

Abstract
UDP-glucose dehydrogenase catalyses the conversion of UDP-glucose into UDP-GlcA, a critical precursor for glycan synthesis across evolution. We have cloned the gene encoding this important enzyme from the opportunistic pathogen Cryptococcus neoformans. In this fungus, UDP-GlcA is required for the synthesis of capsule polysaccharides, which in turn are essential for virulence. The gene was expressed in Escherichia coli and the 51.3-kDa recombinant protein from wild-type and five mutants was purified for analysis. The cryptococcal enzyme is strongly inhibited by UDP-xylose and NADH, has highest activity at pH 7.5 and demonstrates Km (app) values of 0.1 and 1.5 mM for NAD+ and UDP-glucose respectively. Its activity was significantly decreased by mutations in the putative sites of NAD+ and UDP-glucose binding. Unlike previously reported eukaryotic UDP-glucose dehydrogenases, which are hexamers, the cryptococcal enzyme is a dimer.
Keywords: capsule, Cryptococcus neoformans, pathogenic fungi, UDP-GlcA, UDP-glucose dehydrogenase
Abbreviations: DTT, dithiothreitol; UDP-Glc, UDP-glucose; UDP-GlcA, UDP-glucuronic acid; UDP-GlcDH, UDP-glucose dehydrogenase; UDP-Xyl, UDP-xylose
INTRODUCTION
UDP-glucose dehydrogenase (UDP-GlcDH) is a critically important enzyme throughout biology, both for its direct role in generating UDP-GlcA (UDP-glucuronic acid), and because this product can be decarboxylated to yield UDP-Xyl (UDP-xylose). The formation of UDP-GlcA is initiated by NAD+-dependent oxidation of UDP-glucose (UDP-Glc), followed by nucleophilic attack of a catalytic cysteine on the resulting C6 aldehyde, transfer of the remaining hydride to a second molecule of NAD+ to form a thioester intermediate, and hydrolysis of this intermediate to form UDP-GlcA (literature recently reviewed in [1]). The resulting nucleotide sugar is utilized in processes ranging from capsule synthesis in bacteria and fungi to glycosaminoglycan synthesis and the hepatic glucuronidation of potential toxins in mammals. Additionally, UDP-GlcA may be converted into UDP-Xyl by UDP-GlcA decarboxylase [2]. UDP-Xyl is a key metabolite for the synthesis of structures including mammalian proteoglycans, plant xylans and xyloglucans and capsule polysaccharides in pathogenic fungi.
The enzyme responsible for UDP-GlcA synthesis was described and enriched from bovine liver in 1954 [3] and later purified from the same source [4], followed by extensive studies of its sequence [5–10] and unusual mechanism (literature reviewed in [1]). Crystal structure data from Streptococcus pyogenes further support the proposed mechanism of action, and demonstrate how a single active site can perform a 2-fold oxidation [1]. Many examples of the importance of UDP-GlcDH have been explored, including its role in normal embryonic development in Drosophila [7–9], Caenorhabditis elegans [11] and zebrafish [12], its differential expression in various plant tissues [13] and its requirement for virulence in bacterial systems [6,14,15].
Our interest in this enzyme stems from its role in providing biosynthetic precursors for the capsule of Cryptococcus neoformans, an opportunistic fungal pathogen [16,17]. This organism is distinguished among pathogenic yeasts by its polysaccharide capsule, an impressive structure that surrounds the cell wall and aids in thwarting the host immune response [18]. Although the capsule is not required for the growth of C. neoformans in culture, it is essential for its virulence. The more abundant of the two polysaccharides that comprise the capsule contains 15–20% GlcA [19], depending on the specific strain, and both polymers are heavily xylosylated [17]. Thus UDP-GlcDH is required both directly and indirectly to generate capsule precursors, and capsule in cells without this activity, if made at all, should lack both GlcA and xylose. Furthermore, it has been shown that disruption of the gene encoding UDP-glucuronate decarboxylase (UXS1; [2]) results in cells with capsule that lacks xylose; these cells are completely avirulent [20]. To examine further the pathways required for the synthesis of capsular polysaccharides in C. neoformans, we have cloned, expressed and characterized the cryptococcal UDP-GlcDH.
