

Abstract
The advancement of economical and readily available electrocatalysts for the oxygen reduction reaction (ORR) holds paramount importance in the advancement of fuel cells and metal‐air batteries. Recently, 2D non‐metallic materials have obtained substantial attention as viable alternatives for ORR catalysts due to their manifold advantages, encompassing low cost, ample availability, substantial surface‐to‐volume ratio, high conductivity, exceptional durability, and competitive activity. The augmented ORR performances observed in metal‐free 2D materials typically arise from heteroatom doping, defects, or the formation of heterostructures. Here, the authors delve into the realm of electrocatalysts for the ORR, pivoting around metal‐free 2D materials. Initially, the merits of metal‐free 2D materials are explored and the reaction mechanism of the ORR is dissected. Subsequently, a comprehensive survey of diverse metal‐free 2D materials is presented, tracing their evolutionary journey from fundamental concepts to pragmatic applications in the context of ORR. Substantial importance is given on the exploration of various strategies for enhancing metal‐free 2D materials and assessing their impact on inherent material performance, including electronic properties. Finally, the challenges and future prospects that lie ahead for metal‐free 2D materials are underscored, as they aspire to serve as efficient ORR electrocatalysts.
Keywords: 2D materials, electrocatalysts, non‐metal, oxygen reduction reaction
This review presents recent advances in metal‐free 2D materials for the oxygen reduction reaction (ORR), covering mechanistic insights, catalyst development, and performance enhancement. It also addresses challenges and future prospects for the development of high‐performance metal‐free 2D electrocatalysts toward the ORR.
1. INTRODUCTION
The exploration of green and renewable energy generation systems such as fuel cells and metal‐air batteries has become a necessity in recent years due to the rapid growing issues such as climate change, environmental pollution, and energy security.[ 1 , 2 , 3 , 4 , 5 , 6 , 7 ] To promote the performance of these energy systems, it is crucial to understand the fundamental principles of electrocatalysis.[ 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 ] Oxygen reduction reaction (ORR) is an electrochemical process in which O2 is reduced by four electrons at the cathode of fuel cells and metal‐air batteries,[ 16 , 17 , 18 , 19 ] and the sluggish ORR kinetics that limits the overall cell performance has been known as a major problem.[ 20 , 21 , 22 , 23 ]
To tackle this issue, high‐performance Pt‐based catalysts have been adopted,[ 24 , 25 , 26 , 27 , 28 , 29 ] but their scarcity in the earth, high cost, and low stability restrict the large‐scale application.[ 10 , 30 , 31 ] As an alternative, remarkable advances have been achieved in the development of non‐noble metal‐based catalysts, and some of them have demonstrated ORR activity similar to or even superior to the benchmark Pt catalyst.[ 32 , 33 , 34 , 35 , 36 , 37 ] However, non‐noble metal‐based catalysts are much less stable under high operating potentials during ORR. Apart from the development of non‐noble metal‐based catalysts, the recent discovery of metal‐free carbon‐based ORR catalysts has opened up new horizons.[ 38 , 39 , 40 , 41 , 42 ] Metal‐free carbon‐based catalysts boast advantages in tolerance to the fuel, stable chemical structure, and good stability in alkaline media.[ 43 , 44 , 45 ]
During the ORR process, O2 molecules undergo adsorption and diffusion on the catalyst surface.[ 1 , 46 ] Moreover, a change in the catalyst morphology and surface structure often results in variations in active sites, controlling the performance of ORR.[ 47 , 48 ] 2D structured catalysts are highly appealing for ORR electrocatalysis due to their affordability, large surface area, exceptional physicochemical properties, superior electrical conductivity, high mechanical strength, and outstanding chemical stability.[ 49 , 50 , 51 , 52 ] Especially, the electronic structure of active sites and the surface geometric arrangements of 2D electrocatalysts play vital roles in controlling the energy barriers of the reactions, and thereby enhancing the intrinsic activity.[ 53 , 54 , 55 , 56 ] In this regard, metal‐free 2D electrocatalysts have received surging interest owing to their significantly large surface‐to‐volume ratio, allowing the maximum exposure of active sites, and enhanced ORR activity.
Apart from morphology, the surface defect is another factor controlling the number of active sites.[ 57 , 58 ] The abundance of exposed surface atoms on 2D materials facilitates the formation of defect structures, crystal twinning, and corrugated morphologies.[ 59 ] These structural characteristics can significantly impact the material's intrinsic properties and enhance catalytic activities. The number of active sites on a 2D surface can be further increased by considering dopants, edges, twists, and size.[ 59 , 60 , 61 ] Therefore, metal‐free 2D materials offer a versatile platform for tailoring properties to achieve desired functionalities, making it a highly active and dynamic field of research. However, the limited range of 2D materials and challenges in producing them on a large scale gravely hampered their practical application.
Though there are some review articles on the non‐metallic materials for ORR, present articles particularly focused on the 2D structure of metal‐free nanomaterials for ORR. This review aims to comprehensively summarize recent developments in 2D metal‐free ORR catalysts and discuss the mechanistic understanding specific to them, as shown in Figure 1. First, we introduce the fundamental mechanisms of ORR, emphasizing the significance of ORR in fuel cells. Later, we discuss carbon‐based and non‐carbon‐based 2D materials and their ORR activity. We also include different factors, such as heteroatom doping, heterostructure formation, and generation of defects, which impact the ORR performance of the catalysts. Finally, the challenges and perspectives of this rapidly growing topic are discussed.
FIGURE 1.
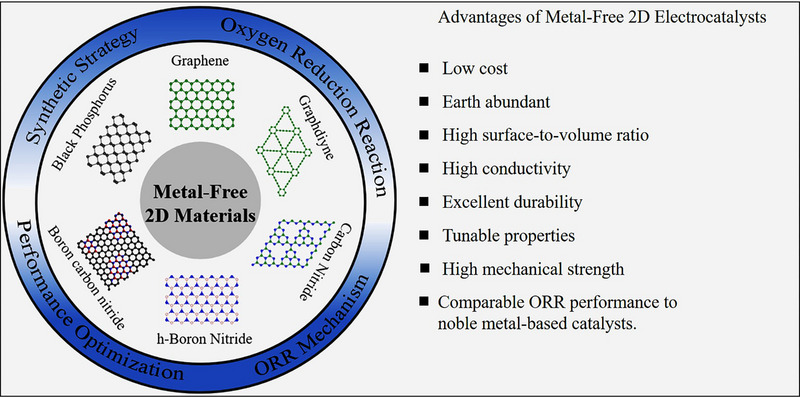
Summary of metal‐free 2D electrocatalysts for ORR.
2. MECHANISTIC UNDERSTANDING OF THE ORR
2.1. A general ORR mechanism for metal‐based catalysts
It has been widely accepted that the cathodic ORR undergoes via either a four‐electron (direct) pathway or a series of two‐electron pathways. The reaction mechanism and the electron transfer process largely depend on the electrolytes (either acidic or alkaline). The overall pathways are as follows (Equations (1)−(4)), where SHE stands for standard hydrogen electrode:[ 62 , 63 ]
Four‐electron pathway
(Alkaline)
| (1) |
(Acidic)
| (2) |
Two‐electron pathway
(Alkaline)
| (3a) |
followed by further two‐electron reduction
| (3b) |
or disproportionation
| (3c) |
(Acidic)
| (4a) |
followed by further two‐electron reduction
| (4b) |
or disproportionation
| (4c) |
For the purpose of the fuel cell or metal‐air battery applications, a direct four‐electron pathway is preferred to a series of indirect two‐electron pathways because the formation of intermediate H2O2 or HO2 – would reduce the overall number of transferred electrons. Moreover, versatile H2O2 and HO2 – would significantly degrade ionomers and electrocatalysts, reducing overall stability.[ 5 , 64 , 65 ]
For the direct four‐electron pathway of the ORR, the reaction proceeds (i) adsorption of O2 molecules, (ii) electron transfer to adsorbed O2, (iii) weakening and scission of O═O bonds, and (iv) desorption of products (H2O or OH–). Under alkaline electrolyte, the associative mechanism goes through the following steps (Equation (5)), where * represents a surface of electrocatalysts and *O2, *OOH, *OH, and *O represent adsorbed reaction intermediates:[ 63 ]
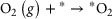 |
(5a) |
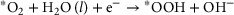 |
(5b) |
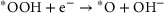 |
(5c) |
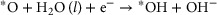 |
(5d) |
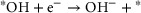 |
(5e) |
Competitive two‐electron mechanism
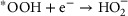 |
(5f) |
In acidic electrolytes, H3O+ rather than H2O acts as the proton donor, and the corresponding reaction steps are as follows:[ 63 ]
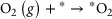 |
(6a) |
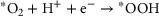 |
(6b) |
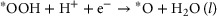 |
(6c) |
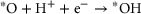 |
(6d) |
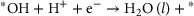 |
(6e) |
Competitive two‐electron mechanism
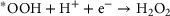 |
(6f) |
The selectivity of four‐ and two‐electron ORR is determined by the competitive reactions after forming the *OOH intermediate, whether the facilitating O─O bond scission occurs (Equations (5f) and (6f)).[ 66 ] Conversely, the dissociative mechanism via direct cleavage of the O═O bond initiates with the following step (Equation (7)), followed by Equations (5d–e) or (6d–e) in alkaline or acidic media, respectively.[ 62 ]
 |
(7a) |
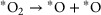 |
(7b) |
However, due to the higher bond dissociation energy of the O═O bond in O2 than O─O bond in superoxides, the associative mechanism is preferred to the dissociative mechanism.[ 62 ] Overall plausible mechanisms of the ORR are summarized in Figure 2.
FIGURE 2.
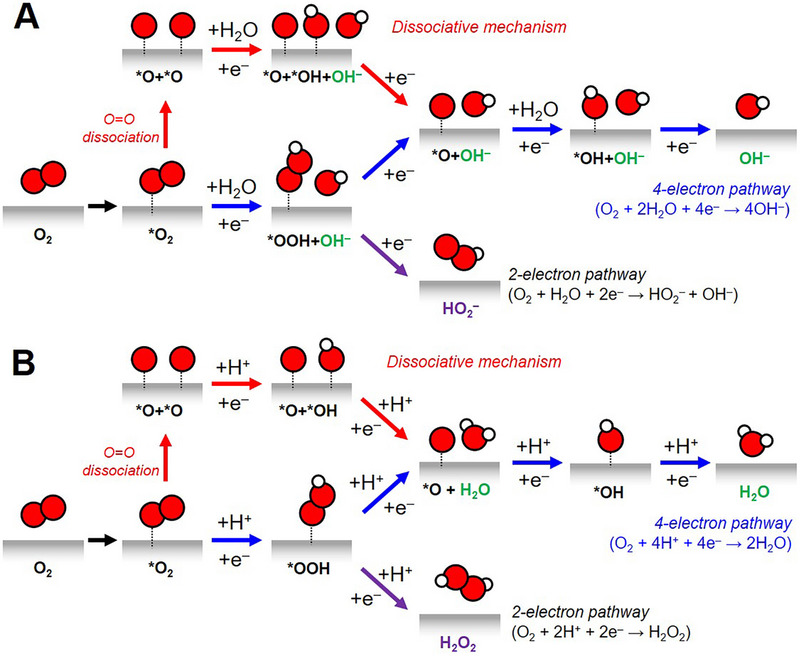
Schematic illustration of potential mechanisms of the ORR under (A) basic and (B) acidic conditions.
The theoretical descriptor for the ORR activity for metal‐based electrocatalysts has been widely explored based on the d‐band theory.[ 67 , 68 ] In 2004, Norskøv et al. employed density functional theory (DFT) and compared free energy landscapes for the binding of the ORR intermediates on the Pt(111) surface.[ 69 ] The DFT study on the model Pt(111) surface suggested that the binding energies of *O, *OH, and *OOH intermediates are strongly correlated, which indicates scaling relations between *OOH and *OH or *OOH and *O.[ 67 ] According to these scaling relations, the limiting potential for the ORR on the metal surface shows Sabatier volcano shape versus the difference of theoretical adsorption free energy of *OH intermediate (ΔG OH), which would be changed by the d‐band center of the metal surface.[ 68 ] The commonly‐accepted rate‐determining step for the ORR is the desorption of *OH to form H2O or the adsorption of O2 to form *OOH.[ 67 , 70 ] Moreover, the selectivity of four‐ and two‐electron ORR is determined by the binding energies of *O and *OOH intermediates.[ 63 , 66 , 71 ] The four‐electron pathway is chosen when the adsorption of *O intermediate from O─O bond dissociation is more favored than *OOH intermediate. On the other hand, more dominant adsorption of *OOH species over *O intermediate facilitates a two‐electron pathway over a four‐electron pathway because the *OOH intermediates are preserved with the suppression of O─O dissociation.[ 66 , 71 ]
2.2. An ORR mechanism for metal‐free catalysts
For the metal‐free electrocatalysts, the overall ORR mechanism also involves the adsorption of O2 and the ORR intermediates, similar to the mechanism in metal‐based electrocatalysts. However, the ORR thermodynamics and kinetics could not be explained by the adsorption energies of reaction intermediates from the d‐band theory in the metal‐free electrocatalysts because they are generally composed of carbon. Moreover, pristine sp2 carbon without any defects or dopants is inactive to the adsorption of O2 and the ORR intermediates.[ 62 , 66 ] Therefore, the outer sphere electron‐transfer mechanism is suggested as the ORR mechanism in metal‐free, carbon‐based electrocatalysts.[ 72 , 73 , 74 ] In this mechanism, O2 molecules do not directly interact with the surface of the electrocatalysts; rather, the electron transfer occurs through a solution‐phase mediator such as an alkali metal cation.[ 75 , 76 , 77 ] During ORR, the solvated molecular O2 cluster, O2(H2O) n , weakly interacts with surface‐adsorbed hydroxyl species to promote the two‐electron pathway, indicating that the outer‐sphere electron transfer occurs with the two‐electron reduction.[ 74 , 78 ]
Therefore, to improve the four‐electron reduction pathway, it is strongly required to regulate the energy states of basal carbon to promote the adsorption of O2 and the ORR intermediates. Particularly in metal‐free, carbon‐based electrocatalysts, hetero‐atom doped sites,[ 79 , 80 ] or defective sites[ 81 , 82 , 83 ] exhibited lower free energy for the adsorption of O2 and the intermediates (*O, *OH, and *OOH) than sp2 carbon atoms in the basal plane. Several recent theoretical and experimental studies revealed that the adsorption free energy of O2 and the ORR intermediates are changed by the local electron density (charge distribution), the electronegativity of adjacent dopants, the electron configuration of dopants, and hybridization (sp2 or sp3).[ 82 , 83 , 84 , 85 , 86 ] However, the exact reaction mechanism and the active sites in metal‐free electrocatalysts toward the ORR are still under debate, and therefore, further thorough investigation on both theoretical and experimental is strongly required.
3. CARBON‐BASED 2D ELECTROCATALYSTS FOR THE ORR
Carbon‐based ORR catalysts started to gain attention after the invention of N‐doped carbon nanotubes as an alternative to Pt catalysts.[ 10 , 38 , 87 ] Since then, enormous progress has been attained in metal‐free 2D catalysts in the ORR process. In this section, we discuss carbon‐based 2D ORR catalysts such as graphene, graphyne, and graphitic carbon nitride.
3.1. Graphene and graphyne
Graphene is composed of hexagonally arranged sp2 carbon atoms, which resemble a honeycomb structure (Figure 3A). For electrocatalysis, the delocalized π bonding network within graphene could induce the endothermic adsorption of reaction intermediates.[ 88 ] Therefore, the basal plane of graphene is electrocatalytically inactive. Defect engineering, heteroatom doping, and heterostructure formation can be conveniently employed to fine‐tune the electronic and surface properties of graphene. High electrical conductivity and large surface area are prerequisites for the successful electrocatalytic applications. As N exhibits higher electron negativity and one more valence electron than C, N has been the favorable choice to tune the overall electronic structure of graphene, to increase its conductivity, as well as to create active sites toward the ORR.[ 89 ] As shown in Figure 3B, doping leads to the formation of different types of N doping such as graphitic N, pyrrolic N, pyridinic N, amide N, and N oxides.[ 90 ] The planar structure of graphene can be disrupted by the inclusion of an sp3‐bonded pyrrolic N atom. However, pyridinic N and graphitic N do not significantly affect the structure of graphene as the C─N bond length is very similar to the C─C bond length.[ 91 ] Furthermore, pyridinic N has demonstrated excellent stability when a single vacancy is present. Interestingly, both Stone–Wales and vacancy defects presented in graphitic N and pyridinic N promoted the ORR performances.[ 92 , 93 ]
FIGURE 3.
