

Abstract
Superoxide reacts rapidly with other radicals, but these reactions have received little attention in the context of oxidative stress. For tyrosyl radicals, reaction with superoxide is 3-fold faster than dimerization, and forms the addition product tyrosine hydroperoxide. We have explored structural requirements for hydroperoxide formation using tyrosine analogues and di- and tri-peptides. Superoxide and phenoxyl radicals were generated using xanthine oxidase, peroxidase and the respective tyrosine derivative, or by γ-radiation. Peroxides were measured using FeSO4/Xylenol Orange. Tyrosine and tyramine formed stable hydroperoxides, but N-acetyltyrosine and p-hydroxyphenylacetic acid did not, demonstrating a requirement for a free amino group. Using [14C]tyrosine, the hydroperoxide and dityrosine were formed at a molar ratio of 1.8:1. Studies with pre-formed hydroperoxides, and measurements of substrate losses, indicated that, in the absence of a free amino group, reaction with superoxide resulted primarily in restitution of the parent compound. With dipeptides, hydroperoxides were formed only on N-terminal tyrosines. However, adjacent lysines promoted hydroperoxide formation, as did addition of free lysine or ethanolamine. Results are compatible with a mechanism [d'Alessandro, Bianchi, Fang, Jin, Schuchmann and von Sonntag (2000) J. Chem. Soc. Perkin Trans. II, 1862–1867] in which the phenoxyl radicals react initially with superoxide by addition, and the intermediate formed either releases oxygen to regenerate the parent compound or is converted into a hydroperoxide. Amino groups favour hydroperoxide formation through Michael addition to the tyrosyl ring. These studies indicate that tyrosyl hydroperoxides should be formed in proteins where there is a basic molecular environment. The contribution of these radical reactions to oxidative stress warrants further investigation.
Keywords: peroxidase, phenoxyl radical, protein hydroperoxide, superoxide, tyrosine hydroperoxide, tyrosyl radical
Abbreviations: SOD, superoxide dismutase; FOX, FeSO4/Xylenol Orange
INTRODUCTION
Oxidative stress is commonly initiated by the one-electron reduction of oxygen to generate the superoxide radical. Protection against superoxide by SOD (superoxide dismutase) is vital for aerobic life. Why superoxide is toxic has long been debated [1–4]. The current view is that inactivation and iron release from iron–sulphur proteins such as aconitase, and secondary products such as hydroxyl radicals and peroxynitrite, all contribute to its toxicity. Peroxynitrite is formed in a very rapid reaction between superoxide and nitric oxide radicals. Other radical–radical reactions are also very fast, but these have received surprisingly little attention. In particular, reactions of superoxide with phenoxyl radicals have rate constants of the order of 109 M−1·s−1 [5], and generate organic peroxides and other toxic products [6–8] that could contribute to oxidative injury.
Phenoxyl radicals are produced by the one-electron oxidation of tyrosine and other phenolic compounds by mechanisms that include oxidation by hydrogen peroxide and peroxidases such as myeloperoxidase (reaction 1, Scheme 1A) [9], hydroxyl radical- and metal-catalysed oxidations, and electron or hydrogen transfer to other radicals [10,11]. In proteins, tyrosyl radicals are part of the catalytic cycle of ribonucleotide reductase, prostaglandin synthase and lactoperoxidase [12,13]. They are also formed on myoglobin and haemoglobin during reaction with hydrogen peroxide [14–16]. Phenoxyl radicals react poorly with oxygen, but combine readily to form dimers (reaction 2, Scheme 1A) [5]. Tyrosyl radical generation by isolated peroxidases and neutrophils leads to 3,3′-dityrosine formation [9,17,18]. Dityrosine has been detected in proteins after exposure to various oxidants [19–22], and is being increasingly analysed in clinical samples as a biomarker of oxidative injury [23–26]. However, in many physiological situations, the reaction of tyrosyl radicals with superoxide may be more significant than dimerization. The rate constant for reaction with superoxide {(1.5–1.7)×109 M−1·s−1 [5,6]} is approx. 3-fold higher than that for dimerization (0.45×109 M−1·s−1 [5]), so when both radicals are formed concurrently, the reaction with superoxide should predominate. This would be the case particularly when the tyrosyl radical is isolated on a protein, and prevented from undergoing dimerization for steric or electronic reasons. Direct experimental evidence for this has been obtained with stimulated neutrophils, where SOD enhanced dityrosine formation several-fold [7,17,27].
Scheme 1. Reactions involved in hydroperoxide formation.

(A) Reactions involved in the formation, dimerization and interaction of phenoxyl radicals with superoxide. Reactions are shown for the o-isomer (I). The p-isomer should also be formed, but cannot undergo rearrangement from the cyclohexadienone to the phenolic hydroperoxide. (B) Reactions proposed for the cyclization and breakdown of tyrosine hydroperoxide to the product (III) characterized by MS and NMR [6].
Until recently, it was assumed that superoxide reacts with phenoxyl radicals by electron transfer, to regenerate oxygen and the parent phenol (reaction 3, Scheme 1A). However, it is now established that the mechanism is more complex, and that addition to form a hydroperoxide also occurs (reaction 4) [6,7,28–30]. The relative contributions of repair and addition vary for different phenols [29]. Formation of a hydroperoxide from tyrosyl radicals and superoxide has been demonstrated in radiolytic systems [6], and biochemically with neutrophils or myeloperoxidase [7] and when tyrosyl radicals are scavenged by GSH [28]. In the GSH system, the thiyl radical (GS•) generated in reaction (7) is the source of superoxide via reactions (8) and (9):
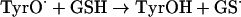 |
(7) |
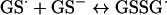 |
(8) |
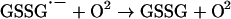 |
(9) |
Reaction products from tyrosine have been characterized. The hydroperoxide gives an HPLC peak [6,7,28] that has been isolated and characterized by MS and NMR. The product identified is III in Scheme 1B (or the equivalent p-hydroxy isomer) [6]. A mechanism was proposed in which I is produced in reaction 4 (Scheme 1A), followed by cyclization through the amine to give II (reaction 5, Scheme 1B), which then undergoes hydrolysis and loss of hydrogen peroxide during analysis to give III (reaction 6, Scheme 1B).
