

Abstract
In the present study, ten new substituted 3-hydroxypyridine-4-one derivatives were synthesized in a four-step method, and their chemical structures were confirmed using various spectroscopic techniques. Subsequently, the inhibitory activities of these derivatives against tyrosinase enzyme and their antioxidant activities were evaluated. Amongest the synthesized compounds, 6b bearing a 4-OH-3-OCH3 substitution was found to be a promising tyrosinase inhibitor with an IC50 value of 25.82 μM, which is comparable to the activity of kojic acid as control drug. Kinetic study indicated that compound 6b is a competitive inhibitor of tyrosinase enzyme, which was confirmed by molecular docking results. The molecular docking study and MD simulation showed that compound 6b was properly placed within the tyrosinase binding pocket and interacted with key residues, which is consistent with its biological activity. The DFT analysis demonstrated that compound 6b is kinetically more stable than the other compounds. In addition, compounds 6a and 6b exhibited the best antioxidant activities. The findings indicate that compound 6b could be a promising lead for further studies.
Keywords: Synthesis, 3-Hydroxypyridine-4-one, Anti-tyrosinase, Antioxidant, Docking study, MD simulation, DFT analysis
Highlights
-
•
Ten substituted 3-hydroxypyridine-4-one derivatives were synthesized, and evaluated as anti-tyrosinase and antioxidant agents.
-
•
Among all the synthesized compounds, 6b bearing 4-OH-3-OCH3 substitution was found to be a promising tyrosinase inhibitor with IC50 value of 25.82 μM.
-
•
The molecular docking studies and MD simulation were carried out to obtain the binding mode of these derivatives in the active site of receptor.
-
•
Compounds 6a and 6b exhibited the best antioxidant activities.
-
•
These results suggest that compound 6b is a promising lead compound for further studies.
1. Introduction
Tyrosinase is a copper-dependent oxidoreductase that plays an essential role in the process of melanin production [1,2]. Tyrosinase catalyzes the conversion of l-tyrosine to 3,4-dihydroxyphenylalanine (l-DOPA) and also the conversion of l-DOPA to dopaquinone, which then turns into melanin [[3], [4], [5]]. Melanin plays an important role in protecting the skin from ultraviolet rays and is also responsible for the browning of fruits and vegetables [6]. Studies show that overproduction of melanin can lead to several skin disorders, such as malignant melanoma, hyperpigmentation, and melasma, and is one of the main causes of reduced quality of fruits and vegetables, which is a serious challenge in the food science industry. In addition, research shows that the tyrosinase enzyme may be associated with Parkinson's and other neurological diseases. Therefore, the development of safe and potent tyrosinase inhibitors is of great importance in the pharmaceutical, agricultural, food, and cosmetic industries [5,7,8].
According to reports, many compounds, such as arbutin, azelaic acid, hydroquinone, and kojic acid have tyrosinase inhibitory activity, but despite their high inhibitory potential, these compounds have various side effects, such as cytotoxicity and instability [[9], [10], [11]]. Therefore, efforts are ongoing to introduce potent tyrosinase inhibitors with fewer side effects.
Studies have shown that most synthesized tyrosinase inhibitors possess additional properties such as anti-inflammatory, antioxidant, and chelating properties [4,12]. It has been found that fragmentation hybridization, which involves combining active fragments, can significantly aid in the discovery of potent and effective tyrosinase inhibitors. Accordingly, in recent decades, many derivatives have been designed and synthesized as tyrosinase inhibitors, including thyazolylhydrazone [13], oxo-2,3-dihydro quinazolinone [14], N-hydroxycinnamoyl amide [15], benzylidene hydrazine [16], quinazolinone benzamides [17], benzopyrimidinone [18], hydroxypyridinone [[19], [20], [21]], etc. Some of these derivatives have demonstrated high activity as tyrosinase inhibitors.
Hydroxypyridinones are one of the attractive structures for researchers due to their distinctive structural features [22]. Due to the presence of carbonyl and hydroxyl groups, this scaffold can be used as a metal chelator, which is effective for many biological activities, such as antimicrobial [23,24], monoamino oxidase B inhibitors [25], cytotoxicity [[26], [27], [28]], anti-Alzheimer's [29,30], and anti-inflammatory [31,32] activities. The combination of hydroxypyridinone with various fragments for anti-tyrosinase activity has been considered in the last decade [19,33,34]. Many derivatives of these compounds exhibited greater tyrosinase inhibitory activity compared to kojic acid. Although the inhibitory mechanism of these compounds is not completely clear, it is likely that the chelation of copper metal present in the tyrosinase active site and competition with the substrate to occupy the active site contribute to the potent inhibition of these compounds [22,35,36]. Some of the evaluated hydroxypyridinone derivatives as tyrosinase inhibitors [19,33,34,37] are represented in Fig. 1.
Fig. 1.

Some of the evaluated hydroxypyridinone derivatives as tyrosinase inhibitors.
In previous studies, we synthesized and investigated a series of 3-hydroxypyridin-4-one hybrids [35,36] as tyrosinase inhibitors, which yielded promising results. The design of this class of compounds is based on the hybridization of two components: 3-hydroxypyridin-4-one and acylhydrazone. The role of each these components in inhibiting tyrosinase enzyme has already been proven. The role of thiosemicarbazone derivatives (Fig. 2, compound I) in tyrosinase inhibition has been proven in past studies due to their metal-chelating properties [[38], [39], [40], [41], [42]]. A group of researchers obtained acylhydrazone derivatives with high tyrosinase inhibitory potential by replacing the oxygen atom instead of sulfur in thiosemicarbazone (compound II) [43]. Furthermore, we designed and synthesized novel derivatives by hybridizing 3-hydroxypyridin-4-ones and acylhydrazones, which provided favorable results [19,34,43]. In our previous studies, phenyl derivatives or specific heterocycles were linked to 3-hydroxypyridin-4-one-acylhydrazone, and their effects on tyrosinase inhibition and antioxidant properties were investigated. The results indicated that the molecule with furan ring substitution exhibited an inhibitory effect with an IC50 value of 8.94 μM (compound IV), which was twice stronger the kojic acid with an IC50 value of 16.68 μM [36]. Additionally, the derivative containing 5-bromo-2-hydroxybenzyl hydrazide (compound III) displayed a promising inhibitory effect (IC50 = 25.29 μM) [35] (Fig. 2).
Fig. 2.

The basis of the design of the studied compounds.
To achieve a comprehensive description of the structure-activity relationship for this class of compounds, in the present study, ten new derivatives of 3-hydroxypyridine-4-one were synthesized and assessed as tyrosinase inhibitors. Additionally, the antioxidant activities of these compounds were investigated using the DPPH method. Molecular docking, MD simulation, and kinetic studies were also carried out to understand the possible interaction of these compounds within the tyrosinase binding pocket. Furthermore, DFT analysis was carried out for the theoretical determination of chemical reactivity and stability of the potent compouds.
2. Results and discussion
2.1. Chemistry
3-Hydroxypyridine-4-one derivatives 6a-j were synthesized by a four-step method shown in Scheme 1. Briefly, maltol (1) was reacted with 3-amino benzoic acid (2) in a mixture of H2O and ethanol at pH = 5.0 to give intermediate 3 as a cream-colored powder in good yield. The second step involves the 4-dimethylaminopyridine (DMAP)-catalyzed conversion of compound 3 to compound 4 in the presence of 1,1′-carbonyldiimidazole (CDI) and methanol. In the third step, the intermediate 5 was synthesized from the reaction of compound 4 with hydrazine hydrate in methanol. Finally, the target derivatives 6a-j were synthesized by coupling intermediate 5 with the corresponding aldehydes in ethanol under reflux conditions. The final compounds were purified through recrystallization from ethyl acetate. Various spectroscopic methods were utilized to confirm the chemical structures of the final compounds.