EXPERIMENTAL
Cloning UGD1
Using TRIzol® reagent (Gibco BRL, Gaithersburg, MD, U.S.A.) as described in [2], total RNA was isolated from a 200 ml culture of C. neoformans Cap67 in YPD medium (10 g/l yeast extract, 20 g/l Bacto-peptone and 20 g/l dextrose). RNA was reverse-transcribed to cDNA using 1 mM Clamp-oligo(dT) primer (BD Biosciences Clontech, Palo Alto, CA, U.S.A.) and 200 units of Superscript II reverse transcriptase (Gibco BRL). The resulting product was incubated with 2 units of RNase H (Gibco BRL), and used as a template for PCR using high-fidelity Platinum Taq DNA polymerase (Gibco BRL), sense primer DH-7 (5′-GGGGATATCATATGCACCATCACCACCATCACGGATCCATGGCCCCCATCACTGTTAAGAAGATCTGC-3′) and antisense primer DH-1 (5′-CAGAAAACTAGCACGAATGTTGGG-3′). The product was gel-purified and cloned into pCR2.1-TOPO (Invitrogen, Carlsbad, CA, U.S.A.). The coding region and 120 nt of the 3′-untranslated region were excised using NcoI and EcoRI, purified, and subcloned into similarly restricted pET24d (Novagen, Madison, WI, U.S.A.) to form pDH11.1. A C-terminal His6 tag was incorporated by PCR using DH-His-1 (5′-GAGTTCCTTGCCGAGGGTACCGC-3′) and DH-His-2 (5′-CCCAAGCTTGGGTTTTAATGATGATGATGATGATGAATACGGTCGCCAGTACC-3′) primers and pDH11.1 as template DNA. The resulting PCR fragment was digested with HindIII and KpnI and used to replace the corresponding fragment of pDH11.1, yielding pDH-His. DNA sequencing (Protein Nucleic Acid Chemistry Laboratory, Washington University School of Medicine, St. Louis, MO, U.S.A.) confirmed correct sequence in each construct, and the genomic and coding sequences were submitted to GenBank® (AAK95561).
Mutagenesis
QuikChange site-directed mutagenesis reagents (Stratagene, La Jolla, CA, U.S.A.) were used to mutate five residues to alanine. Residue positions and primers used are listed in Table 1, and all the mutations were confirmed by DNA sequencing.
Table 1. Primers used to generate mutations to alanine at the indicated positions.
All sequences are listed 5′ to 3′.
| Mutant | 5′-primer | 3′-primer |
|---|---|---|
| C278A | GGCGTCTGTCGGTTTCGGTGGTAGCGCTTTCCAGAAGGACATTCTCAACTTGG | CCAAGTTGAGAATGTCCTTCTGGAAAGCGCTACCACCGAAACCGACAGACCC |
| R43A | GACCTCAACCAGCAGGCAATTCACGCCTGGAAC | GTTCCAGGCGTCAATTGCCTGCTGGTTGAGGTC |
| R262A | GTCGGTAAGGACACGGCAATGGGTTCCAAGTTCC | GGAACTTGGAACCCATTGCCGTGTCCTTACCGAC |
| K341A | CTTGGTTGGGCCTTCGCGAAGGACACTGGTGAC | GTCACCAGTGTCCTTCGCGAAGGCCCAACCAAG |
| R348A | GGACACTGGTGACACTGCAGAGTCTCCTTCCATCGGC | GCCGATGGAAGGAGACTCTGCAGTGTCACCAGTGTCC |
Protein expression and purification
For expression of protein, either pDH-His or the empty pET24d vector was transformed into Escherichia coli strain BL21 Gold (DE3) pLysS (Stratagene). A single colony from each transformation was grown to an A600 of 0.5 at 37 °C in 500 ml Luria–Bertani containing 60 μg/ml kanamycin and 34 μg/ml chloramphenicol. The culture was transferred to 25 °C for 1 h and then induced with 0.1 mM isopropyl β-D-thiogalactoside at 25 °C overnight. Cells were collected by centrifugation, washed once with cold deionized water, resuspended in 15 ml of lysis buffer [50 mM Tris/HCl, pH 8.8/10% (v/v) glycerol/1 mM EDTA/1 mM DTT (dithiothreitol)/0.5 mM PMSF] and stored at −80 °C in 3 ml aliquots for convenient small-scale enzyme preparations. For use, one aliquot of cells was thawed, adjusted to 100 μg/ml lysozyme and supplemented with protease inhibitors (final concentrations of 5 μg/ml antipain, trypsin inhibitor and leupeptin; 1 μg/ml o-phenanthroline; 0.1 mM aminocaproic acid; 1.6 μg/ml benzamidine). The suspension was incubated for 30 min at 30 °C with vortex-mixing every 10 min, and sonicated with a microtip sonicator on ice for 3×1 min intervals alternating with 1 min rest. Whole cells and cell debris were pelleted (SS34 rotor; 13000 rev./min for 30 min at 4 °C), and the supernatant fraction was removed and immediately used for UDP-GlcDH assays or purification.
