

Abstract
Triclosan is a potent inhibitor of FabI (enoyl-ACP reductase, where ACP stands for acyl carrier protein), which catalyses the last step in a sequence of four reactions that is repeated many times with each elongation step in the type II fatty acid biosynthesis pathway. The malarial parasite Plasmodium falciparum also harbours the genes and is capable of synthesizing fatty acids by utilizing the enzymes of type II FAS (fatty acid synthase). The basic differences in the enzymes of type I FAS, present in humans, and type II FAS, present in Plasmodium, make the enzymes of this pathway a good target for antimalarials. The steady-state kinetics revealed time-dependent inhibition of FabI by triclosan, demonstrating that triclosan is a slow-tight-binding inhibitor of FabI. The inhibition followed a rapid equilibrium step to form a reversible enzyme–inhibitor complex (EI) that isomerizes to a second enzyme–inhibitor complex (EI*), which dissociates at a very slow rate. The rate constants for the isomerization of EI to EI* and the dissociation of EI* were 5.49×10−2 and 1×10−4 s−1 respectively. The Ki value for the formation of the EI complex was 53 nM and the overall inhibition constant Ki* was 96 pM. The results match well with the rate constants derived independently from fluorescence analysis of the interaction of FabI and triclosan, as well as those obtained by surface plasmon resonance studies [Kapoor, Mukhi, N. Surolia, Sugunda and A. Surolia (2004) Biochem. J. 381, 725–733].
Keywords: crotonoyl-CoA, enoyl-acyl carrier protein (enoyl-ACP) reductase, fatty acid biosynthesis, Plasmodium falciparum, slow-tight-binding inhibitor, triclosan
Abbreviations: ACP, acyl carrier protein; FabI, enoyl-ACP reductase; FAS, fatty acid synthase
INTRODUCTION
The occurrence and spread of drug-resistant strains of Plasmodium falciparum has led to a resurgence of malaria, which claims 1–3 million lives annually and to which 40% of the world's population remains at risk [1]. P. falciparum malaria has been primarily treated with chloroquine and pyrimethamine-sulpha-doxine. However, the emergence of strains resistant to these drugs along with the reappearance of malaria in well-controlled areas has led to increased efforts towards the development of new antimalarials.
Owing to the basic differences in the structure and organization of enzymes of the fatty acid biosynthesis pathway between humans and bacteria, this pathway has attracted a lot of attention [2,3]. The associative or type I FAS (fatty acid synthase) is present in higher organisms, fungi and many mycobacteria, whereas the dissociative or type II FAS is present in bacteria and plants. In type I FAS, all the enzymes are present as part of a single large homodimeric, multifunctional enzyme containing many domains, each catalysing a separate reaction step of the pathway. Pioneering studies of Rock and co-workers have established the fatty acid biosynthesis pathway as an effective antimicrobial target [2–4]. The FAS-II enzymes have been identified as the targets of several widely used antibacterials including isoniazid [5], diazaborines [6], triclosan [7,8] and thiolactomycin [9].
In the type II system, there are distinct proteins catalysing the various reactions of the pathway. FabI (enoyl-ACP reductase, where ACP stands for acyl carrier protein) catalyses the final step in the sequence of four reactions during fatty acid biosynthesis and has a determinant role in completing cycles of elongation phase of FAS in Escherichia coli [3]. FabI catalyses the NADH/NADPH-dependent reduction of the double bond between C-2 and C-3 of enoyl-ACP. We have recently demonstrated the presence of type II FAS in the malarial parasite Plasmodium falciparum [10]. Triclosan inhibited the growth of P. falciparum cultures with an IC50 of 0.7 μM [10] at 150–2000 ng/ml [11]. Triclosan also inhibited Plasmodium growth in vivo and inhibited the activity of FabI isolated from Plasmodium cultures [10]. FabI has been earlier characterized from E. coli [12], Brassica napus [13], Mycobacterium tuberculosis [14] and Bacillus subtilis [15]. We have also cloned and expressed FabI from P. falciparum and studied its interaction with its substrates and inhibitors [16].