Scheme 1.

Synthesis of 3-hydroxypyridine-4-one derivatives 6a-j. Reagents and conditions: (i) H2O, EtOH, HCl (6.0 M), pH = 5.0, 100 °C, reflux, 72 h; (ii) Acetone, CDI, CH3OH, DMAP, r.t., 24 h; (iii) NH2NH2.H2O, CH3OH, reflux, 24 h; (iv) EtOH, CH3COOH, Aldehydes, reflux, 4h.
2.2. Tyrosinase inhibitory activity
The in vitro anti-tyrosinase activities of the target 3-hydroxypyridine-4-one derivatives 6a-j were assessed according to the previously reported method [36]. The obtained results, including inhibition percentage and IC50, are shown in Table 1. Kojic acid, a widely used tyrosinase inhibitor, was considered as the standard drug.
Table 1.
Tyrosinase inhibitory activity of the tested compounds 6a-j and kojic acid.
| Entry | Ar | Inhibition%a ± SEMb | IC50 (μM)c | Entry | Ar | Inhibition%a ± SEMb | IC50 (μM)c |
|---|---|---|---|---|---|---|---|
| 6a | 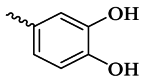 |
31.62 ± 1.23 | >100 | 6g |  |
19.34 ± 2.09 | >100 |
| 6b |  |
71.02 ± 3.88 | 25.82 | 6h |  |
52.6 ± 0.71 | 83.94 |
| 6c |  |
42.75 ± 0.71 | >100 | 6i |  |
13.59 ± 1.38 | >100 |
| 6d |  |
29.30 ± 1.73 | >100 | 6j |  |
30.56 ± 2.23 | >100 |
| 6e |  |
52.24 ± 0.52 | 94.73 | Kojic acid | - | - | 19.14 |
| 6f | 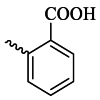 |
51.76 ± 3.71 | 88.30 |

Values for tested compounds and kojic acid were measured at 100 μM.
Values for 3 repetitions of the experiment.
50 % inhibitory concentration (IC50).
As shown in Table 1, the tested compounds indicated variable tyrosinase inhibitory activities. Among all the synthesized compounds, 6b bearing 4-OH-3-OCH3 substitution with an IC50 value of 25.82 μM was found to be a promising tyrosinase inhibitor compared to other compounds. The potency of this compound is comparable to kojic acid, the control drug. Additionally, compounds 6e, 6f, and 6h exhibited IC50 values of 94.73, 88.30, and 83.94 μM, respectively. Other compounds, including 6a, 6c, 6d, 6g, 6i, and 6j exhibited IC50 values greater than 100 μM.
2.3. Structure-activity relationship
The in vitro anti-tyrosinase activities of the target 3-hydroxypyridine-4-one derivatives 6a-j revealed that the aryl group (Ar) played a significant role in determining the anti-tyrosinase activities. It was observed that the introduction of a hydroxyl group at the para position of the phenyl ring (compound 6b) led to a significant increase in anti-tyrosinase activity, and the 4-hydroxyphenyl group appears to be the most favorable functional group for this series of compounds. The better effect of compound 6b can be attributed to its structural similarity to l-tyrosine, the natural substrate of the tyrosinase enzyme. Studies show that there is a long and narrow lipophilic gorge near the active site of the tyrosinase enzyme, which allows compounds with the 4-hydroxyphenyl group to reach it easily [2]. Unexpectedly, compound 6a with 3,4-dihydroxy substitution showed weak anti-tyrosinase activity compared to compound 6b, which is probably due to the different orientation of this compound in the enzyme active site, consistent with the molecular docking results. Molecular docking results indicated that compound 6a interacted with no critical residues of the tyrosinase active site and was located in a different binding pocket compared to compound 6b.
The obtained biological results demonstrated that the 4-hydroxyl group is the optimal functional group for this series of compounds. The introduction of an additional hydroxyl group at the meta-position of the phenyl ring (compound 6a) or shifting the hydroxyl group to the ortho-position of the phenyl ring (compound 6c) significantly decreased the anti-tyrosinase activity. Additionally, the placement of electron-withdrawing groups like Br, COOH, and Cl at the ortho position of the phenyl ring (compounds 6e, 6f, and 6h) is more suitable for inhibitory activity than the electron-donating groups (compound 6d) and the benzyl moiety (compound 6j).
Overall, the biological results revealed that the nature and position of the phenyl-ring substitutions had a significant effect on the tyrosinase inhibitory activity of the synthesized compounds and the presence of a hydroxyl group at the para position of the phenyl ring resulted in significant increase in tyrosinase inhibitory activity.
2.4. Kinetic study of enzyme inhibition
Kinetic study was conducted using compound 6b, the most potent tyrosinase inhibitor, to explore the mechanism of active compounds to inhibit the tyrosinase enzyme. The Lineweaver-Burk double-reciprocal plot of 1/V versus 1/[S], with l-DOPA as the substrate, was utilized to determine the type of inhibition. The results of the kinetic study revealed that with increasing the concentration of 6b, the Km increased, but the Vmax values remained constant, as represented in Fig. 3A. According to these findings, it appears that compound 6b acts as a competitive inhibitor. These findings suggest that probably the structural similarity of compound 6b with l-DOPA, causes competition for the active site of the enzyme. The inhibition constant (Ki) of 0.25 μM was obtained using the slopes of double reciprocal plots versus the different concentrations of compound 6b (Fig. 3B).
Fig. 3.

Kinetic study of tyrosinase inhibition by compounds 6b. (A) Lineweaver-Burk plot in the concentrations of 51.64, 25.82, and without inhibitor (control) in the presence of l-DOPA. (B) Steady-state inhibition constant (Ki) of compound 6b.
2.5. Free radical scavenging activity
The antioxidant activity of all target derivatives 6a-j was evaluated through the DPPH assay, a frequently utilized procedure to determine the antioxidant potential of compounds (Table 2). The screening of all compounds showed that compounds 6a with a 3,4-di-hydroxyphenyl moiety and 6b with a 4-hydroxy-3-methoxyphenyl moiety were the most active compounds, with EC50 values of 2.21 and 17.49 μM, respectively. As shown in Table 2, other compounds displayed lower scavenging activities than quercetin, the reference compound. Compounds 6i and 6j showed EC50 values of 173.78 and 142.23 μM, respectively, while the activities of the remaining compounds, with EC50 > 400 μM, were negligible.
Table 2.
Antioxidant activity of the target compounds 6a-j and quercetin.
| Entry | Ar | Inhibition%a ± SEMb | EC50 (μM) | Entry | Ar | Inhibition%a ± SEMb | EC50 (μM) |
|---|---|---|---|---|---|---|---|
| 6a |  |
90.06 ± 3.75 | 2.21 ± 4.01 | 6g |  |
44.20 ± 2.41 | >400 |
| 6b |  |
84.64 ± 2.68 | 17.49 ± 4.01 | 6h |  |
44.55 ± 0.91 | >400 |
| 6c |  |
48.06 ± 2.25 | >400 | 6i | 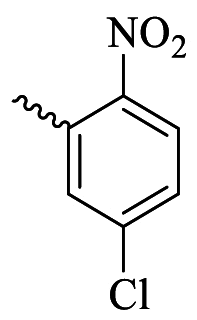 |
67.65 ± 0.85 | 173.78 |
| 6d |  |
35.16 ± 1.37 | >400 | 6j |  |
68.59 ± 1.14 | 142.23 ± 4.01 |
| 6e |  |
19.59 ± 1.76 | >400 | Quercetin | - | – | 8.98 ± 0.05 |
| 6f |  |
35.01 ± 5.71 | >400 |
Values for tested compounds and quercetin were measured at 400 μM.
