

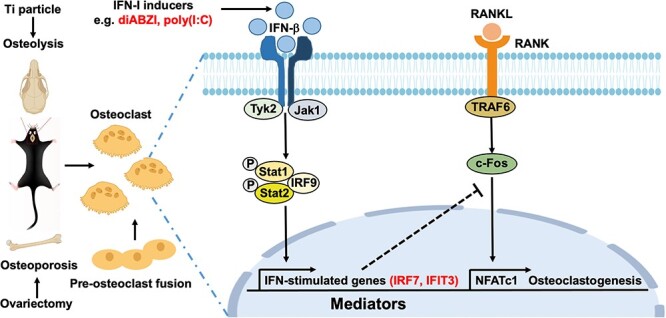
Abstract
Type I interferons (IFN-I) are pleiotropic factors endowed with multiple activities that play important roles in innate and adaptive immunity. Although many studies indicate that IFN-I inducers exert favorable effects on broad-spectrum antivirus, immunomodulation, and anti-tumor activities by inducing endogenous IFN-I and IFN-stimulated genes, their function in bone homeostasis still needs further exploration. Here, our study demonstrates 2 distinct IFN-I inducers, diABZI and poly(I:C), as potential therapeutics to alleviate osteolysis and osteoporosis. First, IFN-I inducers suppress the genes that control osteoclast (OC) differentiation and activity in vitro. Moreover, diABZI alleviates bone loss in Ti particle-induced osteolysis and ovariectomized -induced osteoporosis in vivo by inhibiting OC differentiation and function. In addition, the inhibitory effects of IFN-I inducers on OC differentiation are not observed in macrophages derived from Ifnar1−/−mice, which indicate that the suppressive effect of IFN-I inducers on OC is IFNAR-dependent. Mechanistically, RNAi-mediated silencing of IRF7 and IFIT3 in OC precursors impairs the suppressive effect of the IFN-I inducers on OC differentiation. Taken together, these results demonstrate that IFN-I inducers play a protective role in bone turnover by limiting osteoclastogenesis and bone resorption through the induction of OC-specific mediators via the IFN-I signaling pathway.
Keywords: IFN-I inducer, osteoclast, osteoporosis, osteolysis, bone resorption, ISG mediator, IRF7, IFIT3
Graphical Abstract
Graphical Abstract.
Introduction
Osteoclasts (OCs) are characterized by multinucleated giant cells from myeloid mononuclear/macrophages, which are critical for bone development, bone metabolism, and homeostasis maintenance.1,2 OC are responsible for bone resorption, and their excessive proliferation, differentiation, and enhanced activity will lead to diseases related to bone destruction, such as osteoporosis and osteolysis.3,4 M-CSF and RANKL are necessary for OC differentiation.5 When the bone resorption mediated by OC exceeds the bone formation mediated by osteoblasts, the imbalance in bone metabolism leads to osteoporosis.6,7 Osteoporosis is characterized by reduced bone mass density and destruction of bone microstructure, resulting in increased bone fragility and a higher risk of fractures.8 Osteoporosis and its fractures have become a major public health problem worldwide because of their high disability and mortality.9,10 In addition, the implanted peri-prosthetic wear particles activate the proliferation and differentiation of OC to cause osteolysis.11,12 Excessive proliferation, differentiation, and hyperfunction of OC are the main causes of aseptic loosening of artificial joints.11 Therefore, inhibiting the differentiation and function of OC around the prosthesis and alleviating bone mass loss is an effective way to prevent osteolysis induced by wear particles and prosthesis loosening.13 Although the essential roles of OC have been recognized, the exact processes involved in osteoclastic resorption remain to be fully investigated.
As an important immunomodulatory factor, IFN-I plays an essential role in regulating the differentiation, proliferation, and function of OC, and also participates in the occurrence, development, and prognosis of bone destruction diseases.14,15 IFN-β binds to the IFN-α/β receptor (IFNAR) to activate its downstream JAK–STAT signaling pathway and phosphorylate STAT1 and STAT2, inducing more than 300 IFN-stimulated genes (ISGs).16 Among them, Protein Kinase R (PKR) partially mediates the inhibitory effect of IFN-β on RANKL-induced transcription factor c-Fos and OC differentiation.17 Furthermore, many ISGs have been identified as OC mediators, including CXCL11, ISG15, and USP18.18,19 Therefore, it is of important clinical value to develop small molecular drugs to activate the IFN-I signal pathway and inhibit the excessive activity of OC to prevent and treat osteoporosis and osteolysis. Initially, IFN regulatory factor 7 (IRF7) was believed to be an important transcription factor responsible for producing IFN-I and triggering innate immune responses. Further research has demonstrated that IRF7 has diverse and versatile roles in several biological systems.20 The IFN-inducible protein with tetrapeptide repeats 3 (IFIT3) is a key member of the IFIT family. Recent studies suggest that IFIT3 is involved in cell biology, such as cell growth, cell death, differentiation, and cancer development.21 However, it is unknown whether IRF7 and IFIT3 are involved in osteoclastic bone resorption.
IFN-I inducers are a kind of substance that can stimulate the body to produce endogenous IFN-I. IFN-I inducers have broad-spectrum antiviral, immunomodulatory, and anti-tumor functions.22-24 Swift and thrilling advancements in IFN-I inducers have greatly enhanced the potential for great clinical application in antiviral and anti-tumor treatments.25,26 IFN-I inducers include poly (inosinic acid) (poly(I:C)) and various stimulator of interferon genes (STING) agonists.27 IFN-I inducers can stimulate the body to produce IFN-I through self-regulation and balancing of the immune system, leading to immune suppression and abnormal antibody increases. Compared with IFN-I recombinant protein drugs, most IFN-I inducers are small molecular drugs, which can not only induce the production of IFN-I rapidly and massively in vivo but also avoid most of the problems encountered in the clinical application of recombinant proteins. It has the advantages of high efficiency, low cost, stability, and low drug resistance.28 Poly(I:C) is a synthetic double-stranded RNA analog that induces the production of IFN-I and cytokines by activating Toll-like receptor/retinoic acid-inducible gene I and its downstream TRIF/MAVS-dependent signaling pathways. STING agonists include cyclic dinucleotide (CDN) and their derivatives, non-nucleotide linked dimeric amidobenzimidazole (diABZI) from GlaxoSmithKline, and other small molecular compounds. STING directly recognizes these agonists, or cytoplasmic DNA, combined with the downstream signal of TBK1 activation, NF-κB, and IRF3, which are activated to produce inflammatory cytokines and IFN-I, respectively.29 STING also activates the NIK (NF-κB-induced kinase)-dependent non-classical NF-κB signal pathway and the IKK (IκB kinase)-dependent classical NF-κB signal pathway, resulting in increased expression of pro-inflammatory cytokines.30,31 diABZI is noteworthy as it is the first documented non-nucleotide small-molecule STING agonist that can bind to both hSTING and mSTING, activating anti-tumor immunity and suppressing tumor development when given intravenously.32 In addition, diABZI has the characteristics of high efficiency, stability, economy, and safety, and it can be administered intravenously, which makes it a promising clinical candidate for STING agonists and IFN-I inducers.
Considering the important role of IFN-I on OC formation and differentiation, the prospective functions of broad-spectrum antiviral, immunomodulatory, and anti-tumor functions of IFN-I inducers, we investigated the therapeutic potentials of IFN-I inducers on RANKL-induced osteoclastogenesis in vitro, OVX-induced osteoporosis, and Ti particles-induced osteolysis in vivo. In addition, we focused on determining the anti-OC capacity of the IFN-I inducers as well as elucidating their underlying mechanisms. Thus, we find that diABZI and poly(I:C), as IFN-I inducers, can induce IRF7 and IFIT3 through the IFN-I signaling pathway, inhibit OC, and potentially be used to treat diseases related to osteoclastic bone resorption. Our in-depth and comprehensive discovery of the IFN-I inducers would provide new insight into OC biology and therapeutic targets for osteoclastic bone resorption diseases.
