

Abstract
The SOR (sulphur oxygenase reductase) is the initial enzyme in the sulphur-oxidation pathway of Acidianus ambivalens. Expression of the sor gene in Escherichia coli resulted in active, soluble SOR and in inclusion bodies from which active SOR could be refolded as long as ferric ions were present in the refolding solution. Wild-type, recombinant and refolded SOR possessed indistinguishable properties. Conformational stability studies showed that the apparent unfolding free energy in water is approx. 5 kcal·mol−1 (1 kcal=4.184 kJ), at pH 7. The analysis of the quaternary structures showed a ball-shaped assembly with a central hollow core probably consisting of 24 subunits in a 432 symmetry. The subunits form homodimers as the building blocks of the holoenzyme. Iron was found in the wild-type enzyme at a stoichiometry of one iron atom/subunit. EPR spectroscopy of the colourless SOR resulted in a single isotropic signal at g=4.3, characteristic of high-spin ferric iron. The signal disappeared upon reduction with dithionite or incubation with sulphur at elevated temperature. Thus both EPR and chemical analysis indicate the presence of a mononuclear iron centre, which has a reduction potential of −268 mV at pH 6.5. Protein database inspection identified four SOR protein homologues, but no other significant similarities. The spectroscopic data and the sequence comparison led to the proposal that the Acidianus ambivalens SOR typifies a new type of non-haem iron enzyme containing a mononuclear iron centre co-ordinated by carboxylate and/or histidine ligands.
Keywords: electron paramagnetic resonance (EPR), hyperthermophilic archaea, quaternary structure, reversible denaturation, sulphur oxidation
Abbreviations: BN, Blue-native; GdmCl, guanidinium chloride; KPi buffer, potassium phosphate buffer; SOR, sulphur oxygenase reductase; Sox, sulphur-oxidizing
INTRODUCTION
Elemental sulphur and reduced inorganic sulphur compounds are metabolized by a large number of micro-organisms belonging to the domains of the Bacteria and the Archaea. Elemental sulphur is the electron donor in various sulphur-oxidation pathways in aerobic lithotrophic species (reviewed in, e.g., [1–3]) and is the electron acceptor in respiratory chains of anaerobic heterotrophic and autotrophic micro-organisms [4]. Many studies have been conducted to elucidate the mechanisms of inorganic sulphur compound oxidation. In most cases, water-soluble substrates such as thiosulphate or tetrathionate were used (e.g. [3,5,6]). However, much less is known about the oxidation of the virtually insoluble elemental sulphur (5 μg/l of water at 25 °C [7]).
Soluble glutathione-dependent and periplasmatic sulphur oxygenases have been identified in the mesoacidophilic bacteria such as Acidithiobacillus ferrooxidans and Acidithiobacillus thiooxidans; however, there is not much known about the biochemistry and molecular biology of the enzymes (e.g. [8–10]). More is known about the periplasmatic multi-enzyme Sox (sulphur-oxidizing) system of the neutrophilic, facultatively lithoautotrophic bacterium Paracoccus pantotrophus GB17 and other species [1,5,11,12]. This system catalysed the thiosulphate-, sulphite-, sulphur- and hydrogen sulphide-dependent cytochrome c reduction in vitro. These findings, together with the analysis of genetic and genomic data, led to the suggestion that we observe the “emergence of a common mechanism” [3] of the oxidation of inorganic sulphur compounds by the Sox system in many different neutrophilic bacteria; however, Sox proteins or genes have not been found in the acidophiles. According to this model, the oxidation of these compounds occurs completely in the enzyme-bound state without any free intermediates.
Less is known about sulphur-oxidation pathways in the Archaea. A soluble SOR (sulphur oxygenase reductase) was purified from aerobically growing cells of the thermoacidophilic crenarchaeote Acidianus ambivalens, which catalysed the simultaneous oxidation and reduction of elemental sulphur in the presence of oxygen, with sulphite, thiosulphate and hydrogen sulphide as products [13]. Cofactors or an external electron donor for sulphur reduction were not required, and the two enzyme activities could not be separated. The enzyme consisted of a multimer of identical subunits. Its molecular mass deduced from the gene sequence was 35188 Da (the N-terminal methionine is cleaved off in the wild-type enzyme [14]). As the SOR was the only sulphur-oxidizing enzyme found in Acidianus ambivalens, it was thus assumed that the enzyme catalysed the initial step of the sulphur-oxidizing pathway in this organism. Considering that its cytoplasmic location does not contribute to the formation of a transmembrane proton gradient, it is likely that it provides soluble precursors for membrane-bound enzymes such as a thiosulphate:quinone oxidoreductase [14a].
A similar enzyme had been purified from a phylogenetically uncharacterized isolate tentatively termed “Sulfolobus brierleyi”. A moderate 18O incorporation into sulphite was demonstrated; however, a sulphur reductase activity was not reported [15]. In addition, a sor gene from Acidianus sp. strain S5 was cloned and expressed heterologously in Escherichia coli [16]. The three enzymes had comparable properties [17,18]. So far, little is known about the cofactor content, the reaction mechanism, the number of subunits and the exact quaternary structure of the SOR.
In the present study, we show that a low-potential mononuclear non-haem iron centre is present in the Acidianus ambivalens SOR and that it plays an essential role in catalysis. We also show that the purified complex is a large oligomer in solution, having a ferritin-like assembly. We propose that the sulphur-oxidation pathway in Acidianus ambivalens is distinctly different from the Sox pathway found in numerous mesophilic aerobic and anaerobic oxidizers of sulphur compounds.
EXPERIMENTAL
Heterologous gene expression and purification of recombinant SOR from E. coli
The sor gene was amplified from genomic Acidianus ambivalens DNA using the primers SoroN (5′-TAGACATCTAGATAACGAGGGCAAAAAATGCCGAAACCATACGT-3′) and SoroC (3′-AGAAAAAGCTTAAGCGCTTTGTTCGTTTAAATATTCTC-5′). The product was digested with XbaI and Eco47III and ligated into the pASK 75-plasmid [19] double-digested with the same enzymes, resulting in a gene fusion encoding a C-terminal ten-amino-acid streptavidin tag. The construct was transformed into E. coli BL 21 Codon plus cells (Stratagene). Attempts to construct an expression plasmid with the wild-type sequence failed.
The expression of the sor gene was induced by addition of anhydrotetracycline (200 μg/l of culture) to a 15 l culture growing at 37 °C at a D600 of 0.8 and incubated overnight after induction with vigorous aeration. The harvested cells (77 g wet mass) were resuspended in 770 ml of 100 mM potassium phosphate (KPi) buffer, pH 8, containing 1% (v/v) Tween 20 and 50 μM EDTA. The cells were disrupted in a cell mill (Vibrogen V/3) by shaking with glass beads for 20 min. After removal of the glass beads by filtration, the crude extract was centrifuged at 48000 g for 20 min. The supernatant (700 ml, 3.5 g of protein) containing the soluble SOR was heat-treated for 45 min at 75 °C to precipitate heat-labile E. coli proteins and centrifuged again under the same conditions. The supernatant obtained was dialysed against 20 mM Tris/HCl, pH 7.5. A total volume of 100 ml of the dialysed protein fraction was applied to a DEAE-Sepharose fast-flow column (60 ml bed volume; all columns and column materials were from Amersham Biosciences) and eluted with an increasing gradient of NaCl in the same buffer. The recombinant SOR eluted at 150 mM NaCl. Active fractions were pooled. The protein was sedimented by ultracentrifugation at 42000 rev./min (Beckman 45 Ti rotor) for 16 h. The pellet was resuspended in 50 mM Tris/HCl, pH 7.5, and was frozen at −20 °C.