Values for 3 repetitions of the experiment.
2.6. Molecular docking studies
Molecular docking studies help to design bioactive compounds and justify the biological activity of compounds by examining the interactions of the compounds in the target protein binding site. The molecular docking study was done against the tyrosinase enzyme (PDB code: 2Y9X) for compounds 6a and 6b, which showed the lowest and the best tyrosinase inhibitory activities, respectively. The binding energies for the co-crystal ligand, 6a, and 6b were −7.9, −5.6, and −7.8 kcal mol−1, respectively. The 2D and 3D orientations and interactions for compounds 6a and 6b are shown in Fig. 4. The tyrosinase enzyme's active site is characterized by the presence of two copper atoms surrounded by six key residues, namely, His61, His85, His94, His259, His292, and His296. A potent tyrosinase compound is positioned in the binding pocket containing these residues. The docking results indicated that compound 6b interacted with catalytic residues, including His244 (0.298 nm), Cys83 (0.301 nm), and Arg268 (0.333 nm) through hydrogen bonding, and His85 (0.342 nm), Phe264 (0.371 nm), Glu322 (0.385 nm), and Val248 (0.360 nm) through hydrophobic interactions (Fig. 4b). The 3-hydroxypyridin-4-one scaffold was in contact with Arg268 and Phe264 residues, and the 4-hydroxy-3-methoxy substitution at the phenyl ring made hydrogen bonding interactions with Glu322, His85, and Cys83 residues. In contrast, docking analysis for compound 6a indicated that this compound was surrounded by Glu317 (0.311 nm), Gln48 (0.283 nm), Leu11 (0.301 nm), and Asp10 (0.347 nm) residues (Fig. 4a). The molecular docking results indicated that compound 6a interacted with no critical residues of the active site and was located in a different binding pocket compared to compound 6b, which justifies the weak anti-tyrosinase effect of this compound. In contrast, compound 6b was placed properly into the tyrosinase binding pocket and interacted with key residues, which is consistent with its good biological activity.
Fig. 4.

The 2D and 3D interactions of compounds a) 6a and b) 6b, in the active site of tyrosinase enzyme (PDB: 2Y9X). (Green: van der Waals, light green: carbon-hydrogen bond, dark green: hydrogen bond, dark pink: π-π, light pink: alkyl & π-alkyl, purple: π-sigma, orange: π-sulfur). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
2.7. Molecular dynamic simulation
The ligand-protein interactions of compounds 6a and 6b were subsequently assessed through a 100 ns molecular dynamics simulation. Fig. 5 displays the RMSD values of the protein throughout the simulation. The protein 6a-tyrosinase has exhibited a convergence of RMSD fluctuations at around 2.3 Å, indicating the stability of the system after the simulation period. The recorded value for 6b-tyrosinase stands at approximately 2 Å, denoting a lesser magnitude when compared to 6a-tyrosinase. The ligand-protein interactions between tyrosinase and 6b are illustrated in Fig. 6A, spanning a period of 100 ns. The residues with the highest interaction fractions are His263, Glu256, Met280, Arg268, and Phe264. The interactions exhibited by His263, particularly the hydrophobic and ionic contacts, are the most prominent among the interactions observed in the simulation. This interaction for Glu256, Met280, Arg268, and Phe264 is ionic contact, H-bond, and hydrophobic contacts, respectively. The interactions between 6b and tyrosinase throughout the simulation are visually depicted in Fig. 6A, providing a comprehensive overview. The graphical representation in Fig. 6B illustrates the interactions between 6a and the protein during the entire simulation period. It is apparent that the number of contacts displaying a high interaction fraction between 6b and tyrosinase surpasses those formed between 6a and tyrosinase. This factor could potentially account for the heightened activity observed in 6b in comparison to 6a. For 6a, all of the interaction fractions are lower than 0.8. Fig. 7 depicts the distribution of secondary structure elements (SSE) observed during the molecular dynamics (MD) simulation in the 6b-tyrosinase complex. The provided visual representation demonstrates the consistent stability of the alpha-helices, depicted in red, and the beta-strands, denoted by the blue segments, during the entire simulation. The outcome affirms that the simulation effectively preserves the structural integrity of the protein, thereby validating the precision of the results.
Fig. 5.

The RMSD values of the protein throughout the MD simulation of 6b-tyrosinase and 6a-tyrosinase complexes which are converged at about 2 Å and 2.3 Å, respectively during the simulation.
Fig. 6.

The interactions constructed between the ligands (A: 6b, B: 6a) and tyrosinase during the MD simulation.
Fig. 7.

The percent variation of SSE for tyrosinase during the MD simulation in the presence of 6b. The red regions represent the alpha-helices, while the blue regions represent the beta-strands. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
2.8. DFT analysis
HOMO and LUMO energies determine the stability and reactivity of compounds, which help researchers in designing compounds. In the comparison of several compounds, the compound with lower HOMO and LUMO energy values is more stable. Additionally, by determining the gap between HOMO and LUMO, the reactivity and stability of compounds can be predicted. The compound with a lower energy gap is more reactive, while the compound with a larger energy gap is more stable. The HOMO and LUMO energy values and the energy gap between HOMO and LUMO are shown in Fig. 8. According to Fig. 8, the computed energy values displayed that the energy gap between HOMO and LUMO for compound 6b is greater and it is more stable compared to 6a. The structural difference between compounds 6a and 6b is the presence of a methoxy group, which can be attributed to this variation. For both compounds, 6a and 6b, the LUMO energies are the same, and the LUMO orbitals are distributed throughout the molecule. Furthermore, in both compounds, the HOMO orbital is placed on 2-methyl-3-hydroxypyridine-4-one, which is the electron-donating site of the molecular structure. The electrostatic potential map of compounds 6a and 6b is shown in Fig. 9. As the color changes from blue to green to yellow to orange to red, the electrostatic potential increases. In compounds 6a and 6b, oxygen atoms are placed in the red area, indicating the nucleophilic site, and nitrogen atoms in the blue area, indicating the electrophilic site.
Fig. 8.

The computed HOMO and LUMO and energy gap for 6a and 6b at B3LYP/6-31+G** level of theory.
Fig. 9.

ESP map for 6a and 6b at B3LYP/6-31+G**level of theory.
The IR spectra for compounds 6a and 6b were calculated with the B3LYP/6-31+G** levels and depicted in Fig. 10. The comparison of experimental and theoretical IR spectra confirmed the presence of aromatic and aliphatic C–H and C=O, C–C, C–O, C–N, and O–H groups.
Fig. 10.

Calculated IR spectra for 6a and 6b at B3LYP/6-31+G** level of theory.
The thermochemical parameters, including total, enthalpy, free energy, electron affinity, chemical hardness, and softness were computed with the B3LYP/6-31+G** and presented in Table 3. The calculated energies indicated that compound 6b is more stable than 6a, as inferred from the energy gap. The hardness and softness were calculated using the HOMO and LUMO energies. The smaller the HOMO-LUMO energy gap, the more easily the electrons can be excited from the ground to the excited state. This gap strongly influences the chemical and biological activity, as well as the conductivity of molecules [44]. Molecules with greater hardness are kinetically more stable and less chemically reactive, and vice versa. According to the obtained values, the hardness value for compound 6b is higher than that of compound 6a, which shows its kinetic stability, consistent with the previous results.
Table 3.