Materials and methods
Reagents
diABZI STING agonist-1 trihydrochloride (HY-112921B) was ordered from MedChemExpress and dissolved in DMSO at a concentration of 10 mM for in vitro experiments or in 10% DMSO, 40% co-solvent PEG400, and 50% saline at a concentration of 1 mg/mL for in vivo experiments as a storage solution. Poly(I:C) (P1530) and Tartaric Phosphate Kit (387A-1KT) were from Sigma-Aldrich. Recombinant mouse M-CSF (416-ML-010) and RANKL (462-TEC-010) were from R&D Systems. Primary antibodies against NFATc-1 (ab25916), MMP9 (ab38898), and CTSK (ab19027) were from Abcam, and c-Fos (#2250), STAT1 (#14994), p-STAT1 (#9167), IRF3 (#4032), p-IRF3 (#29047), TBK1 (#51872), p-TBK1 (#5483), p65 (#6956S), p-p65 (#3033S), and GAPDH (#2118) were from Cell Signaling Technology. Osteo Assay Surface Plate (3987) was from Coring. PrimeScript RT Master Mix (RR036B) and TB Green Premix Ex Taq (RR420B) were from Takara. INTERFERin transfection reagent (409-10) was from Polyplus-transfection. Mouse RANKL Quantikine ELISA Kit (MTR00) and Mouse Osteoprotegerin (OPG) Quantikine ELISA Kit (MOP00) were from R&D Systems. CTX-I (EK13528) and PINP (EK16107) ELISA Kits were from Signalway Antibody. BCIP/NBT Alkaline Phosphatase Color Development Kit (C3206) was from Beyotime. Ti particles (00681) and LIVE/DEAD Viability/Cytotoxicity Kit (L3224) were from Thermo Fisher Scientific.
Mice
The Ifnar1−/− mice were purchased from the Jackson Laboratory and bred with C57BL/6J background mice over 20 generations. Ifnar1−/− mice were bred and genotyped as described previously.33 The genotyping primer sequences were 5′-CGAGGCGAAGTGGTTAAAAG-3′ (Common-forward), 5′-ACGGATCAACCTCATTCCAC-3′ (WT-reverse), and 5′-AATTCGCCAATGACAAGACG-3′ (KO-reverse). The PCR products were 155 bp and 255 bp for WT and KO mice, respectively. The WT C57BL/6J mice used in this study were ordered from Vital River Laboratory Animal Technology. All the mice were maintained in the specific pathogen-free environment at Suzhou Institute of Systems Medicine (ISM) under a controlled temperature of 25°C and a 12 h day–night cycle. All animal experiments were performed in accordance with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals and approved by the Animal Service Center of ISM (ISM-IACUC-0009-R).
OC differentiation
Fresh bone marrow monocytes (BMMs) from C57BL/6J WT and Ifnar1−/− mice were flushed from the femoral and tibial BM. After removal of RBCs with RBC lysis buffer, the cells were cultured in α-MEM containing 10% FBS, 1% penicillin/streptomycin, and 5 ng/mL M-CSF in 5% CO2 at 37°C overnight. To obtain pure BMMs, non-adherent cells were then collected and cultured in complete α-MEM containing 30 ng/mL M-CSF. After a further 3 d in culture, the attached cells were used for the following experiments. The cells were subcultured using 0.25% trypsin and cultured with 30 ng/mL M-CSF and 50 ng/mL RANKL for OC differentiation.
TRAP staining
BMMs were cultured in 48-well plates at a density of 105 cells per well in osteoclastogenic induction medium for 6 d. TRAP staining was performed according to the manufacturer’s instructions. Briefly, the cells were fixed with the fixative solution for 30 min and stained using a Tartaric Phosphate Kit for 60 min. TRAP-positive cells were visualized and quantified in each well using microscopy.
ALP staining
Bone marrow mesenchymal stem cells (BMSCs) were cultured in 48-well plates at a density of 105 cells per well in osteogenic induction medium (α-MEM with 50 μg/mL ascorbic acid, 100 nM dexamethasone, and 10 mM β-glycerol phosphate) for 7 d. Alkaline phosphatase (ALP) staining was performed according to the manufacturer’s instructions. Briefly, the cells were fixed with the fixative solution for 30 min and stained using a BCIP/NBT Alkaline Phosphatase Color Development Kit for 30 min. ALP-positive cells were visualized in each well using microscopy.
Mineral resorption assay
The effect of diABZI and poly(I:C) on OC function was measured with the 24-well Osteo Assay Surface multiple well plates. BMMs were induced in osteoclastogenic induction medium for 6 d. Subsequently, the cells were eliminated using ultrasonic washing, and the mineral resorption was examined using bright-field microscopy. The percentage of resorbed area was then calculated using Image J software.
CCK-8 assay and live/dead staining assay for cell viability
Cell viability was assessed by a commercial cell counting kit-8 (CCK-8) assay. Briefly, cells were seeded in 96-well plates, and different concentrations of diABZI and poly(I:C) were directly added to the medium with 5 parallel control wells in each group for 3 d. Afterward, 10 μL of CCK-8 buffer was added to each well and incubated at 37°C for 2 h. The absorbance at 450 nm was determined using an absorbance microplate reader. For the live/dead staining assay, add 5 μL calcein AM (Component A) and 20 μL ethidium homodimer-1 (Component B) to 10 mL PBS to create a staining solution. Afterward, 200 μL of the staining solution was directly added to each well and incubated for 30 min at room temperature. Images were captured using a LEICA TCA SP8 confocal microscope.
Ti particles-induced osteolysis mouse model
Osteolysis mouse model was induced by implanting Ti particles on the mouse calvaria, as mentioned before.34 Briefly, the 8-wk-old male C57BL/6J mice were randomly assigned to 4 groups: saline was injected into the murine calvaria and received an intraperitoneal injection saline (Sham), saline was injected into the murine calvaria and received an intraperitoneal injection with 0.5 mg/kg diABZI (Sham + diABZI), 30 mg of titanium particles dissolved in saline was injected into the murine calvaria and received an intraperitoneal injection saline (Ti), 30 mg of titanium particles dissolved in saline was injected into the murine calvaria and received an intraperitoneal injection with 0.5 mg/kg diABZI (Ti + diABZI). After the mice regained consciousness and were in stable condition following the operation, they received an intraperitoneal injection with 0.5 mg/kg diABZI or saline 3 times a week for 2 wk. All mice were sacrificed 2 wk after the operation.
OVX-induced osteoporosis mouse model
Female C57BL/6J mice (8 wk old) were used to establish the osteoporosis model. The OVX model was developed according to a previously described method.6 In brief, mice were anesthetized with pentobarbital sodium. Bilateral ovariectomy was performed in the OVX group, whereas the sham group only received skin incision after laparotomy. Six weeks later, the mice randomly divided into 4 groups: sham group received an intraperitoneal injection with saline (Sham), sham group received an intraperitoneal injection with 0.5 mg/kg diABZI 3 times a week (Sham + diABZI), OVX group received an intraperitoneal injection with saline (OVX), and OVX group received an intraperitoneal injection with 0.5 mg/kg diABZI 3 times a week (OVX + diABZI). All mice were sacrificed after 2 wk.
HE and TRAP staining
Bones were decalcified in 10% EDTA for 4 wk. After that, the bones were then processed through ethanol and xylene, embedded into paraffin blocks, and sectioned on a microtome at a thickness of 5 μm. Sections were stained with H & E for general histology and with TRAP staining to detect OC. Images for each section were taken by microscopy.
Micro-CT analysis
After mice were sacrificed, the femurs and skulls were collected and fixed in 4% paraformaldehyde. The bones were scanned using a micro-CT (SkyScan1176). Briefly, scanning was performed at 60 kV and 170 mA at a resolution of 9 μm. 3D images were reconstructed using the NRecronServer system. To reduce bias in the 3D analyses, the complete area of bone trabeculae was set as the ROI. Sixty layers on the growth plate of the distal femur were selected as the bottom layer, and 150 layers in the direction of the femoral shaft were selected as the top layer to analyze the bone trabecular area. The parameters of trabecular bone, including BMD (mg/cm3), bone volume to bone volume fraction (BV/TV, %), trabecular separation (Tb.Sp, mm), trabecular number (Tb.N, mm–1), and trabecular thickness (Tb.Th, mm), were calculated and analyzed.
RNA extraction and RT-qPCR
RT-qPCR was performed to evaluate the expression levels of the OC-specific genes and ISGs. BMMs were seeded in 12-well plates at a density of 5 × 105 cells/well in OC induction medium and treated with IFN-I inducers. Then, RNA was extracted using a TRIzol reagent. cDNA was synthesized using PrimeScript RT Master Mix. Real-time PCR amplification was performed using TB Green Premix Ex Taq on a Roche LightCycler 480 II system. GAPDH as an internal control gene used in RT-qPCR. The primer sequences are listed in Table S1.
