

SUMMARY
A DNA double-strand break (DSB) jeopardizes genome integrity and endangers cell viability. Actively transcribed genes are particularly detrimental if broken and need to be repressed. However, it remains elusive how fast the repression is initiated and how far it influences the neighboring genes on the chromosome. We adopt a recently developed, very fast CRISPR to generate a DSB at a specific genomic locus with precise timing, visualize transcription in live cells, and measure the RNA polymerase II (RNAPII) occupancy near the broken site. We observe that a single DSB represses the transcription of the damaged gene in minutes, which coincides with the recruitment of a damage repair protein. Transcription repression propagates bi-directionally along the chromosome from the DSB for hundreds of kilobases, and proteasome is evoked to remove RNAPII in this process. Our method builds a foundation to measure the rapid kinetic events around a single DSB and elucidate the molecular mechanism.
Graphical Abstract
In brief
He et al. apply light-activated CRISPR to induce a DNA double-strand break and observe rapid transcription repression and its propagation along the damaged chromosome. They demonstrate that proteasomes remove RNAPII around the damaged site. Their method builds a foundation to measure the kinetics after DSB and elucidate the molecular mechanism.
INTRODUCTION
DNA double-strand breaks (DSBs) are toxic lesions induced by radiation, chemotherapies, or endogenous processes such as DNA replication and transcription.1-4 The DSB-induced DNA damage response (DDR) and defects in the DDR are linked to various diseases, especially cancers.5-7 After DSB induction, DDR proteins are rapidly deployed to the break site, orchestrated by serine/threonine kinases: ataxia telangiectasia mutated protein (ATM), ataxia telangiectasia and Rad3 related protein (ATR), and DNA-dependent protein kinase (DNAPK), or poly-ADP ribose polymerases (PARPs), to repair the damage.1,2,8-10 During this process, the chromatin around the damaged site is remodeled. For example, H2AX is phosphorylated (γH2AX) for megabases around the DSB, and histones are mono- or poly-ubiquitinated to signal downstream repairing events.11,12
Endogenous DSBs exhibit a bias toward accessible chromatin, especially transcribed genes, possibly due to the exposure of vulnerable single-strand DNA and the accumulation of a G-quadruplex, RNA:DNA hybrid, or other secondary structures.13,14 DSBs in transcribed regions may lead to the accumulation of truncated gene products and clashes between transcription and repairing machinery.15,16 To avoid these side effects, transcription needs to be shut down quickly since RNA polymerases move thousands of base pairs per minute and may quickly run into DSBs.17 Indeed, it was reported that DSB triggers transcription repression on the damaged gene as well as neighboring genes.18-24 However, it remains unclear how quickly transcription is repressed and how far a single DSB influences transcription along the chromosome. The repression was reported as early as 30 min or as late as hours after DSB induction.19,21,22 Besides, some suggested that transcription repression spreads over megabases that largely overlap with the range of γH2AX,23 while others have shown that it was a local effect that happened in the vicinity (10 kb) of DSB sites.18 This controversy is mainly because of the technical limitations of current methods to experimentally induce DNA damage. For example, ion or ultraviolet (UV) laser radiation,25 DNA intercalators,26 and topoisomerase inhibition27 produce non-specific and miscellaneous DNA damages. Tethered nuclease (FokI) may generate a cluster of non-specific DSBs.18 The restriction endonucleases (I-PpoI, AsiSI) can create an isolated single DSB once they translocate into nuclei.19,23,24 But they are inefficient, unsynchronized, and have hundreds of target sites in the genome, leading to global cellular responses like cell-cycle arrest, as was shown for Cas9-generated breaks using a guide RNA targeting ~100 sites.28 Therefore, it remains unclear how a single DSB influences transcription repression and how far it propagates along the chromosome.
Transcription repression after DSB is actively regulated by damage-sensing proteins and their effectors. For example, PARP recruits the negative elongation factor at the damaged site to reduce RNA polymerase II (RNAPII) escape from promoter-proximal pause.22 DNAPK was required to remove RNAPII from genes damaged by I-PpoI endonuclease.19,24 Besides, ATM and PARP induce extensive changes in chromatin around the damaged site for efficient DNA damage repair.18,29 Additionally, in response to DSBs induced by endonuclease FokI or I-SceI, H2AK119ub was deposited around the damaged gene by polycomb-repressive complex 1 (PRC1).20,21 H2AK119ub is a major histone post-translational modification for repressed genes and was also considered as the key player for DSB-induced transcription repression. However, in a separate study, MEL18 of the PRC1 was not enriched at single DSB sites.30 Most of the studies investigated the repression hours after potential DSB because the timing of DSB induction was not precisely synchronized. Therefore, it is challenging to separate the molecular pathways that initiate transcription repression from the those that maintain a repressive environment. Particularly, if multiple DSBs were introduced at the same locus or across the genome, then the repression signals might positively influence one other to reinforce the repressive environment. It is therefore essential to study the early transcription response events from a single DSB.
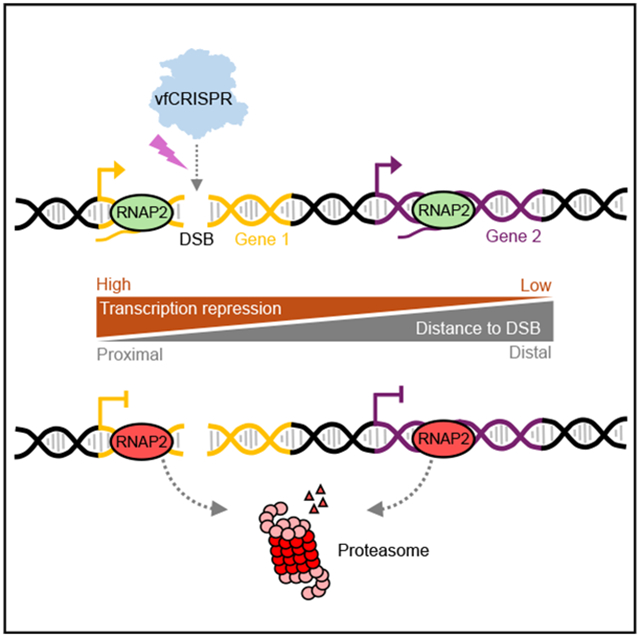
To understand the kinetics of DSB-induced transcription repression and the regulation mechanism, an efficient tool is required to induce single DSBs with precise timing. Recently, we developed a very fast CRISPR-Cas9 (vfCRISPR) that efficiently cleaves target DNA in response to light stimulation.31 A caged guide RNA (cgRNA) allows Cas9 to bind DNA targets without cutting it. Light stimulation releases the caging groups and activates Cas9 in seconds. The synchronous creation of DSBs among cells allows the investigation of early transcription dynamics after damage. To monitor transcriptional dynamics in live cells, we applied the MS2 system.32 MS2 binding sites (MBS) were inserted into the target gene and fluorescently labeled MS2 coat proteins (MCPs) bound to nascent transcripts to label the transcription site (TS). To monitor transcriptome-wide responses, we used chromatin immunoprecipitation (ChIP) with antibodies targeting RNAPII. We discovered rapid transcription repression occurring minutes after inducing a single DSB in the target gene by vfCRISPR. We also confirmed that proteasome-mediated RNAPII removal was essential for this fast repression. By focusing on the early events after DSB, we characterized transcriptional dynamics after DNA damage and elucidated how this process was regulated.
RESULTS
A live-cell reporter reveals rapid transcription repression after DNA damage
To monitor transcription and DDR simultaneously, we employed and optimized a previously published reporter33 (Figure 1A). A cyan fluorescent protein was under the control of an inducible Tet-On promoter, and 24xMBS was inserted in the 3′ untranslated region (UTR). The reporter was integrated into a single flippase recognition target (FRT) site in a U-2 osteosarcoma (U-2 OS) Flp-In cell line. A bright fluorescent punctum appeared when the cell was induced by doxycycline (Dox), representing the TS as stdMCP-stdGFP bound to nascent transcripts32 (Figure 1B). A cgRNA targeting the sequence after the MBS in the 3′ UTR was designed for vfCRISPR to induce DSBs. To monitor DNA damage repair in live cells, the mCherry-tagged 53BP1 Tudor domain (mCherry-53BP1) was stably integrated into the same cell line31 such that mCherry-53BP1 would form a bright focus at the DSB site.
Figure 1. DNA double-strand break induced by vfCRISPR causes rapid transcription repression that coincides with 53BP1 recruitment.

(A) Schematic of live-cell reporter to simultaneously monitor transcription and DNA damage repair. The reporter was under the control of a Tet-On promoter and encoded cyan fluorescent protein (CFP). 24xMBS was inserted in the 3′ UTR. MCP-GFP bound to the MBS and labeled the transcription site (TS). A caged guide RNA (cgRNA) was designed to target a site downstream of the MBS. When stimulated by light, Cas9/cgRNA was activated and created a DSB. mCherry-53BP1 stably expressed in the cell was recruited to the DSB to mark the damaged DNA.
(B) TS of the live-cell reporter was identified before stimulation. Green: MCP-GFP; red: mCherry-53BP1; white arrowhead: TS. Scale bar: 5 μm.
(C) DSB was created by scanning a diffraction-limited 405 nm laser beam surrounding the TS. The transcription and DSB repair were monitored by time-lapse imaging (Video S1). Snapshots showed the TS’s disappearance and 53BP1’s recruitment to the damaged locus. Scale bar: 1 μm.
(D) Fluorescence intensity traces of TS and 53BP1 in (C). Intensities were normalized to the maximum value of each trace.
(E) Heatmaps of TS and 53BP1 intensities. Top: cells with 53BP1 recruitment within 30 min, n = 170 out of 267 total stimulated cells. Bottom: the exact procedure was performed on cells electroporated with a mutant guide RNA that bound to but did not cut the DNA target, n = 68 cells. Each row represents an individual cell, and the color denotes normalized fluorescence intensity. Cells are ranked by the sum of normalized TS intensity.
(F) The averaged TS (top) and 53BP1 (bottom) intensities as shown in (E). The negative control is shown in gray. Shading represents the 95% confidence interval.
(G) Top: fitting of TS intensity trace with a delayed exponential decay model. τ, decay time; δ, response time. Bottom: a representative TS intensity trace (symbol) and the fitting to the model (curve).
(H) Scatterplots of decay (left) and response (right) times from fitting TS traces in (E). Lines: quartiles.