The reaction of a range of phenoxyl radicals with excess superoxide has been studied using γ-radiolysis. The proportion of the reaction resulting in addition products as against restitution of the phenol was found to increase with decreasing reduction potential of the phenoxyl radical [29]. However, tyrosine, which was not studied in that series, does not fit the pattern. Based on reduction potential, it would be expected to react mainly via the restitution reaction, yet the peroxide was the major product [6]. d'Alessandro et al. [30] have sought an explanation. Their proposed mechanism is discussed later, but a key conclusion is that the presence of an amino group explains the anomalous behaviour.
These observations have important implications for biological systems, as they suggest that molecular features, such as the proximity of a free amino group, could determine whether a tyrosyl residue in a peptide forms a stable hydroperoxide. Tyrosyl radicals are readily formed on peptides and proteins [11], but no studies have been undertaken to determine if they react with superoxide to produce hydroperoxides, or whether structural features influence what products are formed. In the present investigation, we have compared tyrosine with tyramine, N-acetyltyrosine and p-hydroxyphenylacetic acid to assess the influence of free amino and carboxyl groups on hydroperoxide formation. We have also studied dipeptides and tripeptides to determine if the position of the amino group affects the reaction.
EXPERIMENTAL
Materials
Tyrosine analogues, peptides (unless noted otherwise) and enzymes were all obtained from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Lys-Tyr-Gly and Gly-Tyr-Lys were custom-synthesized by Auspep (Parkville, Victoria, Australia). Preformed peroxides of tyrosine, N-acetyltyrosine and 3-(p-hydroxyphenyl)propionic acid were kindly prepared by P.E. Morgan (The Heart Research Institute, Sydney, NSW, Australia) by visible light photolysis of 2.5 mM solutions in the presence of Rose Bengal (10 μM), and peroxide content was quantified using the FOX (FeSO4/Xylenol Orange) assay as described previously [31]. Myeloperoxidase was purified from human neutrophils [32].
Peroxidase-mediated oxidation of tyrosine analogues
Reaction mixtures consisted of acetaldehyde (1 mM), xanthine oxidase (0.01 unit/ml), the appropriate phenolic compound (1 mM) and horseradish peroxidase (10 μg/ml) in 50 mM sodium phosphate buffer, pH 7.4. SOD (20 μg/ml) was added where indicated. Reactions were carried out at 20–25 °C for 30–40 min and stopped by adding catalase (20 μg/ml) to remove any residual hydrogen peroxide. Control samples containing all reagents and analysed at zero time were used as blanks. One 0.7 ml portion of reaction mixture was added to 0.25 ml of FOX reagent for peroxide determination, and another was diluted 1:10 (v/v) in 100 mM sodium phosphate buffer, pH 8.0, for fluorescence measurements. The total amount of hydrogen peroxide generated was determined independently by incubating the same concentration of xanthine oxidase with acetaldehyde and SOD for the same length of time, then diluting a portion of the solution 1:20 (v/v) in FOX reagent.
Peroxide quantification
Peroxides were analysed using the FOX method [33], with concentrations modified so that 0.7 ml of sample in 50 mM sodium phosphate buffer could be added to 0.25 ml of reagent. The composition of the FOX reagent was Xylenol Orange (30 mg), ferrous ammonium sulphate (39 mg) and sorbitol (7.3 g) in 100 ml of 0.2 M sulphuric acid. Samples were left at room temperature for at least 45 min before reading absorbances at 560 nm. Concentrations were determined from a standard curve for hydrogen peroxide, calibrated using ε 240=43.6 M−1·cm−1 [34], and results are expressed as hydrogen peroxide equivalents.
To compare the yields of tyrosine hydroperoxide and dityrosine, the reaction was performed using [14C]tyrosine (Amersham Pharmacia Biotech) under standard conditions, except that the tyrosine concentration was 0.4 mM, the reaction time was 15 min and xanthine oxidase was added to generate 15–20 μM hydrogen peroxide. The reaction was stopped by adding catalase, and the reaction mixture (100 μl containing 1 μCi of 14C) was separated by HPLC as described below. Fractions were collected every 0.5 min and counted for radioactivity.
Dimer quantification
Dimers of the tyrosine analogues and peptides were measured fluorimetrically (excitation 325/emission 400 nm) after dilution at pH 8. The method was calibrated by generating the dimer from the relevant phenol with horseradish peroxidase and hydrogen peroxide, and relating the absorbance at its λmax (315–318 nm, depending on the compound) to the fluorescence signal. The published molar absorption coefficient of 5080 M−1·cm−1 for both dityrosine and dityramine [35] was used, and assumed to apply also to N-acetyltyrosine and p-hydroxyphenylacetic acid. For the tyrosyl peptides, calibration was not performed and the results are expressed as relative fluorescence.
Radiolysis of tyrosine analogues
Air-saturated solutions containing 10 mM sodium azide, 20 mM sodium formate and 50 μM of the phenolic reagent under test were irradiated in a 60Co γ-radiation source at 68 Gy/min to the required doses, using the method of Jonsson et al. [29]. The pH was kept constant at 7.4 by 10 mM sodium phosphate or at 9.2 by a mixture of 10 mM sodium phosphate and 10 mM sodium tetraborate. In this system, some of the hydroxyl radicals produce azide radicals, the remainder reacting with formate to produce superoxide. The azide radicals oxidize the phenols to phenoxyl radicals. The ratio of formate to azide is such that more superoxide than phenoxyl radicals is produced [29]. Radiation-generated hydrogen peroxide was removed by a 30 min incubation with 65 units/ml catalase. Energy dose rates were measured by ferrous sulphate dosimetry.