RNA-seq and bioinformatics analysis
Total RNA was extracted from BMMs or OCs using a TRIzol reagent. All samples passed quality control analysis using an Agilent Bioanalyzer 2100. RNA-seq libraries were constructed using an Illumina TruSeq Stranded mRNA Sample Preparation Kit. High-throughput sequencing was performed using the Illumina NextSeq 500 at the Suzhou Institute of Systems Medicine. For RNA-Seq data analysis, Trimmomatic was used to remove Illumina sequencing adapters from the raw reads of every sample and trim the quality bases of both read ends. RNA-seq reads were aligned to the mouse genome (mm10) using the Biomedical Genomics Server (CLC). HTSeq was used to count the read numbers mapped to each gene, including known and novel genes. The resulting P-values were adjusted using the Benjamini–Hochberg approach for controlling the FDR. Genes with a FDR < 0.05 were identified as significantly differentially expressed genes (DEGs) between conditions of 2 RNA-seq biological replicates. To identify IFN-I inducers-regulated pathways, we performed Gene Ontology (GO) analysis with the RANKL-regulated DEGs that were more highly expressed in the IFN-I inducers-treated cells than the control (≥1.5 fold). IFN-I inducers-regulated pathways by the GO analysis were ranked based on the P-values. Gene Set Enrichment Analysis (GSEA) input with the RANKL-regulated DEGs was performed according to the program’s instructions. Hallmark pathways by GSEA were ranked based on the normalized enrichment score (NES). All original RNA-seq raw data were deposited in the Genome Sequence Archive of the National Genomics Data Center (NGDC) under the accession number CRA003715.
Western blotting
BMMs were seeded in a 6-well culture plate at a density of 1 × 106 cells per well. After OC induction, OCs were lysed on ice for 30 min in RIPA lysis buffer. An equal amount of total protein was separated using 10% SDS-PAGE and then transferred to a PVDF membrane. Membranes were blocked with 5% skim milk dissolved in TBST and then incubated with primary antibodies specific to target proteins overnight at 4°C. The HRP-conjugated secondary antibodies were added and visualized by chemiluminescence (ECL, Millipore) using the Bio-Rad ChgmiDoc MP System.
Immunofluorescence staining
OCs were fixed in 4% (v/v) paraformaldehyde for 10 min, permeabilized with 0.1% Triton X-100 for 10 min at room temperature, and blocked with blocking buffer for 1 h at room temperature. After washing with PBS, samples were incubated with the primary antibody against NFATc1 (1:20) overnight at 4°C. After washing, the sections were incubated with the respective Alexa Flour 488 conjugated-donkey anti-rabbit IgG used as the secondary antibody. Then samples were incubated with TRITC Phalloidin at room temperature for 30 min. After washing, nuclei were counterstained with DAPI for 10 min at room temperature. Images were captured using a LEICA TCA SP8 confocal microscope.
Transfection with siRNA
BMMs were seeded for 12 h and then transfected with 20 nM siRNA targeting IRF7 and IFIT3 or non-targeting control siRNA using the OPTI-MEM and INTERFERin transfection reagent according to the manufacturer’s instructions. After 24 h, the cells were differentiated into OC with 30 ng/mL M-CSF and 50 ng/mL RANKL. A list of siRNAs targeting IRF7 and IFIT3 is shown in Table S2.
Retroviral transduction
For ectopic expression, IFIT3 and IRF7 were cloned into the pMSCV-PIG retroviral vector. Retroviral vectors were transfected into 293 T with pCL-Eco using PEI, and retrovirus was collected from the culture medium after 48 h. BMMs were infected with retrovirus in a medium containing 90 ng/mL M-CSF in the presence of 10 μg/mL polybrene for 12 h, and then selected with 2 μg/mL puromycin for 2 d in the presence of 60 ng/mL M-CSF. Puromycin-resistant BMMs were used for the experiments.
Enzyme-linked immunosorbent assay
Serum was obtained from whole blood using centrifugation of 7500 rpm at 4°C for 20 min. RANKL, OPG, CTX-I, and PINP serum concentrations were determined by an ELISA kit following the manufacturer’s guidelines.
Statistical analysis
All data were presented as the mean ± standard deviation and were analyzed statistically using one-way ANOVA. A P-value <0.05 was considered significant.
Study approval
All animal experiments were performed in accordance with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals, and approved by the Animal Service Center of ISM (ISM-IACUC-0009-R).
Results
IFN-I inducers inhibit OC differentiation and function in vitro
To assess whether the distinct IFN-I inducers diABZI and poly(I:C) potently inhibit osoteoclastogenesis, we first checked the safety concentration of them during OC differentiation by using the CCK-8 assay (Figure S1A and B). The viability of the pre-OC, bone marrow-derived macrophages (BMMs), was not affected after the cells were treated with 10–200 nM diABZI or 0.1–1 μg/mL poly(I:C) for 3 d (Figure S1B). In addition, the live/dead staining assay of BMMs treated with diABZI and poly(I:C) did not affect cell proliferation or death (Figure S1C and D). Next, BMMs differentiated into OC by adding M-CSF and RANKL in the absence or presence of the IFN-I inducers. TRAP staining assays were performed 6 d after the induction of OC differentiation. Fewer TRAP-stained cells were observed in the diABZI-treated or poly(I:C)-treated group than the control group (Figure 1A–C). Consistently, the OC number and area were also significantly reduced after the treatment with diABZI and poly(I:C) during osteoclastogenesis (Figure 1D–G). These results indicated that both diABZI and poly(I:C) potently suppressed OC formation. Furthermore, Osteo Assay plates were used to determine whether diABZI and poly(I:C) suppressed the osteoclastic bone resorption activity. The extent of surface resorption was significantly reduced in a dose-dependent manner when the OC differentiation medium was added with diABZI or poly(I:C) (Figure 1B and C, H and I). We next examined whether diABZI and poly(I:C) suppressed the expression of the genes that determine OC differentiation and function, including Oscar, Dc-stamp, Acp5, Mmp9, and Ctsk. These OC-specific genes were dramatically induced when the BMMs were stimulated with RANKL. However, the induction of these genes by RANKL was significantly suppressed by diABZI and poly(I:C) and further suppressed by the higher concentration of them (Figure 1J and K). Furthermore, protein expression of OC-specific proteins in BMMs, including NFATc1, MMP9, and CTSK, was suppressed after 3 d osteoclastogenic induction in the presence of diABZI and poly(I:C) (Figure 1L and M). In addition, we next checked the expression of NFATc1 and the size of OC through immunofluorescence staining, which are found to be significantly suppressed by diABZI and poly(I:C) (Figure S1E–H). As shown in Figure S2, the expression of c-Fos and Nfatc1 was suppressed by diABZI and poly(I:C) at the early stage of OC differentiation. Collectively, these results demonstrated that IFN-I inducers, diABZI and poly(I:C), inhibited OC differentiation and function in vitro, suggesting a potential role of IFN-I inducers in the prevention or treatment of osteoporotic bone diseases such as osteolysis and osteoporosis.
Figure 1.

IFN-I inducers inhibit OC differentiation and function in vitro. (A) The general view of TRAP staining of BMMs after 6 d osteoclastogenic induction in the presence of diABZI and poly(I:C). (B, C) Representative images of TRAP staining of BMMs after 6 d osteoclastogenic induction and the demineralization area after 6 d osteoclastogenic induction in the presence of diABZI and poly(I:C). Scale bar: 100 μm. (D–G) Quantification of number and area of TRAP-positive cells after 6 d osteoclastogenic induction in the presence of diABZI and poly(I:C). TRAP-positive cells with 3 or more nuclei were counted as OC. (H, I) Quantification of the demineralization area after 6 d osteoclastogenic induction in the presence of diABZI and poly(I:C). (J, K) Relative mRNA expression of OC-specific genes in BMMs including Oscar, Dc-stamp, Acp5, Mmp9, and Ctsk in BMMs after 3 d osteoclastogenic induction in the presence of diABZI and poly(I:C). Gapdh as an internal control gene. (L, M) Protein expression of OC-specific proteins in BMMs including NFATc1, MMP9, and CTSK in BMMs after 3 d osteoclastogenic induction in the presence of diABZI and poly(I:C). Data of (A–C) are representative images from 3 independent experiments. Data of (D–K) are shown as the mean ± SD from at least 3 independent experiments. Data of (L) and (M) are shown as representative of 3 independent experiments. *p<.05, **p<.01 by one-way ANOVA.
Bone homeostasis is maintained by osteogenesis and osteoclastogenesis, so we also examined the effect of diABZI on osteoblasts. First, ALP staining assays were performed 7 d after the induction of osteoblast differentiation. ALP-stained cells were not affected by diABZI treatment (Figure S3A and B). We next examined whether diABZI affected the expression of the genes that determine osteoblast differentiation and function, including Bmp2, Col1a1, Ocn, and Alp. The induction of these genes by osteogenic induction was not affected by diABZI (Figure S3C).
diABZI alleviates Ti particle-induced osteolysis in mice
We investigated whether IFN-I inducers could mitigate Ti particle-induced bone damage by using diABZI in an osteolysis model caused by Ti particles (Figure 2A). First, we conducted micro-CT analysis on mouse calvaria obtained from the 4 distinct groups. Compared to the Ti group, the diABZI group showed less bone degradation on the calvaria in the 3D reconstruction figures, with even less observed in the sham group (Figure 2B). In addition, the quantitative results of the 3D analysis of BV/TV were significantly decreased in the Ti group and increased in area of porosity (%) and the number of pores compared to those from the sham group. On the contrary, the diABZI group notably improved bone erosion and effectively reversed the decrease in BV/TV and the rise in porosity. However, diABZI treatment had no effects on bone in sham mice (Figure 2C). The serum was examined for levels of RANKL, a factor that stimulates the formation of OC, and its controlling decoy receptor, OPG. We found that diABZI remarkably reduced the RANKL/OPG ratio in blood serum (Figure 2D). Consistently, histology analysis further confirmed that diABZI alleviated Ti particle-induced osteolysis. HE staining indicated a decrease in the eroded surface in the diABZI group, compared with the Ti group. TRAP staining revealed that the TRAP-positive cells were mostly located around the bone pitting, with fewer TRAP-positive cells in the diABZI group (Figure 2E and Figure S4A). These results suggested that diABZI suppressed Ti particle-induced bone loss in vivo.
