

Abstract
Extracts of frozen rat liver were found to catalyse the formation of 3H2O from DL-2-hydroxy[2-3H]glutarate. Three peaks of enzyme activities were observed on separation by chromatography on DEAE-Sepharose. The first and second peaks corresponded to an enzyme acting on L-2-hydroxyglutarate and the third peak corresponded to an enzyme acting on D-2-hydroxyglutarate, as indicated by competitive inhibition of the detritiation of the racemic radioactive compound by the unlabelled L- and D-isomers respectively. The enzyme acting on the D-form was further characterized. It was independent of NAD or NADP and it converted D-2-hydroxyglutarate into α-ketoglutarate, transferring electrons to artificial electron acceptors. It also oxidized D-lactate, D-malate and meso-tartrate and was stimulated by Zn2+, Co2+ and Mn2+, but not by Mg2+ or Ca2+. Subcellular fractionation indicated that it was present in the mitochondrial fraction. The enzyme was further purified by chromatography on Blue Trisacryl and phenyl-Sepharose, up to a stage where only a few bands were still visible by SDS/ PAGE. Among the four candidate polypeptides that were identified by MS, one corresponded to a predicted mitochondrial protein homologous with FAD-dependent D-lactate dehydrogenase. The corresponding human protein was expressed in HEK-293 cells and it was shown to catalyse the detritiation of DL-2-hydroxy[2-3H]glutarate with similar properties as the purified rat enzyme.
Keywords: dehydrogenase, D-2-hydroxyglutarate, D-2-hydroxyglutaric aciduria, D-lactate, FAD
Abbreviations: ORF, open reading frame; DCIP, dichlorophenol indophenol; ETF, electron transfer flavoprotein; PMS, phenazine methosulphate
INTRODUCTION
D-2-Hydroxyglutaric aciduria is a neurometabolic disease characterized by the accumulation of abnormal amounts of D-2-hydroxyglutarate in cerebrospinal fluid, blood and urine [1,2]. D-2-Hydroxyglutarate is formed as a degradation product of L-hydroxylysine [3] and, possibly, also from aminolaevulinate [1]. The enzyme that catalyses its utilization is not well identified. Wanders and Mooyer [4] reported the presence of a mitochondrial D-2-hydroxyglutarate dehydrogenase independent of NAD+ or NADP+, and transferring electrons to the artificial acceptor, iodotetrazolium chloride. This enzyme is present mainly in liver and kidney and also in other tissues. Neither the molecular identity of this enzyme nor the product of the reaction it catalyses are known to date. This enzyme is possibly related to D-2-hydroxyacid dehydrogenase, a mitochondrial, FAD-dependent enzyme capable of oxidizing a variety of D-2-hydroxyacids, including D-lactate, D-malate and meso-tartrate [5–7], and therefore possibly also D-2-hydroxyglutarate. The purpose of the present study was to characterize and identify the enzyme acting on D-2-hydroxyglutarate.
EXPERIMENTAL
Materials
Blue Trisacryl was obtained from Sigma. Phenyl-Sepharose and tritiated borohydride were obtained from Amersham Biosciences. The source of other materials was as reported previously [8]. DL-2-Hydroxy[2-3H]glutarate was prepared by incubating 1 mM α-ketoglutarate with 3.5×108 c.p.m. of sodium [3H]borohydride (15 Ci/mmol) in a final volume of 200 μl for 90 min at 25 °C. The remaining borohydride was destroyed by the addition of 0.1 ml of 10% (w/v) HClO4. After 60 min at 25 °C, the sample was neutralized with 3 M K2CO3; the precipitate was removed by centrifugation and the supernatant was brought to 3 ml with water before being applied to a Dowex AG1×8 column. The column was washed first with 5 ml of 50 mM NaCl and then with 5 ml of 500 mM NaCl to elute the radiolabelled DL-2-hydroxy[2-3H]glutarate.
Purification of D-2-hydroxyglutarate dehydrogenase
Frozen rat liver (typically approx. 20 g) was homogenized with 2 vol. (v/w) of buffer A (25 mM Hepes, pH 7.1/1 μg/ml leupeptin/1 μg/ml antipain) containing 100 mM KCl. The extract was made to 2% in poly(ethylene glycol) 8000 and centrifuged for 15 min at 15000 g. The supernatant (33 ml) was diluted 2-fold with buffer A and applied to a DEAE-Sepharose column (1.5 cm×20 cm) equilibrated in the same buffer. The column was washed with 30 ml of buffer A and developed with a 0–0.5 M NaCl gradient in 2×100 ml of buffer A. The most active fractions (approx. 13 ml) were pooled, supplemented with 2.5 mM CoCl2, diluted with 2 vol. of a buffer containing 2.5 mM CoCl2 and applied to a Blue Trisacryl column (9 cm3) equilibrated in the same buffer. The column was washed with 25 ml of equilibration buffer and developed with a 0–1 M NaCl gradient in 2×75 ml of buffer A containing 2.5 mM CoCl2. The active fractions from two such preparations were pooled (approx. 30 ml) and supplemented with 0.5 M NaCl. They were then applied to a phenyl-Sepharose column (5 cm3), equilibrated with buffer A containing 2.5 mM CoCl2 and 0.5 M NaCl. The column was then developed with a stepwise decreasing gradient of NaCl (0.5–0 M), followed by a stepwise increasing gradient of ethylene glycol (to 50%, v/v).