For purification, 1.5 ml of the supernatant fraction from sonication was mixed with an equal volume of binding buffer (50 mM NaH2PO4/300 mM NaCl, pH 7.0) and passed over a 2.5 ml column of TALON Superflow metal affinity resin (Clontech #8908-2), pre-equilibrated in the same buffer. The column was washed three times with 3 ml of binding buffer containing 10 mM imidazole, and proteins were eluted with 3×1 ml of binding buffer containing 250 mM imidazole. Most of the protein eluted in the second elution fraction. This was supplemented with 5 mM DTT and stored at 4 °C; the protein and activity were assayed within 48 h. Purity of this fraction was estimated to be >95%.
UDP-GlcDH assay
For assays of UDP-GlcDH, 29 μg of protein was incubated at 25 °C in a final volume of 50 μl of 40 mM Tris/HCl (pH 7.5), 2 mM pyruvate, 4.5 units of L-lactate dehydrogenase (Sigma) and the concentrations of NAD+ and UDP-Glc indicated. In experiments to determine the Km value for UDP-Glc, concentrations of up to 5 mM UDP-Glc were tested in the presence of 2 mM NAD+. Under these conditions, <30% of the UDP-Glc was used in the 10 min incubation period. For experiments to determine the Km value for NAD+, concentrations of up to 2 mM NAD+ were tested in the presence of UDP-Glc at levels 5–7-fold above the Km value for the enzyme preparation being tested: 2 mM UDP-Glc for mutant enzymes and 10 mM UDP-Glc for wild-type enzyme. (Depending on the exact proportion of substrate used at each concentration, the Km and kcat values may be slightly overestimated.) Reactions were stopped by vortex-mixing with 50 μl of cold phenol (pH 4)/chloroform/deionized water (3:3:1, by vol.) followed by centrifugation (16000 g for 5 min at room temperature 25 °C). (Here and elsewhere deionized water indicates 18 MΩ water from a Millipore system.) A 25 μl portion of the upper phase was removed, and the lower phase was re-extracted with 80 μl of deionized water. An 80 μl portion of the second upper phase was combined with the initial 25 ml, and 80 μl of that mixture was analysed by HPLC (Waters, Milford, MA, U.S.A.) on a 250 mm×4.6 mm quaternary amine-silica gel anion-exchange column (Hypersil 5 μ SAX gel; Phenomenex, Torrance, CA, U.S.A.) run at 1.5 ml/min. After injection, the column was washed for 5 min with buffer A (5 mM phosphate buffer, pH 3.2) and eluted with a 20 min linear gradient from 0 to 60% buffer B (0.6 M KH2PO4) as described in [21].
Antibody production and immunoblotting
A large-scale preparation of UDP-GlcDH for antibody production was generated by first inducing a 0.5 litre culture (A600∼0.6) of BL21(De3)pLysS cells harbouring pDH11.1 with 1 mM isopropyl β-D-thiogalactoside for 3 h at 30 °C. Cells were washed with water and resuspended in lysis buffer as described above. The entire quantity of cells was then lysed in a French Pressure cell (7.6×106 Pa). After two successive centrifugation steps (6000 and 20000 g) the pellet, enriched with inclusion bodies, was solubilized in 1 ml of inclusion body extraction buffer (0.1 M Tris/HCl, pH 9.1/2% sarcosyl/0.5 mM PMSF/2 mM DTT; 1 or 2 ml of initial packed cells) and incubated for 3 h at room temperature with slow rocking. The sample was then brought to 0.25 mM L-cystine from a 50 mM stock dissolved in 0.1 M NaOH and incubated for an additional 3.5 h. Protein was dialysed and concentrated by ultrafiltration (Millipore, Billerica, MA, U.S.A.) to 3 mg/ml for use in polyclonal antibody production in rabbits (Covance Research Products, Denver, PA, U.S.A.).
For immunoblotting, C. neoformans, Tremella mesenterica or Saccharomyces cerevisiae were resuspended in lysis buffer, mixed with an equal volume of glass beads (Biospec Products, Bartlesville, OK, U.S.A.) and vortex-mixed vigorously for 3×1 min, alternating with 1 min on ice. Total protein (10 mg) from fungal preparations or E. coli cells (expressing empty vector or pDH11.1) was mixed with an equal volume of 2× SDS/PAGE sample buffer and boiled (5 min) before subjecting to SDS/PAGE (12% gel). Immunoblotting was performed by standard methods using 1:5000 dilutions of both primary antiserum and secondary antibody (anti-rabbit IgG conjugated with horseradish peroxidase; Amersham Biosciences, Piscataway, NJ, U.S.A.), and the Western Lightning Chemiluminescence Reagent (Applied Biosystems, Foster City, CA, U.S.A.) for detection.