Figure 2.

diABZI alleviates Ti particle-induced osteolysis in vivo. (A) Schematic diagram of the titanium particle-induced osteolysis model. (B) Representative micro-CT images of murine calvaria 3D reconstruction figure. (C) Quantitative parameters of morphological bone alterations, including BV/TV, area of porosity, number of pores, Tb.Th, Tb.N, and Tb.Sp (Sham, n=3; Ti, n=7). (D) RANKL and OPG serum concentrations and RANKL/OPG ratio (Sham, n=3; Ti, n=7). (E) Representative images of histological staining with HE and TRAP staining. Arrows indicate OC. Scale bar: 100 μm. Data of (B) and (E) are representative images of one representative experiment from 3 independent experiments. Data of (C) and (D) are shown as the mean ± SD of one representative experiment from 3 independent experiments. *p<.05, **p<.01, n.s.: not significant by one-way ANOVA.
diABZI alleviates OVX-induced osteoporosis in mice
Having established that diABZI effectively inhibited OC differentiation and bone resorption in vitro, the potential of diABZI as a prophylactic agent to prevent OC-related disease was further investigated using an OVX-induced osteoporosis model in vivo. After 6 wk, mice were randomized to the sham and OVX groups then injected with saline or diABZI (0.5 mg/kg) 3 times a week for 2 wk (Figure 3A). First, after the treatment period of 2 wk, the femurs were evaluated by micro-CT. The 2D and 3D reconstruction figures showed that diABZI prevented the bone loss on the femurs in the OVX model (Figure 3B and C). In addition, the values of bone morphometric-related parameters, including BMD, BV/TV, Tb.Th, and Tb.N were all increased in the diABZI-treated than OVX mice. As expected, the diABZI-treated group decreased bone loss, as indicated by an almost 40% decrease in trabecular separation compared with the OVX group (Figure 3D). We next examined the expression of OC-specific genes, including Oscar, Dc-stamp, Acp5, Ctsk, Atp6v0d2, and Ifnb1 of tibia. This observation suggested that OC-specific gene expression was upregulated in the OVX group compared with the sham group. However, the expression was suppressed by diABZI-treated (Figure 3E). However, there were no significant differences in bone parameters or OC-specific genes between the saline-treated and diABZI-treated in the sham group (Figure 3D and E). We found that diABZI significantly reduced the concentration of RANKL, CTX-I, and the RANKL/OPG ratio in blood serum (Figure 3F and Figure S4B). More importantly, HE staining indicated that the bone surface was well maintained in the diABZI treatment group. TRAP staining showed that the OC decreased after diABZI treatment compared with the OVX group (Figure 3G and Figure S4C). Consistently, the diABZI-treated OVX mice presented an elevated bone mass with a tense trabecular architecture in the femurs. It was found that diABZI could effectively rescue the enhanced bone loss in OVX mice.
Figure 3.
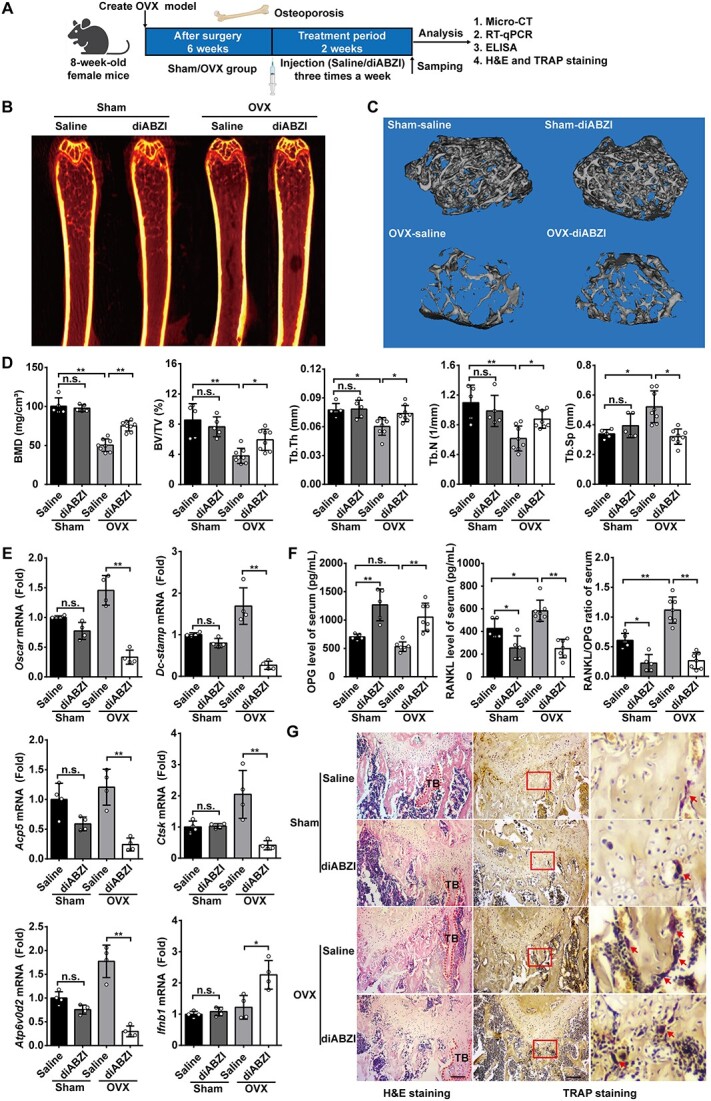
diABZI alleviates OVX-induced osteoporosis in vivo. (A) Schematic diagram of the OVX-induced osteoporosis model. (B, C) Representative micro-CT images of murine femur calvaria showing 2D fault and 3D reconstruction figure (D) Quantitative parameters of morphological bone alterations, including BMD, BV/TV, Tb.Th, Tb.N, and Tb.Sp (Sham, n=5; OVX, n=8). (E) Relative mRNA expression of OC-specific genes, including Oscar, Dc-stamp, Acp5, Ctsk, Atp6v0d2, and Ifnb1 in bone tissues (n=4). Gapdh as an internal control gene. (F) RANKL and OPG serum concentrations and RANKL/OPG ratio (Sham, n=5; OVX, n=8). (G) Representative images of histological staining with HE and TRAP staining. Scale bar: 100 μm. TB: trabecular bone. Arrows indicate OC. Data of (B), (C), and (G) are representative images of one representative experiment from 3 independent experiments. Data of (D–F) are shown as the mean ± SD of one representative experiment from 3 independent experiments. *p<.05, **p<.01, n.s.: not significant by one-way ANOVA.
We also investigated the effect of diABZI on osteoblasts in vivo, the concentration of PINP level in blood serum showed that there was no significant difference between the diABZI-treated group and the OVX group (Figure S4D). We next examined the expression of osteoblast-specific genes, including Bmp2, Col1a1, Ocn, and Alp in the tibia. This observation suggested that osteoblast-specific gene expression was not affected by diABZI (Figure S4E). More importantly, ALP staining showed that osteogenesis was not affected after diABZI treatment when compared with the OVX group (Figure S4F and G). In conclusion, diABZI does not affect the differentiation of osteoblasts in vivo.
