs were lower in infants in the AB+ group, this was not statistically significant (acetate p = 0.08, butyrate p = 0.14, and propionate p = 0.37) (Figure S3B; Table S6A). Interestingly, the levels of these three SCFAs were not significantly associated with time point or feeding mode (Table S6A; Figures S3A and S3C).
Based on growing evidence of the importance of bile acids (BAs) in autoimmune diseases in infants,15 the emerging prominent role of BAs in the establishing and maturation of the early gut microbiome in murine models16 as well as the associations of BA with antibiotic administration in infants,17 we also quantified 18 BAs in the feces of infant samples at weeks 1, 4, and 6. We found that the overall composition of BAs was driven strongly by time point and feeding mode (p = 0.024 and p = 0.001 with 999 permutations; Figures 3A and 3C). Both levels of primary and secondary BAs were strongly influenced by time point (Figure 3A), with levels of glycocholic acid (GCA), glycochenodeoxycholic acid (GCDCA), taurolithocholic acid (TLCA), and TLCA 3-sulfate (TLCA-3S) decreasing significantly with time (false discovery rate [FDR] < 0.05) (Figure 3B). Feeding mode had a strong effect on levels of taurochenodeoxycholic acid (TCDCA), taurocholic acid (TCA) (p = 2.93e−6 and p = 4.16e−3, respectively), cholic acid (CA) (p = 0.02), and the CA-chenodeoxycholic acid (CDCA) ratio (p = 9.67e−3) (FDR < 0.1) (Figure 3D; Table S6B). Levels of GCDCA, glycoursodeoxycholic acid (GUDCA) and glycolithocholic acid (GLCA) were significantly associated with AB use after correction for feeding mode (p = 0.02, p = 0.03, and p = 0.04, respectively), although the association with GLCA was driven by outliers and none were FDR significant (Figures 3E and 3F; Table S6C, results uncorrected for feeding mode shown in Table S6B).
Figure 3.

Infant fecal bile acids in relation to time point, feeding mode, and antibiotic groups
(A) PCoA plot using Canberra distances. Dot color indicates the time point. p value was derived from a PERMANOVA test with 999 permutations without correction for other phenotypes.
(B) Boxplots of the relative abundance of the most significantly associated bile acids with time. p value derived from a linear mixed-effect model without correction for other phenotypes. GCA, glycocholic acid; GCDCA, glycochenodeoxycholic acid; TLCA.3S.quant, taurolithocholic acid 3-sulfate.
(C) PCoA plot using Canberra distances. Dot color indicates the feeding mode of each infant at each time point. p values derived from a PERMANOVA test with 999 permutations without correction for other phenotypes.
(D) Boxplots of the relative abundance of the most significantly associated bile acids with feeding mode (Table S6B). p value derived from a linear mixed-effect model without correction for other phenotypes. CA, cholic acid; TCA, taurocholic acid; TCDCA, taurochenodeoxycholic acid.
(E) PCoA plot using Canberra distances. Dot color indicates the antibiotic group the infant belongs to (AB+ or AB−). p values derived from a PERMANOVA test with 999 permutations after correction for feeding mode.
(F) Boxplots of the relative abundance of the most significantly associated bile acids with AB group after correction for feeding mode (Table S6C). GCDCA, glycochenodeoxycholic acid; GLCA, glycolithocholic acid; GUDCA, glycoursodeoxycholic acid. In (A)–(F), each dot represents a sample, with the following sample sizes: W01 (n = 21), W04 (n = 18), and W06 (n = 20) from a total of 22 unique infants. Boxplots visualize the median, hinges (25th and 75th percentiles), and whiskers extending up to 1.5 times the interquartile range from the hinges.
Combined analysis of CS Baby Biome, Lifelines NEXT, and MAMI trial
Based on the consistent trend of our observations in relation to diversity and AR gene load, we questioned whether our negative results were due to lack of power. We therefore combined our study data with data from the MAMI trial, which had a very similar study design.11,12 In this RCT, the antibiotic cefuroxim (also a cephalosporin group) was given to gravidae. To further increase our sample size, we also included samples obtained from Lifelines NEXT, a longitudinal birth cohort. From Lifelines NEXT, we first selected women who underwent scheduled CS and then measured the concentration of cephalosporin in the cord blood of their infants (n = 13). In 7 of the 13 samples, no detectable cefazolin was identified in cord blood, suggesting that antibiotics were not given to the mother according to current protocols. In total, we had data from 79 infants at month 1 after birth from 3 cohorts (42 AB− and 37 AB+) (Figure 4A). We first observed that cohort had no effect on the overall composition of the gut microbiome of the infants at 1 month (p = 0.45; Figure 4B; Table S5C). Here too the overall composition was driven by feeding mode (Figure 4C) (R2 = 0.05, p = 0.003, Table S5C). No differences in the overall composition were observed between the AB+ and AB− groups (Figure 4D; Tables S5C and S5D). Shannon diversity index was increased with formula feeding compared with breastfeeding (Figure 4E; p = 0.024; Table S5A), but no differences were found in the Shannon diversity index between the two AB groups (Figure 4F; p = 0.747; Tables S5A and S5B). There were also no significant differences in species richness (p = 0.731; Tables S5A and S5B). In the Lifelines NEXT cohort, we further looked at late effects of these AB at months 6, 9, and 12 and here too found no significant differences in species richness or Shannon diversity index (Table S7A).
Figure 4.

Combined results of the CS Baby Biome, MAMI trial, and Lifelines NEXT
(A) Diagram showing the cohorts used in the combined analysis, together with the sample sizes.
(B) PCoA plot showing the overall composition of the gut microbiome of infants of all cohorts. Each dot represents a single infant of either the MAMI trial (red), CS Baby Biome (blue), or Lifelines NEXT (LLNEXT in figure; green) at the age of 1 month. p value derived from a PERMANOVA test with 10,000 permutations without correction for other phenotypes (Table S5C).
(C) PCoA plot showing the overall composition of the gut microbiome of infants of all cohorts. Each dot represents a single infant, and dot color indicates feeding mode (blue, breastfeeding; pink, mixed feeding; and khaki, formula feeding). Feeding mode significantly influences overall gut microbiome composition in infants, with the combined data of all cohorts explaining 5% of the variation of the infant microbiome at 1 month of age. p value derived from a PERMANOVA test with 10,000 permutations without correction for other phenotypes (Table S5C).
