

Abstract
Recent studies in metabolic profiling have underscored the importance of the concept of a metabolic network of pathways with special functional characteristics that differ from those of simple reaction sequences. The characterization of metabolic functions requires the simultaneous measurement of substrate fluxes of interconnecting pathways. Here we present a novel stable isotope method by which the forward and reverse fluxes of the futile cycles of the hepatic glucose metabolic network are simultaneously determined. Unlike previous radio-isotope methods, a single tracer [1,2-13C2]D-glucose and mass isotopomer analysis is used. Changes in fluxes of substrate cycles, in response to several gluconeogenic substrates, in isolated fasted hepatocytes from male Wistar rats were measured simultaneously. Incubation with these substrates resulted in a change in glucose-6-phosphatase/glucokinase and glycolytic/gluconeogenic flux ratios. Different net redistributions of intermediates in the glucose network were observed, resulting in distinct metabolic phenotypes of the fasted hepatocytes in response to each substrate condition. Our experimental observations show that the constraints of concentrations of shared intermediates, and enzyme kinetics of intersecting pathways of the metabolic network determine substrate redistribution throughout the network when it is perturbed. These results support the systems-biology notion that network analysis provides an integrated view of the physiological state. Interaction between metabolic intermediates and glycolytic/gluconeogenic pathways is a basic element of cross-talk in hepatocytes, and may explain some of the difficulties in genotype and phenotype correlation.
Keywords: futile cycles, GC–MS, glucose metabolism, glucose stable isotope tracer, metabolic network
Abbreviations: G6P, glucose-6-phosphate; G6Pase, glucose-6-phosphatase (EC 3.1.3.9); GK, glucokinase (EC 2.7.1.2); MIDA, mass isotopomer distribution analysis; PC, pyruvate carboxylase (EC 6.4.1.1); PDH, pyruvate dehydrogenase (EC 1.2.4.1); PPC, pentose phosphate cycle
INTRODUCTION
Isolated hepatocytes have an impressive capacity for adjusting their intracellular machinery in response to changes in their nutrient and hormonal environment. The high complexity of the glucose metabolic network permits multiple metabolic endpoints in response to changes in environmental factors for a given genomic or proteomic expression. Thus, the physiological state of hepatocytes at a given moment cannot be predicted by the genome alone (or even by the proteins it encodes) [1,2].
The hepatic glucose metabolic network comprises a number of pathways of intersecting substrate cycles, of which the main one is the glycolytic/gluconeogenic pathway. These can operate simultaneously, resulting in futile cycling of substrates with no net flux of metabolites [3–5]. The glycolytic/gluconeogenic cycle intersects with the glycogenesis/glycogenolysis cycle and the pentose phosphate cycle (PPC), being thus embedded in a larger hepatic glucose metabolic network (Scheme 1). Traditionally studies of hepatic glucose metabolism have focused on the regulation of the individual reactions of the glycolytic and gluconeogenic pathways. Few studies have addressed the effect of the impinging pathways of the PPC at the triose phosphate and the hexose phosphate levels on glycolytic/gluconeogenic fluxes. Recent studies in metabolic profiling have underscored the importance of the concept of a metabolic network of pathways with special functional characteristics that differ from those of simple reaction sequences. Under this network the individual substrate concentrations, enzyme activities and cofactor availabilities serve as ‘constraints’, which ultimately determine the performance of the network, as measured by substrate fluxes [6,7]. The effects of genetic or nutrient environment changes can be best understood in terms of an alteration of ‘constraints’ and the resulting response in substrate production and utilization (fluxes). An important implication of the operation of the glucose metabolic network is that for a given change in genomic or proteomic expression, the regulation of flux distribution along the entire network is affected. Therefore, hepatic glucose metabolism can be best studied using techniques that permit the simultaneous characterization of substrate fluxes across the network.
Scheme 1. Scheme of hepatic glucose metabolic network showing the relative location of glycolytic/gluconeogenic substrate cycles.

Metabolic outfluxes (a, glycogen synthase; b, G6Pase; c, glycolysis; and d, PPC) and influxes (e, GK; and f, gluconeogenesis) involved in homoeostasis of metabolites of the network in fasted rat hepatocytes. Central to the regulation of glucose homoeostasis in fasted-refed transition are the futile cycles around the hepatic G6P.
Mass isotopomer distribution analysis (MIDA) using GC coupled to MS (GC–MS) has been widely used in metabolic profiling. This technique has been applied to tumoral metabolic profiling, and has allowed the design of new strategies that target the metabolic network in cancer cells [8–11]. The stable isotope approach of MIDA differs from traditional methods that use multiple radioisotopes, in that a single isotope is used and individual fluxes through all the futile cycles and the PPC and Krebs cycle can be determined simultaneously. Here we apply this novel approach for the first time to provide a metabolic profile of the individual fluxes in response to several substrate inputs using [1,2-13C2]glucose in hepatocytes. This characterization shows that cross talk between pathways and simultaneous regulation of the numerous futile cycles are crucial to understand the high flexibility of the network for a given genomic and proteomic expression. Furthermore, we show that the use of GC–MS together with 13C stable isotope labelling is a robust approach for examining and modelling metabolic pathways and metabolite transformations. The resulting flux map of the metabolic network may be regarded as the phenotypic equivalent of a micro-array gene expression profile.
EXPERIMENTAL
Materials
[1,2-13C2]D-glucose (>99% enriched) was purchased from Isotec (Miamisburg, OH, U.S.A.), and other reagents from Sigma.
Animals
Male Wistar rats (180–200 g) were used. Rats were maintained in a 12 h:12 h light/dark cycle with free access to standard laboratory rat chow pellets (MDS Panlabs, Bothell, WA, U.S.A.) and water. Animals were fasted for 24 h before hepatocyte isolation. Experiments were conducted in accordance with the guidelines of the University Animal Care and Use Committee. Appropriate measures were taken to minimize animal pain or discomfort.
Preparation of cells and incubation
Suspensions of isolated parenchymal liver cells were prepared from 24 h fasted animals as described previously [12]. Cells were resuspended in Krebs–Ringer bicarbonate buffer, pH 7.4. Preparations with viability below 90%, established by the Trypan Blue exclusion method, were not used. Samples (6 ml) of these suspensions, containing 2.3×106 cells/ml, were incubated at 37 °C with gassing and continuous shaking at 160 strokes/min for 2 h, as this was the optimum time to ensure maximum glycogen synthesis without diminishing cell viability. Conditions for cell incubation were: a) 20 mM glucose, b) 20 mM glucose and 10 mM glutamine (glucose+glutamine), c) 20 mM glucose and 10 mM lactate/pyruvate (9:1) (glucose+lactate/pyruvate), and d) 20 mM glucose and 3 mM fructose (glucose+fructose). In all conditions glucose enriched by 50% in [1,2-13C2]glucose was used.