It has been observed that certain enzyme inhibitors do not show their effect instantaneously. Therefore they have been divided into four categories according to the strength of their interaction with the enzyme and the rate at which equilibrium involving enzyme and inhibitor is achieved [17]. The categories are classical, slow-binding, tight-binding and slow-tight-binding inhibitors. Historically, classical inhibitors have been studied in greater detail. Only a few studies have been made on the behaviour of tight-binding inhibitors [18,19]. Some workers have studied the action of compounds that cause time-dependent inhibition of enzymes and have termed them as slow-binding inhibitors [17,18,20]. Recently, cerivastatin has been shown to inhibit 3-hydroxy-3-methylglutaryl-CoA reductase from Trypanosoma cruzi in a biphasic manner and has been characterized as a slow-tight-binding inhibitor [21]. In addition, immucillins have been shown to be slow-onset tight-binding inhibitors of P. falciparum purine nucleoside phosphorylase [22]. Since, in the case of tight-binding inhibitors, there is a reduction in the concentration of the free inhibitor, Sculley et al. [23,24] have proposed ways for analysing such data by using a pair of parametric equations that describe the progress curves at different inhibitor concentrations.
Considering the importance of the fatty acid biosynthesis pathway and its inhibition by triclosan, it is imperative to study the inhibition kinetics of triclosan in greater detail. Triclosan follows tight-binding kinetics, as the concentration of binding sites is similar to the concentration of compound added to the assay. In the present study, we have characterized the inhibition of FabI by triclosan as a slow-tight-binding mechanism. The results are consistent with a two-step time-dependent inhibition.
MATERIALS AND METHODS
β-NADH, β-NAD+, crotonoyl-CoA, imidazole and SDS/PAGE reagents were obtained from Sigma (St. Louis, MO, U.S.A.). Triclosan was obtained from Kumar Organic Products (Bangalore, India). All other chemicals used were of analytical grade.
Expression and purification of FabI
FabI was expressed and purified as described earlier [16]. Briefly, the plasmid containing PffabI was transformed into BL21(DE3) cells. Cultures were grown at 37 °C for 12 h, followed by subsequent purification of the His-tagged FabI on a Ni2+-nitrilotriacetate agarose column using an imidazole gradient. PfFabI was eluted at 400 mM imidazole concentration. The purity of the protein was confirmed by SDS/PAGE. Protein concentration was determined from the absorbance A280 and the molar absorption coefficient ε=39560 M−1·cm−1, using the formula given in [25].
Enzyme assay
All experiments were performed on a UV–Vis spectrophotometer at 25 °C in 20 mM Tris/HCl (pH 7.4) and 150 mM NaCl. The standard reaction mixture in a total volume of 100 μl contained 20 mM Tris/HCl (pH 7.4), 150 mM NaCl, 200 μM crotonoyl-CoA, 100 μM NADH and 1% DMSO. The initial kinetic analysis for the inhibition of FabI by triclosan was performed using Dixon plots. The activity of FabI was measured in the presence of 100 μM NADH and 200 μM crotonoyl-CoA as a function of triclosan concentration at two concentrations of NAD+, and the Ki value was determined from the x-intercept of the Dixon plot.
The rate constant for association of triclosan with FabI was estimated in experiments where the onset of inhibition was monitored. The assay was started by the addition of 0.2 μM enzyme (subunit concentration) to various concentrations of triclosan (0–800 nM), containing 100 μM NADH, 200 μM crotonoyl-CoA and 50 μM NAD+.
For the calculation of dissociation rate constant, experiments were conducted in which 10 μM enzyme was preincubated with 10 μM triclosan and 2 mM NAD+ for 30 min before 200-fold dilution into a solution of competing NADH and crotonoyl-CoA. The dissociation of triclosan was monitored by following the enzyme activity during the initial part of the time course when the concentrations of substrate and NAD+ are relatively constant. The data were analysed by fitting the amount of product formed as a function of time.
Evaluation of kinetic parameters
Initial rate studies were analysed assuming uncompetitive kinetics in the Dixon plot:
 |
(1) |
where Km is the Michaelis constant, Vmax the maximal catalytic rate at saturating substrate concentration [S] in the absence of inhibitor, Ki the dissociation constant for the enzyme–inhibitor complex and [I] the inhibitor concentration.
Three basic kinetic mechanisms have been described to account for the slow-binding inhibition of the enzyme-catalysed reaction [26]. Mechanism A involves a single slow bimolecular interaction of an inhibitor with the enzyme leading to the formation of the enzyme–inhibitor complex:
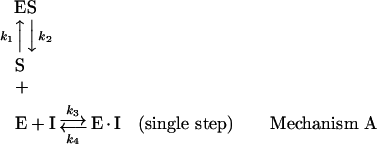 |
where E stands for the free enzyme, I for the free inhibitor, EI for the rapidly forming pre-equilibrium complex, S for the free substrate and ES for the enzyme–substrate complex. This mechanism assumes that the magnitude of k3I is very small relative to the rate constants for the conversion of substrate into product [27]. In mechanism B, there is an initial rapid binding of the inhibitor to the enzyme forming the initial complex EI, followed by a slow isomerization of EI to the stable enzyme–inhibitor complex EI*:
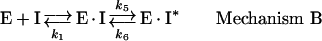 |
where Ki is the equilibrium inhibition constant for the formation of the initial complex EI and k5 and k6 are respectively the forward and reverse rate constants for the slow conversion of initial EI complex into a tight complex EI*.
