

Abstract
Acute myeloid leukemia (AML) cells resist differentiation stimuli despite high expression of innate immune receptors, such as Toll-like receptor 9 (TLR9). We previously demonstrated that targeting Signal Transducer and Activator of Transcription 3 (STAT3) using TLR9-targeted decoy oligodeoxynucleotide (CpG-STAT3d) increases immunogenicity of human and mouse AML cells. Here, we elucidated molecular mechanisms of inv(16) AML reprogramming driven by STAT3-inhibition/TLR9-activation in vivo. At the transcriptional levels, AML cells isolated from mice after intravenous administration of CpG-STAT3d or leukemia-targeted Stat3 silencing and TLR9 co-stimulation, displayed similar upregulation of myeloid cell differentiation (Irf8, Cebpa, Itgam) and antigen-presentation (Ciita, Il12a, B2m)-related genes with concomitant reduction of leukemia-promoting Runx1. Single-cell transcriptomics revealed that CpG-STAT3d induced multilineage differentiation of AML cells into monocytes/macrophages, erythroblastic and B cell subsets. As shown by an inducible Irf8 silencing in vivo, IRF8 upregulation was critical for monocyte-macrophage differentiation of leukemic cells. TLR9-driven AML cell reprogramming was likely enabled by downregulation of STAT3-controlled methylation regulators, such as DNMT1 and DNMT3. In fact, the combination of DNA methyl transferase (DNMT) inhibition using azacitidine with CpG oligonucleotides alone mimicked CpG-STAT3d effects, resulting in AML cell differentiation, T cell activation, and systemic leukemia regression. These findings highlight immunotherapeutic potential of bi-functional oligonucleotides to unleash TLR9-driven differentiation of leukemic cells by concurrent STAT3 and/or DNMT inhibition.
Keywords: MT: Oligonucleotides: Therapies and Applications, Oligonucleotide therapeutics, decoy DNA, CpG deoxynucleotides, STAT3, TLR9, acute myeloid leukemia, cancer immunotherapy
Graphical abstract
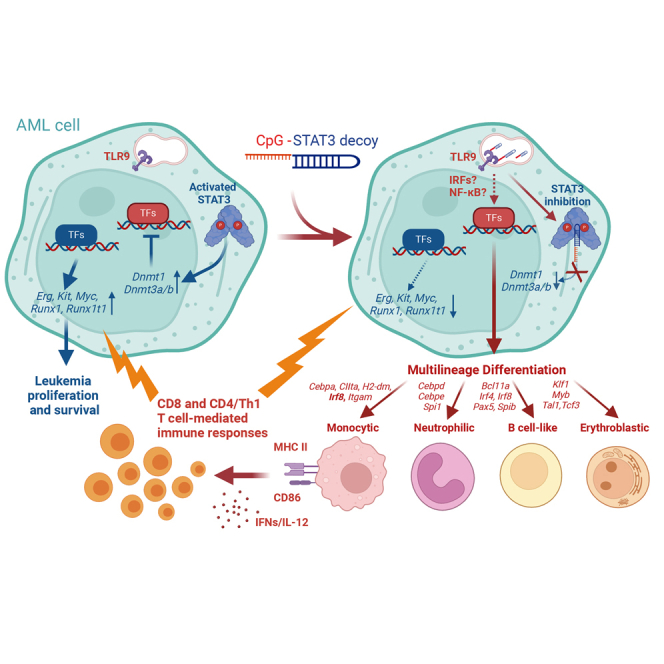
Kortylewski and colleagues developed a decoy oligonucleotide-based strategy to induce multilineage differentiation and regression of acute myeloid leukemia in mice. Here, they demonstrated that such therapeutic effect requires inhibition of STAT3 and/or DNMT1 epigenetic control to enable TLR9-driven reprogramming of leukemic cells with expression of lineage-specific transcription factors and markers.
Introduction
Acute myeloid leukemia (AML) is a genetically heterogeneous malignancy characterized by the accumulation of immature myeloid cells.1 Inv(16) leukemia represents one of the most common AML subtypes but despite relatively favorable prognosis, 50%–60% of patients relapse within 5 years after standard chemotherapy.2 The development of AML immunotherapies using immune adjuvants, leukemia-specific antibodies, immune checkpoint blockade, or cellular strategies proved challenging.3 Growing evidence suggests that AML immune tolerance is partly driven by leukemia cell-intrinsic mechanisms, such as low mutational burden and active immunosuppression through programmed death-ligand 1 (PD-L1) or galectin-9 (Gal-9) expression.4,5 Patients’ age and leukemia genetic subtype alter the immune landscape of AML and thus response to chemo- or immunotherapy.6
Signal Transducer and Activator of Transcription 3 (STAT3) is an oncogenic transcription factor (TF) commonly activated in leukemic blasts that correlates with worse patient outcomes.7,8,9 While STAT3 attracted attention as a driver of AML cell proliferation and survival,10 more recent evidence points to the role of STAT3 as a master regulator of immune evasion in both AML cells and also in leukemia-associated myeloid cells, such as dendritic cells (DCs) and macrophages.4,11 Direct, pharmacologic STAT3 inhibition proved difficult due to lack of any enzymatic activity. Synthetic oligonucleotides provide alternative strategies for targeting STAT3 expression, using small interfering RNA (siRNA) or antisense molecules, or transcriptional activity using decoy oligodeoxynucleotides (ODNs), which are competitive inhibitors of STAT3 and STAT1 DNA-binding.12,13 Both STAT3 antisense and decoy molecules were tested in clinical trials and well tolerated, although their efficacy as naked oligonucleotides lacking targeted delivery strategy was limited.14,15 We previously developed a strategy for the delivery of oligonucleotide therapeutics specifically into AML cells using cytosine-guanine dinucleotide (CpG) ODNs to target an innate immune receptor, Toll-like receptor (TLR9), commonly expressed in all cellular compartments of AML, including leukemia stem cells (LSCs).16,17,18,19 Repeated intravenous administration of CpG-STAT3d, but not CpG or the unconjugated STAT3d ODN, which is not internalized by AML cells, showed efficacy against human and mouse models of AML.16 Importantly, therapeutic effects of the combined TLR9-immunostimulation and STAT1/3-inhibition in syngeneic AML harboring CBFB/MYH11 gene fusion relied primarily on the enhanced immunogenicity of leukemic cells, thereby leading to CD8 and CD4 T cell-mediated leukemia eradication and long-term animal survival. In this study, we used transcriptomic and functional analyses to elucidate the molecular impacts of AML cell-intrinsic TLR9 and STAT3 signaling for driving leukemic cell differentiation, immunogenicity, and antigen presentation.
Results
CpG-STAT3d-induced leukemia regression corresponds to myeloid differentiation of AML cells
We previously demonstrated that targeted STAT3 inhibition together with TLR9 activation not only alleviates immunosuppression by Cbfb/MYH11/Mpl (CMM) AML but also stimulates T cell-mediated immune responses, thereby resulting in leukemia eradication in mice.16 Here, we dissected molecular mechanisms underlying AML cell immunogenicity and the induction of anti-leukemic immune responses after intravenous (i.v.) administration of bi-functional CpG-STAT3d compared with a matched control CpG-scrODN (a TLR9-activating CpG-conjugate with a scrambled decoy sequence). As shown in Figure 1A, the majority of mice treated with CpG-STAT3d rejected AML and survived for >80 days, whereas mice receiving CpG-scrODN or PBS succumbed to leukemia within 3 weeks. STAT3-inhibiting/TLR9-activating oligonucleotide provided protective immunity to rechallenge with the same CMM leukemic cells but not with an unrelated, virally induced FBL3 leukemia (Figure 1B). As shown before,16 only CpG-STAT3d treatments reduced percentage of c-Kit+ CMM AML cells in various organs, including spleen (Figure 1C) and bone marrow (Figures S1A and S1B). The negative control CpG-scrODN did not show any significant anti-leukemic effect compared with vehicle-treated mice. The therapeutic effect of CpG-STAT3d was associated with the strong infiltration by macrophages and neutrophils together with CD8+ T cells (Figure 1C; Figure S1A) and the elevation of markers of Th1-specific immune response (Figures S1C and S1D). The cytofluorimetric analysis confirmed that reduction of proliferating GFP+/c-Kit+ AML cells inversely correlated with the significant expansion of leukemic cells expressing myeloid differentiation markers (CD11b/MHC-II/CD86) after CpG-STAT3d but not after control CpG-scrODN treatment that failed to induce AML cell differentiation (Figure 1D). CpG-STAT3d-induced GFP+/CD11b+ AML cells showed significantly reduced proliferation (Figure 1E) and augmented phagocytic activity (Figure 1F) compared with the bulk GFP+/CD11b− leukemic cells or to the small percentage of GFP+/CD11b+ AML cells detectable in control-treated mice. Finally, CpG-STAT3d but not CpG-scr induced a cytomorphologically distinct leukemic cell subset with off-center nuclei, increased cytoplasm volume, and larger and elongated mitochondria consistent with features of differentiating leukemic cells (Figure 1G).20 Importantly, CpG-STAT3d showed similar anti-leukemic effects against another model of mouse C1498 leukemia representing myelomonocytic AML,16 accompanied by an upregulation of CD11b differentiation marker (Figure S2). Overall, these findings suggested that in the absence of immunosuppressive STAT3 activity, TLR9 stimulation can drive differentiation of CMM and potentially other myelomonocytic AML cells into immunogenic and antigen-presenting myeloid cells.