HPLC analysis
Losses of tyrosine analogues following γ-radiolysis were determined by measuring concentrations before and after treatment by reverse-phase HPLC (Phenomenex C18, 5 μm, 250 mm×4.6 mm column) with diode array detection (Waters 2690 pump, 996 diode array and Millenium32© software). The solvent systems used were as follows. Tyrosine: 25 mM acetate buffer, pH 4 (buffer A), 0.8 ml/min, elution time 13 min; tyramine: buffer A containing 4% (v/v) methanol, 0.8 ml/min, elution time 8 min; N-acetyltyrosine and p-hydroxyphenylacetic acid: 100% buffer A for 9 min, then a gradient of 0–50% (v/v) methanol in buffer A for 10 min, 0.7 ml/min, elution times 13 and 14 min respectively.
RESULTS
Hydroperoxide and dimer formation from tyrosine analogues
A biochemical system consisting of acetaldehyde and xanthine oxidase as a source of superoxide and hydrogen peroxide, and horseradish peroxidase to generate phenoxyl radicals, was used to investigate reactions between phenoxyl radicals and superoxide. It has been established previously that tyrosine reacts in this system to produce dityrosine, which is detectable by fluorescence, and a hydroperoxide that can be detected by the FOX assay [7]. Other evidence for the superoxide-dependent formation of tyrosine hydroperoxide was the detection of an HPLC peak that gave a positive response in the FOX assay and was not present if the reaction was carried out in the presence of SOD or if the sample was treated with sodium borohydride prior to chromatography [7,28]. Initial experiments reproduced these findings, and confirmed that SOD enhanced dimer formation (Figure 1A), but inhibited hydroperoxide formation (Figure 1B). Formation of both products required tyrosine, acetaldehyde, xanthine oxidase and peroxidase, and was inhibited by catalase (results not shown).
Figure 1. Formation of (A) dimers and (B) peroxides from tyrosine, tyramine, N-acetyltyrosine (NAT) and p-hydroxyphenylacetic acid (pOH-PA).

Substrates were incubated with horseradish peroxidase, acetaldehyde and xanthine oxidase, as described in the Experimental section, without (▪) or with (□) SOD. The total amount of hydrogen peroxide generated was 54 μM. Dimers were determined by fluorescence and peroxides by the FOX assay. In the FOX assay, zero-time blanks (typically equivalent to approx. 0.2 μM hydrogen peroxide) have been subtracted. Results are means and S.D. for triplicate analyses from one experiment. Similar results were obtained in two other independent experiments.
Tyrosine was compared with tyramine, N-acetyltyrosine and p-hydroxyphenylacetic acid. Fluorescence measurements showed that all four compounds were substrates for the peroxidase, generating phenoxyl radicals and forming dimers (Figure 1A). Dimer yields were all substantially increased in the presence of SOD. This indicates that in the absence of SOD a major reaction of the phenoxyl radicals was with superoxide rather than dimerization. Peroxide measurements (Figure 1B) showed that, like tyrosine, tyramine gave superoxide-dependent formation of a hydroperoxide. However, for the other two compounds, responses in the FOX assay were very low and no different in the presence and absence of SOD. These products are unlikely, therefore, to have arisen from a reaction of superoxide. These results suggest that the presence of a free amino group is critical for the formation of detectable hydroperoxides.
Experiments carried out with myeloperoxidase gave essentially the same results as with horseradish peroxidase, with only tyrosine and tyramine forming detectable hydroperoxides. 3-Chlorotyrosine and 3-nitrotyrosine were also investigated: the former gave a similar hydroperoxide yield as tyrosine, but no 3-nitrotyrosine peroxide was detected.
Quantification of tyrosine hydroperoxide yield
When measured as hydrogen peroxide equivalents, yields of hydroperoxides appeared much lower than those of dimers. However, it was recognized in previous studies [28] that the FOX assay as used in the present work gives a lower response for tyrosine hydroperoxide than for hydrogen peroxide. Therefore, to measure relative yields of the two products directly, the reaction was performed with [14C]tyrosine and the products were separated by HPLC. Compared with the tyrosine control, UV detection gave two major product peaks: one at 17.5 min which was identified as dityrosine, and one at 6.9 min which was in the same position as that shown previously [7,28] for the hydroperoxide (Figure 2A). Only a trace of trityrosine (retention time 23 min) was formed under these conditions. As already reported [7], the 6.9 min peak was absent and the area of the dityrosine peak was approx. 2.5-fold greater in the presence of SOD (results not shown). In the control, approx 90% of the counts eluted with the tyrosine, with a small amount of dimer and background levels in the hydroperoxide position (Figure 2B). Treatment with the xanthine oxidase/peroxidase system resulted in loss of approx. 10% of the tyrosine and increased counts in both product peaks. The ratio of the increase in counts in the hydroperoxide compared with the dimer peak was 0.91±0.02 (n=2), which corresponds to a molar ratio of tyrosine peroxide to dityrosine formation of 1.82:1. In the presence of SOD, no radiolabelled peak corresponding to the hydroperoxide was generated (results not shown).
Figure 2. HPLC separation and quantification of products formed from [14C]tyrosine.