In mechanism C, the enzyme exists in two states undergoing a reversible, slow interconversion between two forms E and E*, of which only E* is capable of binding the inhibitor:
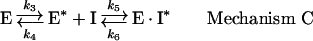 |
where k3 and k4 stand for the rate constants for the forward and backward reactions respectively for the conversion of the enzyme into a form competent to bind the inhibitor. Various studies have attempted to distinguish between the different inhibition mechanisms by steady-state kinetic techniques. In each of the mechanisms, the initial rate of substrate hydrolysis has a characteristic dependence on the inhibitor concentration, which can be used to distinguish them experimentally.
The progress curves for the interaction between triclosan and FabI were non-linear least-squares-fitted to the equation
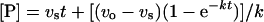 |
(2) |
where [P] is the product concentration at time t, voand vs are the initial and final steady-state rates and k is the apparent first-order rate constant for the establishment of the final steady-state equilibrium. The relationship between k, the rate constant and the kinetic constant is given by the following equation:
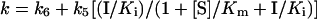 |
(3) |
The progress curves were fitted to eqns (2) and (3) using non-linear least-squares parameter estimation to determine the best-fit values. The overall inhibition constant Ki* is then defined as
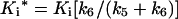 |
(4) |
where Ki=k4/k3.
Fluorescence analysis
Fluorescence measurements were performed on a computer-controlled JobinYvon Horiba fluorimeter. The excitation and emission monochromator slit widths were 3 nm. Measurements were performed at 25 °C in a 3 ml quartz cuvette and the solutions were mixed continuously with a magnetic stirrer. For fluorescence studies, solutions containing FabI were excited at 295 nm and the emission was recorded from 300 to 500 nm.
For inhibitor binding studies, FabI (4 μM) in 20 mM Tris and 150 mM NaCl (pH 7.4) was titrated with different concentrations of triclosan. Time courses of the protein fluorescence after the addition of the inhibitor were measured for 20 min with excitation and emission wavelengths of 295 and 340 nm respectively. The magnitude of rapid fluorescence decrease (F0−F), subsequent to the addition of triclosan, was fitted to the equation
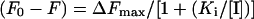 |
(5) |
to determine the value of Ki.
For a tight-binding inhibitor, k6 can be considered negligible at the onset of the slow loss of fluorescence and, hence, k5 was determined from the equation
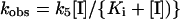 |
(6) |
where kobs is the rate constant for the loss of fluorescence at each inhibitor concentration [I].
Corrections for the inner filter effect were performed according to the equation [28]:
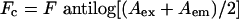 |
(7) |
where Fc and F are the corrected and measured fluorescence intensities respectively and Aex and Aem are the solution absorbances at the excitation and emission wavelengths respectively.
RESULTS
Inhibition of FabI by triclosan
During the course of the assay of FabI, there is an increase in the concentration of NAD+ due to the oxidation of NADH (the cofactor of FabI) and it is known that NAD+ potentiates inhibition by triclosan [12,16]. Thus NAD+ was included in all the assays so that the concentration of NAD+ does not change significantly during the course of the assay to maintain it close to its steady-state levels that are achieved during the course of the reaction. Triclosan inhibited FabI with an IC50 of 66 nM (Figure 1). Triclosan appears to act at approximately stoichiometric concentrations to that of the enzyme, thus classifying it as a tight-binding inhibitor. Examination of progress curves revealed that the steady-state rate was reached in the absence of triclosan, whereas the rate decreased in a time-dependent manner in its presence (Figure 2). We also observed a time range where the conversion of EI into EI* was minimal and the Dixon plot could be used to determine the Ki value of triclosan with respect to FabI. In the Dixon plot, the enzyme activity was determined at two concentrations of NAD+ as a function of inhibitor concentration. NAD+ was maintained at a high initial concentration so that its concentration does not vary during the time course of measurement. From such experiments, the Ki value was determined to be 14 nM (Figure 3). The uncompetitive kinetics with respect to NAD+ shows that prior binding of the oxidized coenzyme promotes the association of the inhibitor.