Protein identification by MS
In-gel tryptic digestions of protein bands and desalting of the peptides were performed as described in [9]. Peptides were analysed by nanoelectrospray ionization–tandem MS in an LCQ Deca XP Plus ion-trap mass spectrometer (ThermoFinnigan, San Jose, CA, U.S.A.) fitted with a nanoelectrospray probe. The results were analysed using the X-calibur software (ThermoFinnigan) and the proteins were identified using TurboSEQUEST of the BioWorks software suite (ThermoFinnigan).
Enzyme assays
In the radiochemical assay, the preparation to be assayed (usually 25 μl) was incubated for 20 min at 30 °C in the presence of 20 mM Hepes (pH 7.1), 25 mM KCl, 1 mM MgCl2 and 300000 c.p.m. DL-2-hydroxy[2-3H]glutarate (approx. 0.1 nmol) in a final volume of 250 μl. Where indicated, non-labelled D-2-hydroxyglutarate (2 μM in the experiments shown in Figures 2–4 and 7), salts of bivalent cations or other compounds were also present. The reaction was stopped by the addition of 125 μl of 10% HClO4. After neutralization with 3 M K2CO3 and elimination of the salt precipitate by centrifugation, the samples were diluted 3-fold with water and applied to 1 ml Dowex AG1×8 columns, which were washed with 2 ml of water to elute 3H2O. The eluate was mixed with 5 vol. of scintillation cocktail (Ultima Gold; Packard, Zürich, Switzerland) and its radioactivity was counted. The Km value for D-2-hydroxyglutarate was determined by this method using a constant amount of radiolabelled substrate and variable amounts of unlabelled D-2-hydroxyglutarate. It can be demonstrated that the Km value determined in this way corresponds to the Km value for unlabelled D-2-hydroxyglutarate, whereas, due to the primary kinetic isotope effect, the Vmax value corresponds to the Vmax value for unlabelled D-2-hydroxyglutarate divided by a factor corresponding to the ratio of the catalytic efficiencies for unlabelled and tritiated D-2-hydroxyglutarate. It was checked that the activity was linear with time and the concentration of the enzyme at least up to a substrate consumption of approx. 25%.
Figure 2. Co-elution of the enzyme utilizing radiolabelled D-2-hydroxyglutarate with a dehydrogenase acting on D-2-hydroxyglutarate, D-malate and D-lactate.

A portion of the elution profile of a DEAE-Sepharose column is shown similar to the one shown in Figure 1. The activity was determined by the radiochemical assay in the presence of 2 μM unlabelled D-2-hydroxyglutarate (▴) or through the oxidation of DCIP in the presence of 1 mM D-2-hydroxyglutarate (▵), D-lactate (□) or D-malate (○). The approx. 50-fold difference in activity between the two conditions is due to a combination of several factors such as the use of subsaturating concentrations of substrate in the radiochemical assay, a primary isotopic effect and the stimulation exerted by electron acceptors (see the Discussion section).
Figure 4. Effect of bivalent cations and EGTA on the activity of D-2-hydroxyglutarate dehydrogenase.

Radiochemical assay was performed on the enzyme in the presence of 2 μM unlabelled D-2-hydroxyglutarate and the indicated concentrations of CoCl2 (○), MnCl2 (▵), MgCl2 (□), ZnCl2 (▿), CaCl2 (⋄) and EGTA (•).
Figure 7. Effect of overexpression of the putative human D-2-hydroxyglutarate on the detritiation of radiolabelled 2-hydroxyglutarate.

Cells were transfected with a vector containing the ORF of the putative D-2-hydroxyglutarate dehydrogenase (D-OHGD) or with an empty vector (control). The enzyme activity was determined by the radiochemical assay in the presence of 2 μM unlabelled D-2-hydroxyglutarate; 50 μM CoCl2 or ZnCl2 was present where indicated. Results are the means±S.E.M. for four independent transfections.
Enzyme activity was also determined spectrophotometrically at 600 nm via the reduction of DCIP (dichlorophenol indophenol) in the presence of PMS (phenazine methosulphate) [6,7]. The assay mixture (1 ml) comprised 40 μM DCIP, 11 μM PMS, 50 mM Tris/HCl (pH 8) and 250 μM D-2-hydroxyglutarate or other substrates. In some experiments, such incubations were stopped after 180 min by the addition of activated charcoal to a final concentration of 2% to remove PMS and DCIP. The samples were centrifuged and the concentration of α-ketoglutarate in the resulting supernatant was determined with a spectrophotometric assay using glutamate dehydrogenase [10]. H2O2 formation was determined spectrophotometrically at 440 nm in an assay mixture containing 50 mM Tris/HCl (pH 8), 0.4 mM o-dianisidine, 0.5 mM NaN3 and 2 units/ml horseradish peroxidase [11]; 1 unit is the amount of enzyme catalysing the oxidation of 1 μmol of substrate per min under the specified conditions.