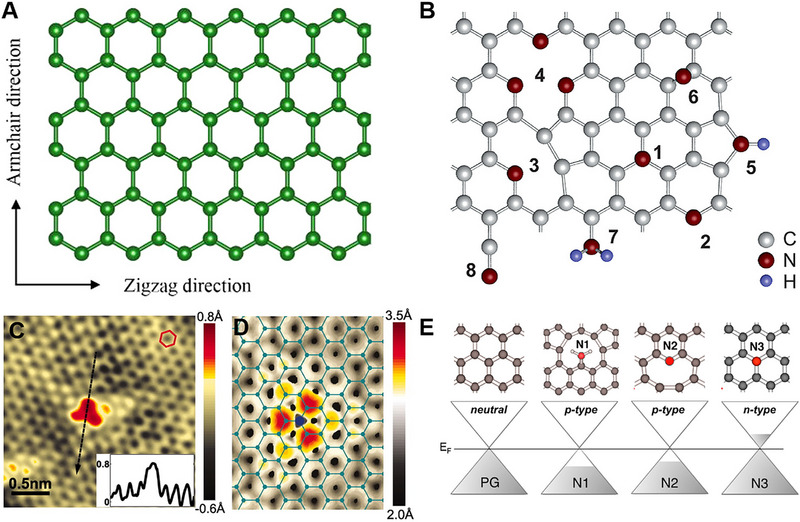
(A) Schematic molecular configuration of graphene. Reproduced with permission.[ 1 ] Copyright 2018, American Chemical Society. (B) Different types of N impurities in graphene (1) substitutional or graphitic N, (2) pyridine‐like N, (3) single N pyridinic vacancy, (4) triple N pyridinic vacancy, (5) pyrrole‐like, (6) interstitial N or adatom, (7) amine, (8) nitrile. Reproduced with permission.[ 90 ] Copyright 2011, American Chemical Society. (C) STM image of most common doping form observed on N‐doped graphene, corresponding to a single graphitic N dopant. The inset showing the atomic corrugation and apparent height of the dopant. (D) Simulated STM image of graphitic N dopant based on DFT calculations. Reproduced with permission.[ 94 ] Copyright 2011, Science. (E) Computed doping effect of N dopant. Reprinted with permission.[ 95 ] Copyright 2012, American Chemical Society.
Scanning tunnelling microscopy (STM) analysis can reveal the exact type of N doping in the graphene. Figure 3C,D show the STM and simulated STM images, respectively.[ 94 ] Investigating the authentic catalytic site within N‐doped graphene holds significance due to the ambiguity surrounding whether the pyridinic N or graphitic N is the true active site. Anderson and co‐workers discovered that the graphitic N, which is far from the sheet edge, is more active than N atoms close to the edge.[ 96 ] Miyata and co‐workers also reported that graphene with graphitic N performs better ORR activity than that with pyridinic N.[ 97 ] In another work, Miyata and co‐workers showed that graphitic N atoms adjacent to the zigzag edge of graphene enhance the process of adsorption of O2 and subsequent ORR, resulting in the conversion of two molecules of H2O with a low activation barrier of approximately 5 kcal mol‐1.[ 98 ] However, they suggested that the graphitic N atoms located on the inner side of the graphene sheet do not take part in the ORR. The conductive properties of N‐doped graphene are also influenced by its configuration. Every N atom forms three σ bonds with neighboring carbon atoms in graphitic N (N3) (Figure 3E).[ 95 ] Additionally, one of the nitrogen's valence electrons forms a π bond with the neighboring carbon atoms, while the fifth electron contributes in the π* state of the junction band. The π network of the graphene lattice can receive 0.5 electrons from each N3. In contrast, pyridinic N (N2), and nitrilic N (N1) have the opposite electronic effect. They withdraw electrons from the π network of the graphene lattice.
Lazaro and co‐workers prepared N‐doped graphene by annealing graphene and urea at different temperatures ranging from 500–800°C.[ 99 ] Lower temperature synthesis (500°C and 600°C) produced a small amount of carbon nitride (CN) along with N‐doped graphene, while higher temperatures led to higher purity of N‐doped graphene. It was observed that CN does not show promising ORR performance as a catalyst. A uniform 2D laminar structure was formed in all the cases. The N‐doped graphene synthesized at 800°C, which lacks CN, showed the highest ORR performance among all samples, with a halfwave potential (E 1/2) of 0.76 V versus RHE, similar to commercial Pt/C. Guo and co‐workers adopted a pyrolysis process to synthesize N‐doped graphene, taking polyaniline as a nitrogen source.[ 100 ] A typical 2D graphene morphology was generated with stacked and interlaced ultrathin nanosheets. In addition, a large number of pores were observed, which are believed to play a significant role in transporting the electron, thus improving the ORR activity. This N‐rich graphene showed an onset potential (E onset) of 0.99 V versus RHE and E 1/2 of 0.84 V versus RHE. Ruoff and co‐workers investigated the impact of N precursor and temperature on the performance of the ORR.[ 101 ] Figure 4A shows a schematic for the synthesis process for different N‐doped reduced graphene oxide (N‐RG‐O). The adoption of nitrogen atom‐containing aniline, pyrrole, and ammonia gas as precursors gave rise to the N‐doping of carbon materials. Aniline precursor created pyridinic N‐doped RGO, whereas pyrrole created pyrrolic N‐doped RGO. In the case of ammonia, the type of N‐doping was determined by the annealing temperature. At 550°C, only pyridinic N‐doped RGO was obtained, whereas at 850°C, a small amount of graphitic N‐doped RGO was formed along with pyridinic N‐doped RGO. In the case of 1000°C, mostly graphitic N‐doped RGO was produced with a small amount of pyridinic N‐doped RGO. Graphitic N containing RG‐O prepared at 1000°C (N‐RG‐O 1000°C) showed higher ORR performance than pyridinic N and pyrrolic N species (Figure 4B,C). The 2D structure of both N‐RG‐O 850°C and N‐RG‐O 1000°C have more exposed edges compared to pristine G‐O. It is observed that the total percentage of N in the system does not affect the ORR performance. Graphitic N controls the limiting diffusion current (J L), whereas pyridinic N controls the E onset. In all these cases, graphitic N is believed to play a major role in enhancing ORR performance.
FIGURE 4.
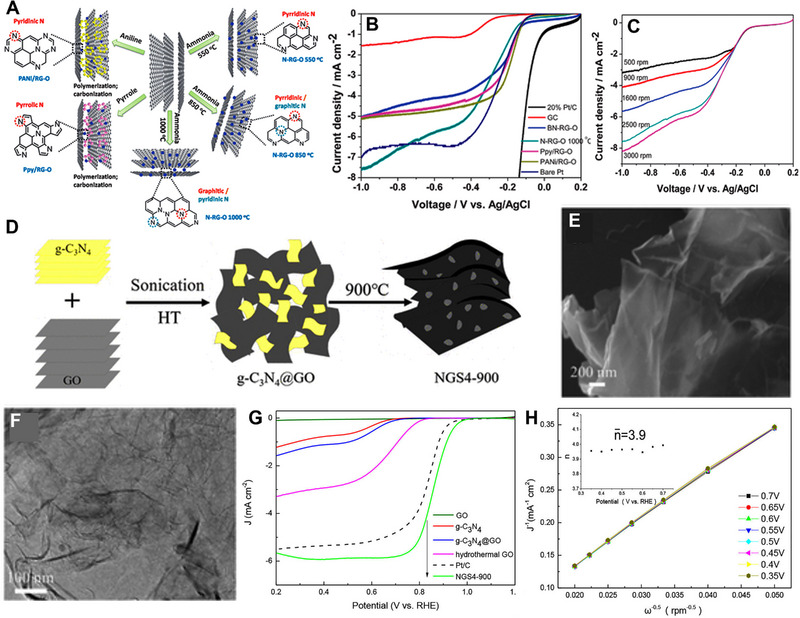
(A) Schematic representation of N‐doped graphene synthesis with different N states. (B) Linear sweep voltammetry (LSV) plot of N‐RG‐O 1000°C, bare GC electrode, PANi/RG‐O, Ppy/RG‐O, BN‐RG‐O, bare Pt electrode, and 20% Pt/C in O2 saturated 0.1 m KOH solution (scan rate: 10 mV s‐1, rotation rate: 2500 rpm). (C) LSV of N‐RG‐O 1000°C with a rotation rate from 500 to 3000 rpm. Reproduced with permission.[ 101 ] Copyright 2012, Royal Society of Chemistry. (D) Schematic representation of the synthesis process of NGS4−900. TEM images of (E) g‐C3N4@GO and (F) NGS4−900. (G) LSV plots of GO, g‐C3N4, g‐C3N4@GO, hydrothermal GO, Pt/C, and NGS4−900 obtained on 1600 rpm RDE in O2 saturated 0.1 m KOH with 10 mV s‐1. (H) K–L plots of NGS4−900 at different potentials. Reproduced with permission.[ 102 ] Copyright 2018, American Chemical Society.
Gupta and co‐workers reported that solvent treatment could alter the ORR performance of graphene oxide (GO).[ 102 ] The solvent treatment was performed before the N doping process. Different solvents, such as water, ethanol, toluene, pyridine, and diethyl ether, were utilized for this treatment. They prepared an N‐doped GO catalyst in the presence of ammonia at 850°C. The ORR activity was more significantly affected in cyclic solvents such as toluene and pyridine than in non‐cyclic solvents like water, ethanol, and diethyl ether. In the case of water, E 1/2 was 0.77 V versus RHE, which changed to 0.81 V in ethanol and diethyl ether. E 1/2 increased to 0.80 V and 0.84 V versus RHE in the case of pyridine and toluene, respectively. Samples treated with pyridine and diethyl ether exhibited superior selectivity and performance in an alkaline medium. Chen and co‐workers prepared N‐doped graphene nanosheets (NGS4‐900) by pyrolysis (900°C) of g‐C3N4@GO gel‐like hybrid.[ 103 ] First, the g‐C3N4@GO was prepared via a hydrothermal method by exfoliating g‐C3N4 sheets on GO sheets, which resulted in the 3D hybrid film structure. Then the g‐C3N4@GO was annealed at the temperature range of 700–1000°C under vacuum for 1 h. During the thermal expansion process, GOs are typically assembled into a fluffy porous 3D structure. This design improved the ease of accessing exfoliated g‐C3N4 sheets and promoted more efficient interactions between the two materials. During the annealing process of the g‐C3N4@GO composite, uniform distribution of nitrogen atoms occurred within the graphene matrix. Figure 4D shows a schematic diagram of the synthesis process. TEM images of g‐C3N4@GO and NGS4−900 are shown in Figure 4E,F, where ultra‐thin 2D morphology of NGS4‐900 is observed. The NGS4−900 showed a higher ORR activity than pure GO, g‐C3N4, and g‐C3N4@GO due to the doping of N atoms in the pyrolysis process, which forms abundant defect structure in the edge of graphene skeleton (Figure 4G). NGS4−900 exhibited E onset (0.984 V), E 1/2 (0.859 V), and J L (5.98 mA cm‐ 2), which were higher than those of commercial Pt/C catalyst (0.971 V, 0.848 V, and 5.41 mA cm‐ 2), respectively. The calculated number of electrons per O2 molecule was 3.9, suggesting a four‐electron reaction pathway (Figure 4H). However, it still is difficult to conclude which, graphitic N or pyridinic N, is more responsible for the enhanced ORR performance.
In addition to N, elements from the p‐block like B, S, and P can be incorporated into the graphene matrix to enhance the ORR performance. This doping can be done either by chemical or physical approaches. As with N, the difference in size and electronegativity of these atoms to C can cause electron modulation, resulting in changes in the electronic properties of pristine graphene. The common precursors to dope these elements are boric acid or boron oxide for B; CS2, phenyl disulfide, or benzyl disulfide for S; triphenylphosphine for P.[ 1 ] Theoretical calculations indicated that among the potential heteroatom dopants, B notably enhances the local spin density on the basal plane of graphene. This results in enhanced oxygen adsorption kinetics due to the presence of inherent acidic B‐C active sites, which promote hydroxyl adsorption.[ 104 , 105 ] Considerable electronegativity difference between B (2.04) and C (2.55) is the main reason behind this phenomenon. As a result, the paired covalent electrons of the B─C bonds experience a slight polarization, resulting in the formation of a localized positive charge density on the B atoms. These sites behave as strong acidic defects, providing better adsorption sites for O2 and its intermediates.[ 106 ] The efficiency of electron transfer from the catalytic sites to the reaction intermediates is a pivotal factor that impacts the electrocatalytic activity of B‐doped graphene. It has been reported that during the ORR transfer process, the B sites behave as electron donors, where p‐electrons of graphene form a bonding pz orbital that includes an unpaired electron.[ 106 , 107 , 108 ] Incorporating B atoms into the graphene lattice is a challenging process, mainly because of the exceptional stability of the C─C bonds. However, because of their comparable atomic dimensions, B can replace C in the graphene lattice.[ 108 , 109 , 110 , 111 ]
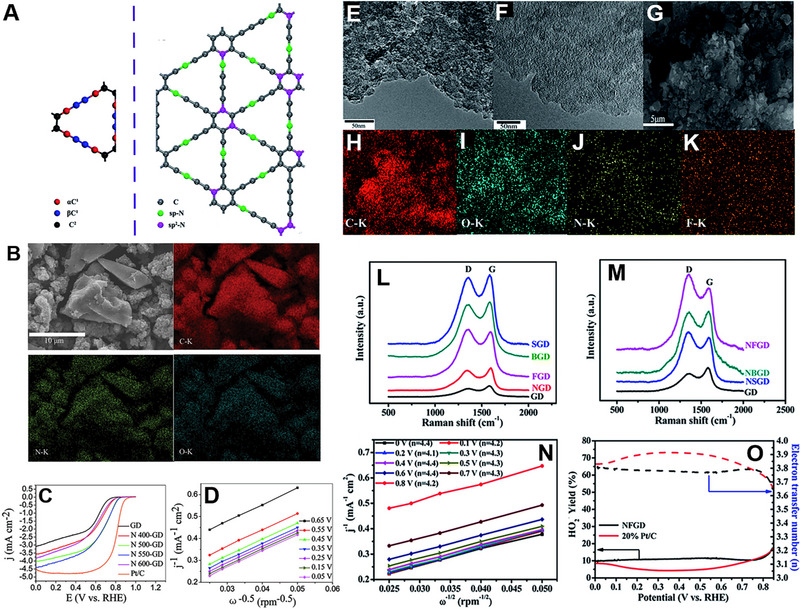
Four different kinds of B doping are possible: substitutional, boronic, borinic, and organo‐borane (Figure 5A).[ 112 ] In the case of boronic doping, a carbon atom is attached to the B(OH)2 group, while in the case of borinic doping, two carbon atoms are connected to the B(OH) group. In the case of organo‐borane, the BH2 group is attached to the carbon atom of the graphene layer. The substitutional B doping has been predominantly attributed to electrocatalytic activity in most cases.[ 108 , 109 , 110 , 111 ] Sobrido and co‐workers proposed the increased B doping level by applying ultrasonication before hydrothermal treatment, leading to a higher ORR activity.[ 112 ] They first sonicated GO solution for different time durations. Then, they added H3BO3 to the solution and heated the mixture in an autoclave at 180°C for 12 h. It is observed that the sample prepared with 90 min of ultrasonication showed a large surface area (438 m2 g‐1) compared to samples prepared with 60 min (301 m2 g‐1) and 30 min (211 m2 g‐1) of ultrasonication. It is also observed that the pore size of the B‐doped reduced graphene oxide (rGO) decreases with the ultrasonication time. B‐doped rGO prepared with 90 min of ultrasonication showed E onset of 0.833 V versus RHE, which is 67 mV lower than the commercial Pt/C (Figure 5B). It also exhibits J L of 4.1 mA cm‐ 2 (at 0.5 V vs RHE) and E 1/2 of 0.671 V versus RHE. B‐doped rGO prepared in 90 min showed a calculated electron transfer number (n) of 3.7 under 0.5 V versus RHE, suggesting a four‐electron pathway. The higher performance of B‐rGO was attributed to B‐substitution doping, defect concentration, and surface area. Wang and co‐workers improved the activity of B‐doped graphene (BG) by depositing polynitrogen (N8 −, PN).[ 113 ] NaBH4 solution was added to the colloidal GO solution, which was heated in a Teflon‐lined stainless steel autoclave. By controlling the amount of initial boron precursor, three samples (B X G, X = 1, 2, 3) containing different amounts of B were prepared in the first step using a hydrothermal treatment method. Figure 5C,D shows the FESEM and TEM images of the B1G sample, confirming the 2D structure. PN‐BG was prepared using an electrochemical deposition technique to deposit N8 − on BG. A larger amount of N8 − were deposited on B1G compared to pure graphene. B doping enhanced the charge transfer from BG to PN, forming more PN on BG. PN‐B1G, which had a boron content of 1.23%, showed the highest J L compared to PN‐B2G (2.01%), PN‐B3G (2.78%), and PN‐G (0%) (Figure 5E,G). The calculated electron transfer number for PN‐B1G was in the range of 3.8–4.0, suggesting a 4‐electron transfer process in ORR (Figure 5F,H). Larger amount of N8 − species were observed on B1G, which provided more active sites for ORR, leading to superior ORR performance.
FIGURE 5.