Figure 1. Inhibition of FabI by triclosan.

The activity of FabI was determined in the presence of 100 μM NADH, 50 μM NAD+, 0.2 μM enzyme in 20 mM Tris/HCl (pH 7.4), 150 mM NaCl and increasing concentrations of triclosan (0–150 nM).
Figure 2. Progress curves for the inhibition of FabI by triclosan.

The reaction mixture contained 100 μM NADH, 200 μM crotonoyl-CoA, 50 μM NAD+, 0.2 μM enzyme in 20 mM Tris/HCl (pH 7.4) and different concentrations of triclosan (0, 800 nM; from top to bottom) at 25 °C. The data were fitted to eqn (2) and the lines indicate the best fits of the data.
Figure 3. Initial rate of FabI reaction in the presence of triclosan.

Enzyme activity was determined in the presence of 100 μM NADH, 200 μM crotonoyl-CoA and (•) 100 μM and (▪) 150 μM NAD+. The Ki value was determined from the x-intercept of Dixon plot assuming uncompetitive inhibition.
The apparent rate of reaction kapp, from the progress curves, when plotted versus the inhibitor concentration, followed a hyperbolic curve (Figure 4), indicating a two-step mechanism. In agreement with mechanism B, the rate increased linearly with the inhibitor concentration and became saturated as the inhibitor concentration increased from a value much lower than Ki to a concentration greater than it (see Mechanism B above). Therefore the data were fitted to eqn (3), and the Ki value of 53 nM and an overall inhibition constant Ki* of 96 pM were calculated using the following equation:
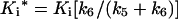 |
where Ki=k4/k3.
Figure 4. Dependence of the initial rate of FabI reaction on triclosan concentration.

The apparent rate constant k was calculated from an analysis of progress curves. The data fit well to eqn (3), demonstrating a two-step mechanism for the inhibition of FabI by triclosan.
The rate constant for the dissociation of triclosan from FabI was determined in an independent experiment, wherein high concentrations of the enzyme and inhibitor were preincubated for sufficient time to allow the system to reach equilibrium. This was followed by 200-fold dilution of the enzyme–inhibitor mix into a solution of crotonoyl-CoA and NADH and the regeneration of enzyme activity was studied (Figure 5). The k6 value as determined using eqn (2) was 1×10−4 s−1. The final steady-state rate was determined from the control that was preincubated without the inhibitor. The rate constant k5, related to the isomerization of EI to EI*, was 5.49×10−2 s−1 as obtained by fitting eqn (3) to the onset of inhibition data using the experimentally determined values of Ki and k6. On the basis of the various kinetic parameters (Table 1), we can rule out a kinetic model for the inhibition of FabI with triclosan in which a single slow step leads to the slow, tight binding of the inhibitor. Thus FabI binds to triclosan in two steps, where the first step involves a rapid formation of an initial enzyme–inhibitor complex EI, which slowly isomerizes to form a tightly bound complex EI*, from which the inhibitor dissociates in a very slow manner.
Figure 5. Determination of the dissociation rate constant k6 for the FabI–triclosan complex.

FabI was preincubated with or without equimolar concentrations of triclosan and 2 mM NAD+ for 30 min in Tris/HCl (pH 7.4) at 25 °C. The preincubated sample was then diluted 200-fold into a solution of competing NADH and crotonoyl-CoA and the dissociation of triclosan was monitored by following the enzyme activity.
Table 1. Inhibition constants of triclosan against FabI.
The rate constants for the inhibition of FabI by triclosan were calculated at 25 °C in 20 mM Tris/HCl buffer (pH 7.4) as described in the text.
| Inhibition constant | Value |
|---|---|
| IC50 | 66 nM |
| Ki | 53 nM |
| Ki* | 96 pM |
| k5 | 5.49×10−2 s−1 |
| k6 | 1×10−4 s−1 |
Fluorescence analysis
The excitation of FabI at 295 nm, where tryptophan has maximum absorption, resulted in an emission maximum at 340 nm. We have followed the intrinsic fluorescence of tryptophan to analyse the FabI–triclosan interactions. The binding of triclosan to FabI resulted in a concentration-dependent quenching of fluorescence; however, no red or blue shift was observed. The magnitude of rapid fluorescence decrease (F0−F) after the addition of various concentrations of triclosan followed a hyperbola. This is consistent with the earlier observation of a two-step mechanism as evident by enzyme inhibition studies. The Ki value estimated from the results was 45 nM (Figure 6). The effect of triclosan on FabI fluorescence is both concentration- and time-dependent (Figure 7). After the addition of 20 μM triclosan to a solution of FabI, there was an immediate decrease in fluorescence followed by a slow decrease to a final stable value. It would appear that the initial rapid and subsequent slow decrease in intrinsic FabI fluorescence induced by triclosan corresponds to a two-step mechanism for inhibition of FabI. The k5 value determined from the slow decrease in fluorescence was 7×10−2 s−1. These values match well with those obtained from the analyses of enzyme-inhibition studies. Thus the initial rapid decrease in fluorescence corresponds to the formation of the reversible FabI–triclosan complex. The time-dependent slow decrease reflects the formation of a tightly bound slowly dissociating EI* complex.