Unfolding and refolding of SOR from inclusion bodies
The pellet of the crude E. coli extract containing large amounts of inclusion bodies was resuspended in KPi buffer, pH 8, containing 1.5% sodium deoxycholate, 1 M urea, 10% (v/v) glycerol and 2 mM EDTA, by sonication for 5 min (Branson Sonifier II) and was washed four times in the same buffer. The washed inclusion body fraction (approx. 5 g) was resuspended in 50 ml of 8 M urea containing 100 μM ferric citrate and 30 mM dithiothreitol. After denaturation at 60 °C for 1 h, the solution was centrifuged at 48000 g for 30 min to remove particulate material. Refolding of the SOR was performed aerobically at 4 °C by a two-step dialysis against 50 mM Tris/HCl, pH 7.2, containing 2 mM 2-mercaptoethanol and 100 μM ferric citrate, with 2 M urea in the first step, and no urea in the second step. Residual ferric citrate and 2-mercaptoethanol were removed from the refolded enzyme preparation in another dialysis step against 50 mM Tris/HCl, pH 7.5. The protein solution was centrifuged under the same conditions to remove precipitated protein and subjected to the SOR activity assay.
Alternatively, unfolded inclusion bodies were dialysed three times against 8 M urea without ferric citrate to remove residual iron from the samples. The samples were subsequently refolded with 0, 10 or 100 μM ferric citrate as described above.
Denaturant-induced protein unfolding
Stock solutions of GdmCl (guanidinium chloride) and urea for protein unfolding of the soluble recombinant SOR were prepared following published recommendations [20], and final molarities were calculated from refractive index measurements. Denaturation curves were determined at 25 °C using wild-type SOR in 50 mM Tris/HCl buffer, pH 7, at a final concentration of 0.01 mg/ml. The unfolding process was monitored by fluorescence spectroscopy monitoring tryptophan emission (300–450 nm; excitation, 292 nm; 1 cm cell) on a Varian Eclipse fluorimeter. The spectroscopic data were processed in order to determine the average emission wavelength at each denaturant concentration [21], and the resulting unfolding transitions were analysed using a two-state model, allowing for the calculation of thermodynamic parameters [22]. Direct fits to the experimental unfolding curves were performed using the following expression:
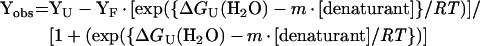 |
where Yobs, YU and YF are the observed spectroscopic signal, denaturant protein baseline and folded protein baseline respectively, m is a constant of proportionality, R is the gas constant and T is the absolute temperature (in K). The transition midpoints were calculated as ΔGU(H2O)/m or by direct inspection of the transitions.
For the determination of the molecular masses of unfolding intermediates and of the partially unfolded proteins, 200 μl portions of a 1 mg/ml solution of recombinant SOR were dialysed against 0–8 M urea in 20 mM KPi buffer, pH 6.5, at room temperature (22–25 °C) for 48–72 h. Dialysed protein fractions (200 μl) were applied to a Superose 6 HR 10/30 gel-permeation column equilibrated with 20 mM Tris/HCl buffer, pH 7.5, and 150 mM NaCl not containing any urea. Identically treated samples (45 μg each) were separated by Blue-native (BN) PAGE [23] by applying a 4–15% polyacrylamide gradient in 14 cm×14 cm×0.15 cm gels, run in an SE400 vertical electrophoretic chamber. The molecular masses of the SOR bands were calculated from retardation coefficients after electrophoresis on BN gels of different polyacrylamide concentrations (Ferguson plot; 5.5, 6.5, 7.5, 8.5, 9.5 and 10.5% polyacrylamide; Sigma Technical Bulletin No. MKR-137 available at http://www.sigmaaldrich.com/sigma/bulletin/mwnd500bul.pdf). The MW ND-500 kit, containing lactalbumin, carbonic anhydrase, ovalbumin, BSA and urease, was used for standards, in addition to catalase, ferritin and thyroglobulin (all from Sigma).
Purification of wild-type SOR from Acidianus ambivalens
Acidianus ambivalens DSM 3772 was grown aerobically according to published procedures [24,25]. A novel protocol for the preparation of wild-type SOR from Acidianus ambivalens was developed. Crude-extract preparation and sucrose density-gradient centrifugation were performed as described previously [13]. Active fractions were combined and dialysed against 0.5 M (NH4)2SO4 and 20 mM Tris/HCl, pH 7.5. The protein solution was loaded on a phenyl-Sepharose column, equilibrated with the same buffer, and eluted with a decreasing (NH4)2SO4 gradient in a HiLoad system 26/10. The SOR was eluted at around 50 mM (NH4)2SO4 and was pooled according to activity. The active fractions were combined, dialysed against 20 mM Tris/HCl, pH 7.5, applied to a DEAE-Sepharose fast-flow column as described above, and eluted with an increasing NaCl gradient in the same buffer. The SOR eluted at around 150 mM NaCl. The active fractions were pooled and concentrated using a Jumbosep ultrafiltration unit (cut-off size 30 kDa) and stored at −20 °C.
The purified proteins from the purification and refolding protocols were finally run over a Superose 6 HR 10/30 gel-permeation column in 20 mM Tris/HCl, pH 7.5, and 150 mM NaCl. Thyroglobulin, ferritin, catalase, aldolase, chicken albumin and carbonic anhydrase (Sigma) were used for calibration and determination of the holoenzyme and subunit masses.
Biochemical and analytical methods
The SOR activity assay was performed aerobically at 85 °C in 70 mM Tris/HCl, pH 7.2, containing 0.1% (v/v) Tween 20, as previously described [13]. After incubation for different time intervals, the products were determined colorimetrically. It was assumed that sulphite and sulphide are the sole primary products and that thiosulphate resulted from a non-enzymic reaction between sulphite and sulphur [13]. One unit of enzyme activity was defined as the formation of 1 μmol of sulphite plus thiosulphate (oxygenase reaction) or 1 μmol of hydrogen sulphide (reductase reaction) per min. The reaction velocity was calculated from the linear increase of product concentrations.
A concentration of 0.5% (v/v) CHAPS was used for the Km and kcat determinations, and the assay temperature was lowered to 65 °C to slow down the reaction velocity. The determinations of the two Km values for sulphur were performed using the rate of formation of the products sulphite and sulphide separately.
Enzyme assays for inhibition studies were usually performed with EDTA-washed sulphur prepared as follows: 50 g of sulphur (Merck) was dispersed in 500 ml of enzyme buffer with 10 mM EDTA by sonication for 5 min with a Branson Sonifier (II) followed by centrifugation at 48000 g for 15 min three times each. EDTA was removed by washing the sulphur over a filter with 5 litres of double-deionized water. Potassium tetrathionate and unwashed sulphur were alternatively used as substrates. The following reagents were tested for potentially inhibitory effects: EDTA, ZnCl2, CaCl2, MgCl2, Fe(III) citrate, (NH4)2Fe(SO4)2 and sodium citrate. The reagents were added to the enzyme-containing assay mixture. After 30 min of incubation at room temperature, the pH was readjusted to 7.2 with a solution of 1 M Tris base. Raising the temperature to 85 °C started the enzymic reaction. An enzymic reaction without inhibitor and a non-enzymic reaction with inhibitor, but without SOR, were used as controls. The interference of the substances with the colorimetric assays was determined by adding mixtures of sodium sulphite, sodium thiosulphate and sodium sulphide (50 μM each) in enzyme buffer to the colorimetric assay reactions with inhibitor concentrations equal to the ones used in the enzymic assays.