(D) PCoA plot showing the overall composition of the gut microbiome of infants of all cohorts. Each dot represents a single infant, and dot color indicates AB+ or AB− group. p value derived from a PERMANOVA test with 10,000 permutations without correction for other phenotypes (Table S5C).
(E) Shannon diversity index at 1 month was higher in formula-fed infants than breastfed infants (p = 0.024, Table S5A).
(F) No difference was found in the Shannon diversity index between AB+ and AB− groups (p = 0.747, Tables S5A and S5B).
(G) No significant difference was found in total AR load between the AB+ and AB− groups (p = 0.080, Table S5G).
In (E)–(G), boxplots visualize the median, hinges (25th and 75th percentiles), and whiskers extending up to 1.5 times the interquartile range from the hinges.
We then looked at microbiome composition at species level and observed that the abundance of Bifidobacterium dentium was nominally significantly associated with antibiotic use, with this bacterium decreased in the AB+ group (p = 0.009) (Table S5E). This association was retained after correction for feeding mode (Table S5F). No significant associations were found when correcting for multiple testing. The total AR load of the AB+ group was greater than that of the AB− group, in line with CS Baby Biome findings, but this difference was not statistically significant (p = 0.08) (Figure 4G; Table S5G). Our combined cross-sectional cohort data therefore suggest that the effect of timing of prophylactic antibiotic administration on the microbiome of infants is minor compared with the effect of feeding mode.
Discussion
In this RCT, we investigated the effect of the timing of maternal prophylactic antibiotic administration during CS on the early-life microbiome. Our findings suggest that fetal exposure to antibiotics, due to maternal antibiotic administration prior to CS, has only a limited effect on infant early-life gut microbiome composition and function. Moreover, our findings show that that feeding mode is by far the most defining factor for microbial composition and functionality in early life in the group of infants born via scheduled CS.
Two 16S studies previously investigated the influence of antibiotics given during CS on the gut microbiome. Most recently, Bossung et al.18 showed in 16 AB− and 20 AB+ meconium samples that the microbiome detected in meconium was different but that these strong differences disappeared by the age of 1 month. In our previous experiments with meconium, we observed that these samples show remarkably low biomass and produce insufficient amounts of microbial reads for metagenomic sequencing, in line with the prevailing belief in the field that the gut ecosystem remains sterile before birth. Hence, meconium samples were not included in our analysis. The second study, by Kamal et al.,19 in 42 pregnant women undergoing scheduled CS in Denmark, investigated the infant microbiome at two time points (10 days and 9 months) in two groups of infants with gravidae randomly assigned to receive cefuroxim before skin incision or immediately after clamping of the umbilical cord. In accordance with our results, these authors concluded that the timing of cefuroxim administration to gravidae undergoing a CS does not have a major effect on the gut microbiota and bacterial AR traits in infants.
To our knowledge, this is one of the only studies to characterize the infant SCFA and BA profile in the first few weeks of life in relation to antibiotic administration to mothers during delivery. Our results show that the levels of acetate, butyrate, and propionate in the first 4 weeks of life remained relatively stable and were not influenced by feeding mode or antibiotic use in CS infants. Based on growing evidence of the importance of BAs in autoimmune diseases,15 the emerging prominent role of BAs in the establishing and maturation of the early gut microbiome in murine models,16 as well as associations of BA with antibiotic administration in infants,17 we also quantified fecal BAs in infants. We found that the overall composition of BAs was driven strongly by time point and feeding. In addition, our data show some indications that AB exposure could influence BA production in infants. As our sample size was low, these results warrant further follow-up in larger studies.
It was previously shown in the MAMI trial, using both 16S and metagenomics, that maternal antibiotic (cefuroxim) administration prior to CS did not have an effect on the infant gut microbiome colonization. The MAMI trial was the first RCT to evaluate the effects of exposure to maternal antibiotics during CS on infant microbiota colonization in a randomized design using metagenomics. However, despite being one of the largest RCTs in the field, the sample size was relatively small (n = 40), and there was an absence of dense longitudinal sampling during the first weeks of life. By combining data from CS Baby Biome, Lifelines NEXT, and the MAMI trial, we were able to substantially increase the sample size. We further corrected for an important covariate, feeding mode, given its known substantial effect on the gut microbiome. The conclusion remained the same in the combined analysis: antibiotic administration prior to CS has a limited effect on the infant early-life gut microbiome composition. Despite the increase in power to study the microbial associations, our power to draw firm conclusions is still limited as the combined sample size of 79 still seems insufficient to answer these questions. Considering the trend of increased AR gene load when combining all 3 cohorts, we carried out a formal power analysis. Our power analysis suggests we would need a sample size of 109 infants to discern a difference in the total AR gene load. To detect a difference in alpha diversity, data from 887 infants would be necessary. Therefore, to draw robust conclusions, significantly larger studies with detailed longitudinal data are needed. In countries with high CS rates, gathering this data could involve routinely assessing cord blood for antibiotics and examining its impact on the infant gut microbiome. Finally, our study could not link the gut microbiome to long-term health outcomes like asthma, allergies, or obesity, making larger future studies essential for such insights.