(A) UV traces at 224 nm for tyrosine control (broken line) and, after incubation with horseradish peroxidase, acetaldehyde and xanthine oxidase (solid line). (B) Radioactivity in tyrosine control (broken line) and reaction mixture from two experiments (lines labelled ‘1’ and ‘2’ respectively). Fractions in the vicinity of all of the UV peaks were counted. Reaction and HPLC conditions are described in the Experimental section. Dityrosine (17.5 min elution) was identified on the basis of its λmax of 284 nm and fluorescence (375 nmex/410 nmem). The 6.9 min peak was not fluorescent and had a λmax of 224 nm. Trityrosine eluted at 23.0 min. There is a lag of approx. 0.5 min between the UV and radiolabelled peaks because of the time between detection and collection of the fractions. A small amount of labelled dityrosine was present in the [14C]tyrosine obtained from the manufacturer, but was not evident in the UV trace because it was diluted out with unlabelled material.
The radiolabelling results indicate that, in the experiments shown in Figure 1(B), the actual concentration of tyrosine hydroperoxide formed was approx. 13 μM, i.e. about 6 times higher than when expressed as hydrogen peroxide equivalents in the FOX assay.
Stability of pre-formed hydroperoxides
Superoxide can react with phenoxyl radicals to regenerate the parent phenol and oxygen (reaction 3, Scheme 1A), or by addition (reaction 4). Therefore a lack of detectable peroxides with two of the tyrosine analogues could be due either to reaction 3 predominating, or to hydroperoxide generation followed by rapid decomposition. To test their stability, hydroperoxides of tyrosine, N-acetyltyrosine and 3-(p-hydroxyphenyl)propionic acid were produced by treating the parent compounds with singlet oxygen [31]. This procedure generates predominately para hydroperoxides (with respect to the phenolic hydroxy group, i.e. at C-1), whereas the peroxidase/superoxide pathway should generate both ortho and para isomers. Previous studies have shown that the para hydroperoxides decay slowly at neutral pH, with those containing amine groups being less stable [31]. We observed that incubation in pH 7.4 buffer for 45 min at 37 °C (at initial concentrations of 10 μM hydrogen peroxide equivalents in the FOX assay) resulted in hydroperoxide losses of 35% for tyrosine, 13% for N-acetyltyrosine and 9% for p-hydroxyphenylpropionic acid. No decreases in stability were observed in the presence of myeloperoxidase (200 nM) or horse-radish peroxidase (20 μg/ml), with or without homovanillic acid added as a peroxidase substrate. These results suggest that rapid breakdown was not responsible for the lack of detectable hydroperoxides with N-acetyltyrosine and p-hydroxyphenylacetic acid.
Superoxide-dependent regeneration of parent compounds
We tested whether the lack of an amino group favours regeneration of the parent phenol by superoxide, by measuring loss of the parent phenol. For these experiments, superoxide and phenoxyl radicals were generated by γ-irradiation, to avoid complications due to superoxide reacting with the peroxidase. Formate was present to give conditions whereby sufficient superoxide was generated to react with most of the phenoxyl radicals and very little dimer was produced (black bars in Figure 3). Experiments carried out in the presence of SOD (results not shown) gave substantial dimer formation, confirming that phenoxyl radicals were generated. Irradiation of the four compounds under identical conditions resulted in a dose-dependent loss of tyrosine and tyramine, and much smaller losses of p-hydroxyphenylacetic acid and N-acetyltyrosine (Figure 3). The reaction was also studied at pH 9.2, where losses were greater than at pH 7.4 for the aminophenols, but not for p-hydroxyphenylacetic acid or N-acetyltyrosine (results not shown). These results indicate that there is more regeneration of the parent for the compounds lacking a free amino group.
Figure 3. Loss of substrate on γ-irradiation of tyrosine, tyramine, N-acetyltyrosine (NAT) and p-hydroxyphenylacetic acid (pOH-PA).

γ-Irradiation was performed at pH 7.4 under the conditions described in the Experimental section. □, Parent compound measured by HPLC; ▪, dimer measured by fluorescence. Results represent means and S.E.M. from four independent experiments.
Amine requirement for hydroperoxide formation on tyrosyl peptides
Whether dipeptides with tyrosine at the N- or C-terminus could form stable hydroperoxides in a radical reaction with superoxide was investigated using Gly-Tyr, Ala-Tyr and Tyr-Gly. All of the peptides were confirmed as substrates for horseradish peroxidase by measuring dimer formation, and the 2–3-fold increase in fluorescence on adding SOD indicated that each of the radicals reacted with superoxide in preference to forming the dimer (Figure 4A). Tyr-Gly, with a free amino group on an N-terminal tyrosine, gave more hydroperoxide formation than tyrosine (Figure 4B). However, Ala-Tyr and Gly-Tyr gave superoxide-dependent responses in the FOX assay that were only marginally greater than in the presence of SOD.
Figure 4. Formation of fluorescent dimers (A) and peroxides (B) from tyrosyl peptides.

Experimental conditions were the same as for Figure 1 ▪, No SOD; □, with SOD. The amount of hydrogen peroxide generated was 55 μM. Results are means and S.D. for triplicate analyses from one experiment. Similar results were obtained in two other independent experiments. Differences between with and without SOD are significant for Y and YG at P<0.001, and for GY and AY at P<0.05 (Student's t test).
Tripeptides with lysine groups adjacent to the tyrosine also underwent radical addition to form long-lived hydroperoxides. Figure 5 shows that although responses were lower than for tyrosine, lysine at the C- or N-terminus was able to promote the reaction, and two lysine residues adjacent to the tyrosine had a greater effect than either one alone. Arginine was unable to substitute for lysine, as no superoxide-dependent peroxide formation was seen with Gly-Gly-Tyr-Arg (results not shown). Exogenous amine groups were also able to promote hydroperoxide formation on N-terminally blocked tyrosyl residues. Addition of ethanolamine to Gly-Tyr (Figure 6A) or of lysine to N-acetyltyrosine (Figure 6B) gave a concentration-dependent increase in hydroperoxides in the absence but not in the presence of SOD.
Figure 5. Effects of lysine residues on dimer fluorescence (A) and peroxide formation (B) from tyrosyl peptides.