Subcellular fractionation
Fresh liver from an overnight fasted rat was homogenized in isotonic sucrose containing 25 mM Hepes (pH 7.1), 1 μg/ml leupeptin and 1 μg/ml antipain, and the homogenate was submitted to differential centrifugation [12]. The fractions were stored at −80 °C. D-2-Hydroxyglutarate dehydrogenase was partially purified from 0.5 ml of each fraction by chromatography on 1 cm3 DEAE-Sepharose columns, from which the protein was eluted with a stepwise NaCl gradient (successively, 50, 100, 150, 200, 300 and 500 mM NaCl in 25 mM Hepes, pH 7.1). D-2-Hydroxyglutarate dehydrogenase activity was determined by the radiochemical assay in the presence of 2 μM D-2-hydroxyglutarate and 50 μM ZnCl2. It was essentially present in the fractions eluted with 200–500 mM NaCl.
Overexpression of human D-2-hydroxyglutarate dehydrogenase
The open reading frame of D-2-hydroxyglutarate dehydrogenase (accession no. BC036604) was amplified by PCR using Pwo DNA polymerase, human liver cDNA as the template and the primers 5′-AGGAGGAGCCCGAGGTCTCC-3′ and 5′-CCTTGGCAGCAGCAGGAGTG-3′. The PCR product was cloned into pBluescript (restricted with EcoRV) and checked by sequencing. The ORF was excised from this plasmid by digestion with SacI and HindIII and inserted into the SacI and HindIII sites of pCMV5 (Novagen), a mammalian expression vector.
HEK-293 cells were plated on day 0 in Petri dishes (100 mm diameter) at a cell density of 3.6×106 cells/plate in DMEM, supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin and 10% (v/v) foetal bovine serum. After 16 h, the medium was replaced by a fresh medium, supplemented with 1% foetal bovine serum. The cells were then transfected with 8 μg of pCMV5 vector containing the D-2-hydroxyglutarate dehydrogenase ORF or empty vector, using high-molecular-mass polyethyleneimine [13] obtained from Aldrich. The medium was replaced by fresh medium containing 10% foetal bovine serum 16 h after transfection and the culture was continued for 48 h. The cells were then washed with PBS and harvested in 500 μl of 20 mM Hepes (pH 7.1) containing 5 μg/ml leupeptin, 5 μg/ml antipain and 0.5 mM PMSF. Cells were frozen in liquid nitrogen, thawed and lysed by mixing. The lysates were clarified by centrifugation at 2000 g for 5 min.
RESULTS
Separation of enzymes acting on L- and D-2-hydroxyglutarate
Extracts of frozen rat liver catalysed the detritiation of [2-3H]DL-2-hydroxyglutarate, the reaction reaching approx. 40% completion after 20 min incubation of a 1/40 homogenate at 30 °C. This reaction was inhibited by both unlabelled L- and D-2-hydroxyglutarate with an apparent Ki value of approx. 20 μM (results not shown).
The enzymes acting on radioactive DL-2-hydroxy[2-3H]glutarate were separated by chromatography of a frozen liver extract on DEAE-Sepharose. As shown in Figure 1, three peaks were observed. The first peak corresponded to the flow-through fraction the second one, which was quite small, eluted at approx. 100 mM NaCl; the third one eluted at approx. 250 mM NaCl. Competition experiments indicated that the enzyme in the first and second peaks was inhibited more powerfully by L-2-hydroxyglutarate than by D-2-hydroxyglutarate. In peak 1, 50% inhibition of the formation of radiolabelled water was reached at 3.5 μM L-2-hydroxyglutarate and 180 μM D-2-hydroxyglutarate. The opposite was true for the enzyme present in the third peak (50% inhibition at 65 and 4 μM L- and D-2-hydroxyglutarate respectively). Allowing for small reciprocal contaminations of the two enantiomers, these results indicated that the enzyme(s) present in the first two peaks acted on L-2-hydroxyglutarate, whereas the enzyme of the third peak acted on D-2-hydroxyglutarate. Further work was focused on the characterization of the latter enzyme.
Figure 1. Separation of the enzymes acting on D- and L-2-hydroxyglutarate by chromatography on DEAE-Sepharose.

Activity (•) was measured with radiolabelled DL-2-hydroxy[2-3H]glutarate on 25 μl of each fraction. The absorbance A280 was also measured (▵). The oblique line indicates the NaCl gradient.