To the best of our knowledge, this study represents the most extensive metagenomic investigation to date to explore the influence of the timing of antenatal antibiotic administration during CS on the gut microbiome of infants and the only study to consider bacterial strain phylogeny, bacterial key metabolites, and specific groups of AR genes. Our results indicate that maternal prophylactic antibiotic use during CS has minimal impact on the early-life composition and functionality of the infant gut microbiome. Notably, our findings emphasize that feeding mode significantly outweighs other factors in shaping the gut microbial and BA composition during early life in CS-born infants. However, larger-scale and long-term studies are necessary to validate these observations and to assess the potential long-term health implications for both pregnant women and their infants.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Infant and maternal fecal samples | This study | N/A |
| Cord blood samples | This study | N/A |
| Critical Commercial Assays | ||
| QIAamp Fast DNA Stool Mini kit | QIAGEN | Cat#51604 |
| NEBNext® Ultra™ II DNA Library Prep Kit | New England Biolabs | Cat # E7370L |
| Deposited Data | ||
| Metagenomic sequencing data of CS Baby Biome | This study | European Genomics-Phenome Archive:EGAC00001003433 |
| Original code for microbiome, SCFA and BA analysis | This study | https://zenodo.org/records/11537223https://github.com/GRONINGEN-MICROBIOME-CENTRE/CS_BABY_BIOME |
| Original code for microbiome profiling | Gascesa et al.20 | https://github.com/GRONINGEN-MICROBIOME-CENTRE/DMP/tree/main/microbiome_profiling |
| Software and Algorithms | ||
| REDCap | Harris et al.21 | http://www.sciencedirect.com/science/article/pii/S1532046408001226 |
| blockrand | Snow22 | https://CRAN.R-project.org/package=blockrand t |
| KneadData | Huttenhower Lab | https://github.com/biobakery/kneaddata |
| Bowtie2 (version 2.4.2) | Langmead and Salzberg et al.23 | https://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| FastQC toolkit (version 0.11.9) | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| MetaPhlAn4 | Blanco-Míguez et al.24 | https://github.com/biobakery/MetaPhlAn/tree/8338b344239a296e6654f8770d012e250106ab70 |
| StrainPhlAn4 | Truong et al.25 | https://github.com/biobakery/MetaPhlAn/wiki/StrainPhlAn-4 |
| HUMAnN (version 3.6) | Beghini et al.26 | https://github.com/biobakery/humann |
| ShortBRED | Kaminski et al.27 | https://github.com/biobakery/shortbred |
| metaWRAP (version 1.3.2) | Uritskiy et al.28 | https://github.com/bxlab/metaWRAP |
| dRep (version 3.4.2) | Olm et al.29 | https://github.com/MrOlm/drep |
| CoverM (version 0.6.1) | Woodcroft, B30 | https://github.com/wwood/CoverM |
| RGI (version 6.0.3) | Alcock et al.31 | https://github.com/arpcard/rgi |
| lmerTest (version 3.1-3) | Kuznetsova et al.32 | https://CRAN.R-project.org/package=lmerTest |
| vegan (version 2.6-2) | Oksanen et al.33 | https://github.com/vegandevs/vegan/ |
| TCAM | Mor et al.14 | https://github.com/UriaMorP/tcam_analysis_notebooks |
| MCMCglmm (version 2.34) | Hadfield34 | https://github.com/jarrodhadfield/MCMCglmm |
| Phangorn (version 2.11.1) | Schliep35 | https://CRAN.R-project.org/package=phangorn |
| pwrss (version 0.3.1) | Bulus, M36 | https://CRAN.R-project.org/package=pwrss |
| R (version 4.3.1) | R Core Team37 | https://www.r-project.org/ |
| Other | ||
| Clincial Trial Registration | https://clinicaltrials.gov/study/NCT06030713?cond=NCT06030713&rank=1 | NCT06030713 |
Resource availability
Lead contact
Further information and requests for resources, software, reagents and data sharing should be directed to the lead Contact, Trishla Sinha (t.sinha@rug.nl)
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Sample metadata and quality-trimmed sequencing reads have been deposited at European Genome-Phenome Archive and are publicly available as of the date of publication. Accession numbers are listed in the key resources table (EGAC00001003433).
-
•
All original code has been deposited on Zenodo and Github and is publicly available as of the date of publication. DOIs are listed in the key resources table (https://doi.org/10.5281/zenodo.11537223 and https://github.com/GRONINGEN-MICROBIOME-CENTRE/CS_BABY_BIOME).
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
Experimental model and subject details
Study design and oversight
The CS Baby Biome RCT was conducted in the Department of Obstetrics and Gynecology and the Department of Genetics at the University Medical Center Groningen (UMCG), a tertiary referral center. Recruitment took place between 2019 and 2022. The study protocol (CCMO: NL61493.042.17) was approved by the medical ethics committee of the University Medical Center Groningen (2017/240). Written informed consent for participation was obtained from all mothers. It was further registered as NCT06030713 on ClinicalTrials.gov: https://clinicaltrials.gov/study/NCT06030713?cond=NCT06030713&rank=1
Participants
During the inclusion period we counseled 161 women scheduled for delivery via CS ≥38 weeks of gestation, and 33 (20.5%) agreed to participate in the study and signed the informed consent. Among the participants, the most common indication for scheduled CS was previous CS (85%) with a few other indications like previous uterus surgery, previous shoulder dystocia in infant and placenta previa. After signing informed consent and randomization, there were 5 dropouts (4 cases of no fecal sample collection and 1 emergency CS). A flowchart of the recruitment process is presented in Figure S1. Exclusion criteria were maternal cephalosporin allergy, emergency CS, maternal exposure to any antibiotic agent in the 2 weeks before CS, maternal temperature >37.5°C before CS, pre-labor rupture of membranes, not being fluent in the Dutch language and not having a freezer at home to store fecal samples. Study data were collected and managed using REDCap electronic data capture tools hosted at the UMCG.21,38 Given the geographical focus in the northern region of the Netherlands, the majority of participants were of European descent, which may limit the generalizability of our findings. Ethnicity of the participants were not collected due to restrictions imposed by the medical ethical committee. The median age of the included pregnant women was 34.0 years (range 27.0 to 42.0 years). Among the infants, 60.7% were female and 39.3% were male.
Outcomes
The primary endpoint of this study was to compare the composition of the infant gut microbiome in the group of infants whose mothers were given antibiotic prophylaxis before skin incision (AB+) with the infants whose mothers were given antibiotic prophylaxis after umbilical cord clamping (AB-). The composition of the gut microbiome was assessed by the alpha diversity, beta diversity, species level abundances, strain variability and the abundances of antibiotic resistance genes. In addition to this, the functionality of the infant gut microbiome between the two groups was also compared. The functionality was assessed by the abundances of bacterial pathways, levels of short chain fatty acids and bile acids in infant feces. Secondary outcomes in this study were to compare the rates of wound infection and endometritis in women in both groups. To define Early Wound Infections the following variables were extracted from the hospital records: “Fever during first 3 days”, “Leukocyte and / or C-reactive protein (CRP) increased in blood tests”, “Erythema of wound site”, “Purulent discharge from wound”, “Induration of surgical wound site”, “Uterine tenderness” and “Tachycardia” in the first 3 days after CS. Early wound infection was defined based on a positive measurement to any of the above criteria, with the absence of another explanation of the symptoms. Furthermore, if a doctor defined an Early Wound Infection this was sufficient to give the diagnosis “Early Wound infection” even in the absence of the above-mentioned factors. We further assessed Late Wound Infection at 6 weeks extracting from the hospital records whether the mother reported having a late infection at the 6 weeks follow-up with the obstetrician.