Gel filtration
For gel filtration, purified His-tagged protein at 0.4, 2 or 4 mg/ml was analysed on a Sephacryl S300 HR column equilibrated with 20 mM Tris/HCl (pH 7.0), 100 mM NaCl, 1 mM DTT and 10% glycerol. The column was calibrated with samples from a molecular-mass standard kit (Sigma), run individually; elution volumes for the standards fit well (R2=0.99) when plotted against molecular mass.
RESULTS AND DISCUSSION
Conserved peptide domains from plant UDP-GlcDH were used to identify the corresponding partial sequences in translations of C. neoformans expressed sequence tag and genomic databases. These sequences guided the primer design for amplification of a cryptococcal cDNA, which encoded a 51.3 kDa protein with extensive sequence homology with known UDP-GlcDH enzymes (Figure 1). Protein alignment indicated that critical amino acids, such as the catalytic cysteine (residue 278 in the C. neoformans sequence) and the N-terminal GXGXXG NAD+-binding motif, were strictly conserved. Examination of the genomic DNA sequence from C. neoformans showed that the corresponding gene was composed of 15 exons. No other homologues of the protein were detected in cryptococcal genome sequence.
Figure 1. Alignment of sequences encoding UDP-GlcDH.

An alignment of sequences from S. pyogenes (GenBank® accession number Q07172), Arabidopsis thaliana (NP_197053), Homo sapiens (AAC36095) and C. neoformans (AAK95561) is shown, with filled boxes indicating identical residues. Asterisks under the cryptococcal sequence indicate positions that were mutagenized to alanine.
To facilitate purification of the putative UDP-GlcDH, the gene was modified to incorporate the sequence encoding a C-terminal His6 tag. Comparison of enzyme activity in crude preparations of cells expressing either tagged or untagged protein showed no detectable effect of the His tag. For further studies, the recombinant protein was expressed at high levels in E. coli and purified on cobalt resin (Figure 2). In the presence of NAD+, the purified protein readily converted UDP-Glc into UDP-GlcA as judged by an HPLC assay (Figure 3), and the gene encoding the enzyme was correspondingly named UGD1 (for UDP-Glc dehydrogenase). Time-course experiments (results not shown) indicated that the pure enzyme was progressively inhibited by the accumulation of NADH formed during conversion of UDP-Glc into UDP-GlcA. To avoid this and to facilitate kinetic analysis, the assay was modified by the inclusion of pyruvate and lactate dehydrogenase to consume the NADH.
Figure 2. UDP-GlcDH expression and purification.
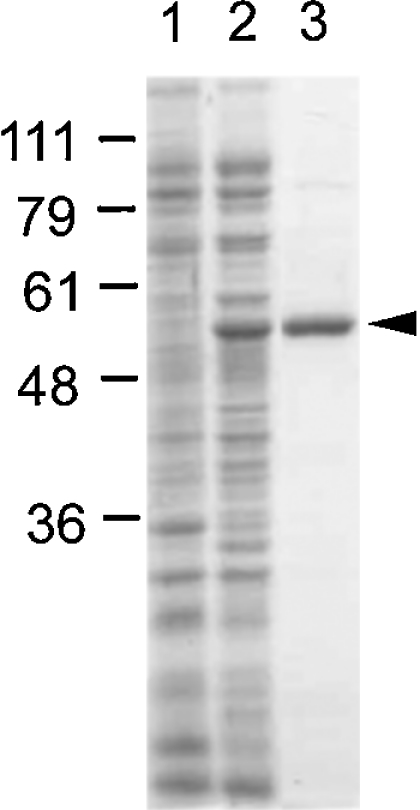
Bacterial proteins analysed by SDS/PAGE and stained with Coomassie Blue. Standards (in kDa) are shown on the left. Lane 1, 20 μg of soluble protein from E. coli expressing plasmid only; lane 2, 20 μg of soluble protein from E. coli expressing His-tagged UDP-GlcDH; lane 3, 2 μg of UDP-GlcDH purified by cobalt chromatography. A minor contaminant is barely visible at approx. 90 kDa.
Figure 3. UDP-GlcDH activity of purified protein.

Assays were performed and products were analysed by HPLC as described in the Experimental section using 1 mM UDP-Glc and 2 mM NAD+. ----, 0 min incubation; —, 10 min incubation. Elution position of standards is indicated above the profile.