We investigated the transcriptional dynamics of the reporter in response to DSB. Cells were electroporated with a Cas9/cgRNA ribonucleoprotein (RNP) complex and incubated overnight to allow it to bind to the target. Single cells containing a pre-existing TS were stimulated by raster scanning a focused 405 nm laser beam in a 5 × 5 μm region of interest surrounding the TS to uncage Cas9/cgRNA. Two-color time-lapse z stack images were acquired for 30 min after stimulation. The TS disappeared rapidly within minutes after uncaging. Simultaneously, mCherry-53BP1 was recruited and colocalized with the diminishing TS (Figures 1C and 1D; Video S1). vfCRISPR efficiently induced DSB, and 170 out of 267 cells showed 53BP1 foci within 30 min after uncaging. We used 53BP1 foci formation as a proxy to DSB creation and chose cells with 53BP1 foci whenever possible to select damaged ones. We quantified TS and 53BP1 foci intensities in over 170 cells and summarized them in heatmaps (Figure 1E). Due to the bursting dynamics,34 transcription may stochastically turn off. To control the potential side effect of laser illumination and stochastic transcription turning off, we used mutated guide RNA with mismatches in the PAM distal region that bound to the same target site but did not induce Cas9 cleavage. Laser stimulation did not induce noticeable transcription repression or 53BP1 foci formation, indicating that the observed transcription repression depended on vfCRISPR-generated DSBs (Figures 1E and 1F).
To capture the heterogeneity in DDR and transcription repression, we fit a delayed exponential decay model to the single-cell TS intensities (Figure 1G). We assumed that TS intensity remained constant until the detection of Cas9 cleavage, after which it underwent exponential decay. Two kinetic parameters were extracted from the fitting. First, the response time δ was defined as the time lag between laser stimulation and the moment when the TS started to decay. It encapsulated the time for DSB detection and steps leading to the initiation of transcription repression. The second parameter was the decay time τ of the exponential decay function, which is inversely proportional to the rate of nascent RNA removal from the damaged locus. We fit TS intensity traces in Figure 1E to extract response and decay times (Figure 1H). The median response time is 2.3 min, and the decay time is 3.0 min. These two parameters were statistically independent of one other (Pearson's correlation coefficient −0.076), suggesting that transcription decay kinetics is intrinsic and does not depend on DSB detection (Figure S1). Together, these results demonstrated that transcription of the cut gene is rapidly repressed, which coincides with the recruitment of repair protein 53BP1. The decay can be described by a simple exponential function and is completed within minutes after the detection of the single DSB.
Transcription repression propagates bi-directionally along the chromosome from DSBs for hundreds of kilobases in an attenuated manner
In addition to the damaged gene, previous research indicated that the neighboring undamaged genes may also be repressed.13,18,23 However, how transcription repression propagates along the chromosome remains unclear. To address this question, we used vfCRISPR with cgRNA targeting the endogenous ACTB locus. After electroporating Cas9/cgRNA RNP into HEK293T cells and incubating overnight, we activated the whole plate of cells by shining a UV light-emitting diode lamp (Figure S2A). Efficient light-dependent Cas9 cleavage on ACTB was confirmed, while no DSB was detected on the non-targeting control locus (Figure S2B). After incubating the activated cells for a defined time, we harvested them and performed ChIP with antibodies targeting endogenous RNAPII. We measured the occupancy of RNAPII on a gene by next-generation sequencing (ChIP-seq) or quantitative PCR (ChIP-qPCR) (Figure S2A).
At 1 h after light stimulation, RNAPII occupancy drastically decreased along the whole damaged ACTB gene. Besides, transcription was also repressed on the nearby gene FSCN1 ~70 kb from the break site, although not as much as ACTB. The PMS2 gene that is ~500 kb away did not show a difference (Figures 2A and 2B). We quantified the change of RNAPII occupancy upon light stimulation for all expressed genes around the ACTB locus. For each biological replicate, we summed all reads falling in the annotated range of a gene and divided it by gene length, which we defined as RNAPII occupancy for this gene. To account for the potential perturbation on global transcription by light exposure, we compared the RNAPII occupancy between light-stimulated and no-light samples on actively transcribed genes not on chromosome 7, where the targeted ACTB is located (STAR Methods; Figure S2C). We assumed that these non-damaged genes were not influenced by light-induced Cas9 cleavage. A linear fit revealed that the RNAPII occupancy in the light-stimulated sample is slightly less than in the no-light control (Figures S2C and S2D). We used the slope as the calibration factor (0.75 at 60 min). We divided the RNAPII occupancy of the light-stimulated sample by the calibration factor to eliminate the difference in sample preparation and global transcription. For each gene within 1.5 Mb around the DSB, we measured its transcription repression index (TRI) by dividing the calibrated RNAPII occupancy in the light-stimulated sample by that in the no-light control sample. This value excluded the global effect of UV exposure and represented the local RNAPII occupancy change resulting from Cas9 cleavage. We plotted the TRI of a gene as a function of its distance to the DSB site. TRI had the minimum value at the damaged gene and increased bi-directionally as a function of the distance (Figure 2C). These results were recapitulated by ChIP-qPCR experiments with primers targeting the selected genes (Figure S2E). Overall, we observed a 75%–85% reduction of RNAPII occupancy for ACTB after Cas9 cleavage, compared with a 40%–50% reduction for the neighboring gene FSCN1. We use ChIP-qPCR to quantify RNAPII occupancy on genes of interest when possible, as it is more convenient and cost effective, while keeping in mind that the choice of qPCR amplicons may introduce bias when comparing different genes.
Figure 2. DSB-induced transcription repression propagates bi-directionally along the chromosome for hundreds of kilobases in an attenuated manner.

ChIP-seq experiments with RNAPII antibody were performed with vfCRISPR targeting endogenous ACTB locus in HEK293T cells to measure genome-wide transcription responses.
(A) Configuration of human endogenous ACTB locus and two nearby genes FSCN1 and PMS2 with different distances to the DSB site. Red arrowhead: cutting site.
(B) RNAPII ChIP-seq profile for representative genes before and 60 min after vfCRISPR activation. Red arrowhead: cutting site. Red bars: qPCR amplicons.
(C) Transcription repression index (TRI; STAR Methods) for expressed genes as a function of their distances to DSB. The sign of distance was determined with respect to the direction of ACTB gene transcription. TRI measured the change of RNAPII occupancy on a gene in response to vfCRISPR activation and was defined as /calibration factor. The cross-sample calibration factor was calculated by assuming that the transcription of genes on non-damaged chromosomes did not change (STAR Methods; Figure S2C). Cells were fixed at 1 h after light stimulation. Error bar: SEM (n = 2 biological replicates).
(D) TRI traces for representative genes. Numbers in titles denote genes’ distances to the DSB site or the chromosome number for GAPDH. Error bar: SEM (n = 2 biological replicates).
(E) Time-resolved TRI as a function of absolute distance to the DSB. ChIP-seq experiments were conducted on cells fixed at 5, 30, and 60 min after light stimulation. Solid lines denote the fitting of repression propagation with an exponential model: . Error bar: SEM (n = 2 biological replicates).
(F) Estimation of repression propagation speed from fitting in (E). The repression range extracted from (E) scales linearly as a function of time, and the slope of fitted line measures the speed of repression propagation along the chromosome: 4.5 kb/min.
To characterize transcription repression propagation in time, we performed time-resolved ChIP-seq experiments. Cells were harvested at 5, 30, and 60 min after light stimulation. GAPDH on a different chromosome showed constant RNAPII occupancy after light stimulation. The damaged gene ACTB and the neighbor FSCN1 exhibited RNAPII removal by 5 and 30 min, respectively, while distant PMS2 showed minor change even after 60 min (Figure 2D). We plotted the TRI for genes within 1.5 Mb from the DSB (Figure 2E, symbols). Transcription began to be repressed for genes close to the DSB within 5 min. By 30 min after DSB induction, distant genes were repressed, and this repression was further expanded by 60 min post-DSB. We fit an exponential association model to the measured TRI of genes as a function of their distances to the DSB (Figure 2E, curves). We extracted the characteristic repression range, which increased linearly with time. By fitting the repression range as a linear function of time, we estimated the repression propagation speed along the chromosome as 4.5 kb/min (Figure 2F). The observed transcription repression is not unique to the ACTB locus. We targeted the MYC locus with vfCRISPR and observed a similar repression phenomenon after light stimulation (Figure S2F). Together, we concluded that transcription repression induced by vfCRISPR spreads bi-directionally from the cleavage site along the chromosome for hundreds of kilobases, and this repression is negatively correlated with the distance of a gene to the break site.
DSB-induced rapid transcription repression does not depend on PRC1- or RNF8/RNF168-mediated histone ubiquitination
We further explored the mechanism of DSB-induced transcription repression by perturbing the potential regulators. Upstream damage signaling factors, such as ATM and DNAPK, orchestrate various damage repair pathways, and their perturbation may lead to diverse repairing defects.9,35,36 Thus, we sought downstream effectors that repress transcription directly. Histone post-translational modifications, such as methylation, phosphorylation, and mono- and poly-ubiquitination, are important for chromosome structure and transcriptional regulation. Particularly, histone ubiquitination is involved in DSB repair and transcription repression.37-39 We, therefore, focused on the ubiquitination pathway, especially during the early repression stage.
PRC1-mediated H2AK119ub is a common repressive marker and was shown to play a role in DSB-induced transcription repression.20,21,40-42 Besides, RNF8/RNF168, the two E3 ubiquitin ligases that mostly contribute to repair protein recruitment, could be used to study if transcriptional regulation is correlated with repair signaling after damage. Moreover, K48-linked poly-ubiquitination-mediated protein degradation by proteasome was shown to play a role in transcription repression and repair protein recruitment.19,24,39 To distinguish their contributions to transcription repression, we perturbed these ubiquitination markers through chemical or genetic methods and observed the effect of transcription by live-cell imaging and ChIP, respectively.