Measurement of metabolites
At the end of incubations, cells were centrifuged (3000 g, 20 s), and incubation medium and cell pellets were obtained. For glycogen determination, pellets were immediately homogenized with 30% (w/v) KOH using a modification of the methodology described by Chan et al. [13], using 3 MM paper to precipitate glycogen. Intracellular glucose-6-phosphate (G6P) levels, and incubation medium concentrations of glucose and lactate were determined as described previously [14–16].
GC–MS sample processing and analysis
At the end of incubations, cells were centrifuged (3000 g, 20 s), thereby separating the incubation medium from cell pellet, and all fractions were frozen in liquid nitrogen and stored at −80 °C until processing. Incubation media were processed to isolate lactate, glucose and glutamate, using methods established previously [17,18]. Glycogen was isolated from cell pellets after ethanol precipitation over 3 MM paper. It was then treated with amyloglucosidase, and the hydrolysed glucose was isolated using ion exchange chromatography [18]. Immediately after, glucose from the medium or from hydrolysed glycogen, as well as lactate and glutamate, were derivatized for GC–MS analysis [17,19,20]. A mass selective detector HP 5973 equipment coupled to a gas-liquid chromatograph HP 6890, using previously described GC conditions [17,19,20], was used for all the metabolites. Chemical ionization was used to give the C1–C6 molecular ion of the glycogen or glucose molecules in medium at m/z 328, and the same for the lactate molecule (C1–C3) at m/z 328. Electron impact ionization was used to characterize the isotopomers of the C1–C4 glycogen glucose fragment at m/z 242, and C2–C4 (m/z 152) and C2–C5 (m/z 198) glutamate fragments.
Results of the mass isotopomers in glucose, lactate and glutamate are reported as molar fractions, where m0, m1, m2 etc. indicate the number of 13C atoms in the molecule [21]. M0, M1, M2 etc. are used for glycogen or medium glucose isotopomers, whereas m0, m1, and so on, are for lactate and glutamate isotopomers. The enrichment, Σmn, is the weighted sum of the labelled species (Σmn=m1×1+m2×2+m3×3…) and is equivalent to the specific activity of a compound in radioactive tracer experiments.
Data interpretation
Formulae used to obtain the parameters expressed in results, such as G6P outfluxes (to glycogen synthesis, to glucose incubation medium, to lactate through glycolysis, and to lactate through PPC), G6P influxes (from glucose incubation medium and from lactate through gluconeogenesis), and Krebs cycle evaluation parameters, are described in the Appendix.
Statistical analysis
All data are expressed as mean±S.E.M. Statistical significance was determined using the Student’s t test. P≤0.01 was considered statistically significant.
RESULTS
The metabolism of [1,2-13C2]glucose results in the rearrangement, exchange, or loss of the 13C label, which is incorporated into glucose metabolic intermediates in specific patterns. The enrichment of isotopomers of these intermediates also depends on the dilution by their unlabelled counterparts. Thus, the specific isotopomer distribution and their respective enrichment provide information on the flow of substrates along the forward and reverse pathways of substrate cycles. Here we analysed the flexibility of the glucose metabolic network to re-organize flux distribution in response to changes in carbon inputs, in incubation conditions with no changes at proteomic or genomic levels.
Effect of gluconeogenic substrates on the re-distribution of fluxes involved in the glucose metabolic network
Before estimating the fluxes involved in the hepatic glucose metabolic network (Scheme 1), we analysed glycogen glucose and lactate isotopomers. Isolated rat hepatocytes from fasted rats were incubated for 2 h with 20 mM glucose in the presence or absence of distinct gluconeogenic substrates [10 mM glutamine, 10 mM lactate/pyruvate (9:1), or 3 mM fructose]. The distribution of isotopomers in C1–C6 and C1–C4 fragments of glucose from the glycogen pellet was practically equal, indicating that little randomization of the 13C on C1 and C2 via triose phosphates occurred (Table 1). Moreover, between 1–2% of glycogen glucose contained one 13C (M1), representing recycled species. Since the recycling of 13C from the Krebs cycle and phosphoenolpyruvate typically leads to almost symmetrically labelled glucose molecules, the lack of symmetry of the 13C distribution in glycogen indicates that the formation of M1 glucose is primarily by the action of G6P dehydrogenase and the recycling of 13C from the PPC. The mixing of G6P from glucose uptake and gluconeogenesis (direct/indirect pathways) in glycogen synthesis was estimated. In spite of the large changes in substrate environment, the ratio of glucose uptake to gluconeogenesis was only modestly changed.
Table 1. Mass isotopomer distribution in C1–C6 and C1–C4 fragments of glycogen glucose.
Hepatocytes were incubated for 2 h in Krebs–Ringer buffer with 20 mM glucose (glucose), 20 mM glucose+,10 mM glutamine (glucose+,glutamine), 20 mM glucose+,10 mM lactate/pyruvate (9:1) [glucose+lactate/pyruvate (9:1)] or 20 mM glucose+,3 mM fructose (glucose+,fructose). C1–C6 and C1–C4 fragments of glycogen glucose were measured as described in the Experimental section. M4, M5 and M6 were not detected. M2(glycogen)/M2(medium) reflects the contribution of medium glucose to glycogen (direct pathway). Means±S.E.M. of several experiments (n=6) are provided.