As tested by the modified assay, recombinant UDP-GlcDH had an optimum pH of 7.5, with a broad optimum temperature of 25–37 °C. Moderate inhibition of activity was observed in the presence of UDP-GlcA, the immediate product of the enzyme, but there was no inhibition by UDP, UTP or UDP-Gal. In contrast, the enzyme was strongly inhibited by NADH and by the downstream metabolite, UDP-Xyl (Table 2); these effects were similar to those observed in cytosolic extracts of C. neoformans [22]. Mn2+ at high concentrations was inhibitory, and EDTA slightly enhanced the activity, presumably by chelating bivalent cations.
Table 2. Inhibitors of UDP-GlcDH.
Assays were performed in the presence of the indicated compounds, as described in the Experimental section using 1 mM UDP-glucose. The amount of UDP-GlcA formed is expressed as a percentage of an assay with no additions. Results are expressed as means±S.D. for triplicate samples and are representative for at least four independent experiments.
| Addition | Concentration (mM) | % of control |
|---|---|---|
| 0 time control | – | 0 |
| – | – | 100±4 |
| NADH* | 1 | 1.3±0.2 |
| UDP-Xyl | 1 | 0.5±0.1 |
| UDP-GlcA† | 1 | 75±5 |
| UDP | 1 | 111±2 |
| UDP-Gal | 1 | 98±6 |
| Mn2+ | 1 | 95±5 |
| Mn2+ | 10 | 19±2 |
| EDTA | 1 | 110±4 |
* Lactate dehydrogenase and pyruvate were omitted from this assay.
† The product peak in this reaction was corrected for the amount of UDP-GlcA added.
Biochemical and biophysical studies from Feingold and others (reviewed in [1]) indicate that the functional unit of UDP-GlcDH is a dimer, a conclusion supported by X-ray structure analysis, which indicates that a residue required for the activity (Arg-262) actually occurs in a loop of the second molecule of the enzyme. However, in bacteria the dimer is the basic structural unit of the protein [1,23], whereas in the higher eukaryotes studied the protein is hexameric, occurring as a ‘trimer of dimers’ [24–27]. Gel-filtration studies of recombinant cryptococcal UDP-GlcDH at various concentrations showed that it elutes in all cases between the expected masses for a monomer and a dimer, with higher concentrations yielding elution volumes closer to the value predicted for a dimer (results not shown). In no case did we observe data consistent with a higher-order oligomer. The most probable interpretation of these results is that the fungal protein occurs in a monomer–dimer equilibrium, unlike the hexamers of other eukaryotic proteins studied.
To compare the cryptococcal enzyme with UDP-GlcDH of other organisms, we examined its kinetic properties (Figure 4). The Km (app) of 1.5 mM for UDP-Glc was slightly higher than that reported for other organisms, whereas the Km value for NAD+ was in the middle of the reported range; in general, the enzyme appears to function at a similar range of concentrations (Table 3). These values were also close to those measured for this enzyme in the cytosol of C. neoformans [22].
Figure 4. Cryptococcal UDP-GlcDH product formation.

Assays were performed in the presence of 2 mM NAD+ and the indicated concentrations of UDP-Glc. Results were analysed by non-linear least-squares curve-fitting using Kaleidograph software.
Table 3. Km values of UDP-GlcDH from selected prokaryotic and eukaryotic organisms.
To assess whether amino acids implicated in binding and catalysis by the known X-ray structure were critical for cryptococcal UDP-GlcDH activity, we generated site-specific mutations of the residues indicated in Figure 1 converting each into an alanine residue. Mutation of the catalytic Cys-278 yielded a completely non-functional enzyme, as expected (Table 4). Mutation of two residues that are suggested to bind the cofactor (Arg-43 and Arg-348) yielded severely impaired enzymes, with the expected increase in the Km value for NAD+. Mutation of two residues with roles in binding UDP-Glc (Arg-262 and Lys-341) caused an almost complete loss of activity. Residue 262, as mentioned above, occurs in a loop of one member of a dimer, yet contributes to the UDP-Glc-binding site of the second. The fact that mutating this amino acid abrogates activity of the protein supports the conclusion from gel-filtration studies that the functional unit of the cryptococcal enzyme is a dimer.
Table 4. Kinetic parameters of mutant and wild-type UDP-GlcDH from C. neoformans.