H2AK119ub has been reported to be enriched at the damaged sites and repress transcription. However, the experiments were performed with gene arrays containing multiple endonuclease cutting sites or UV laser-irradiation-generated complex DNA lesions.20,21 In another study, MEL18, a component of PRC1, was not enriched at single damage sites created by the endonuclease AsiSI.30 To test the effect of H2AK119ub, we applied small interfering RNAs (siRNAs) to knock down RNF2, a key catalytic subunit of the PRC1, and measured transcriptional dynamics after Cas9 cleavage. The efficient depletion of RNF2 protein was confirmed by western blot assay (Figure S3A). RNF2 knockdown did not perturb light-stimulated DSB creation by vfCRISPR (Figure S3B). We measured the 53BP1 recruitment ratio and confirmed that it was not influenced by RNF2 knockdown compared to the negative control (Figure S3C). We then monitored the dynamics of transcription and 53BP1 with the live-cell reporter (Figure 1A) after vfCRISPR-induced cleavage (Figure 3A). Among cells with 53BP1 recruitment, the percentage of cells in which transcription was repressed did not show a difference between RNF2 knockdown samples and non-targeting siRNA control (Figure 3B). Similar results were obtained when all stimulated cells were analyzed, though the fraction of repressed cells was around 60% (Figure S3D). Furthermore, neither transcription decay time nor response time from fitting single TS intensity traces changed significantly (Figure 3C). To study whether H2AK119ub influences damaged and neighboring genes, we applied vfCRISPR to the endogenous ACTB locus and performed ChIP-qPCR experiments to assess RNAPII occupancy on ACTB and two representative neighboring genes with different distances to the break site. There was no significant difference between RNF2 knockdown samples and non-targeting siRNA control (Figure 3D). Besides applying siRNAs to deplete RNF2, we also performed ChIP experiments in non-treated cells to measure the endogenous H2AK119ub enrichment level at several positions within the ACTB gene as well as the promoter region of FSCN1 after Cas9 cleavage. We confirmed that the H2AK119ub ChIP-qPCR agreed with previous ChIP-seq data at H2AK119ub-positive sites (Figures S3E and S3F).43 Similarly, we did not observe enrichment of this histone modification at 1 h after DSB induction (Figure S3G). Together, these results suggested that PRC1-mediated H2AK119ub did not significantly contribute to the rapid transcription repression after Cas9 cleavage.
Figure 3. DSB-induced transcription repression does not depend on PRC1- or RNF8/168-mediated ubiquitination.

Cells were treated with siRNAs to knock down RNF2 (A–D) or RNF8/168 (E–H). The DSB was induced by vfCRISPR in the live-cell transcription reporter (Figure 1A) stably integrated in U-2 OS cells (A–C and E–G) or endogenous ACTB in HEK293T cells (D and H).
(A and E) Heatmaps of TS and 53BP1 intensities were measured from live-cell imaging in the same way as in Figure 1. (A) RNF2 knockdown samples (siRNF2, n = 89 cells) and negative control (siNeg, n = 91 cells). Cells with recruited 53BP1 were selected. (E) RNF8 and RNF168 knockdown samples (siRNF8/168, n = 94 cells) and negative control (siNeg, n = 80 cells). All stimulated cells were analyzed since there was no 53BP1 recruitment.
(B and F) Percentages of repressed cells were measured for knockdown samples and control samples during 30 min tracking. Cells were deemed repressed if the averaged TS intensities in the last 4 frames were lower than a threshold value determined from siNeg control cells with 53BP1 recruitment (STAR Methods). (B) RNF2 knockdown samples and negative control (n = 3 biological replicates). (F) RNF8/168 knockdown samples and negative control (n = 2 biological replicates). Error bar: SEM.
(C and G) Transcription decay and response times were obtained by fitting the delayed decay model (Figure 1G) to the repressed TS intensities in (B) and (F). (C) RNF2 knockdown samples (n = 70 cells) and negative control (n = 62 cells). (G) RNF8/168 knockdown samples (n = 48 cells) and negative control (n = 44 cells). Error bar: quartiles. Unpaired Mann-Whitney test was performed.
(D and H) ChIP-qPCR experiments of RNAPII were performed in HEK293T cells when the ACTB locus was damaged by vfCRISPR. Change of RNAPII occupancy (measured in reads per million [RPM]) upon light stimulation on the ACTB locus and nearby genes was measured. GAPDH on a different chromosome served as a control gene to indicate global transcriptome change upon UV exposure. (D) RNF2 knockdown samples and negative control (n = 3 biological replicates). (H) RNF8/168 knockdown samples and negative control (n = 3 biological replicates). FSCN1: 70 kb from DSB. PMS2: 487 kb from DSB. Error bar: SEM. Unpaired t test was performed.
We tested the effect of RNF8 and RNF168 in transcription repression. These two E3 ubiquitin ligases are downstream of ATM-phosphorylated MDC1 and regulate the recruitment of 53BP1.37 siRNAs were applied to knock down both RNF8 and RNF168, as confirmed by western blot (Figure S3H). RNF8/168 knockdown compromised the formation of nuclear 53BP1 foci (Figures S3I and S3J) but did not influence Cas9 cleavage efficiency (Figure S3K). We then conducted live-cell imaging with the transcription and DDR reporter (Figure 1A). In agreement with previous studies,37,44 the depletion of RNF8 and RNF168 abolished 53BP1 recruitment to the DSB site (Figure 3E). Due to the lack of 53BP1 recruitment to indicate damaged cells, all stimulated cells were analyzed. Nevertheless, transcription of the damaged gene was similarly repressed between RNF8/RNF168-depleted cells and non-targeting control (Figure 3E). Quantitative analysis showed that the percentage of repressed cells, transcription decay time, and response time did not change significantly (Figures 3F and 3G). The lower percentage of repressed cells at the baseline (Figures 3B and 3F) was because we analyzed all stimulated cells, including those without DNA damage, for the RNF8/RNF168 knockdown experiment. This conclusion was further confirmed by RNAPII ChIP after damaging the endogenous ACTB locus by vfCRISPR. There was no significant difference in the reduction of RNAPII occupancy on the damaged ACTB gene between knockdown samples and controls. However, after Cas9 cleavage, the nearby gene FSCN1 was not repressed anymore when RNF8 and RNF168 were depleted (Figure 3H). These results suggested that these two E3 ubiquitin ligases may contribute to repression propagation beyond the damaged gene. Together, we found that the RNF8/RNF168/53BP1 repair pathway is decoupled with transcription repression of the damaged gene but may contribute to repression propagation.
DSB-induced rapid transcription repression is regulated by proteasome-mediated RNAPII removal
Since PRC1-mediated H2AK119ub and RNF8/RNF168-dependent ubiquitination did not seem to control rapid transcription repression, we investigated other histone ubiquitination pathways. K48-linked poly-ubiquitination-dependent proteasome degradation has been reported to degrade RNAPII when DNA damage occurred due to UV radiation or endonuclease cleavage.19,24,45-47 We therefore investigated whether the proteasome was directly involved in DSB-induced transcription repression.
To test the hypothesis, we applied the drug MG132 to inhibit proteasome37,48 (Figure S4A). The drug did not influence the normal transcriptional dynamics (Figures S4D and S4E) and did not influence Cas9 cleavage efficiency or DNA damage sensing (Figures S4B and S4E). U-2 OS cells with the transcription reporter (Figure 1A) were electroporated with Cas9/cgRNA RNP and treated with 20 μM MG132 for 1 h. 53BP1 was no longer recruited to the damaged site (Figure 4A), consistent with the report that 53BP1 recruitment required proteasomal degradation of L3MBTL1, JMJD2A, and JMJD2B to unmask methylated Lys20 on histone 4.38,44,49 Transcription imaging and DSB induction by vfCRISPR were performed as before. Due to lack of 53BP1 recruitment to indicate damaged cells, all stimulated cells were analyzed. Proteasome inhibition significantly compromised transcription repression on the damaged gene (Figure 4A; Video S2). Fewer cells were repressed when treated with MG132 (Figure 4B). The transcription decay time increased significantly upon MG132 treatment, while the response time did not (Figure 4C), consistent with the observation that proteasome inhibition did not influence DSB detection (Figures S4B and S4E). This indicated that the compromised transcription repression by proteasome inhibition was mostly due to the slower rate but not the delayed onset of RNAPII removal by the proteasome. These results were confirmed by RNAPII ChIP experiments after damaging the endogenous ACTB locus. The DSB-induced reduction of RNAPII occupancies on both ACTB and FSCN1 was compromised in the presence of MG132 (Figure 4D). Taking these results together, we concluded that the proteasome mediates the efficient removal of RNAPII after DSB, essential for rapid transcription repression after DNA damage.
Figure 4. DSB-induced transcription repression is regulated by proteasome-mediated RNAPII removal.

Cells were treated with 20 μM MG132 for 1 h to inhibit proteasome. The DSB was induced by vfCRISPR in a live-cell transcription reporter (Figure 1A) stably integrated in U-2 OS cells (A–C) or endogenous ACTB in HEK293T cells (D).
(A) Heatmaps of TS and 53BP1 intensities were measured from live-cell imaging as in Figure 1 (MG132, n = 119 cells; negative control, n = 110 cells). All stimulated cells were analyzed since there was no 53BP1 recruitment.
(B) Percentages of repressed cells were measured for drug treatment and control samples during 30 min tracking. Cells were considered repressed if the averaged TS intensities in the last 4 frames were lower than a threshold determined from control cells with 53BP1 recruitment. Error bar: SEM (n = 3 biological replicates).
(C) Transcription decay and response times were obtained by fitting the delayed decay model (Figure 1G) to the repressed TS intensities in (B) (MG132, n = 59 cells; negative control, n = 94 cells). Error bar: quartiles. An unpaired Mann-Whitney test was performed.
(D) ChIP-qPCR experiments of RNAPII were performed in HEK293T cells when the ACTB locus was damaged by vfCRISPR. Change of RNAPII occupancy (measured in RPM) upon light stimulation on the ACTB locus and nearby genes were measured in MG132-treated and control samples. GAPDH on a different chromosome served as a control gene to indicate global transcriptome change upon UV exposure. FSCN1: 70 kb from DSB. PMS2: 487 kb from DSB. Error bar: SEM (n = 2 biological replicates). Unpaired t test was performed.
To confirm the effect of the proteasome on transcription repression, we further evaluated the upstream valosin-containing protein (VCP) ATPase, an enzyme that translocates poly-ubiquitinated proteins to the proteasome for further degradation. VCP has been reported to regulate genome-wide degradation of promoter-bound RNAPII after UV exposure.45-47 Here, we used VCP inhibitor (VCPi) CB5083,50 which was effective and did not influence normal transcription kinetics (Figures S4C-S4E) or perturb DSB creation by vfCRISPR (Figure S4G). We applied VCPi to the live-cell reporter (Figure 1A). Similar to MG132, transcription repression was slowed down and 53BP1 recruitment was compromised on the damaged gene upon VCP inhibition (Figure S4H). Due to the lack of 53BP1 recruitment to indicate damaged cells, all stimulated cells were analyzed here. Quantitative analysis showed that fewer cells were repressed at 30 min upon drug treatment (Figure S4I), which was due to the slower decay instead of later response (Figure S4J). These results further confirmed that rapid transcription repression is regulated by proteasome-mediated RNAPII removal.