Sample size estimation
At the start of the trial no formal power analysis could be performed due to the lack of relevant data regarding the effect of maternal prophylactic antibiotics on infant gut microbial species. According to previous studies of Yassour et al., a sample size of 39 was sufficient to observe differences in the infant gut microbiome due to oral antibiotics.2 Furthermore, in the study of Bokulich et al., a sample size of 43 infants was sufficient to observe differences.3 Expecting a drop-out rate of 20% an initial sample size of 50 was chosen for this study, with 25 in each group.
Based on the results of this study, a formal power analysis was conducted showing that much larger sample sizes were required (see discussion). To test the sample size needed to see the effect on total AR gene load and alpha diversity, we first derived the estimate from the linear model from the analysis of all three cohorts in this study. We further derived the standard deviation and then computed the sample size required using the pwrss.t.reg function from R package pwrss (v 0.3.1) using a power of 0.9 and an alpha of 0.05. Our power analysis suggests we would need a sample size of 109 infants to discern a difference in the total AR gene load. To detect a difference in alpha diversity, data from 887 infants would be necessary.
Randomization and blinding
Randomization was executed using REDCap by the research nurse one day prior to the CS using IDs were generated using the “blockrand” package (v1.4).22 Following randomization, the research nurse informed the operating team of the group assignments, which were as follows: 1) administration of 2000 mg cefazolin to the mother 20 minutes before skin incision, adhering to current guidelines, and 2) administration of 2000 mg cefazolin to the mother after umbilical cord clamping. The final randomization group was recorded in REDCap post-surgery. Parents were blinded to the group assignments.
Method details
Collection of biological samples
Meconium from neonates was collected at the maternity ward and stored at -20°C at the Department of Genetics. Subsequent stool samples from the infant taken at weeks 1, 2, 3, 4, 5 and 6 were collected at home by the parents, following written and oral instructions provided by the research nurse, and immediately stored in their home freezer at -20°C. In addition, women collected their stool soon after birth. At 6 weeks, stool samples were collected, transported under frozen conditions and stored at the -80°C until DNA extraction. Stool was collected the same way at the 6 and 12 month timepoints.
Collection of phenotypes
Relevant information regarding the indication for CS, pre-pregnancy BMI, weight gain during pregnancy, gestational age, para, gravida, infant sex, birthweight and Apgar scores were collected from the hospital record and stored in REDCap. The rates of pregnancy complications of participants were relatively low with 2 participants with gestational diabetes mellitus, 3 participants with anemia and 3 with hyperemesis gravidarum. Each week, parents also answered a digital questionnaire about feeding mode, medication use, exposure to pets, living environment (city, village, farm) and general infant health during the week. Feeding mode per timepoint was derived from the questionnaires sent to parents, supplemented by patient records. For the data in Table 1, participants were assigned an early life feeding mode that was calculated as the most frequent feeding mode (in number of weeks) in the first 6 weeks of life. None of the women reported smoking or consuming alcohol. In addition, none of the families indicated partner smoking inside the household. In the first 6 weeks of life, no infants consumed medications apart from oral vitamin D and K supplements and no infants consumed any antibiotics or had an infectious disease, so these phenotypes are not listed in Table 1.
Although our power to evaluate maternal health outcomes was limited, our secondary outcome was to compare the rates of wound infection and endometritis in women in both groups. We therefore also collected information regarding these outcomes from the hospital records.
Umbilical cord blood collection
To confirm that the study protocol was indeed being followed and determine the infant’s exposure to cefazolin, umbilical cord vein blood was collected from infants after clamping of the umbilical cord and delivery of the placenta. Blood samples were collected in an EDTA tube and directly transported to the laboratory. Samples were centrifuged and plasma stored at −80°C until the cefazolin concentration was determined. Cefazolin plasma concentrations (mg/L) were determined using validated high performance liquid chromatography–ultraviolet detection analysis at the Department of Clinical Pharmacy and Pharmacology, UMCG.
Total microbial DNA extraction
Microbial DNA was isolated using the QIAamp Fast DNA Stool Mini Kit (Qiagen, Germany) using the QIAcube (Qiagen) from 0.2–0.5 g fecal material, with a final elution volume of 100 μl. DNA eluates were stored at -20°C.
Genomic library preparation and sequencing
Fecal microbial DNA and DNA samples were sent to Novogene, Cambridge, UK for genomic library preparation and shotgun metagenomics sequencing. Sequencing libraries were prepared using the NEBNext® Ultra™ DNA Library Prep Kit or the NEBNext® Ultra™ II DNA Library Prep Kit, depending on the sample DNA concentration, and sequenced using HiSeq 2000 sequencing with 2 × 150 bp paired-end chemistry (Illumina), as previously described.20
Measurements of short chain fatty acids (SCFAs) and bile acids (BAs)
Stool samples from infants from weeks 1 and 4 were prioritized for short chain fatty acid measurements, resulting in a total of 37 samples from 20 unique infants. Methods to prepare SCFA has been described previously.39 Concentrations of the different SCFA were measured using an Agilent 5975C series gas chromatography/mass spectrometry (GC–MS) (Agilent Technologies, Santa Clara, USA). The GC was equipped with a ZB-1 column (Phenomenex, Torrance, USA). Mass spectrometry analysis was performed by positive electron ionization. Ions monitored were m/z 117 for acetate, m/z 131 for propionate and m/z 145 for butyrate.
Stool samples from infants from weeks 1, 4 and 6 were prioritized for bile acid measurements, resulting in a total of 59 samples from 22 unique infants. BAs were quantified from freeze-dried and thoroughly ground stool samples as described before,40,41 with minor modifications. Briefly, 2 mL alkaline methanol was added to approximately 30 mg of freeze-dried feces and the sample was heated at 80 oC for 180 minutes. Samples were cooled down and ultrasonicated for 15 minutes, followed by vigorous mixing for an additional 15 minutes and centrifugation at 3000 x g for 10 minutes. Deuterium-labeled internal standards were then added to 25 μL of the supernatant and mixed. BAs were subsequently extracted with Oasis HLB Cartridges (Waters, Milford, MA), using 2 mL methanol as eluent. Samples were dried under a stream of nitrogen and dissolved in 200 μL 50% methanol. BAs were then quantified by ultra-high performance liquid chromatography with tandem mass spectrometry (UHPLC-MS/MS) using a ACQUITY Premier UPLC system (Waters) coupled to a Xevo TQ-S mircro triple quadrupole mass spectrometer (Waters). Fecal BA contents were calculated using the deuterium-labelled internal standards.