Figure 1.

CpG/TLR9 stimulation results in regression of Cbfb/Myh11/Mpl leukemia in mice only when combined with STAT3 inhibition
C57BL/6 mice were intravenously injected with 1 × 106Cbfb/Myh11/Mpl (CMM) leukemia cells. After tumors were established (1%–2% of AML cells in blood), mice were treated i.v. using CpG-STAT3d, control CpG-scr oligonucleotides (5 mg/kg), or PBS every other day for six times (days 6–16). (A) CpG-STAT3d, but not control CpG-scr treatment, resulted in survival of the majority of mice. The presented result represents one of two independent experiments with similar outcome (n = 7–8 mice/group). (B) CpG-STAT3d-treated mice that rejected CMM leukemia were rechallenged with CMM cells or with an unrelated FBL3 leukemia. Shown are survival curves for rechallenged mice as well as naive mice engrafted with both AML types in parallel (n = 5–10 mice/group). (C) Mice were euthanized 1 day after the last treatment to assess leukemia burden, cell morphology, and immune infiltration. The percentages of c-Kit+ leukemic cells, CD8+ T cells, F4/80+ macrophages, and Ly6B.2+ neutrophils were assessed using immunohistochemical staining in spleens from mice treated as indicated above; scale bar, 200 μm. Shown are data representative of one of three independent experiments. (D–F) Treatment with CpG-STAT3d triggers AML cell differentiation to the antigen-presenting phenotype. CMM leukemia-bearing mice were treated three times every other day using CpG-STAT3d, control CpG-scr (5 mg/kg), or PBS and euthanized 1 day later. Data are representative of 1 of 2 independent experiments. (D) Reduction of leukemia burden in spleen after CpG-STAT3d treatment is inversely correlated with AML cell differentiation. The percentages of GFP+c-Kit+ AML cells and differentiated GFP+CD11b+/MHC-II+/CD86+ AML cells were assessed using flow cytometry; shown are means ± SEM (n = 4 mice/group). (E and F) The cytofluorimetric analysis of intracellular staining for proliferation marker Ki-67 (E) and phagocytosis assay using pHrodo-E.coli-BioParticles (F) on freshly isolated GFP+CD11b+ vs. GFP+CD11b− leukemic cells after in vivo treatments as indicated earlier; shown as means ± SEM (n = 3–6 mice/group). (G) CpG-STAT3d induces morphological changes in ultrastructure of the differentiating AML cell subset. Representative TEM images of splenic AML cell cytomorphology from two independent experiments; scale bar, 2 μm. Statistically significant p values were indicated as follows: ∗∗∗, p < 0.001; ∗∗, p < 0.01; and ∗, p < 0.05
Blocking STAT3 orchestrates TLR9-dependent leukemic cell differentiation
To unravel the molecular mechanism underlying leukemic cell differentiation in response to combined TLR9-activation/STAT3-inhibition, we compared global gene expression in sorted GFP+/c-Kit+ CMM cells derived from mice treated three times every other day using CpG-STAT3d, CpG-scr oligonucleotides or PBS. Such short-term, 5-day treatment allowed for harvesting differentiating AML cells before the onset of T cell immune responses and leukemia regression occurring after 12–14 days.16 RNA-sequencing (RNA-seq) analysis and hierarchical clustering of the top 2,000 of differentially regulated transcripts in individual mice revealed specific gene signatures of AML cells derived from CpG-STAT3d-treated mice compared with both control treatments (Figure 2A). Transcriptional signature of CpG-STAT3d covered 95% of targets induced by CpG-scr control while activating numerous unique transcripts (Figure S3A). The CpG-STAT3d-upregulated genes were key markers of myeloid cell differentiation (Ciita, Cebpa, Irf8, Itgam), antigen-presentation (Cd86, H2-Eb2, Il12), and interferon responses (Irf1, Stat1), while certain leukemogenesis-associated regulators (Mycl, Runx1t1) were suppressed (Figure 2B; Figure S3B). Irf8 was the most significantly and consistently CpG-STAT3d-upregulated TF gene compared with both control treatments (PBS and CpG-scr) (Figures 2B and 2E; Figure S3B). Correspondingly, gene set enrichment analysis (GSEA) of the top differentially expressed genes identified monocyte/macrophage differentiation (Figures 2C–2E) and antigen-presentation (Figure S3C)-related genes among the most significantly enriched functional groups in the CpG-STAT3d-treated CMM cells. This is consistent with the well-established function of STAT3 as a transcriptional master regulator of oncogenesis and immunosuppression in many human cancers.21,22
Figure 2.

The combination of STAT3 inhibition and TLR9 stimulation is required to transcriptionally reprogram CMM leukemic cells toward myeloid cell differentiation and antigen presentation
(A–D) CMM-bearing mice were treated i.v. using CpG-STAT3d or control CpG-scr oligonucleotides (5 mg/kg) every other day for three times as in Figure 1A. RNA-seq analysis was performed using magnetically enriched leukemic cells. (A) The heatmap of hierarchical clustering of differentially expressed genes (DEGs), demonstrating distinct separation between treatment groups. (B) Volcano plots of DEGs with a log2 fold change (log2FC) greater than 2 and an adjusted p value (padj) less than 0.05 shown in red, highlighting a significant majority of upregulated genes following CpG-STAT3d treatment compared with both CpG-scr and PBS treatments. (C–E) Gene set enrichment analysis (GSEA) of the KEGG database-derived gene sets indicating the top-scoring immune signaling pathways for all three treatment groups (C), the gene enrichment score indicating the differentiation of AML cells primarily into macrophage-like cells (D), and the heatmap (E) of key myeloid cell differentiation gene expression in all three treatment groups. (F–J) Mice engrafted with 1 × 106 CMM-tetON-shStat3 cells expressing doxycycline-inducible STAT3shRNA (tetON-shStat3) were treated daily for six times with PBS, CpG, Dox (100 mg/kg), or CpG plus Dox. Compared with CpG or Dox treatments alone, the combined CpG/Dox treatment was the most effective in reducing splenic GFP+/mCherry+ AML cells (F) and in the upregulation of Irf8 expression in leukemic cells (G) as assessed using flow cytometry or qPCR, respectively. (H) RNA-seq analysis and hierarchical clustering showing pattern of gene expression in CMM-tetON-shStat3 cells treated with CpG+Dox as observed in the parental CMM treated with CpG-STAT3d (see A). (I and J) GSEA results (I) along with (J) a heatmap of differentially expressed genes associated with myeloid cell differentiation most robustly activated by the combined CpG+Dox treatment. All the presented data are representative of the results obtained in two independent experiments, n = 3–5 mice/group, and the quantification results were shown as means ± SEM. Statistically significant differences are indicated by asterisks: ∗∗∗∗, p < 0.0001; ∗∗, p < 0.01.