Equal amounts of recombinant protein were used in all assays. Km values for UDP-Glc were determined in the presence of 2 mM NAD+, with UDP-Glc varied from 0 to 5 mM; Km for NAD+ was determined in the presence of 10 mM UDP-Glc for wild-type and 2 mM UDP-Glc for mutants, with NAD varied from 0 to 2 mM. Results are expressed as means±S.E.M.
| Km (mM) | |||
|---|---|---|---|
| Mutation | kcat (s−1) | UDP-Glc | NAD+ |
| None | 2.7±0.1×10−1 | 1.5±0.1 | 0.1±0.01 |
| C278A | – | – | – |
| R43A | 5.5±0.2×10−3 | 0.4±0.03 | 0.8±0.2 |
| R348A | 2.8±0.1×10−2 | 0.4±0.04 | 0.4±0.04 |
| R262A | – | – | – |
| K341A | – | – | – |
We used polyclonal antiserum raised against recombinant Ugd1p to investigate the protein expression in several fungi. UDP-GlcDH is clearly expressed in C. neoformans (Figure 5), and in T. mesenterica, a related non-pathogenic fungus, which produces a similar but more highly xylosylated capsule polysaccharide [28–30]. There was no evidence of cross-reacting material in the model yeast S. cerevisiae, consistent with the absence of any related protein sequence encoded by the S. cerevisiae genome. Wild-type C. neoformans cells were also grown under low-iron conditions that induce the formation of larger capsules [31], but there was no apparent alteration of protein expression (results not shown). Presumably, levels of UDP-Glc are sufficiently high to support increased levels of capsule synthesis without additional induction.
Figure 5. UDP-GlcDH is expressed in several basidiomycetes but not in S. cerevisiae.

Total cell protein was separated by SDS/PAGE and immunoblotted with polyclonal anti-UDP-GlcDH as described in the Experimental section. –, E. coli expressing plasmid alone; +, E. coli expressing UDP-GlcDH; C, C. neoformans; T, T. mesenterica; S, S. cerevisiae. Standards (in kDa) are shown on the left. Regions of the gel not shown displayed no specific bands corresponding to UDP-GlcDH.
UDP-GlcDH in C. neoformans shares most of the characteristics of the enzymes from higher eukaryotes, in terms of mass and composition (e.g. amount of cysteine and tryptophan residues [23]). However, unlike the other eukaryotic enzymes that have been studied, it appears to function as a dimer rather than a hexamer. We expect that cryptococcal cells with impaired or absent UDP-GlcDH will be unable to generate UDP-GlcA and UDP-Xyl. The overall effects on cell physiology will depend on which cellular components are modified by these sugars, but the polysaccharide capsule will certainly be greatly altered, as it will lack both GlcA and xylose. On the basis of the results when only xylose is absent from the capsule [20], cells lacking UDP-GlcDH activity are probably avirulent. It remains to be seen whether the difference between the cryptococcal enzyme and its counterpart in the mammalian host can be exploited for antifungal therapy.
Acknowledgments
We appreciate helpful discussions with members of the Doering laboratory, including cloning advice from H. Liu and comments on the paper from S. Klutts. This work was supported by NIH R01 GM66303, an Andrew and Virginia Craig Faculty Research Award and a Burroughs Wellcome Fund Junior Investigator in Molecular Pathogenic Mycology Award to T.L.D.
References
- 1.Campbell R. E., Mosimann S. C., van De Rijn I., Tanner M. E., Strynadka N. C. The first structure of UDP-glucose dehydrogenase reveals the catalytic residues necessary for the two-fold oxidation. Biochemistry. 2000;39:7012–7023. [PubMed] [Google Scholar]
- 2.Bar-Peled M., Griffith C. L., Doering T. L. Functional cloning and characterization of a UDP-glucuronic acid decarboxylase: the pathogenic fungus Cryptococcus neoformans elucidates UDP-xylose synthesis. Proc. Natl. Acad. Sci. U.S.A. 2001;98:12003–12008. doi: 10.1073/pnas.211229198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strominger J. L., Kalckar H. M., Axelrod J., Maxwell E. S. Enzymatic oxidation of uridine diphosphate glucose to uridine diphosphate glucuronic acid. J. Am. Chem. Soc. 1954;76:6411–6412. [Google Scholar]