| M0 | M1 | M2 | M3 | ΣMn | M2(glycogen)/M2(medium) | ||
|---|---|---|---|---|---|---|---|
| C1–C6 | Glucose | 0.601±0.011 | 0.017±0.001 | 0.368±0.010 | 0.004±0.000 | 0.806±0.021 | 0.753±0.022 |
| Glucose+,glutamine | 0.593±0.010 | 0.023±0.001 | 0.374±0.009 | 0.004±0.0004 | 0.805±0.021 | 0.785±0.018 | |
| Glucose+,lactate/pyruvate (9:1) | 0.681±0.012 | 0.012±0.001 | 0.302±0.011 | 0.002±0.0004 | 0.635±0.024 | 0.640±0.023 | |
| Glucose+,fructose | 0.655±0.005 | 0.011±0.001 | 0.327±0.004 | 0.002±0.0006 | 0.691±0.011 | 0.710±0.009 | |
| C1–C4 | Glucose | 0.601±0.013 | 0.024±0.003 | 0.367±0.014 | 0.005±0.000 | 0.785±0.025 | 0.769±0.040 |
| Glucose+,glutamine | 0.581±0.003 | 0.028±0.003 | 0.383±0.004 | 0.005±0.001 | 0.821±0.007 | 0.805±0.009 | |
| Glucose+,lactate/pyruvate (9:1) | 0.679±0.013 | 0.016±0.002 | 0.301±0.013 | 0.003±0.0003 | 0.632±0.027 | 0.640±0.067 | |
| Glucose+,fructose | 0.664±0.005 | 0.016±0.0003 | 0.315±0.004 | 0.003±0.001 | 0.665±0.012 | 0.701±0.009 |
Isotopomer distribution in lactate from the incubation medium was also determined after 2 h incubation (Table 2). The Σmn of 13C in lactate at the end of the incubation was maximum when hepatocytes were incubated with glucose only. The dilution of lactate enrichment when gluconeogenic substrates were added resulted from the mixing of m2 lactate from G6P with unlabelled lactate produced from these substrates. To calculate the fractional contribution of G6P to lactate, isotopic equilibrium between G6P and glycogen was assumed. This assumption is based on previous observations by Katz et al. [22] that when hepatocytes were incubated with [2-3H]- and [U-14C]glucose, the specific activity and the 3H/14C ratio in glycogen glucose and G6P equalled each other. We observed previously that G6P produced by overexpressed hexokinase does not trigger glycogen synthesis while G6P produced by glucokinase (GK) does [23,24]. Since the amount of G6P produced by hexokinase is minimal at a medium glucose concentration of 20 mM, the assumption of a single pool of G6P is essentially valid. Thus, the fractional contribution from G6P was decreased in the presence of glutamine, lactate or fructose, indicating the conversion of glutamine and fructose to lactate without suppression of glycolysis (Table 2).
Table 2. Mass isotopomer distribution in lactate from incubation medium.
Hepatocytes were incubated as in Table 1. m0, m1, m2 and m3 were calculated as described in the Experimental section. The ratio m2(lactate)×2/M2(glycogen) reflects the contribution of G6P to lactate. One minus this ratio should be the contribution of pyruvate from alanine or Krebs cycle, or other contributions such as glycerol to the triose phosphate pool. Means±S.E.M. of several experiments (n=6) are provided.
| m0 | m1 | m2 | m3 | Σmn | m2(lactate)×2/M2(glycogen) | |
|---|---|---|---|---|---|---|
| Glucose | 0.850±0.006 | 0.029±0.001 | 0.118±0.006 | 0.003±0.0012 | 0.275±0.012 | 0.641±0.026 |
| Glucose+,glutamine | 0.929±0.004 | 0.006±0.001 | 0.065±0.003 | −0.001±0.0010 | 0.135±0.006 | 0.345±0.014 |
| Glucose+,lactate/pyruvate (9:1) | 0.982±0.004 | 0.002±0.001 | 0.009±0.001 | 0.006±0.0028 | 0.039±0.010 | 0.059±0.008 |
| Glucose+,fructose | 0.917±0.006 | 0.015±0.001 | 0.067±0.005 | 0.001±0.0001 | 0.152±0.011 | 0.410±0.027 |
Effect of gluconeogenic substrates on glucose metabolic intermediates
Despite their similarity as gluconeogenic substrates, glutamine, lactate and fructose evoked different responses in substrate redistributions, resulting in distinct levels of metabolic intermediates, which were also necessary to calculate the absolute values of fluxes. Isolated rat hepatocytes from fasted rats contained 3.6 μg of glycogen glucose and 0.1 nmol of G6P per million cells. Under all incubation conditions, there was a substantial lactate formation, accompanied by an increase in the glycogen content and intracellular G6P levels of hepatocytes, compared with values obtained in cells incubated with Krebs–Ringer buffer only (Table 3). Incubations with glucose and gluconeogenic precursors resulted in net glycogen synthesis. However, this synthesis was further stimulated by media containing glutamine or fructose. Of these two treatments, G6P concentration was increased 2-fold only in the former. G6P concentration was also increased 35% when cells were treated with glucose+lactate/pyruvate, although the amount of glycogen synthesized did not differ from that of the glucose only control. Lactate concentration in the incubation medium increased significantly when fructose was added, whereas it decreased significantly (from 9 mM to 6.2 mM) in the presence of lactate/pyruvate, demonstrating net lactate uptake. Furthermore, glucose released to the medium was analysed. While in glucose only and glucose+glutamine conditions, an absolute uptake of glucose was observed, for glucose+lactate/pyruvate or glucose+ fructose an absolute glucose production of around 4% was detected. Thus, gluconeogenic precursors had distinct effects on the substrate cycles and redistribution of intermediates of hepatic glucose metabolic network.
Table 3. Lactate concentration from incubation medium, glycogen content and intracellular G6P levels in hepatocytes treated with gluconeogenic substrates.
Hepatocytes were incubated as in Table 1. Lactate concentration from incubation medium, glycogen content and G6P intracellular levels in all these incubation conditions and in hepatocytes incubated only with Krebs–Ringer buffer, were determined at the end of the incubation as described in the Experimental section. Glucose released determined from subtraction of absolute glucose consumption [column (e) from Table 4] over the absolute glucose release (column (b) from Table 4) was also estimated in all the different incubation conditions at the end of incubation. Values are expressed as means±S.E.M. *P≤0.01 compared with glucose condition (n=6).
| Lactate (mM in incubation medium) | Glycogen (μg/million cells) | G6P (nmol/million cells) | Glucose release (μg/million cells) | |
|---|---|---|---|---|
| Krebs | 0.02±0.01 | 2.8±0.2 | 0.2±0.1 | − |
| Glucose | 0.8±0.1 | 24.1±2.0 | 0.8±0.1 | −14.3±3.3 |
| Glucose+glutamine | 1.0±0.1 | 49.2±3.1* | 1.7±0.2* | −22.7±4.0 |
| Glucose+lactate/pyruvate (9:1) | 6.2±0.2* | 27.7±2.7 | 1.1±0.1* | 47.4±11.8* |
| Glucose+fructose | 2.2±0.1* | 47.6±3.7* | 0.9±0.1 | 64.8±10.6* |
Effect of gluconeogenic substrates on G6P outfluxes and influxes
From the amount of glucose and lactate released into the medium, and the intracellular glycogen content, several components of outflux and influx of intracellular G6P were calculated after considering the dilution at the level of G6P and triose phosphate (Table 4). The calculations of these fluxes assume that intracellular G6P is in isotopic equilibrium with glycogen (described above), and that the system is in a metabolic steady state. The constancy of G6P levels at 2 h incubation was accepted after verification that G6P from cells incubated from 1 h 30 min until 2 h 30 min was constant in all the incubation conditions tested (results not shown). These metabolic and isotopic steady states allowed the calculation of outfluxes and influxes of G6P using the equations in the Appendix. The pathways leading to the substrate outflux from the G6P pool (see Scheme 1) are: (a) glycogen synthesis, (b) glucose-6-phosphatase (G6Pase) releasing glucose into the medium, (c) glycolytic pathway, and (d) the oxidative PPC, which also contributes to lactate production. Only two pathways led to substrate influx into the G6P pool under glycogen-depletion, (e) glucose phosphorylation by GK and (f ) gluconeogenic flux.