The protein concentration was determined by the Bradford [26] or BCA (bicinchoninic acid) methods [27]. The protein concentration of pure SOR preparations was determined by measuring the A280 using the calculated molar absorption coefficient, ε=57840 M−1·cm−1 (from sequence). Sample purity was assessed using SDS/10% polyacrylamide gels [28] and visualized with colloidal Coomassie Blue (Roti-Blue; Roth, Karlsruhe, Germany). The iron content was determined by the 2,4,6-tripyridyl-1,3,5-triazine method [29].
Spectroscopic methods and redox titration
UV/visible spectra were recorded at room temperature in a Shimadzu spectrophotometer. CD spectra were obtained by use of a Jasco J810 spectropolarimeter. The CD spectra were obtained using a cuvette of 0.2 cm path length and at a scanning speed of 50 nm/min and a bandwidth of 2 nm. Scans were collected at 1 nm intervals with a response time of 0.25 s and accumulated ten times. The CD spectra were corrected by subtracting the spectrum of the buffer solution (20 mM KPi, pH 6.5). The spectra between 187 and 250 nm were used to calculate the α-helical and β-sheet content of the native, recombinant and refolded SOR preparations using the programs CONTINLL and SELCON3 included in the CDPro software package and a reference protein set containing 37 proteins [30].
EPR spectra were recorded on a Bruker ESP380 spectrometer, equipped with an ESR 900 continuous flow helium cryostat. Acidianus ambivalens wild-type SOR was titrated anaerobically in 50 mM KPi, pH 6.5, by sequential addition of alkaline buffered sodium dithionite and continuously flushing the cell with argon. EPR samples were withdrawn at the desired potential and were immediately frozen in liquid nitrogen. The intensity of the g=4.3 resonance was followed as a function of the reduction potential of the solution. The following compounds were used as redox mediators (40 μM each, E′0 in parentheses): 1,2-naphthoquinone-4-sulphonic acid (215 mV); 1,2-naphthoquinone (180 mV); trimethylhydroquinone (115 mV); phenazine methosulphate (80 mV); phenazine ethosulphate (55 mV); Methylene Blue (11 mV); Indigo Tetrasulphonate (−30 mV); resorufin (−51 mV); Indigo Trisulphonate (−70 mV); Indigo Disulphate (−182 mV); 2,5-hydroxy-p-benzoquinone (−130 mV); 2-hydroxy-1,4-naphthoquinone (−152 mV); anthraquinone-2-sulphonate (−225 mV); phenosafranine (−255 mV); safranine (−280 mV); Neutral Red (−325 mV); Benzyl Viologen (−359 mV); and Methyl Viologen (−446 mV). A platinum and a silver/silver chloride electrode were used, calibrated against a saturated quinhydrone solution at pH 7.0. The reduction potentials are quoted in respect to the standard hydrogen electrode.
Electron microscopy
SOR samples were diluted to a protein concentration of 0.1 mg·ml−1, applied to glow-discharged carbon-coated copper grids and negatively stained with 2% uranyl acetate. Electron micrographs were taken using a slow-scan CCD (charge-coupled device) Tietz camera mounted to a Philips CM 12 electron microscope operated at 120 kV. Image analysis by single particle averaging was performed as described previously [31,32] using the EM program [33] and the SEMPER software [34].
Sequence analysis
The amino acid sequence of the Acidianus ambivalens SOR was compared with sequences in the public databases using different BLAST tools for the identification of homologues. SOR sequences were retrieved and aligned using the alignment tools PileUp, ClustalW and HmmerAlign included in the Wisconsin package (Accelrys). The PredictProtein server and the programs within were used to predict secondary-structure elements (http://cubic.bioc.columbia.edu/predictprotein/). The number of atoms was calculated using ProtParam (http://www.expasy.org/tools/protparam.html). The Acidithiobacillus ferrooxidans genome was queried at the TIGR web server (http://www.tigr.org).
RESULTS
Heterologous gene expression in E. coli and purification of recombinant and wild-type SOR
A sor gene expression plasmid was constructed with the open reading frame fused to the streptavidin tag at the 3′ end. Soluble E. coli extracts harvested 20 h after induction of sor gene expression showed an enzyme activity of 464 m-units of oxidase/mg of protein and 145 m-units of reductase/mg of protein. From the extract (3.5 g of protein), 120 mg of SOR was isolated. The specific activity of the purified recombinant soluble enzyme was 2.82 units of oxygenase/mg of protein and 0.66 unit of reductase/mg of protein, at 85 °C.
At 2 h after induction, the occurrence of the first inclusion bodies was observed microscopically. Approx. 80% of the protein in the pellet fraction after cell lysis consisted of SOR (results not shown). After extensive washing, the yield was 2 g of inclusion body fraction (wet mass)/l of E. coli culture. The inclusion body fraction was unfolded in 8 M urea and refolded in a two-step dialysis. The activities of the recombinant enzyme from inclusion bodies were up to 5.98 units of oxygenase/mg of protein and 1.35 units of reductase/mg of protein, when refolded in the presence of ferric iron (see below).
Wild-type SOR (10.5 mg) was purified using a novel protocol from 175 mg of soluble Acidianus ambivalens cell extract via sucrose density-gradient centrifugation and two chromatographic steps. The specific activities were 2.96 units of oxygenase/mg of protein and 0.60 unit of reductase/mg of protein. SDS/PAGE showed that the different procedures yielded pure enzyme (Figure 1). Apart from the major 36 kDa band, two faint low-molecular-mass bands were observable, but N-terminal sequencing revealed that they were proteolysis products of the 36 kDa subunit (results not shown).
Figure 1. SDS/10% polyacrylamide gel stained with colloidal Coomassie Blue with purified wild-type and recombinant SOR.

(A) Wild-type SOR (6.3 μg of protein), (B) recombinant SOR refolded from inclusion bodies (6.3 μg of protein) and (C) recombinant SOR purified from the soluble E. coli fraction (4.2 μg of protein). M, molecular mass markers (in kDa).
SOR is a multimeric protein composed of dimer building blocks
The apparent molecular masses of wild-type, recombinant soluble and refolded holoenzymes determined by gel-permeation chromatography were approx. 550 kDa, showing that the enzyme preparations from E. coli adopted a near-native state. Recombinant SOR preparations were dialysed extensively against different urea concentrations and were analysed with gel-permeation chromatography. An additional peak corresponding to a molecular mass of 72 kDa equivalent to a SOR dimer appeared in preparations containing 3 M urea or above, whereas the holoenzyme peak decreased with increasing urea concentration (Figure 2A). A peak corresponding to 35 kDa or the SOR monomer appeared in preparations dialysed against 6 M urea or above, whereas the dimer and holoenzyme peaks diminished. The 72 kDa peak was also observed in varying intensities in samples of refolded and wild-type SOR not treated with any urea (results not shown).
Figure 2. Gel permeation chromatography, enzyme activity and BN gels of recombinant soluble SOR dialysed against various concentrations of urea as described in the Experimental section.

(A) Merged gel permeation chromatograms given with the respective urea concentration. The protein peaks were monitored by measuring the A280. The elution volumes of thyroglobulin, ferritin, aldolase, chicken albumin and carbonic anhydrase are given with their molecular masses, together with the holoenzyme, dimer and monomer peaks. (B) Relative specific activities of the SOR fractions after dialysis against different urea concentrations as indicated below the gel (C). The oxygenase (black) and reductase activities (grey) of the 0 M urea control were each set to 100%. (C) BN gradient gel of SOR preparations dialysed against different urea concentrations re-stained with colloidal Coomassie Blue after electrophoresis. (D) As in (C) showing a ladder of oligomers of the partially denatured enzyme. H, holoenzyme; D*, dimer denatured; D, dimer; M, monomer; F, migration front.