Characterization of the enzyme acting on the D-isomer
As shown in Figure 2, fractions containing activity towards the D-isomer reduced DCIP enzymically in the presence of D-2-hydroxyglutarate, indicating that the enzyme is a D-2-hydroxyglutarate dehydrogenase. The total D-2-hydroxyglutarate dehydrogenase activity recovered in the column amounted to the equivalent of 17 m-units/g liver. The enzyme acted also on D-lactate, D-malate (Figure 2) and meso-tartrate (results not shown) with approx. 2-, 4- and 3-fold lower Vmax values respectively. Assays with different concentrations of these dicarboxylic acids indicated that the enzyme was saturated with 0.25 mM D-2-hydroxyglutarate, D-malate and meso-tartrate, and the Km value for D-lactate was 0.25 mM. Figure 3 shows that meso-tartrate, D-malate and D-lactate inhibited the detritiation of DL-2-hydroxy[2-3H]glutarate, indicating that the same enzyme is involved in all cases. The L forms of malate and lactate and both D- and L-tartrate were poor inhibitors. Some activity was observed in the spectrophotometric assay with L-2-hydroxyglutarate, but the change in absorption rapidly levelled off with time, indicating that it was due to the presence of a substrate, presumably D-2-hydroxyglutarate, contaminating the L-2-hydroxyglutarate preparation. This contamination was estimated at approx. 6% by comparing the activity with ‘L-2-hydroxyglutarate’ with a series of low concentrations of D-2-hydroxyglutarate.
Figure 3. Sensitivity of D-2-hydroxyglutarate dehydrogenase to several 2-hydroxy carboxylic acids.

The partially purified enzyme was assayed through the release of tritium from DL-2-hydroxy[2-3H]glutarate in the presence of 2 μM unlabelled D-2-hydroxyglutarate, 50 μM CoCl2 and the indicated concentrations of D-malate (○), mesotartrate (▵), D-lactate (□), L-malate (•), L-tartrate (▴), L-lactate (▪) and D-tartrate (▵).
The detritiation of DL-2-hydroxyglutarate by purified D-2-hydroxyglutarate dehydrogenase was unaffected by FAD, FMN, NAD+ or NADP+. However, this does not exclude the possibility that the enzyme has tightly bound FAD, as indeed known for D-2-hydroxyacid dehydrogenase [7]. Since D-lactate dehydrogenase from several species [14] and mammalian D-2-hydroxy acid dehydrogenase [5] appear to be metalloenzymes, we tested the effect of bivalent cations on the activity of D-2-hydroxyglutarate dehydrogenase using the radiochemical assay. As shown in Figure 4, Zn2+, Co2+ and Mn2+ increased the activity by 2.5– 4.5-fold, whereas Ca2+ and Mg2+ had no effect and EGTA decreased the activity. Other experiments showed that 50 μM Zn2+ or Co2+ decreased the Km value for D-2-hydroxyglutarate from 3.2±0.2 (mean±S.E.M.; n=3) to 2.1±0.4 and 1.0±0.2 μM respectively, whereas 1 mM EGTA increased it to 9.9±0.8 μM. Zn2+ increased the Vmax value by approx. 4-fold and Co2+ increased it by only 35%, whereas EGTA had no significant effect. It should be noted that the effect of metals tended to decrease as the enzyme became older. This loss of effect resulted from both an increase in the basal activity and a decrease in the activity with Zn2+ or Co2+. It could be partly restored by preincubating the ‘desensitized’ enzyme with mercaptoethanol (results not shown).
To compare the radiochemical assay with the spectrophotometric assay, we tested the effect of the electron acceptors used in this assay on the activity as determined by the radiochemical assay. In the absence of metal, Vmax increased from a value of 0.31 m-unit/ml (i.e. 0.16 m-unit/mg of protein) to 0.91 and 1.07 m-units/ml in the presence of 11 μM PMS + 40 μM DCIP at pH 7.0 and 8.0 respectively. The latter value corresponded to 22% of the rate of the reaction determined spectrophotometrically in the same fraction and at the same pH (results not shown). As mentioned in the Discussion section, this effect is most probably explained by a primary kinetic isotopic effect. Using the radiochemical assay, we also noted that detritiation of the radiolabelled substrate was stimulated up to 2.5-fold by DCIP (at 300 μM), up to 5-fold by PMS (at 1 mM) and up to 4-fold by potassium ferricyanide (at 1 mM).
We also measured the ability of the enzyme to transfer its reducing equivalent to O2 by measuring the formation of H2O2 in the peak fractions of the DEAE-Sepharose column. This rate was estimated to be approx. 20% of the rate of reduction of DCIP and PMS (results not shown), indicating that the enzyme is capable of transferring its electrons to PMS better than to O2.
To determine the identity of the product formed from D-2-hydroxyglutarate, the purified enzyme was incubated in the presence of PMS and DCIP together with 250 μM D-hydroxyglutarate. The amount of α-ketoglutarate formed at the end of the 3 h incubation (91 μM) amounted to 84% of the quantity expected from spectrophotometric assays of the D-2-hydroxyglutarate activity. Allowing for a probable time-dependent decrease in the activity, these results indicate that α-ketoglutarate is the reaction product. No α-ketoglutarate was formed in the absence of D-2-hydroxyglutarate or enzyme.
Subcellular distribution
Fractionation of a rat liver homogenate by differential centrifugation [12] indicated that D-2-hydroxyglutarate dehydrogenase was essentially present in the heavy and light mitochondrial fractions in which specific activities of 2.1 and 0.5 pmol·min−1·(mg of protein)−1 were found, compared with 0.1 pmol·min−1·(mg of protein)−1 in the microsomal fraction and the final supernatant.