Experimental conditions were the same as for Figure 1 ▪, No SOD; □, with SOD. The amount of hydrogen peroxide generated was 34 μM. Results are means and S.D. for triplicate analyses from one experiment. Similar results were obtained in two other independent experiments. Differences between with and without SOD are all significant at P<0.001 (Student's t test).
Figure 6. Enhancement of hydroperoxide formation by exogenous amines.

(A) Gly-Tyr incubated with ethanolamine without (•) and with (○) SOD. (B) N-Acetyltyrosine incubated with lysine without (•) and with (○) SOD. Experimental conditions were similar to those in Figure 1 with 37 μM hydrogen peroxide generated. Each point represents the mean of duplicate measurements from one experiment, with ranges indicated. Similar results were obtained on two other occasions, and also with Gly-Tyr incubated with lysine or N-acetyltyrosine incubated with ethanolamine.
DISCUSSION
It is well established from chemical and biochemical studies that tyrosyl radicals react with superoxide to form a hydroperoxide [6,7,28,30]. Measurements of substrate loss indicated that hydroperoxide was a major product [6,30], and with excess superoxide there was little dityrosine formation. However, concerns about quantification and assay specificity, and apparently low concentrations measured in the FOX assay (as observed in the present study), have meant that the hydroperoxide is not widely considered as a major biological product of tyrosine oxidation. By quantifying product yields directly using [14C]tyrosine, we have shown that tyrosine oxidation by xanthine oxidase/acetaldehyde/peroxidase produces almost twice as many moles of hydroperoxide as of dityrosine. Thus in this system tyrosine hydroperoxide is not a minor product. For the experiment shown in Figure 1, a yield of 13 μM can be calculated, i.e. approx. 6 times that measured as hydrogen peroxide equivalents. This confirms previous indirect evidence that the version of the FOX assay used is less sensitive for tyrosine peroxide than for hydrogen peroxide [28], and in fact a 6-fold difference was also estimated in that work.
Only 40% of the oxygen consumed by xanthine oxidase and acetaldehyde results in superoxide, with the remainder being reduced to hydrogen peroxide directly [36]. Therefore oxidation of tyrosine by hydrogen peroxide would give twice as many tyrosyl radicals as superoxide, and some dimer formation would be inevitable. Physiologically, the most likely situation whereby both tyrosyl radicals and superoxide would be generated simultaneously is in the surroundings of stimulated neutrophils. In contrast with xanthine oxidase, the neutrophil NADPH oxidase only reduces oxygen univalently to superoxide. Therefore the ratio of superoxide to tyrosyl radicals would be higher, and formation of the hydroperoxide should be even more favoured. Applying the radiolabelling evidence that the FOX assay is 6-fold less sensitive for tyrosine peroxide than for hydrogen peroxide to data obtained previously for neutrophils [7] shows that this is the case. With 2×106 neutrophils stimulated with PMA in the presence of 0.5 mM tyrosine, approx. 25 nmol of tyrosine hydroperoxide was formed, compared with 10 nmol of dityrosine. At lower tyrosine concentrations, the relative yield of the hydroperoxide was even more favourable.
The main focus of the present study was defining the molecular characteristics of tyrosine analogues that favour hydroperoxide formation. We found that the phenoxyl radicals of tyrosine, tyramine, N-acetyltyrosine and p-hydroxyphenylacetic acid all reacted readily with superoxide, but only tyrosine and tyramine produced stable hydroperoxides. Dimerization was slightly more favoured for the the tyrosyl analogues lacking free amino groups, but the substantial increase in dimer formation in the presence of SOD indicates that reaction of the radicals with superoxide was still a major pathway. It appears that, with these compounds, repair of the radical and release of oxygen is favoured over superoxide addition. This was evident with the radiolytic system, where substrate losses were substantially less than for the aminophenols. The stabilities of hydroperoxides pre-formed from the reaction of N-acetyltyrosine or p-hydroxyphenylacetic acid with singlet oxygen in our experimental system also suggest that rapid breakdown does not explain the lack of detectable hydroperoxides in the radical system. (A proviso with this explanation is that the photochemical system produces only para isomers, whereas the radical mechanism should give both ortho and para forms. If ortho isomers were much less stable, and were formed preferentially from phenols lacking amino groups, this might partially explain our results.)
The extent of superoxide addition to a range of phenoxyl radicals has been shown to vary with reduction potential [29]. All of the tyrosine analogues have similar values [30], so differences in their behaviour are not explicable on this basis. However, these differences are consistent with the mechanism proposed by d'Alessandro et al. [30] to explain the anomalously high peroxide yield seen with tyrosine in comparison with other phenols (adapted in Scheme 2). These authors concluded that electron transfer (reaction 3 in Scheme 1A) is of minor importance and that, regardless of the final products, the initial step is superoxide addition (reaction 10, Scheme 2). This is followed either by elimination of bound oxygen to regenerate the phenol (reaction 11 or 12, Scheme 2), or by protonation and stabilization of the hydroperoxide (reaction 13), with the contributions of the two processes varying depending on the substituent groups. d'Alessandro et al. [30] propose that, for compounds such as tyrosine with nucleophilic groups, cyclization due to Michael addition (reaction 14 or 15) favours peroxide formation over dissociation of oxygen. Our smaller losses of N-acetyltyrosine and p-hydroxyphenylacetic acid, in which the nucleophilic amine is blocked or absent, and the findings of d'Alessandro et al. [30] with tyrosol, support this concept. It is noteworthy that, although this mechanistic explanation was proposed on the basis of radiolytic studies at pH 11, it also holds for our enzymic system at pH 7.4.
Scheme 2. Proposed mechanism for generation of an intermediate addition product from phenoxyl radicals and superoxide, with subsequent breakdown to regenerate the phenol or conversion into the stable hydroperoxide.