Cohesin regulates repression propagation along the chromosome but does not contribute to transcription repression of the damaged gene
Cohesin is an essential factor for the establishment and maintenance of the 3D genomic structure. Recently, its role in DNA damage has been studied intensively. Cohesin was required for transcription repression of a gene array damaged by tethered nuclease FokI.51 It was also proposed that cohesin is anchored at the DSB site and loops distal chromatin to spread phosphorylation signals.29 Therefore, we investigated how cohesin contributes to rapid transcription repression around a single DSB.
We applied siRNA targeting SCC1, an essential component in the mammalian cohesin complex, to knock down its activity. Efficient SCC1 protein depletion was confirmed with western blot assay (Figure S5A). We induced a DSB and performed transcription imaging in the U-2 OS reporter cells as before (Figure 1A). SCC1 knockdown did not influence DSB creation by vfCRISPR (Figure S5B); however, it compromised 53BP1 recruitment to the DSB site (Figure S5C), consistent with a previous report using irradiation to induce DNA damage.52 Depletion of SCC1 led to a small decrease in the percentage of repressed cells while not significantly perturbing the decay and response times (Figures S5D-S5F). Besides, among damaged cells with 53BP1 recruitment, transcription repression was similar between SCC1 knockdown and non-target control samples (Figure 5A). The percentages of repressed cells, as well as transcription decay and response times, were not significantly different between the knockdown and control samples (Figures 5B and 5C). We further tested the function of cohesin in transcription repression at the damaged endogenous ACTB locus. In agreement with live-cell imaging, the damaged ACTB gene was still repressed after SCC1 knockdown (Figure 5D). Together, these results indicated that cohesin has a minor effect on the transcription regulation of the damaged gene. However, the RNAPII occupancy on the neighboring gene FSCN1 was restored upon SCC1 depletion (Figure 5D). Therefore, we concluded that cohesin may contribute more to repression propagation along the chromosome while exhibiting a modest effect on the damaged gene.
Figure 5. Cohesin contributes to transcription repression propagation but does not regulate the repression of the damaged gene.

Cells were treated with siRNAs to knock down SCC1 of cohesin. The DSB was induced by vfCRISPR in a live-cell transcription reporter (Figure 1A) stably integrated in U-2 OS cells (A–C) or endogenous ACTB in HEK293T cells (D).
(A) Heatmaps of TS and 53BP1 intensities were measured from live-cell imaging as in Figure 1 (siSCCI, n = 74 cells; siNeg, n = 98 cells). Cells with 53BP1 recruitment were selected.
(B) Percentages of repressed cells were measured for knockdown samples and control samples during 30 min tracking. Cells were considered repressed if the averaged TS intensities in the last 4 frames were lower than a threshold determined from siNeg control cells with 53BP1 recruitment. Error bar: SEM (n = 3 biological replicates).
(C) Transcription decay and response times were obtained by fitting the delayed decay model (Figure 1G) to the repressed TS intensities in (B) (siSCCI, n = 49 cells; siNeg, n = 73 cells). Error bar: quartiles. Unpaired Mann-Whitney test was performed.
(D) ChIP-qPCR experiments of RNAPII were performed in HEK293T cells when ACTB locus was damaged by vfCRISPR. Change of RNAPII occupancy (measured in RPM) upon light stimulation on ACTB locus and nearby genes were measured in siSCCI and siNeg samples. GAPDH on a different chromosome served as a control gene to indicate global transcriptome change upon UV exposure. FSCN1: 70 kb from DSB. PMS2: 487 kb from DSB. Error bar: SEM (n = 2 biological replicates).
(E) Working model: DSB induces rapid transcription repression, regulated by proteasome-mediated RNAPII removal on the damaged and surrounding genes.
DISCUSSION
Transcription repression is an important step to prepare the damaged genomic locus for proper repair. However, the kinetics of repression and its molecular mechanism have not been thoroughly investigated in real time. In this study, we applied the recently developed vfCRISPR to induce a single DSB at a specific locus with precise timing. We applied single-molecule transcription imaging in live cells and time-resolved ChIP to measure the transcriptional kinetics after vfCRISPR-induced DSB. We found that transcription was rapidly repressed on the damaged gene. Transcription started to decline in around 2 min, and the decay of TS intensity was described by a simple exponential function with a median decay time of 3 min (Figures 1E-1H). The transcription repression coincided with repair protein 53BP1 recruitment but was not completely coupled with it since inhibition of the 53BP1 signaling pathway did not compromise the repression (Figures 3E-3G). Additionally, transcription was not only repressed on damaged genes but also propagated bi-directionally along the chromosome for hundreds of kilobases in an attenuated manner (Figure 2). With genetic and pharmacological perturbation, we demonstrated that the rapid transcription repression was regulated by proteasome-mediated RNAPII removal instead of PRC1-mediated H2A K119 ubiquitination (Figures 3A-3D and 4). In addition, the propagation of repression may depend on RNF8/RNF168-mediated repair signaling and cohesin-mediated chromatin reorganization (Figures 3H and 5).
As RNAP moves at a speed of kilobases per minute, it is advantageous for cells to rapidly repress polymerase to avoid its collision with repair proteins and interference with the repairing process. However, rapid kinetics have not been characterized previously due to a lack of precise temporal induction of a DSB at a specific site. Other techniques, such as translocating endonucleases into nuclei, have been used. However, we have shown previously that it may take tens of minutes for Cas9 to find its target.31 It is plausible that a similar amount of time is needed for the endonuclease to find the target. In this study, we have shown that it only takes a few minutes for cells to repress transcription. Therefore, it is crucial to generate DSBs rapidly and synchronously to observe the minute timescale repression kinetics, which would not be obscured by tens of minutes timescale for DSB induction.
We demonstrated that the transcription propagates bidirectionally from a DSB with a speed of 4.5 kb/min, and it depends on RNF8/RNF168-mediated repair signaling and cohesin-mediated chromatin reorganization. Previously, it was proposed that cohesin dwells at the DSB and propagates the γH2AX signal through loop extrusion.29 The γH2AX signal spread along the chromosome rapidly, at a speed measured to be from 36 to 150 kb/min.29,31 This is much faster than the transcription repression propagation, suggesting that additional molecular processes are involved for transcription repression. Ubiquitination and subsequent proteasome-mediated protein degradation are the plausible rate-limiting steps that slow down the kinetics.
The post-translational modifications of histone, especially ubiquitination, play important roles in regulating transcription as well as the DDR. H2AK119ub was reported to be required for damage-induced transcription repression. However, we found that this PRC1-mediated histone modification was dispensable in this process (Figures 3A-3D). This inconsistency could be due to the following reasons. First, there could be differences in DNA damage induction methods. The recruitment of H2AK119ub was mostly reported at clustered endonuclease-cutting sites or with UV radiation methods,20,21,40-42 where multiple DSBs or a variety of damages were created. It remains unclear whether H2AK119ub was enriched at a single DSB site.30 One possibility is that damage at multiple sites may be required for large-scale chromatin change to allow chromatin remodeler factor BAF180 and BRG1, or transcription elongation factor ENL, to recruit PRC1 to the damage sites.20,21 Second, the time to measure transcriptional kinetics after DNA damage may also contribute to this inconsistency. Traditional methods like inducible endonuclease cleavage require 30 min to hours of drug treatment for enzyme translocation, leading to unsynchronized cleavage and delayed detection of transcription dynamics.18,20,21,42 The delayed measurement may miss the early transcriptional kinetics but capture long-term effects. Taken together, H2AK119ub may not play an important role in early transcription repression but may contribute to the maintenance of a transcription inactive state that facilitates damage repair.
The position of DNA damage with respect to a gene may influence its transcription repression. Two mechanisms have been proposed depending on whether the DSB occurs within the gene body or outside: ubiquitin/VCP/proteasome-mediated protein degradation and ATM/PARP-mediated chromatin remodeling.24 We found that different mechanisms are used to repress the damaged locus and neighboring genes. VCP/proteasome rapidly removes RNAPII on the damaged gene (Figures 4 and S4F-S4H). Repression of the neighboring genes requires proteasomal activity too. But, in addition, they need repair-signal propagation such as cohesin-mediated chromatin looping and RNF8/RNF168-mediated ubiquitination (Figures 3H and 5D). We estimated the propagation speed to be about 4.5 kb/min, and the response amplitude is attenuated. As a result, the response to repress the damaged locus is almost immediate (2 min) but that to repress the neighboring genes is delayed. The final effective signal should be K48-linked poly-ubiquitination, which leads to proteasomal removal of RNAPII. It would be interesting to unravel how these signaling pathways are orchestrated and interplay with each other in the future.
Limitations of the study
Although we showed that the proteasome was required for removing RNAPII after Cas9 cleavage, indicating that the signal propagating along chromatin could be K48 poly-ubiquitination that labeled protein for degradation, the potential upstream E3 ubiquitin ligase for this modification is still not clear. Previous research suggested that DNAPK signaled the E3 ligase WWP2 to poly-ubiquitinate RNAPII after endonuclease I-PpoI cleavage,24 but it was also reported that WWP2 did not poly-ubiquitinate RNAPII after UV radiation.53 Instead, NEDD4 and the Elongin-Cullin complex played a significant role in UV-irradiation-induced RNAPII proteolysis.53-55 NEDD4, one homolog to Rsp5 in Saccharomyces cerevisiae, serves as the E3 ubiquitin ligase that directly targets elongating RNAPII.53 The Elongin complex assembles with Cul5/Rbx modules and efficiently ubiquitinates Ser5 phosphorylated Rpb1 for proteolysis after UV irradiation.54,55 These two ubiquitin ligases acted sequentially to add a K48-linked poly-ubiquitin chain to the Rpb1 subunit of RNAPII for degradation.56 Additionally, the E3 ubiquitin ligases CSA and CSB also facilitated the formation of K48- and K63-linked ubiquitin chains on the Rpb1 K1268 residue in transcription-coupled nucleotide excision repair.57 These findings were based on UV irradiation that produced bulky DNA lesions. Whether these E3 ubiquitin ligases contribute to repression after single DSB cleavage requires further investigation.
In this study, we used CRISPR-Cas9 to generate a DSB. There is concern about whether Cas9 binding influences DSB detection and subsequent damage repair. SpCas9 is considered a single-turnover enzyme in vitro. Upon DNA cleavage, SpCas9 remains bound to the DNA target for over 5 h, resulting in extremely slow product release and ultimately inhibiting enzymatic turnover.58,59 In eukaryotic cells, the Cas9-generated DSB is rapidly exposed, possibly facilitated by transcription or chromatin remodeling activity,60,61 but there could still be subtle differences between Cas9-generated DSBs and other methods. The generality of our conclusion should be validated in other systems.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
All material requests should be directed to Bin Wu (bwu20@jhmi.edu).