The calculated total energy (Etot), Enthalpy (H), Gibbs free energy (G), hardness (ɳ), softness (σ), and electron affinity (A) of 6a and 6b at B3LYP/6-31+G**level of theory.
| Entry | Etota | Ha | Ga | Sb | ɳc | σd | Ac |
|---|---|---|---|---|---|---|---|
| 6a | −1305.10 | −1305.10 | −1305.18 | 172.98 | 1.50 | 0.33 | 1.86 |
| 6b | −1344.17 | −1344.16 | −1344.25 | 180.303 | 1.63 | 0.30 | 1.86 |
in Hartree/particle.
in cal/mol.K.
in ev.
in ev−1.
3. Material and method
3.1. Chemistry
All chemical reagents were obtained from the Sigma-Aldrich and Merck companies. 1H NMR and 13C- NMR spectra were recorded on a Bruker 300 and 400 MHz spectrophotometer, with DMSO‑d6 as the solvent. Chemical shifts are reported in δ units (ppm) relative to tetramethylsilane (TMS) as the internal standard for NMR, and coupling constant (J) values are given in hertz (Hz). The splitting patterns were described as s (singlet), d (doublet), t (triplet), dd (doublet of doublet), m (multiplet), and br (broad). Infrared (IR) spectra of the synthesized compounds were obtained on a PerkinElmer IR instrument. Melting points were determined using an electrothermal IA 9100 apparatus. Mass spectra (MS) were recorded on Agilent Technologies (HP) MS. All reactions were monitored by thin layer chromatography (TLC) using silica gel 60 F254 TLC plates from Merck.
3.2. Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)benzoic acid (3)
A mixture of maltol (1) (50 mmol) and 3-amino benzoic acid (2) (50 mmol) in 98 mL H2O and 10 mL of ethanol at pH = 5.0 was refluxed for 72 h. Thin-layer chromatography (TLC) was employed to monitor the progress of the reaction. Upon the completion of the reaction, the reaction mixture was cooled to room temperature and the resulting precipitate was filtered and washed with acetone to obtain the intermediate 3 as a cream-colored powder with 82 % yield.
3.3. Synthesis of methyl 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)benzoate (4)
A solution of DMAP (0.5 mmol) in 7 mL methanol was added dropwise to a solution of intermediate 3 (5 mmol) and CDI (9 mmol) in dry acetone (15 mL). After stirring overnight at room temperature, the solvent was removed under reduced pressure. The residue was extracted with dichloromethane three times, and the organic layer was separated, dried using Na2SO4, evaporated and dried at room temperature to obtain the compound 4 as a white-colored powder with 22 % yield.
3.4. Synthesis of 3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)benzohydrazide (5)
A mixture of compound 4 (0.5 mmol) and the appropriate hydrazine hydrate (1 mmol) in 5 mL methanol was refluxed for 24 h. Thin-layer chromatography (TLC) was employed to monitor the progress of the reaction. The resulting participate was then filtered and washed with cool methanol to produce compound 5 as a white-colored powder with 80 % yield.
3.5. Synthesis of compounds 6a-j
For the synthesis of target derivatives 6a-j, a mixture of intermediate 5 (1.0 mmol), corresponding aldehydes (2.0 mmol), 10 mL ethanol, and glacial acetic acid (4 drops) was refluxed at 65 °C for 4 h. Upon the completion of the reaction, the reaction mixture was cooled to room temperature, and the precipitate was filtered and purified through recrystallization from ethyl acetate.
3.5.1. (E)-N'-(3,4-dihydroxybenzylidene)-3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl) benzohydrazide (6a)
Cream powder; Yield: 80 %; m.p.: 167–169 °C; IR (KBr, /cm−1): 3374 (N–H), 3191 (OH), 1634 (C=O, ketone), 1566 (C=O, amide), 1254 (C=N). 1H NMR (300 MHz, DMSO) δ (ppm): 11.74 (s, 1H, NH), 8.30 (s, 1H), 8.11 (br, 1H), 8.01 (s, 1H), 7.76 (d, J = 4.5 Hz, 2H), 7.69 (d, J = 7.2 Hz, 1H), 7.31 (d, J = 1.2 Hz, 1H), 7.00 (dd, J = 8.1, 1.5 Hz, 1H), 6.84 (d, J = 8.1 Hz, 1H), 6.31 (d, J = 7.5 Hz, 1H), 2.05 (s, 3H, methyl). 13C NMR (75 MHz, DMSO) δ (ppm): 170.23, 161.77, 149.31, 148.67, 146.24, 145.61, 141.98, 138.42, 135.40, 130.61, 130.40, 129.16, 128.87, 126.31, 125.97, 121.20, 116.04, 113.15, 111.53, 13.90. Mass m/z (%): 377.3 (100), 348.2 (9.83), 243.2 (18.03), 226.1 (15.57), 199.2 (45.08), 170.1 (5.73), 154.1 (6.55), 133.1 (7.37), 77.1 (6.55).
3.5.2. (E)-3-(3-Hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N'-(4-hydroxy-3-methoxybenzylidene)benzohydrazide (6b)
Cream powder; Yield: 74 %; m.p.: 282–284 °C; IR (KBr, /cm−1): 3440 (N–H), 2995 (OH), 1641 (C=O, ketone), 1580 (C=O, amide), 1227 (C=N). 1H NMR (300 MHz, DMSO) δ (ppm): 11.81 (s, 1H, NH), 8.34 (s, 1H), 8.07 (br, 1H), 7.97 (s, 1H), 7.71 (d, J = 4.2 Hz, 2H), 7.64 (d, J = 7.2 Hz, 1H), 7.32 (s, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.86 (d, J = 7.8 Hz, 1H), 6.26 (d, J = 7.2 Hz, 1H), 3.83 (s, 3H, methoxy), 2.00 (s, 3H, methyl).13C NMR (75 MH, DMSO): 170.24, 161.90, 149.66, 149.39, 148.54, 145.69, 141.99, 138,38, 135.36, 130.65, 130.42, 129.25, 128.91, 126.31, 125.95, 122.88, 115.90, 111.53, 109.30, 55.99, 13.92. Mass m/z (%): 393.2 (24.54), 300.1 (100), 258.1 (25.17), 222.1 (31.82), 199.1 (23.82), 177.1 (53.37), 149.1 (21.29), 121.1 (25.32), 91.1 (12.44), 64 (43.18).
3.5.3. (E)-3-(3-Hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N'-(2-hydroxy-3-methoxybenzylidene) benzohydrazide (6c)
White powder; Yield: 68 %; m.p.: 188–190 °C; IR (KBr, /cm−1): 3384 (N–H), 3213 (OH), 1669 (C=O, ketone), 1570 (C=O, amide), 1251 (C=N). 1H NMR (300 MHz, DMSO) δ (ppm): 12.15 (s, 1H, NH), 8.67 (s, 1H), 8.11 (br, 1H), 8.00 (s, 1H), 7.74 (d, J = 4.5 Hz, 2H), 7.64 (d, J = 7.2 Hz, 1H), 7.19 (d, J = 7.8 Hz, 1H), 7.05 (d, J = 8.1 Hz, 1H), 6.87 (t, J = 7.8 Hz, 1H), 6.26 (d, J = 7.2 Hz, 1H), 3.82 (s, 3H, methoxy), 2.01 (s, 3H, methyl). 13C NMR (75 MHz, DMSO) δ (ppm): 170.25, 161.86, 148.65, 148.42, 147.55, 145.61, 142.04, 138.42, 134.70, 130.99, 130.53, 129.12, 129.00, 126.45, 120.85, 119.58, 114.28, 111.56, 56.27, 13.89. Mass m/z (%): 390.3 (100), 361.2 (27.04), 245.2 (12.29), 227.1 (10.65), 199.1 (25.40), 146.2 (17.21), 118.1 (12.29), 91.1 (7.37), 77.1 (6.53).