Figure 6. Effect of triclosan concentration on the tryptophan fluorescence of FabI.

FabI (4 μM) was treated with increasing concentrations of triclosan and the changes were measured at 25 °C. The change in fluorescence (F0−F) was plotted against triclosan concentrations. The hyperbola indicates the best fit of the data. a.u., arbitrary units.
Figure 7. Time-dependent quenching of FabI fluorescence by triclosan.

Triclosan (20 μM) was added to 4 μM FabI and the fluorescence emission was followed for 20 min at 25 °C. The excitation wavelength was fixed at 295 nm, whereas the emission wavelength was at 340 nm; •, absence of triclosan; and ▪, presence of triclosan. In the presence of triclosan, a rapid decrease in fluorescence was followed by a slow change in the fluorescence intensity. a.u., arbitrary units.
DISCUSSION
The reaction catalysed by FabI in the fatty acid elongation pathway has been validated as an antimicrobial drug target. Triclosan is a potent FabI inhibitor and we have previously reported the apparent inhibition parameters for the inhibition of Plasmodium FabI by triclosan [16].
In the case of classical inhibitors, the attainment of equilibrium between the enzyme, the inhibitor and the enzyme–inhibitor complexes is rapid and requires an excess of the inhibitor. In contrast, in tight-binding inhibitors, the attainment of equilibrium might be rapid, but the total concentration of the inhibitor required is similar to the total concentration of the enzyme [20]. Triclosan demonstrates a high potency against FabI and its 1:1 molar ratio for the inhibition of the enzyme indicates its tight-binding nature.
As reported in the literature, certain enzymes do not show the effect of inhibitor instantaneously and inhibitor complexes take a long time to form (seconds to minutes) relative to the catalytic rate of the enzyme. This class of inhibitors are classified as slow-binding inhibitors. This is due to the slow conformational isomerization of the enzyme–inhibitor complex from a state where the enzyme and drug are in rapid equilibrium to a state where the enzyme–inhibitor complex undergoes very slow dissociation.
As discussed in the Materials and methods section, three basic kinetic mechanisms have been described to account for the slow inhibition of an enzyme-catalysed reaction According to mechanism A, the rate of inhibition would increase linearly with inhibitor concentration. However, in mechanism B, the inhibition rate would increase linearly with the inhibitor concentration but tends to saturate as the inhibitor concentration increases from a value much lower than Ki to a concentration greater than that. Thus the plot of rate versus inhibitor concentration would be a hyperbola. In mechanism C, the inhibition rate would decrease with increase in the inhibitor concentration. An examination of Figure 4 shows that PfFabI–triclosan interaction follows mechanism B. Therefore the kinetic data were analysed assuming a two-step mechanism for binding and equilibrium constants for the formation of both the initial (Ki) and the final (Ki*) complexes. The slow-binding nature of triclosan was observed when the triclosan concentration was varied from 0 to 800 nM. A time-dependent decrease in the rate was seen, which varied as a function of triclosan concentration. The kinetics was characteristic of enzyme–inhibitor interactions, where the initial step involves the rapid formation of a weak complex, followed by a slow conversion into the tight-binding complex. The rate constant of this slow-binding process was determined as it was noted to be analogous to enzyme inactivation by a slow-tight-binding inhibitor [29].