Materials availability
Reagents and materials produced in this study are available from Bin Wu pending a completed Materials Transfer Agreement.
Data and code availability
The accession number for the ChIP-seq data reported in this paper is NCBI: PRJNA1005701. The Original imaging and Western blot data have been deposited to Mendeley Data: 10.17632/c4t3sss9r7.1].
This paper does not report the original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell culture and treatment
Human embryonic kidney 293 cell line (HEK293T) and human U-2 osteosarcoma cell line (U-2 OS) were cultured at 37°C with 5% CO2 in Dulbecco’s Modified Eagle’s Medium (DMEM, Corning), supplemented with 10% FBS (Corning), 100 units/mL penicillin and 100 μg/mL streptomycin (DMEM complete). For drug inhibition, cells were treated with 20 μM MG132 (Sigma-Aldrich), or 100 μM VCP inhibitor CB5083 (MedChem) for 1 h before imaging or ChIP. siRNA treatment was denoted in “RNA interference” section.
Construction of stable U-2 OS cell line
The reporter U-2 OS cell line stably integrated with transcription reporter was a generous gift from Sergio F. de Almeida lab. To make lentivirus, stdMCP-stdGFP32 and mCherry-53BP1 (Addgene #19835) in the lentiviral vector were transfected into HEK293T cells with viral packaging components. After 48 h, the supernatant was collected, centrifuged to remove cell debris (1000 rpm, 5 min), and filtered through 0.45 μm PVDF membrane (Millipore Sigma, SLHV013SL). The supernatant was aliquoted and stored in a −80°C freezer. To infect cells, the aliquots were thawed and diluted with 1 mL serum-free DMEM media before adding to cells. After 16 h, the DMEM-lentivirus mixture was replaced with DMEM complete. U-2 OS cells with the integrated transcription reporter were infected with RNA coat proteins and 53BP1 together before being sorted for median GFP and mCherry-expressing cells.
METHOD DETAILS
Cas9 protein purification
The protocol for Cas9 protein purification was adapted from Y. Liu and R. Zou.31 Briefly, BL21-CodonPlus (DE3)-RIL competent cells (Agilent Technologies 230245) were transformed with Cas9-His tag plasmid (Addgene #67881). One single colony was picked and inoculated in 5 mL of LB media supplemented with ampicillin. The bacteria culture grew overnight (37°C, 220 rpm) before being transferred to 1 L of LB media supplemented with ampicillin and 0.1% glucose, in which the culture grew until OD600 of ~0.5. Then, 0.2 mM IPTG was applied to induce protein expression. The culture was incubated at 18°C overnight. Afterward, bacteria were harvested by centrifugation at 4500g, 4°C for 15 min. The cell pellet was resuspended in 20 mL lysis buffer (20 mM Tris pH 8.0, 250 mM KCl, 20 mM imidazole, 10% glycerol, 1 mM TCEP, 1 mM PMSF, and cOmplete EDTA-free protease inhibitor tablet (Sigma-Aldrich 11836170001)). The cell suspension was then lysed with a microfluidizer. The cell debris was removed by centrifugation at 16,000 g, 4°C for 40 min. The supernatant containing Cas9 protein was collected and filtered with 0.2 μm syringe filters (Thermo Scientific F25006). Ni-NTA agarose beads (Qiagen 30210) were equilibrated with 5 column volumes of lysis buffer at 4°C before the supernatant with Cas9 was loaded. The protein-bound Ni-NTA beads were washed with 15 column volumes wash buffer (20 mM Tris pH 8.0, 800 mM KCl, 20 mM imidazole, 10% glycerol, and 1 mM TCEP). Cas9 protein was then eluted with gradient elution buffers (20 mM HEPES pH 8.0, 500 mM KCl, 10% glycerol, and varying concentrations of imidazole (100, 150, 200, and 250 mM)). The eluate was tested on an SDS-PAGE gel and imaged by Coomassie blue (Bio-Rad 1610400) staining. To avoid potential DNA contamination, 1 mL Q Sepharose column (GE Healthcare 17051005) was charged with 1M KCl and equilibrated with the previous elution buffer with 250 mM imidazole. The protein eluate from Ni-NTA beads was passed over the Q column at 4°C. The flow-through was dialyzed in a 10 kDa SnakeSkin dialysis tubing (Thermo Fisher Scientific 68100) immersed in 2 L dialysis buffer (20 mM HEPES pH 7.5, and 500 mM KCl, 20% glycerol) at 4°C overnight. Next day, the protein in the tubing was dialyzed for another 3 h in fresh buffer. The final Cas9 protein was concentrated to a final concentration of 10 μg/μL with Amicon Ultra 10 kDa centrifugal filter unit (Millipore UFC801024) before being aliquoted, flash-frozen and stored at −80°C.
Electroporation of Cas9 RNP into human cells
This protocol was modified from Y. Liu and R. Zou.31 To anneal cgRNA and tracrRNA, equal volumes of 100 μM cgRNA (Bio-Synthesis Inc) and tracrRNA (Integrated DNA Technologies) were mixed and heated to 95°C for 3 min in a thermocycler. The mixture was allowed to slowly cool down at room temperature for >5 min. For live cell imaging, cgRNA/Cas9 RNP complex was formed by mixing 17 μg of purified Cas9 with 125 pmol cgRNA:tracrRNA at a ratio of 1:1.2. The mixture was then incubated for an additional 30 min at room temperature. U-2 OS cells were harvested at a confluency of ~80% prior to electroporation. 800 thousand cells were centrifuged in DMEM and DPBS (Millipore Sigma, D8537-500ML) sequentially (each 5 min, 1000 rpm). 20 μL of nucleofection solution (Lonza) was used to suspend the cell pellet and mixed thoroughly. The pre-incubated RNP solution and 1 μL of Cas9 Electroporation Enhancer (Integrated DNA Technologies) were added to the cell-nucleofection solution mixture. For ChIP experiments, RNP was formed by mixing 45 μg of Cas9 with 300 pmol annealed cgRNA:tracrRNA. 18 million HEK293T cells for 2 samples were prepared and re-suspended in 90 μL of nucleofection solution (Lonza). The prepared RNP and 2 μL of Electroporation Enhancer were added to the cells. Electroporation was performed according to the manufacturer’s instructions on the 4D-NucleofectorTM Core Unit (Lonza). SF Cell Line 4D-Nucleofector X Kit L with code CA189 was used for HEK293T cells in ChIP. SE Cell Line 4D-Nucleofector X Kit S with code DN100 was used for U-2 OS cells in live-cell imaging. DMEM complete was added to the electroporated cells before seeding in culture dishes.
Sequences of cgRNA and tracrRNA were listed in Table S1.
Fluorescence imaging of transcription in living cells
The reporter U-2 OS cells were seeded into an imaging chamber (Cellvis, D35-20-1.5-N) after electroporation with Cas9 RNP and incubated for 20–22 h before being loaded onto the microscope. To activate cgRNA/Cas9, we scanned a 5 × 5 μm region of interest surrounding the transcription site with a focused 405 nm laser beam. We used a Nikon Ti-E fluorescence microscope equipped with two Andor EMCCDs for simultaneous tracking of transcription sites (stdMCP-stdGFP) and 53BP1 foci (mCherry-53BP1) with 488 nm and 561 nm excitation lasers respectively. The time-lapse movies were acquired for 30 min at an interval of 1 min. For FRAP, the transcription site of a cell was first imaged before bleached with a focused 488 nm laser beam in a 2 μm-diameter circle. After bleaching, a time-lapse video of the cell was acquired immediately with a 30-s interval for 15 min. All live-cell imaging experiments were performed with ± 3.5 μm z stack in 15 steps. Cells were incubated at 37°C with 5% CO2 in FluoroBrite DMEM media (Gibco) supplemented with 10% FBS and 1% Penicillin/Streptomycin.
Chromatin-immunoprecipitation (4 samples)
Electroporation was performed as described in the “Electroporation of Cas9 RNP” section. Afterward, 18 million cells were split into 2 samples and seeded in 6-well plates. After 20–22 h, cells were stimulated with a 365 nm flashlight and harvested at certain time points after light exposure.
The ChIP protocol was adapted from Y. Liu and R. Zou.31 Briefly, cells were washed once with warmed DPBS, then scrapped off the plate in 10 mL serum-free DMEM and transferred to 15 mL tubes. 721 μL of 16% methanol-free formaldehyde (Pierce 28908) in 1xPBS was added and the tubes were rotated for 15 min at room temperature on a rotator (Thermo Scientific, 11-676-341). 750 μL of 2 M glycine in 1xPBS was added to quench the formaldehyde. Cells were spun down at 2000 g and 4°C for 3 min and washed twice with ice-cold DPBS using the same centrifugation condition. Cells were then resuspended in 4 mL lysis buffer LB1 (50 mM HEPES, 140 mM NaCl, EDTA, 10% glycerol, 0.5% Igepal CA-630, 0.25% Triton X-100, pH to 7.5 using KOH, add 1x protease inhibitor right before use) and incubated for 10 min at 4°C on a rotator, then spun down with the same condition. The supernatant was discarded. The pellets were then resuspended in 4 mL LB2 (10 mM Tris-HCl pH 8, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, pH adjusted to 8.0 using HCl, and adding 1x protease inhibitor right before use) for 5 min at 4°C on a rotator, spun down with the same protocol. The pellets were then resuspended in 1.5 mL LB3 (10 mM Tris-HCl pH 8, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.1% Na-Deoxycholate, 0.5% N-lauroylsarcosine, pH adjusted to 8.0 using HCl, adding 1x protease inhibitor right before use) and transferred to 2 mL DNA low-bind Eppendorf tubes (Eppendorf, 022-43-104-8) for sonication with 50% amplitude, 30s ON, 30s OFF for 12 min total time (Qsonica, Q125-110). Samples were spun down with 20000 g at 4°C for 10 min, and the supernatant was carefully transferred to 1.5 mL LB3 + 300 μL of 10% Triton X-100 in a 5 mL DNA low-bind tube (Eppendorf, 0030108310).