3.5.4. (E)-3-(3-Hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N'-(2,4,6-trimethoxybenzylidene)benzohydrazide (6d)
Brown powder; Yield: 75 %; m.p.: 274–276 °C; IR (KBr, /cm−1): 3329 (OH), 3219 (N–H), 1666 (C=O, ester), 1632 (C=O, ketone), 1576 (C=C), 1524 (C=O, amide).1H NMR (300 MHz, DMSO) δ (ppm): 11.55 (s, 1H, NH); 8.58 (s, 1H); 8.26–8.20 (m, 1H); 7.97 (s, 1H); 7.69 (d, J = 5.4 Hz, 2H), 7.63 (d, J = 7.2 Hz, 1H), 6.30 (s, 2H), 6.26 (d, J = 3.3 Hz, 1H), 3.84 (s, 3H, methoxy), 3.81 (s, 6H, methoxy), 2.00 (s, 3H, methyl). 13C NMR (75 MH, DMSO): 170.25, 162.27, 145.60, 144.15, 142.04, 140.12, 138.40, 134.75, 134.41, 131.80, 131.11, 130.49, 129.59, 129.10, 126.58, 111.54, 59.95, 59.85, 59.74, 13.89.
3.5.5. (E)-N'-(2-bromobenzylidene)-3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)benzohydrazide (6e)
Cream powder; Yield: 60 %; m.p.: 219–221 °C; IR (KBr, /cm−1): 3248 (N–H, type two amine), 3063 (OH), 1649 (C=O, ketone), 1567 (C=O, amide), 1255 (C=N), 702 (C–Br). 1H NMR (300 MHz, DMSO) δ (ppm): 12.25 (s, 1H, NH), 8.90 (s, 1H), 8.20 (br, 1H), 8.05–8.01 (m, 2H), 7.75–7.64 (m, 4H), 7.49 (t, J = 7.5 Hz, 1H), 7.39 (t, J = 7.5 Hz, 1H), 6.24 (d, J = 7.2 Hz, 1H), 2.11 (s, 3H, methyl).13C NMR (75 MH, DMSO): 170.35, 162.24, 146.90, 145.69, 142.08, 138.44, 134.79, 133.69, 133.42, 132.55, 131.17, 130.48, 129.08, 128.65, 127.79, 126.51, 124.15, 111.54, 13.94. Mass m/z (%): 427.2 (81.96), 366.0 (8.19), 243.1 (18.02), 226.1 (32.78), 199.1 (100), 170.1 (12.29), 154.1 (13.93), 118.1 (12.29), 103.1 (8.59), 89.1 (21.31).
3.5.6. (E)-2-((2-(3-(3-Hydroxy-2-methyl-4-oxopyridin-1(4H)-yl) benzoyl) hydrazono) methyl) benzoic acid (6f)
White powder; Yield: 85 %; m.p.: 244–246 °C; IR (KBr, /cm−1): 3415 (N–H), 3065 (OH), 1694 (C=O, carboxylic acid), 1634 (C=O, ketone), 1576 (C=O, amide), 1206 (C=N). 1H NMR (400 MHz, DMSO) δ (ppm): 12.15 (s, 1H, NH), 9.20 (s, 1H), 8.10–8.08 (m, 2H), 8.02 (s, 1H), 7.92 (d, J = 7.6 Hz, 1H), 7.73 (d, J = 4.0 Hz, 2H), 7.66–7.64 (m, 2H), 7.54 (t, J = 7.6 Hz, 1H), 6.27 (d, J = 7.2 Hz, 1H), 2.01 (s, 3H, methyl).13C NMR (100 MHz, DMSO) δ (ppm):169.72, 168.01, 161.83, 147.05, 145.13, 141.48, 137.93, 134.55, 134.42, 131.97, 130.82, 130.38, 130.32, 129.89, 129.74, 128.68, 128.63, 126.67, 126.02, 111.02, 13.41. Mass m/z (%): 373.1 (2.55), 244.1 (90.23), 226.1 (12.41), 199.1 (43.2), 170.1 (7.43), 146.1 (100), 118.1 (16.82), 89.1 (51.46), 63.1 (18.76).
3.5.7. (E)-N'-(3-chlorobenzylidene)-3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl) benzohydrazide (6g)
White powder; Yield: 77 %; m.p.: 271–273 °C; IR (KBr, /cm−1): 3430 (N–H, type two amine); 3061 (OH); 1662 (C=O, ketone); 1571 (C=O, Amide); 1206 (C=N). 1H NMR (400 MHz, DMSO) δ (ppm): 12.13 (s, 1H, NH), 8.48 (s, 1H), 8.14 (br, 1H), 8.04 (s, 1H), 7.85 (s, 1H), 7.79–7.76 (m, 3H), 7.69 (d, J = 7.2 Hz, 1H), 7.57 (br, 2H), 6.32 (d, J = 7.2 Hz, 1H), 2.06 (s, 3H, methyl).13C NMR (100 MHz, DMSO) δ (ppm): 170.25, 162.34, 147.08, 145.59, 142.01, 138.41, 136.82, 134.95, 134.14, 131.29, 130.97, 130.50, 130.37, 129.19, 129.02, 126.83, 126.48, 126.37, 111.58, 13.90. Mass m/z (%): 381.2 (100), 243.2 (14.63), 226.1 (26.02), 199.2 (79.67), 170.1 (9.75), 154.1 (11.38), 137.1 (4.91), 117.1 (5.37), 89.1 (10.56), 76.1 (8.14).
3.5.8. N'-(2,6-dichlorobenzylidene)-3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)benzohydrazide (6h)
White powder; Yield: 74 %; m.p.: 200–204 °C; IR (KBr, /cm−1): 3519 (N–H), 3269 (OH), 1647 (C=O, ketone), 1562 (C=O, amide), 1198 (C=N). 1H NMR (300 MHz, DMSO) δ (ppm): 12.26 (s, 1H, NH), 8.65 (s, 1H), 8.10 (br, 1H), 8.02 (s, 1H), 7.75 (br, 2H), 7.65 (d, J = 7.2 Hz, 1H), 7.58 (d, J = 7.8 Hz, 2H), 7.45 (t, J = 7.8 Hz, 1H), 6.27 (d, J = 7.2 Hz, 1H), 2.01 (s, 3H, methyl). 13C NMR (75 MHz, DMSO) δ (ppm): 170.24, 162.29, 145.61, 144.17, 142.02, 138.40, 134.74, 134.42, 131.79, 131.11, 130.93, 130.49, 129.21, 129.12, 129.12, 126.56, 111.57, 13.90. Mass m/z (%): 415 (42.46), 364.2 (5.55), 243.1 (38.79), 226.1 (41.81), 199.1 (100), 171 (11.27), 149.1 (17.16), 109 (12.68), 77.1 (16.43).
3.5.9. (E)-N'-(5-chloro-2-nitrobenzylidene)-3-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)benzohydrazide (6i)
Cream powder; Yield: 48 %; m.p.: 290–293 °C; IR (KBr, /cm−1): 3296 (N–H), 3053 (OH), 1659 (C=O, ketone), 1574 (C=O, amide), 1243 (C=N). 1H NMR (300 MHz, DMSO) δ (ppm): 12.39 (s, 1H, NH), 8.86 (s, 1H), 8.16 (d, J = 8.7 Hz, 1H), 8.10–8.08 (m, 2H), 8.01 (s, 1H), 7.80–7.76 (m, 3H), 7.65 (d, J = 7.5 Hz, 1H), 6.26 (d, J = 7.2 Hz, 1H), 2.00 (s, 3H, methyl).13C NMR (75 MHz, DMSO) δ (ppm): 170.26, 162.50, 147.10, 145.62, 142.90, 138.92, 138.40, 134.52, 131.30, 130.86, 130.55, 129.54, 129.17, 129.07, 127.58, 126.59, 114.35, 112.66, 111.58, 13.91.