Decoy sequence within CpG-STAT3d inhibits activity of STAT3 as well as closely related STAT1 if they are coactivated.16 To verify that observed effects are STAT3-specific, we generated CMM cells expressing Stat3 short hairpin RNA (shRNA) under doxycycline (Dox)-regulated promoter to allow inducible and dose-dependent Stat3 knockdown (CMM-tetON-shStat3) (Figure 2F; Figures S4A and S4B). Mice engrafted with CMM-tetON-shStat3 cells (AML cells >1% in blood) were treated i.v. using PBS, an unconjugated CpG oligonucleotide, doxycycline (Dox) alone to induce Stat3 silencing, or a combination of CpG plus doxycycline (CpG+Dox). Similar to CpG-STAT3d (Figure 1D), the combined Stat3 silencing and TLR9 stimulation using CpG+Dox co-treatment reduced leukemia burden more significantly than either treatment alone (Figure 2F). Treatment efficacy was correlated with the upregulation of Irf8 expression as a marker of AML cell differentiation, reaching maximum after the combined CpG+Dox treatment (Figure 2G) consistently with our earlier observation in CpG-STAT3d-treated CMM cells (Figure 2E). Next, we sorted CMM-tetON-shStat3 leukemic cells (GFP+mCherry+) from mice of all four treatment groups for a whole transcriptome analysis as done before. The clustering of differentially expressed transcripts in individual mice revealed gene signature unique for the combined TLR9-activation/STAT3-silencing (CpG+Dox) treatment and not overlapping with CpG alone or Stat3 silencing alone treatment groups (Figure 2H; Figure S4C). Similar to CpG-STAT3d effect, the combined CpG+Dox treatment induced maximal expression of innate immunity (Figure 2I) and myeloid differentiation-related genes such as Itgam and TFs, Irf8, and Cebpa (Figure 2J; Figures S4D–S4F) as well as antigen-presentation regulators (Figure S4G). Both CpG/TLR9 stimulation alone and STAT3 silencing alone had minimal to moderate effects on the expression of myeloid differentiation genes. These results suggest that the combination of TLR9 triggering with STAT3 inhibition is required for an effective transcriptomic reprogramming of CMM cells as neither of single treatments achieved comparable therapeutic efficacy. In contrast to other studies,23,24 these results emphasized the role of STAT3 as a suppressor of myeloid cell differentiation and immune activation rather than as a driver of leukemia proliferation or survival.
CpG-STAT3d induced AML cell differentiation into phenotypically diverse immune-activated cell subsets
To gain insights into potential heterogeneity of AML cell differentiation after the combined TLR9-activation/STAT3-inhibition, we employed single-cell transcriptomic analysis (scRNA-seq). Splenic GFP+ CMM cells were sorted from mice treated with CpG-STAT3d or PBS for only 1 week, before immune-mediated AML regression, similarly as done earlier for bulk RNA-seq analysis. The successful differentiation of AML cells was verified by estimating levels of CD11b myeloid cell marker, which increased from <1% in controls to ∼30% of GFP+ leukemic cells after CpG-STAT3d (Figure S5A). Unsupervised, graph-based clustering revealed nine major cell populations (Figures 3A–3C), four of which were bulk AML cells, namely proliferating AML (Mki67, Cdk1), LSCs (Kit, Runx1), transitory AML-erythroblasts (Gata1, Hba-a1), and erythroblast-like AML (Hmbs, Hbb-bs). The remaining five populations exhibited signatures of differentiated cells: monocyte-like AML (Ccr2, Ly6c2), macrophage-like AML (Adgre1, Cd14), neutrophil-like AML (Ly6g, Ngp), B-cell-like AML (Ms4a1, Cd19), and erythroblasts (Klf1, Epor) (Figure 3B) and expanded in CpG-STAT3d-treated samples compared with controls dominated by proliferating AML cells (Figure 3C). We created module scores to compare the potential antigen-presenting (MHC-I and MHC-II), immunostimulatory (interferon [IFN] response) and cell cycle activities of various AML cell subsets (Figure 3D). CpG-STAT3d-treated animals showed a decline in the proliferating AML cells paralleled by an expansion of the potentially immunogenic cell subsets with the IFN-related and antigen-presenting functions such as the macrophage-like cell cluster (Figure 3D). Correspondingly, GSEA indicated that the leukemia treatment promoted the shift from cell cycle-related gene activities to immune activation with strong indication of immune signaling via TLRs, IFNs, cytokines, and Jak/STAT pathway (Figure 3E). These changes were also reflected by the pattern of TF expression across AML subsets. The differentiated AML clusters showed reduced levels of oncogenic regulators (Runx1, Myb, Erg) or c-Kit specifically in LSCs (Figures 3F and 3G), whereas the levels of cell differentiation-related (Mafb, Cebpa for monocytes/macrophages, Pax5 for B cells) or antigen-presentation-related TFs (Irf8, Ciita, or Stat1) were elevated (Figures 3F and 3H). Overall, the scRNA-seq and flow cytometry results (Figure S5B) were consistent with our earlier observations and underscored the effect of TLR9-activation/STAT3-inhibition on CMM differentiation not only into myeloid but also neutrophil, B cell, and other cellular subsets. Given that Irf8 upregulation was consistently among top TF genes induced by CpG-STAT3d treatment (Figures 2B and 2E; Figure S3B) and Irf8 expression was common for CMM-derived monocytes/macrophages and B cells (Figure 3H), we next assessed the role of IRF8 in the effect of CpG-STAT3d. We generated CMM-tetON-shIrf8 cells to allow for a doxycycline-inducible Irf8 silencing in leukemic cells in vivo. The doxycycline significantly reduced IRF8 mRNA (Figure 3I) and protein upregulation (Figure 3J) in CMM-tetON-shIrf8 leukemia treated with CpG-STAT3d in vivo. As a result, Irf8 silencing abrogated CpG-STAT3d-induced differentiation of AML cells to CD11b+ myeloid cells (Figure 3K). These results support the role of IRF8 as a downstream mediator of CpG-STAT3d-induced myeloid differentiation of CMM leukemic cells.
Figure 3.

Single-cell transcriptomic analysis of CMM cell differentiation into immunogenic myeloid cells in vivo
CMM leukemia-bearing mice were treated using i.v. injections of CpG-STAT3d (5 mg/kg) or PBS three times every other day. (A–H) Single-cell RNA-seq analysis was performed on the sorted GFP+ leukemic cells from four individually treated mice or PBS control (88%–94% purity). (A) UMAP plots of AML cells in control (top) and CpG-STAT3d-treated (bottom) mice, clustered using the Leiden algorithm, revealing nine distinct populations. (B) Cell clusters were labeled based on the expression of lineage and immune marker genes. Among these, four populations represented CMM cells, while the remaining five exhibited signatures of differentiated immune cell types. (C) Stacked bar plots depict percentages of various cell clusters to highlight the enrichment of differentiated cell subsets in CpG-STAT3d-treated mice. (D) Module score plots indicating a correlation between reduced proliferative potential of macrophage and B cell-like clusters together with augmented antigen-presenting potential. (E) The upregulation of antigen-presentation and immune signaling gene sets coupled with a decrease in DNA synthesis and proliferation-related genes in CMM cells from treated vs. control mice as assessed using GSEA. (F) Heatmap of transcription factor expression levels revealing the downregulation of pro-tumor TFs (e.g., Runx1, Myb, Myc) and the upregulation of TFs driving cell differentiation, antigen-presentation and immune activity (e.g., Cebpa, Irf8, Stat1, Ciita). (G) UMAP plot of Kit expression levels in the LSC subset of AML cells. (H) UMAP plot of Irf8 expression levels overlapping with monocyte/macrophage clusters. (I–K) To achieve inducible Irf8 silencing in AML cells, we generated CMM-tetON-shIrf8 cells expressing doxycycline (Dox)-inducible Irf8shRNA (tetON-shIrf8). Mice engrafted with CMM-tetON-shIrf8 cells were treated three times daily with PBS, CpG-STAT3d, or Dox+CpG-STAT3d. Dox-inducible IRF8 knockdown decreased CpG-STAT3d-driven IRF8 upregulation in splenic CMM cells at mRNA (I) and protein (J) levels. (K) Inducible Irf8 knockdown abrogates differentiation of AML cells after CpG-STAT3d treatment. The data (I–K) are representative of the results obtained in three independent experiments. Statistically significant differences were indicated by asterisks: ∗∗, p < 0.01; ∗, p < 0.05; shown are means ± SEM (n = 3–5 mice/group).