The progress curves were analysed by assuming that the rates of inactivation reflected a pseudo-first-order process. The pseudo-first-order rate constant when plotted as a function of triclosan concentration fitted well to a hyperbolic equation. On the basis of this kinetic analysis of the inhibition data, one can conclude that triclosan follows biphasic kinetics for its binding to FabI. This is also reflected in the fluorescence analysis of the interaction. Triclosan induced a rapid fluorescence quenching that followed a slower decrease to a constant final value. The magnitude of initial rapid fluorescence quenching increased with the inhibitor concentration, which tended to reach saturation. That an isomerization step in the interaction of triclosan with PfFabI occurs is demonstrated when the change in the intrinsic fluorescence of the protein is followed as a function of time. As shown in Figure 7, a rapid loss in fluorescence resulting from the formation of a reversible EI complex is observed initially, followed by a much slower decrease, which corresponds to the isomerization of EI to the EI* complex consistent with the above kinetic model. The kinetic constants (Ki and k5), derived for the binding of triclosan to FabI from the fluorescence changes, are in good agreement with those obtained from the steady-state kinetic analyses of the inhibition results.
Thus the formation of a ternary complex of FabI–NAD+–triclosan and the slow transition of this complex to a stable form appear to be the factors determining the highly potent inhibition of FabI by triclosan. In this model (Scheme 1), triclosan forms a complex with NAD+-bound FabI, with the complex being in rapid equilibrium with the free enzyme. This complex undergoes a slow conformational change to a final stable form, which dissociates very slowly. Such tight-binding inhibitors of FabI have important implications in the development of antimalarial drugs.
Scheme 1. Formation of a ternary complex of FabI–NAD+–triclosan, and the slow transition of this complex to a stable form.

Triclosan binds to FabI more potently in the presence of NAD+, leading to the formation of the ternary complex. This complex undergoes a slow transformation to a final slowly dissociating complex. Thus the formation of a ternary complex and the slow conversion of this complex into a final stable form make triclosan a potent inhibitor of FabI.
In conclusion, we demonstrate that triclosan follows a two-step inhibition mechanism as shown by equilibrium binding studies of the enzyme and inhibitor. It has been proposed earlier that the ability of FabI inhibitors to form stable ternary complexes with the enzyme is the critical feature required for antibacterial activity [30]. The inhibition of FabI by triclosan becomes progressively stronger with time and is essentially irreversible after several minutes. Indeed, this irreversible inhibition in the case of FabI can be correlated with the formation of a stable FabI–NAD+–triclosan ternary complex that was shown to be accompanied by a conformational change in the flexible loop in FabI in the case of E. coli FabI. The structure of the triclosan–NAD+–FabI complex has been solved from E. coli [31]. The diazaborines are another class of potent FabI inhibitors that act via the formation of a tight-binding bi-substrate complex [32,33]. In the case of plasmodial FabI, superposition of binary (FabI–NAD+) and ternary (FabI–NAD+–triclosan) complex structures revealed subtle conformational changes in the protein after inhibitor binding [34,35]. These studies also set the stage for analysing the interactions of the mutants of Pf ENR to bind to triclosan [36].
Acknowledgments
N. S. is supported by a grant from the Department of Biotechnology, Government of India.
References
- 1.WHO. The World Health Report. 1999:49–63.
- 2.Rock C. O., Cronan J. E. Escherichia coli as a model for the regulation of dissociable (type II) fatty acid biosynthesis. Biochim. Biophys. Acta. 1996;1302:1–16. doi: 10.1016/0005-2760(96)00056-2. [DOI] [PubMed] [Google Scholar]