Quantification and statistical analysis
Statistical details of all experiments and data analyses are provided in this section, as well as in Figure legends and in the results section.
Profiling of total gut microbiome composition
All bioinformatic analysis was carried out in-house using our bioinformatic pipeline (https://github.com/GRONINGEN-MICROBIOME-CENTRE/DMP/tree/main/microbiome_profiling ).20 In short, total microbiome sequencing reads were trimmed by quality, and Illumina sequence adaptor sequences were removed using KneadData (v0.7.4). Following trimming, the KneadData-integrated Bowtie2 tool (v2.4.2)23 was used to remove reads that aligned to the human genome (GRCh37/hg19), and the quality of the processed data was examined using the FastQC toolkit (v0.11.9). Taxonomic composition of metagenomes was profiled using the MetaPhlAn4 tool with the MetaPhlAn database of marker genes and the ChocoPhlAn species-level genomic bin (SGB) database (vOct2022).24 Bacterial strain SNP haplotypes were generated using StrainPhlAn4.25 This method is based on reconstructing consensus sequence variants within species-specific marker genes and using them to estimate strain-level phylogenies. It only considers the dominant strain of species and thus misses overlap in secondary strains. We profiled the abundance of microbial metabolic pathways using HUMAnN (v3.6).26
Profiling of antibiotic resistance genes
The abundance of antibiotic resistance (AR) proteins was detected and quantified in each sample using two different methods. On one hand, ShortBRED was used with the default parameters.27 This database was provided with the AR-marker sequences created from the Comprehensive Antibiotic Resistance Database (CARD), which was used as a reference using the latest version of the database (June 2023). In addition to quantifying total AR gene load, we also looked at genes specifically known to confer resistance to cephazolin (NmcA, CMY-2, CTX-M-3, CTX-M-27, TEM-1, CTX-M-15, PDC-3, PDC-5, PRC-1 and LAQ-1). Additionally, a binning-based approach was used. Each sample was binned independently using metaWRAP (v1.3.2).28 Bacterial bins were then dereplicated at 98% whole-genome average nucleotide identity (gANI) with dRep (v3.4.2),29 selecting those with a minimum completeness of 75% and maximum contamination of 10%. Abundance of metagenome-assembled bacterial genomes (MAGs) in each sample was estimated using CoverM (v0.6.1). AR genes were then annotated in the MAGs by using the Resistance Gene Identifier (RGI) software (v6.0.3) with the CARD database.31 Only perfect or strict RGI matches were retained. Abundance of each AR gene per sample was calculated as the sum of the abundance of all MAGs containing that gene, accounting for the number of gene copies present within each MAG. Gene abundances were grouped into antimicrobial class abundances according to RGI “Drug Class” annotation.
Association analysis between timing of antibiotics during CS and infant gut microbiome in CS Baby Biome
To test the effect of AB group on alpha diversity and species richness during the first 6 weeks of life, we tested this as a fixed effect in a mixed model using the package lmerTest (v3.1-3),32 adding read depth and sample DNA concentration as covariates and sample ID as a random effect. Alpha diversity was calculated using the Shannon diversity index in R package vegan (v2.6-2).33
Given that pre-pregnancy BMI differed between the two AB groups, we also tested this factor in relation to alpha diversity and richness (ST1A). This was not significant, and we therefore did not add it to our model. Based on prior knowledge of the large effect of feeding mode on the gut microbiome, we also tested the association of AB group with and without correction for feeding mode (ST1B). To test the differences in alpha diversity and species richness at the 6- and 12-month timepoints, we performed the Wilcoxon rank sum test at each timepoint.
We then filtered our species for those with a relative abundance (RA) >0.05% that were present in at least two infants, leaving 89 species. On these we performed a centered log ratio (CLR) transformation. We used the vegdist function from R package vegan to calculate the distance matrix using the method "aitchison", using half of the minimum value of all bacteria as pseudocount. As we had multiple samples from each infant, we used TCAM,14 a method for unsupervised dimensionality reduction for trajectory analysis of longitudinal ’omics data. This allowed us to combine the first 6 weekly timepoints into one representation that mainly reflects inter-individual differences. We then performed PERMANOVA analysis on the resulting distance matrix. Given the strong effect of feeding mode on gut microbiome composition, we corrected for feeding mode in our analysis of association. Furthermore, as pre-pregnancy BMI had a significant effect on overall composition, we also added this to our model. The adonis2 function from R package vegan was used with 10,000 permutations using “margin”, the marginal effects of the terms (each marginal term analysed in a model with all other variables)
For associations with specific bacterial species, we associated AB group with the CLR-transformed RA of species as outcome using linear mixed models (using the lmer function from the lmerTest package) including participant ID as a random effect. Given the strong effect of feeding mode on bacterial composition, we also corrected for the effect of feeding mode.
For association with metabolic pathways, we used the same approach as for species, using filtering based on a prevalence of more than 15% and a minimum relative abundance of 0.001.
Association of AB group and feeding mode with the genetic diversity of microbial strains was performed using Markov Chain Monte Carlo generalized linear mixed models (MCMCGLMM) implemented in the R package MCMCglmm (v.2.34),34 treating traits as outcome using ordinal distribution family. Feeding mode was binarized to two categories: exclusive breast feeding versus mixed or formula feeding. SGB-level phylogenetic trees were reconstructed in the R package Phangorn (v.2.11.1)35 using the maximum likelihood method, generalized time reversible substitution model, nearest neighbor interchange and a requirement for ultrametric topology. The scaled inverse relatedness matrix from the phylogenetic tree was used as a random factor in MCMCGLMM fit. To account for multiple sampling per individual, infant IDs were added to the model as an additional random factor. In MCMC chain, residual variance was fixed, and χ2 prior distribution with one degree of freedom was used for random effects. 10,000 burn-in, and 600,000 MCMC iterations with thinning interval of 100 was used. Posterior distribution of strain-associated variance component of the trait was scaled to its sum with residual variance. Species was included in the analysis, if samples from at least two infants for each group (AB+/AB-, of BF/MF+FF) are present in a dataset. Given the inclusion cutoffs, 26 SGBs were used in analysis for association to SGBs and 28 SGBs were included in the analysis of association to feeding mode. Species strain composition was assumed to be associated with a trait, if effective sample size of MCMC exceeds 1000, and posterior mode of scaled strain-driven trait variance is higher that 0.5.