3.5.10. (E)-3-(3-Hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)-N'-(2-phenylethylidene)benzohydrazide (6j)
Yellow powder; Yield: 60 %; m.p.: 324–326 °C; IR (KBr, /cm−1): 3224 (N–H), 3063 (OH), 1638 (C=O, ketone), 1572 (C=O, amide), 1305 (C=N). 1H NMR (300 MHz, DMSO) δ (ppm): 11.55 (s, 1H, NH), 8.02–7.96 (m, 1H), 7.89 (s, 1H), 7.77–7.73 (m, 1H), 7.69–7.58 (m, 3H), 7.28–7.15 (m, 5H), 6.23 (d, J = 7.2 Hz, 1H), 2.89–2.81 (dd, J = 15.3, 7.5 Hz, 1H), 2.63–2.57 (dd, J = 12.3, 7.5 Hz, 1H), 1.97 (s, 3H. methyl). 13C NMR (75 MHz, DMSO) δ (ppm): 178.22, 170.22, 145.58, 141.98, 138.39, 135.68, 135.25, 135.16, 130.35, 130.29, 129.06, 128.86, 128.74, 128.70, 126.47, 126.27, 111.49, 30.71, 13.84.
3.6. Tyrosinase inhibition assay
The tyrosinase inhibitory potential of the tested 3-hydroxypyridine-4-one derivatives was measured based on the previously reported method [36]. In this assay, the conversion of l-DOPA (L-3,4-dihydroxyphenylalanine) as the substrate to dopachrome is detected at 492 nm.
3.7. Kinetic study of enzyme inhibition
Kinetic analyses were conducted to identify the tyrosinase inhibition type by the most effective tyrosinase inhibitor, compound 6b, based on previously reported methods [45]. The initial linear portion of absorbance up to 10 min after the addition of l-DOPA, was used to measure the maximum initial velocity. The Lineweaver-Burk plot at various concentrations of l-DOPA, was employed to calculate the Michaelis constant (Km), maximal velocity (Vmax), and Ki (inhibition constant). The tyrosinase inhibition mechanism was determined by Lineweaver-Burk plots that plotted the inverse of velocities (1/V) versus the inverse of substrate concentrations 1/[S].
3.8. DPPH radical scavenging assay
The DPPH assay was utilized to evaluate the radical scavenging potential of the target compounds. Each well consisted of 180 μL of DPPH solution (110 μM) and 20 μL of various concentrations of samples. Subsequently, the plate was incubated in the dark condition for 30 min. The absorption of the reaction mixture was measured at 517 nm using a microplate reader Synrgy HTX Reader-BioTek. Finally, the percent inhibition and EC50 were calculated from the absorption changes. The analysis was carried out in triplicates for each concentration.
3.9. Molecular docking study
The molecular docking study was conducted using Autodock Tools 1.5.6 software [46]. The tyrosinase enzyme's crystal structure with PDB code of 2Y9X was obtained from the protein data bank [47]. The self-docking was performed for validation of docking protocol. Subsequently, compounds 6a and 6b was optimized using Chem3D 16.0 and converted to pdbqt format. Finally, compounds 6a and 6b were docked into the tyrosinase active site. The visualization of 2D and 3D interactions of compounds 6a and 6b was performed using Discovery Studio 2016 64-bit Client.
3.10. Molecular dynamic simulation
Further assessment was conducted on the interactions between the top-ranked synthesized compounds and the compound with the least activity against tyrosinase (PDB ID: 2Y9X) in a dynamic context. The MD simulation was conducted utilizing the Desmond software program [48]. The system employed in the molecular dynamics simulation was acquired from the docking calculation. The simulation was carried out in an orthorhombic box, employing the transferable intermolecular potential with 3 points (TIP3P) solvent model [49]. Through the implementation of Schrödinger's system setup, a state of balance was reached in the system by introducing sodium and chloride ions at a concentration of 0.15 M [50]. Utilizing the default configurations, the simulation was executed for a duration of 100 ns under the NPT ensemble. The NPT ensemble preserved a constant count of atoms, pressure, and temperature for the complete duration of the simulation [51]. The temperature of the system was successfully set at 310.15 K (37 °C) through the implementation of the Nose-Hoover protocol, with the pressure being controlled at 1 atm using isotropic scaling.
3.11. DFT analysis
The DFT analysis was carried out for compounds 6a and 6b by using the Gaussian 09W package and GaussView 5.0 molecular visualization programs. The derivatives were optimized with Density Functional Theory (DFT), and Becke's Three-Parameter Hybrid Functional using the Lee, Yang, and Parr correlation (B3LYP) method with 6-311G(d,p) basis set in the ground state. The HOMO and LUMO energies, electrostatic surface potential (ESP), IR spectroscopy, and thermochemistry parameters including electron affinity (A = -ELUMO), η; chemical hardness (η=(I-A)/2), σ; chemical softness (σ = 1/2η), and μ; chemical potential (μ = −(I + A)/2) were determined using HOMO-LUMO energy values.
4. Conclusions
In conclusion, ten substituted 3-hydroxypyridine-4-one derivatives were synthesized, their structures confirmed by various spectroscopic techniques, and subsequently investigated as tyrosinase inhibitors and antioxidant agents. The biological results revealed that compound 6b with a 4-OH-3-OCH3 substitution, exhibited high tyrosinase inhibitory activity, with an IC50 value of 25.82 μM, which is comparable to the activity of kojic acid, the reference compound. According to the structure-activity relationship, the nature and position of the phenyl-ring substitutions significantly influenced the inhibitory potency of the synthesized compounds towards the tyrosinase enzyme, and the presence of a hydroxyl group at the para position of the phenyl ring resulted in a significant increase in tyrosinase inhibitory activity. The kinetic study showed that compound 6b acts as a competitive inhibitor. The molecular docking study and MD simulation indicated that compound 6b, as the most potent inhibitor, placed properly into the tyrosinase binding pocket and interacted with key residues, which is consistent with its good biological results. Furthermore, the antioxidant screening of the synthesized compounds revealed that compound 6b has an acceptable antioxidant effect. These findings provide valuable insight for the design and development of tyrosinase inhibitors in the future.
Data availability statement
The data sets used and analyzed during the current study are available from the corresponding author upon reasonable request. We have presented all data in the form of Tables, Figures and supplementary file.
CRediT authorship contribution statement
Sara Sadeghian: Writing – review & editing, Writing – original draft, Data curation. Fateme Zare: Software. Mehdi Khoshneviszadeh: Project administration, Investigation. Arian Fathi Hafshejani: Methodology, Data curation. Farhang Salahshour: Methodology. Ahmadreza Khodabakhshloo: Methodology, Data curation. Lotfollah Saghaie: Methodology. Ghazal Goshtasbi: Writing – original draft. Zahra Sarikhani: Data curation. Alireza Poustforoosh: Software. Razieh Sabet: Supervision, Project administration. Hossein Sadeghpour: Supervision, Project administration.
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Razieh Sabet reports financial support was provided by Shiraz University of Medical Sciences. Razieh Sabet reports a relationship with Shiraz University of Medical Sciences that includes: funding grants. If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Financial assistance from Shiraz University of Medical Sciences (grant no. 25952) is gratefully acknowledged.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.heliyon.2024.e35281.
Contributor Information
Razieh Sabet, Email: sabet_r@sums.ac.ir.