- 3.Heath R. J., Rock C. O. Enoyl-acyl carrier protein reductase (FabI) plays a determinant role in completing cycles of fatty acid elongation in Escherichia coli. J. Biol. Chem. 1995;270:26538–26542. doi: 10.1074/jbc.270.44.26538. [DOI] [PubMed] [Google Scholar]
- 4.Marrakchi H., Zhang Y.-M., Rock C. O. Mechanistic diversity and regulation of Type II fatty acid synthesis. Biochem. Soc. Trans. 2002;30:1050–1055. doi: 10.1042/bst0301050. [DOI] [PubMed] [Google Scholar]
- 5.Parikh S. L., Xiao G., Tonge P. J. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochemistry. 2000;39:7645–7650. doi: 10.1021/bi990529c. [DOI] [PubMed] [Google Scholar]
- 6.Roujeinikova A., Sedelnikova S., de Boer G. J., Stuitje A. R., Slabas A. R., Rafferty J. B., Rice D. W. Inhibitor binding studies on enoyl reductase reveal conformational changes related to substrate recognition. J. Biol. Chem. 1999;274:30811–30817. doi: 10.1074/jbc.274.43.30811. [DOI] [PubMed] [Google Scholar]
- 7.Heath R. J., Yu Y. T., Shapiro M. A., Olson E., Rock C. O. Broad spectrum antimicrobial biocides target the FabI component of fatty acid synthesis. J. Biol. Chem. 1998;273:30316–30320. doi: 10.1074/jbc.273.46.30316. [DOI] [PubMed] [Google Scholar]
- 8.Levy C. W., Roujeinikova A., Sedelnikova S., Baker P. J., Stuitje A. R., Slabas A. R., Rice D. W., Rafferty J. B. Molecular basis of triclosan activity. Nature (London) 1999;398:383–384. doi: 10.1038/18803. [DOI] [PubMed] [Google Scholar]
- 9.Tsay J. T., Rock C. O., Jackowski S. Overproduction of β-ketoacyl-acyl carrier protein synthase I imparts thiolactomycin resistance to Escherichia coli K-12. J. Bacteriol. 1992;174:508–513. doi: 10.1128/jb.174.2.508-513.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Surolia N., Surolia A. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat. Med. (N.Y.) 2001;7:167–173. doi: 10.1038/84612. [DOI] [PubMed] [Google Scholar]
- 11.McLeod R., Muench S. P., Rafferty J. B., Kyle D. E., Mui E. J., Kirisits M. J., Mack D. G., Roberts C. W., Samuel B. U., Lyons R. E., et al. Triclosan inhibits the growth of Plasmodium falciparum and Toxoplasma gondii by inhibition of apicomplexan Fab I. Int. J. Parasitol. 2001;31:109–113. doi: 10.1016/s0020-7519(01)00111-4. [DOI] [PubMed] [Google Scholar]
- 12.Ward W. H. J., Holdgate G. A., Rowsell S., McLean E. G., Pauptit R. A., Clayton E., Nichols W. W., Colls J. G., Minshull C. A., Jude D. A., et al. Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochemistry. 1999;38:12514–12525. doi: 10.1021/bi9907779. [DOI] [PubMed] [Google Scholar]
- 13.Fawcett T., Copse C. L., Simon J. W., Slabas A. R. Kinetic mechanism of NADH-enoyl-ACP reductase from Brassica napus. FEBS Lett. 2000;484:65–68. doi: 10.1016/s0014-5793(00)02128-1. [DOI] [PubMed] [Google Scholar]
- 14.Parikh S., Moynihan D. P., Xiao G., Tonge P. J. Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochemistry. 1999;38:13623–13634. doi: 10.1021/bi990529c. [DOI] [PubMed] [Google Scholar]
- 15.Heath R. J., Su N., Murphy C. K., Rock C. O. The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J. Biol. Chem. 2000;275:40128–40133. doi: 10.1074/jbc.M005611200. [DOI] [PubMed] [Google Scholar]
- 16.Kapoor M., Dar M. J., Surolia A., Surolia N. Kinetic determinants of the interaction of enoyl-ACP reductase from Plasmodium falciparum with its substrates and inhibitors. Biochem. Biophys. Res. Commun. 2001;289:832–837. doi: 10.1006/bbrc.2001.6061. [DOI] [PubMed] [Google Scholar]
- 17.Morrison J. F. The slow-binding and slow, tight-binding inhibition of enzyme catalyzed reactions. Trends Biochem. Sci. 1982;7:102–105. [Google Scholar]
- 18.Williams J. W., Morrison J. F. The kinetics of reversible tight-binding inhibition. Methods Enzymol. 1979;63:437–467. doi: 10.1016/0076-6879(79)63019-7. [DOI] [PubMed] [Google Scholar]
- 19.Greco W. R., Hakala M. T. Evaluation of methods for estimating the dissociation constant of tight binding enzyme inhibitors. J. Biol. Chem. 1979;254:12104–12109. [PubMed] [Google Scholar]
- 20.Morrison J. F., Walsh C. T. The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- 21.Hurtado-Guerrero R., Pena-Diaz J., Montalvetti A., Ruiz-Perez L. M., Gonzalez-Pacanowska D. Kinetic properties and inhibition of Trypanosoma cruzi 3-hydroxy-3-methylglutaryl CoA reductase. FEBS Lett. 2002;510:141–144. doi: 10.1016/s0014-5793(01)03238-0. [DOI] [PubMed] [Google Scholar]