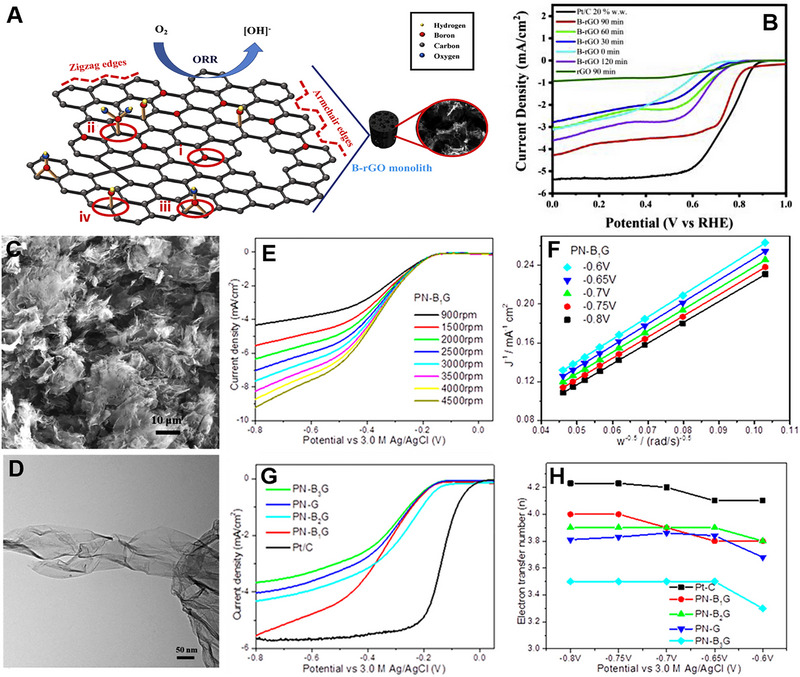
(A) Schematic representation of (i) B substitutional doping on graphene lattice, (ii) boronic, (iii) borinic, (iv) organo‐borane, and typical defects found in B‐doped rGO. (B) LSV plots of B‐doped rGO samples in 0.1 m KOH solution. Reproduced with permission.[ 112 ] Copyright 2022, Elsevier. (C) FESEM and (D) TEM images of B1G. (E) LSV of PN‐B1G sample measured in an O2 saturated 0.1 m KOH solution with different rotation speeds at a scan rate of 5 mV s‐1. (F) K–L plots of PN‐B1G at different potentials. (G) LSV curves of different PN‐BG samples and Pt/C with a rotation speed of 1500 rpm at a scan rate of 5 mV s‐1. (H) Electron‐transfer number of different PN‐BG samples and Pt/C, calculated from the K–L equation. Reproduced with permission.[ 113 ] Copyright 2020, American Chemical Society.
Two or three elements doping on graphene were also studied to enhance the ORR activity compared to single‐element doped graphene. The DFT study suggested that introducing two dopants can modify the electron‐donor characteristics of adjacent carbon atoms, enhancing adsorption of intermediate.[ 114 ] Shin and co‐workers synthesized a B and N co‐doped graphene (BNG) using a microwave hydrothermal treatment using ammonium biborate as a precursor.[ 115 ] Later, the BNG was reduced by hydrazine hydrate using hydrothermal treatment to remove the excess oxygen in the graphene layer. A lowly reduced BN co‐doped graphene (LRBNG) and a highly reduced BN co‐doped graphene (HRBNG) were prepared at 393 and 423 K, respectively. Figure 6A shows the HRTEM image of the HRBNG, showing different graphene layers. It is observed that the morphology of the sample does not alter after the reduction process. XPS analysis confirmed the doping of 3.55% B and 4.43% N in the HRBNG. Figure 6B shows the XPS survey spectra of GO, BNG, LRBNG, and HRBNG. A deconvoluted N1s XPS peak showed four different peaks for pyridinic, pyrrolic, quaternary, and oxidized nitrogen functionalities, as seen in Figure 6C. A B1s XPS peak was deconvoluted into two peaks; one for B─N, BC3, and the other for BC2O, BCO2 components (Figure 6D). CVs of different samples in O2 saturated 0.1 m KOH solutions confirmed the positive effect of B and N doping in graphene for ORR (Figure 6E). Further, LSV curves demonstrated the highly active nature of HRBNG with E onset of 0.93 V, which is higher than LRBNG (0.84 V) and BNG (0.73 V) (Figure 6F). HRBNG showed a constant E onset and the J L increases with increase in rotation speed (Figure 6G). K–L plot suggested that HRBNG followed a 4‐electron pathway for the ORR process (Figure 6H).
FIGURE 6.
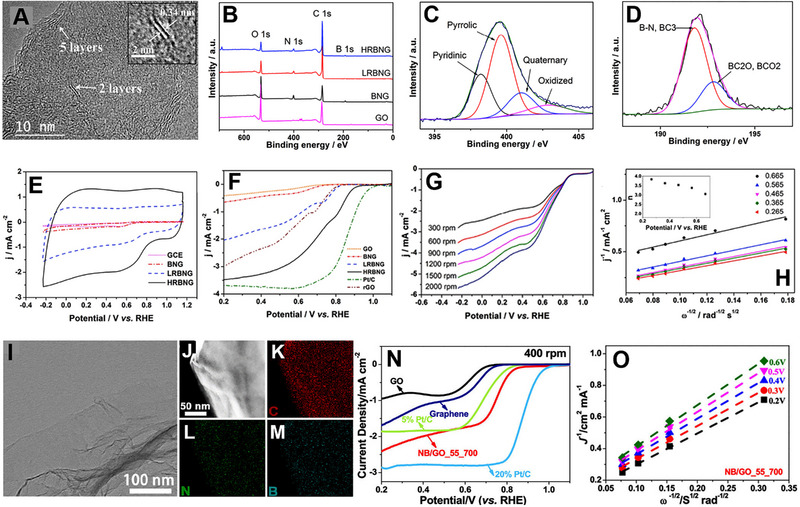
(A) HRTEM image of HRBNG. (B) XPS survey spectra of GO, BNG, LRBNG, and HRBNG. XPS region spectra of (C) N1s and (D) B1s. (E) CVs of GCE, BNG, LRBNG, and RHBNG in O2 saturated 0.1 M KOH solutions at a scan rate of 100 mV‐1. (F) LSV curves of GO, BNG, LRBNG, HRBNG, Pt/C, and rGO in O2 saturated 0.1 m KOH solution at a scan rate of 10 mV s‐1 and electrode rotating rate of 1500 rpm. (G) LSV curves of HRBNG at different rotation speeds. (H) K–L plot for the HRBNG at different potentials. Reproduced with permission.[ 115 ] Copyright 2016, Elsevier. (I) TEM and (J) STEM images of NB/GO_55_700 with corresponding EDX mapping of (K) C, (L) N, and (M) B. (N) LSV curves of graphene, GO, NB/GO_55_700, 5% Pt/C, and 20% Pt/C in O2 saturated 0.1 m KOH solution at a scan rate of 5 mV s‐1 and electrode rotating rate of 400 rpm. (O) K–L plots of NB/GO_55_700 derived from LSVs recorded at different rotation speeds. Reproduced with permission.[ 116 ] Copyright 2019, American Chemical Society.
Sun and co‐workers prepared N and B co‐doped graphene using ultrasonication treatment.[ 116 ] Ammonia solution and boric acid solution were employed as precursors for N and B doping. The doping amount of N and B can be tuned by ultrasonication and annealing temperatures. The co‐doped sample, which was ultrasonicated at 55°C and annealed at 700°C (termed as NB/GO_55_700), exhibited a 2D layer morphology as confirmed by the TEM analysis (Figure 6I ). Scanning TEM (STEM) and EDX analysis suggested that B and N are equally distributed throughout GO (Figure 6J–M). NB/GO_55_700 exhibited higher ORR performance in 0.1 m KOH solution than B/GO_55_700 and N/GO_55_700 (Figure 6N). NB/GO_55_700 showed E onset of 0.82 V versus RHE and J L of 2.43 mA cm‐ 2. Graphitic N, Pyridinic N, and BC3 acted as active ORR sites and followed a 4‐electron transfer process generating OH− as the main product (Figure 6O). The synergistic interaction of B and N plays a crucial role in enhancing ORR activity.
Surface defects can also play a role in increasing the catalytic performance of carbon‐based 2D materials. In the case of graphene, some intrinsic or extrinsic defects can be introduced during synthesis or post‐treatment processes.[ 117 ] These defects help to adsorb the reactant during the catalytic process. In graphene, defects are always associated with heteroatom doping. However, it is very hard to identify the actual active site, even in the case of a well‐studied N‐doped graphene system.[ 118 ] Theoretical studies suggested that intrinsic defects in graphene play a huge role in electrocatalytic activity, but it is a big challenge to distinguish the individual defect types and associated roles and characterize experimentally.[ 1 ] Li and co‐workers introduced defects in N‐doped carbon nanosheets (NCN) using a pyrolysis method.[ 119 ] The material was synthesized by a simple one‐step carbonization of citric acid and NH4Cl. NH4Cl acted as a nitrogen source by generating NH3 during its thermal decomposition. From TEM analysis, it is observed that the NCN contains mesopores and micropores (Figure 7A). HRTEM further confirmed that the NCN contained four graphitic layers (Figure 7B). The sample prepared at 1000°C possessed a large surface area (1793 m2 g‐1) and edge defects. Near edge X‐ray absorption fine structure (NEXAFS) study confirmed that in the case of C K‐edge spectra, the defects observed at 284.0 eV were due to the low‐coordination carbon atoms located at the edges of NCNs (Figure 7C). N K‐edge NEXAFS spectra confirmed the presence of graphitic N, pyrrolic N, pyridinic N, C─N, and C─N─C (Figure 7D). DFT calculation suggested that the carbon atoms positioned at the armchair edge and next to the graphitic N dopants act as intrinsic active sites for ORR. The as‐synthesized sample showed E onset of 0.95 V versus RHE, E 1/2 of 0.82 V versus RHE, and a high J L of 6.43 mA cm‐ 2 (Figure 7E). The catalyst also showed a long‐term durability toward ORR (Figure 7F). Ultra‐thin sheet‐like structure, high surface area, and rich in edge defects promoted the ORR activity of NCN. Wang and co‐workers established a defect‐activity relationship for ORR.[ 120 ] A pyridinic‐N‐doped defective graphene was synthesized via a pyrolysis process. First, a black hydrogel was prepared by hydrothermal treatment of g‐C3N4 and GO mixture, which was then dried and heated at a temperature range of 600–900°C under N2 flow. Thin layers of graphene were obtained along with uniform N distribution confirmed from STEM and elemental mapping (Figure 7G). The sample synthesized at 800°C (NDGs‐800) displayed a maximum percentage of pyridinic N content confirmed from the XPS analysis (Figure 7H). In Raman spectra, the peaks at ≈1350 cm‐1 (D band) and ≈1580 cm‐1 (G band) correspond to the disorder and the vibration of sp2‐bonded carbon atoms, respectively. All the samples showed an I D/I G ratio in a range of 1.09–1.23, confirming highly defective structures (Figure 7I). NDGs‐800 delivered the highest ORR activity among all the catalyst exhibiting E onset of 0.98 V versus RHE, E 1/2 of 0.85 V versus RHE, and J L of 5.6 mA cm‐ 2 (Figure 7J). NDGs‐800 followed a 4‐electron ORR pathway confirmed from K–L plot (Figure 7K). DFT study further confirmed the synergy between pyridinic N‐doped carbon site and a vacancy defect to lower the ORR overpotential of the material.
FIGURE 7.
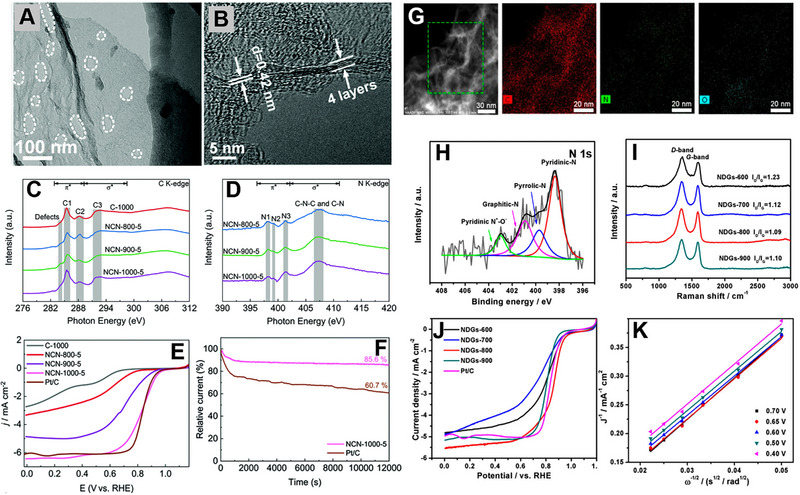
(A) TEM and (B) HRTEM images of NCN‐1000‐5. (C) C K‐edge and (D) N K‐edge NEXAFS spectra of C‐1000, NCN‐800‐5, NCN‐900‐5, and NCN‐1000‐5. (E) LSV of C‐1000, NCN‐800‐5, NCN‐900‐5, NCN‐1000‐5, and Pt/C at a scan rate of 5 mV s‐1 and electrode rotating rate of 1600 rpm. (F) Durability tests of NCN‐1000‐5 and Pt/C at 0.67 V (1600 rpm). Reproduced with permission.[ 119 ] Copyright 2019, Royal Society of Chemistry. (G) STEM and corresponding elemental analysis of NDGs‐800. (H) High‐resolution N 1s XPS spectrum of the NDGs‐800. (I) Raman spectra of NDGs prepared at different temperatures. (J) LSV curves of NDGs prepared at different temperatures and Pt/C catalyst for ORR in 0.1 m KOH at a scan rate of 5 mV s‐1. (K) K–L plots of NDGs‐800 at different potential values. Reproduced with permission.[ 120 ] Copyright 2018, American Chemical Society.
Graphynes (GYs) represent a novel class of carbon allotropes characterized by a 2D planar network structure that combines sp‐ and sp2‐hybridized carbon atoms, achieved by linking acetylene bonds with benzene rings. The graph‐n‐yne (where n = 1, 2, 3, and so on) can be named based on the number of acetylenic chains present between the neighboring benzene rings within a GYs unit.[ 121 ] The typical structure of GYs is shown in Figure 8A. The primary approach for synthesizing 2D GYs involves bottom‐up methods, like chemical vapor deposition (CVD) and liquid/liquid or gas/liquid processes taking hexaethynyl benzene as a precursor.[ 122 , 123 ] It was suggested that certain GY allotropes, characterized by non‐hexagonal symmetry and the presence of two self‐doped non‐equivalent distorted Dirac cones, might display superior electronic properties compared to graphene.[ 1 ] Furthermore, due to the presence of additional alkyne units between the benzene rings in graphdiyne (GD, n value is 2), the network's pore size is enlarged to around 2.5 Å. This enlargement enables adsorption of air into the pores when the sample is introduced in atmosphere.[ 124 ]
FIGURE 8.
(A) Types of carbons in pristine GY (red: αC1; blue: βC1; black: C2) and position distribution of different N doping in GY. Reproduced with permission.[ 121 ] Copyright 2020, Royal Society of Chemistry. (B) FESEM and EDS elemental mapping images of the prepared N 550‐GD powder. (C) LSV curves for GD, N‐doped GDs, and Pt/C in O2‐saturated 0.1 m KOH solution at a scan rate of 10 mV s‐1 with a rotation of 1600 rpm. (D) Koutecky–Levich plots for the N 550‐GD. Reproduced with permission.[ 124 ] Copyright 2014, Royal Society of Chemistry. TEM images of (E) GD and (F) NFGD. (G) FESEM and (H‐K) EDS elemental mapping images of NFGD. Raman spectra of (L) single elemental doped GD and (M) N, X (X = S, B, F)‐co‐doped GD materials. (N) K–L plots of NFGD calculated at different potentials. (O) HO2 − yields and electron transfer number of NFGD and 20% Pt/C at various disk electrode potentials obtained from the rotating ring‐disk electrode tests. Reproduced with permission.[ 125 ] Copyright 2016, Royal Society of Chemistry.
Like graphene, the electronic structure of GYs can be tuned via heteroatom doping, resulting in high positive charges on carbon atoms that can enhance the electrocatalytic properties of GYs.[ 125 ] Zhang and co‐workers reported N‐doped GD as a metal‐free catalyst for the first time.[ 124 ] To dope the N atom, GD was heated at different temperatures under high‐purity NH3 mixed with Ar. Figure 8B shows the SEM and corresponding elemental mapping of the sample prepared at 550°C. XPS analysis suggested that N atom have two different kinds of bonding characteristics, imine 1 N and imine 2 N. Sample prepared at 550°C showed the best performance among all the synthesized materials, having E onset of 0.899 V versus RHE (Figure 8C). From the K–L plot, the number of electrons transferred per O2 molecule was calculated to be about 3.8 (Figure 8D). The DFT calculation indicated that interaction between O2 and imine N‐doped GD is a chemical adsorption, with a significantly stronger binding energy than O2 on pyridinic N‐doped GD, which can be categorized as a physical adsorption. The significant elongation of O2 molecule bond length from 1.210 Å in pristine O2 molecules to 1.376 Å upon adsorption on imine N‐doped GD suggested that parallel diatomic adsorption can efficiently weaken the O─O bonding, facilitating the ORR at the imine N‐doped GD/GC electrodes. Large pore size of the N‐doped GD network also enhances the oxygen adsorption and reduction process, making the material more stable than Pt/C in ORR.