Identification of the sequence encoding D-2-hydroxyglutarate dehydrogenase
D-2-Hydroxyglutarate dehydrogenase was further purified by chromatography on Blue Trisacryl and eluted from the column at a concentration of approx. 150 mM NaCl (results not shown). Further purification was obtained by chromatography on phenyl-Sepharose, where the enzyme eluted at the start of the ethylene glycol gradient (Figure 5, upper panel). The overall yield of these two steps was approx. 20% as estimated by the radiochemical assay, and the purification factor over the homogenate was approx. 1000-fold, assuming complete recovery of the enzyme after the DEAE-Sepharose column. SDS/PAGE indicated the presence of about 15 bands in the most purified fractions. Four of these, which co-purified with the D-2-hydroxyglutarate dehydrogenase activity in both the Blue Trisacryl (results not shown) and the phenyl-Sepharose columns (Figure 5), were cut from the gel, digested with trypsin and analysed by electrospray ionization–tandem MS. The upper three bands were identified as rat α1-macroglobulin, rat aldehyde oxidase and rat sulphite oxidase. The fourth gave a high score (five identical peptides; Figure 6) with a 536-residue mouse putative protein (gi/23271107) that displays approx. 30% sequence identity with D-lactate dehydrogenase from Kluyveromyces lactis (results not shown). It also matched (four identical peptides) a rat sequence (gi/27686389) corresponding to the last 150 residues of the above-mentioned mouse protein. As shown in Figure 6, the mouse protein was homologous with human hypothetical protein gi/22477764 (85% identity in the last 480 residues), as well as with Saccharomyces cerevisiae ‘actin-interacting protein 2’ (P46681). These sequences are more distantly related to D-lactate dehydrogenases, including the human enzyme (gi/29126992) [15].
Figure 5. Purification by chromatography on phenyl-Sepharose.

Upper panel: a preparation (9 mg of protein) purified by chromatography on DEAE-Sepharose and Blue Trisacryl was applied on to a phenyl-Sepharose column and eluted with a decreasing gradient of NaCl and an increasing gradient of ethylene glycol. Fractions of 30 ml (fraction 1) and 2 ml (fractions 2–13) were collected. D-2-Hydroxyglutarate dehydrogenase (•) was measured by the radiochemical assay on 10 μl of each fraction. Lower panel: SDS/PAGE of the fractions. The indicated bands were found to co-elute with the activity both in the Blue Trisacryl (results not shown) and the phenyl-Sepharose columns. They were cut from the gel, digested with trypsin and their identity was determined by electrospray ionization–tandem MS.
Figure 6. Sequence alignment of human D-2-hydroxyglutarate dehydrogenase (HsHd) and mouse D-2-hydroxyglutarate dehydrogenase (MmHd) with S. cerevisiae actin-interacting protein 2 (CsD2) and human D-lactate dehydrogenase (HsLd).

The peptidic sequences identified in the rat protein are underlined in the mouse sequence. Except for the last peptide (NVLGYSKPPVAVK in the rat sequence), they are identical in the sequences from both species. The predicted cleavage site for the mitochondrial presequence is double underlined.
Using the TargetP prediction program [16], the human and mouse putative D-2-hydroxyglutarate dehydrogenases were predicted to be mitochondrial proteins with scores of 0.781 and 0.916 respectively. This is in agreement with the subcellular fractionation mentioned above. The predicted mitochondrial presequence cleavage sites (RRG/CC in the human sequence, HRAY/S in the mouse sequence) are at similar positions in the two sequences. The N-terminal presequences were found to be poorly conserved (<20% identity) when the mouse and human proteins were compared. Removal of the prepeptide results in polypeptide chains with a predicted mass of approx. 53 kDa, in agreement with the molecular mass of the purified rat protein as determined by SDS/PAGE.
Expression of D-2-hydroxyglutarate dehydrogenase in HEK-293 cells
To confirm the identity of the protein, we overexpressed the human sequence encoding the putative D-2-hydroxyglutarate dehydrogenase in HEK-293 cells. As shown in Figure 7, overexpression of the protein significantly increased the conversion of radiolabelled DL-2-hydroxyglutarate. As expected, the activity on radiolabelled DL-2-hydroxyglutarate was stimulated by Zn2+ and Co2+ and was inhibited by unlabelled D-2-hydroxyglutarate at concentrations lower than 10-fold when compared with L-2-hydroxyglutarate (50% inhibition of the detritiation at 0.8 and 10 μM respectively). Allowing for a contamination of L-2-hydroglutarate by the D-isomer, this indicated that the sequence encoded D-2-hydroxyglutarate dehydrogenase.
DISCUSSION
Identification of D-2-hydroxyglutarate dehydrogenase
In the present study, we report the identification of an enzyme catalysing the following reaction:
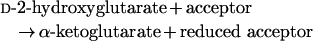 |
This enzyme also catalyses the oxidation of other D-2-hydroxy acids, particularly D-malate, D-lactate and mesotartrate, although at lower rates or with lower affinities than observed with D-2-hydroxyglutarate. Therefore this enzyme is best named as ‘D-2-hydroxyglutarate dehydrogenase’.