IFNAR is required for the suppressive effect of IFN-I inducers on osteoclastogenesis
Although IFN-I inducers activate their downstream signaling via different pathways, the IFNAR signaling pathway is the dominant one. To further confirm, BMMs from Ifnar1−/− mice were treated with diABZI and poly(I:C) in the presence of M-CSF and RANKL. Both diABZI and poly(I:C) failed to inhibit the differentiation of Ifnar1−/− BMMs into OC (Figure 4A–C). In addition, as shown in Figure 4D and E, the OC number and area were not suppressed by diABZI and poly(I:C) in Ifnar1−/−BMMs. As shown in Figure 4B–E, the extent of resorption was not affected by diABZI and poly(I:C). Furthermore, neither diABZI nor poly(I:C) influenced the protein and mRNA expression of OC-specific markers in Ifnar1−/− BMMs with OC induction (Figure 4F–H). We next checked the size of OC through immunofluorescence staining, and found that they were not affected by diABZI (Figure S5A). STAT1 and TBK1 are recognized for their role in transmitting signals when IFNAR is activated by IFN-I. Stimulation with diABZI and poly(I:C) induced the phosphorylation of STAT1 and TBK1, but such effects were not observed in Ifnar1−/−BMMs under the same condition (Figure 4I). Moreover, diABZI and poly(I:C) also suppressed the protein expression levels of OC-specific protein in WT BMMs, all of which are important for OC formation and differentiation in RANKL-stimulated. In contrast, the protein expression levels of OC-specific protein were not suppressed by diABZI and poly(I:C) in RANKL-stimulated Ifnar1−/− BMMs (Figure S5B). These results indicated that IFN-I inducers primarily inhibited OC differentiation via the IFNAR signaling pathway. In addition, some results suggested that other pathways also contributed to the induction of these mediators affecting OC differentiation via IFN-I inducers.
Figure 4.

IFNAR is required for the suppressive effect of IFN-I inducers on osteoclastogenesis. (A) The general view of TRAP staining in Ifnar1−/− BMMs after 6 d osteoclastogenic induction in the presence of diABZI and poly(I:C). (B, C) Representative images of TRAP staining after 6 d osteoclastogenic induction and the demineralization area after 6 d osteoclastogenic induction in Ifnar1−/− BMMs in the presence of diABZI and poly(I:C). (D, E) Quantification of number and area of TRAP-positive cells, and the demineralization area in the presence of diABZI and poly(I:C). TRAP-positive cells with 3 or more nuclei were counted as OC. (F) Protein expression of OC-specific proteins in Ifnar1−/− BMMs including NFATc1, MMP9, and CTSK after 3 d osteoclastogenic induction in the presence of diABZI and poly(I:C). (G, H) Relative mRNA expression of OC-specific genes in Ifnar1−/− BMMs including Oscar, Dc-stamp, Acp5, Mmp9, and Ctsk after 3 d osteoclastogenic induction in the presence of diABZI and poly(I:C). Gapdh as an internal control gene. (I) Protein expression of IFN-I signaling in WT BMMs including p-TBK1, TBK1, p-IRF3, IRF3, p-STAT1, and STAT1 after 3 h and 6 h osteoclastogenic induction in the presence of diABZI and poly(I:C). Protein expression of IFN-I signaling in Ifnar1−/− BMMs including p-p65, p65, p-TBK1, TBK1, p-STAT1, and STAT1 after 3 h and 6 h osteoclastogenic induction in the presence of diABZI and poly(I:C). Scale bar: 100 μm. Data of (A–C) are representative images from 3 independent experiments. Data of (D–E) and (G–H) are shown as the mean ± SD from at least 3 independent experiments. Data of (F) and (I) are shown as representative of 3 independent experiments. *p<.05 by one-way ANOVA.
IRF7 and IFIT3 are common mediators during IFN-I inducers inhibiting osteoclastogenesis
We next sought to further investigate the mechanisms by which diABZI and poly(I:C) function as osteoclastogenic regulators. First, we performed gene expression profiling using high-throughput RNA-seq with the control after RANKL stimulation and in the presence of diABZI and poly(I:C) to identify genes regulated by IFN-I inducers during osteoclastogenesis. An RNA-seq analysis displayed a heatmap illustrating the expression of osteoclastic genes affected by diABZI and poly(I:C). The results revealed that the IFN-I inducers notably inhibited the expression of OC marker genes such as Oscar, Dc-stamp, Mmp9, Acp5, Ctsk, and Atp6v0d2 (Figure 5A). GO analysis of the upregulated genes by diABZI and poly(I:C) during RANKL-induced osteoclastogenesis showed a considerable activation of genes related to defense response, including the IFN-β response and bone resorption (Figure 5B). GSEA showed that the diABZI and poly(I:C) regulated genes were significantly enriched in IFN-I response genes (Figure 5C and D). We extracted gene expression levels of the enriched IFN-I response genes from our RNA-seq data and confirmed RANKL-regulated IFN-I response genes that showed significant elevation with diABZI and poly(I:C), such as Usp18, Ifit3, Irf7, and Cxcl11 (Figure 5E). The significant enrichment in IFN-I response genes with the presence of diABZI and poly(I:C) suggested a significant augment in the IFN-I pathway. We overlapped these IFN-I response genes with DEGs, then found the 9 genes as follows: Irf7, Ifit3, Cxcl11, Usp18, Ifitm3, Ly6e, Cmpk2, Mx1, and Ifi44 (Figure 5F). We next checked the mRNA expression of the possible OC-related mediators (Cxcl11, Usp18, Irf7, and Ifit3) during RANKL-induced osteoclastogenesis in the presence of diABZI and poly(I:C). As expected, diABZI and poly(I:C) enhanced the mRNA expression of the possible mediators (Figure 5G and H). Consistently, IRF7 and IFIT3 are common mediators of IFN-I inducers inhibiting osteoclastogenesis.
Figure 5.
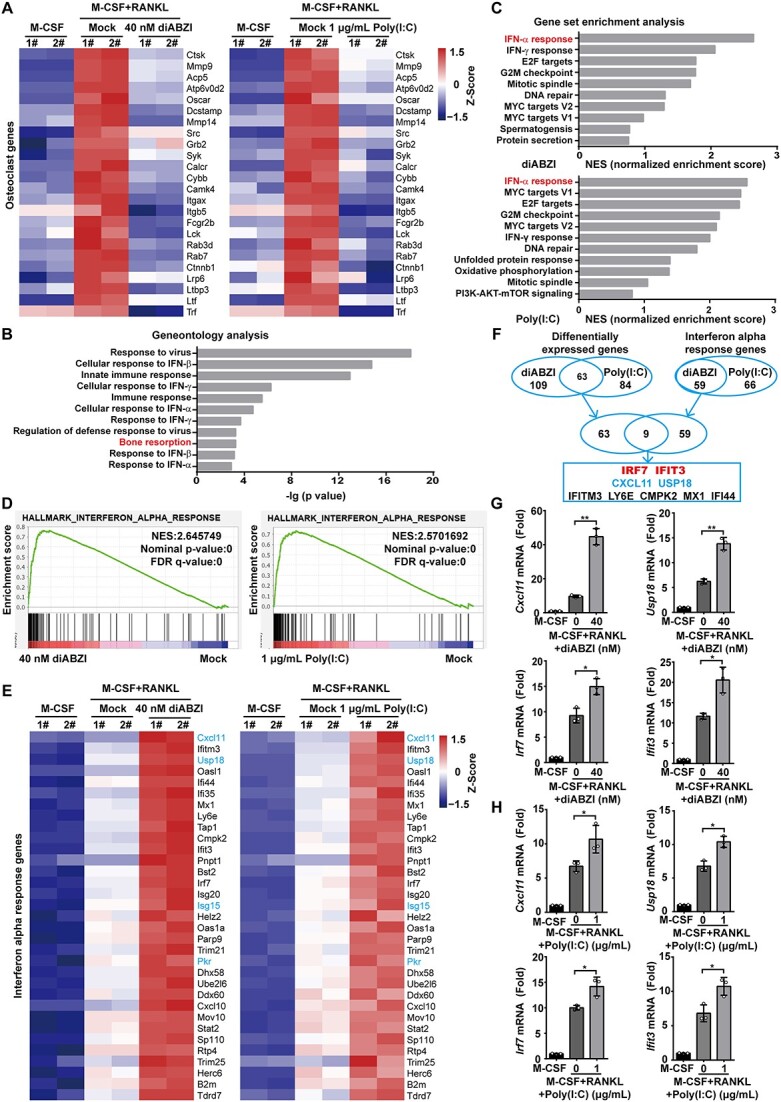
IRF7 and IFIT3 are common mediators during IFN-I inducers inhibiting osteoclastogenesis. (A) Heatmap of OC marker genes in BMMs after 3 d osteoclastogenic induction in the presence of diABZI and poly(I:C) (B) Geneontology analysis of RANKL-inducible genes regulated by diABZI and poly(I:C). (C, D) GSEA of RANKL-inducible genes regulated by diABZI and poly(I:C) ranked by NES of Hallmark. (E) Heatmap of the enriched interferon alpha response genes in BMMs after 3 d osteoclastogenic induction in the presence of diABZI and poly(I:C). (F) Left Venn diagram showing the overlap of the DEGs between diABZI and poly(I:C); right Venn diagram showing the overlap of the interferon alpha response genes between diABZI and poly(I:C); finally overlap the DEGs and interferon alpha response genes. (G, H) The mRNA expression of the possible OC-related ISGs including Cxcl11, Usp18, Irf7, and Ifit3 during RANKL-induced osteoclastogenesis in the presence of diABZI and poly(I:C). Gapdh as an internal control gene. Data of (G) and (H) are shown as the mean ± SD from 3 independent experiments. *p<.05, **p<.01 by one-way ANOVA.