Li and co‐workers doped elements such as B, N, S, and F in GD, and found that N and F co‐doped GD (NFGD) performed similarly to Pt/C in half‐cell and full‐cell.[ 126 ] The doping was accomplished via a pyrolysis using ammonia, boron oxide, thiourea, and ammonium fluoride as precursors for N, B, S, and F, respectively. TEM, FESEM, and EDS elemental analysis confirmed the uniform doping of N and F in the GD layers (Figure 8E–K). Raman spectra revealed that the I D/I G increases from 0.74 to 0.90 for N doping and further increases to 1.22 for dual atom doping, suggesting the increase of defects in the GD system (Figure 8L,M). NFGD exhibited an average electron transfer number of 4.2 across the reaction, spanning a broad potential range from 0 V to 0.8 V versus RHE, signifying full selectivity toward total oxygen reduction (Figure 8N). HO2 − yield for NFGD was about 10% in the potential range of 0–0.85 V versus RHE, suggesting the potential of cell application (Figure 8O). DFT calculation suggested that the pure GD does not have catalytic properties, but after doping, the number of active sites increases, promoting the ORR performance. Considering all the examples discussed in this section, it can be concluded that doping heteroatoms and creating defects in the graphene and graphyne greatly enhanced the ORR activity in alkaline media. The enhanced ORR activity parameters of graphene and graphyne catalysts are listed in Table 1.
TABLE 1.
Summary of N‐doped graphene catalysts for the ORR.
| Catalyst | Synthetic method | Electrolyte | E onset (vs RHE) | E 1/2 (vs RHE) | Electron transfer (n) | Tafel slope (mV dec‐1) | J L (mA cm‐1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| NrGO800 | Thermal annealing | 0.1 m NaOH | 0.88 V | 0.76 V | 3.66 | 65.4 | [99] | |
| NrGO‐900 | Pyrolysis | 0.1 m KOH | 0.99 V | 0.84 V | 4 | – | [100] | |
| NrGO | Thermal annealing | 0.1 m NaOH | 1.03 V | 0.84 V | 3.5 | – | [103] | |
| NGS4‐900 | Pyrolysis | 0.1 m KOH | 0.984 V | 0.859 V | 3.9 | 72 | 5.98 | [102] |
| PCNs | Thermal annealing | 0.1 m KOH | −0.02 V (vs Ag/AgCl) | – | 3.49–3.67 | – | 4.6 | [127] |
| N‐doped graphene | Hydrothermal and microwave treatments | 0.1 m KOH | −0.056 V (vs Ag/AgCl) | – | ≈4 | – | 3.91 | [128] |
| 1‐NGF‐9 | Thermal annealing | 0.1 m KOH | 0.89 V | – | ≈4 | – | 5.30 | [129] |
| 10N‐G‐800 | Pyrolysis | 0.1 m KOH | 0.930 V | – | 3.99 | 60 | 5.13 | [130] |
| N‐G‐1000 | Pyrolysis | 0.1 m KOH | 0.982 V | 0.862 V | 3.92 | 72 | 5.48 | [131] |
| B‐rGO | Hydrothermal | 0.1 m KOH | 0.833 V | 0.671 V | 3.7 | 76 | 4.1 | [112] |
| PN‐BG | Hydrothermal treatment and electrochemical deposition | 0.1 m KOH | – | – | 3.8–4.0 | – | – | [113] |
| S‐doped graphene nanosheet | Electrochemical exfoliation | 0.1 m NaOH | 0.810 V | – | 3.93 | −50.8 | – | [132] |
| F‐doped graphene | Thermal treatment | 0.1 m KOH | −0.28 V (vs Ag/AgCl) | – | 3.9–4.05 | 55 | [133] | |
| N, S co‐doped Graphene | Three‐step pyrolysis | 0.1 m KOH | 1.01 V | 0.870 V | 3.975 | – | 5.99 | [134] |
| B&N‐rGO | Pyrolysis | 0.1 m KOH | 0.850 V | – | 3.5 | 61.8 | – | [135] |
| B,N‐Graphene | Two‐step pyrolysis | 0.1 m KOH | 0.06 V (vs Ag/AgCl) | – | – | – | 13.87 | [114] |
| HRBNG | Microwave hydrothermal | 0.1 m KOH | 0.93 V | – | 3.84 | – | – | [115] |
| NB/GO_55_700 | Ultrasonication and thermal annealing | 0.1 m KOH | 0.82 V | – | 3.98 | 43 | 2.43 | [116] |
| NBC‐1000 | Thermal annealing | 0.1 m KOH | 0.97 V | 0.84 V | 3.93 | – | 5.4 | [136] |
| P‐N‐Gr | Two step thermal annealing | 0.1 m KOH | 1.005 V | 0.82 V | 3.95 | 67 | 5.98 | [137] |
| NP‐PrGO‐1100 | Pyrolysis | 0.1 m KOH | 0.913 V | 0.819 V | 4 | 78 | 5.48 | [138] |
| NCN‐1000‐5 | Pyrolysis | 0.1 m KOH | 0.95 V | 0.82 V | 3.92 | 86 | 6.43 | [119] |
| NDGs‐800 | Pyrolysis | 0.1 m KOH | 0.98 V | 0.85 V | ≈4 | 81 | 5.6 | [120] |
| N‐doped carbon nanoribbon | Pyrolysis | 0.1 m KOH | 0.99 V | 0.870 V | 3.9 | 78 | 5.8 | [139] |
| N and S co‐doped carbon | Wet chemical | 50 mm PBS | 0.769 V | 0.613 V | 3.76–4.02 | – | 4.773 | [140] |
| N and P co‐doped porous carbon | Pyrolysis | 0.1 m KOH | 0.98 V | 0.870 V | 3.93 | – | 6.24 | [141] |
| N‐doped carbon/graphene | Pyrolysis | 0.1 m KOH | 0.922 V | 0.524 V | – | – | 1.85 | [142] |
| N‐doped graphitic porous carbon | Carbonization | 0.1 m KOH | −0.04 V (vs Ag/AgCl) | −0.18 V (vs Ag/AgCl) | 3.92 | – | 5.7 | [143] |
| S, N dual‐doped graphene‐like carbon nanosheets | Pyrolysis | 0.1 m KOH | 0.950 V | 0.830 V | 3.6–3.9 | – | 4.86 | [144] |
| N‐doped graphdiyne | Pyrolysis | 0.1 m KOH | 0.899 V | – | 3.8 | – | −4.5 | [124] |
| N, F co‐doped graphdiyne | Pyrolysis | 0.1 m KOH | 1.0 V | – | 4.2 | – | – | [125] |
| N‐doped few‐layer graphdiyne | Pyrolysis | 0.1 m KOH | – | 0.87 V | 3.9 | 60 | – | [145] |
| N‐doped porous graphdiyne | Pyrolysis | 0.1 m KOH | 0.98 V | 0.83 V | 3.5 | 74 | 5.1 | [146] |
| N, P‐co‐doped graphdiyne | Thermal annealing | 0.1 m KOH | – | – | 3.62–3.68 | 58 | – | [147] |
3.2. Graphitic carbon nitride (g‐C3N4)
The g‐C3N4 is widely acknowledged for its exceptional stability as a stable allotrope, making it a remarkable 2D material with significant potential in energy‐related applications. The generation of a 2D framework with N heteroatoms substituted into graphite occurs through the sp2 hybridization of N and C atoms.[ 148 ] Previous studies have identified tri‐s‐triazine as the fundamental unit of the g‐C3N4 network. Different types of graphene nitride structures are shown in Figure 9A–C. In theory, g‐C3N4 showed an abundance of pyridine‐like N atoms within the heptazine heteroring, offering an ample supply of electron lone pairs, making them active sites for ORR. However, the limited conductivity posed a significant obstacle to its practical application in electrocatalysis.[ 149 ] Free energy calculation indicated that the restricted electron transfer capacity of g‐C3N4 results in a build‐up of OOH− intermediates on the catalyst surface, primarily due to an ineffective 2e− reduction process (Figure 9D).[ 150 ] In the case of path I, oxygen reduction cannot occur naturally on the g‐C3N4 surface in the absence of electron involvement because of the presence of two high energy barriers in the free energy plot corresponding to intermediate and final products (Figure 9E). Upon the introduction of electrons (via path II), the free energy of the intermediate OOH@g‐C3N4 was reduced to a level comparable to that of the initial state of O2@g‐C3N4. This suggests that the initial 2e− reaction can proceed unhampered. However, an evident barrier remains at the last state of OH−/g‐C3N4, preventing the progression of the second 2e− reaction (Figure 9F). Incorporating a conducting material with g‐C3N4 increased the electron transfer efficiency and facilitates a 4e− transfer process (Figure 9G). Various types of carbon materials have been incorporated to increase the electron transfer efficiency and thus the ORR efficiency of g‐C3N4.[ 151 , 152 , 153 , 154 ]
FIGURE 9.
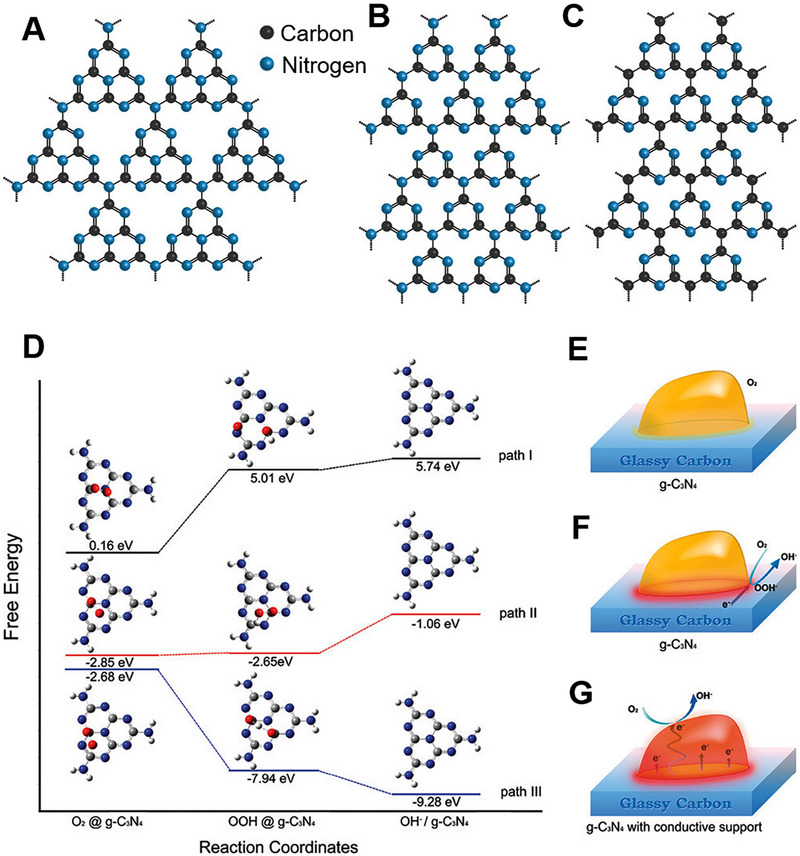
Different structural motifs of (A) polyheptazine g‐C3N4, (B) polytriazine g‐C3N4, (C) polytriazine g‐C4N3. Reproduced with permission.[ 155 ] Copyright 2018, American Chemical Society. (D) Free energy plots of ORR and optimized configurations of adsorbed species on the g‐C3N4 surface with zero, two, and four electron participation demonstrated as paths I, II, and III. Gray, blue, red, and white small spheres representing C, N, O, and H. Schematics of ORR pathways on (E) pristine g‐C3N4 without electron participation, (F) pristine g‐C3N4 with 2e– participation, and (G) g‐C3N4 and conductive support composite with 4e– participation. Reproduced with permission.[ 150 ] Copyright 2011, American Chemical Society.
Qu and co‐workers synthesized a heterostructure by combining the basal plane of the graphene sheet with monolayer g‐C3N4 dots (MTCG).[ 156 ] Figure 10A shows the schematic of the synthesis process. The g‐C3N4 was oxidatively exfoliated by a modified Hummers method and was then mixed with GO. The overall solution was hydrothermally treated to obtain MTCG. STEM elemental mappings showed that the C and N elements are uniformly distributed all over the material, confirming the even distribution of g‐C3N4 across the graphene surface. XPS analysis further confirmed the formation of MTCG (Figure 10B,C). In the case of C1 spectra, the peak at 288 eV was assigned to sp2 carbon atoms within the triazine rings, which are connected to nitrogen atoms within the aromatic structure group or the ‐NH2 group arising from g‐C3N4. On the other hand, the peak at 284.6 eV was designated to graphitic carbon originating from graphene. The deconvoluted N1s spectra show two individual peaks, one for s‐triazine rings (C─N─C, 398.8 eV) and another for bridging N atoms in N─(C)3 (400.1 eV). The ORR performance of MTCG was carried out in 0.1 m KOH solution, and results were compared with commercial Pt/C. MTCG showed an oxygen reduction peak at −0.13 versus Ag/AgCl, close to the Pt/ C value (Figure 10D). MTCG showed E onset of −0.02 V versus Ag/AgCl and E 1/2 of −0.09 V versus Ag/AgCl, both lower than the commercial Pt/C (Figure 10E). Both MTCG and Pt/C showed a two‐stage feature in overall reaction kinetics. In the lower potential, a small slope was observed, which is controlled by the surface reaction rate. However, a steeper curve was observed in the higher potential because the ORR is more relied on oxygen diffusion. Tafel slopes of MTCG are 59 and 240 mV dec‐1, which are close to 65 and 210 mV dec‐1 values of Pt/C. Monolayer g‐C3N4 dots (MTC) exhibited a reduced onset potential, half‐wave potential, and limiting current density when compared to MTCG, confirming the beneficial impact of the 2D graphene layer structure on ORR.
FIGURE 10.
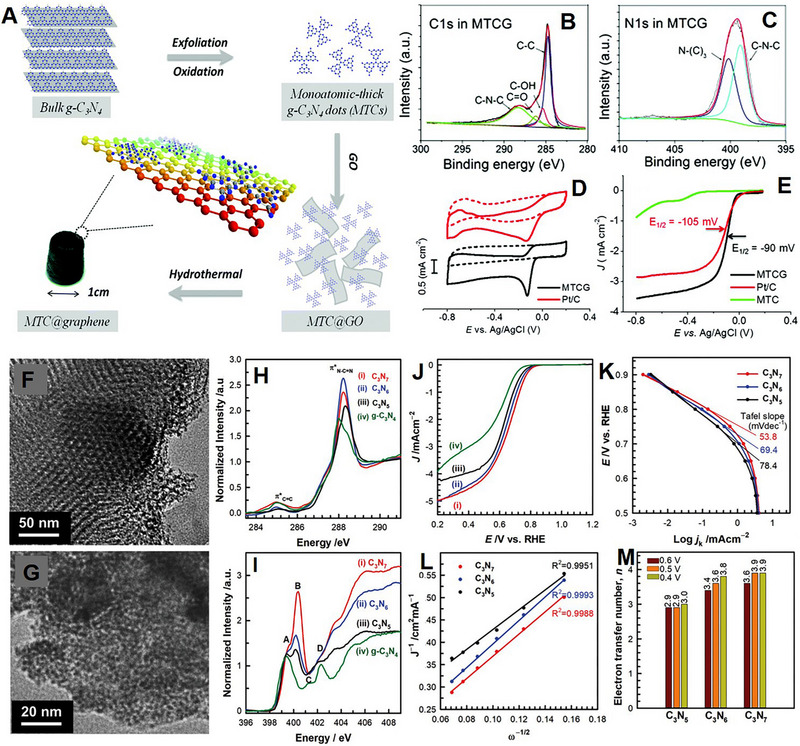
(A) Schematic of monoatomic‐thick g‐C3N4 dots@graphene (MTCG) synthesis procedure. XPS spectra of MTCG (B) C1s and (C) N1s. (D) CVs of MTCG and Pt/C catalysts at a scan rate of 10 mV s‐1 in O2‐saturated (solid lines) or N2‐saturated (dashed lines) 0.1 m KOH solution. (E) RRDE polarization curves of MTCG and Pt/C catalysts with a scan rate of 10 mV s‐1 at 1200 rpm in O2‐saturated 0.1 m KOH solution. Reproduced with permission.[ 156 ] Copyright 2015, Royal Society of Chemistry. HRTEM images of (F) C3N7 and (G) C3N6. (H) C K‐ and (I) N K‐edge NEXAFS spectra of C3N7 and C3N6 with reference materials. (J) LSV of (i) C3N7, (ii) C3N6, (iii) C3N5, and (iv) g‐C3N4 at 1600 rpm. (K) Tafel plots of C3N7, C3N6, and C3N5 at 1600 rpm. (L) K–L plots at 0.6 V versus RHE, and (M) electron transfer number of the C3N7, C3N6, and C3N5 calculated by K–L plots at 0.6, 0.5, and 0.4 V versus RHE. Reproduced with permission.[ 157 ] Copyright 2021, Wiley‐VCH.