Beads pre-loaded with antibodies were prepared before cell harvesting as follows. 50 μL Protein A beads (Thermo Fisher) were used per IP. 200 μL beads (for 4 IPs) were transferred to a 2 mL DNA low-bind Eppendorf tube on a magnetic stand. Beads were washed twice with blocking buffer (0.5% BSA in 1xPBS), then resuspended in 400 μL blocking buffer (100 μL per IP). 14 μL of antibodies (3.5μL per IP for RNAP2, Abcam 5095) were added to the beads and placed on a rotator for 2–3 h. Right before IP, the beads in the 2 mL tube were washed 3 times with the blocking buffer on a magnetic rack, and then resuspended in 200 μL blocking buffer (50 μL per IP). 50μL of beads-antibody and 3 mL of sample were combined and placed on a rotator for thorough mixing at 4°C overnight (25 rpm).
After overnight incubation, samples were transferred to 2 mL DNA low-bind tubes on a magnetic stand, washed 6 times with 1 mL RIPA buffer (50 mM HEPES, 500 mM LiCl, 1 mM EDTA, 1% Igepal CA-630, 0.7% Na-Deoxycholate, pH adjusted to 7.5 using KOH), then washed once with 1 mL TBE buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl), Afterward, beads with bound DNA were mixed with 50 μL ChIP elution buffer (50 mM Tris-HCl, 10 mM EDTA, 1% SDS, pH 8.0) and incubated at 65°C overnight. After incubation, 30 μL of TE buffer (IDT, pH = 8.0) was added followed by 2 μL of 20 mg/mL RNaseA (New England BioLabs) for 15 min at 37°C and 4 μL of 800 unit/ml Proteinase K (New England BioLabs) for 30 min at 55°C. The genomic DNA was column purified and eluted in 41 μL EB buffer supplemented with the kit (Qiagen, 28204) according to the manufacturer’s protocol.
Oligo sequences for library construction are in Table S3.
RNA interference
Three small interference RNAs (siRNAs) were designed online (IDT) per targeting gene. Each siRNA was annealed in duplex buffer (IDT) for 3 min at 95°C. The three siRNAs were combined and delivered into cells through two transfections. Briefly, for live-cell imaging, 900 thousand U-2 OS cells were seeded in a 6-well plate, incubated overnight, then transfected with 40 pmol siRNA mixture in Lipofectamine RNAiMAX Reagent (ThermoFisher) according to the manufacturer’s protocol. The transfected cells were incubated for 22–24 h and harvested for counting. 800 thousand of them were electroporated with 20 pmol siRNA mixture together with Cas9 RNP as described in the “Electroporation of Cas9 RNP” section. For ChIP experiments, 22–24 million HEK293T cells were electroporated with 200 pmol siRNA mixture. On the second day, 18 million treated cells were electroporated again with 200 pmol siRNA mixture together with Cas9 RNP as described in the “Electroporation of Cas9 RNP” section.
Transient transfection of caged guide RNA
Transfection protocol to deliver cgRNA:tracr RNA was adapted from Y. Liu and R. Zou.31 Briefly, 60–80 thousand U-2 OS cells stably expressing Cas9-EGFP were seeded on 12 mm coverslips (Electron Microscopy, 72290-03). To target a single cleavage site, we transfected each coverslip with a mixture of 2.5 pmol of Ch3Rep1 guide (truncated 11mer) targeting the repetitive region and ~5 pmol of cgRNA targeting PPP1R2 gene with 1.5 μL Lipofectamine RNAiMAX Transfection Reagent. After 24-h incubation, Cas9-EGFP foci were imaged with epi-fluorescence microscopy.
Immunofluorescence microscopy
U-2 OS cells stably expressing Cas9-EGFP cells were seeded on a 12 mm coverslip and transfected with guide RNAs for 22–24 h. After stimulation with 365 nm flashlight, cells were fixed with 4% of paraformaldehyde (Electron Microscopy, 15713) in 1x PBS for 10 min at room temperature and then quenched by 1x PBS supplemented with 0.1 M glycine for 10 min at room temperature. After washing with 1x PBS, 0.5% Triton X- in PBS was applied to permeabilize the cell membrane for 10 min at room temperature. 2% w/v protease/nuclease-free BSA (Sigma-Aldrich A7030-100G) in 1x PBS was used to block the sample for 1 h and at room temperature. The primary antibody was diluted in 1x PBS and directly added into the chamber for 1 h at room temperature. Afterward, the primary antibody was removed, and the sample was washed with 1x PBS three times. The secondary antibody was diluted in 1x PBS as 1:1000 and applied to the sample for 45 min at room temperature. Finally, the sample was rinsed three times with 1x PBS and mounted with Prolong Diamond mounting media (Thermo Fisher Scientific) overnight.
Alexa 647 conjugated Goat anti-rabbit IgG (H + L) antibody (A21245, A21039) was purchased from Thermo Fisher. Anti-RNF2 (HPA026803) and anti-FK2 (04–263) antibodies were purchased from Sigma-Aldrich (Cambridge, MA). Anti-53BP1 (NB100-304) was purchased from Novus Biologicals (Centennial, CO). Dilution of the primary antibody was based on the recommended ratio from the manufacturers.
Immunoblotting
U2-OS cells were gently washed with PBS and lysed in ice-cold RIPA buffer containing 50 mM Tris (pH 7.5), 150 mM NaCl, 0.1% w/v SDS, 1% v/v NP40, 100 mM NaF, 17.5 mM b-glycerophosphate, 0.5% v/v Sodium deoxycholate, 10% v/v glycerol, 2 mM Na3VO4, supplemented with fresh 1 mM PMSF, and Protease Inhibitor Cocktail (0.5% v/v, Millipore Sigma, P8340). After sonication on ice, the samples were centrifuged at 12,000 g for 10 min at 4°C, and supernatant were collected and suspended in 1x SDS loading buffer. Samples were further incubated on a thermal mixer at 95°C, 600 rpm for 10 min. Following SDS-PAGE and western blotting, membranes were incubated with primary antibodies at 4°C overnight, including anti-actin (Santa Cruz, sc-47778), anti-RNF2 (Proteintech, 16031-1-AP), anti-RNF168 (EMD Millipore, ABE367), anti-Scc1 (Abcam, ab217678). After thorough washes with TBST, the membranes were either incubated with a fluorescence secondary antibody at room temperature for 2 h (anti-actin) or with an HRP-linked antibody at 4°C overnight (anti-RNF2, anti-RNF168, anti-Scc1). Western blot images were either captured by an Odyssey scanner (LI-COR) for fluorescence imaging or by an GeneGnome XRQ scanner (SYNGENE) for chemiluminescence imaging. Protein expression levels were normalized with actin protein level, and the images were analyzed with ImageJ (version 2.3.0).
QUANTIFICATION AND STATISTICAL ANALYSIS
Image analysis of transcriptional kinetics
For each cell, time-lapse z stack images were maximum intensity projected. Image shift between 488 nm and 561 nm channels due to chromatic aberration was corrected through TetraSpeck Fluorescent Microspheres as previously described70. To account for cell movement during tracking, we first extracted coordinates of transcription sites manually using the FIJI plugin PointPicker, and then cropped around these coordinates with 3x3 μm. These cropped images were then loaded into ‘u-track’, a single-particle tracking program developed by Danuser lab64 (https://www.utsouthwestern.edu/labs/danuser/software/), to identify transcription sites and extract their coordinates. Coordinates of the previous frame would be used if no spot was found in the current frame. The intensity trace of this transcription site was then estimated with a custom MATLAB script based on the Gaussian Mask algorithm71 . Intensities within a trace were normalized by the maximum value to calibrate cell-to-cell heterogeneity. The normalized transcription intensity traces for all cells were summarized into a heatmap together with that of 53BP1.
We then identified repressed transcription traces by looking at if the mean of normalized intensities in the last 4 frames within a trace, which we termed final intensity, was below the repression threshold. For the control dataset in each experiment, cells were categorized into cut or un-cut groups by 53BP1 recruitment. Final intensities were calculated for all cells in the control dataset. The repression threshold was determined by maximizing the percentage of repressed cells in the cut group while minimizing this value in the un-cut group. After determining the repression threshold, the percentage of repressed cells was calculated for the drug or siRNA-treated dataset and compared with that of the control dataset.
To further extract the transcription decay and response times, we fit TS intensity traces with a delayed exponential decay model in a custom MATLAB script. Traces with R Square larger than 0.75 were used to extract the decay and response times and shown as scatterplots in the main context. Traces with R2 less than 0.75 are typically unrepressed cells.
qPCR assay for measuring double-strand break percentage
To measure the DSB level, two amplicons were designed for each locus of interest. One amplicon (target1) includes the Cas9 cleavage site; the other (target2) is a nearby sequence that is not cleaved. 50 ng of purified genomic DNA was added to a 10 μL reaction with a final primer concentration of 250 nM in LunaR qPCR Master Mix. Genomic DNA from wild-type HEK293T cells was used as blank control to calibrate the differences in amplification efficiency among loci. PCR amplification was performed with the following conditions: 95°C for 1min, 40 cycles of {95°C for 15sec, 60°C for 30sec with plate read}, 60°C–95°C for melting curve. DSB percentage for each sample was calculated as .
Primer sequences for qPCR were listed in Table S2.
High-throughput sequencing and data processing for ChIP samples
The protocol for sequencing library construction was adapted from Y. Liu and R. Zou 2020. Briefly, end-repair/A-tailing was performed on 16.67 μL of genomic DNA from ChIP using NEB Next Ultra II End Repair/dA-Tailing Module (New England BioLabs), followed by ligation of annealed adaptor MNase_F/MNase_R with T4 DNA Ligase (New England BioLabs). 13 cycles of PCR using PE_i5 and PE_i7XX primer pairs were performed to amplify libraries. Samples were pooled and quantified with Qubit Fluorometer (Thermo Fisher) and tested by qPCR (Bio-Rad, CFX96), then sequenced on a NextSeq 500 (Illumina) using high-output paired 2x36 bp reads.
The pipeline for NGS data analysis was modified from Y. Liu and R. Zou.31 Briefly, reads from a fastq file were demultiplexed after sequencing using bcl2fastq. Paired-end reads were aligned to hg38 with bowtie2. Samtools was used to filter for mapping quality ≥25, remove singleton reads, convert to BAM format, remove potential PCR duplicates, index reads, count the number of mapped reads, and subset to generate 3 technical replicates with 10 million reads each. The steps above were performed with a custom Shell script.
Gene coordinates were obtained from refGene.txt.gz downloaded from the UCSC database (https://hgdownload.cse.ucsc.edu/goldenpath/hg38/database/). For a certain gene, all reads falling in the annotated range were counted and normalized to gene length with a custom Python script, defined as RNAP2 occupancy. Results for all genes were exported in a spreadsheet and further analyzed in MATLAB.