Induction of IRF7 and IFIT3 mediates the suppressive effect of IFN-I inducers on OC differentiation and function
We thought that IRF7 and IFIT3 in OC precursors were involved in the inhibition of osteoclastogenesis. First, we validated the efficiency of gene knock-down by investigating the expression of BMMs transfected with specific si-IRF7 and si-IFIT3 and with non-targeting as a negative control. Then, we chose the highest knock-down efficiency as si-IRF7-1 and si-IFIT3-3 for the following experiments (Figure 6A and B). Furthermore, we found that IRF7 and IFIT3 expression were also decreased during RANKL-induced osteoclastogenesis, although with slower kinetics (Figure 6A and B). After TRAP staining, more TRAP-stained cells were present in the silencing of the IRF7 and IFIT3 groups than in the si-NC group with diABZI and poly(I:C) (Figure 6C–F). We next examined the OC number and area, which were found to be significantly increased by the silencing of IRF7 and IFIT3 groups than in the si-NC group with diABZI and poly(I:C) (Figure 6G and H). These results indicated that IRF7 and IFIT3 mediated the suppressive effect of IFN-I inducers on OC differentiation and function. In addition, the silencing of the IRF7 and IFIT3 groups reversed the suppressive effect of OC-specific genes of the IFN-I inducer (Figure 6I and J). We also found the TRAP-stained OC and the mRNA expression of OC-specific genes, which were found to be significantly promoted by the silencing of IRF7 and IFIT3 without diABZI and poly(I:C) (Figure S6). These results suggested that silencing of IRF7 and IFIT3 mRNA in OC precursors by siRNAs resulted in enhanced OC differentiation, indicating the function of IRF7 and IFIT3 in osteoclastogenesis. Novel mediators during IFN-I suppressed osteoclastogenesis, which were potential targets for the treatment of osteolysis and osteoporosis, and developing drugs to induce these mediators.
Figure 6.

Induction of IRF7 and IFIT3 mediates the suppressive effect of IFN-I inducers on OC differentiation and function. (A, B) Efficiency of silencing of IRF7 and IFIT3 and kinetics of IRF7 and IFIT3 mRNA expression during osteoclastogenic induction at indicated time points. (C, D) The general view of TRAP staining of BMMs after 6 d osteoclastogenic induction in the presence of si-IRF7 and si-IFIT3 with diABZI and poly(I:C). (E, F) Representative images of TRAP staining of BMMs after 6 d osteoclastogenic induction in the presence of si-IRF7 and si-IFIT3 with diABZI and poly(I:C). (G, H) Quantification of number and area of TRAP-positive cells after 6 d osteoclastogenic induction in the presence of si-IRF7 and si-IFIT3 with diABZI and poly(I:C). TRAP-positive cells with 3 or more nuclei were counted as OC (I, J) Relative mRNA expression of OC-specific genes including Oscar, Dc-stamp, Acp5, and Mmp9 in BMMs after 3 d osteoclastogenic induction in the presence of si-IRF7 and si-IFIT3 with diABZI and poly(I:C). Gapdh as an internal control gene. Scale bar: 100 μm. Data of (A) and (B) and (G–J) are shown as the mean ± SD from at least 3 independent experiments. Data of (C–F) are representative images from 3 independent experiments. *p<.05, **p<.01 by one-way ANOVA.
We further investigated the effect of overexpression of IRF7 and IFIT3 via the retrovirus delivery system in osteoclast precursors on osteoclastogenesis. First, a schematic diagram of the overexpression of IRF7 and IFIT3 with retroviruses was shown (Figure S7A). After TRAP staining, fewer TRAP-stained cells were observed in the overexpression of IRF7 and IFIT3 groups than in the pMSCV group (Figure S7B–E). Consistently, the OC number and area were also significantly reduced after the overexpression of IRF7 and IFIT3 during osteoclastogenesis (Figure S7F and G). In addition, the overexpression of IRF7 and IFIT3 groups suppressed the expression of the genes that determine OC differentiation and function (Figure S7H and I). At the same time, the effect of inhibition was further amplified with the treatment of diABZI and poly(I:C).
Discussion
One main cause of aseptic loosening and revision surgery is excessive bone resorption determined by wear particles.35 In addition, osteoporosis is a prevalent metabolic bone disease that causes an imbalance between bone formation and resorption, placing a substantial financial and health burden on the aging society.36 Osteoclastogenesis has been the focus of therapeutic strategies to alleviate osteolysis and osteoporosis.37 In this study, we focused on the IFN-I inducers diABZI and poly(I:C) to inhibit osteoclastic bone resorption in vitro. Furthermore, we established Ti particle-induced osteolysis and OVX-induced osteoporosis mouse model to investigate the effect of IFN-I inducers on OC differentiation in vivo. More importantly, diABZI did not affect the differentiation of osteoblasts in vitro or in vivo. Our current findings offer therapeutic potential for osteolysis and osteoporosis and describe the unique role of IFN-I inducers in OC differentiation via IRF7 and IFIT3. However, diABZI treatment had no effects on bone in the sham group of osteolysis and osteoporosis models. IFN-I/IFNAR expression is at a low level under physiological conditions and only increases after inflammatory stimulation, thus only diABZI-treatment is not sufficient to produce a distinct bone phenotype.
In humans, the cGAS-STING pathway plays significant roles in innate and adaptive immunity.38 Therefore, STING is the focus of drug development and immunological research. The previous study discovered that CDN can inhibit the differentiation and function of OC by activating STING and downstream the IFN-β signal pathway. CDN can be used as a biocompatibility therapy to treat bone destruction caused by OC overactivation.39 STING agonists based on CDN need to be administered by intratumorally injection because of their low metabolic stability, which brings many limitations to clinical application. Although recent study reported that diABZI from GlaxoSmithKline elicited anti-tumor treatment following systemic administration in mice.32 In addition, several STING agonists are currently in clinical trials and navigating the pharmaceutical pipeline for cancer therapy due to their enhanced drug properties.40 To our knowledge, diABZI is the first non-CDN molecule to show high STING selectivity and represent a milestone in the cancer therapies, although subsequent studies have reported STING agonists such as SR-717 and MSA-2.28,41 Furthermore, the latest studies have shown that diABZI inhibits viral replication in mice and primary human bronchial epithelial cells, indicating its potential in treating COVID-19 and future pandemics caused by human respiratory pathogens.42,43 More importantly, STING activation, due to its unique synergistic effects on nociceptors, immune cells, and OCs, provides specific advantages in treating bone cancer-related pain.44 Therefore, we propose that diABZI, activated by the STING and IFN-I signal pathways, may represent a promising therapeutic strategy to alleviate osteolysis and osteoporosis.
Compared to recombinant IFN-I, poly(I:C) is characterized by less costly, simpler administration, and increased stability. Poly(I:C) has been used in cancer treatment to induce tumor cell apoptosis through both IFN-I-dependent and independent mechanisms.45 Based on our work, RANKL-induced OC-specific genes and proteins were not fully reversed in Ifnar1−/− BMMs with diABZI and poly(I:C). Therefore, a further investigation of whether IFN-I inducers regulate OC in IFN-I-independent pathways by using MAVS-TRIF-DKO or STING-KO mice and cell lines is required. More importantly, a recent study systematically summarized the mechanism and adverse effects related to the pharmacologic therapies for postmenopausal osteoporosis.46 Small-molecule drugs have the advantages of low cost, convenient transportation, excellent stability and solubility, easy delivery, low drug resistance, and few side effects. With the development of aging and the increased incidence of osteoporosis, more effective therapeutic drugs should be developed.
IFN-I triggers downstream ISGs to perform various biological functions in immunity, cell differentiation, and other aspects. Based on our data, we overlapped these IFN-I response genes with DEGs in the presence of diABZI and poly(I:C) and found the 9 genes as follows: Irf7, Ifit3, Cxcl11, Usp18, Ifitm3, Ly6e, Cmpk2, Mx1, and Ifi44. Consistent with our results, the previous study has shown that recombinant CXCL11 inhibits osteoclastic differentiation, and autocrine CXCL11 is involved in regulating osteoclastogenesis via IFN-I.18 Furthermore, another report demonstrates that uncontrolled stimulation of IFN signaling in USP18-knockout mice accelerates OC development.47 Interestingly, we found that ISG15 and PKR increased in the presence of diABZI and poly(I:C). Meanwhile, the prior study demonstrated that overexpression of ISG15 can hinder the formation of OC. Since RANKL stimulates IFN-β expression that restricts excessive osteoclastogenesis, ISG15 may have the potential to assist IFN-β in regulating OC formation.48 Moreover, the capacity of IFN-β to inhibit RANKL-driven osteoclastogenesis in mice lacking PKR is attenuated through suppressing RANKL-induced c-Fos protein expression.17 In the present study, IFN-I inducers inhibited BMMs differentiation into OC through IRF7 and IFIT3. It is tempting to surmise from our observations that the IFN-induced IRF7 and IFIT3 play important roles in OC differentiation. However, the precise mechanism between activation of IRF7 and IFIT3 and inhibition of OC differentiation remains unknown and requires further exploration. In the present study, our data illustrate the inhibition of osteoclastogenesis by IFITM3 and LY6E and the possibility that IFITM3 and LY6E might target OC fusion, which is critical in bone resorption activity. Interestingly, tracking OC during fission and fusion cycles revealed distinct populations of “osteomorph” that are capable of fusing, motile, and able to generate bone-resorbing.49 A better understanding of the role of IFN-I and other mediators in bone density regulation is crucial to demonstrating the correlation between the immune system and bone homeostasis.