The activity of this enzyme could be measured in several ways, e.g. through the formation of a reduced acceptor or α-ketoglutarate or through the detritiation of DL-2-hydroxy[2-3H]glutarate. The first two types of assays were not very sensitive and could be used only on a partially purified enzyme, due to interferences in cruder material (presence of compounds reacting spontaneously with artificial electron acceptors; presence of substrates and enzymes, leading to the formation and/or the consumption of α-ketoglutarate).
The principle of the radiochemical assay is based on the fact that the reduced function in FADH2 [17] or in artificial acceptors (unlike in NAD or NADP) is a nitrogen or an oxygen, which easily exchanges protons with water. Since the tritiated substrate was a racemic mixture, this assay could be used to measure D-2-hydroxyglutarate dehydrogenase only after it had been separated from an enzyme acting on the L-form. This difficulty should be overcome by preparing the pure radiolabelled D-isomer. An advantage of this radiochemical assay is its high sensitivity, allowing us indeed to detect D-2-hydroxyglutarate dehydrogenase and to determine its very low Km value. The activity was less with DL-2-hydroxy[2-3H]glutarate compared with unlabelled substrate. The value of the primary kinetic isotopic effect (approx. 4.5) indicates that the breaking of the C2–H bond is probably a rate-limiting step in the reaction. This is reminiscent of the situation observed with L-2-hydroxyacid oxidase, for which a large (approx. 8) deuterium isotopic effect has been observed using L-lactate as a substrate [18].
Extensive purification of the enzyme allowed us to obtain sequence information on several candidate polypeptides. Three of them could be excluded because of their function (aldehyde oxidase 1; sulphite oxidase) or extracellular localization (α1-macroglobulin). A fourth protein, a hypothetical protein of unknown function, homologous with D-lactate dehydrogenase, was a logical candidate. Its identity with D-2-hydroxyglutarate dehydrogenase was confirmed by overexpression in HEK-293 cells. Extracts from these cells indeed catalysed the detritiation of DL-2-hydroxy[2-3H]glutarate with characteristics (activation by Zn2+ and Co2+, inhibition by unlabelled D-2-hydroxyglutarate at concentrations in the μmol range) similar to that of the enzyme partially purified from rat liver.
The sequence D-2-hydroxyglutarate dehydrogenase belongs to a family of enzymes (including D-lactate dehydrogenases and vanillyl-alcohol oxidase) that use FAD as a cofactor. Search of the pfam database (http://www.sanger.ac.Uk/Software/Pfam/) indicates the presence of a FAD-binding domain type 4 (residues 100–261) and a FAD-oxidase C-terminal domain (residues 275–516). The absence of any detectable effect of FAD on the enzyme activity was presumably due to a tight binding of this prosthetic group to the enzyme, as observed with many flavoproteins. Several D-lactate dehydrogenases appear to be Zn2+-dependent enzymes [14,19,20]. Similarly, D-2-hydroxyglutarate dehydrogenase is stimulated by Zn2+, Co2+ and Mn2+, and inhibited by the chelating agent EDTA.
Comparison with previous work
The enzyme that we have identified shares many of the properties of a D-2-hydroxyglutarate dehydrogenase described by Wanders and Mooyer [4]. The latter enzyme is indeed able to transfer electrons from its substrate to artificial acceptors, but not to NAD or NADP. Furthermore, it is found in several tissues, particularly liver and kidney. Our preliminary results indicate a similar tissue distribution for the low-Km enzyme described in the present study (E. van Schaftingen, unpublished work). However, neither the substrate specificity nor the identity of the reaction product has been reported by Wanders and Mooyer [4]. Furthermore, these authors mention that their enzyme has a Km value of 15 mM, which is more than three orders of magnitude higher than our estimate. One explanation for the discrepancy is that there are two distinct enzymes acting on D-2-hydroxyglutarate. The low concentration of D-2-hydroxyglutarate found in the plasma of control subjects (<1 μM according to Cremona and Singer [20]) indicates that the low-Km dehydrogenase that we describe would be of more importance to account for the physiological metabolism of this compound.
The D-2-hydroxyglutarate dehydrogenase described in the present study also shares several properties with a rabbit kidney D-2-hydroxyacid dehydrogenase characterized approx. 40 years ago [5–7]. Both enzymes are mitochondrial proteins that contain FAD, are inhibited by metal-chelating agents and reduce O2 at only a fraction of the rate at which they reduce DCIP in the presence of PMS [6,7]. Both of them act on D-malate, D-lactate and meso-tartrate, but either do not act or act poorly on L-lactate and L-malate [6]. Unfortunately, D-2-hydroxyglutarate was not tested on D-2-hydroxyacid dehydrogenase. A major difference between the two enzymes again appears to be in the affinity for the substrates. The Km value of 1 mM for D-malate found by Tubbs and Greville [6] is much higher than that found for D-2-hydroxyglutarate dehydrogenase, since, using the same type of assay as these authors, we found that the rate was already maximal with 0.25 mM D-malate. Furthermore, the competition experiments (see Figure 3) suggest that the Km value is probably lower than 10 μM. This large difference again suggests the existence of two different FAD-dependent dehydrogenases acting on D-2-hydroxy acids. One of them, described in the present study, acts best on D-2-hydroxyglutarate. The other would act best on D-lactate [6] and may therefore correspond to the sequence reported as D-lactate dehydrogenase [15].