We estimated the change of RNAP2 occupancy after Cas9 cleavage for individual genes. First, to calibrate the heterogeneity between light-stimulated and no-light samples, we calculated RNAP2 occupancy for all individual genes that are not on chromosome 7, where the target gene ACTB locates. We assumed these genes would not be influenced by light-induced Cas9 cleavage. RNAP2 occupancy for all these genes from 2 samples was fitted with a linear model and the slope was used as the calibration factor between these 2 samples (Figure S2C). Then, for each gene, the transcription repression index (TRI) was calculated as RNAP2 occupancy in the light-stimulated sample over that in no-light control, further divided by the cross-sample calibration factor. The scripts for the described ChIP analysis are available.
Oligo sequences for library preparation are in Table S3. ChIP-qPCR primers are in Table S2.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-RNA polymerase II CTD repeat YSPTSPS (Phospho S2) | Abcam | ab5095; RRID:AB_304749 |
| Anti-RNF2 | Sigma-Aldrich | HPA026803; RRID:AB_10602086 |
| Anti-RNF2 | Proteintech | 16031-1-AP; RRID:AB_2180459 |
| Anti-FK2 | Sigma-Aldrich | 04-263 |
| Anti-53BP1 | Novus Biologicals | NB100-304; RRID:AB_10003037 |
| Anti-53BP1 | Novus Biologicals | NB100-304; RRID:AB_10003037 |
| Anti-RNF168 | EMD Millipore | ABE367; RRID:AB_11205761 |
| Anti-SCC1 | Abcam | ab217678; RRID:AB_2920658 |
| Anti-ACTB | Santa Cruz | sc-47778; RRID:AB_626632 |
| Bacterial and virus strains | ||
| BL21-CodonPlus (DE3)-RIL competent cells | Agilent | 230245 |
| NEB Stable Competent E.coli | New England Biolabs | C3040I |
| Chemicals, peptides, and recombinant proteins | ||
| SpCas9 | This study | N/A |
| MG132 | Sigma-Aldrich | M7449 |
| CB5083 (VCPi) | MedChem | HY-12861 |
| Dulbecco’s Modified Eagle Medium | Corning | 10-017-CV |
| Fetal Bovine Serum | Millipore Sigma | F4135-500ML |
| FluoroBrite DMEM | Gibco | A1896701 |
| PBS | Corning | 21-031-CV |
| BSA | VWR | VWRV0332-25G |
| ProLong Diamond antifade reagent with DAPI | Invitrogen | P36962 |
| Rat tail collagen I | Gibco | A1048301 |
| Fibronectin bovine plasma solution | Sigma-Aldrich | F1141-2MG |
| Electroporation Enhancer | Integrated DNA Technologies | N/A |
| Luna Universal qPCR Master Mix | New England Biolabs | M3003X |
| Deposited data | ||
| Imaging and Western Blot data | This paper | Mendeley: 10.17632/c4t3sss9r7.1 |
| NGS raw data | This paper | PRJNA1005701 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | RRID: CVCL_0063 |
| U-2 OS cells with Cas9-EGFP | Yang et al.31 | N/A |
| U-2 OS cells with EX2 reporter | Vitor et al.33 | N/A |
| Oligonucleotides | ||
| crRNA and tracrRNA sequences, see Table S1 | GenScript | N/A |
| Primers for qPCR, see Table S2 | Integrated DNA Technologies | N/A |
| Oligos for ChIP-seq library, see Table S3 | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| Plasmid: stdMCP-stdGFP | Wu et al.32 | N/A |
| Plasmid: mCherry-BP1-2 pLPC-Puro | Dimitrova et al.62 | Addgene #19835 |
| Plasmid: pHO4d-Cas9 | Hua Fu et al.63 | Addgene #67881 |
| Software and algorithms | ||
| u-track Tracking Algorithm | Jaqaman et al.64 | N/A |
| ImageJ | Schneider et al.65 | https://imagej.nih.gov/ij/ |
| Imaging analysis codes | This paper | N/A |
| MATLAB | MathWorks | N/A |
| Visual Studio Code | Microsoft | N/A |
| Integrative Genomics Viewer (IGV) | Robinson et al.66 | N/A |
| stdshade function | Simon Musall (2024) | Simon Musall (2024). stdshade (https://www.mathworks.com/matlabcentral/fileexchange/29534-stdshade), MATLAB Central File Exchange. |
| SAMtools | Li et al.67 | http://samtools.sourceforge.net/ |
| bowtie2 | Langmead et al.68 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Adapted ChIP-seq analysis codes | Zou et al.69 | https://github.com/rogerzou/chipseq_pcRNA |
| ChIP-seq analysis codes | This paper | N/A |
Highlights.
Single double-strand break causes transcription repression of broken genes in a few minutes
Transcription repression propagates along the chromosome for hundreds of kilobases
Rapid transcription repression is not regulated by PRC1-mediated H2A K119 ubiquitination
Proteasome-mediated RNAPII removal contributes to transcription repression and propagation
ACKNOWLEDGMENTS
This study is supported by the National Institutes of Health (R01GM136897) and the Pew Charitable Trust (00030601) to B.W. and HHMI to T.H. S.H. and Z.H. are supported by NIH training grant T32 GM007445. We thank Dr. Mihoko Kai for requesting the U-2 OS reporter cell lines from Dr. Sérgio F. de Almeida.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114420.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Jackson SP (2002). Sensing and repairing DNA double-strand breaks. Carcinogenesis 23, 687–696. 10.1093/carcin/23.5.687. [DOI] [PubMed] [Google Scholar]
- 2.Chapman JR, Taylor MRG, and Boulton SJ (2012). Playing the End Game: DNA Double-Strand Break Repair Pathway Choice. Mol. Cell 47, 497–510. 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- 3.Mehta A, and Haber JE (2014). Sources of DNA Double-Strand Breaks and Models of Recombinational DNA Repair. Cold Spring Harbor Perspect. Biol 6, a016428. 10.1101/cshperspect.a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cannan WJ, and Pederson DS (2016). Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin: DOUBLE-STRAND DNA BREAK FORMATION IN CHROMATIN. J. Cell. Physiol 231, 3–14. 10.1002/jcp.25048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helleday T, Petermann E, Lundin C, Hodgson B, and Sharma RA (2008). DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 8, 193–204. 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 6.O’Driscoll M. (2012). Diseases associated with defective responses to DNA damage. Cold Spring Harbor Perspect. Biol 4, a012773. 10.1101/cshperspect.a012773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li L-Y, Guan Y.d., Chen X-S, Yang J-M, and Cheng Y (2020). DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol 11, 629266. 10.3389/fphar.2020.629266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lieber MR (2010). The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem 79, 181–211. 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maréchal A, and Zou L (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harbor Perspect. Biol 5, a012716. 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ray Chaudhuri A, and Nussenzweig A (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol 18, 610–621. 10.1038/nrm.2017.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Price BD, and D’Andrea AD (2013). Chromatin remodeling at DNA double-strand breaks. Cell 152, 1344–1354. 10.1016/j.cell.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uckelmann M, and Sixma TK (2017). Histone ubiquitination in the DNA damage response. DNA Repair 56, 92–101. 10.1016/j.dnarep.2017.06.011. [DOI] [PubMed] [Google Scholar]
- 13.Lesage E, Clouaire T, and Legube G (2021). Repair of DNA double-strand breaks in RNAPI- and RNAPII-transcribed loci. DNA Repair 104, 103139. 10.1016/j.dnarep.2021.103139. [DOI] [PubMed] [Google Scholar]
- 14.Puget N, Miller KM, and Legube G (2019). Non-canonical DNA/RNA structures during Transcription-Coupled Double-Strand Break Repair: Roadblocks or Bona fide repair intermediates? DNA Repair 81, 102661. 10.1016/j.dnarep.2019.102661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bader AS, Hawley BR, Wilczynska A, and Bushell M (2020). The roles of RNA in DNA double-strand break repair. Br. J. Cancer 122, 613–623. 10.1038/s41416-019-0624-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guha S, and Bhaumik SR (2022). Transcription-coupled DNA double-strand break repair. DNA Repair 109, 103211. 10.1016/j.dnarep.2021.103211. [DOI] [PubMed] [Google Scholar]
- 17.Muniz L, Nicolas E, and Trouche D (2021). RNA polymerase II speed: a key player in controlling and adapting transcriptome composition. EMBO J. 40, e105740. 10.15252/embj.2020105740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, and Greenberg RA (2010). ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981. 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pankotai T, Bonhomme C, Chen D, and Soutoglou E (2012). DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat. Struct. Mol. Biol 19, 276–282. 10.1038/nsmb.2224. [DOI] [PubMed] [Google Scholar]
- 20.Kakarougkas A, Ismail A, Chambers AL, Riballo E, Herbert AD, Künzel J, Löbrich M, Jeggo PA, and Downs JA (2014). Requirement for PBAF in transcriptional repression and repair at DNA breaks in actively transcribed regions of chromatin. Mol. Cell 55, 723–732. 10.1016/j.molcel.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ui A, Nagaura Y, and Yasui A (2015). Transcriptional elongation factor ENL phosphorylated by ATM recruits polycomb and switches off transcription for DSB repair. Mol. Cell 58, 468–482. 10.1016/j.molcel.2015.03.023. [DOI] [PubMed] [Google Scholar]
- 22.Awwad SW, Abu-Zhayia ER, Guttmann-Raviv N, and Ayoub N (2017). NELF-E is recruited to DNA double-strand break sites to promote transcriptional repression and repair. EMBO Rep. 18, 745–764. 10.15252/embr.201643191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iannelli F, Galbiati A, Capozzo I, Nguyen Q, Magnuson B, Michelini F, D’Alessandro G, Cabrini M, Roncador M, Francia S, et al. (2017). A damaged genome’s transcriptional landscape through multilayered expression profiling around in situ-mapped DNA double-strand breaks. Nat. Commun 8, 15656. 10.1038/ncomms15656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caron P, Pankotai T, Wiegant WW, Tollenaere MAX, Furst A, Bonhomme C, Helfricht A, de Groot A, Pastink A, Vertegaal ACO, et al. (2019). WWP2 ubiquitylates RNA polymerase II for DNA-PK-dependent transcription arrest and repair at DNA breaks. Genes Dev. 33, 684–704. 10.1101/gad.321943.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dmitrieva NI, Cui K, Kitchaev DA, Zhao K, and Burg MB (2011). DNA double-strand breaks induced by high NaCl occur predominantly in gene deserts. Proc. Natl. Acad. Sci. USA 108, 20796–20801. 10.1073/pnas.1114677108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mischo HE, Hemmerich P, Grosse F, and Zhang S (2005). Actinomycin D induces histone gamma-H2AX foci and complex formation of gamma-H2AX with Ku70 and nuclear DNA helicase II. J. Biol. Chem 280, 9586–9594. 10.1074/jbc.M411444200. [DOI] [PubMed] [Google Scholar]