In this study, we have demonstrated that IFN-I inducers alleviate osteolysis and osteoporosis by inhibiting osteoclastogenesis, which is IFNAR-dependent. In addition, deficiencies in IFN-I production may contribute to deficiencies in IFNAR signaling, as IFN-I enhances the expression of key mediators (IRF7 and IFIT3) of the signaling pathway (Figure S8). Additionally, IFN-I inducers might serve as biocompatible treatments for bone disorders resulting from increased OC activity.
Supplementary Material
Acknowledgments
We appreciate the excellent technical supports from RNA technology platform of Suzhou Institute of Systems Medicine.
Contributor Information
Yingkang Huang, National Key Laboratory of Immunity and Inflammation, and CAMS Key Laboratory of Synthetic Biology Regulatory Elements, Chinese Academy of Medical Sciences & Peking Union Medical College, Suzhou Institute of Systems Medicine, Suzhou 215123, Jiangsu, China.
Mingchao Zhang, National Key Laboratory of Immunity and Inflammation, and CAMS Key Laboratory of Synthetic Biology Regulatory Elements, Chinese Academy of Medical Sciences & Peking Union Medical College, Suzhou Institute of Systems Medicine, Suzhou 215123, Jiangsu, China; Department of Orthopedics, The Second Affiliated Hospital of Soochow University, Suzhou 215004, Jiangsu, China.
Jun Zhang, Department of Orthopedics, Zhejiang Provincial People’s Hospital, Hangzhou 310014, Zhejiang, China.
Siying Liu, National Key Laboratory of Immunity and Inflammation, and CAMS Key Laboratory of Synthetic Biology Regulatory Elements, Chinese Academy of Medical Sciences & Peking Union Medical College, Suzhou Institute of Systems Medicine, Suzhou 215123, Jiangsu, China; Department of Orthopedics, The Second Affiliated Hospital of Soochow University, Suzhou 215004, Jiangsu, China.
Dapei Li, National Key Laboratory of Immunity and Inflammation, and CAMS Key Laboratory of Synthetic Biology Regulatory Elements, Chinese Academy of Medical Sciences & Peking Union Medical College, Suzhou Institute of Systems Medicine, Suzhou 215123, Jiangsu, China.
Zigang Qiao, National Key Laboratory of Immunity and Inflammation, and CAMS Key Laboratory of Synthetic Biology Regulatory Elements, Chinese Academy of Medical Sciences & Peking Union Medical College, Suzhou Institute of Systems Medicine, Suzhou 215123, Jiangsu, China.
Haiping Yao, National Key Laboratory of Immunity and Inflammation, and CAMS Key Laboratory of Synthetic Biology Regulatory Elements, Chinese Academy of Medical Sciences & Peking Union Medical College, Suzhou Institute of Systems Medicine, Suzhou 215123, Jiangsu, China.
Qin Shi, Department of Orthopedics, The First Affiliated Hospital of Soochow University, Suzhou 215006, Jiangsu, China.
Xiaozhong Zhou, Department of Orthopedics, The Second Affiliated Hospital of Soochow University, Suzhou 215004, Jiangsu, China.
Feng Ma, National Key Laboratory of Immunity and Inflammation, and CAMS Key Laboratory of Synthetic Biology Regulatory Elements, Chinese Academy of Medical Sciences & Peking Union Medical College, Suzhou Institute of Systems Medicine, Suzhou 215123, Jiangsu, China.
Author contributions
Yingkang Huang (Formal analysis, Investigation, Validation, Visualization, Writing—original draft, Writing—review & editing [equal]), Mingchao Zhang (Formal analysis, Investigation, Validation, Visualization, Writing—original draft, Writing—review & editing [equal]), Jun Zhang (Formal analysis, Investigation [equal]), Siying Liu (Data curation, Formal analysis, Investigation), Dapei Li (Formal analysis, Investigation), Zigang Qiao (Formal analysis, Investigation), Haiping Yao (Formal analysis, Investigation), Qin Shi (Methodology, Resources), Xiaozhong Zhou (Methodology, Resources), and Feng Ma (Conceptualization, Formal analysis, Funding acquisition, Investigation, Supervision, Validation, Writing—original draft, Writing—review & editing)
Funding
This work was supported by the National Key Research and Development Program of China (2018YFA0900803), National Natural Science Foundation of China (82301982, 82271804, 32270924, and 32170880), Natural Science Foundation of Jiangsu Province (BK20230281, BK20221256, and BK20200004), CAMS Initiation Fund for Medical Sciences (CIFMS, 2021-I2M-1-041, 2023-I2M-2-010, and 2022-I2M-2-004), Non-profit Central Research Institute Fund of CAMS (2019PT310028), the Special Research Fund for Central Universities, Peking Union Medical College (3332022160), the Suzhou Municipal Key Laboratory (SZS2023005), the NCTIB Fund for R&D Platform for Cell and Gene Therapy, and the 333 High-level Talent Training Project.
Conflicts of interest
All authors state that they have no conflicts of interest.
Data availability
The raw RNA-Seq data generated in this study have been deposited in the Genome Sequence Archive in the NGDC, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences, under accession number CRA003715 that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Du T, Yan Z, Zhu S, et al. QKI deficiency leads to osteoporosis by promoting RANKL-induced osteoclastogenesis and disrupting bone metabolism. Cell Death Dis. 2020;11(5):330. 10.1038/s41419-020-2548-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423(6937):337–342. 10.1038/nature01658 [DOI] [PubMed] [Google Scholar]
- 3. Wang R, Yang Y, Zhang Z, et al. Major vault protein (MVP) suppresses aging- and estrogen deficiency-related bone loss through Fas-mediated apoptosis in osteoclasts. Cell Death Dis. 2023;14(9):604. 10.1038/s41419-023-05928-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adam S, Simon N, Steffen U, et al. JAK inhibition increases bone mass in steady-state conditions and ameliorates pathological bone loss by stimulating osteoblast function. Sci Transl Med. 2020;12(530):eaay4447. 10.1126/scitranslmed.aay4447 [DOI] [PubMed] [Google Scholar]
- 5. Teitelbaum SL. Bone resorption by osteoclasts. Science (New York, NY). 2000;289(5484):1504–1508. 10.1126/science.289.5484.1504 [DOI] [PubMed] [Google Scholar]
- 6. Huang Y, Yin Y, Gu Y, et al. Characterization and immunogenicity of bone marrow-derived mesenchymal stem cells under osteoporotic conditions. Sci China Life Sci. 2020;63(3):429–442. 10.1007/s11427-019-1555-9 [DOI] [PubMed] [Google Scholar]
- 7. Kim BJ, Lee YS, Lee SY, et al. Osteoclast-secreted SLIT3 coordinates bone resorption and formation. J Clin Invest. 2018;128(4):1429–1441. 10.1172/JCI91086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Little EA, Eccles MP. A systematic review of the effectiveness of interventions to improve post-fracture investigation and management of patients at risk of osteoporosis. Implement Sci. 2010;5(1):80. 10.1186/1748-5908-5-80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhu L, Tang Y, Li XY, et al. Osteoclast-mediated bone resorption is controlled by a compensatory network of secreted and membrane-tethered metalloproteinases. Sci Transl Med. 2020;12(529):eaaw6143. 10.1126/scitranslmed.aaw6143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fu Q, Bustamante-Gomez NC, Reyes-Pardo H, et al. Reduced osteoprotegerin expression by osteocytes may contribute to rebound resorption after denosumab discontinuation. JCI Insight. 2023;8(18):e167790. 10.1172/jci.insight.167790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gallo J, Goodman SB, Konttinen YT, Raska M. Particle disease: biologic mechanisms of periprosthetic osteolysis in total hip arthroplasty. Innate Immunity. 2013;19(2):213–224. 10.1177/1753425912451779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li Y, Lin S, Liu P, et al. Carnosol suppresses RANKL-induced osteoclastogenesis and attenuates titanium particles-induced osteolysis. J Cell Physiol. 2020;236(3):1950–1966. 10.1002/jcp.29978 [DOI] [PubMed] [Google Scholar]