- 4.Zalitis J., Feingold D. S. Purification and properties of UDPGDH from beef liver. Arch. Biochem. Biophys. 1969;132:457–465. doi: 10.1016/0003-9861(69)90389-0. [DOI] [PubMed] [Google Scholar]
- 5.Hempel J., Perozich J., Romovacek H., Hinich A., Kuo I., Feingold D. S. UDP-glucose dehydrogenase from bovine liver: primary structure and relationship to other dehydrogenases. Protein Sci. 1994;3:1074–1080. doi: 10.1002/pro.5560030710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dougherty B. A., van de Rijn I. Molecular characterization of hasB from an operon required for hyaluronic acid synthesis in group A streptococci. Demonstration of UDP-glucose dehydrogenase activity. J. Biol. Chem. 1993;268:7118–7124. [PubMed] [Google Scholar]
- 7.Binari R. C., Staveley B. E., Johnson W. A., Godavarti R., Sasisekharan R., Manoukian A. S. Genetic evidence that heparin-like glycosaminoglycans are involved in wingless signaling. Development. 1997;124:2623–2632. doi: 10.1242/dev.124.13.2623. [DOI] [PubMed] [Google Scholar]
- 8.Haerry T. E., Heslip T. R., Marsh J. L., O'Connor M. B. Defects in glucuronate biosynthesis disrupt Wingless signaling in Drosophila. Development. 1997;124:3055–3064. doi: 10.1242/dev.124.16.3055. [DOI] [PubMed] [Google Scholar]
- 9.Hacker U., Lin X., Perrimon N. The Drosophila sugarless gene modulates Wingless signaling and encodes an enzyme involved in polysaccharide biosynthesis. Development. 1997;124:3565–3573. doi: 10.1242/dev.124.18.3565. [DOI] [PubMed] [Google Scholar]
- 10.Spicer A. P., Kaback L. A., Smith T. J., Seldin M. F. Molecular cloning and characterization of the human and mouse UDP-glucose dehydrogenase genes. J. Biol. Chem. 1998;273:25117–25124. doi: 10.1074/jbc.273.39.25117. [DOI] [PubMed] [Google Scholar]
- 11.Hwang H. Y., Horvitz H. R. The Caenorhabditis elegans vulval morphogenesis gene sqv-4 encodes a UDP-glucose dehydrogenase that is temporally and spatially regulated. Proc. Natl. Acad. Sci. U.S.A. 2002;99:14224–14229. doi: 10.1073/pnas.172522499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walsh E. C., Stainier D. Y. UDP-glucose dehydrogenase required for cardiac valve formation in zebrafish. Science. 2001;293:1670–1673. doi: 10.1126/science.293.5535.1670. [DOI] [PubMed] [Google Scholar]
- 13.Seitz B., Klos C., Wurm M., Tenhaken R. Matrix polysaccharide precursors in Arabidopsis cell walls are synthesized by alternate pathways with organ-specific expression patterns. Plant J. 2000;21:537–546. doi: 10.1046/j.1365-313x.2000.00696.x. [DOI] [PubMed] [Google Scholar]
- 14.Arrecubieta C., Lopez R., Garcia E. Molecular characterization of cap3A, a gene from the operon required for the synthesis of the capsule of Streptococcus pneumoniae type 3: sequencing of mutations responsible for the unencapsulated phenotype and localization of the capsular cluster on the pneumococcal chromosome. J. Bacteriol. 1994;176:6375–6383. doi: 10.1128/jb.176.20.6375-6383.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang K. W., Weng S. F., Tseng Y. H. UDP-glucose dehydrogenase gene of Xanthomonas campestris is required for virulence. Biochem. Biophys. Res. Commun. 2001;287:550–555. doi: 10.1006/bbrc.2001.5591. [DOI] [PubMed] [Google Scholar]
- 16.Casadevall A., Perfect J. R. Washington, DC: American Society for Microbiology; 1998. Cryptococcus neoformans. [Google Scholar]
- 17.Bose I., Reese A. J., Ory J. J., Janbon G., Doering T. L. A yeast under cover: the capsule of Cryptococcus neoformans. Eukaryotic Cell. 2003;2:655–663. doi: 10.1128/EC.2.4.655-663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buchanan K. L., Murphy J. W. What makes Cryptococcus neoformans a pathogen? Emerg. Infect. Dis. 1998;4:71–83. doi: 10.3201/eid0401.980109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cherniak R., Valafar H., Morris L. C., Valafar F. Cryptococcus neoformans chemotyping by quantitative analysis of 1H nuclear magnetic resonance spectra of glucuronoxylomannans with a computer-simulated artificial neural network. Clin. Diagn. Lab. Immunol. 1998;5:146–159. doi: 10.1128/cdli.5.2.146-159.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moyrand F., Klaproth B., Himmelreich U., Dromer F., Janbon G. Isolation and characterization of capsule structure mutant strains of Cryptococcus neoformans. Mol. Microbiol. 2002;45:837–849. doi: 10.1046/j.1365-2958.2002.03059.x. [DOI] [PubMed] [Google Scholar]