Synthesis of carbon nitride with an N:C atomic ratio > 2 is challenging due to substituting C─N bonds with thermodynamic less stable N─N bonds. A higher ratio is believed to offer superior basicity and unique electronic properties. Vinu and co‐workers synthesized C3N6 and C3N7 using a low‐temperature pyrolysis method.[ 157 ] 5‐amino‐1H‐tetrazole and KIT‐6 were mixed together and heated at 160°C for 6 h. The resulting product was further calcined at 250 and 300°C under N2 atmosphere for 4 h to form mesoporous C3N7 and C3N6, respectively. TEM analysis suggested that the materials have highly ordered 2D porous structures (Figure 10F,G). The N─N bonds were stabilized by forming tetrazine and/or triazole moieties, confirmed by near‐edge X‐ray absorption fine structure (NEXAFS) analysis (Figure 10H,I). C3N7 and C3N6 showed better ORR performance and lower Tafel slopes than C3N5 and g‐C3N4 (Figure 10J,K). The K‐L plots of the materials implied that the material with higher N content showed a higher n number, suggesting an effective reduction of O2 to 4OH− (Figure 10L,M). According to DFT calculation, C3N7 contained a higher amount of cyclic N─N bonds, which effectively absorb O2 and OH species, making it more ORR active. The enhanced ORR activity parameters of g‐C3N4 catalysts are listed in Table 2. Therefore, a heterostructure of g‐C3N4 is much more electrocatalytically active than pure g‐C3N4 in ORR.
TABLE 2.
Summary of g‐C3N4 for the ORR.
| Catalyst | Synthetic method | Electrolyte | E onset (vs RHE) | E 1/2 (vs RHE) | Electron transfer (n) | Tafel slope (mV dec‐1) | J L (mA cm‐1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Graphitic‐C3N4@Carbon | Template method | 0.1 m KOH | −0.10 V versus Ag/AgCl | – | 3.8 | −113 | 11.3 | [150] |
| C3N7 | Pyrolysis | 0.1 m KOH | 0.81 V | – | 3.9 | 53.8 | 8.2 | [157] |
| g‐C3N4 dots@graphene | Hydrothermal | 0.1 m KOH | −0.02 V versus Ag/AgCl | 0.09 V versus Ag/AgCl | 3.72 | 59, 240 | – | [156] |
| g‐C3N4@N‐G | Ball milling | 0.1 m KOH | −0.02 V versus SCE | −0.22 V versus SCE | 4 | – | – | [158] |
| Graphene platelet decorated carbon nitride | Pyrolysis | 0.1 m KOH | 0.87 V | – | 3.8 | 63 | 5.1 | [159] |
| g‐C3N4@rGO | Semi‐closed pyrolysis | 0.1 m KOH | −0.248 V versus Ag/AgCl | −0.281 V versus Ag/AgCl | 3.21 | – | 3.36 | [160] |
| g‐C3N4 QD/g‐C3N4 sheet/rGO | Ultrasonication | 0.1 m KOH | −0.075 V versus Ag/AgCl | −0.210 V versus Ag/AgCl | 3.9–4.2 | – | −0.491 | [161] |
| s‐g‐C3N4@GQDs | Hydrothermal | 0.1 m KOH | −0.07 V versus Ag/AgCl | – | 3.5 | – | – | [162] |
| S‐doped C3N4‐mesoporous carbon | Polycondensation | 0.1 m KOH | −0.11 V versus Ag/AgCl | – | 4 | – | 30.9 | [163] |
4. NON‐CARBON‐BASED 2D ELECTROCATALYSTS FOR THE ORR
As seen in the previous section, carbon‐based materials, mainly graphene, have been extensively studied for the ORR. In this section, the advancements of other metal‐free 2D nanomaterials, such as hexagonal boron nitride (h‐BN), boron carbon nitride (BCN), and black phosphorus (BP) for the ORR are discussed.
4.1. h‐BN
Recently, BN has gained significant attention for green energy conversion due to its remarkable optoelectrical properties, thermal stability, mechanical durability, and chemical inertness. Four types of BN have been reported in the literature, including amorphous BN (a‐BN), h‐BN, cubic BN (c‐BN), and wurtzite BN (w‐BN).[ 164 ] h‐BN exhibits a layered structure similar to graphene, composed of sp2 hybridized B─N bonds that are highly polarized and possess a strong covalent character within the plane. However, unlike graphene, h‐BN displays significant polarization due to the electronegativity of N atoms, leading to the potential for anisotropic properties. Due to the huge gap in electronegativity between B and N, h‐BN behaves as an insulator, and the band gap depends on the thickness of the material. Although h‐BN has been used in optical and electrical devices as a dielectric substrate, h‐BN remains inert toward electrocatalysis.[ 1 , 164 , 165 ] Utilization of pristine h‐BN for the ORR in fuel cells is impractical due to its inherent limitations in electronic conductivity and its tendency to form H2O2 through a two‐electron process.[ 166 ] To activate the h‐BN, various methods have been adopted, such as physical methods and chemical methods. There are reports of adding non‐metal nanostructures with h‐BN to increase its electrocatalytic activity. Doping carbon into the h‐BN matrix or forming a heterostructure between graphene and h‐BN might resolve this issue.[ 167 , 168 ]
Utilizing a CVD method, Kakade and co‐workers prepared a one‐step carbon‐doped h‐BN (BNC).[ 167 ] Figure 11A represents the schematic of BNC for ORR. Boric acid and hexamethylenetetramine were mixed in a solid‐state condition and then heated for 2 h under an N2 atmosphere. TEM analysis of the sample prepared at 850°C confirmed that the rice‐shaped BNC nanocrystals were impregnated into the carbon matrix (Figure 11B,C). XPS region spectra of B1s and N1s suggested the formation of B─N bond and a small amount of B─C and N─C bonds (Figure 11D,E). C1s XPS spectrum also indicated the formation of C─B, C─O, and C─N bond formation along with the C─C bond (Figure 11F). A strong cathodic peak at 0.76 V versus RHE in the presence of O2 was observed, but absence in N2, confirms the potential of BCN for ORR (Figure 11G). An E onset of 0.83 V versus RHE and J L of 4.6 mA cm‐ 2 were observed at 1600 rpm (Figure 11H). The number of electrons per O2 molecule was ≈3.6, calculated from K–L plots at different potentials (Figure 11I). They further confirmed that the carbon coating on BNC increases the active sites, making it more ORR active compared to pure h‐BN. A similar kind of ORR enhancement was observed when rGO was added to h‐BN to make a composite.[ 168 ] Multiple contacts between rGO and h‐BN act as an oxygen adsorption site and enhance the performance. In another work, Kakade and co‐workers synthesized a heterostructure combining rGO and h‐BN using a single‐step hydrothermal method.[ 169 ] Initially, GO was mechanically activated via a ball milling process and later mixed with h‐BN heated at 180°C for 6 h in an autoclave. HAADF‐STEM study suggested the generation of a homogeneous composite (Figure 11J–N). The amount of h‐BN was varied to obtain different compositions of heterostructures. Out of all the samples, the heterostructure containing 2% h‐BN (GOBN2‐BM) showed maximum performance toward ORR (Figure 11O). The average number of electrons per O2 molecule was 3.89, with a peroxide yield below 10% (Figure 11P). The addition of h‐BN decreased the crystallite size of the GO, subsequently enhancing the active surface area of the material, resulting in higher electrocatalytic activity. The enhanced ORR activity parameters of h‐BN catalysts are listed in Table 3. From the above discussion and results, it is clearly visible that the ORR performance of h‐BN is enhanced either by doping or by forming a heterostructure.
FIGURE 11.
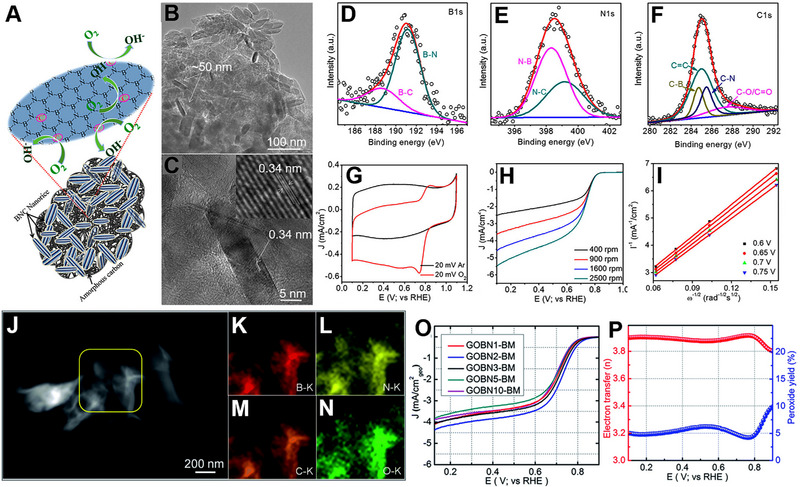
(A) Schematic illustration for ORR of carbon‐doped h‐BN (BNC) catalyst. (B) TEM and (C) HRTEM images of BNC catalyst. Deconvoluted XPS spectra of (D) B1s, (E) N1s, and (F) C1s. (G) CVs of BNC catalyst, recorded in Ar‐ and O2‐saturated 0.1 m KOH at a scan rate of 20 mV s‐1. (H) ORR polarization curves of BNC catalyst under O2‐saturated 0.1 m KOH recorded at various rotation speeds (I) K‐L plot of BNC catalyst at different potentials. Reproduced with permission.[ 167 ] Copyright 2018, American Chemical Society. (J) HAADF‐STEM image of the GOBN2–BM catalyst and (K–N) corresponding elemental mapping of B, N, C, and O. (O) Comparison of the ORR LSV curves of electrocatalysts with varied h‐BN amount in O2‐saturated 0.1 m KOH at 1600 rpm with a scan rate of 10 mV s‐1. (P) Extent of peroxide yield and number of electrons transferred (n) of the GOBN2–BM catalyst in the potential range of 0.1 to 0.9 V versus RHE. Reproduced with permission.[ 169 ] Copyright 2018, Royal Society of Chemistry.
TABLE 3.
Summary of h‐BN for the ORR.
| Catalyst | Synthetic method | Electrolyte | E onset (vs RHE) | E 1/2 (vs RHE) | Electron transfer (n) | Tafel slope (mV dec‐1) | J L (mA cm‐1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| C‐doped h‐BN | CVD | 0.1 m KOH | 0.83 V | – | 3.6 | – | 1.58 | [167] |
| rGO/h‐BN | Hydrothermal and annealing | 0.1 m KOH | 0.798 V | 0.64 V | 3.7 | – | – | [168] |
| rGO/h‐BN | Hydrothermal | 0.1 m KOH | 0.98 V | 0.74 V | 3.89 | 102 | 4.4 | [169] |
| CNT/h‐BN | Hydrothermal and annealing | 0.1 m KOH | 0.86 V | 0.72 V | 3.9 | – | 5.78 | [170] |
| BN‐Graphene | EDC Coupling | 0.1 m KOH | – | – | 3.8 | – | −4.6 | [171] |
| Graphene‐hBN | ball milling | 0.1 m KOH | 0.79 V | – | 2.23 | – | – | [172] |
4.2. BCN
Recently, BCN has been getting much attention due to its diverse composition. BCN has a bandgap of 1.18 eV, much lower than h‐BN (5 eV), which is useful for optoelectronic applications.[ 173 ] BCN is a ternary 2D compound with the advantages of graphene, BN nanosheets, and CN analogs.[ 174 ] Figure 12A,B shows phase diagram and crystal structure of BCN monolayer, respectively. Like graphene and h‐BN, BCN is also explored for electrocatalytic applications. Lei and co‐workers prepared porous BCN nanosheets using a polymer sol‐gel method.[ 175 ] Figure 12C shows the schematic of the synthesis process. Initially, a polymeric gel was formed by hydroxyl and amino group cross‐linking between polyvinyl alcohol (PVA), boric acid, and guanidine carbonate salt. The polymeric precursor was subjected to additional curing after the introduction of P123. Following heat treatment at 900°C in an N2 atmosphere, the architecture of the precursor gel was gradually transformed into a 2D porous layered structure, resulting in the bonding of B, N, and C atoms. From XRD and FT‐IR spectra, it is noticed that the peak position of BCN is different from the graphene and h‐BN. Raman analysis showed that BCN exhibits D and G bands at ≈1360 and 1600 cm‐1 (Figure 12D). Graphene generally shows a G band at 1580 cm‐1, which confirms the structural distortion of BCN due to the existence of different bond lengths of B─N and C─N. TEM and EDX showed that all the B, N, and C atoms are uniformly distributed in the 2D BCN nanosheets (Figure 12E). XPS results confirmed the presence of B─O and B─N─C bonds in B1s region spectra, whereas sp2 C═C bonds, C═N bonds, C─O/C─N bonds were observed in C1s region spectra (Figure 12F,G). N1s XPS region spectrum showed four peaks corresponding to pyrrolic N, pyridinic N, quaternary N, and C─N─B (Figure 12H). A higher amount of pyridinic N presence in the material is believed to be an active site for ORR. BCN showed E onset of 0.940 V versus RHE, which is 13 mV less than commercial Pt/C (Figure 12I). K–L plots suggested that the ORR process followed a four‐electron pathway (Figure 12J). In RRDE measurement, the calculated H2O2 yield is below 6%, confirming the 4e− transfer process in the alkaline ORR process. The BCN material also showed excellent methanol tolerance and high durability compared to commercial Pt/C. It is believed that the abundant B─N─C bonding in the material is readily available to OH adsorption and O protonation in the graphitic carbon edge area, resulting in higher ORR activity.
FIGURE 12.
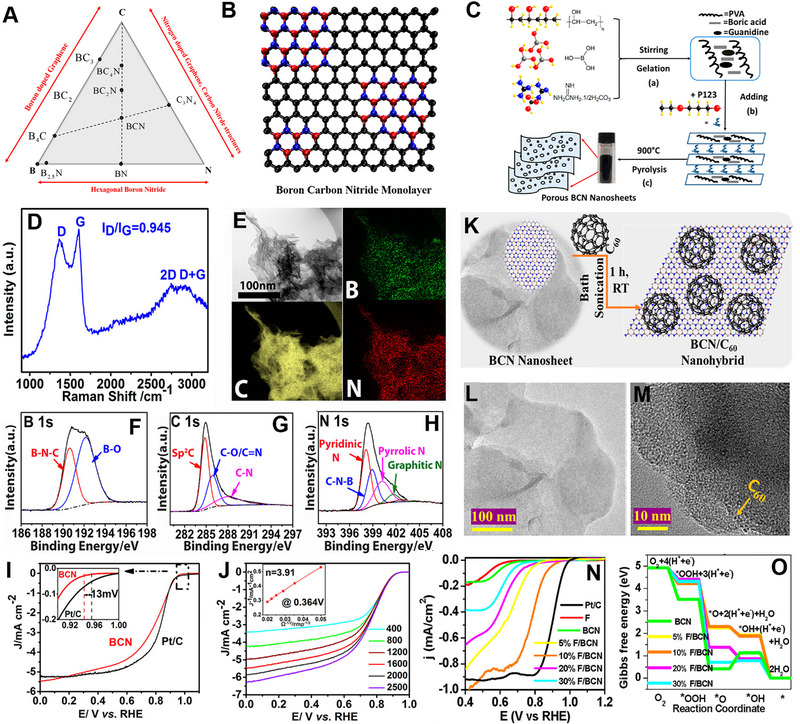
(A) Phase diagram of B–C–N showing possible stable intermediate phases. (B) Schematic illustration of boron carbon nitride crystal structure. Reproduced with permission.[ 176 ] Copyright 2020, The Electrochemical Society. (C) Schematic of porous BCN nanosheet synthesis. (D) Raman spectra of porous BCN nanosheet. (E) TEM and corresponding elemental mapping of B, C, and N elements. XPS region spectra of (F) B1s, (G) C1s, and (H) N1s. (I) LSVs of BCN and Pt/C in 0.1 m KOH at a rotation rate of 1600 rpm and a scan rate of 5 mV s‐1. The inset showing the higher magnified LSV curves between 0.9 and 1.0 V versus RHE. (J) LSVs of BCN at different rotation rates. The inset showing the corresponding K–L plot. Reproduced with permission.[ 175 ] Copyright 2017 American Chemical Society. (K) Schematic representation of the synthesis of fullerene/BCN nanohybrids. TEM images of (L) BCN nanosheets and (M) 10% Fullerene/BCN nanohybrids. (N) LSV curves of F, BCN, 5% fullerene/BCN, 10% fullerene/BCN, 20% fullerene/BCN, 30% fullerene/BCN, and Pt/C for ORR at 2 mV s‐1 in 0.5 m NaOH. (O) Gibbs free energy diagram showing the differences of each elementary ORR catalytic step for both BCN and fullerene/BCN nanohybrids. Reproduced with permission.[ 177 ] Copyright 2021 American Chemical Society.