TLR9-stimulated leukemia cell differentiation is facilitated by the inhibition of DNMTs
The differentiation and activation of non-malignant as well as leukemic myeloid cells is a complex epigenetically regulated process related to the aberrant activity of DNA methyl transferases (DNMTs) in AML.25 STAT3 is well known for regulating expression and interacting with DNMT1 to silence tumor suppressor genes in cancer cells.26,27 Dnmt1 was expressed in actively proliferating CMM cells but not in the differentiated myeloid cell subsets in our scRNA-seq analysis. Thus, we assessed whether levels of DNMTs present in CMM cells would differ between CD11b+ CMM cell subset and the remaining bulk of CD11b– CMM cells in CpG-STAT3d-treated mice (Figures 4A and 4B) or when comparing to control CpG-scr and PBS-treated mice (Figure 4C). Like an oncogenic Runx1, also Dnmt1, Dnmt3a, and Dnmt3b were downregulated specifically in CD11b+ but not in CD11b– CMM cells or in control-treated CMM cells (Figure 4B). In contrast, we observed strong induction of myeloid differentiation-related Cebpa and Irf8 in CD11b+ CMM cells compared with undetectable and low levels of both transcripts in CD11b– CMM cells (Figure 4B). We further validated these results at protein levels of DNMT1, DNMT3b, and IRF8. In fact, DNMT1 and DNMT3b expression was inversely correlated with IRF8 upregulation in CMM cells in mice treated with CpG-STAT3d compared with both negative controls CpG-scr and PBS treatments (Figure 4C).
Figure 4.

CpG-STAT3d promotes downregulation of methyl transferases with concomitant induction of IRF8 expression in differentiating CMM cells
(A) C57BL/6 mice were intravenously injected with 1 × 106 CMM leukemia cells. Mice with established leukemia (1%–2% of AML cells in blood), were treated i.v. using CpG-STAT3d, CpG-scr (5 mg/kg), or PBS every other day for three times. qPCR and western blot were performed on sorted leukemic cells from spleen. (B) GFP+CD11b− or GFP+CD11b+ leukemic cells were sorted from CpG-STAT3d-treated mice to determine the expression of certain genes. CpG-STAT3d upregulated Cebpa and Irf8 transcription factors that regulate myeloid cell differentiation, while it downregulated DNA methyltransferases (Dnmt1, Dnmt3a, Dnmt3b) in differentiated CMM cells (CD11b+) compared with non-differentiated CMM cells (CD11b−). (C) The representative western blot (left) and the results quantification (right) showing decreased protein levels of DNMT1, DNMT3b, and elevated IRF8 in sorted GFP+ CMM cells after treatment using CpG-STAT3d, CpG-scr, or PBS. The presented data are representative of three independent experiments; shown are means ± SEM (n = 3–5). Statistically significant p values were indicated with asterisks: ∗∗∗∗, p < 0.0001; ∗∗∗, p < 0.001; ∗∗, p < 0.01 and ∗, p < 0.05
To assess the potential role of DNMT1 in STAT3-mediated checkpoint control of AML cell differentiation, we used azacitidine as a clinically relevant hypomethylation agent and DNMT1 inhibitor. As expected, systemic administration of azacitidine alone reduced CMM leukemia burden in spleens and bone marrows of treated mice while CpG stimulation alone (without STAT3 inhibition) did not show a significant effect (Figures 5A–5C). Importantly, the co-injection of azacitidine with CpG oligonucleotide had improved anti-leukemic efficacy and resulted in almost complete elimination of GFP+/c-Kit+ CMM cells in both spleen and bone marrow (Figures 5D and 5E). The anti-leukemic effect of the azacitidine/CpG combination correlated with over 3-fold increase in the percentage of IRF8-positive cells (Figure 5F) as well as IRF8 surface expression in spleens (Figure 5G). The small percentage of GFP+ AML cells remaining in spleen or in bone marrow after the combination treatment showed upregulation of CD11b+ and antigen-presentation-related markers (MHC-II/CD86) compared with both control groups (Figure 5H; Figures S6A and S6B). Finally, the combination treatment led to maximal accumulation of CD8 T cells, which outnumbered regulatory T cells, in the spleen and bone marrow compared with each treatment alone (Figure 5I; Figures S6C and S6D). Overall, our results suggest that when combined with CpG/TLR9 immunostimulation, small molecule DNMT1 inhibitors mimic the effect of the bi-functional CpG-STAT3d oligonucleotide and unlock the potential of AML cell differentiation and immunogenicity.
Figure 5.

DNMT1 inhibition and CpG/TLR9 stimulation synergize to stimulate CMM cell differentiation and leukemia regression in vivo
C57BL/6 mice with established, disseminated CMM leukemia were treated i.v. using azacitidine (1 mg/kg), CpG oligonucleotide (1 mg/kg), a combination thereof, or PBS every day for six times. Two days after the last treatment, mice were euthanized to analyze AML burden by measuring spleen weight (A) and size (B), bone marrow appearance (C), and the percentages of proliferating GFP+/c-Kit+ leukemic cells in spleen and bone marrow (D and E). (F) The combined azacitidine/CpG treatment induced expression of IRF8 in splenic leukemic cells as assessed using flow cytometry (F) and western blotting (G). (H and I) The combination of DNMT1 inhibitor and TLR9 stimulation promoted (H) differentiation and maturation of CMM cells to antigen-presenting phenotype (CD11b+/MHC-II+/CD86+) with (I) the increased recruitment of predominantly CD8 T cells with smaller percentages of regulatory T cells; means ± SEM (n = 5). Shown are results representative of two independent experiments. Statistically significant p values were indicated as follows: ∗∗∗∗, p < 0.0001; ∗∗∗, p < 0.001; ∗∗, p < 0.01 and ∗, p < 0.05.
Discussion
Despite being a genetically highly heterogeneous disease, AML is characterized by the lowest mutational burden compared with other tumors. The rare gene mutations found in AML often occur in epigenetic regulators, while AML-specific gene fusions, such as CBFB-MYH11, often alter transcriptional programs, the epigenome, and/or chromatin activity.25,28 Thus, epigenetic dysregulation contributes to AML heterogeneity and offers an attractive therapeutic target. Here, we described the novel role of STAT3 in maintaining stability of the AML cell epigenome, preventing terminal differentiation and immunogenicity, thereby promoting therapeutic resistance. In vivo targeting of STAT3 in leukemic cells using gene silencing or decoy strategy proved essential for facilitating TLR9-induced myeloid cell differentiation. While STAT3 inhibition was required, it was not sufficient for AML cell reprogramming or for any significant effect on leukemia burden or on the expression of myeloid differentiation markers such as IRF8.29 As shown by the inducible STAT3 silencing with or without CpG/TLR9 stimulation, the actual trigger of myeloid differentiation and immunogenicity was likely delivered by TLR9 signaling in AML cells. In the absence of STAT3, CpG stimulation resulted in transition from the AML/LSC phenotype to diverse subsets of proinflammatory and antigen-presenting myeloid cells as revealed by single-cell transcriptomic analysis. We previously demonstrated that CpG-STAT3d treatments eliminated LSCs in CMM-bearing mice as a result of CD8-/CD4-mediated T cell immune responses.16,30 The new macrophage-like and/or Bc-like cell subsets that were induced by CpG-STAT3d are the most likely to drive anti-leukemic T cell immunity due to highly elevated expression of MHC class I/II complexes and IFNs. We will dissect the specific contributions of CMM-derived immune cell subsets for the anti-leukemic immunity in further studies.