- 13. Sun Z, Zeng J, Wang W, et al. Magnoflorine suppresses MAPK and NF-κB signaling to prevent inflammatory osteolysis induced by titanium particles in vivo and osteoclastogenesis via RANKL in vitro. Front Pharmacol. 2020;11:389. 10.3389/fphar.2020.00389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wong S-W, Huang B-W, Hu X, et al. Global deletion of optineurin results in altered type I IFN signaling and abnormal bone remodeling in a model of Paget’s disease. Cell Death Differ. 2020;27(1):71–84. 10.1038/s41418-019-0341-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takayanagi H, Sato K, Takaoka A, Taniguchi T. Interplay between interferon and other cytokine systems in bone metabolism. Immunol Rev. 2005;208(1):181–193. 10.1111/j.0105-2896.2005.00337.x [DOI] [PubMed] [Google Scholar]
- 16. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49. 10.1038/nri3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takayanagi H, Kim S, Matsuo K, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-β. Nature. 2002;416(6882):744–749. 10.1038/416744a [DOI] [PubMed] [Google Scholar]
- 18. Coelho LF, de Freitas Almeida GM, Mennechet FJD, Blangy A, Uzé G. Interferon-alpha and -beta differentially regulate osteoclastogenesis: role of differential induction of chemokine CXCL11 expression. Proc Natl Acad Sci U S A. 2005;102(33):11917–11922. 10.1073/pnas.0502188102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. MacLauchlan S, Kushwaha P, Tai A, et al. STING-dependent interferon signatures restrict osteoclast differentiation and bone loss in mice. Proc Natl Acad Sci U S A. 2023;120(15):e2210409120. 10.1073/pnas.2210409120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma W, Huang G, Wang Z, Wang L, Gao Q. IRF7: role and regulation in immunity and autoimmunity. Front Immunol. 2023;14:1236923. 10.3389/fimmu.2023.1236923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang W, Li Y, Xin S, et al. The emerging roles of IFIT3 in antiviral innate immunity and cellular biology. J Med Virol. 2023;95(1):e28259. 10.1002/jmv.28259 [DOI] [PubMed] [Google Scholar]
- 22. Kane M, Zang TM, Rihn SJ, et al. Identification of interferon-stimulated genes with antiretroviral activity. Cell Host Microbe. 2016;20(3):392–405. 10.1016/j.chom.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luo M, Wang H, Wang Z, et al. A STING-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol. 2017;12(7):648–654. 10.1038/nnano.2017.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12(2):125–135. 10.1038/nri3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang H, You Q-D, Xu X-L. Targeting stimulator of interferon genes (STING): a medicinal chemistry perspective. J Med Chem. 2019;63(8):3785–3816. 10.1021/acs.jmedchem.9b01039 [DOI] [PubMed] [Google Scholar]
- 26. Benmerzoug S, Ryffel B, Togbe D, Quesniaux VFJ. Self-DNA sensing in lung inflammatory diseases. Trends Immunol. 2019;40(8):719–734. 10.1016/j.it.2019.06.001 [DOI] [PubMed] [Google Scholar]
- 27. Matsumoto M, Seya T. TLR3: interferon induction by double-stranded RNA including poly (I: C). Adv Drug Deliv Rev. 2008;60(7):805–812. 10.1016/j.addr.2007.11.005 [DOI] [PubMed] [Google Scholar]
- 28. Pan BS, Perera SA, Piesvaux JA, et al. An orally available non-nucleotide STING agonist with antitumor activity. Science. 2020;369(6506):eaba6098. 10.1126/science.aba6098 [DOI] [PubMed] [Google Scholar]
- 29. Dou Z, Ghosh K, Vizioli MG, et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. 2017;550(7676):402–406. 10.1038/nature24050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Flood BA, Higgs EF, Li S, Luke JJ, Gajewski TF. STING pathway agonism as a cancer therapeutic. Immunol Rev. 2019;290(1):24–38. 10.1111/imr.12765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS–STING pathway in health and disease. Nat Rev Genet. 2019;20(11):657–674. 10.1038/s41576-019-0151-1 [DOI] [PubMed] [Google Scholar]
- 32. Ramanjulu JM, Pesiridis GS, Yang J, et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature. 2018;564(7736):439–443. 10.1038/s41586-018-0705-y [DOI] [PubMed] [Google Scholar]
- 33. Müller U, Steinhoff U, Reis LF, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264(5167):1918–1921. 10.1126/science.8009221 [DOI] [PubMed] [Google Scholar]
- 34. Gu Y, Wang Z, Shi J, et al. Titanium particle-induced osteogenic inhibition and bone destruction are mediated by the GSK-3β/β-catenin signal pathway. Cell Death Dis. 2017;8(6):e2878. 10.1038/cddis.2017.275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liao S, Feng W, Liu Y, et al. Inhibitory effects of biochanin A on titanium particle-induced osteoclast activation and inflammatory bone resorption via NF-κB and MAPK pathways. J Cell Physiol. 2021;236(2):1432–1444. 10.1002/jcp.29948 [DOI] [PubMed] [Google Scholar]
- 36. Jiang M, Liu R, Liu L, et al. Identification of osteogenic progenitor cell-targeted peptides that augment bone formation. Nat Commun. 2020;11(1):4278. 10.1038/s41467-020-17417-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Harre U, Lang SC, Pfeifle R, et al. Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat Commun. 2015;6(1):6651. 10.1038/ncomms7651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Z, Zhou H, Ouyang X, et al. Multifaceted functions of STING in human health and disease: from molecular mechanism to targeted strategy. Signal transduction and targeted therapy. 2022;7(1):394. 10.1038/s41392-022-01252-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kwon Y, Park OJ, Kim J, Cho JH, Yun CH, Han SH. Cyclic dinucleotides inhibit osteoclast differentiation through STING-mediated interferon-β Signaling. J Bone Miner Res. 2019;34(7):1366–1375. 10.1002/jbmr.3701 [DOI] [PubMed] [Google Scholar]
- 40. Garland KM, Sheehy TL, Wilson JT. Chemical and biomolecular strategies for STING pathway activation in cancer immunotherapy. Chem Rev. 2022;122(6):5977–6039. 10.1021/acs.chemrev.1c00750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chin EN, Yu C, Vartabedian VF, et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science. 2020;369(6506):993–999. 10.1126/science.abb4255 [DOI] [PubMed] [Google Scholar]
- 42. Humphries F, Shmuel-Galia L, Jiang Z, et al. A diamidobenzimidazole STING agonist protects against SARS-CoV-2 infection. Science Immunol. 2021;6(59):eabi9002. 10.1126/sciimmunol.abi9002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li M, Ferretti M, Ying B, et al. Pharmacological activation of STING blocks SARS-CoV-2 infection. Sci Immunol. 2021;6(59):eabi9007. 10.1126/sciimmunol.abi9007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang K, Donnelly CR, Jiang C, et al. STING suppresses bone cancer pain via immune and neuronal modulation. Nat Commun. 2021;12(1):4558. 10.1038/s41467-021-24867-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang C, Chen S, Zhang T, et al. TLR3 ligand PolyI:C prevents acute pancreatitis through the interferon-β/interferon-α/β receptor signaling pathway in a caerulein-induced pancreatitis mouse model. Front Immunol. 2019;10:980. 10.3389/fimmu.2019.00980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Walker MD, Shane E. Postmenopausal osteoporosis. N Engl J Med. 2023;389(21):1979–1991. 10.1056/NEJMcp2307353 [DOI] [PubMed] [Google Scholar]
- 47. Yim HY, Park C, Lee YD, et al. Elevated response to type I IFN enhances RANKL-mediated osteoclastogenesis in Usp18-knockout mice. J Immunol. 2016;196(9):3887–3895. 10.4049/jimmunol.1501496 [DOI] [PubMed] [Google Scholar]
- 48. Takeuchi T, Shimakawa G, Tamura M, Yokosawa H, Arata Y. ISG15 regulates RANKL-induced osteoclastogenic differentiation of RAW264 cells. Biol Pharm Bull. 2015;38(3):482–486. 10.1248/bpb.b14-00410 [DOI] [PubMed] [Google Scholar]
- 49. McDonald MM, Khoo WH, Ng PY, et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell. 2021;184(5):1330–1347.e13. 10.1016/j.cell.2021.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw RNA-Seq data generated in this study have been deposited in the Genome Sequence Archive in the NGDC, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences, under accession number CRA003715 that are publicly accessible at https://ngdc.cncb.ac.cn/gsa. The data that support the findings of this study are available from the corresponding author upon reasonable request.