- 21.Bar-Peled M., Lewinsohn E., Fluhr R., Gressel J. UDP-rhamnose:flavanone-7-O-glucoside-2″-O-rhamnotransferase, purification and characterization of an enzyme catalyzing the production of bitter compounds in citrus. J. Biol. Chem. 1991;266:20953–20959. [PubMed] [Google Scholar]
- 22.Jacobson E. S. Cryptococcal UDP-glucose dehydrogenase: enzymic control of capsular biosynthesis. J. Med. Vet. Mycol. 1987;25:131–135. doi: 10.1080/02681218780000201. [DOI] [PubMed] [Google Scholar]
- 23.Schiller J. G., Lamy F., Frazier R., Feingold D. S. UDP-glucose dehydrogenase from Escherichia coli. Purification and subunit structure. Biochim. Biophys. Acta. 1976;453:418–425. doi: 10.1016/0005-2795(76)90137-9. [DOI] [PubMed] [Google Scholar]
- 24.Gainey P. A., Pestell T. C., Phelps C. F. A study of the subunit structure and the thiol reactivity of bovine liver UDP glucose dehydrogenase. Biochem. J. 1972;129:821–830. doi: 10.1042/bj1290821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franzen J. S., Ashcom J., Marchetti P., Cardamone J. J., Feingold D. S. Induced vs. preexisting asymmetry models for half-of-the-sites reactivity in bovine liver uridine diphosphoglucose dehydrogenase. Biochim. Biophys. Acta. 1980;614:242–255. doi: 10.1016/0005-2744(80)90214-4. [DOI] [PubMed] [Google Scholar]
- 26.Jaenicke R., Rudolph R. Dissociation and in vitro reconstitution of bovine liver uridine diphosphoglucose dehydrogenase. The paired subunit nature of the enzyme. Biochemistry. 1986;25:7283–7287. doi: 10.1021/bi00371a006. [DOI] [PubMed] [Google Scholar]
- 27.Franzen J. S., Marchetti P. S., Lockhart A. H., Feingold D. S. Special effects of UDP-sugar binding to bovine liver uridine diphosphoglucose dehydrogenase. Biochim. Biophys. Acta. 1983;746:146–153. doi: 10.1016/0167-4838(83)90068-7. [DOI] [PubMed] [Google Scholar]
- 28.Slodki M. E., Wickerham L. J., Bandoni R. J. Extracellular heteropolysaccharides from Cryptococcus and Tremella. A possible taxonomic relationship. Can. J. Microbiol. 1966;12:489–494. doi: 10.1139/m66-071. [DOI] [PubMed] [Google Scholar]
- 29.Cherniak R., Jones R. G., Slodki M. E. Type-specific polysaccharides of Cryptococcus neoformans. N.m.r.-spectral study of a glucuronomannan chemically derived from a Tremella mesenterica exopolysaccharide. Carbohydr. Res. 1988;182:227–239. doi: 10.1016/0008-6215(88)84005-9. [DOI] [PubMed] [Google Scholar]
- 30.Fraser C. G., Jennings H. J., Moyna P. Structural analysis of an acidic polysaccharide from Tremella mesenterica NRRL Y-6158. Can. J. Biochem. 1973;51:219–224. doi: 10.1139/o73-027. [DOI] [PubMed] [Google Scholar]
- 31.Pierini L. M., Doering T. L. Spatial and temporal sequence of capsule construction in Cryptococcus neoformans. Mol. Microbiol. 2001;41:105–115. doi: 10.1046/j.1365-2958.2001.02504.x. [DOI] [PubMed] [Google Scholar]
- 32.Schiller J. G., Bowser A. M., Feingold D. S. Partial purification and properties of UDPG dehydrogenase from Escherichia coli. Biochim. Biophys. Acta. 1973;293:1–10. doi: 10.1016/0005-2744(73)90369-0. [DOI] [PubMed] [Google Scholar]
- 33.Campbell R. E., Sala R. F., van de Rijn I., Tanner M. E. Properties and kinetic analysis of UDP-glucose dehydrogenase from group A streptococci. Irreversible inhibition by UDP-chloroacetol. J. Biol. Chem. 1997;272:3416–3422. doi: 10.1074/jbc.272.6.3416. [DOI] [PubMed] [Google Scholar]
- 34.Turner W., Botha F. C. Purification and kinetic properties of UDP-glucose dehydrogenase from sugarcane. Arch. Biochem. Biophys. 2002;407:209–216. doi: 10.1016/s0003-9861(02)00500-3. [DOI] [PubMed] [Google Scholar]
- 35.Bdolah A., Feingold D. S. Decarboxylation of uridine diphosphate-D-glucuronic acid by an enzyme preparation from hen oviduct. Biochem. Biophys. Res. Commun. 1965;21:543–546. doi: 10.1016/0006-291x(65)90519-x. [DOI] [PubMed] [Google Scholar]