Hossein Sadeghpour, Email: sadeghpurh@sums.ac.ir.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.He M., et al. Novel kojic acid-1, 2, 4-triazine hybrids as anti-tyrosinase agents: synthesis, biological evaluation, mode of action, and anti-browning studies. Food Chem. 2023;419 doi: 10.1016/j.foodchem.2023.136047. [DOI] [PubMed] [Google Scholar]
- 2.Peng Z., et al. Synthesis, antioxidant and anti-tyrosinase activity of 1, 2, 4-triazole hydrazones as antibrowning agents. Food Chem. 2021;341 doi: 10.1016/j.foodchem.2020.128265. [DOI] [PubMed] [Google Scholar]
- 3.Darroudi M., et al. Synthesis of novel triazole incorporated thiazolone motifs having promising antityrosinase activity through green nanocatalyst CuI‐Fe3O4@ SiO2 (TMS‐EDTA) Appl. Organomet. Chem. 2020;34(12) [Google Scholar]
- 4.Lu L., Zhang X., Kang Y., Xiong Z., Zhang K., Xu X., Bai L., Li H. Novel coumarin derivatives as potential tyrosinase inhibitors: synthesis, binding analysis and biological evaluation. Arab. J. Chem. 2023;16(6) [Google Scholar]
- 5.Boateng S.T., et al. Synthesis, in silico modelling, and in vitro biological evaluation of substituted pyrazole derivatives as potential anti-skin cancer, anti-tyrosinase, and antioxidant agents. J. Enzym. Inhib. Med. Chem. 2023;38(1) doi: 10.1080/14756366.2023.2205042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng Z., et al. Anti-browning and antibacterial dual functions of novel hydroxypyranone-thiosemicarbazone derivatives as shrimp preservative agents: synthesis, bio-evaluation, mechanism, and application. Food Chem. 2023;419 doi: 10.1016/j.foodchem.2023.136106. [DOI] [PubMed] [Google Scholar]
- 7.Liu J., Wu F., Chen C. Design and synthesis of aloe-emodin derivatives as potent anti-tyrosinase, antibacterial and anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2015;25(22):5142–5146. doi: 10.1016/j.bmcl.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Dong H., Liu J., Liu X., Yu Y., Cao S. Combining molecular docking and QSAR studies for modeling the anti-tyrosinase activity of aromatic heterocycle thiosemicarbazone analogues. J. Mol. Struct. 2018;1151:353–365. [Google Scholar]
- 9.Wang G., et al. Synthesis and biological evaluation of new kojic acid-1, 3, 4-oxadiazole hybrids as tyrosinase inhibitors and their application in the anti-browning of fresh-cut mushrooms. Food Chem. 2023;409 doi: 10.1016/j.foodchem.2022.135275. [DOI] [PubMed] [Google Scholar]
- 10.Mughal E.U., et al. Design, synthesis, and structural characterization of thioflavones and thioflavonols as potential tyrosinase inhibitors: in vitro and in silico studies. ACS Omega. 2022;7(20):17444–17461. doi: 10.1021/acsomega.2c01841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu T.-H., Cao S.-W., Yu Y.-Y. Synthesis, characterization and biological evaluation of paeonol thiosemicarbazone analogues as mushroom tyrosinase inhibitors. Int. J. Biol. Macromol. 2013;62:589–595. doi: 10.1016/j.ijbiomac.2013.09.056. [DOI] [PubMed] [Google Scholar]
- 12.Cariola A., et al. Anti-tyrosinase and antioxidant activity of meroterpene bakuchiol from Psoralea corylifolia (L.) Food Chem. 2023;405 doi: 10.1016/j.foodchem.2022.134953. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y., et al. Microwave‐assisted synthesis and biological evaluation of new thiazolylhydrazone derivatives as tyrosinase inhibitors and antioxidants. J. Heterocycl. Chem. 2020;57(3):991–1002. [Google Scholar]
- 14.Sepehri N., et al. Design, synthesis, biological evaluation, and molecular docking study of thioxo-2, 3-dihydroquinazolinone derivative as tyrosinase inhibitors. J. Mol. Struct. 2022;1253 [Google Scholar]
- 15.Wang D., et al. Synthesis of N-hydroxycinnamoyl amide derivatives and evaluation of their anti-oxidative and anti-tyrosinase activities. Bioorg. Med. Chem. 2019;27(20) doi: 10.1016/j.bmc.2019.05.031. [DOI] [PubMed] [Google Scholar]
- 16.Iraji A., et al. Synthesis, biological evaluation and molecular docking analysis of vaniline–benzylidenehydrazine hybrids as potent tyrosinase inhibitors. BMC Chem. 2020;14(1):1–11. doi: 10.1186/s13065-020-00679-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahajan P.G., Dige N.C., Vanjare B.D., Raza H., Hassan M., Seo S.Y., Kim C.H., Lee K.H. Facile synthesis of new quinazolinone benzamides as potent tyrosinase inhibitors: comparative spectroscopic and molecular docking studies. J. Mol. Struct. 2019;1198 [Google Scholar]
- 18.Chortani S., et al. Synthesis, biological evaluation and molecular docking analysis of novel benzopyrimidinone derivatives as potential anti-tyrosinase agents. Bioorg. Chem. 2019;92 doi: 10.1016/j.bioorg.2019.103270. [DOI] [PubMed] [Google Scholar]
- 19.Zhao D.-Y., et al. Design and synthesis of novel hydroxypyridinone derivatives as potential tyrosinase inhibitors. Bioorg. Med. Chem. Lett. 2016;26(13):3103–3108. doi: 10.1016/j.bmcl.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 20.Zhu Y.-Z., et al. Design and synthesis of novel stilbene-hydroxypyridinone hybrids as tyrosinase inhibitors and their application in the anti-browning of freshly-cut apples. Food Chem. 2022;385 doi: 10.1016/j.foodchem.2022.132730. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X., et al. Preparation, antioxidant and tyrosinase inhibitory activities of chitosan oligosaccharide-hydroxypyridinone conjugates. Food Chem. 2023;420 doi: 10.1016/j.foodchem.2023.136093. [DOI] [PubMed] [Google Scholar]
- 22.He M., et al. An overview of hydroxypyranone and hydroxypyridinone as privileged scaffolds for novel drug discovery. Eur. J. Med. Chem. 2021;221 doi: 10.1016/j.ejmech.2021.113546. [DOI] [PubMed] [Google Scholar]
- 23.Liu J., et al. Design, synthesis and biological evaluation of novel 3-hydroxypyridin-4 (1H)-ones based hybrids as Pseudomonas aeruginosa biofilm inhibitors. Eur. J. Med. Chem. 2023;259 doi: 10.1016/j.ejmech.2023.115665. [DOI] [PubMed] [Google Scholar]
- 24.Xiao W.-L., et al. Exploiting natural maltol for synthesis of novel hydroxypyridone derivatives as promising anti-virulence agents in bactericides discovery. J. Agric. Food Chem. 2023;71(17):6603–6616. doi: 10.1021/acs.jafc.3c00465. [DOI] [PubMed] [Google Scholar]
- 25.Zhang C., et al. Design, synthesis and biological evaluation of hydroxypyridinone-coumarin hybrids as multimodal monoamine oxidase B inhibitors and iron chelates against Alzheimer's disease. Eur. J. Med. Chem. 2019;180:367–382. doi: 10.1016/j.ejmech.2019.07.031. [DOI] [PubMed] [Google Scholar]
- 26.Sadeghi-Aliabadi H., et al. Evaluation of the cytotoxic effect of hydroxypyridinone derivatives on HCT116 and SW480 colon cancer cell lines. Pharm. Chem. J. 2019;53:388–391. [Google Scholar]