Table 4. G6P influx and outflux in hepatocytes treated with gluconeogenic substrates.
Hepatocytes were incubated as described in Table 1. (a) Is the outflux from G6P to glycogen, (b) is the outflux from G6P to glucose incubation medium (hepatic glucose output or G6P action), (c) is the outflux from G6P to lactate (glycolysis), and (d) is the outflux from G6P through pentose phosphate cycle (G6P dehydrogenase action), (e) is the influx from glucose incubation medium to G6P (GK action), and (f) is the influx from lactate to G6P (gluconeogenesis). All these fluxes were determined as described in the Appendix. The total G6P flux was determined from the addition of all G6P outfluxes or all G6P influxes. Furthermore, under each flux the percentage that this flux represents over the total G6P flux is expressed. Values are expressed as means±S.E.M. *P≤0.01 compared with glucose condition (n=6).
| G6P outflux (μg glucose/million cells) | G6P influx (μg glucose/million cells) | ||||||
|---|---|---|---|---|---|---|---|
| (a) | (b) | (c) | (d) | (e) | (f) | Total G6P flux (μg glucose/million cells) | |
| Glucose | 24.1±2.0 | 97.8±9.0 | 25.0±2.0 | 1.8±0.2 | 112.1±5.9 | 36.5±2.1 | 148.6±8.0 |
| 16.2% | 65.8% | 16.8% | 1.2% | 75.4% | 24.6% | ||
| Glucose+glutamine | 49.2±3.1* | 144.7±9.4* | 13.8±1.5* | 0.4±0.1* | 167.4±13.5 | 40.2±3.7 | 207.6±16.2 |
| 23.6% | 69.5% | 6.6% | 0.2% | 80.6% | 19.4% | ||
| Glucose+lactate/pyruvate (9:1) | 27.7±2.7 | 234.5±24.3* | 19.6±2.5 | 1.9±0.4 | 187.1±35.2 | 92.6±16.5 | 279.7±51.7 |
| 9.8% | 82.7% | 6.9% | 0.7% | 66.9% | 33.1% | ||
| Glucose+fructose | 47.6±3.7* | 442.7±52.3* | 36.9±4.9 | 2.5±0.3 | 377.8±42.5* | 152.3±12.2* | 530.2±54.2* |
| 9.0% | 83.6% | 7.0% | 0.5% | 71.0% | 29.0% | ||
G6P outflux to glycogen synthesis [column (a), Table 4]
In the absence of glycogenolysis, as in hepatocytes from fasted rats, G6P outflux to glycogen synthesis coincided with the net flux of the pathway from G6P to glycogen. This G6P outflux was doubled when cells were incubated with glucose+glutamine or glucose+fructose compared with glucose only (Table 4). The effect of fructose and glutamine is probably mediated by a number of metabolic cross-talk mechanisms since the same increase in glycogen synthesis was associated with G6P concentrations that differed greatly (Table 3).
G6P outflux to glucose incubation medium [recycling through G6Pase; column (b), Table 4]
G6P outflux to glucose incubation medium, i.e. futile cycling G6P↔glucose, was calculated from the dilution of M2 glucose in the medium using the tracer dilution principle. Significant futile cycling was detected under all experimental conditions as glucose was continuously released into the medium. Recycling through G6Pase increased substantially with the addition of gluconeogenic precursors. The increase in glucose outflow was balanced by an increase in glucose uptake, such that the change in glucose concentration in the incubation medium was small under all conditions (Table 3).
G6P outflux to lactate formation through glycolysis [column (c), Table 4]
Multiplying the absolute lactate concentration by the lactate fractional contribution from G6P (Table 2) provided the absolute glycolytic flux (from G6P to lactate). In the glucose+glutamine treatment, the amount of G6P that was directed to lactate though glycolysis was significantly less than in the control, indicating that the balance of fluxes through the glycolytic/gluconeogenic pathways favours gluconeogenesis (Table 4). In contrast, the effect of either lactate or fructose differed considerably, as the glycolytic flux was not suppressed in either of these incubation conditions. These results indicate that the same metabolic endpoints were achieved through differential response of substrate cycles to gluconeogenic substrate inputs.
G6P outflux to lactate formation through the PPC [column (d),Table 4]
The percentage of the oxidative PPC flux with regard to glycolysis was estimated using the m1/m2 13C ratio in lactate isotopomers, as described previously [20,25]. Glucose+glutamine and glucose+ fructose resulted in a significantly lower percentage of PPC (decreases of PPC being 60% and 13% respectively) compared with the glucose only treatment, where this percentage was equal to 7.7±0.3%. No change was observed in the glucose+lactate/ pyruvate condition. Furthermore, the oxidative PPC flux [column (d), Table 4] did not differ significantly from that of the control condition in glucose+lactate/pyruvate and glucose+fructose conditions, whereas in the presence of glutamine it was significantly lower than that of the control, as a result of lower lactate production and lower PPC activity.
G6P influx [columns (e and f), Table 4]
Since the G6P concentration was constant during the period of observation, the sum of all G6P outfluxes must be equal to that of all influxes. The relative contribution from glucose uptake (GK) to G6P would be the same as the contribution of glucose uptake to glycogen synthesis (Table 1). The magnitude of GK and gluconeogenesis was calculated by multiplying the total outflow with the respective relative contributions. The contribution of GK to G6P influx (or G6P influx from glucose incubation medium) fluctuated between 68% and 79%, depending on the incubation condition, whereas the remaining 21% to 32% was for the contribution attributed to gluconeogenic flux (or gluconeogenesis). Of all the conditions tested, fructose was found to stimulate a 3-fold increase in glucose uptake and in gluconeogenesis.