Noveron and co‐workers developed a BCN‐based heterostructure catalysis for ORR.[ 177 ] BCN was prepared through carbonization of the mixture of urea, boric acid, and PEG‐2000 precursors. Later, they added varied amounts of fullerene to BCN to form the heterostructure in an ultrasonication method (Figure 12K). Pure BCN exhibited a smooth 2D structure (Figure 12L), whereas fullerene was uniformly dispersed in the BCN sheet in the case of fullerene/BCN composite (Figure 12M). Higher amount of pyridinic N and C─N─B were detected in the 10% fullerene/BCN composite, implying a higher number of active ORR sites. 10% fullerene/BCN composite showed E onset of 0.920 V versus RHE in 0.5 m NaOH solution, which is higher compared to other composites and pristine BCN (Figure 12N). DFT calculation predicted that the *O → *OH step is the rate‐limiting step for all the fullerene/BCN composite. The composite containing 10% fullerene/BCN displayed the least rate‐limiting potential (0.81 eV), leading to a reduction in the energy barrier for the rate‐determining catalytic step, and subsequently, an enhancement in the reaction rate (Figure 12O). The enhanced ORR activity parameters of BCN catalysts are listed in Table 4. Although pure BCN or heterostructure of BCN showed promising results, further study is needed to explore its full potential.
TABLE 4.
Summary of BCN for the oxygen reduction reaction.
| Catalyst | Synthetic method | Electrolyte | Onset potential (vs RHE) | E 1/2 (vs RHE) | Electron transfer (n) | Tafel slope (mV dec‐1) | J L (mA cm‐1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| BCN | Pyrolysis | 0.1 m KOH | 0.940 V | 0.820 V | 3.91 | – | – | [175] |
| BCN/C60 | Pyrolysis | 0.5 m NaOH | 0.920 V | 0.790 V | 3.85 | 87 | – | [177] |
| BCN/C | Pyrolysis and solvothermal | 0.1 m KOH | 1.01 V | 0.860 V | ≈4 | 72.4 | – | [178] |
| NBC nanosheets | Pyrolysis | 0.1 m KOH | 0.87 V | – | ≈4 | 79 | 4.18 | [179] |
4.3. Black phosphorus
The BP is a layered semiconductor material that is artificially synthesized, belonging to the class of monoatomic 2D van der Waals materials.[ 180 , 181 ] The individual layers of BP, known as phosphorene, exhibit a puckered honeycomb structure and are attached together by a combination of van der Waals and ionic forces.[ 182 , 183 ] Monolayer phosphorene has emerged as an exceptionally promising candidate among various 2D electronic materials due to its bandgap comparable to silicon and high hole mobility.[ 184 , 185 ] Phosphorene has a bandgap of ≈2 eV, which can be adjusted by precisely regulating the thickness.[ 186 ] BP exhibits three crystal structures: orthorhombic, simple cubic, and rhombohedral. Figure 13A,B shows the crystal lattice of orthorhombic BP. A single layer of BP contains two different types of P─P bonds and two different atomic layers. Until now, very few reports have been reported on the electrocatalytic performance of BP, which could be attributed to its inherent challenges, such as poor electrical conductivity and low stability under electrocatalytic conditions. Theoretical study suggested that pure phosphorene exhibits inferior ORR performances due to the adsorption of O* on the phosphorene surface. However, local oxidation can reduce the adsorption strength of O* on the phosphorene and increase the ORR performance.[ 187 ] In another work, Feng and co‐workers reported that Te doping in BP can enhance the ORR performance of BP.[ 188 ] Instead of occurring as isolated Te defects, the doped Te atoms tend to form clusters by bonding with each other. Through the synergistic utilization of intrinsic defects and Te dopants, Te cluster's catalytic activity can be precisely adjusted across a broad range. O* exhibit a moderate binding strength at Te sites, further enhances the performance of Te clusters in ORR compared to pristine phosphorene.
FIGURE 13.
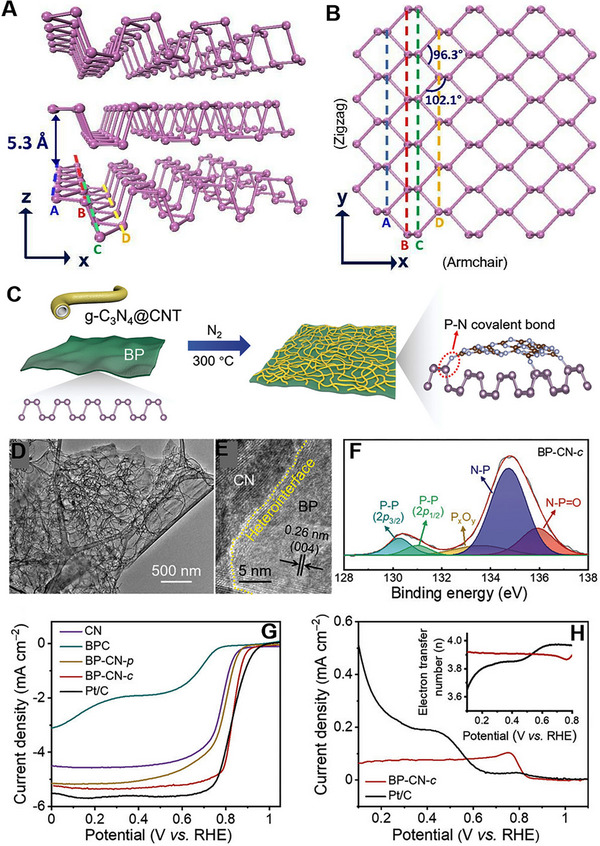
Crystal structure of BP (A) side view and (B) top view. Reproduced with permission.[ 186 ] Copyright 2015, National Academy of Sciences. (C) Schematic representation of BP‐CN‐c synthesis. (D, E) TEM and HRETM images of BP‐CN‐c (F) P 2p region XPS spectrum (G) ORR polarization curves of CN, BPC, BP‐CN‐p, BP‐CN‐c, and Pt/C at 5 mV s‐1 with a rotating speed of 1600 rpm. (H) Ring current densities and the calculated electron transfer numbers (inset) for BP‐CN‐c and Pt/C. Reproduced with permission.[ 189 ] Copyright 2021, Wiley‐VCH.
Feng and co‐workers first reported a 2D BP‐based metal‐free catalyst for ORR.[ 189 ] BP was prepared by adopting an electrochemical exfoliation method, whereas g‐C3N4@CNT (CN) was made by mixing g‐C3N4 and CNT and annealing at 550°C. A covalently bonded BP with g‐C3N4@CNT (BP‐CN‐c) was prepared by physically mixing both the materials and heating at 300°C for 2 h under N2 atmosphere (Figure 13C). TEM analysis suggested that the CN is uniformly distributed on BP nanosheets, showing abundant heterointerface (Figure 13D,E). Along with the P─P bond, N─P and N─P═O bonds also formed, confirmed by XPS analysis (Figure 13F). The as‐synthesized BP‐CN‐c catalyst showed a similar ORR performance to the commercial Pt/C catalyst (Figure 13G). BP‐CN‐c catalyst showed E 1/2 of 0.84 V versus RHE and J L of 5.34 mA cm‐ 2, close to Pt/C (0.85 V vs RHE, 5.60 mA cm‐ 2). RRDE study revealed that the number of electrons per O2 molecule was 3.87–3.92 for BP‐CN‐c catalyst (Figure 13H). The enhanced ORR performance of the BP‐CN‐c catalyst can be ascribed to two main factors: the faster electron transport facilitated by the P─N bonded BP/g‐C3N4 hybrid and the lower energy barriers for ORR steps taking place on the carbon atoms of g‐C3N4.
Yu and co‐workers prepared a composite of exfoliated BP (EBP) and amine‐functionalized EBP (N‐EBP), both derived from bulk BP.[ 190 ] The iso‐type heterostructure (Iso‐EBP) was generated by ultrasonically mixing EBP and N‐EBP in a 1:1 ratio. Iso‐EBP exhibited E onset of 0.88 V versus RHE and J L of 3 mA cm‐ 2, higher compared to EBP and N‐EBP. The electron transfer number of Iso‐EBP was 3.75, which is much higher than EBP (2.92) and N‐EBP (3.18). The amine‐functionalization serves two purposes. First, it accelerates intramolecular electron transfer (P → N) within N‐EBP, generating positively charged P atoms on N‐EBP, which act as efficient active sites for the ORR. Second, it lowers the work function of N‐EBP compared to EBP through chemical modification. Additionally, creating a distinct Iso‐type heterostructure allows directional interfacial electron transfer (N‐EBP → EBP) due to the work function disparity between EBP and N‐EBP. To further improve the activity of iso‐EBP, poly acrylic acid (PAA) was added, which increases the active surface area and shielding effect, increasing stability. Iso‐EBP/PAA exhibited E 1/2 of 0.80 V and J L of 5.1 mA cm‐ 2, much higher than Iso‐EBP, EBP, and N‐EBP samples. Based on these works, it is clear that a heterostructure formation of BP can enhance the ORR performance of the overall catalyst. Until now, very few studies have been reported on BP as an ORR catalyst. Further study is needed to understand the electrocatalytic properties of BP.
5. CHALLENGES AND PERSPECTIVES
This review summarized the recent developments of 2D metal‐free electrocatalysts toward ORR. First, we discussed the reaction mechanism of the ORR process. Since the metal‐free electrocatalysts are composed of carbon, the ORR mechanism could not be explained by the d‐band theory. Instead, the outer sphere electron‐transfer mechanism is suggested, in which the adsorption of O2 molecules and the ORR intermediates is preferred on the hetero‐atom doped sites or defective sites. Later, we discussed the metal‐free 2D materials electrocatalysts in two parts; carbon‐based and non‐carbon‐based 2D materials. For each material class, the doping and defect introduction strategies were discussed in detail. First, a systematic description of the ORR catalysts was provided from the viewpoint of 2D material doped with heteroatoms, along with a comprehensive analysis and detailed discussion of the impact of elemental doping on catalytic performances. Then, heterostructure formation and vacancy generation were also discussed for the improvement of the ORR performances.
Despite significant advances in this fledgling field, the study of metal‐free 2D materials has encountered numerous new challenges. First, the production of ultrathin metal‐free 2D materials with high quality and quantity remains a challenging task. Although the strategies for introducing doping and defects have been well‐studied, syntheses that allow quantitative control of the doping level or degree of defects, and syntheses that generate active sites uniformly throughout the 2D materials are very difficult. In addition, existing synthesis methods need to be optimized and scaled to meet the commercial and industrial requirements.
Second, understanding the growth mechanism is vital for the precise design of new 2D electrocatalysts and increasing their intrinsic activity. For example, in the case of 2D carbon materials, the ORR activity depends largely on where the doping elements are substituted. Therefore, understanding the formation of these active sites and being able to selectively generate only those that are favorable to the catalysis would allow us to take synthesis strategies to the next level. To facilitate this understanding, there is a significant need to develop low cost, efficient, and advanced in situ characterization techniques.
Third, most of the metal‐free 2D materials showed similar ORR performance to Pt/C, but were only tested in alkaline solutions. PEMFCs operating in acidic media are a more economical and mature technology.[ 191 ] Therefore, if the development of acid‐resistant metal‐free 2D materials can be achieved, ORR catalysts in acidic media are expected to be in great demand.
In conclusion, the key strategies for developing new catalysts involve augmenting the number of active centers and enhancing the intrinsic activity of the catalysts. The potential to tailor specific properties for ORR electrocatalytic processes makes developing 2D materials highly promising. Future research will combine experimental electrochemical measurements, theoretical calculations, and advanced spectral characterization as a vital approach for designing and developing novel metal‐free 2D electrocatalysts. The pursuit is to forge 2D electrocatalysts exhibiting exceptional performance and broad utility, aligned with the demands of practical applications. As such, the rapid accomplishments achieved in the realm of metal‐free 2D materials are poised to make substantive contributions to both fundamental research and tangible real‐world implementations in the foreseeable future.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
ACKNOWLEDGEMENTS
This work was financially supported by the National Research Foundation of Korea (NRF‐2021R1A2C4001411, RS‐2023‐00207831, RS‐2023‐00256106, NRF‐2019R1A6A1A11044070, NRF‐2022R1C1C2004703).
Biographies
Joyjit Kundu is currently a postdoctoral researcher at Kyungpook National University, South Korea, under the supervision of Prof. Sang‐Il Choi. He received his Ph.D. degree in materials science from Indian Institute of Technology Kharagpur, India, in 2021. His research interests include electrochemical water splitting, CO/CO2 reduction, and anion‐exchange‐membrane‐water‐electrolyzer.

Taehyun Kwon obtained his Ph.D. degree (2021) in inorganic chemistry from Korea University. After working as a post‐doctoral researcher at the Korea Institute of Science and Technology (KIST), he joined the Department of Chemistry at Incheon National University (INU) as an assistant professor. His current research interests include the design of nanostructured electrocatalysts and radical scavengers for electrochemical energy conversion devices including fuel cells and water electrolyzers.

Kwangyeol Lee obtained his Ph.D. degree (1997) in chemistry from the University of Illinois at Urbana–Champaign. After fulfilling his military obligation, he joined Korea University in 2003 as a chemistry faculty member before being appointed as professor. His current research interests lie in the development of synthetic methods for catalytic nanoscale materials to support the environment by creating sustainable energy sources.

Sang‐Il Choi received his Ph.D. degree in inorganic chemistry from the KAIST (2011). After his postdoctoral research in the Xia group at the Georgia Institute of Technology, USA, he joined the Kyungpook National University in 2015 as an associate professor. He is the recipient of the 2018 POSCO TJ Park Science Fellowship and the 2023 Prime Minister's Citation of Korea. His research interests include the design and synthesis of nanomaterials and exploration of their applications in electro‐catalysis.

Kundu J., Kwon T., Lee K., Choi S.‐I., Exploration 2024, 4, 20220174. 10.1002/EXP.20220174
Joyjit Kundu and Taehyun Kwon contributed equally to this work.
Contributor Information
Kwangyeol Lee, Email: kylee1@korea.ac.kr.
Sang‐Il Choi, Email: sichoi@knu.ac.kr.