SOR activity was observed with up to 5 M urea, but not above (Figure 2B). Comparative analysis of all peak fractions by SDS/PAGE under reducing and non-reducing conditions showed only the 36 kDa band; the absence of a dimer band indicated that intersubunit disulphide bridges were not present (results not shown).
Identically treated samples separated with non-denaturing BN gels showed that the holoenzyme band dominated in samples dialysed against 0–3 M urea (Figures 2C and 2D). Intensely stained bands corresponding to monomer and dimer sizes and in lesser intensity to higher oligomers were seen in samples dialysed against 6 M urea and above. In the 2–4 M urea lanes, a faint ladder of partially denatured protein was observed. A dominant dimer band appeared in these lanes, which had a different migration pattern compared with those of the fully denatured samples (Figure 2D). The determination of the apparent molecular mass of the holoenzyme by BN PAGE gave a value of 732 kDa.
Conformational stability of Acidianus ambivalens SOR
The conformational stability of wild-type Acidianus ambivalens SOR was investigated by inducing unfolding at neutral pH using the denaturants GdmCl and urea. Protein unfolding was monitored by following the emission fluorescence spectrum of tryptophan residues, which were used as intrinsic probes for protein stability. The reversible denaturation curves were found to have midpoints at 3.4 M GdmCl and 5.9 M urea (Figure 3), when analysed using a two-state model for the folding/unfolding reaction. The protein apparent folding free energy in water was determined to be approx. 5 kcal·mol−1 (1 kcal=4.184 kJ), at pH 7 (Figure 3, see legend for details). This low value is explained by the fact that, although the native conformation is essential for activity, its stability in respect to the unfolded state is very low, typically ranging from 5 to 15 kcal·mol−1 [22]. Dialysis of the unfolded protein in the presence of high denaturant concentrations restored the initial emission spectra, indicating that the observed chemical-denaturant-induced unfolding transition is reversible. This was corroborated further by the fact that active SOR could be obtained from inclusion bodies dissolved in 8 M urea.
Figure 3. GdmCl (A) and urea (B) denaturation curves.

The solid lines are a least-squares analysis fit of the experimental data to a two-state F↔U model yielding the following parameters: ΔG(H2O)=5.04 kcal/mol, m=1.49 kcal/mol per M and [GdmCl]1/2=3.38 M (A); ΔG(H2O)=4.13 kcal/mol, m=0.7 kcal/mol per M and [urea]1/2=5.89 M (B). It should be noted that m depends on the amount of polypeptide chain that is exposed to solvent upon unfolding, and thus varies with denaturants [22]. The insets are plots of the variation of ΔGU in the transition region as a function of denaturant concentration, according to the ΔGU=ΔG(H2O)−m·[denaturant] equation.
Electron microscopy reveals a globular multimer
In transmission electron micrographs of negatively stained wild-type and SOR preparations, globular particles of 15.5 nm diameter were visible [13]. The inner core appeared densely stained. Electron micrographs of recombinant protein gave essentially the same results (Figures 4A and 4B). Single particle averaging of individual wild-type particles revealed two distinct subpopulations occurring in similar amounts. One subpopulation showed a 4-fold (Figure 4C), the other a 2- or 3-fold symmetry axis (Figure 4D), indicating that the holoenzyme is a highly symmetrical particle.
Figure 4. Electron micrographs of negatively stained SOR, and image reconstruction.

(A, B) Micrographs of wild-type and recombinant SOR respectively; the bar represents 100 nm; (C, D) results of single particle averaging of SOR particles with 2-fold and 3-fold symmetry axis respectively.
Secondary-structure analysis reveals an almost equal distribution of α-helices and β-sheets
CD spectra of the different preparations were rather similar, with minima around 220 and 207 nm, and a maximum around 195 nm, and minor differences in the molar ellipticity at 187–197 nm and at 220 nm (Figure 5). Secondary-structure analysis of the wild-type SOR spectrum resulted in 23.8±1.5% α-helix and 23.4±0.8% β-sheet content. The recombinant and refolded preparations showed an α-helical content of 25.2±0.6% and 25.7±0.9% and a β-sheet content of 26.2±0.5% and 24.2±0.2% respectively. The average length of the helices predicted was nine amino acids. Secondary-structure prediction programs gave 29% α-helical and 27% β-sheet content, which is in good agreement with those calculated from the CD spectra.
Figure 5. CD spectra of wild-type (light grey), recombinant soluble (dark grey) and recombinant refolded (black) SOR.

All the CD spectra were displayed relative to the buffers used. The y-axis represents the mean residue molar ellipticity obtained using the protein concentration, the path length and the number of protein residues for calculation (wild-type, 308; recombinant, 318).
Iron is essential for SOR activity
The determination of the iron content of wild-type SOR preparations resulted in a ratio of 1.0 iron atom per subunit of SOR. For the two recombinant SOR preparations, ratios of 2.2 (soluble) and 1.4 (refolded) iron were calculated per subunit. When no ferric citrate was added to the unfolding and refolding buffers and the unfolded protein solution was extensively dialysed against 8 M urea before refolding to remove residual iron, the specific activities were 0.006 units of oxygenase/mg of protein and 0 unit of reductase/mg of protein. A parallel sample, refolded in the presence of 10 μM ferric citrate, had specific activities of 0.29 unit of oxygenase/mg of protein and 0.06 unit of reductase/mg of protein, whereas the activities were 5.98 units of oxygenase/mg of protein and 1.35 units of reductase/mg of protein, when refolded in the presence of 100 μM ferric citrate.
EPR spectroscopy reveals a mononuclear non-haem iron centre
The UV/visible spectra of SOR did not show any feature besides the protein absorption at 280 nm in the reduced or oxidized state (results not shown). EPR spectroscopy performed at liquid-helium temperature of wild-type SOR showed an isotropic resonance at g=4.3 in the ‘as prepared’ state, consistent with the presence of a mononuclear non-haem iron centre in the ferric high-spin state and a rhombicity of 0.33 (Glu/Asp; Figure 6). Both recombinant and refolded proteins showed the same type of signal, although with a higher intensity. Adequate controls showed that the observed signal was from the enzyme and not from the buffers used.
Figure 6. EPR spectrum of wild-type Acidianus ambivalens SOR.

Temperature, 4.6 K; protein concentration, 226 μM; microwave power, 2.4 mW; modulation amplitude, 1 mT. Inset, redox titration of the SOR mononuclear iron centre. The normalized intensities of the g=4.3 resonance are plotted against the reduction potential. The line represents a Nernst equation for a one-electron step transition, with an Em=−268 mV.
The intensity of the g=4.3 signal decreased upon incubation with elemental sulphur at 80 °C up to 20 min. The signal disappearance was not reversible after the SOR had been washed with oxidized buffer following the reaction. This observation suggests that an intermediate may remain bound to the metal reaction site upon catalysis. The E′0 of the mononuclear iron centre was determined in an EPR-monitored redox titration experiment to be −268 mV (Figure 6, inset).
Catalytic properties of wild-type and recombinant SOR
The apparent Km values for sulphur, determined with the oxygenase and the reductase reaction separately at 65 °C, were 23 mM and 13 mM respectively, calculated on the basis of S°, not S8. The kcat values were 2.2 s−1 and 0.1 s−1 (oxygenase and reductase respectively).
Several substrates and potential inhibitors were tested for their effects on SOR activity (Table 1). The SOR was not active with tetrathionate. Metal chelators, such as EDTA and sodium citrate, were not inhibitory even in concentrations as high as 100 mM in the case of EDTA. In contrast, EDTA showed a slight stimulatory effect (Table 1), which was also seen in enzymic assays with partially denatured SOR (results not shown).