While TLR9 is commonly expressed by human and mouse AML, little is known about its potential role in leukemogenesis. The TLR9 gene variant with reduced expression compared with wild-type was associated with the post-transplant AML relapse rate reduced by >50%.31 These beneficial effects of TLR9 polymorphism are also associated with a lower accumulation of T regulatory cells (Tregs), suggesting a relationship between basal TLR9 activity and AML immune evasion. We did not observe Treg expansion in CMM-bearing mice after CpG stimulation alone in this or in our earlier study.16 However, Tregs are often expanded by TLR9 triggering in non-malignant myeloid cells as a result of CpG ODN treatment or radiation therapy unless STAT3 is inhibited.32,33 A recent study suggested that TLR9 signaling was coupled through Bruton tyrosine kinase (BTK) to NF-κB and STAT5 survival signaling in FLT3-ITD-negative AML.34 Our study suggests the potential role of tonic TLR9 signaling for AML cell survival and therapeutic resistance, also indicated by our inability to generate CMM leukemic cells lacking TLR9 expression. It is also consistent with a broad expression of TLR9 in all AML cell subsets, including LSCs (CD34+/CD38–) as reported previously.16 Based on our current results, the outcome of CpG/TLR9 signaling is defined by the status of STAT3 activity in AML cells. These results underscore the need for cell-selective and bi-functional oligonucleotide to concomitantly inhibit STAT3 while unleashing TLR9 in the same target cell compartment.
STAT3 attracted attention as a therapeutic target in AML, primarily due to its contribution to leukemia cell survival and strong association with multiple key drivers of leukemogenesis, such as EZH2, FLT3, KRAS, TP53, or PTPN1.9,12,23,35 Our current study supports the notion that STAT3 plays a role of key epigenetic checkpoint in CBFB/MYH11 and potentially in other acute myelomonocytic leukemia cells. STAT3 activity is required to maintain high expression of genes controlling cell cycle, survival, and immune evasion, while repressing genes related to cell differentiation, antigen presentation, and immune activation. This is consistent with the well-established role of STAT3 in myeloid cells as a master regulator of tumor immune evasion.21,36 STAT3 was originally described as an epigenetic regulator in stem cell reprograming.37 Little is known about STAT3 as a regulator of DNA methylation in myeloid leukemia.38 However, STAT3 was shown to recruit DNMT1 to silence tumor suppressor genes in T cell lymphomas and in solid tumors.26,39,40 In addition, STAT3 directly regulates expression of DNMT1 as well as histone methyltransferase EZH2 in cancer cells.26 This is consistent with our data showing that in vivo STAT3-inhibition/TLR9-triggering in CMM cells drastically reduced DNMT1 as well as DNMT3a/3b proteins with concomitant induction of IRF8 TF. Similar inverse relationship between STAT3-dependent upregulation of DNMT1 and DNMT3b and low expression of IRF8 due to promoter hypermethylation was reported before in non-malignant, myeloid-derived suppressor cells (MDSCs) in colorectal cancers.41 Intriguingly, both AML and tolerogenic MDSCs rely on STAT3-mediated epigenetic control of IRF8. The role of IRF8 in myeloid cell differentiation, M1 macrophage polarization, and production of type-I IFNs and interleukin (IL)-12 is well known.42 The contribution from other differentiation-related TFs such as C/EBPα,43 also upregulated by CpG-STAT3d ODNs, cannot be excluded. Recent report demonstrated that forced expression of IRF8 together with two other myeloid cell-specific TFs, PU.1 and BATF3, can reprogram human and mouse cancer cells into antigen-presenting phenotype while impairing their tumorigenicity.44
The common epigenome deregulation in AML stimulated development of epigenetic leukemia therapies. While the primary goal of these strategies was to interfere with leukemic cell proliferation and survival, the immunogenic consequences of AML rewiring would effectively address key therapeutic hurdles such as leukemia heterogeneity and clonal evolution. Differentiated APC-like leukemia cells could generate polyclonal, AML-specific T cell-mediated immune responses, at the same time reducing tolerogenic effects of leukemic cells and the AML-associated myeloid cells. Our earlier study demonstrated increased immunogenicity of patient-derived AML blasts and T cell activation after CpG-STAT3d ODNs.16 Here, we demonstrated that hypomethylating agent (azacitidine) targeting DNMT1 synergizes with CpG/TLR9 stimulation in triggering AML differentiation and regression associated with CD8 T cell recruitment. To our knowledge this is the first report of a synergistic immunogenic effect between DNMT1 inhibitor and TLR9 agonist on AML cells, although others showed IFN-I responses and/or enhanced MHC class II expression on AML cells treated with HDAC or EZH2 inhibitors.45,46 Recent clinical study suggested improved overall response of AML patients to the combination of azacitidine and immune checkpoint blockade (anti-PD1) compared with monotherapies.47 Despite these promising results, it is important to note concerns about off-target toxicities of broadly acting epigenetic drugs to non-malignant cells.25 In fact, azacitidine therapy enhanced T cell exhaustion in treatment-refractory AML patients, thus limiting therapeutic outcomes.47 The use of myeloid cell-targeted CpG-STAT3 inhibitors or other CpG-conjugates alleviates such safety concerns and maximizes immunotherapeutic efficacy against AML. CpG-STAT3d ODNs were well tolerated in preclinical studies in rodents.16,48 With the common TLR9 expression and a broad role of STAT3 in leukemogenesis and immune evasion, our strategy may be relevant to treatment of other than inv(16) AML subtypes, which will be verified in our further studies.
Materials and methods
Animal studies
All animal experiments followed established institutional guidance from the institutional animal care and use committee (City of Hope [COH]). C57BL/6 mice (6–8 weeks old, male and female) were purchased from the National Cancer Institute (NCI). Male and female NOD/SCID/Il2γgKO (NSG; Jackson Laboratory), were housed under pathogen-free conditions in the COH-Animal Facility in accordance with approved protocols from Institutional Animal Care and Use Committee. CBFB/MYH11/Mpl mouse leukemia (CMM) and C1498 cells were previously described.30 Briefly, cells were cultured in RPMI 1640 with 10% FBS, 1% penicillin/streptomycin, 1% GlutaMAX (Thermo Fisher, 35050061), 50 μM β-mercaptoethanol (Sigma-Aldrich, 440203), 10 mM HEPES (Sigma-Aldrich, H0887), and 0.25 mg/mL G418 sulfate (Thermo Fisher, 10-131-035). Cells were cultured for <1 month prior to experiments and tested to be free of Mycoplasma infection. CMM-tetON-shStat3 and CMM-tetON-shIrf8 cells were generated by transducing parental CMM cells using tetON-shRNA/mCherry-expressing lentiviruses selected from three to four shRNA sequences (supplemental methods). Transduced cells expressed mCherry reporter gene and doxycycline-inducible Stat3 or Irf8 shRNA. NSG mice were used specifically for mechanistic studies on myeloid differentiation using tetON-shRNA engineered CMM cells to improve their engraftment/progression. Mouse FBL3 leukemia cells were obtained from Dr. J. Kline (University of Chicago). Mice were injected into the tail vein using 1 × 106 of CMM cells or variants thereof without prior irradiation. After successful leukemia engraftment (>1% of AML cells in blood corresponding to 10%–20% in the bone marrow), mice were treated using i.v. injections of oligonucleotide reagents (5–10 mg/kg) or 5′-azacytidine/azacitidine (1 mg/kg; Sigma-Aldrich, A2385) or intraperitoneal injections of doxycycline (100 mg/kg; Sigma-Aldrich A9891).
Transmission electron microscopy
CMM cells sorted from spleens of mice treated by various oligonucleotides were pelleted and cryo-fixed in a Leica EM PACT2 high-pressure freezer (Leica). Specimens were then freeze-substituted in anhydrous acetone containing 0.5% glutaraldehyde as described before.49 Ultra-thin sections (∼70 nm) cut using an Ultra Cut UCT microtome (Leica) were placed on 200-mesh nickel grids and imaged using a Tecnai-12 transmission electron microscope with a CCD camera (FEI).