The Scheme is based on the results obtained by d'Alessandro et al. [30] and the mechanism that they proposed. For simplicity, only reactions of the p-isomer are shown. Equivalent reactions of the o-isomer are proposed, except that formation of the endoperoxide and reaction 12 would not be relevant. In accordance with the spin conservation rule, the oxygen released from the intermediate (in the reverse of the reaction that forms the hydroperoxide) is represented as singlet oxygen, although this has not been demonstrated experimentally. The amine group in R′NH2 could be the α-amino group of tyrosine, in which case cyclization as in Scheme 1(B) would occur.
The question arises as to where in the reaction sequence a free amino group (or alternative nucleophile) acts to stabilize the hydroperoxide. The stability of the pre-formed para hydroperoxides of N-acetyltyrosine and p-hydroxyphenylacetic acid (with structures equivalent to I in Scheme 1) [31] suggests that the amine must interact with an earlier intermediate in the radical sequence. A possible mechanism could involve an initial reaction (reaction 10) to give a complex of superoxide and the phenoxyl radical (designated IV and shown in Scheme 2 as a peroxyl anion, but not necessarily of this form). Compound IV could release oxygen (either directly or via an endoperoxide; reactions 11 and 12) or be converted into the protonated hydroperoxide, I (reaction 13). In the absence of a nucleophile, reactions 13 and 15 would be slow and oxygen release favoured. However, with an amino group or other nucleophile present, reaction 14 could compete favourably with reactions 11 and 12, and produce more hydroperoxide. (We are assuming that the stability of the ortho isomers is similar. However, as they are secondary peroxides, and the para isomers are tertiary peroxides, differences in behaviour might be expected.)
A free tyrosyl amino group was also important for hydroperoxide formation on dipeptides. Very little of this product was formed with Gly-Tyr or Ala-Tyr, but Tyr-Gly gave a good response. However, adjacent lysines enabled some hydroperoxide formation. These could be positioned on either side of the tyrosine, and their effects were additive. Notably, free lysine and ethanolamine were also able to invoke concentration-dependent hydroperoxide formation on N-blocked tyrosines. With 10–20 mM amine, yields approximated that with free tyrosine. Presumably this enhancement is also attributable to the nucleophilicity of the amine and its ability to undergo Michael addition. Whereas with tyrosine this leads to cyclization, adjacent lysine residues or free amines should form cross-links with the aromatic ring. This could constitute a novel mechanism for intra- or inter-molecular cross-linking of proteins following oxidative stress.
These findings have implications for the reaction of superoxide with tyrosyl radicals on proteins. This reaction should occur, provided that the radical is accessible, and particularly where the protein structure does not favour phenoxyl radical dimerization. However, the products will differ depending on the molecular environment. We predict that hydroperoxides would be formed on N-terminal tyrosines and those with lysine residues either adjacent or in close proximity. Tyrosyl radicals in more neutral environments might be more likely to undergo repair.
If the mechanism of repair occurs as in Scheme 2 rather than by electron transfer, there is a twist to the story. Provided that the spin conservation rule is obeyed, the oxygen should be released in its singlet state (as represented in Scheme 2) [30]. If so, phenoxyl-radical–superoxide reactions could be a biological source of singlet oxygen. Repair of tyrosyl radicals would then result in localized singlet oxygen generation, and hence would not be benign.
Amino acid and protein hydroperoxides are gaining increasing attention as common products of oxidative stress [10,37–40]. They can be formed from hydroxyl radicals reacting with aliphatic residues with larger numbers of available C–H bonds or from the reaction of singlet oxygen with Tyr, Trp and His residues. Reaction of superoxide with tyrosyl radicals must be considered as another likely route for protein peroxide formation. Apart from modifying the structure of the protein, these hydroperoxides can oxidize GSH and ascorbate, inactivate thiol enzymes (e.g. glyceraldehyde-3-phosphate dehydrogenase and caspase 3) and induce DNA damage [31,41–;45].
Superoxide also reacts with radical forms of other phenols and indoles to give diverse, although not well characterized, products [29,46]. These include drugs and other xenobiotics, and physiological compounds such as α-tocopherol, 5-hydroxytryptamine (serotonin) and melatonin. Depending on the molecular structure, stable hydroperoxides may be formed, or they may act as intermediates in the production of o-quinones [30]. Conditions should be favourable for this radical chemistry to occur widely in biological systems, but as yet it is largely unexplored.
Acknowledgments
This work was supported by the Health Research Council of New Zealand. We are grateful to Philip Morgan for preparing the photolytically generated hydroperoxides, and to Tony Kettle for helpful discussion.