Effect of gluconeogenic substrates on G6Pase/GK and glycolysis/gluconeogenesis futile cycles
The substrate cycles of the glucose metabolic network showed distinct reponses to gluconeogenic inputs. As expected, incubation with gluconeogenic precursors increased the rate of reaction in the direction of gluconeogenesis and glucose release. Interestingly, these changes were often associated with similar increases in the reverse pathways of these substrate cycles. The change in the ratio of the forward and reverse reactions of a substrate cycle is the key determinant of net flow into glycogen, glucose release, glycolysis and PPC (Table 4). While the G6Pase/GK ratio was <1 in glucose only and glucose+glutamine conditions, this ratio was reversed in the presence of lactate and fructose, being approximately 40% higher than the ratio of the glucose control (Table 5). The former conditions resulted in net glucose uptake and the latter conditions were accompanied by a 2- to 4-fold increase in glucose release into the medium (Table 4). A different relationship was found between glycolytic and gluconeogenic flux. Except in the case of fructose, an increase in gluconeogenic flux was not associated with increased glycolytic flux (Table 4). Thus, an increase in gluconeogenic flux bore a roughly inverse relationship with the decrease in the glycolysis/gluconeogenesis ratio of the same incubation condition. However, the magnitude of change of absolute flux was not reflected by the magnitude of change in the glycolysis/gluconeogenesis ratio as in glucose+glutamine and glucose+fructose conditions compared with the glucose only condition. GK/G6Pase and glycolytic/gluconeogenic futile cycles are known to result in isotope recycling, thereby complicating the estimation of production or turnover of substrates with tracers. The glucose to G6P isotope recycling ratio has been previously estimated using radio-isotopes. The value equivalent to isotope recycling is provided in Table 5. Under all experimental conditions futile cycling was high. Thus, 66% to 83% of isotope was recycled. The implication of this recycling is that the estimation of glucose production using non-recyclable tracer overestimates the true glucose production rate by 17% to 34%.
Table 5. Evaluation of G6P/GK and glycolysis/gluconeogenesis flux ratios in hepatocytes treated with gluconeogenic substrates.
Hepatocytes were incubated as in Table 1. G6Pase, GK, glycolysis and gluconeogenesis fluxes were evaluated as described in the Appendix. G6Pase/GK flux ratio was given by dividing values of column (b) by the corresponding value of column (e) of Table 4. Glycolytic/gluconeogenic flux ratio was given by dividing values of column (c+,d) by the corresponding value of column (f) of Table 4. Glucose to G6P isotope recycling is given by G6Pase/GK flux ratio multiplied by the direct pathway from Table 1. Means±S.E.M. of several experiments (n=6) are provided. *P≤0.01 compared with glucose condition.
| G6Pase/GK flux ratio | Glucose to G6P isotope recycling | Glycolytic/gluconeogenic flux ratio | |
|---|---|---|---|
| Glucose | 0.87±0.02 | 0.66±0.02 | 0.73±0.02 |
| Glucose+glutamine | 0.86±0.01 | 0.68±0.02 | 0.35±0.05* |
| Glucose+lactate/pyruvate (9:1) | 1.25±0.02* | 0.80±0.02 | 0.23±0.05* |
| Glucose+fructose | 1.17±0.02* | 0.83±0.01 | 0.26±0.01* |
Effect of gluconeogenic substrates on the Krebs cycle
Isotopomeric analysis of C2–C5 and C2–C4 fragments of glutamate in the medium was performed to estimate anaplerotic flux and the relative contributions of pyruvate carboxylase (PC) and pyruvate dehydrogenase (PDH) to the Krebs cycle. Anaplerotic flux was estimated from m1/m2 ratio of the C2–C5 fragment [20] and was between 1 and 1.5 times that of the Krebs cycle flux. Except for the condition glucose+glutamine, where anaplerotic flux could not be computed because of the small enrichment detected, no significant differences were observed between the conditions tested. With regard to the relative contribution of PC and PDH as possible carbon entries to the Krebs cycle, the distinct labelling pattern in glutamate molecules was analysed (Table 6). Enrichment of m2 from the C2–C5 fragment was highest in the glucose only condition and lowest when lactate/pyruvate was added. Therefore the label from glucose was greatly diluted by fructose and lactate, indicating the use of the carbon from these sugars in the Krebs cycle. PC was 3-fold higher than PDH flux when glucose was the sole substrate, whereas, in the other conditions the contributions of PC and PDH were similar, indicating the activation of PDH by lactate and by fructose (Figure 1). Activation in PDH was from 15% (1−m2C2-C4/m2C2-C5) in the glucose only treatment to 62% and 44% in glucose+lactate and glucose+fructose conditions respectively.
Table 6. Mass isotopomer distribution in C2–C5 and C2–C4 fragments of glutamate from incubation medium.
Hepatocytes were incubated as in Table 1. C2–C5 and C2–C4 fragments of glutamate were measured as described in the Experimental procedures. Means±S.E.M. of several experiments (n=6) are provided.
| m0 | m1 | m2 | m3 | m4 | Σmn | |||
|---|---|---|---|---|---|---|---|---|
| C2–C5 | Glucose | 0.901±0.008 | 0.034±0.003 | 0.059±0.006 | 0.000±0.001 | 0.006±0.002 | 0.178±0.015 | |
| Glucose+lactate/pyruvate (9:1) | 0.985±0.003 | 0.005±0.003 | 0.008±0.001 | 0.000±0.000 | 0.003±0.000 | 0.03±0.003 | ||
| Glucose+fructose | 0.926±0.006 | 0.024±0.002 | 0.046±0.005 | 0.001±0.000 | 0.004±0.000 | 0.133±0.011 | ||
| C2–C4 | Glucose | 0.909±0.009 | 0.042±0.005 | 0.050±0.004 | 0.000±0.001 | 0.141±0.015 | ||
| Glucose+lactate/pyruvate (9:1) | 0.984±0.006 | 0.011±0.002 | 0.003±0.002 | 0.003±0.003 | 0.025±0.013 | |||
| Glucose+fructose | 0.932±0.007 | 0.042±0.003 | 0.026±0.004 | 0.000±0.000 | 0.094±0.010 |
Figure 1. Relative contribution of [1,2-13C2]glucose to m2 of glutamate via PC and PDH in hepatocytes treated with gluconeogenic substrates.

Hepatocytes were incubated as in Table 1. Relative contributions of [1,2-13C2]glucose to m2 of glutamate via PC and PDH relative fluxes were calculated using glutamate GC–MS values; they were calculated using the C2–C5 and C2–C4 fragments of glutamate isotopomers as described in the Appendix. Light and dark bars are PC and PDH contributions respectively. Values are expressed as means±S.E.M.*P≤0.01 compared with the related enzyme under glucose condition, and #P≤0.01 between PC and PDH contributions in the same group (n=6).