We used linear mixed models to associate total AR gene load, AB classes and individuals ARGs using the same approach for both our ways of annotating ARGs.
As feeding mode did not significantly impact AR gene load, we did not perform any correction for this factor.
Association analysis between timing of antibiotics during CS and infant gut metabolites in CS Baby Biome
To check the association between AB group and each SCFA we used linear mixed models, correcting for timepoint.
To quantify the overall compositional differences of the fecal BA profiles, the Canberra distance between all samples was calculated based on the relative proportions of the individual BA species present in the stool samples. Distance matrices were computed using the vegdist() function from R package vegan. Principal coordinates analysis (PCoA) were performed on Canberra distance matrices that were calculated based on the relative proportions of BAs using the cmdscale() function from vegan, while the proportions of BA pool variance explained by timepoint, feeding mode and AB usage was estimated by permutational multivariate analysis of variance (PERMANOVA) using the adonis2() function (with 999 permutations) from vegan. To assess the impact of AB group and each BA species we used linear mixed models, correcting for timepoint and performing the analysis with and without correction for feeding mode.
For all analyses, we defined a nominal significance at p<0.05 and statistical significance at FDR<0.05. FDR was controlled using the Benjamini-Hochberg procedure.
List of abbreviations of bile acids
CA: Cholic Acid, TCA: Taurocholic Acid, GCA: Glycocholic Acid, CDCA: Chenodeoxycholic Acid, TCDCA: Taurochenodeoxycholic Acid, GCDCA: Glycochenodeoxycholic Acid, DCA: Deoxycholic Acid, TDCA: Taurodeoxycholic Acid, GDCA: Glycodeoxycholic Acid, UDCA: Ursodeoxycholic Acid, TUDCA: Tauroursodeoxycholic Acid, GUDCA: Glycoursodeoxycholic Acid, LCA: Lithocholic Acid, TLCA: Taurolithocholic Acid, GLCA: Glycolithocholic Acid, LCA.3S: Lithocholic Acid 3-Sulfate, TLCA.3S.quant: Taurolithocholic Acid 3-sulfate, GLCA.3S.quant: Glycolithocholic acid 3-sulfate
Replication in the MAMI trial and Lifelines NEXT
To increase the sample size, we performed a combined analysis of the gut microbiome of infants at the age of one month. For this we included all samples of CS Baby Biome and samples from the timepoint M1 (n=27) of the MAMI trial for which metagenomic sequencing data were available (n=39)11,12,42 In the MAMI trial RCT, the antibiotic cefuroxim (also a cephalosporin group) was given to gravidae prior to incision or after clamping of the umbilical cord. Additional fecal samples were obtained from the Lifelines NEXT cohort (n=13),13 a birth cohort designed to study the effects of intrinsic and extrinsic determinants on health and disease. From Lifelines NEXT, we selected women who underwent a scheduled CS and then measured cefazolin in the cord blood of their infants (n=13). In seven samples, no detectable cefazolin levels were observed, reflecting that the antibiotic was not given to the mother according to current protocols. In total we collected data from 79 infants at one month after birth from the three unique cohorts, with 42 infants not exposed to antenatal antibiotics (AB- group) and 37 infants exposed to antibiotics (AB+ group).
To test the effect of phenotypes on the overall composition, we used the adonis2 function from R package vegan with 10,000 permutations using “margin”, which assessed the marginal effects of the terms (each marginal term analysed in a model with all other variables). We thereby confirmed that the cohort had no effect on the overall microbiome composition and therefore only corrected for feeding mode when assessing the effect of AB group on the overall microbiome composition.
To test the effect of the AB group on Shannon diversity index, species richness and individual CLR-transformed species, we used a linear model correcting for cohort, with and without correction for feeding mode.
To test the effect of the AB group on ARG load, we used a linear model correcting for cohort, without correction for feeding mode (as there were no significant associations of ARG load with feeding mode).
Additional resources
Data and samples were collected as part of the study registered at ClinicalTrials.gov (accession number: NCT06030713) available at: https://clinicaltrials.gov/study/NCT06030713?cond=NCT06030713&rank=1
Acknowledgments
The authors would like to thank all patients for their active and enthusiastic participation in the trial. We thank the staff at the Department of Obstetrics and Gynecology, UMCG, for their contributions to this trial. We thank Kate Mc Intyre for editing the manuscript and Kateryna Onistrat for assisting with the design of the graphical abstract. We also thank the Genomics Coordination Center and the Center for Information Technology of the University of Groningen for their support and for providing access to the Gearshift and Peregrine high-performance computing clusters. This work was supported by funds from the EASI-Genomics grant (PID7780) to T.S. and A.Z. T.S. holds a scholarship from the Junior Scientific Masterclass, University of Groningen, and a De Cocks-Hadders Stichting grant (Winston Bakker Fonds WB-08). A.Z. is supported by the Dutch Heart Foundation IN-CONTROL (CVON2018-27), European Reserach Council (ERC) Starting Grant 715772, Nederlandse Organisatie voor Wetenschappelijk Onderzoek-VIDI (NWO-VIDI) grant 016.178.056, EU Horizon Europe Program grant INITIALISE (101094099), and Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) Gravitation grant Exposome-NL (024.004.017).
Author contributions
T.S., S.S. and A.Z. conceived the project. T.S., S.S., and A.Z. designed the randomized controlled trial. T.S. wrote the trial protocol. T.S. and A.Z. acquired funding for the trial and experiments. T.S., S.S., and J.R.P. carried out the randomized controlled trial together with the research team of the Department of Obstetrics and Gynecology. T.S. and M.K. performed lab experiments. J.F.d.B. facilitated the bile acid experiments and data interpretation. T.S. performed the bulk of the bioinformatic analyses in this study, A.K. performed the phylogenetics analysis, and T.S. and A.F.-P. performed the analysis of AR genes. T.S. carried out the statistical analysis supervised by A.K. T.S., J.R.P., A.K., and A.Z. were involved in data interpretation. T.D., T.d.M., and M.d.B. were involved in the design and data collection of the MAMI trial. T.S. wrote the manuscript. All authors discussed the data and assisted in writing the manuscript. All authors have read and agreed to the published version of the manuscript.