- 22.Kicska G. A., Tyler P. C., Evans G. B., Furneaux R. H., Kim K., Schramm V. L. Transition state analogue inhibitors of purine nucleoside phosphorylase from Plasmodium falciparum. J. Biol. Chem. 2001;277:3219–3225. doi: 10.1074/jbc.M105905200. [DOI] [PubMed] [Google Scholar]
- 23.Sculley M. J., Morrison J. F. The determination of kinetic constants governing the slow, tight-binding inhibition of enzyme-catalysed reactions. Biochim. Biophys. Acta. 1986;874:44–53. [Google Scholar]
- 24.Sculley M. J., Morrison J. F., Cleland W. W. Slow-binding inhibition: the general case. Biochim. Biophys. Acta. 1996;1298:78–86. doi: 10.1016/s0167-4838(96)00118-5. [DOI] [PubMed] [Google Scholar]
- 25.Mulvey R. S., Gualtieri R. J., Beychok S. Composition, fluorescence, and circular dichroism of rat lysozyme. Biochemistry. 1974;13:782–787. doi: 10.1021/bi00701a021. [DOI] [PubMed] [Google Scholar]
- 26.Cha S. Tight-binding inhibitors – III. A new approach for the determination of competition between tight-binding inhibitors and substrates-inhibition of adenosine deaminase by coformycin. Biochem. Pharmacol. 1976;25:2695–2702. doi: 10.1016/0006-2952(76)90259-8. [DOI] [PubMed] [Google Scholar]
- 27.Dash C., Vathipadiekal V., George S. P., Rao M. Slow-tight binding inhibition of xylanase by an aspartic protease inhibitor: kinetic parameters and conformational changes that determine the affinity and selectivity of the bifunctional nature of the inhibitor. J. Biol. Chem. 2002;277:17978–17986. doi: 10.1074/jbc.M111250200. [DOI] [PubMed] [Google Scholar]
- 28.Lakowicz J. R. New York: Plenum Press; 1983. Principles of Fluorescence Spectroscopy. [Google Scholar]
- 29.Schloss J. V. Significance of slow-binding enzyme inhibition and its relationship to reaction-intermediate analogs. Acc. Chem. Res. 1988;21:348–353. [Google Scholar]
- 30.Heath R. J., Li J., Roland G. E., Rock C. O. Inhibition of the Staphylococcus aureus NADPH-dependent enoyl-acyl carrier protein reductase by triclosan and hexachlorophene. J. Biol. Chem. 2000;275:4654–4659. doi: 10.1074/jbc.275.7.4654. [DOI] [PubMed] [Google Scholar]
- 31.Roujeinikova A., Levy C. W., Rowsell S., Sedelnikova S., Baker P. J., Minshull C. A., Mistry A., Colls J. G., Camble R., Stuitje A. R., et al. Crystallographic analysis of triclosan bound to enoyl reductase. J. Mol. Biol. 1999;294:527–535. doi: 10.1006/jmbi.1999.3240. [DOI] [PubMed] [Google Scholar]
- 32.Baldock C., Rafferty J. B., Sedelnikova S. E., Baker P. J., Stuitje A. R., Slabas A. R., Hawkes T. R., Rice D. W. A mechanism of drug action revealed by structural studies of enoyl reductase. Science. 1996;274:2107–2110. doi: 10.1126/science.274.5295.2107. [DOI] [PubMed] [Google Scholar]
- 33.Levy C. W., Baldock C., Wallace A. J., Sedelnikova S., Viner R. C., Clough J. M., Stuitje A. R., Slabas A. R., Rice D. W., Rafferty J. B. A study of the structure–activity relationship for diazaborine inhibition of Escherichia coli enoyl-ACP reductase. J. Mol. Biol. 2001;309:171–180. doi: 10.1006/jmbi.2001.4643. [DOI] [PubMed] [Google Scholar]
- 34.Perozzo R., Kuo M., Sidhu A. S., Valiyaveettil J. T., Bittman R., Jacobs W. R., Jr, Fidock D. A., Sacchettini J. C. Targeting tuberculosis and malaria through inhibition of enoyl reductase: compound activity and structural data. J. Biol. Chem. 2002;277:13106–13114. [Google Scholar]
- 35.Kapoor M., Mukhi P. L. S., Surolia N., Sugunda K., Surolia A. Kinetic and structural analysis of the increased affinity of enoyl-ACP (acyl-carrier protein) reductase for triclosan in the presence of NAD+ Biochem. J. 2004;381:725–733. doi: 10.1042/BJ20040228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kapoor M., Gopalakrishnapai J., Surolia N., Surolia A. Mutational analysis of the triclosan-binding region of enoyl-ACP (acyl-carrier protein) reductase from Plasmodium falciparum. Biochem. J. 2004;381:735–741. doi: 10.1042/BJ20040302. [DOI] [PMC free article] [PubMed] [Google Scholar]