Table 1. Effects of various substrates and reagents on SOR activity.
The substrate (sulphur) concentration was 78 mM.
| Activity (%) | ||||
|---|---|---|---|---|
| Substrate | Reagent | Reagent concentration (mM) | Oxygenase | Reductase |
| Sulphur (S8) | – | – | 100 | 100 |
| EDTA | 1 | 127 | 143 | |
| EDTA | 10 | 150 | 141 | |
| EDTA | 50 | 160 | 108* | |
| EDTA | 100 | 134 | 13* | |
| Ca2+ | 1 | 82 | 101 | |
| Mg2+ | 1 | 110 | 92 | |
| Sodium citrate | 1 | 103 | 85 | |
| Sulphur (S8), EDTA-washed | – | – | 104 | 94 |
| EDTA | 10 | 147 | 124 | |
| Zn2+ | 0.5 | 5 | 16 | |
| EDTA/Zn2+ | 1/0.5 | 114 | 86 | |
| Fe(III) citrate | 0.1 | 102 | 19* | |
| Fe(III) citrate | 0.01 | 96 | 1* | |
| (NH4)2Fe(SO4)2 | 0.1 | 105 | 150 | |
| (NH4)2Fe(SO4)2 | 0.01 | 110 | 90 | |
| No sulphur | K2 Tetrathionate | 10 | 0 | 0 |
* Interference with the colorimetric assays.
We tried to determine whether or not the EDTA effect was due to the binding of potentially inhibitory bivalent cations. Ca2+ and Mg2+ had no effect on the enzyme activity (Table 1), whereas Zn2+ showed a strong inhibition, as observed previously [13]. The effect of Zn2+ was abolished when EDTA was also added to the assay mixture. To determine whether inhibitory traces of transition metals in the substrate sulphur caused the stimulatory effect of EDTA, sulphur was washed with EDTA-containing buffer. The incubation of washed sulphur with additional EDTA in the assay buffer resulted in the same increase in activity as determined with the unwashed sulphur (Table 1). The addition of low amounts of Fe2+ or Fe3+ neither inhibited nor increased the activity in low concentrations, but interfered with the colorimetric assays, thus preventing the use of higher concentrations.
Comparison of SOR amino acid sequences
Homologous sor genes were also found in BLAST searches in the genomes of the archaea Sulfolobus tokodaii, Ferroplasma acidarmanus and the hyperthermophilic bacterium Aquifex aeolicus; in addition, the sor gene from Acidianus sp. strain S5 had already been described [16]. The pairwise similarities of the deduced amino acid sequences compared with the Acidianus ambivalens enzyme ranged from 39% (Aquifex aeolicus) to 88% (Acidianus S5; Figure 7). Extensive database searches using PSIBLAST and other search tools did not provide any further motifs or significant similarities to other proteins. No similar gene was found when searching the genome sequence of the sulphur- and iron-oxidizing mesophilic bacterium Acidithiobacillus ferrooxidans.
Figure 7. Multiple alignment of SOR sequences.

#, conserved cysteine residues; *, His-Glu-Xaa-Xaa-His motif known from proteins with mono- and di-iron oxo clusters; †, putative iron-binding residue found in biopterin-dependent tyrosine hydroxylases [35]; cylinders, predicted α-helical regions; open arrows, β-sheet regions for the Acidianus ambivalens SOR. Ac.ambi., Acidianus ambivalens (GenBank® accession number X56616); Ac. S5, Acidianus sp. strain S5 (GenBank® accession number AF267286); Su.toko., Sulfolobus tokodaii (GenBank® accession number NC_003106); Fe.acid., Ferroplasma acidarmanus (Faci1674; GenBank® accession number NZ_AABC02000001); Aq.aeol., Aquifex aeolicus (GenBank® accession number NC_000918). Consens., consensus sequence. Immediately upstream of the Ferroplasma acidarmanus Faci1674 open reading frame, an in-frame stop codon was found (X, position 49). It was treated as an unknown residue, because of the similarity of the deduced amino acid sequences of the upstream region to the other SOR sequences. It is not known whether this represents a pseudogene or whether Ferroplasma acidarmanus produces active SOR under suitable conditions. Black shading indicates identical residues, and grey shading indicates conservative exchanges in at least four of the sequences.
DISCUSSION
In the present paper, we report a biochemical and functional characterization of the recombinant and the wild-type Acidianus ambivalens SOR, focusing on the enzymic activity, on the first description of one of the redox active centres, and on evidence regarding the quaternary structure of the enzyme.
The specific activities of several preparations of recombinant, refolded and wild-type SOR were comparable, as well as the results of the electron microscopy, the gel-permeation chromatography, and the CD and EPR spectra, suggesting that: (i) the recombinant enzyme possess similar properties to those of the wild-type enzyme, (ii) the purification and refolding procedures gave reliable results, and (iii) the C-terminal ten-amino-acid streptavidin tag did not impair the function of the enzyme.
The Km of 18 mM (average of the oxygenase and reductase tests), and the kcat of 2.2 s−1 and 0.1 s−1 respectively, suggest a low substrate affinity and enzyme velocity. A similar Km of 50 mM had been reported for the oxygenase reaction of the “Sulfolobus brierleyi” enzyme [15]. A possible explanation for the unfavourable values lies in the poor solubility of sulphur [7] and, therefore, low substrate availability, despite the large excess in the assay thus preventing the enzyme from reaching maximal velocity.
The results of the refolding experiments with different iron concentrations showed that its presence was essential for the catalytic activity of the refolded SOR. Slightly higher iron/subunit ratios were observed in the recombinant enzyme preparations as compared with the wild-type enzyme; however, the strong EPR signal of the oxidized ‘as prepared’ SOR showed that iron is probably present in a mononuclear centre in each subunit. The lack of any absorbance of the enzyme in the UV/visible spectrum, besides the absorption at 280 nm, points to histidine and/or carboxylate, but not cysteine, ligation of the iron, as observed in many oxygenases with mononuclear or binuclear non-haem iron centres (e.g. [35]).
The reduction potential of the iron is, to our knowledge, the lowest observed for mononuclear centres (Em=−268 mV). Other proteins known to harbour colourless mononuclear non-haem iron centres show clearly positive E′0 values, typically ranging from +200 to +600 mV (e.g. [36–39]). This low reduction potential most probably originates from a different protein environment around the iron centre. Nevertheless, the reduction potential is low enough to explain the observed reductase activity of the enzyme (E′0 for H2S/S°=−270 mV [40]).
A tight binding of the iron is suggested by the observation that EDTA was not inhibitory at all; in contrast, it stimulated the enzyme activity even in assays with partially denatured enzyme (results not shown). The removal of potentially inhibitory traces of transition metals bound to the SOR in the ‘as isolated’ state could be a reason for this effect. This explanation is supported further by the observations that: (i) the specific activities of the enzyme refolded from inclusion bodies were often higher than those obtained after regular purification, (ii) the inhibitory effect of Zn2+ could be reversed by the addition of EDTA to the reaction mixture, and (iii) the EDTA effect could not be explained by the presence or absence of Mg2+ and Ca2+, since those metals did not have any effect (Table 1).