References
- 1.Halliwell B., Gutteridge J. M. C. Oxford: Clarendon Press; 1989. Free Radicals in Biology and Medicine. [Google Scholar]
- 2.Beckman J. S., Koppenol W. H. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol. 1996;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 3.Liochev S. I., Fridovich I. Superoxide and iron: partners in crime. IUBMB Life. 1999;48:157–161. doi: 10.1080/713803492. [DOI] [PubMed] [Google Scholar]
- 4.Winterbourn C. C. Superoxide as an intracellular radical sink. Free Radical Biol. Med. 1993;14:85–90. doi: 10.1016/0891-5849(93)90512-s. [DOI] [PubMed] [Google Scholar]
- 5.Hunter E. P. L., Desrosiers M. F., Simic M. G. The effect of oxygen, antioxidants, and superoxide radical on tyrosine phenoxyl radical dimerization. Free Radical Biol. Med. 1989;6:581–585. doi: 10.1016/0891-5849(89)90064-6. [DOI] [PubMed] [Google Scholar]
- 6.Jin F., Leitich J., von Sonntag C. The superoxide radical reacts with tyrosine-derived phenoxyl radicals by addition rather than by electron transfer. J. Chem. Soc. Perkin Trans. II. 1993:1583–1588. [Google Scholar]
- 7.Winterbourn C. C., Pichorner H., Kettle A. J. Myeloperoxidase-dependent generation of a tyrosine peroxide by neutrophils. Arch. Biochem. Biophys. 1997;338:15–21. doi: 10.1006/abbi.1996.9773. [DOI] [PubMed] [Google Scholar]
- 8.Kochman A., Koska C., Metodiewa D. Submolecular adventures of brain tyrosine: what are we searching for now? Amino Acids. 2002;23:95–101. doi: 10.1007/s00726-001-0114-6. [DOI] [PubMed] [Google Scholar]
- 9.Marquez L. A., Dunford H. B. Kinetics of oxidation of tyrosine and dityrosine by myeloperoxidase compounds I and II. J. Biol. Chem. 1996;270:30434–30440. doi: 10.1074/jbc.270.51.30434. [DOI] [PubMed] [Google Scholar]
- 10.Dean R. T., Fu S., Stocker R., Davies M. J. Biochemistry and pathology of radical-mediated protein oxidation. Biochem. J. 1997;324:1–18. doi: 10.1042/bj3240001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prütz W. A., Butler J., Land E. J. Phenol coupling initiated by one-electron oxidation of tyrosine in peptides and histone. Int. J. Radiat. Biol. 1983;44:183–196. doi: 10.1080/09553008314550981. [DOI] [PubMed] [Google Scholar]
- 12.Hsi L. C., Hoganson C. W., Babcock G. T., Smith W. L. Characterization of a tyrosyl radical in prostaglandin endoperoxide synthase-2. Biochem. Biophys. Res. Commun. 1994;202:1592–1598. doi: 10.1006/bbrc.1994.2114. [DOI] [PubMed] [Google Scholar]
- 13.Gaudu P., Niviere V., Petillot Y., Kauppi B., Fontecave M. The irreversible inactivation of ribonucleotide reductase from Escherichia coli by superoxide radicals. FEBS Lett. 1996;387:137–140. doi: 10.1016/0014-5793(96)00480-2. [DOI] [PubMed] [Google Scholar]
- 14.Tew D., Ortiz de Montellano P. R. The myoglobin protein radical. Coupling of Tyr-103 to Tyr-151 in the H2O2-mediated cross-linking of sperm whale myoglobin. J. Biol. Chem. 1988;263:17880–17886. [PubMed] [Google Scholar]
- 15.McArthur K. M., Davies M. J. Detection and reactions of the globin radical in haemoglobin. Biochim. Biophys. Acta. 1993;1202:173–181. doi: 10.1016/0167-4838(93)90002-9. [DOI] [PubMed] [Google Scholar]
- 16.Giulivi C., Cadenas E. Heme protein radicals: formation, fate, and biological consequences. Free Radical Biol Med. 1998;24:269–279. doi: 10.1016/s0891-5849(97)00226-8. [DOI] [PubMed] [Google Scholar]
- 17.Jacob J. S., Cistola D. P., Hsu F. F., Muzaffar S., Mueller D. M., Hazen S. L., Heinecke J. W. Human phagocytes employ the myeloperoxidase-hydrogen peroxide system to synthesize dityrosine, trityrosine, pulcherosine, and isodityrosine by a tyrosyl radical-dependent pathway. J. Biol. Chem. 1996;271:19950–19956. doi: 10.1074/jbc.271.33.19950. [DOI] [PubMed] [Google Scholar]
- 18.Heinecke J. W. Tyrosyl radical production by myeloperoxidase: a phagocyte pathway for lipid peroxidation and dityrosine cross-linking of proteins. Toxicology. 2002;177:11–22. doi: 10.1016/s0300-483x(02)00192-0. [DOI] [PubMed] [Google Scholar]
- 19.Aeschbach R., Amado R., Neukom H. Formation of dityrosine crosslinks in proteins by oxidation of tyrosine residues. Biochim. Biophys. Acta. 1976;439:292–301. doi: 10.1016/0005-2795(76)90064-7. [DOI] [PubMed] [Google Scholar]
- 20.De Cristofaro R., Landolfi R. Oxidation of human alpha-thrombin by the myeloperoxidase-H2O2-chloride system: structural and functional effects. Thromb. Haemostasis. 2000;83:253–261. [PubMed] [Google Scholar]
- 21.Giulivi C., Davies K. J. Mechanism of the formation and proteolytic release of H2O2-induced dityrosine and tyrosine oxidation products in hemoglobin and red blood cells. J. Biol. Chem. 2001;276:24129–24136. doi: 10.1074/jbc.M010697200. [DOI] [PubMed] [Google Scholar]
- 22.Heinecke J. W., Li W., Francis G. A., Goldstein J. A. Tyrosyl radical generated by myeloperoxidase catalyzes the oxidative cross-linking of proteins. J. Clin. Invest. 1993;91:2866–2872. doi: 10.1172/JCI116531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ushijima Y., Nakano M., Goto T. Production and identification of bityrosine in horseradish peroxidase-hydrogen peroxide-tyrosine system. Biochem. Biophys. Res. Commun. 1984;125:916–918. doi: 10.1016/0006-291x(84)91370-6. [DOI] [PubMed] [Google Scholar]