Oligonucleotide design and synthesis
The CpG oligonucleotides and conjugates thereof were synthesized in the DNA/RNA Synthesis Core (COH) by linking CpG-1668 ODNs to STAT3 decoy similarly as previously described.16 The resulting oligonucleotides are shown below (x indicates a single C3 unit; asterisk indicates phosphothioation site).
CpG-STAT3d ODN
5′ T∗C∗C∗A∗T∗G∗A∗C∗G∗T∗T∗C∗C∗T∗G∗A∗T∗G∗C∗T-xxxxx-C∗A∗T∗TTCCCGTAAATC-xxxx-GATTTACGGGAA∗A∗T∗G-xxxxx 3′.
CpG-scrambled ODN (scr ODN)
5′ T∗C∗C∗A∗T∗G∗A∗C∗G∗T∗T∗C∗C∗T∗G∗A∗T∗G∗C∗T-xxxxx-A∗C∗T∗CTTGCCAATTAC-xxxx-GTAATTGGCAAG∗A∗G∗T-xxxxx 3′.
CpG1668 alone
5′ T∗C∗C∗A∗T∗G∗A∗C∗G∗T∗T∗C∗C∗T∗G∗A∗T∗G∗C∗T 3′.
Flow cytometry analysis and cell sorting
For mouse samples, single-cell suspensions were prepared by enzymatic and mechanic dispersion of spleen and bone marrow, or from circulating blood leukocytes as described before.49 Cell surface staining was performed using fluorochrome-labeled antibodies specific to CD11b (47011282), CD19 (17532182), CD8 (47008182), CD4 (17004282), B220 (553090), Gr-1 (108415), c-Kit (25117182), F4/80 (123141), MHC-II (205321), and CD86 (48086282) after anti-FcγIII/IIR Block (Thermo Fisher) after LIVE/DEAD staining (Invitrogen, L34962). For intracellular cell staining we used Fix/Perm staining kit (Thermo Fisher, GAS004) and fluorochrome-labeled antibodies to Ki-67, FoxP3, IRF8 (Thermo Fisher, 14569882, 12577382, IRF8M-FITC), and pSTAT3 (BD, 557815). For cell sorting, GFP+c-Kit+ or GFP+CD11b− vs. GFP+CD11b+ leukemic cells were sorted using BD FACSAria III. Phagocytosis into endo-/lysosomes was assessed using in vivo-treated, freshly sorted GFP+CD11b− vs. GFP+CD11b+ leukemic cells incubated with pHrodo-E.coli-BioParticles (Thermo Fisher, P36600) according to the manufacturer’s protocol. Fluorescence data were analyzed on BD LSRFortessa, NovoCyte Quanteon (Agilent) or Attune NxT (Thermo Fisher) flow cytometers using FlowJo v.10 software (TreeStar, Ashland, OR).
Quantitative real-time PCR
Total RNA was extracted from CMM cells using the Maxwell RSC simplyRNA tissue kit in combination with the Maxwell system (Promega). The RNA was then transcribed into cDNAs using an iScript cDNA Synthesis kit (Bio-Rad) according to the manufacturer’s protocol. Quantitative PCR was carried out using SYBR Green on CFX96 Real-Time PCR Detection System (Bio-Rad). Actin b was used as an internal control.
PCR primers(5′ to 3′).
Dnmt1 F: GGGTCTCGTTCAGAGCTG.
R: GCAGGAATTCATGCAGTAAG.
Dnmt3a F: CCGCCTCTTCTTTGAGTTCTAC.
R: AGATGTCCCTCTTGTCACTAACG.
Dnmt3b F: ATGGAGATCAGGAGGGTATGGA.
R: GTCGCTTGGAGGTGGCTTTC.
Runx1 F: CCTCCTTGAACCACTCCACT.
R: CTGGATCTGCCTGGCATC.
Cebpa F: CAGTTTGGCAAGAATCAGAGCA.
R: GGGTGAGTTCATGGAGGAATGG.
Irf8 F: CGTGGAAGACGAGGTTACGCTG.
R: GCTGAATGGTGTGTGTCATAGGC.
Actb F: GTGGGCCGCCCTAGGCACCAG.
R: TTGGCCTTAGGGTTCAGGGGGG.
Western blot and immunohistochemical staining
Total cellular lysates were prepared as previously described30 and analyzed using antibodies specific to DNMT1, DNMT3b (Abcam: ab305429, ab122932), IRF8, mouse anti-rabbit immunoglobulin (Ig)G, and β-actin (Cell Signaling: 5628, 93702, 5125). For immunohistochemical staining, the formalin-fixed paraffin-embedded spleen sections were stained using primary and horseradish peroxidase-conjugated secondary antibodies and then analyzed on the Observer Z1 microscope (Zeiss) as reported before.50
Bulk transcriptomics
RNA was extracted using the mirVana Isolation Kit (Ambion) or Maxwell RSC simplyRNA tissue kit (Promega) from magnetically enriched c-Kit+ AML cells using PE-positive selection (EasySep) with >90% purity. RNA-seq libraries were prepared with mRNA-HyperPrep kit (Kapa Biosystems). The final libraries were validated with the Agilent Bioanalyzer DNA High Sensitivity Kit and quantified with Qubit. Sequencing was performed on HiSeq2500 (Illumina). RNA-seq reads were aligned to the mouse reference genome (mm10) using the TopHat (v.1.3.1) or STAR (v2.7.9) software. Low-expressed genes were excluded and expression data were normalized using DESeq2. Differentially expressed genes were identified at log2FC >1 and padj <0.05. Gene set enrichment was computed using fGSEA.
Single-cell transcriptomics
Total RNA was sequenced and analyzed at COH’s Integrative Genomics Core. A total of 2,300 to 19,000 cells were captured on a 10×Genomics Chromium controller using a Single Cell-3 Solution kit v3.1 following the manufacturer’s protocol. The libraries were sequenced on NovaSeq6000 platform (Illumina). Raw sequencing data were analyzed using the 10×Genomics Cell Ranger (v.7.1.0) for sample demultiplexing, barcode processing, alignment with mm10 mouse genome, filtering, and UMI counting. The scRNA-seq data were analyzed using the R Seurat Package (v4). The doublets were removed using DoubletFinder and low-quality cells were eliminated by control filters: >10% mitochondrial reads, <5% ribosomal reads, or <200 detected features. The normalization and variance stabilization were performed with SCTransform (v2). A total of 2,000 of the most variable genes were selected to calculate the first 40 principal components for the downstream analysis. The expression profiles clustered using the Leiden algorithm were visualized using two-dimensional UMAP. The cell type of each cell cluster was identified and annotated using known maker genes. The differentially expressed genes of each cell cluster were found with pseudo-bulk method using Deseq2. The pathway enrichment analysis was done using fGSEA with KEGG, Reactome, and GO-BP databases.
Statistics
Unpaired t test was used to calculate two-tailed p value to estimate statistical significance of differences between two treatment groups. One- or two-way ANOVA plus Bonferroni post-test were applied to assess differences between multiple groups or in tumor growth kinetics experiments, respectively. Statistically significant p values were indicated in figures as ∗∗∗p < 0.001; ∗∗p < 0.01; and ∗p < 0.05 compared with untreated or PBS-treated groups. Data were analyzed using Prism software v.7 (GraphPad).
Data and code availability
Genomic results are available on the gene omnibus database: GSE233099.
Acknowledgments
This work was supported by the NCI/NIH award R01CA213131 and R01CA284593 (to M.K.). Research reported here included work performed in the Analytical Cytometry, Integrative Genomics/Bioinformatics, Electron Microscopy, Light Microscopy/Digital Imaging, DNA/RNA Synthesis, Research Pathology Core Laboratories and in the Animal Facility Shared Resources supported by the NCI/ NIH grant number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The authors are grateful to Dr. James Moon (University of Michigan Medical School, MI) for sharing C1498-luc-mCherry cells and also to Drs Priyanka Duttagupta and Tomasz Adamus, Szymon Baluszek, Pawel Segit, and Duotian Qin for support, suggestions and/or critical reading.