SOR sequence analysis and active-site predictions
Four of the five SOR sequences had been tentatively grouped in a Pfam B domain (Pfam-B_28360; http://www.sanger.ac.uk/Software/Pfam). No overlap with other domains was detected. Similarly, our analyses did not provide evidence that the SORs are phylogenetically related to any other enzyme family, regardless of whether they are known to harbour mononuclear Fe centres or not. In addition, the identification of sor genes in other Archaea and in Aquifex aeolicus, but not in Acidithiobacillus ferrooxidans, Paracoccus pantotrophus and other mesophilic bacteria, points to a novel pathway of sulphur oxidation in Archaea and in thermophilic bacteria. It differs both from the Sox pathway found in neutrophilic bacteria [1–3] and the glutathione-dependent sulphur oxygenases of the mesoacidophiles [8–10]. Interestingly, Aquifex aeolicus possesses both sor and sox genes (e.g. [3]), suggesting that elemental sulphur is oxidized by the SOR, whereas the products of the reaction are oxidized by the Sox system. Sox and Soxrelated proteins or genes have not been found in Acidianus ambivalens so far, which has a membrane-bound thiosulphate:quinone oxidoreductase [14a] and a sulphite acceptor oxidoreductase activity [41].
A multiple alignment of the five SORs showed a His-Xaa3-His site in the sequence His86-Glu-Glu/Asp-Met-His90-Glu/Asp-Xaa38-Glu/Asp129 as a putative metal-binding sequence motif (Figure 7), which is similar to the one found in tyrosine hydroxylases containing a mononuclear non-haem iron centre (TyrH; [42,43]). These residues form the structural motif of the 2-His-1-carboxylate facial triad, which anchors the iron in the active site. Secondary-structure prediction identified a putative extensive α-helix including the His-Xaa3-His box in the SOR sequences (Figure 7). Helix projection suggested that both histidines are facing the same side as in TyrH (results not shown) and that the SORs may adopt a similar structural motif for the iron binding. However, the tentative identification of the possible glutamic acid ligand (Figure 7) is preliminary without additional evidence.
Each SOR has three conserved cysteine residues in common, two of which are arranged as a Cys-Xaa-Xaa-Cys motif (Figure 7). The activity of purified SOR was inhibited by thiol-binding reagents such as N-ethylmaleimide and Zn2+ ions ([13] and the present study). In consequence, the cysteine residues, or some of them, must be essential, although it is improbable that they might co-ordinate the metal site as explained above. It is more likely that they play a role in sulphur binding and activation, either as enzyme-bound polysulphide in a similar way as has been described for reduced glutathione [8,9] and for the periplasmatic bacterial sulphur oxygenase [10], or with a disulphide redox chemistry as described for the sulphide:quinone oxidoreductase [44].
The SOR reaction involves complex electron transitions, and it is most likely that more than one redox-active centre is participating in catalysis. The SOR activity is strictly oxygen-dependent. The use of complexed Zn2+ ions resulted in a 1:1 stoichiometry of the oxidized and reduced products [13]. The observations can be summed up in two partial reactions consisting formally of an oxygenase reaction (eqn 1) and a sulphur-disproportionation reaction (eqn 2), the latter of which is known to occur non-enzymically in the presence of hydroxyl ions at pH values of 7.4 and above [13,45]:
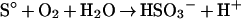 |
(1) |
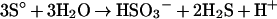 |
(2) |
The sum of eqns (1) and (2) gives:
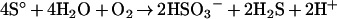 |
(3) |
These interpretations are supported by the rather modest 18O incorporation observed in the case of the “Sulfolobus brierleyi” sulphur oxygenase [15]. It is probable, although it has not yet been demonstrated, that an initial oxygen reduction and activation with sulphur as the electron donor takes place at the iron site. One of the major questions is whether the sulphur reduction or sulphur disproportionation is an indispensable part of the reaction mechanism itself, or whether it is a competing side reaction, i.e. that sulphur can replace oxygen at the iron site. This and the exact reaction mechanism are unanswered at present and will be the object of future studies.
A shell-like quaternary structure with a central hollow cavity
The well-contrasted dot in the middle of the spherical SOR particles observed in EM images (Figure 4) point to a hollow interior filled with uranyl acetate used for negative staining. Other classes of proteins are known to have this structural feature, most notably the ferritins and the ferritin-like DNA-binding Dps proteins (Figure 4) [46–49]. Electron micrographs of these particles resemble SOR particles, thus strengthening the hypothesis that SOR is a shell-like multimeric protein complex with a hollow core [46,47,49]. The Desulfovibrio desulfuricans bacterioferritin is slightly smaller in diameter than the SOR (13.0 nm compared with 15.5 nm, internal hollow core: 8.5 nm). It consists of 24 subunits of a 179-amino-acid protein arranged in dimers and possesses a 432 symmetry (500 kDa molecular mass [48]), whereas Dps proteins are homododecamers with external and internal diameters of 9.0 nm and 4.5 nm respectively, and have a 23 symmetry [50].
It is so far unknown of how many subunits the SOR holoenzyme is actually composed. The molecular mass of the SOR determined by gel-permeation chromatography would point to a 15-mer (550 kDa, 308 amino acid residues [13,14]), whereas the results of the BN PAGE point to a higher molecular mass (732 kDa), equivalent to 20 subunits. A structural model explaining either number of subunits would require the presence of a 5-fold symmetry axis, which was not seen. We calculated the number of atoms fitting into a sphere of the observed size (1950 nm3), equalling 170% of the total Desulfovibrio desulfuricans bacterioferritin volume (1150 nm3) not considering its hollow core (322 nm3). The number of atoms in the holoenzyme would be 178% of that of the ferritins when assuming 24 subunits. Thus the existence of a 4-fold axis as well as the size of the SOR particles point to a quaternary structure being an icosatetramer with the same overall symmetry as the ferritins, although this conclusion is in contrast with the apparent molecular masses. Preliminary X-ray crystallographic data suggested the presence of an icosatetramer as well (T. Urich and C. Frazão, unpublished work).
CD spectra and secondary-structure-prediction programs agreed well that the SOR should form similar percentages of α-helices and β-sheets of approx. 25% each. Thus the SOR exhibited a different pattern of secondary-structure elements from the ferritins, which are almost completely made of a four-helix bundle [48], suggesting a different folding pattern.
The tryptophan-fluorescence-monitored experiments showed that the SOR is relatively resistant to unfolding at concentrations of urea up to 4 M, whereas at urea concentrations above 7 M, the fluorescence reached its minimum, indicating a complete denaturation of the enzyme (Figure 3). Monitoring by gel-permeation chromatography showed that a predominant dimer peak was observed up to 5 M urea besides the holoenzyme peak (Figure 2A), whereas the monomer peak appeared at higher urea concentrations. The signal intensities of the holoenzyme and the monomer peaks in the gel-permeation experiment could be superimposed without significant differences to the denaturation curve, which points to a dimer being the building block of the holoenzyme (Figure 2D). This is supported by the results of the BN PAGE showing the dimer band being most prominent in the 2 M urea sample. The fully denatured protein migrated and stained differently in the BN PAGE, suggesting that the denatured enzyme monomers expose more of the hydrophobic residues and aggregate non-specifically, whereas the partially denatured enzyme forms a ladder of bands, presumably showing the holoenzyme in various states of disintegration.
Taking the experiments together, the dimeric state observed in the partially denatured samples in BN gels is properly folded, whereas the complete unfolding of SOR is in good correlation with the appearance of the monomeric state. Furthermore, the data indicate that the assembly of the holoenzyme may proceed via oligomerization of dimers as the building blocks.
Conclusions
We show that a low-potential mononuclear non-haem iron centre is present in the Acidianus ambivalens SOR and that this centre is essential for catalytic activity. We also show that the protein is a large oligomer in solution, having a shell-like assembly made of dimers surrounding a central cavity. The data suggest that the iron core and cysteine residues are essential for the enzyme activity, and that iron is co-ordinated by N and O ligands and not by cysteine residues. The most puzzling observation is the sulphur reductase activity of the SOR, although the low reduction potential of the iron core explains the ability to perform this reaction. It cannot yet be decided which of the two possible alternative hypotheses on enzyme activity will prove true: (i) the SOR is a true sulphur-disproportionating enzyme, with the sulphur reductase activity as an inseparable property of the reaction mechanism; or (ii) sulphur competes for the oxygen-binding site and is reduced as a by-product of the enzyme reaction.