The enzyme that we have isolated is probably also distinct from the trans-hydrogenase identified by Kaufman et al. [21]. This enzyme catalyses the reversible transfer of reducing equivalents from γ-hydroxybutyrate to α-ketoglutarate, leading to the formation of succinate semialdehyde and D-2-hydroxyglutarate. We have indeed observed that the activity of D-2-hydroxyglutarate dehydrogenase, measured by the radiochemical assay, was not affected by up to 5 mM γ-hydroxybutyrate (results not shown).
Physiological and pathophysiological implications
Since D-2-hydroxyglutarate dehydrogenase is most probably the main enzyme that consumes D-2-hydroxyglutarate, mutations in its gene are probably responsible for primary D-2-hydroxyglutaric aciduria [22]. D-2-Hydroxyglutarate also accumulates in glutaric aciduria type II [23] due to a deficiency in ETF (electron transfer flavoprotein) or in ETF dehydrogenase [24]. ETF and ETF dehydrogenase serve to transfer the electrons collected by dehydrogenases that are soluble in the mitochondrial matrix to the respiratory chain. The accumulation of D-2-hydroxyglutarate in glutaric aciduria type II therefore suggests that the ETF–ETF-dehydrogenase system also serves to transfer electrons from D-2-hydroxyglutarate dehydrogenase. It is worth stressing, in this respect, that D-2-hydroxyglutarate dehydrogenase appears to be easily released from mitochondria by freezing–thawing, indicating that it is not tightly bound to membranes but is probably well soluble in the mitochondrial matrix.
Assuming that the activity of the dehydrogenase is the same in both human liver and rat liver (approx. 15 m-units/g) and the activity at 37 °C is two times higher than that at 30 °C, it can be calculated that the liver can metabolize at least 40 mmol of D-2-hydroxyglutarate per day in a 70 kg adult. This value has to be compared with excretions of D-2-hydroxyglutaric acid at the rate of 4 mmol/mmol of creatinine [22], i.e. approx. 40 mmol/day. Therefore the enzyme that we have identified could account for the rate of disposal of D-2-hydroxyglutarate, particularly if one takes into account the fact that D-2-hydroxyglutarate dehydrogenase is also present in tissues other than liver.
Presence of an enzyme acting on L-2-hydroxyglutarate
Our findings also confirm the existence of an enzyme that utilizes L-2-hydroxyglutarate. Although we have not formally proven that it is a dehydrogenase, it is tempting to speculate that it is responsible for the oxidation of L-2-hydroxyglutarate by tissues observed more that 60 years ago by Weil-Malherbe [25] and its deficiency is responsible for L-2-hydroxyglutaric aciduria [26], a disease for which the enzyme defect is hitherto unknown. Further work will be devoted to its characterization and identification.
Acknowledgments
This work was supported by the Concerted Research Action programme of the Communauté Française de Belgique, the Interuniversity Attraction Poles Program–Belgian Science Policy and by the Fonds de la recherche Scientifique Médicale.
References
- 1.Chalmers R. A., Lawson A. M., Watts R. W., Tavill A. S., Kamerling J. P., Hey E., Ogilvie D. D-2-hydroxyglutaric aciduria: case report and biochemical studies. J. Inherit. Metab. Dis. 1980;3:11–15. doi: 10.1007/BF02312516. [DOI] [PubMed] [Google Scholar]
- 2.van der Knaap M. S., Jakobs C., Hoffmann G. F., Duran M., Muntau A. C., Schweitzer S., Kelley R. I., Parrot-Roulaud F., Amiel J., de Lonlay P., et al. D-2-hydroxyglutaric aciduria: further clinical delineation. J. Inherit. Metab. Dis. 1999;22:404–413. doi: 10.1023/a:1005548005393. [DOI] [PubMed] [Google Scholar]
- 3.Lindahl G., Lindstedt G., Lindstedt S. Metabolism of 2-amino-5-hydroxyadipic acid in the rat. Arch. Biochem. Biophys. 1967;119:347–352. doi: 10.1016/0003-9861(67)90463-8. [DOI] [PubMed] [Google Scholar]
- 4.Wanders R. J., Mooyer P. D-2-hydroxyglutaric acidaemia: identification of a new enzyme, D-2-hydroxyglutarate dehydrogenase, localized in mitochondria. J. Inherit. Metab. Dis. 1995;18:194–196. doi: 10.1007/BF00711764. [DOI] [PubMed] [Google Scholar]
- 5.Tubbs P. K. Effects of metal-complexing agents on mitochondrial D-α-hydroxy acid dehydrogenase. Biochem. Biophys. Res. Commun. 1960;3:513–517. doi: 10.1016/0006-291x(60)90166-2. [DOI] [PubMed] [Google Scholar]