- 27.Liu X., et al. New polyazamacrocyclic 3-hydroxy-4-pyridinone based ligands for iron depletion antitumor activity. Bioorg. Chem. 2020;96 doi: 10.1016/j.bioorg.2020.103574. [DOI] [PubMed] [Google Scholar]
- 28.Shirvani P., et al. Design, synthesis, in silico studies, and antiproliferative evaluations of novel indolin-2-one derivatives containing 3-hydroxy-4-pyridinone fragment. Bioorg. Med. Chem. Lett. 2022;70 doi: 10.1016/j.bmcl.2022.128784. [DOI] [PubMed] [Google Scholar]
- 29.Jiang X., et al. Discovery of benzamide-hydroxypyridinone hybrids as potent multi-targeting agents for the treatment of Alzheimer's disease. J. Enzym. Inhib. Med. Chem. 2021;36(1):2045–2054. doi: 10.1080/14756366.2021.1978081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu X., et al. Exploration of the novel phthalimide-hydroxypyridinone derivatives as multifunctional drug candidates against Alzheimer's disease. Bioorg. Chem. 2023;141 doi: 10.1016/j.bioorg.2023.106817. [DOI] [PubMed] [Google Scholar]
- 31.Li T.-X., et al. Novel kojic acid derivatives with anti-inflammatory effects from Aspergillus versicolor. Fitoterapia. 2021;154 doi: 10.1016/j.fitote.2021.105027. [DOI] [PubMed] [Google Scholar]
- 32.Liu X., et al. Novel neuroprotective pyromeconic acid derivatives with concurrent anti-Aβ deposition, anti-inflammatory, and anti-oxidation properties for treatment of Alzheimer's disease. Eur. J. Med. Chem. 2023;248 doi: 10.1016/j.ejmech.2023.115120. [DOI] [PubMed] [Google Scholar]
- 33.Li D.-F., Hu P.-P., Liu M.-S., Kong X.-L., Zhang J.-C., Hider R.-C., Zhou T. Design and synthesis of hydroxypyridinone-L-phenylalanine conjugates as potential tyrosinase inhibitors. J. Agric. Food Chem. 2013;61(27):6597–6603. doi: 10.1021/jf401585f. [DOI] [PubMed] [Google Scholar]
- 34.Shao L.-L., et al. Novel hydroxypyridinone derivatives containing an oxime ether moiety: synthesis, inhibition on mushroom tyrosinase and application in anti-browning of fresh-cut apples. Food Chem. 2018;242:174–181. doi: 10.1016/j.foodchem.2017.09.054. [DOI] [PubMed] [Google Scholar]
- 35.Hassani B., et al. Synthesis of 3-hydroxypyridin-4-one derivatives bearing benzyl hydrazide substitutions towards anti-tyrosinase and free radical scavenging activities. RSC Adv. 2023;13(46):32433–32443. doi: 10.1039/d3ra06490e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fazel R., et al. Design, synthesis, in silico ADME, DFT, molecular dynamics simulation, anti-tyrosinase, and antioxidant activity of some of the 3-hydroxypyridin-4-one hybrids in combination with acylhydrazone derivatives. J. Biomol. Struct. Dyn. 2023:1–11. doi: 10.1080/07391102.2023.2252087. [DOI] [PubMed] [Google Scholar]
- 37.Singh L.R., et al. Functionality study of chalcone-hydroxypyridinone hybrids as tyrosinase inhibitors and influence on anti-tyrosinase activity. J. Enzym. Inhib. Med. Chem. 2020;35(1):1562–1567. doi: 10.1080/14756366.2020.1801669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai P., et al. Synthesis, screening and biological activity of potent thiosemicarbazone compounds as a tyrosinase inhibitor. New J. Chem. 2019;43(35):14102–14111. [Google Scholar]
- 39.Hałdys K., Latajka R. Thiosemicarbazones with tyrosinase inhibitory activity. MedChemComm. 2019;10(3):378–389. doi: 10.1039/c9md00005d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen L.-H., Hu Y.-H., Song W., Song K.-K., Liu X., Jia L., Zhuang J.-X., Chen Q.-X. Synthesis and antityrosinase mechanism of benzaldehyde thiosemicarbazones: novel tyrosinase inhibitors. J. Agric. Food Chem. 2012;60(6):1542–1547. doi: 10.1021/jf204420x. [DOI] [PubMed] [Google Scholar]
- 41.Soares M.A., et al. Thiosemicarbazones as inhibitors of tyrosinase enzyme. Bioorg Med Chem Lett. 2017;27(15):3546–3550. doi: 10.1016/j.bmcl.2017.05.057. [DOI] [PubMed] [Google Scholar]
- 42.Song S., et al. Study on the design, synthesis and structure-activity relationships of new thiosemicarbazone compounds as tyrosinase inhibitors. Eur. J. Med. Chem. 2017;139:815–825. doi: 10.1016/j.ejmech.2017.08.033. [DOI] [PubMed] [Google Scholar]
- 43.Dehghani Z., Khoshneviszadeh M., Khoshneviszadeh M., Ranjbar S. Veratric acid derivatives containing benzylidene-hydrazine moieties as promising tyrosinase inhibitors and free radical scavengers. Bioorg. Med. Chem. 2019;27(12):2644–2651. doi: 10.1016/j.bmc.2019.04.016. [DOI] [PubMed] [Google Scholar]
- 44.Arshad R., et al. DFT, molecular docking and ADME studies of thiazolidinones as tyrosinase inhibitors. Polycycl. Aromat. Comp. 2023;43(8):6750–6765. [Google Scholar]
- 45.Mahdavi M., et al. Synthesis of new benzimidazole‐1, 2, 3‐triazole hybrids as tyrosinase inhibitors. Chem. Biodivers. 2018;15(7) doi: 10.1002/cbdv.201800120. [DOI] [PubMed] [Google Scholar]
- 46.Bitencourt-Ferreira G., Azevedo W.F. Molegro virtual docker for docking. Docking screens for drug discovery. 2019:149–167. doi: 10.1007/978-1-4939-9752-7_10. [DOI] [PubMed] [Google Scholar]
- 47.Emir C., Coban G., Emir A. Metabolomics profiling, biological activities, and molecular docking studies of elephant garlic (Allium ampeloprasum L.) Process Biochem. 2022;116:49–59. [Google Scholar]
- 48.Poustforoosh A. Investigation on the structural and dynamical properties of cationic, anionic, and catanionic niosomes as multifunctional controlled drug delivery system for cabozantinib. Colloids Surf. A Physicochem. Eng. Asp. 2024;687 [Google Scholar]
- 49.Poustforoosh A., Moosavi F. Evaluation of the FDA-approved kinase inhibitors to uncover the potential repurposing candidates targeting ABC transporters in multidrug-resistant cancer cells: an in silico approach. J. Biomol. Struct. Dyn. 2023:1–13. doi: 10.1080/07391102.2023.2277848. [DOI] [PubMed] [Google Scholar]
- 50.Poustforoosh A., et al. Tracing the pathways and mechanisms involved in the anti-breast cancer activity of glycyrrhizin using bioinformatics tools and computational methods. J. Biomol. Struct. Dyn. 2024;42(2):819–833. doi: 10.1080/07391102.2023.2196347. [DOI] [PubMed] [Google Scholar]
- 51.Poustforoosh A., et al. Investigation on the mechanisms by which the herbal remedies induce anti-prostate cancer activity: uncovering the most practical natural compound. J. Biomol. Struct. Dyn. 2024;42(7):3349–3362. doi: 10.1080/07391102.2023.2213344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets used and analyzed during the current study are available from the corresponding author upon reasonable request. We have presented all data in the form of Tables, Figures and supplementary file.