Acknowledgments
We thank Felicitas Pfeifer (Darmstadt, Germany) for her generosity and encouragement. We acknowledge the help of V. Yatseev and W. Haase (Eduard Zintl Institute of Inorganic and Physical Chemistry, Darmstadt) for support in recording the CD spectra, and A. Skerra (Technische Universität München) for the vector. This work has been supported by grants from the Deutsche Forschungsgemeinschaft to A.K. and T.U. (Kl885/3-1, 3-2 and 3-3), grants from the Fundação Ciência e Tecnologia (FCT), Portugal, to M.T. (BIO99/36560) and C.M.G. (Qui/37521/2001), and by travel grants from the Deutscher Akademischer Austauschdienst (DAAD) (to A.K.) and the Instituto Cooperação Científica Internacional (to C.M.G.). T.M.B. is a recipient of an FCT Ph.D. fellowship (BD 3133/00). S.S.L. is a recipient of an FCT Bolserio Investigação Cientifica fellowship (017/BIC/2002).
References
- 1.Kelly D. P., Shergill J. K., Lu W. P., Wood A. P. Oxidative metabolism of inorganic sulfur compounds by bacteria. Antonie van Leeuwenhoek. 1997;71:95–107. doi: 10.1023/a:1000135707181. [DOI] [PubMed] [Google Scholar]
- 2.Friedrich C. G. Physiology and genetics of sulfur-oxidizing bacteria. Adv. Microb. Physiol. 1998;39:235–289. doi: 10.1016/s0065-2911(08)60018-1. [DOI] [PubMed] [Google Scholar]
- 3.Friedrich C. G., Rother D., Bardischewsky F., Quentmeier A., Fischer J. Oxidation of reduced inorganic sulfur compounds by bacteria: emergence of a common mechanism? Appl. Environ. Microbiol. 2001;67:2873–2882. doi: 10.1128/AEM.67.7.2873-2882.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hedderich R., Klimmek O., Kröger A., Dirmeier R., Keller M., Stetter K. O. Anaerobic respiration with elemental sulfur and with sulfides. FEMS Microbiol. Rev. 1999;22:353–381. [Google Scholar]
- 5.Kappler U., Friedrich C. G., Trüper H. G., Dahl C. Evidence for two pathways of thiosulfate oxidation in Starkeya novella (formerly Thiobacillus novellus) Arch. Microbiol. 2001;175:102–111. doi: 10.1007/s002030000241. [DOI] [PubMed] [Google Scholar]
- 6.Nakamura K., Nakamura M., Yoshikawa H., Amano Y. Purification and properties of thiosulfate dehydrogenase from Acidithiobacillus thiooxidans JCM7814. Biosci. Biotechnol. Biochem. 2001;65:102–108. doi: 10.1271/bbb.65.102. [DOI] [PubMed] [Google Scholar]
- 7.Boulegue J. Solubility of elemental sulfur in water at 298 K. Phosphorus Sulfur. 1978;5:127–128. [Google Scholar]
- 8.Suzuki I. Oxidation of elemental sulfur by an enzyme system of Thiobacillus thiooxidans. Biochim. Biophys. Acta. 1965;104:359–371. doi: 10.1016/0304-4165(65)90341-7. [DOI] [PubMed] [Google Scholar]
- 9.Silver M., Lundgren D. G. Sulfur-oxidizing enzyme of Ferrobacillus ferrooxidans (Thiobacillus ferrooxidans) Can. J. Biochem. 1968;46:457–461. doi: 10.1139/o68-069. [DOI] [PubMed] [Google Scholar]
- 10.Rohwerder T., Sand W. The sulfane sulfur of persulfides is the actual substrate of the sulfur-oxidizing enzymes from Acidithiobacillus and Acidiphilium spp. Microbiology. 2003;149:1699–1710. doi: 10.1099/mic.0.26212-0. [DOI] [PubMed] [Google Scholar]
- 11.Friedrich C. G., Quentmeier A., Bardischewsky F., Rother D., Kraft R., Kostka S., Prinz H. Novel genes coding for lithotrophic sulfur oxidation of Paracoccus pantotrophus GB17. J. Bacteriol. 2000;182:4677–4687. doi: 10.1128/jb.182.17.4677-4687.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rother D., Henrich H. J., Quentmeier A., Bardischewsky F., Friedrich C. G. Novel genes of the sox gene cluster, mutagenesis of the flavoprotein SoxF, and evidence for a general sulfur-oxidizing system in Paracoccus pantotrophus GB17. J. Bacteriol. 2001;183:4499–4508. doi: 10.1128/JB.183.15.4499-4508.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kletzin A. Coupled enzymatic production of sulfite, thiosulfate, and hydrogen sulfide from sulfur: purification and properties of a sulfur oxygenase reductase from the facultatively anaerobic archaebacterium Desulfurolobus ambivalens. J. Bacteriol. 1989;171:1638–1643. doi: 10.1128/jb.171.3.1638-1643.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kletzin A. Molecular characterization of the sor gene, which encodes the sulfur oxygenase reductase of the thermoacidophilic archaeon Desulfurolobus ambivalens. J. Bacteriol. 1992;174:5854–5859. doi: 10.1128/jb.174.18.5854-5859.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14a.Müller F., Bandeiras T., Urich T., Teixeira M., Gomes C. M., Kletzin A. Coupling of the pathway of sulfur oxidation to dioxygen reduction: characterization of a novel membrane-bound thiosulfate:quinone oxidoreductase. Mol. Microbiol. 2004 doi: 10.1111/j.1365-2958.2004.04193.x. in the press. [DOI] [PubMed] [Google Scholar]
- 15.Emmel T., Sand W., König W. A., Bock E. Evidence for the existence of a sulphur oxygenase in Sulfolobus brierleyi. J. Gen. Microbiol. 1986;132:3415–3420. [Google Scholar]
- 16.He Z., Li Y., Zhou P., Liu S. Cloning and heterologous expression of a sulfur oxygenase/reductase gene from the thermoacidophilic archaeon Acidianus sp. S5 in Escherichia coli. FEMS Microbiol. Lett. 2000;193:217–221. doi: 10.1111/j.1574-6968.2000.tb09427.x. [DOI] [PubMed] [Google Scholar]
- 17.Kletzin A. Sulfur oxidation and reduction in Archaea: sulfur-oxygenase/-reductase and hydrogenases from the extremely thermophilic and facultatively anaerobic Archaeon Desulfurolobus ambivalens. Syst. Appl. Microbiol. 1994;16:534–543. [Google Scholar]
- 18.Sun C. W., Chen Z. W., He Z. G., Zhou P. J., Liu S. J. Purification and properties of the sulfur oxygenase/reductase from the acidothermophilic archaeon, Acidianus strain S5. Extremophiles. 2003;7:131–134. doi: 10.1007/s00792-002-0304-5. [DOI] [PubMed] [Google Scholar]
- 19.Skerra A. Use of the tetracycline promoter for the tightly regulated production of a murine antibody fragment in Escherichia coli. Gene. 1994;151:131–135. doi: 10.1016/0378-1119(94)90643-2. [DOI] [PubMed] [Google Scholar]
- 20.Nozaki Y. The preparation of guanidine hydrochloride. Methods Enzymol. 1972;26:43–50. doi: 10.1016/s0076-6879(72)26005-0. [DOI] [PubMed] [Google Scholar]
- 21.Royer C. A. Fluorescence spectroscopy. In: Shirley B. A., editor. Protein Stability and Folding: Theory and Practice. vol. 40. Totowa: Humana Press Inc.; 1995. pp. 65–89. [Google Scholar]
- 22.Shirley B. A. Urea and guanidine hydrochloride denaturation curves. In: Shirley B. A., editor. Protein Stability and Folding: Theory and Practice. vol. 40. Totowa: Humana Press Inc.; 1995. pp. 177–190. [DOI] [PubMed] [Google Scholar]