DISCUSSION
Metabolic network analysis provides an integrated view of the physiological state of a cell at a given moment, while radio-isotope studies of the effects of gluconeogenic precursors on individual biochemical reactions of the network [26–28] do not provide this integrated view. Although glutamine, lactate and fructose are gluconeogenic precursors, their impact on the glucose metabolic network differs, which is not explained by a simple precursor–product concentration gradient. Our results show for the first time how each of the precursors simultaneously affects G6P flux to glucose release, glycogen synthesis, glycolysis, and the PPC.
Newsholme and co-workers [29,30] proposed that futile cycles provide an excellent mechanism for the control of the direction and magnitude of substrate flux across the substrate cycles. Our results clearly demonstrate that the operation of the glucose metabolic network is much more complex. In all situations, we found that both forward and reverse reactions of the GK/G6Pase and the glycolytic/gluconeogenic cycles were affected to different degrees. Although changes in the substrate cycle resulted in directional changes in substrate flux, the magnitude of change in G6P utilization is a functional property of the network that is not reflected in the accumulation of products because of the compensating changes in the reverse reaction of these futile cycles. Whether the effects on the forward and reverse reactions of the futile cycles are the result of regulation of individual enzyme reactions by allosteric or covalent modification or through the regulation of metabolic intermediates, remains to be elucidated. Moreover, the effects of gluconeogenic precursors on substrate cycles were not easy to predict. These effects were clearly demonstrated in the cases of glutamine and fructose. For example, the addition of glutamine resulted in a reduction in the glycolytic/gluconeogenic ratio but not in net gluconeogenesis; and the effect on the GK/G6Pase cycle was much greater than that on the glycolytic/gluconeogenic cycle. The addition of 3 mM fructose was a more effective gluconeogenic precursor than 10 mM lactate/pyruvate; and the increase in gluconeogenic flux was accompanied by increased glucose release. These examples illustrate that futile cycling not only changes the direction of substrate flux over the cycle but also minimizes the impact of acute changes in substrate concentration or flux of intracellular intermediates. Thus, the change in the G6Pase/GK ratio from values lower than 1 to values higher than 1, caused by fructose or lactate supplementation, resulted in the reversal of the direction of net flow of glucose in and out of hepatocytes. However, because of the presence of G6Pase/GK futile cycling, the net flux in or out of the hepatocytes was only a small fraction of the total flux.
The concept of a glucose metabolic network that links glycolysis and gluconeogenesis with glycogen synthesis and energy substrate production by the PPC and Krebs cycle has important implications. It implies that the induction of enzyme expression by hormones is further modulated by substrate cycles of the glucose metabolic network. The complex cross-talk between substrate cycles makes the simple definition of insulin sensitivity or resistance difficult, if not impossible. Metabolic networks function under constraints from known stoichiometry and reaction energetics, and genetic or regulatory modifications of enzyme activities in the time-frame of the experiment. These constraints delimit the space where experimental observations must lie [6,7]. In our study of fasted rat hepatocytes, the profiles of substrate cycles and the ratio of the forward and reverse reactions were functional characteristics of a metabolic network that constitute the metabolic phenotype of a cell. On the basis of our results, we conclude that the use of stable isotope and MIDA is a more effective and efficient methodology than traditional radio-isotope approaches. This metabolomic analysis in response to various precursor substrate inputs, provides the functional characterization of the glucose metabolic network in fasted rat hepatocytes, and is a rich source of information for the characterization of cross talk with hormone signalling pathways related to hepatic glucose metabolism. These concepts offer new approaches for the study of network states, rather than of individual proteins, as a strategy to improve our understanding of complex metabolic diseases and to develop drugs for their treatment [31].
APPENDIX
Formulae used to obtain the parameters included in the text are given below:
G6P outflux evaluation
As shown in Scheme 1, under our incubation conditions G6P outflux is the sum of four contributions: (a) outflux to glycogen synthesis, (b) outflux to glucose incubation medium, (c) outflux to lactate through glycolysis, and (d) outflux to lactate through PPC.
(a) G6P outflux to glycogen synthesis
This flux corresponds to the biochemical glycogen content, measured as described in the Experimental section.
(b) G6P outflux to glucose incubation medium (G6Pase recycling)
This was measured using glucose and G6P isotopomeric patterns. Assuming isotopic equilibrium between G6P and glycogen, G6P isotopomeric distribution would be the same as that observed in glycogen isolated from cell pellets. To quantify G6P recycling, the fraction of glucose medium that is released from cells (X) was calculated:
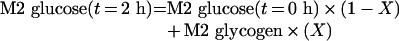 |
(A1) |
This formula considers that M2 labelling in glucose medium at the end of the incubation can be from initial glucose or from recycled G6P. When multiplying this fraction (X) by the biochemical glucose concentration in the medium at the end of incubation {[glucose](t=2h)}, the G6P outflux to glucose incubation medium is obtained:
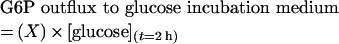 |
(A2) |
(c) G6P outflux to lactate formation through glycolysis
G6P metabolized directly through glycolysis gives two triose phosphate molecules (glyceraldehyde phosphate and dihydroxyacetone phosphate), which can be directed to lactate formation. Thus, when one molecule of G6P, with a 13C label on each of the first two carbons, enters glycolysis, only one of the two newly formed lactate molecules will have two 13C, while the second will have no label. In this way, the rate of lactate synthesized from G6P through the glycolytic pathway can be measured from:
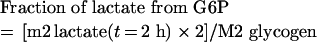 |
(A3) |
and the absolute glycolytic flux (from G6P to lactate):
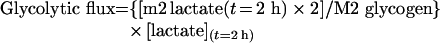 |
(A4) |
where [lactate](t=2h) is the biochemical concentration of lactate in the incubation medium at the end of incubation.
(d) G6P outflux to lactate formation through the PPC
Lactate directly formed from G6P through glycolysis will have m0 labelling when it comes from the bottom of the [1,2-13C2]glucose molecule, or m2 labelling, when it comes from the top of the same molecule. Despite these two possibilities, m1 labelling in triose phosphate may also arise because of the action of PPC on the metabolism of [1,2-13C2]glucose molecules [25]. The PPC contribution can be quantified compared with glycolysis (PPC) using these m1 and m2 values and the formula described previously [25]:
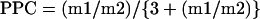 |
(A5) |
Combining the value of PPC and that estimated for the glycolytic flux [point (c) of this Appendix], the oxidative PPC flux (G6P outflux to lactate through PPC) was calculated:
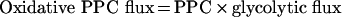 |
(A6) |
G6P influx evaluation
G6P influxes considered in our network were as described in Scheme 1: (e) influx from glucose incubation medium, and (f ) influx from lactate through gluconeogenesis. G6P influx from glycogenolysis was discarded for all our incubation conditions as glycogen phosphorylase (one of the enzymes involved in glycogen degradation) is inhibited in hepatocytes from fasted rats incubated with high glucose levels [32].