Author contributions
Study design: D.W., Y.-L.S., M.K. Data acquisition: D.W., Y.-L.S., D.K.. Data analysis and interpretation: D.W., K.J., D.K., Y.-L.S., H.L., M.K. Administrative, technical, or material support: Y.-H.K., M.F., B.Z., B.K., S.H., G.M., M.K. Manuscript writing: D.W., Y.-L.S., K.J., M.K.
Declaration of interests
M.K. is an inventor on the patents that cover the design of CpG-STAT3d ODNs. M.K. serves on the Scientific Advisory Board of Scopus Biopharma and its subsidiary Duet Biotherapeutics with stock options and sponsored research.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102268.
Supplemental information
References
- 1.Khwaja A., Bjorkholm M., Gale R.E., Levine R.L., Jordan C.T., Ehninger G., Bloomfield C.D., Estey E., Burnett A., Cornelissen J.J., et al. Acute myeloid leukaemia. Nat. Rev. Dis. Prim. 2016;2 doi: 10.1038/nrdp.2016.10. [DOI] [PubMed] [Google Scholar]
- 2.Kantarjian H.M., Short N.J., Fathi A.T., Marcucci G., Ravandi F., Tallman M., Wang E.S., Wei A.H. Acute myeloid leukemia: historical perspective and progress in research and therapy over 5 decades. Clin. Lymphoma, Myeloma & Leukemia. 2021;21:580–597. doi: 10.1016/j.clml.2021.05.016. [DOI] [PubMed] [Google Scholar]
- 3.Damiani D., Tiribelli M. Present and future role of immune targets in acute myeloid leukemia. Cancers. 2022;15 doi: 10.3390/cancers15010253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Curran E., Corrales L., Kline J. Targeting the innate immune system as immunotherapy for acute myeloid leukemia. Front. Oncol. 2015;5:83. doi: 10.3389/fonc.2015.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyiadzis M., Bishop M.R., Abonour R., Anderson K.C., Ansell S.M., Avigan D., Barbarotta L., Barrett A.J., Van Besien K., Bergsagel P.L., et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of hematologic malignancies: multiple myeloma, lymphoma, and acute leukemia. J. Immunother. Cancer. 2016;4:90. doi: 10.1186/s40425-016-0188-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vadakekolathu J., Minden M.D., Hood T., Church S.E., Reeder S., Altmann H., Sullivan A.H., Viboch E.J., Patel T., Ibrahimova N., et al. Immune landscapes predict chemotherapy resistance and immunotherapy response in acute myeloid leukemia. Sci. Transl. Med. 2020;12 doi: 10.1126/scitranslmed.aaz0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bar-Natan M., Nelson E.A., Xiang M., Frank D.A. STAT signaling in the pathogenesis and treatment of myeloid malignancies. JAKSTAT. 2012;1:55–64. doi: 10.4161/jkst.20006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Benekli M., Baumann H., Wetzler M. Targeting signal transducer and activator of transcription signaling pathway in leukemias. J. Clin. Oncol. 2009;27:4422–4432. doi: 10.1200/JCO.2008.21.3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kramer M.H., Zhang Q., Sprung R., Day R.B., Erdmann-Gilmore P., Li Y., Xu Z., Helton N.M., George D.R., Mi Y., et al. Proteomic and phosphoproteomic landscapes of acute myeloid leukemia. Blood. 2022;140:1533–1548. doi: 10.1182/blood.2022016033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakamoto K.M., Grant S., Saleiro D., Crispino J.D., Hijiya N., Giles F., Platanias L., Eklund E.A. Targeting novel signaling pathways for resistant acute myeloid leukemia. Mol. Genet. Metabol. 2015;114:397–402. doi: 10.1016/j.ymgme.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kortylewski M., Moreira D. Myeloid cells as a target for oligonucleotide therapeutics: turning obstacles into opportunities. Cancer Immunol. Immunother. 2017;66:979–988. doi: 10.1007/s00262-017-1966-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moser B., Edtmayer S., Witalisz-Siepracka A., Stoiber D. The ups and downs of STAT inhibition in acute myeloid leukemia. Biomedicines. 2021;9 doi: 10.3390/biomedicines9081051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson D.E., O’Keefe R.A., Grandis J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018;15:234–248. doi: 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong D., Kurzrock R., Kim Y., Woessner R., Younes A., Nemunaitis J., Fowler N., Zhou T., Schmidt J., Jo M., et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015;7 doi: 10.1126/scitranslmed.aac5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sen M., Thomas S.M., Kim S., Yeh J.I., Ferris R.L., Johnson J.T., Duvvuri U., Lee J., Sahu N., Joyce S., et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. 2012;2:694–705. doi: 10.1158/2159-8290.CD-12-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Q., Hossain D.M.S., Duttagupta P., Moreira D., Zhao X., Won H., Buettner R., Nechaev S., Majka M., Zhang B., et al. Serum-resistant CpG-STAT3 decoy for targeting survival and immune checkpoint signaling in acute myeloid leukemia. Blood. 2016;127:1687–1700. doi: 10.1182/blood-2015-08-665604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su Y.-L., Wang X., Mann M., Adamus T.P., Wang D., Moreira D.F., Zhang Z., Ouyang C., He X., Zhang B., et al. Myeloid cell-targeted miR-146a mimic inhibits NF-κB-driven inflammation and leukemia progression in vivo. Blood. 2020;135:167–180. doi: 10.1182/blood.2019002045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Su Y.-L., Swiderski P., Marcucci G., Kortylewski M. Targeted Delivery of miRNA Antagonists to Myeloid Cells In Vitro and In Vivo. Methods Mol. Biol. 2019;1974:141–150. doi: 10.1007/978-1-4939-9220-1_10. [DOI] [PubMed] [Google Scholar]
- 19.Zhang B., Nguyen L.X.T., Li L., Zhao D., Kumar B., Wu H., Lin A., Pellicano F., Hopcroft L., Su Y.-L., et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat. Med. 2018;24:450–462. doi: 10.1038/nm.4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Machado E.A., Gerard D.A., Lozzio C.B., Lozzio B.B., Mitchell J.R., Golde D.W. Proliferation and differentiation of human myeloid leukemic cells in immunodeficient mice: electron microscopy and cytochemistry. Blood. 1984;63:1015–1022. [PubMed] [Google Scholar]
- 21.Yu H., Kortylewski M., Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007;7:41–51. doi: 10.1038/nri1995. [DOI] [PubMed] [Google Scholar]
- 22.Li Y.-J., Zhang C., Martincuks A., Herrmann A., Yu H. STAT proteins in cancer: orchestration of metabolism. Nat. Rev. Cancer. 2023;23:115–134. doi: 10.1038/s41568-022-00537-3. [DOI] [PubMed] [Google Scholar]
- 23.Kanna R., Choudhary G., Ramachandra N., Steidl U., Verma A., Shastri A. STAT3 inhibition as a therapeutic strategy for leukemia. Leuk. Lymphoma. 2018;59:2068–2074. doi: 10.1080/10428194.2017.1397668. [DOI] [PubMed] [Google Scholar]
- 24.Venugopal S., Bar-Natan M., Mascarenhas J.O. JAKs to STATs: A tantalizing therapeutic target in acute myeloid leukemia. Blood Rev. 2020;40 doi: 10.1016/j.blre.2019.100634. [DOI] [PubMed] [Google Scholar]
- 25.Gambacorta V., Gnani D., Vago L., Di Micco R. Epigenetic Therapies for Acute Myeloid Leukemia and Their Immune-Related Effects. Front. Cell Dev. Biol. 2019;7:207. doi: 10.3389/fcell.2019.00207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wingelhofer B., Neubauer H.A., Valent P., Han X., Constantinescu S.N., Gunning P.T., Müller M., Moriggl R. Implications of STAT3 and STAT5 signaling on gene regulation and chromatin remodeling in hematopoietic cancer. Leukemia. 2018;32:1713–1726. doi: 10.1038/s41375-018-0117-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Q., Wang H.Y., Woetmann A., Raghunath P.N., Odum N., Wasik M.A. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood. 2006;108:1058–1064. doi: 10.1182/blood-2005-08-007377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Surapally S., Tenen D.G., Pulikkan J.A. Emerging therapies for inv(16) AML. Blood. 2021;137:2579–2584. doi: 10.1182/blood.2020009933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salem S., Salem D., Gros P. Role of IRF8 in immune cells functions, protection against infections, and susceptibility to inflammatory diseases. Hum. Genet. 2020;139:707–721. doi: 10.1007/s00439-020-02154-2. [DOI] [PubMed] [Google Scholar]