- 23.Schägger H. Native gel electrophoresis. In: von Jagow G., Schägger H., editors. A Practical Guide to Membrane Protein Purification. San Diego: Academic Press; 1994. pp. 81–106. [Google Scholar]
- 24.Zillig W., Yeats S., Holz I., Böck A., Rettenberger M., Gropp F., Simon G. Desulfurolobus ambivalens gen. nov., sp. nov., an autotrophic archaebacterium facultatively oxidizing and reducing sulfur. Syst. Appl. Microbiol. 1986;8:197–203. [Google Scholar]
- 25.Teixeira M., Batista R., Campos A. P., Gomes C., Mendes J., Pacheco I., Anemüller S., Hagen W. R. A seven-iron ferredoxin from the thermoacidophilic Desulfurolobus ambivalens. Eur. J. Biochem. 1995;227:322–327. doi: 10.1111/j.1432-1033.1995.tb20392.x. [DOI] [PubMed] [Google Scholar]
- 26.Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 27.Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., Klenk D. C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 28.Schägger H., von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 29.Fischer D. S., Price D. C. Simple serum iron method using new sensitive chromogen tripyridyl-s-triazine. Clin. Chem. 1964;10:21–25. [PubMed] [Google Scholar]
- 30.Sreerama N., Woody R. W. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000;287:252–260. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 31.Schurig H., Rutkat K., Rachel R., Jaenicke R. Octameric enolase from the hyperthermophilic bacterium Thermotoga maritima: purification, characterization, and image processing. Protein Sci. 1995;4:228–236. doi: 10.1002/pro.5560040209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt M., Rutkat K., Rachel R., Pfeifer G., Jaenicke R., Viitanen P., Lorimer G., Buchner J. Symmetric complexes of GroE chaperonins as part of the functional cycle. Science. 1994;265:656–659. doi: 10.1126/science.7913554. [DOI] [PubMed] [Google Scholar]
- 33.Hegerl R. The EM program package: a platform for image processing in biological electron microscopy. J. Struct. Biol. 1996;116:30–34. doi: 10.1006/jsbi.1996.0006. [DOI] [PubMed] [Google Scholar]
- 34.Saxton W. O. Semper: distortion compensation, selective averaging, 3-D reconstruction, and transfer function correction in a highly programmable system. J. Struct. Biol. 1996;116:230–236. doi: 10.1006/jsbi.1996.0035. [DOI] [PubMed] [Google Scholar]
- 35.Lange S. J., Que L., Jr Oxygen activating non-heme iron enzymes. Curr. Opin. Chem. Biol. 1998;2:159–172. doi: 10.1016/s1367-5931(98)80057-4. [DOI] [PubMed] [Google Scholar]
- 36.Nelson M. Catecholate complexes of ferric soybean lipoxygenase 1. Biochemistry. 1988;27:4273–4278. [Google Scholar]
- 37.Hagedoorn P. L., Schmidt P. P., Andersson K. K., Hagen W. R., Flatmark T., Martinez A. The effect of substrate, dihydrobiopterin, and dopamine on the EPR spectroscopic properties and the midpoint potential of the catalytic iron in recombinant human phenylalanine hydroxylase. J. Biol. Chem. 2001;276:22850–22856. doi: 10.1074/jbc.M009458200. [DOI] [PubMed] [Google Scholar]
- 38.Abreu I. A., Xavier A. V., LeGall J., Cabelli D. E., Teixeira M. Superoxide scavenging by neelaredoxin: dismutation and reduction activities in anaerobes. J. Biol. Inorg. Chem. 2002;7:668–674. doi: 10.1007/s00775-002-0363-1. [DOI] [PubMed] [Google Scholar]
- 39.Vance C. K., Miller A. F. Novel insights into the basis for Escherichia coli superoxide dismutase’s metal ion specificity from Mn-substituted FeSOD and its very high Em. Biochemistry. 2001;40:13079–13087. doi: 10.1021/bi0113317. [DOI] [PubMed] [Google Scholar]
- 40.Thauer R. K., Jungermann K., Decker K. Energy conservation in chemotrophic anaerobic bacteria. Bacteriol. Rev. 1977;41:100–180. doi: 10.1128/br.41.1.100-180.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zimmermann P., Laska S., Kletzin A. Two modes of sulfite oxidation in the extremely thermophilic and acidophilic archaeon Acidianus ambivalens. Arch. Microbiol. 1999;172:76–82. doi: 10.1007/s002030050743. [DOI] [PubMed] [Google Scholar]
- 42.Costas M., Mehn M. P., Jensen M. P., Que L., Jr Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem. Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 43.Hegg E. L., Que L., Jr The 2-His-1-carboxylate facial triad–an emerging structural motif in mononuclear non-heme iron(II) enzymes. Eur. J. Biochem. 1997;250:625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- 44.Griesbeck C., Schutz M., Schodl T., Bathe S., Nausch L., Mederer N., Vielreicher M., Hauska G. Mechanism of sulfide-quinone reductase investigated using site-directed mutagenesis and sulfur analysis. Biochemistry. 2002;41:11552–11565. doi: 10.1021/bi026032b. [DOI] [PubMed] [Google Scholar]
- 45.Roy A. B., Trudinger P. A. Cambridge: Cambridge University Press; 1970. The Biochemistry of Inorganic Compounds of Sulphur. [Google Scholar]
- 46.Ratnayake D. B., Wai S. N., Shi Y., Amako K., Nakayama H., Nakayama K. Ferritin from the obligate anaerobe Porphyromonas gingivalis: purification, gene cloning and mutant studies. Microbiology. 2000;146:1119–1127. doi: 10.1099/00221287-146-5-1119. [DOI] [PubMed] [Google Scholar]
- 47.Kim B. S., Lee C. S., Yun C. Y., Yeo S. M., Park W. M., Kim H. R. Characterization and immunological analysis of ferritin from the hemolymph of Galleria mellonella. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2001;129:501–509. doi: 10.1016/s1095-6433(01)00287-2. [DOI] [PubMed] [Google Scholar]
- 48.Macedo S., Romao C. V., Mitchell E., Matias P. M., Liu M. Y., Xavier A. V., LeGall J., Teixeira M., Lindley P., Carrondo M. A. The nature of the di-iron site in the bacterioferritin from Desulfovibrio desulfuricans. Nat. Struct. Biol. 2003;10:285–290. doi: 10.1038/nsb909. [DOI] [PubMed] [Google Scholar]
- 49.Tonello F., Dundon W. G., Satin B., Molinari M., Tognon G., Grandi G., Del Giudice G., Rappuoli R., Montecucco C. The Helicobacter pylori neutrophil-activating protein is an iron-binding protein with dodecameric structure. Mol. Microbiol. 1999;34:238–246. doi: 10.1046/j.1365-2958.1999.01584.x. [DOI] [PubMed] [Google Scholar]
- 50.Roy S., Gupta S., Das S., Sekar K., Chatterji D., Vijayan M. Crystallization and preliminary X-ray diffraction analysis of Mycobacterium smegmatis Dps. Acta Crystallogr. D Biol. Crystallogr. 2003;59:2254–2256. doi: 10.1107/s0907444903018742. [DOI] [PubMed] [Google Scholar]