(e) G6P influx from glucose incubation medium (through GK action)
G6P synthesized from glucose incubation medium through GK action will have the same labelling as glucose from the medium. Thus, since cells were incubated with [1,2-13C2]glucose (M2 glucose), it was expected that G6P synthesized through this pathway would show the same M2 labelling. As G6P is in isotopic equilibrium with glycogen, and assuming little randomization of M2 in glycogen (which was verified comparing the labelling pattern of the C1–C4 fragment with that of the C1–C6 in the glycogen glucose molecule), the rate of G6P synthesized through GK was calculated by:
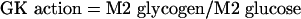 |
(A7) |
where M2 glucose is the average between M2 glucose at the beginning and at the end of incubation. And G6P influx from glucose incubation medium will be:
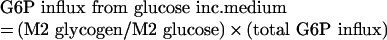 |
(A8) |
Here, total G6P influx has the same value as the total G6P outflux, calculated from the addition of all G6P outfluxes (a+b+c+d). This statement was accepted as all experiments were performed under steady state conditions for G6P intracellular content. Furthermore, the change in medium glucose concentration was not large, and did not affect mass balance, and consequently G6P flux underestimation caused by loss into pyruvate or the Krebs cycle is assumed to be slight.
(f) G6P influx from lactate (through gluconeogenic flux)
As the only G6P influxes considered are influx from glucose incubation medium and influx from lactate through gluconeogenesis, the remaining G6P influx that has not been attributed to GK must go through this pathway, its contribution being:
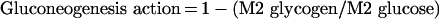 |
(A9) |
and the G6P influx from lactate (through gluconeogenesis) will be:
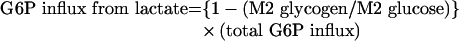 |
(A10) |
Krebs Cycle evaluation
The Krebs cycle can be analysed comparing the m2 enrichment in C2–C4 to the m2, m3 and m4 enrichment in C2–C5 of glutamate. As the m2 label in C2–C3 represents the relative contribution of [1,2-13C2]glucose via PC, and the m2 label in C4–C5 the relative contribution of the same isotope via PDH [8,11], they were quantified using:
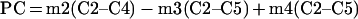 |
(A11) |
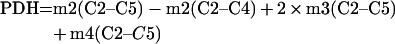 |
(A12) |
In addition, anaplerotic flux, which is the measure of substrate flux of the Krebs cycle that generates (or conserves) three-carbon compounds, was calculated as described in [20].
Acknowledgments
This study was supported by the Ministerio de Ciencia y Tecnología of the Spanish Government (SAF2002-02785 to M.C. and BMC2002-00705 to J.J.G.), by the Fundació La Marató TV3 (992310 to J.J.G.), and a grant from the NIH DK56090-A1 (W.-N.P.L.). The GC–MS Facility is supported by PHS grants P01-CA42710 to the UCLA Clinical Nutrition Research Unit, Stable Isotope Core and M01-RR00425 to the General Clinical Research Center. S.M. was supported by a grant from the Comissionat per a Universitats i Recerca de la Generalitat de Catalunya (2000FI 00050). The authors thank Anna Adrover for her excellent technical assistance.
References
- 1.Frazier M. E., Johnson G. M., Thomassen D. G., Oliver C. E., Patrinos A. Realizing the potential of the genome revolution: the genomes to life program. Science (Washington, D.C.) 2003;300:290–293. doi: 10.1126/science.1084566. [DOI] [PubMed] [Google Scholar]
- 2.Oltvai Z. N., Barabási A.-L. Systems biology. Life’s complexity pyramid. Science (Washington, D.C.) 2002;298:763–764. doi: 10.1126/science.1078563. [DOI] [PubMed] [Google Scholar]
- 3.Hue L. The role of futile cycles in the regulation of carbohydrate metabolism in the liver. Adv. Enzymol. Relat. Areas Mol. Biol. 1981;52:247–331. doi: 10.1002/9780470122976.ch4. [DOI] [PubMed] [Google Scholar]
- 4.Katz J., Rognstad R. Futile cycles in the metabolism of glucose. Curr. Top. Cell Regul. 1976;10:237–289. doi: 10.1016/b978-0-12-152810-2.50013-9. [DOI] [PubMed] [Google Scholar]
- 5.McGarry J. D., Kuwajima M., Newgard C. B., Foster D. W., Katz J. From dietary glucose to liver glycogen: the full circle round. Annu. Rev. Nutr. 1987;7:51–73. doi: 10.1146/annurev.nu.07.070187.000411. [DOI] [PubMed] [Google Scholar]
- 6.Price N. D., Papin J. A., Schilling C. H., Palsson B. O. Genome-scale microbial in silico models: the constraints-based approach. Trends Biotechnol. 2003;21:162–169. doi: 10.1016/S0167-7799(03)00030-1. [DOI] [PubMed] [Google Scholar]
- 7.Reed J. L., Palsson B. O. Thirteen years of building constraint-based in silico models of Escherichia coli. J. Bacteriol. 2003;185:2692–2699. doi: 10.1128/JB.185.9.2692-2699.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boros L. G., Steinkamp M. P., Fleming J. C., Lee W. N., Cascante M., Neufeld E. J. Defective RNA ribose synthesis in fibroblasts from patients with thiamine-responsive megaloblastic anemia (TRMA) Blood. 2003;102:3556–3561. doi: 10.1182/blood-2003-05-1537. [DOI] [PubMed] [Google Scholar]
- 9.Comin-Anduix B., Boros L. G., Marin S., Boren J., Callol-Massot C., Centelles J. J., Torres J. L., Agell N., Bassilian S., Cascante M. Fermented wheat germ extract inhibits glycolysis/pentose cycle enzymes and induces apoptosis through poly(ADP-ribose) polymerase activation in Jurkat T-cell leukemia tumor cells. J. Biol. Chem. 2002;277:46408–46414. doi: 10.1074/jbc.M206150200. [DOI] [PubMed] [Google Scholar]
- 10.Boros L. G., Cascante M., Lee W. N. Metabolic profiling of cell growth and death in cancer: applications in drug discovery. Drug Discov. Today. 2002;7:364–372. doi: 10.1016/s1359-6446(02)02179-7. [DOI] [PubMed] [Google Scholar]