- 24.Bhattacharjee S., Pennathur S., Byun J., Crowley J., Mueller D., Gischler J., Hotchkiss R. S., Heinecke J. W. NADPH oxidase of neutrophils elevates o,o'-dityrosine cross-links in proteins and urine during inflammation. Arch. Biochem. Biophys. 2001;395:69–77. doi: 10.1006/abbi.2001.2557. [DOI] [PubMed] [Google Scholar]
- 25.Fu S., Davies M. J., Stocker R., Dean R. T. Evidence for roles of radicals in protein oxidation in advanced human atherosclerotic plaque. Biochem. J. 1998;333:519–525. doi: 10.1042/bj3330519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leeuwenburgh C., Hansen P. A., Holloszy J. O., Heinecke J. W. Oxidized amino acids in the urine of aging rats: potential markers for assessing oxidative stress in vivo. Am. J. Physiol. 1999;276:R128–R135. doi: 10.1152/ajpregu.1999.276.1.R128. [DOI] [PubMed] [Google Scholar]
- 27.Kettle A. J., Carr A. C., Winterbourn C. C. Assays using horseradish peroxidase and phenolic substrates require superoxide dismutase for accurate determination of hydrogen peroxide production by neutrophils. Free Radical Biol. Med. 1994;17:161–164. doi: 10.1016/0891-5849(94)90111-2. [DOI] [PubMed] [Google Scholar]
- 28.Pichorner H., Metodiewa D., Winterbourn C. C. Generation of superoxide and tyrosine peroxide as a result of tyrosyl radical scavenging by glutathione. Arch. Biochem. Biophys. 1995;323:429–437. doi: 10.1006/abbi.1995.0064. [DOI] [PubMed] [Google Scholar]
- 29.Jonsson M., Lind T., Reitberger T. E., Eriksen T. E., Merenyi G. Free radical combination reactions involving phenoxyl radicals. J. Phys. Chem. 1993;97:8229–8233. [Google Scholar]
- 30.d'Alessandro N., Bianchi G., Fang X., Jin F., Schuchmann H.-P., von Sonntag C. Reactions of superoxide with phenoxyl-type radicals. J. Chem. Soc. Perkin Trans. II. 2000:1862–1867. [Google Scholar]
- 31.Wright A., Bubb W. A., Hawkins C. L., Davies M. J. Singlet oxygen-mediated protein oxidation: evidence for the formation of reactive side chain peroxides on tyrosine residues. Photochem. Photobiol. 2002;76:35–46. doi: 10.1562/0031-8655(2002)076<0035:sompoe>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 32.Kettle A. J., Winterbourn C. C. Superoxide modulates the activity of myeloperoxidase and optimizes the production of hypochlorous acid. Biochem. J. 1988;252:529–536. doi: 10.1042/bj2520529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wolff S. P. Ferrous ion oxidation in presence of ferric ion indicator xylenol orange for measurement of hydroperoxides. Methods Enzymol. 1994;233:182–189. [Google Scholar]
- 34.Beers R. J., Sizer I. W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 1952;195:133–140. [PubMed] [Google Scholar]
- 35.Valoti M., Tipton K. F., Sgaragli G. P. Oxidative ring-coupling of tyrosine and its derivatives by purified rat intestinal peroxidase. Biochem. Pharmacol. 1992;43:945–951. doi: 10.1016/0006-2952(92)90597-c. [DOI] [PubMed] [Google Scholar]
- 36.Nagano T., Fridovich I. Superoxide radical from xanthine oxidase acting upon lumazine. J. Free Radical Biol. Med. 1985;1:39–42. doi: 10.1016/0748-5514(85)90027-3. [DOI] [PubMed] [Google Scholar]
- 37.Davies M. J., Truscott R. J. Photo-oxidation of proteins and its role in cataractogenesis. J. Photochem. Photobiol. B. 2001;63:114–125. doi: 10.1016/s1011-1344(01)00208-1. [DOI] [PubMed] [Google Scholar]
- 38.Fu S., Hick L. A., Sheil M. M., Dean R. T. Structural identification of valine hydroperoxides and hydroxides on radical-damaged amino acid, peptide, and protein molecules. Free Radical Biol. Med. 1995;19:281–292. doi: 10.1016/0891-5849(95)00021-o. [DOI] [PubMed] [Google Scholar]
- 39.Gebicki S., Gebicki J. M. Formation of peroxides in amino acids and proteins exposed to oxygen free radicals. Biochem. J. 1993;289:743–749. doi: 10.1042/bj2890743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gieseg S., Duggan S., Gebicki J. M. Peroxidation of proteins before lipids in U937 cells exposed to peroxyl radicals. Biochem. J. 2000;350:215–218. [PMC free article] [PubMed] [Google Scholar]
- 41.Fu S., Gebicki S., Jessup W., Gebicki J. M., Dean R. T. Biological fate of amino acid, peptide and protein hydroperoxides. Biochem. J. 1995;311:821–827. doi: 10.1042/bj3110821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morgan P. E., Dean R. T., Davies M. J. Inhibition of glyceraldehyde-3-phosphate dehydrogenase by peptide and protein peroxides generated by singlet oxygen attack. Eur. J. Biochem. 2002;269:1916–1925. doi: 10.1046/j.1432-1033.2002.02845.x. [DOI] [PubMed] [Google Scholar]
- 43.Hampton M. B., Morgan P. E., Davies M. J. Inactivation of cellular caspases by peptide-derived tryptophan and tyrosine peroxides. FEBS Lett. 2002;527:289–292. doi: 10.1016/s0014-5793(02)03240-4. [DOI] [PubMed] [Google Scholar]
- 44.Luxford C., Dean R. T., Davies M. J. Induction of DNA damage by oxidised amino acids and proteins. Biogerontology. 2002;3:95–102. doi: 10.1023/a:1015228001561. [DOI] [PubMed] [Google Scholar]
- 45.Gebicki S., Gebicki J. M. Crosslinking of DNA and proteins induced by protein hydroperoxides. Biochem. J. 1999;338:629–636. [PMC free article] [PubMed] [Google Scholar]
- 46.Winterbourn C. C., Kettle A. J. Radical-radical reactions of superoxide: a potential route to toxicity. Biochem. Biophys. Res. Commun. 2003;305:729–736. doi: 10.1016/s0006-291x(03)00810-6. [DOI] [PubMed] [Google Scholar]