Declaration of interests
T.d.M. has served as a speaker for Danone Nutricia Research and Mead Johnson. The funders had no role in study design, data analysis, data interpretation, writing of the manuscript, and the decision to publish.
Published: August 14, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chom.2024.07.010.
Supplemental information
References
- 1.Neuman H., Forsythe P., Uzan A., Avni O., Koren O. Antibiotics in early life: dysbiosis and the damage done. FEMS Microbiol. Rev. 2018;42:489–499. doi: 10.1093/femsre/fuy018. [DOI] [PubMed] [Google Scholar]
- 2.Yassour M., Vatanen T., Siljander H., Hämäläinen A.-M., Härkönen T., Ryhänen S.J., Franzosa E.A., Vlamakis H., Huttenhower C., Gevers D., et al. Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med. 2016;8 doi: 10.1126/scitranslmed.aad0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bokulich N.A., Chung J., Battaglia T., Henderson N., Jay M., Li H., D Lieber A., Wu F., Perez-Perez G.I., Chen Y., et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci. Transl. Med. 2016;8 doi: 10.1126/scitranslmed.aad7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Korpela K., Salonen A., Virta L.J., Kekkonen R.A., Forslund K., Bork P., de Vos W.M. Intestinal microbiome is related to lifetime antibiotic use in Finnish pre-school children. Nat. Commun. 2016;7 doi: 10.1038/ncomms10410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.National Institute for Health and Clinical Excellence . 2021. NICE guideline [NG192]https://www.nice.org.uk/guidance/ng192 [PubMed] [Google Scholar]
- 6.Committee on Practice Bulletins-Obstetrics ACOG Practice Bulletin No. 199:Use of Prophylactic Antibiotics in Labor and Delivery. Obstet. Gynecol. 2018;132:e103–e119. doi: 10.1097/AOG.0000000000002833. [DOI] [PubMed] [Google Scholar]
- 7.World Health Organization . 2021. WHO recommendation on prophylactic antibiotics for women undergoing caesarean section.https://iris.who.int/bitstream/handle/10665/341865/9789240028012-eng.pdf [PubMed] [Google Scholar]
- 8.Jyothirmayi C.A., Halder A., Yadav B., Samuel S.T., Kuruvilla A., Jose R. A randomized controlled double blind trial comparing the effects of the prophylactic antibiotic, Cefazolin, administered at caesarean delivery at two different timings (before skin incision and after cord clamping) on both the mother and newborn. BMC Pregnancy Childbirth. 2017;17:340. doi: 10.1186/s12884-017-1526-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun J., Ding M., Liu J., Li Y., Sun X., Liu T., Chen Y., Liu J. Prophylactic Administration of Cefazolin Prior to Skin Incision versus Antibiotics at Cord Clamping in Preventing Postcesarean Infectious Morbidity: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Gynecol. Obstet. Invest. 2013;75:175–178. doi: 10.1159/000346458. [DOI] [PubMed] [Google Scholar]
- 10.Baaqeel H., Baaqeel R. Timing of administration of prophylactic antibiotics for caesarean section: a systematic review and meta-analysis. BJOG. 2013;120:661–669. doi: 10.1111/1471-0528.12036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dierikx T.H., Berkhout D.J.C., Visser L., Benninga M.A., Roeselers G., de Boer N.K.H., de Vries J.I.P., de Meij T.G.J. The influence of timing of Maternal administration of Antibiotics during cesarean section on the intestinal Microbial colonization in Infants (MAMI-trial): study protocol for a randomised controlled trial. Trials. 2019;20:479. doi: 10.1186/s13063-019-3552-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dierikx T., Berkhout D., Eck A., Tims S., van Limbergen J., Visser D., de Boer M., de Boer N., Touw D., Benninga M., et al. Influence of timing of maternal antibiotic administration during caesarean section on infant microbial colonisation: a randomised controlled trial. Gut. 2022;71:1803–1811. doi: 10.1136/gutjnl-2021-324767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warmink-Perdijk W.D.B., Peters L.L., Tigchelaar E.F., Dekens J.A.M., Jankipersadsing S.A., Zhernakova A., Bossers W.J.R., Sikkema J., de Jonge A., Reijneveld S.A., et al. Lifelines NEXT: a prospective birth cohort adding the next generation to the three-generation Lifelines cohort study. Eur. J. Epidemiol. 2020;35:157–168. doi: 10.1007/s10654-020-00614-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mor U., Cohen Y., Valdés-Mas R., Kviatcovsky D., Elinav E., Avron H. Dimensionality reduction of longitudinal ’omics data using modern tensor factorizations. PLoS Comput. Biol. 2022;18 doi: 10.1371/journal.pcbi.1010212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamichhane S., Sen P., Dickens A.M., Alves M.A., Härkönen T., Honkanen J., Vatanen T., Xavier R.J., Hyötyläinen T., Knip M., et al. Dysregulation of secondary bile acid metabolism precedes islet autoimmunity and type 1 diabetes. Cell Rep. Med. 2022;3 doi: 10.1016/j.xcrm.2022.100762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Best N., Rolle-Kampczyk U., Schaap F.G., Basic M., Olde Damink S.W.M., Bleich A., Savelkoul P.H.M., von Bergen M., Penders J., Hornef M.W. Bile acids drive the newborn’s gut microbiota maturation. Nat. Commun. 2020;11 doi: 10.1038/s41467-020-17183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patton L., Li N., Garrett T.J., Ruoss J.L., Russell J.T., de la Cruz D., Bazacliu C., Polin R.A., Triplett E.W., Neu J. Antibiotics Effects on the Fecal Metabolome in Preterm Infants. Metabolites. 2020;10:331. doi: 10.3390/metabo10080331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bossung V., Lupatsii M., Dashdorj L., Tassiello O., Jonassen S., Pagel J., Demmert M., Wolf E.A., Rody A., Waschina S., et al. Timing of antimicrobial prophylaxis for cesarean section is critical for gut microbiome development in term born infants. Gut Microbes. 2022;14 doi: 10.1080/19490976.2022.2038855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamal S.S., Hyldig N., Krych Ł., Greisen G., Krogfelt K.A., Zachariassen G., Nielsen D.S. Impact of Early Exposure to Cefuroxime on the Composition of the Gut Microbiota in Infants Following Cesarean Delivery. J. Pediatr. 2019;210:99–105.e2. doi: 10.1016/j.jpeds.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 20.Gacesa R., Kurilshikov A., Vich Vila A., Sinha T., Klaassen M.A.Y., Bolte L.A., Andreu-Sánchez S., Chen L., Collij V., Hu S., et al. Environmental factors shaping the gut microbiome in a Dutch population. Nature. 2022;604:732–739. doi: 10.1038/s41586-022-04567-7. [DOI] [PubMed] [Google Scholar]