REFERENCES
- 1. Jin H., Guo C., Liu X., Liu J., Vasileff A., Jiao Y., Zheng Y., Qiao S.‐Z., Chem. Rev. 2018, 118, 6337. [DOI] [PubMed] [Google Scholar]
- 2. Tian X., Lu X. F., Xia B. Y., (David) Lou X. W., Joule 2020, 4, 45. [Google Scholar]
- 3. Shih C. F., Zhang T., Li J., Bai C., Joule 2018, 2, 1925. [Google Scholar]
- 4. Chatenet M., Pollet B. G., Dekel D. R., Dionigi F., Deseure J., Millet P., Braatz R. D., Bazant M. Z., Eikerling M., Staffell I., Balcombe P., Shao‐Horn Y., Schäfer H., Chem. Soc. Rev. 2022, 51, 4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Y., Ruiz Diaz D. F., Chen K. S., Wang Z., Adroher X. C., Mater. Today 2020, 32, 178. [Google Scholar]
- 6. Wang H.‐F., Xu Q., Matter 2019, 1, 565. [Google Scholar]
- 7. Kim H. J., Kim H. Y., Joo J., Joo S. H., Lim J. S., Lee J., Huang H., Shao M., Hu J., Kim J. Y., Min B. J., Lee S. W., Kang M., Lee K., Choi S., Park Y., Wang Y., Li J., Zhang Z., Ma J., Choi S.‐I., J. Mater. Chem. A 2022, 10, 50. [Google Scholar]
- 8. Stamenkovic V. R., Strmcnik D., Lopes P. P., Markovic N. M., Nat. Mater. 2017, 16, 57. [DOI] [PubMed] [Google Scholar]
- 9. Sun Y., Gao S., Lei F., Xie Y., Chem. Soc. Rev. 2015, 44, 623. [DOI] [PubMed] [Google Scholar]
- 10. Dai L., Xue Y., Qu L., Choi H.‐J., Baek J.‐B., Chem. Rev. 2015, 115, 4823. [DOI] [PubMed] [Google Scholar]
- 11. Kim H. Y., Jun M., Lee K., Joo S. H., ACS Catal. 2023, 13, 355. [Google Scholar]
- 12. Kim H. Y., Jun M., Joo S. H., Lee K., ACS Nanosci. Au 2023, 3, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tomboc G. M., Kim T., Jung S., Yoon H. J., Lee K., Small 2022, 18, 2105680. [DOI] [PubMed] [Google Scholar]
- 14. Sun Y., Jiang T., Duan J., Jiang L., Hu X., Zhao H., Zhu J., Chen S., Wang X., ACS Catal. 2020, 10, 11371. [Google Scholar]
- 15. Sun Y., Xu S., Ortíz‐Ledón C. A., Zhu J., Chen S., Duan J., Exploration 2021, 1, 20210021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhoyate S. D., Kim J., de Souza F. M., Lin J., Lee E., Kumar A., Gupta R. K., Coord. Chem. Rev. 2023, 474, 214854. [Google Scholar]
- 17. Kabiraz M. K., Kim H. J., Hong Y., Chang Q., Choi S.‐I., Bull. Korean Chem. Soc. 2022, 43, 1093. [Google Scholar]
- 18. Kabiraz M. K., Ruqia B., Kim J., Kim H., Kim H. J., Hong Y., Kim M. J., Kim Y. K., Kim C., Lee W.‐J., Lee W., Hwang G. H., Ri H. C., Baik H., Oh H.‐S., Lee Y. W., Gao L., Huang H., Paek S. M., Jo Y.‐J., Choi C. H., Han S. W., Choi S.‐I., ACS Catal. 2022, 12, 3516. [Google Scholar]
- 19. Kim J., Hong Y., Lee K., Kim J. Y., Adv. Energy Mater. 2020, 10, 2002049. [Google Scholar]
- 20. Jiao Y., Zheng Y., Jaroniec M., Qiao S. Z., Chem. Soc. Rev. 2015, 44, 2060. [DOI] [PubMed] [Google Scholar]
- 21. Yin Q., Tan J. M., Besson C., Geletii Y. V, Musaev D. G., Kuznetsov A. E., Luo Z., Hardcastle K. I., Hill C. L., Science 2010, 328, 342. [DOI] [PubMed] [Google Scholar]
- 22. Kabiraz M. K., Kim J., Choi S.‐I., Bull. Korean Chem. Soc. 2021, 42, 802. [Google Scholar]
- 23. Kabiraz M. K., Choi S.‐I., ChemCatChem 2023, 15, e202300454. [Google Scholar]
- 24. Su J., Ge R., Jiang K., Dong Y., Hao F., Tian Z., Chen G., Chen L., Adv. Mater. 2018, 30, 1801351. [DOI] [PubMed] [Google Scholar]
- 25. Tao H., Gao Y., Talreja N., Guo F., Texter J., Yan C., Sun Z., J. Mater. Chem. A 2017, 5, 7257. [Google Scholar]
- 26. Zhang Y., Gao F., You H., Li Z., Zou B., Du Y., Coord. Chem. Rev. 2022, 450, 214244. [Google Scholar]
- 27. Lang P., Yuan N., Jiang Q., Zhang Y., Tang J., Energy Technol. 2020, 8, 1900984. [Google Scholar]
- 28. Wang X., Li Z., Qu Y., Yuan T., Wang W., Wu Y., Li Y., Chem 2019, 5, 1486. [Google Scholar]
- 29. Wang Y., Wang D., Li Y., SmartMat 2021, 2, 56. [Google Scholar]
- 30. Seh Z. W., Kibsgaard J., Dickens C. F., Chorkendorff I., Nørskov J. K., Jaramillo T. F., Science 2017, 355, eaad4998. [DOI] [PubMed] [Google Scholar]
- 31. Kwag J., Kim S., Kang S., Park J., Bull. Korean Chem. Soc. 2023, 44, 488. [Google Scholar]
- 32. Cui J., Chen Q., Li X., Zhang S., Green Chem. 2021, 23, 6898. [Google Scholar]
- 33. Zhang Y.‐L., Goh K., Zhao L., Sui X.‐L., Gong X.‐F., Cai J.‐J., Zhou Q.‐Y., Zhang H.‐D., Li L., Kong F.‐R., Gu D.‐M., Wang Z.‐B., Nanoscale 2020, 12, 21534. [DOI] [PubMed] [Google Scholar]
- 34. Ren X., Liu B., Liang X., Wang Y., Lv Q., Liu A., J. Electrochem. Soc. 2021, 168, 044521. [Google Scholar]
- 35. Zheng C., Zhang X., Zhou Z., Hu Z., eScience 2022, 2, 219. [Google Scholar]
- 36. Yang H., Liu Y., Liu X., Wang X., Tian H., Waterhouse G. I. N., Kruger P. E., Telfer S. G., Ma S., eScience 2022, 2, 227. [Google Scholar]
- 37. Wang Y., Wu J., Tang S., Yang J., Ye C., Chen J., Lei Y., Wang D., Angew. Chem., Int. Ed. 2023, 62, e202219191. [DOI] [PubMed] [Google Scholar]
- 38. Gong K., Du F., Xia Z., Durstock M., Dai L., Science 2009, 323, 760. [DOI] [PubMed] [Google Scholar]
- 39. Yang L., Shui J., Du L., Shao Y., Liu J., Dai L., Hu Z., Adv. Mater. 2019, 31, 1804799. [DOI] [PubMed] [Google Scholar]
- 40. Quílez‐Bermejo J., Morallón E., Cazorla‐Amorós D., Carbon 2020, 165, 434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bouleau L., Pérez‐Rodríguez S., Quílez‐Bermejo J., Izquierdo M. T., Xu F., Fierro V., Celzard A., Carbon 2022, 189, 349. [Google Scholar]
- 42. Singh H., Zhuang S., Ingis B., Nunna B. B., Lee E. S., Carbon 2019, 151, 160. [Google Scholar]
- 43. Paul R., Zhu L., Chen H., Qu J., Dai L., Adv. Mater. 2019, 31, 1806403. [DOI] [PubMed] [Google Scholar]
- 44. Hu C., Dai L., Adv. Mater. 2019, 31, 1804672. [DOI] [PubMed] [Google Scholar]
- 45. Paul R., Dai Q., Hu C., Dai L., Carbon Energy 2019, 1, 19. [Google Scholar]
- 46. Niu W.‐J., He J.‐Z., Gu B.‐N., Liu M.‐C., Chueh Y.‐L., Adv. Funct. Mater. 2021, 31, 2103558. [Google Scholar]
- 47. Yang X.‐F., Wang A., Qiao B., Li J., Liu J., Zhang T., Acc. Chem. Res. 2013, 46, 1740. [DOI] [PubMed] [Google Scholar]
- 48. Lili J., Shuaishuai X., Baokai X., Sheng C., Junwu Z., J. Inorg. Mater. 2022, 37, 215. [Google Scholar]
- 49. Yu M., Yuan X., Guo J., Tang N., Ye S., Liang J., Jiang L., Chemosphere 2021, 284, 131254. [DOI] [PubMed] [Google Scholar]
- 50. Zhao Y., Zhang S., Shi R., Waterhouse G. I. N., Tang J., Zhang T., Mater. Today 2020, 34, 78. [Google Scholar]
- 51. Yang W., Zhang X., Xie Y., Nano Today 2016, 11, 793. [Google Scholar]
- 52. Chen D., Zou Y., Wang S., Mater. Today Energy 2019, 12, 250. [Google Scholar]
- 53. Li Z., Chen Y., Ma T., Jiang Y., Chen J., Pan H., Sun W., Adv. Energy Mater. 2021, 11, 2101202. [Google Scholar]
- 54. Yuan K., Lützenkirchen‐Hecht D., Li L., Shuai L., Li Y., Cao R., Qiu M., Zhuang X., Leung M. K. H., Chen Y., Scherf U., J. Am. Chem. Soc. 2020, 142, 2404. [DOI] [PubMed] [Google Scholar]
- 55. Zhou S., Yang X., Xu X., Dou S. X., Du Y., Zhao J., J. Am. Chem. Soc. 2020, 142, 308. [DOI] [PubMed] [Google Scholar]
- 56. Wu Y., Cai J., Xie Y., Niu S., Zang Y., Wu S., Liu Y., Lu Z., Fang Y., Guan Y., Zheng X., Zhu J., Liu X., Wang G., Qian Y., Adv. Mater. 2020, 32, 1904346. [DOI] [PubMed] [Google Scholar]
- 57. Zheng X., Cui P., Qian Y., Zhao G., Zheng X., Xu X., Cheng Z., Liu Y., Dou S. X., Sun W., Angew. Chem., Int. Ed. 2020, 59, 14533. [DOI] [PubMed] [Google Scholar]
- 58. Kim J., Yang Y., Seong A., Noh H.‐J., Kim C., Joo S., Cho A., Zhang L., Zhou J., Wang J.‐Q., Han J. W., Mahmood J., Baek J.‐B., Kim G., J. Mater. Chem. A 2020, 8, 14927. [Google Scholar]
- 59. Liu C., Tian A., Li Q., Wang T., Qin G., Li S., Sun C., Adv. Funct. Mater. 2023, 33, 2210759. [Google Scholar]
- 60. Deng D., Yu L., Pan X., Wang S., Chen X., Hu P., Sun L., Bao X., Chem. Commun. 2011, 47, 10016. [DOI] [PubMed] [Google Scholar]
- 61. Li X.‐F., Li Q.‐K., Cheng J., Liu L., Yan Q., Wu Y., Zhang X.‐H., Wang Z.‐Y., Qiu Q., Luo Y., J. Am. Chem. Soc. 2016, 138, 8706. [DOI] [PubMed] [Google Scholar]
- 62. Ma R., Lin G., Zhou Y., Liu Q., Zhang T., Shan G., Yang M., Wang J., npj Comput. Mater. 2019, 5, 78. [Google Scholar]
- 63. Ge X., Sumboja A., Wuu D., An T., Li B., Goh F. W. T., Hor T. S. A., Zong Y., Liu Z., ACS Catal. 2015, 5, 4643. [Google Scholar]
- 64. Kwon T., Lim Y., Cho J., Lawler R., Min B. J., Goddard W. A., Jang S. S., Kim J. Y., Mater. Today 2022, 58, 135. [Google Scholar]
- 65. Zatoń M., Rozière J., Jones D. J., Sustainable Energy Fuels 2017, 1, 409. [Google Scholar]
- 66. Woo J., Lim J. S., Kim J. H., Joo S. H., Chem. Commun. 2021, 57, 7350. [DOI] [PubMed] [Google Scholar]
- 67. Kulkarni A., Siahrostami S., Patel A., Nørskov J. K., Chem. Rev. 2018, 118, 2302. [DOI] [PubMed] [Google Scholar]
- 68. Sui S., Wang X., Zhou X., Su Y., Riffat S., Liu C., J. Mater. Chem. A 2017, 5, 1808. [Google Scholar]
- 69. Nørskov J. K., Rossmeisl J., Logadottir A., Lindqvist L., Kitchin J. R., Bligaard T., Jónsson H., J. Phys. Chem. B 2004, 108, 17886. [Google Scholar]
- 70. Montoya J. H., Seitz L. C., Chakthranont P., Vojvodic A., Jaramillo T. F., Nørskov J. K., Nat. Mater. 2017, 16, 70. [DOI] [PubMed] [Google Scholar]
- 71. Guo X., Lin S., Gu J., Zhang S., Chen Z., Huang S., ACS Catal. 2019, 9, 11042. [Google Scholar]
- 72. Meng Y., Huang X., Lin H., Zhang P., Gao Q., Li W., Front. Chem. 2019, 7, 759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Li Y., Tong Y., Peng F., J. Energy Chem. 2020, 48, 308. [Google Scholar]
- 74. Gueskine V., Vagin M., Berggren M., Crispin X., Zozoulenko I., Electrochem. Sci. Adv. 2023, 3, e2100191. [Google Scholar]
- 75. Ramaswamy N., Mukerjee S., J. Phys. Chem. C 2011, 115, 18015. [Google Scholar]
- 76. Wan K., Yu Z., Li X., Liu M., Yang G., Piao J., Liang Z., ACS Catal. 2015, 5, 4325. [Google Scholar]
- 77. Choi C. H., Lim H.‐K., Chung M. W., Park J. C., Shin H., Kim H., Woo S. I., J. Am. Chem. Soc. 2014, 136, 9070. [DOI] [PubMed] [Google Scholar]
- 78. Ramaswamy N., Mukerjee S., Adv. Phys. Chem. 2012, 2012, 491604. [Google Scholar]
- 79. Yu L., Pan X., Cao X., Hu P., Bao X., J. Catal. 2011, 282, 183. [Google Scholar]
- 80. Zhou X., Qiao J., Yang L., Zhang J., Adv. Energy Mater. 2014, 4, 1301523. [Google Scholar]
- 81. Li D., Jia Y., Chang G., Chen J., Liu H., Wang J., Hu Y., Xia Y., Yang D., Yao X., Chem 2018, 4, 2345. [Google Scholar]
- 82. Ortiz‐Medina J., Wang Z., Cruz‐Silva R., Morelos‐Gomez A., Wang F., Yao X., Terrones M., Endo M., Adv. Mater. 2019, 31, 1805717. [DOI] [PubMed] [Google Scholar]
- 83. Choi K., Kim S., ACS Nano 2022, 16, 16394. [DOI] [PubMed] [Google Scholar]
- 84. Wu K.‐H., Wang D.‐W., Su D.‐S., Gentle I. R., ChemSusChem 2015, 8, 2772. [DOI] [PubMed] [Google Scholar]
- 85. Ji Y., Dong H., Liu C., Li Y., J. Mater. Chem. A 2018, 6, 13489. [Google Scholar]
- 86. An F., Bao X., Deng X., Ma Z., Wang X., New Carbon Mater. 2022, 37, 338. [Google Scholar]
- 87. Liu X., Dai L., Nat. Rev. Mater. 2016, 1, 16064. [Google Scholar]
- 88. Jiao Y., Zheng Y., Davey K., Qiao S.‐Z., Nat. Energy 2016, 1, 16130. [Google Scholar]
- 89. Zhang M., Dai L., Nano Energy 2012, 1, 514. [Google Scholar]
- 90. Usachov D., Vilkov O., Grüneis A., Haberer D., Fedorov A., Adamchuk V. K., Preobrajenski A. B., Dudin P., Barinov A., Oehzelt M., Laubschat C., Vyalikh D. V., Nano Lett. 2011, 11, 5401. [DOI] [PubMed] [Google Scholar]
- 91. Yang M., Zhou L., Wang J., Liu Z., Liu Z., J. Phys. Chem. C 2012, 116, 844. [Google Scholar]
- 92. Shao Y., Jiang Z., Zhang Q., Guan J., ChemSusChem 2019, 12, 2133. [DOI] [PubMed] [Google Scholar]
- 93. Hou Z., Wang X., Ikeda T., Terakura K., Oshima M., Kakimoto M., Phys. Rev. B 2013, 87, 165401. [DOI] [PubMed] [Google Scholar]
- 94. Zhao L., He R., Rim K. T., Schiros T., Kim K. S., Zhou H., Gutiérrez C., Chockalingam S. P., Arguello C. J., Pálová L., Nordlund D., Hybertsen M. S., Reichman D. R., Heinz T. F., Kim P., Pinczuk A., Flynn G. W., Pasupathy A. N., Science 2011, 333, 999. [DOI] [PubMed] [Google Scholar]
- 95. Schiros T., Nordlund D., Pálová L., Prezzi D., Zhao L., Kim K. S., Wurstbauer U., Gutiérrez C., Delongchamp D., Jaye C., Fischer D., Ogasawara H., Pettersson L. G. M., Reichman D. R., Kim P., Hybertsen M. S., Pasupathy A. N., Nano Lett. 2012, 12, 4025. [DOI] [PubMed] [Google Scholar]
- 96. Sidik R. A., Anderson A. B., Subramanian N. P., Kumaraguru S. P., Popov B. N., J. Phys. Chem. B 2006, 110, 1787. [DOI] [PubMed] [Google Scholar]
- 97. Niwa H., Horiba K., Harada Y., Oshima M., Ikeda T., Terakura K., Ozaki J., Miyata S., J. Power Sources 2009, 187, 93. [Google Scholar]
- 98. Huang S.‐F., Terakura K., Ozaki T., Ikeda T., Boero M., Oshima M., Ozaki J., Miyata S., Phys. Rev. B 2009, 80, 235410. [Google Scholar]
- 99. Lemes G., Sebastián D., Pastor E., Lázaro M. J., J. Power Sources 2019, 438, 227036. [Google Scholar]