- 11.Boren J., Cascante M., Marin S., Comin-Anduix B., Centelles J. J., Lim S., Bassilian S., Ahmed S., Lee W. N., Boros L. G. Gleevec (STI571) influences metabolic enzyme activities and glucose carbon flow toward nucleic acid and fatty acid synthesis in myeloid tumor cells. J. Biol. Chem. 2001;276:37747–37753. doi: 10.1074/jbc.M105796200. [DOI] [PubMed] [Google Scholar]
- 12.Fernández-Novell J. M., Ariño J., Guinovart J. J. Effects of glucose on the activation and translocation of glycogen synthase in diabetic rat hepatocytes. Eur. J. Biochem. 1994;226:665–671. doi: 10.1111/j.1432-1033.1994.tb20094.x. [DOI] [PubMed] [Google Scholar]
- 13.Chan T. M., Exton J. H. A rapid method for the determination of glycogen content and radioactivity in small quantities of tissue or isolated hepatocytes. Anal. Biochem. 1976;71:96–105. doi: 10.1016/0003-2697(76)90014-2. [DOI] [PubMed] [Google Scholar]
- 14.Fernández-Novell J. M., Ariño J., Vilaró S., Bellido D., Guinovart J. J. Role of glucose 6-phosphate in the translocation of glycogen synthase in rat hepatocytes. Biochem. J. 1992;288:497–501. doi: 10.1042/bj2880497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kunst A., Draeger B., Ziegenhorn J. D-Glucose; UV-methods with hexolinase and glucose 6-phosphate dehydrogenase. In: Bergmeyer H. U., editor. Methods of Enzymatic Analysis. vol. VI. Verlag Chemie: Weinheim; 1984. pp. 163–172. [Google Scholar]
- 16.Gutmann I., Wahlefeld A. W. L-(+)-Lactate. In: Bergmeyer H. U., editor. Methods of Enzymatic Analysis. vol. 3. NY: Academic Press; 1974. pp. 1464–1468. [Google Scholar]
- 17.Tserng K. Y., Gilfillan C. A., Kalhan S. C. Determination of carbon-13 labeled lactate in blood by gas chromatography/mass spectrometry. Anal. Chem. 1984;56:517–523. doi: 10.1021/ac00267a049. [DOI] [PubMed] [Google Scholar]
- 18.Katz J., Lee W. N., Wals P. A., Bergner E. A. Studies of glycogen synthesis and the Krebs cycle by mass isotopomer analysis with [U-13C]glucose in rats. J. Biol. Chem. 1989;264:12994–13004. [PubMed] [Google Scholar]
- 19.Kurland I. J., Alcivar A., Bassilian S., Lee W. N. P. Loss of [13C]glycerol carbon via the pentose cycle. Implications for gluconeogenesis measurement by mass isotoper distribution analysis. J. Biol. Chem. 2000;275:36787–36793. doi: 10.1074/jbc.M004739200. [DOI] [PubMed] [Google Scholar]
- 20.Lee W. P., Edmond J., Bassilian S., Morrow J. Mass isotopomer study of glutamine oxidation and synthesis in primary culture of astrocytes. Develop. Neurosci. 1996;18:469–477. doi: 10.1159/000111442. [DOI] [PubMed] [Google Scholar]
- 21.Lee W.-N. P., Byerley L. O., Bergner E. A., Edmond J. Mass isotopomer analysis: theoretical and practical considerations. Biol. Mass Spectrom. 1991;20:451–458. doi: 10.1002/bms.1200200804. [DOI] [PubMed] [Google Scholar]
- 22.Katz J., Wals P. A., Rognstad R. Glucose phosphorylation, glucose 6-phosphatase, and recycling in rat hepatocytes. J. Biol. Chem. 1978;253:4530–4536. [PubMed] [Google Scholar]
- 23.Gomis R. R., Favre C., Garcia-Rocha M., Fernández-Novell J. M., Ferrer J. C., Guinovart J. J. Glucose 6-phosphate produced by gluconeogenesis and by glucokinase is equally effective in activating hepatic glycogen synthase. J. Biol. Chem. 2003;278:9740–9746. doi: 10.1074/jbc.M212151200. [DOI] [PubMed] [Google Scholar]
- 24.Seoane J., Gomez-Foix A. M., O’Doherty R. M., Gomez-Ara C., Newgard C. B., Guinovart J. J. Glucose 6-phosphate produced by glucokinase, but not hexokinase I, promotes the activation of hepatic glycogen synthase. J. Biol. Chem. 1996;271:23756–23760. doi: 10.1074/jbc.271.39.23756. [DOI] [PubMed] [Google Scholar]
- 25.Lee P. W. N., Boros L. G., Puigjaner J., Bassilian S., Lim S., Cascante M. Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2-13C2]glucose. Am. J. Physiol. 1998;274:E843–E851. doi: 10.1152/ajpendo.1998.274.5.E843. [DOI] [PubMed] [Google Scholar]
- 26.Shiota M., Galassetti P., Monohan M., Neal D. W., Cherrington A. D. Small amounts of fructose markedly augment net hepatic glucose uptake in the conscious dog. Diabetes. 1998;47:867–873. doi: 10.2337/diabetes.47.6.867. [DOI] [PubMed] [Google Scholar]
- 27.Salhanick A. I., Chang C. L., Amatruda J. M. Hormone and substrate regulation of glycogen accumulation in primary cultures of rat hepatocytes. Biochem. J. 1989;261:985–992. doi: 10.1042/bj2610985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Schaftingen E., Vandercammen A. Stimulation of glucose phosphorylation by fructose in isolated rat hepatocytes. Eur. J. Biochem. 1989;179:173–177. doi: 10.1111/j.1432-1033.1989.tb14537.x. [DOI] [PubMed] [Google Scholar]
- 29.Newsholme E. A., Stanley J. C. Substrate cycles: their role in control of metabolism with specific references to the liver. Diabetes Metab. Rev. 1987;3:295–305. doi: 10.1002/dmr.5610030113. [DOI] [PubMed] [Google Scholar]
- 30.Newsholme E. A., Arch J. R., Brooks B., Surholt B. The role of substrate cycles in metabolic regulation. Biochem. Soc. Trans. 1983;11:52–56. doi: 10.1042/bst0110052. [DOI] [PubMed] [Google Scholar]
- 31.Huang S. Rational drug discovery: what can we learn from regulatory networks? Drug Discov. Today. 2002;7:S163–S169. doi: 10.1016/s1359-6446(02)02463-7. [DOI] [PubMed] [Google Scholar]
- 32.Stalmans W., Laloux M., Hers H. G. The interaction of liver phosphorylase A with glucose and AMP. Eur. J. Biochem. 1974;49:415–427. doi: 10.1111/j.1432-1033.1974.tb03847.x. [DOI] [PubMed] [Google Scholar]