- 6.Tubbs P. K., Greville G. D. The oxidation of D-α-hydroxy acids in animal tissues. Biochem. J. 1961;81:104–114. doi: 10.1042/bj0810104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cammack R. Assay, purification and properties of mammalian D-2-hydroxy acid dehydrogenase. Biochem. J. 1969;115:55–64. doi: 10.1042/bj1150055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delpierre G., Rider M. H., Collard F., Stroobant V., Vanstapel F., Santos H., van Schaftingen E. Identification, cloning and heterologous expression of a mammalian fructosamine-3-kinase. Diabetes. 2000;49:1627–1634. doi: 10.2337/diabetes.49.10.1627. [DOI] [PubMed] [Google Scholar]
- 9.Foultier B., Troisfontaines P., Vertommen D., Marenne M. N., Rider M. H., Parsot C., Cornelis G. R. Identification of substrates and chaperone from the Yersinia enterocolitica 1B Ysa type III secretion system. Infect. Immun. 2003;71:242–253. doi: 10.1128/IAI.71.1.242-253.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burlina A. Bergmeyer H. U., Bergmeyer J., Grassl M. Methods of Enzymatic Analysis. 3rd edn. vol. VII. Germany: Verlag Chemie, Weinheim; 1986. 2-Oxoglutarate; pp. 20–24. [Google Scholar]
- 11.Duley J., Holmes R. S. A spectrophotometric procedure for determining the activity of various rat tissue oxidases. Anal. Biochem. 1975;69:164–169. doi: 10.1016/0003-2697(75)90577-1. [DOI] [PubMed] [Google Scholar]
- 12.de Duve C., Pressman B. C., Gianetto R., Wattiaux R., Appelmans F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat liver tissue. Biochem. J. 1955;60:604–617. doi: 10.1042/bj0600604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boussif O., Lezoualc’h F., Zanta M. A., Mergny M. D., Scherman D., Demeneix B., Behr J. P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethyleneimine. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reed D. W., Hartzell P. L. The Archaeoglobus fulgidus D-lactate dehydrogenase is a Zn2+ flavoprotein. J. Bacteriol. 1999;181:7580–7587. doi: 10.1128/jb.181.24.7580-7587.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flick M. J., Konieczny S. F. Identification of putative mammalian D-lactate dehydrogenase enzymes. Biochem. Biophys. Res. Commun. 2002;295:910–916. doi: 10.1016/s0006-291x(02)00768-4. [DOI] [PubMed] [Google Scholar]
- 16.Emanuelsson O., Nielsen H., Brunak S., von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 17.Walsh C. W. H. New York: Freeman and Co.; 1979. Enzyme Reaction Mechanism; p. 376. [Google Scholar]
- 18.Cromartie T. H., Walsh C. Mechanistic studies on the rat kidney flavoenzyme L-α-hydroxy acid oxidase. Biochemistry. 1975;14:3482–3489. doi: 10.1021/bi00686a030. [DOI] [PubMed] [Google Scholar]
- 19.Gregolin C., Singer T. P. The lactic dehydrogenase of yeast. III. D(−)Lactic cytochrome c reductase, a zinc-flavoprotein from aerobic yeast. Biochim. Biophys. Acta. 1963;67:201–218. doi: 10.1016/0006-3002(63)91818-3. [DOI] [PubMed] [Google Scholar]
- 20.Cremona T., Singer T. P. The lactic dehydrogenases of yeast. V. Chemical properties and function of the zinc component of D-lactic cytochrome reductase. J. Biol. Chem. 1964;239:1466–1473. [PubMed] [Google Scholar]
- 21.Kaufman E. E., Nelson T., Fales H. M., Levin D. M. Isolation and characterization of a hydroxyacid-oxoacid transhydrogenase from rat kidney mitochondria. J. Biol. Chem. 1988;263:16872–16879. [PubMed] [Google Scholar]
- 22.Geerts Y., Renier W. O., Bakkeren J., de Jong J. 2-Hydroxyglutaric aciduria: a case report on an infant with the D-isomeric form with review of the literature. J. Neurol. Sci. 1996;143:166–169. doi: 10.1016/s0022-510x(96)00179-7. [DOI] [PubMed] [Google Scholar]
- 23.Watanabe H., Yamaguchi S., Saiki K., Shimizu N., Fukao T., Kondo N., Orii T. Identification of the D-enantiomer of 2-hydroxyglutaric acid in glutaric aciduria type II. Clin. Chim. Acta. 1995;238:115–124. doi: 10.1016/0009-8981(95)06074-n. [DOI] [PubMed] [Google Scholar]
- 24.Frerman F. E., Goodman S. I. Defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: glutaric acidemia type II. In: Scriver C. R., et al., editors. The Metabolic and Molecular Bases of Inherited Disease. 8th edn. vol. II. New York: McGraw-Hill Medical Publishing Division; 2001. pp. 2357–2365. [Google Scholar]
- 25.Weil-Malherbe H. The oxidation of L(−)α-hydroxyglutaric acid in animal tissues. Biochem. J. 1937;31:2080–2094. doi: 10.1042/bj0312080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duran M., Kamerling J. P., Bakker H. D., van Gennip A. H., Wadman S. K. L-2-Hydroxyglutaric aciduria: an inborn error of metabolism? J. Inherit. Metab. Dis. 1980;3:109–112. doi: 10.1007/BF02312543. [DOI] [PubMed] [Google Scholar]