- 100. Guo J., Zhang S., Zheng M., Tang J., Liu L., Chen J., Wang X., Int. J. Hydrogen Energy 2020, 45, 32402. [Google Scholar]
- 101. Lai L., Potts J. R., Zhan D., Wang L., Poh C. K., Tang C., Gong H., Shen Z., Lin J., Ruoff R. S., Energy Environ. Sci. 2012, 5, 7936. [Google Scholar]
- 102. Dumont J. H., Martinez U., Artyushkova K., Purdy G. M., Dattelbaum A. M., Zelenay P., Mohite A., Atanassov P., Gupta G., ACS Appl. Nano Mater. 2019, 2, 1675. [Google Scholar]
- 103. Xiang Q., Liu Y., Zou X., Hu B., Qiang Y., Yu D., Yin W., Chen C., ACS Appl. Mater. Interfaces 2018, 10, 10842. [DOI] [PubMed] [Google Scholar]
- 104. Wang L., Dong H., Guo Z., Zhang L., Hou T., Li Y., J. Phys. Chem. C 2016, 120, 17427. [Google Scholar]
- 105. Fazio G., Ferrighi L., Di Valentin C., J. Catal. 2014, 318, 203. [Google Scholar]
- 106. Agnoli S., Favaro M., J. Mater. Chem. A 2016, 4, 5002. [Google Scholar]
- 107. Yang L., Jiang S., Zhao Y., Zhu L., Chen S., Wang X., Wu Q., Ma J., Ma Y., Hu Z., Angew. Chem., Int. Ed. 2011, 50, 7132. [DOI] [PubMed] [Google Scholar]
- 108. Jorge A. B., Jervis R., Periasamy A. P., Qiao M., Feng J., Tran L. N., Titirici M.‐M., Adv. Energy Mater. 2020, 10, 1902494. [Google Scholar]
- 109. Shao M., Chang Q., Dodelet J.‐P., Chenitz R., Chem. Rev. 2016, 116, 3594. [DOI] [PubMed] [Google Scholar]
- 110. Kakaei K., Esrafili M. D., Ehsani A., in Graphene Surfaces: particles and catalyst. Elsevier, Amsterdam: 2019, 203. [Google Scholar]
- 111. Qin L., Wang L., Yang X., Ding R., Zheng Z., Chen X., Lv B., J. Catal. 2018, 359, 242. [Google Scholar]
- 112. Tafoya J. P. V., Doszczeczko S., Titirici M. M., Sobrido A. B. J., Int. J. Hydrogen Energy 2022, 47, 5462. [Google Scholar]
- 113. Yao Z., Hu M., Iqbal Z., Wang X., ACS Catal. 2020, 10, 160. [Google Scholar]
- 114. Zheng Y., Jiao Y., Ge L., Jaroniec M., Qiao S. Z., Angew. Chem., Int. Ed. 2013, 52, 3110. [DOI] [PubMed] [Google Scholar]
- 115. Kim I. T., Song M. J., Kim Y. B., Shin M. W., Int. J. Hydrogen Energy 2016, 41, 22026. [Google Scholar]
- 116. Zhang M., Tao H., Liu Y., Yan C., Hong S., Masa J., Robertson A. W., Liu S., Qiu J., Sun Z., ACS Sustainable Chem. Eng. 2019, 7, 3434. [Google Scholar]
- 117. Banhart F., Kotakoski J., Krasheninnikov A. V, ACS Nano 2011, 5, 26. [DOI] [PubMed] [Google Scholar]
- 118. Tang C., Zhang Q., Adv. Mater. 2017, 29, 1604103. [DOI] [PubMed] [Google Scholar]
- 119. Jiang H., Gu J., Zheng X., Liu M., Qiu X., Wang L., Li W., Chen Z., Ji X., Li J., Energy Environ. Sci. 2019, 12, 322. [Google Scholar]
- 120. Wang Q., Ji Y., Lei Y., Wang Y., Wang Y., Li Y., Wang S., ACS Energy Lett. 2018, 3, 1183. [Google Scholar]
- 121. Gong Y., Shen L., Kang Z., Liu K., Du Q., Ye D., Zhao H., Sun X. A., Zhang J., J. Mater. Chem. A 2020, 8, 21408. [Google Scholar]
- 122. Liu R., Gao X., Zhou J., Xu H., Li Z., Zhang S., Xie Z., Zhang J., Liu Z., Adv. Mater. 2017, 29, 1604665. [DOI] [PubMed] [Google Scholar]
- 123. Matsuoka R., Sakamoto R., Hoshiko K., Sasaki S., Masunaga H., Nagashio K., Nishihara H., J. Am. Chem. Soc. 2017, 139, 3145. [DOI] [PubMed] [Google Scholar]
- 124. Liu R., Liu H., Li Y., Yi Y., Shang X., Zhang S., Yu X., Zhang S., Cao H., Zhang G., Nanoscale 2014, 6, 11336. [DOI] [PubMed] [Google Scholar]
- 125. Yu H., Xue Y., Li Y., Adv. Mater. 2019, 31, 1803101. [DOI] [PubMed] [Google Scholar]
- 126. Zhang S., Cai Y., He H., Zhang Y., Liu R., Cao H., Wang M., Liu J., Zhang G., Li Y., Liu H., Li B., J. Mater. Chem. A 2016, 4, 4738. [Google Scholar]
- 127. Wang Y., Jiang X., ACS Appl. Mater. Interfaces 2013, 5, 11597. [DOI] [PubMed] [Google Scholar]
- 128. Rahsepar M., Nobakht M. R., Kim H., Pakshir M., Appl. Surf. Sci. 2018, 447, 182. [Google Scholar]
- 129. Skorupska M., Ilnicka A., Lukaszewicz J. P., Sci. Rep. 2021, 11, 23970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ou C., Chen H., Wang H., Liao Y., Li R., Liu H., Electrochim. Acta 2021, 380, 138256. [Google Scholar]
- 131. Lu X., Wang D., Ge L., Xiao L., Zhang H., Liu L., Zhang J., An M., Yang P., New J. Chem. 2018, 42, 19665. [Google Scholar]
- 132. Lee J., Noh S., Pham N. D., Shim J. H., Electrochim. Acta 2019, 313, 1. [Google Scholar]
- 133. Kakaei K., Balavandi A., J. Colloid Interface Sci. 2017, 490, 819. [DOI] [PubMed] [Google Scholar]
- 134. Zhang X., Wen X., Pan C., Xiang X., Hao C., Meng Q., Tian Z. Q., Shen P. K., Jiang S. P., Chem. Eng. J. 2022, 431, 133216. [Google Scholar]
- 135. Nazer E. A. A., Muthukrishnan A., Catal. Sci. Technol. 2020, 10, 6659. [Google Scholar]
- 136. Zhou Y., Sun Y., Wang H., Zhu C., Gao J., Wu D., Huang H., Liu Y., Kang Z., Inorg. Chem. Front. 2018, 5, 2985. [Google Scholar]
- 137. Liao Y., Chen H., Ou C., Bao L., Li R., Liu H., J. Electroanal. Chem. 2022, 921, 116560. [Google Scholar]
- 138. Cheng C., Li Y., Maouche C., Li B., Zhou Y., Wang S., Cheng X., Yang J., J. Electroanal. Chem. 2021, 883, 115058. [Google Scholar]
- 139. Huang J., Han J., Gao T., Zhang X., Li J., Li Z., Xu P., Song B., Carbon 2017, 124, 34. [Google Scholar]
- 140. Liu J., Wei L., Wang H., Lan G., Yang H., Shen J., Electrochim. Acta 2020, 364, 137335. [Google Scholar]
- 141. Ren G., Huang B., Li C., Lin C., Qian Y., J. Electroanal. Chem. 2020, 877, 114732. [Google Scholar]
- 142. Qiao M., Tang C., He G., Qiu K., Binions R., Parkin I. P., Zhang Q., Guo Z., Titirici M. M., J. Mater. Chem. A 2016, 4, 12658. [Google Scholar]
- 143. Li Q., Xu D., Ou X., Yan F., Chem. Asian J. 2017, 12, 1816. [DOI] [PubMed] [Google Scholar]
- 144. Li J., Zhang Y., Zhang X., Huang J., Han J., Zhang Z., Han X., Xu P., Song B., ACS Appl. Mater. Interfaces 2017, 9, 398. [DOI] [PubMed] [Google Scholar]
- 145. Zhao Y., Wan J., Yao H., Zhang L., Lin K., Wang L., Yang N., Liu D., Song L., Zhu J., Gu L., Liu L., Zhao H., Li Y., Wang D., Nat. Chem. 2018, 10, 924. [DOI] [PubMed] [Google Scholar]
- 146. Lv Q., Si W., Yang Z., Wang N., Tu Z., Yi Y., Huang C., Jiang L., Zhang M., He J., Long Y., ACS Appl. Mater. Interfaces 2017, 9, 29744. [DOI] [PubMed] [Google Scholar]
- 147. Li M., Wang K., Lv Q., Chem. Res. Chin. Univ. 2021, 37, 1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Besharat F., Ahmadpoor F., Nezafat Z., Nasrollahzadeh M., Manwar N. R., Fornasiero P., Gawande M. B., ACS Catal. 2022, 12, 5605. [Google Scholar]
- 149. Zheng Y., Liu J., Liang J., Jaroniec M., Qiao S. Z., Energy Environ. Sci. 2012, 5, 6717. [Google Scholar]
- 150. Zheng Y., Jiao Y., Chen J., Liu J., Liang J., Du A., Zhang W., Zhu Z., Smith S. C., Jaroniec M., (Max) Lu G. Q., Qiao S. Z., J. Am. Chem. Soc. 2011, 133, 20116. [DOI] [PubMed] [Google Scholar]
- 151. Liang J., Zheng Y., Chen J., Liu J., Hulicova‐Jurcakova D., Jaroniec M., Qiao S. Z., Angew. Chem., Int. Ed. 2012, 51, 3892. [DOI] [PubMed] [Google Scholar]
- 152. Fu X., Hu X., Yan Z., Lei K., Li F., Cheng F., Chen J., Chem. Commun. 2016, 52, 1725. [DOI] [PubMed] [Google Scholar]
- 153. Li Q., Song S., Mo Z., Zhang L., Qian Y., Ge C., Appl. Surf. Sci. 2022, 579, 152006. [Google Scholar]
- 154. Mrinalini Kalyani A. K., Rajeev R., Benny L., Cherian A. R., Varghese A., Mater. Today Chem. 2023, 30, 101523. [Google Scholar]
- 155. Suter T., Brázdová V., McColl K., Miller T. S., Nagashima H., Salvadori E., Sella A., Howard C. A., Kay C. W. M., Corà F., McMillan P. F., J. Phys. Chem. C 2018, 122, 25183. [Google Scholar]
- 156. Wang X., Wang L., Zhao F., Hu C., Zhao Y., Zhang Z., Chen S., Shi G., Qu L., Nanoscale 2015, 7, 3035. [DOI] [PubMed] [Google Scholar]
- 157. Kim I. Y., Kim S., Premkumar S., Yang J.‐H., Umapathy S., Vinu A., Small 2020, 16, 1903572. [DOI] [PubMed] [Google Scholar]
- 158. Wang M., Wu Z., Dai L., J. Electroanal. Chem. 2015, 753, 16. [Google Scholar]
- 159. Selvarajan P., Fawaz M., Sathish C. I., Li M., Chu D., Yu X., Breesec M. B. H., Yi J., Vinu A., Adv. Energy Sustainable Res. 2021, 2, 2100104. [Google Scholar]
- 160. Garcia J. L., Miyao T., Inukai J., Tongol B. J. V., Mater. Chem. Phys. 2022, 288, 126415. [Google Scholar]
- 161. Li C., Li X., Zhang X., Yang X., Wang L., Lü W., J. Electrochem. Soc. 2020, 167, 100534. [Google Scholar]
- 162. Xu C., Han Q., Zhao Y., Wang L., Li Y., Qu L., J. Mater. Chem. A 2015, 3, 1841. [Google Scholar]
- 163. Pei Z., Zhao J., Huang Y., Huang Y., Zhu M., Wang Z., Chen Z., Zhi C., J. Mater. Chem. A 2016, 4, 12205. [Google Scholar]
- 164. Roy S., Zhang X., Puthirath A. B., Meiyazhagan A., Bhattacharyya S., Rahman M. M., Babu G., Susarla S., Saju S. K., Tran M. K., Sassi L. M., Saadi M. A. S. R., Lai J., Sahin O., Sajadi S. M., Dharmarajan B., Salpekar D., Chakingal N., Baburaj A., Shuai X., Adumbumkulath A., Miller K. A., Gayle J. M., Ajnsztajn A., Prasankumar T., Harikrishnan V. V. J., Ojha V., Kannan H., Khater A. Z., Zhu Z., S. A. Iyengar, P. A. d. S. Autreto, E. F. Oliveira, G. Gao, A. G. Birdwell, M. R. Neupane, T. G. Ivanov, J. Taha‐Tijerina, R. M. Yadav, S. Arepalli, R. Vajtai, P. M. Ajayan, Adv. Mater. 2021, 33, 2101589. [Google Scholar]
- 165. Rafiq M., Hu X., Ye Z., Qayum A., Xia H., Hu L., Lu F., Chu P. K., Nano Energy 2022, 91, 106661. [Google Scholar]
- 166. Khan A. F., Randviir E. P., Brownson D. A. C., Ji X., Smith G. C., Banks C. E., Electroanalysis 2017, 29, 622. [Google Scholar]
- 167. Marbaniang P., Patil I., Lokanathan M., Parse H., Catherin Sesu D., Ingavale S., Kakade B., ACS Sustainable Chem. Eng. 2018, 6, 11115. [Google Scholar]
- 168. Patil I. M., Lokanathan M., Kakade B., J. Mater. Chem. A 2016, 4, 4506. [Google Scholar]
- 169. Patil I. M., Jijil C. P., Lokanathan M., Swami A., Kakade B., Sustainable Energy Fuels 2018, 2, 252. [Google Scholar]
- 170. Patil I. M., Lokanathan M., Ganesan B., Swami A., Kakade B., Chem. Eur. J. 2017, 23, 676. [DOI] [PubMed] [Google Scholar]
- 171. Kumar R., Gopalakrishnan K., Ahmad I., Rao C. N. R., Adv. Funct. Mater. 2015, 25, 5910. [Google Scholar]
- 172. Fan M., Wang Z., Zhao Y., Yuan Q., Cui J., Raj J., Sun K., Wang A., Wu J., Sun H., Li B., Wang L., Jiang J., Carbon Energy 2023, 5, e309. [Google Scholar]
- 173. Thomas S., Manju M. S., Ajith K. M., Lee S. U., Asle Zaeem M., Phys. E 2020, 123, 114180. [Google Scholar]
- 174. Recepoglu Y. K., Goren A. Y., Vatanpour V., Yoon Y., Khataee A., Desalination 2022, 533, 115782. [Google Scholar]
- 175. Wang J., Hao J., Liu D., Qin S., Portehault D., Li Y., Chen Y., Lei W., ACS Energy Lett. 2017, 2, 306. [Google Scholar]
- 176. Angizi S., Akbar M. A., Darestani‐Farahani M., Kruse P., ECS J. Solid State Sci. Technol. 2020, 9, 83004. [Google Scholar]
- 177. Ahsan M. A., He T., Eid K., Abdullah A. M., Curry M. L., Du A., Puente Santiago A. R., Echegoyen L., Noveron J. C., J. Am. Chem. Soc. 2021, 143, 1203. [DOI] [PubMed] [Google Scholar]
- 178. Marbaniang P., Ingavale S., Catherin D., Ramgir N., Swami A., Kakade B., J. Catal. 2019, 378, 104. [Google Scholar]
- 179. Qu L., Zhang Z., Zhang H., Zhang H., Dong S., Appl. Surf. Sci. 2018, 448, 618. [Google Scholar]
- 180. Koleśnik‐Gray M., Meingast L., Siebert M., Unbehaun T., Huf T., Ellrott G., Abellán G., Wild S., Lloret V., Mundloch U., Schwarz J., Niebauer M., Szabo M., Rommel M., Hutzler A., Hauke F., Hirsch A., Krstić V., npj 2D Mater. Appl. 2023, 7, 21. [Google Scholar]
- 181. Xia F., Wang H., Hwang J. C. M., Neto A. H. C., Yang L., Nat. Rev. Phys. 2019, 1, 306. [Google Scholar]
- 182. Qiao J., Kong X., Hu Z.‐X., Yang F., Ji W., Nat. Commun. 2014, 5, 4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Shulenburger L., Baczewski A. D., Zhu Z., Guan J., Tománek D., Nano Lett. 2015, 15, 8170. [DOI] [PubMed] [Google Scholar]
- 184. Xiao J., Long M., Zhang X., Ouyang J., Xu H., Gao Y., Sci. Rep. 2015, 5, 9961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Long G., Maryenko D., Shen J., Xu S., Hou J., Wu Z., Wong W. K., Han T., Lin J., Cai Y., Lortz R., Wang N., Nano Lett. 2016, 16, 7768. [DOI] [PubMed] [Google Scholar]
- 186. Ling X., Wang H., Huang S., Xia F., Dresselhaus M. S., Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Xue X.‐X., Shen S., Jiang X., Sengdala P., Chen K., Feng Y., J. Phys. Chem. Lett. 2019, 10, 3440. [DOI] [PubMed] [Google Scholar]
- 188. Zhu J., Jiang X., Yang Y., Chen Q., Xue X.‐X., Chen K., Feng Y., Phys. Chem. Chem. Phys. 2019, 21, 22939. [DOI] [PubMed] [Google Scholar]
- 189. Wang X., Raghupathy R. K. M., Querebillo C. J., Liao Z., Li D., Lin K., Hantusch M., Sofer Z., Li B., Zschech E., Weidinger I. M., Kühne T. D., Mirhosseini H., Yu M., Feng X., Adv. Mater. 2021, 33, 2008752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Yuan Z., Li J., Fang Z., Yang M., Mai K., Yu D., J. Energy Chem. 2021, 63, 468. [Google Scholar]
- 191. Zhang H., Shen P. K., Chem. Rev. 2012, 112, 2780. [DOI] [PubMed] [Google Scholar]