- 30.Hossain D.M.S., Dos Santos C., Zhang Q., Kozlowska A., Liu H., Gao C., Moreira D., Swiderski P., Jozwiak A., Kline J., et al. Leukemia cell-targeted STAT3 silencing and TLR9 triggering generate systemic antitumor immunity. Blood. 2014;123:15–25. doi: 10.1182/blood-2013-07-517987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Elmaagacli A.H., Steckel N., Ditschkowski M., Hegerfeldt Y., Ottinger H., Trenschel R., Koldehoff M., Beelen D.W. Toll-like receptor 9, NOD2 and IL23R gene polymorphisms influenced outcome in AML patients transplanted from HLA-identical sibling donors. Bone Marrow Transplant. 2011;46:702–708. doi: 10.1038/bmt.2010.166. [DOI] [PubMed] [Google Scholar]
- 32.Oweida A.J., Darragh L., Phan A., Binder D., Bhatia S., Mueller A., Court B.V., Milner D., Raben D., Woessner R., et al. STAT3 modulation of regulatory T cells in response to radiation therapy in head and neck cancer. J. Natl. Cancer Inst. 2019;111:1339–1349. doi: 10.1093/jnci/djz036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moreira D., Sampath S., Won H., White S.V., Su Y.-L., Alcantara M., Wang C., Lee P., Maghami E., Massarelli E., Kortylewski M. Myeloid cell-targeted STAT3 inhibition sensitizes head and neck cancers to radiotherapy and T cell-mediated immunity. J. Clin. Invest. 2021;131 doi: 10.1172/JCI137001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oellerich T., Mohr S., Corso J., Beck J., Döbele C., Braun H., Cremer A., Münch S., Wicht J., Oellerich M.F., et al. FLT3-ITD and TLR9 use Bruton tyrosine kinase to activate distinct transcriptional programs mediating AML cell survival and proliferation. Blood. 2015;125:1936–1947. doi: 10.1182/blood-2014-06-585216. [DOI] [PubMed] [Google Scholar]
- 35.Xiang M., Kim H., Ho V.T., Walker S.R., Bar-Natan M., Anahtar M., Liu S., Toniolo P.A., Kroll Y., Jones N., et al. Gene expression-based discovery of atovaquone as a STAT3 inhibitor and anticancer agent. Blood. 2016;128:1845–1853. doi: 10.1182/blood-2015-07-660506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su Y.-L., Banerjee S., White S.V., Kortylewski M. STAT3 in Tumor-Associated Myeloid Cells: Multitasking to Disrupt Immunity. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang Y., Luo Y., Jiang Z., Ma Y., Lin C.-J., Kim C., Carter M.G., Amano T., Park J., Kish S., Tian X.C. Jak/Stat3 signaling promotes somatic cell reprogramming by epigenetic regulation. Stem Cell. 2012;30:2645–2656. doi: 10.1002/stem.1225. [DOI] [PubMed] [Google Scholar]
- 38.Zeinalzadeh E., Valerievich Yumashev A., Rahman H.S., Marofi F., Shomali N., Kafil H.S., Solali S., Sajjadi-Dokht M., Vakili-Samiani S., Jarahian M., Hagh M.F. The role of janus kinase/stat3 pathway in hematologic malignancies with an emphasis on epigenetics. Front. Genet. 2021;12 doi: 10.3389/fgene.2021.703883. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 39.Zhang Q., Wang H.Y., Marzec M., Raghunath P.N., Nagasawa T., Wasik M.A. STAT3- and DNA methyltransferase 1-mediated epigenetic silencing of SHP-1 tyrosine phosphatase tumor suppressor gene in malignant T lymphocytes. Proc. Natl. Acad. Sci. USA. 2005;102:6948–6953. doi: 10.1073/pnas.0501959102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee H., Zhang P., Herrmann A., Yang C., Xin H., Wang Z., Hoon D.S.B., Forman S.J., Jove R., Riggs A.D., Yu H. Acetylated STAT3 is crucial for methylation of tumor-suppressor gene promoters and inhibition by resveratrol results in demethylation. Proc. Natl. Acad. Sci. USA. 2012;109:7765–7769. doi: 10.1073/pnas.1205132109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ibrahim M.L., Klement J.D., Lu C., Redd P.S., Xiao W., Yang D., Browning D.D., Savage N.M., Buckhaults P.J., Morse H.C., Liu K. Myeloid-Derived Suppressor Cells Produce IL-10 to Elicit DNMT3b-Dependent IRF8 Silencing to Promote Colitis-Associated Colon Tumorigenesis. Cell Rep. 2018;25:3036–3046.e6. doi: 10.1016/j.celrep.2018.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jefferies C.A. Regulating irfs in IFN driven disease. Front. Immunol. 2019;10:325. doi: 10.3389/fimmu.2019.00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pulikkan J.A., Tenen D.G., Behre G. C/EBPα deregulation as a paradigm for leukemogenesis. Leukemia. 2017;31:2279–2285. doi: 10.1038/leu.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zimmermannova O., Ferreira A.G., Ascic E., Velasco Santiago M., Kurochkin I., Hansen M., Met Ö., Caiado I., Shapiro I.E., Michaux J., et al. Restoring tumor immunogenicity with dendritic cell reprogramming. Sci. Immunol. 2023;8 doi: 10.1126/sciimmunol.add4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salmon J.M., Todorovski I., Stanley K.L., Bruedigam C., Kearney C.J., Martelotto L.G., Rossello F., Semple T., Arnau G.M., Zethoven M., et al. Epigenetic Activation of Plasmacytoid DCs Drives IFNAR-Dependent Therapeutic Differentiation of AML. Cancer Discov. 2022;12:1560–1579. doi: 10.1158/2159-8290.CD-20-1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gambacorta V., Beretta S., Ciccimarra M., Zito L., Giannetti K., Andrisani A., Gnani D., Zanotti L., Oliveira G., Carrabba M.G., et al. Integrated multiomic profiling identifies the epigenetic regulator PRC2 as a therapeutic target to counteract leukemia immune escape and relapse. Cancer Discov. 2022;12:1449–1461. doi: 10.1158/2159-8290.CD-21-0980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Daver N., Garcia-Manero G., Basu S., Boddu P.C., Alfayez M., Cortes J.E., Konopleva M., Ravandi-Kashani F., Jabbour E., Kadia T., et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019;9:370–383. doi: 10.1158/2159-8290.CD-18-0774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao X., Zhang Z., Moreira D., Su Y.-L., Won H., Adamus T., Dong Z., Liang Y., Yin H.H., Swiderski P., et al. B Cell Lymphoma Immunotherapy Using TLR9-Targeted Oligonucleotide STAT3 Inhibitors. Mol. Ther. 2018;26:695–707. doi: 10.1016/j.ymthe.2018.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nechaev S., Gao C., Moreira D., Swiderski P., Jozwiak A., Kowolik C.M., Zhou J., Armstrong B., Raubitschek A., Rossi J.J., Kortylewski M. Intracellular processing of immunostimulatory CpG-siRNA: Toll-like receptor 9 facilitates siRNA dicing and endosomal escape. J. Contr. Release. 2013;170:307–315. doi: 10.1016/j.jconrel.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moreira D., Adamus T., Zhao X., Su Y.-L., Zhang Z., White S.V., Swiderski P., Lu X., DePinho R.A., Pal S.K., Kortylewski M. STAT3 Inhibition Combined with CpG Immunostimulation Activates Antitumor Immunity to Eradicate Genetically Distinct Castration-Resistant Prostate Cancers. Clin. Cancer Res. 2018;24:5948–5962. doi: 10.1158/1078-0432.CCR-18-1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomic results are available on the gene omnibus database: GSE233099.