- 27.Huang X, Okafuji M, Traganos F, Luther E, Holden E, and Darzynkiewicz Z (2004). Assessment of histone H2AX phosphorylation induced by DNA topoisomerase I and II inhibitors topotecan and mitoxantrone and by the DNA cross-linking agent cisplatin. Cytometry A. 58, 99–110. 10.1002/cyto.a.20018. [DOI] [PubMed] [Google Scholar]
- 28.Zou RS, Marin-Gonzalez A, Liu Y, Liu HB, Shen L, Dveirin RK, Luo JXJ, Kalhor R, and Ha T (2022). Massively parallel genomic perturbations with multi-target CRISPR interrogates Cas9 activity and DNA repair at endogenous sites. Nat. Cell Biol 24, 1433–1444. 10.1038/s41556-022-00975-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnould C, Rocher V, Finoux A-L, Clouaire T, Li K, Zhou F, Caron P, Mangeot PE, Ricci EP, Mourad R, et al. (2021). Loop extrusion as a mechanism for formation of DNA damage repair foci. Nature 590, 660–665. 10.1038/s41586-021-03193-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chandler H, Patel H, Palermo R, Brookes S, Matthews N, and Peters G (2014). Role of polycomb group proteins in the DNA damage response–a reassessment. PLoS One 9, e102968. 10.1371/journal.pone.0102968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Zou RS, He S, Nihongaki Y, Li X, Razavi S, Wu B, and Ha T (2020). Very fast CRISPR on demand. Science 368, 1265–1269. 10.1126/science.aay8204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu B, Miskolci V, Sato H, Tutucci E, Kenworthy CA, Donnelly SK, Yoon YJ, Cox D, Singer RH, and Hodgson L (2015). Synonymous modification results in high-fidelity gene expression of repetitive protein and nucleotide sequences. Genes Dev. 29, 876–886. 10.1101/gad.259358.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vitor AC, Sridhara SC, Sabino JC, Afonso AI, Grosso AR, Martin RM, and de Almeida SF (2019). Single-molecule imaging of transcription at damaged chromatin. Sci. Adv 5, eaau1249. 10.1126/sciadv.aau1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez J, and Larson DR (2020). Transcription in Living Cells: Molecular Mechanisms of Bursting. Annu. Rev. Biochem 89, 189–212. 10.1146/annurev-biochem-011520-105250. [DOI] [PubMed] [Google Scholar]
- 35.Mohiuddin IS, and Kang MH (2019). DNA-PK as an Emerging Therapeutic Target in Cancer. Front. Oncol 9, 635. 10.3389/fonc.2019.00635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiu S, and Huang J (2021). MRN complex is an essential effector of DNA damage repair. J. Zhejiang Univ. - Sci. B 22, 31–37. 10.1631/jzus.B2000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, and Lukas J (2007). RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell 131, 887–900. 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 38.Acs K, Luijsterburg MS, Ackermann L, Salomons FA, Hoppe T, and Dantuma NP (2011). The AAA-ATPase VCP/p97 promotes 53BP1 recruitment by removing L3MBTL1 from DNA double-strand breaks. Nat. Struct. Mol. Biol 18, 1345–1350. 10.1038/nsmb.2188. [DOI] [PubMed] [Google Scholar]
- 39.Mallette FA, and Richard S (2012). K48-linked ubiquitination and protein degradation regulate 53BP1 recruitment at DNA damage sites. Cell Res. 22, 1221–1223. 10.1038/cr.2012.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chagraoui J, Hébert J, Girard S, and Sauvageau G (2011). An anticlastogenic function for the Polycomb Group gene Bmi1. Proc. Natl. Acad. Sci. USA 108, 5284–5289. 10.1073/pnas.1014263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanchez A, De Vivo A, Uprety N, Kim J, Stevens SM, and Kee Y (2016). BMI1-UBR5 axis regulates transcriptional repression at damaged chromatin. Proc. Natl. Acad. Sci. USA 113, 11243–11248. 10.1073/pnas.1610735113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fitieh A, Locke AJ, Mashayekhi F, Khaliqdina F, Sharma AK, and Ismail IH (2022). BMI-1 regulates DNA end resection and homologous recombination repair. Cell Rep. 38, 110536. 10.1016/j.cel-rep.2022.110536. [DOI] [PubMed] [Google Scholar]
- 43.Zhou H, Stein CB, Shafiq TA, Shipkovenska G, Kalocsay M, Paulo JA, Zhang J, Luo Z, Gygi SP, Adelman K, and Moazed D (2022). Rixosomal RNA degradation contributes to silencing of Polycomb target genes. Nature 604, 167–174. 10.1038/s41586-022-04598-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, Sixma TK, and Richard S (2012). RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 31, 1865–1878. 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verma R, Oania R, Fang R, Smith GT, and Deshaies RJ (2011). Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol. Cell 41, 82–92. 10.1016/j.molcel.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.He J, Zhu Q, Wani G, and Wani AA (2017). UV-induced proteolysis of RNA polymerase II is mediated by VCP/p97 segregase and timely orchestration by Cockayne syndrome B protein. Oncotarget 8, 11004–11019. 10.18632/oncotarget.14205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steurer B, Janssens RC, Geijer ME, Aprile-Garcia F, Geverts B, Theil AF, Hummel B, van Royen ME, Evers B, Bernards R, et al. (2022). DNA damage-induced transcription stress triggers the genome-wide degradation of promoter-bound Pol II. Nat. Commun 13, 3624. 10.1038/s41467-022-31329-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dantuma NP, Groothuis TAM, Salomons FA, and Neefjes J (2006). A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J. Cell Biol 173, 19–26. 10.1083/jcb.200510071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fradet-Turcotte A, Canny MD, Escribano-Díaz C, Orthwein A, Leung CCY, Huang H, Landry M-C, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, and Durocher D (2013). 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature 499, 50–54. 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kilgas S, Singh AN, Paillas S, Then C-K, Torrecilla I, Nicholson J, Browning L, Vendrell I, Konietzny R, Kessler BM, et al. (2021). p97/VCP inhibition causes excessive MRE11-dependent DNA end resection promoting cell killing after ionizing radiation. Cell Rep. 35, 109153. 10.1016/j.celrep.2021.109153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meisenberg C, Pinder SI, Hopkins SR, Wooller SK, Benstead-Hume G, Pearl FMG, Jeggo PA, and Downs JA (2019). Repression of Transcription at DNA Breaks Requires Cohesin throughout Interphase and Prevents Genome Instability. Mol. Cell 73, 212–223.e7. 10.1016/j.molcel.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watrin E, and Peters J-M (2009). The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. EMBO J. 28, 2625–2635. 10.1038/emboj.2009.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anindya R, Aygün O, and Svejstrup JQ (2007). Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol. Cell 28, 386–397. 10.1016/j.molcel.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 54.Yasukawa T, Kamura T, Kitajima S, Conaway RC, Conaway JW, and Aso T (2008). Mammalian Elongin A complex mediates DNA-damage-induced ubiquitylation and degradation of Rpb1. EMBO J. 27, 3256–3266. 10.1038/emboj.2008.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuznetsova AV, Meller J, Schnell PO, Nash JA, Ignacak ML, Sanchez Y, Conaway JW, Conaway RC, and Czyzyk-Krzeska MF (2003). von Hippel-Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc. Natl. Acad. Sci. USA 100, 2706–2711. 10.1073/pnas.0436037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harreman M, Taschner M, Sigurdsson S, Anindya R, Reid J, Somesh B, Kong SE, Banks CAS, Conaway RC, Conaway JW, and Svejstrup JQ (2009). Distinct ubiquitin ligases act sequentially for RNA polymerase II polyubiquitylation. Proc. Natl. Acad. Sci. USA 106, 20705–20710. 10.1073/pnas.0907052106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakazawa Y, Hara Y, Oka Y, Komine O, van den Heuvel D, Guo C, Daigaku Y, Isono M, He Y, Shimada M, et al. (2020). Ubiquitination of DNA Damage-Stalled RNAPII Promotes Transcription-Coupled Repair. Cell 180, 1228–1244.e24. 10.1016/j.cell.2020.02.010. [DOI] [PubMed] [Google Scholar]
- 58.Richardson CD, Ray GJ, DeWitt MA, Curie GL, and Corn JE (2016). Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat. Biotechnol 34, 339–344. 10.1038/nbt.3481. [DOI] [PubMed] [Google Scholar]
- 59.Sternberg SH, Redding S, Jinek M, Greene EC, and Doudna JA (2014). DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67. 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang AS, Chen LC, Wu RA, Hao Y, McSwiggen DT, Heckert AB, Richardson CD, Gowen BG, Kazane KR, Vu JT, et al. (2020). The Histone Chaperone FACT Induces Cas9 Multi-turnover Behavior and Modifies Genome Manipulation in Human Cells. Mol. Cell 79, 221–233.e5. 10.1016/j.molcel.2020.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Clarke R, Heler R, MacDougall MS, Yeo NC, Chavez A, Regan M, Hanakahi L, Church GM, Marraffini LA, and Merrill BJ (2018). Enhanced Bacterial Immunity and Mammalian Genome Editing via RNA-Polymerase-Mediated Dislodging of Cas9 from Double-Strand DNA Breaks. Mol. Cell 71, 42–55.e8. 10.1016/j.molcel.2018.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dimitrova N, Chen Y-CM, Spector DL, and De Lange T (2008). 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456, 524–528. 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu BXH, Hansen LL, Artiles KL, Nonet ML, and Fire AZ (2014). Landscape of target:guide homology effects on Cas9-mediated cleavage. Nucleic Acids Res. 42, 13778–13787. 10.1093/nar/gku1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, and Danuser G (2008). Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 5, 695–702. 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson JT, Thorvaldsdottir H, Turner D, and Mesirov JP (2023). igv.js: an embeddable JavaScript implementation of the Integrative Genomics Viewer (IGV). Bioinformatics 39, btac830. 10.1093/bioinformatics/btac830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zou RS, Liu Y, Wu B, and Ha T (2021). Cas9 deactivation with photocleavable guide RNAs. Mol. Cell 81, 1553–1565.e8. 10.1016/j.molcel.2021.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the ChIP-seq data reported in this paper is NCBI: PRJNA1005701. The Original imaging and Western blot data have been deposited to Mendeley Data: 10.17632/c4t3sss9r7.1].
This paper does not report the original code.
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.