- 21.Harris P.A., Taylor R., Thielke R., Payne J., Gonzalez N., Conde J.G. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J. Biomed. Inform. 2009;42:377–381. doi: 10.1016/j.jbi.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Snow G. 2019. Randomization for Block Random Clinical Trials.https://cran.r-project.org/web/packages/blockrand/index.html R package version 1.3. [Google Scholar]
- 23.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Blanco-Míguez A., Beghini F., Cumbo F., McIver L.J., Thompson K.N., Zolfo M., Manghi P., Dubois L., Huang K.D., Thomas A.M., et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 2023;41:1633–1644. doi: 10.1038/s41587-023-01688-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Truong D.T., Tett A., Pasolli E., Huttenhower C., Segata N. Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 2017;27:626–638. doi: 10.1101/gr.216242.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beghini F., McIver L.J., Blanco-Míguez A., Dubois L., Asnicar F., Maharjan S., Mailyan A., Manghi P., Scholz M., Thomas A.M., et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife. 2021;10 doi: 10.7554/eLife.65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaminski J., Gibson M.K., Franzosa E.A., Segata N., Dantas G., Huttenhower C. High-Specificity Targeted Functional Profiling in Microbial Communities with ShortBRED. PLoS Comput. Biol. 2015;11 doi: 10.1371/journal.pcbi.1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uritskiy G.V., DiRuggiero J., Taylor J. MetaWRAP-a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome. 2018;6:158. doi: 10.1186/s40168-018-0541-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olm M.R., Brown C.T., Brooks B., Banfield J.F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017;11:2864–2868. doi: 10.1038/ismej.2017.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woodcroft B., Newell R. CoverM: Read coverage calculator for metagenomics. https://github.com/wwood/CoverM
- 31.Alcock B.P., Huynh W., Chalil R., Smith K.W., Raphenya A.R., Wlodarski M.A., Edalatmand A., Petkau A., Syed S.A., Tsang K.K., et al. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023;51:D690–D699. doi: 10.1093/nar/gkac920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuznetsova A., Brockhoff P.B., Christensen R.H.B. lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017;82:1–26. doi: 10.18637/jss.v082.i13. [DOI] [Google Scholar]
- 33.Oksanen J., Blanchet F.G., Friendly M., Kindt R., Legendre P., McGlinn D., Minchin P.R., O’Hara R.B., Simpson G.L., Solymos P., et al. 2020. vegan: Community Ecology Package.http://CRAN.Rproject.org/package=vegan R package version 2.5-7. [Google Scholar]
- 34.Hadfield J.D. MCMC Methods for Multi-Response Generalized Linear Mixed Models: The MCMCglmm R Package. J. Stat. Softw. 2010;33:1–22. doi: 10.18637/jss.v033.i02. [DOI] [Google Scholar]
- 35.Schliep K.P. phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bulus M. Pwrss: Statistical power and sample size calculation tools. R Package Version 0.3.1. 2023 [Google Scholar]
- 37.R Core Team R: A language and environment for statistical computing. R Foundation for Statistical Computing. 2021 https://www.R-project.org/ [Google Scholar]
- 38.Harris P.A., Taylor R., Minor B.L., Elliott V., Fernandez M., O’Neal L., McLeod L., Delacqua G., Delacqua F., Kirby J., et al. The REDCap consortium: Building an international community of software platform partners. J. Biomed. Inform. 2019;95 doi: 10.1016/j.jbi.2019.103208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rios-Morales M., van Trijp M.P.H., Rösch C., An R., Boer T., Gerding A., de Ruiter N., Koehorst M., Heiner-Fokkema M.R., Schols H.A., et al. A toolbox for the comprehensive analysis of small volume human intestinal samples that can be used with gastrointestinal sampling capsules. Sci. Rep. 2021;11 doi: 10.1038/s41598-021-86980-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verkade E., Shen W., Hovingh M.V., Mulder N.L., de Bruyn K., Koehorst M., de Vries H.D., Bloks V.W., Kuipers F., de Boer J.F. Gut microbiota depletion aggravates bile acid-induced liver pathology in mice with a human-like bile acid composition. Clin. Sci. (Lond) 2023;137:1637–1650. doi: 10.1042/CS20230812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eggink H.M., Oosterman J.E., de Goede P., de Vries E.M., Foppen E., Koehorst M., Groen A.K., Boelen A., Romijn J.A., la Fleur S.E., et al. Complex interaction between circadian rhythm and diet on bile acid homeostasis in male rats. Chronobiol. Int. 2017;34:1339–1353. doi: 10.1080/07420528.2017.1363226. [DOI] [PubMed] [Google Scholar]
- 42.Dierikx T., Berkhout D., Eck A., Tims S., van Limbergen J., Visser D., de Boer M., de Boer N., Touw D., Benninga M., et al. Influence of timing of maternal antibiotic administration during caesarean section on infant microbial colonisation: a randomised controlled trial. Gut. 2022;71:1803–1811. doi: 10.1136/gutjnl-2021-324767. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Sample metadata and quality-trimmed sequencing reads have been deposited at European Genome-Phenome Archive and are publicly available as of the date of publication. Accession numbers are listed in the key resources table (EGAC00001003433).
-
•
All original code has been deposited on Zenodo and Github and is publicly available as of the date of publication. DOIs are listed in the key resources table (https://doi.org/10.5281/zenodo.11537223 and https://github.com/GRONINGEN-MICROBIOME-CENTRE/CS_BABY_BIOME).
-
•
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.