

Abstract
Triclosan, a known antibacterial, acts by inhibiting enoyl-ACP (acyl-carrier protein) reductase (ENR), a key enzyme of the type II fatty acid synthesis (FAS) system. Plasmodium falciparum, the human malaria-causing parasite, harbours the type II FAS; in contrast, its human host utilizes type I FAS. Due to this striking difference, ENR has emerged as an important target for the development of new antimalarials. Modelling studies, and the crystal structure of P. falciparum ENR, have highlighted the features of ternary complex formation between the enzyme, triclosan and NAD+ [Suguna, A. Surolia and N. Surolia (2001) Biochem. Biophys. Res. Commun. 283, 224–228; Perozzo, Kuo, Sidhu, Valiyaveettil, Bittman, Jacobs, Fidock, and Sacchettini (2002) J. Biol. Chem. 277, 13106–13114; and Swarnamukhi, Kapoor, N. Surolia, A. Surolia and Suguna (2003) PDB1UH5]. To address the issue of the importance of the residues involved in strong specific and stoichiometric binding of triclosan to P. falciparum ENR, we mutated the following residues: Ala-217, Asn-218, Met-281, and Phe-368. The affinity of all the mutants was reduced for triclosan as compared with the wild-type enzyme to different extents. The most significant mutation was A217V, which led to a greater than 7000-fold decrease in the binding affinity for triclosan as compared with wild-type PfENR. A217G showed only 10-fold reduction in the binding affinity. Thus, these studies point out significant differences in the triclosan-binding region of the P. falciparum enzyme from those of its bacterial counterparts.
Keywords: enoyl-ACP reductase, FabI, fatty acid biosynthesis (FAS), inhibitor, triclosan
Abbreviations: ACP, acyl-carrier protein; ENR (or FabI), enoyl-ACP reductase; FAS, fatty acid biosynthesis; Ki, inhibition constant; Kii, inhibitor dissociation constant for an uncompetitive inhibitor; Kis, inhibitor dissociation constant for a competitive inhibitor; Ki′, apparent inhibition constant; MIC, minimal inhibitory concentration; PfENR, Plasmodium falciparum ENR
INTRODUCTION
The human malaria-causing parasite, Plasmodium falciparum, has been shown to harbour type II fatty acid biosynthesis (FAS) enzymes. In contrast, its human host synthesizes fatty acids via the type I pathway [1,2]. The realization that the FAS pathway of the malaria parasite could be a potential target of antimalarials, has led to renewed research in this area, as apparent from the recent research and review articles published [1,3–8]. Triclosan has been shown to be effective against a broad spectrum of bacteria including Escherichia coli [9], Staphylococcus aureus [10] and Mycobacterium smegmatis [11]. Triclosan was found to inhibit the growth of P. falciparum in red blood cell cultures (with an IC50 of 0.7 μM) and its efficacy was demonstrated in a mouse model of Plasmodium berghei [1].
It was shown previously that triclosan blocks lipid synthesis in E. coli, and that mutations in enoyl-ACP (acyl-carrier protein) reductase (ENR) prevent this blockage [9]. A series of E. coli mutants were isolated that were resistant to triclosan. Minimal inhibitory concentration (MIC) is defined as the minimum concentration of the drug that inhibits more than 99% of the bacterial population. MIC ratio refers to the ratio of MIC of the drug for a mutant as compared with the wild-type. The MIC ratio of the various mutants with respect to the wild-type was calculated as 95 (G93V; where Gly-93 is replaced by valine using single-letter symbols), 12.2 (M159T) and 6.1 (F203L). ENR, catalysing the last step in the elongation cycle of FAS, reduces a carbon–carbon double bond in an enoyl moiety that is covalently linked to an acyl carrier protein. The enzyme has been studied from various sources [1,6–8,11–15]. The recent investigation into the mechanism of triclosan inhibition and selectivity in E. coli FabI (ENR), in which three mutations were characterized, namely G93V, M159T and F203L, correlate well with the MIC data [9,14]. Also, the mutation G93S leads to diazaborine resistance, while the mutation of the analogous residue in InhA (S94A) leads to isoniazid resistance [16]. These results also correlate with the crystal data of E. coli FabI protein, which shows that all the three residues line a cleft at which NADH binds [15].
In P. falciparum ENR (PfENR), alanine is present at the position corresponding to Gly-93. The other two residues (Met-159 and Phe-203) are conserved. Thus, the residues in PfENR corresponding to Gly-93, Met-159 and Phe-203 of E. coli FabI are Ala-217, Met-281 and Phe-368. On the basis of modelling studies, the residues Ala-217, Met-281 and Phe-368 were implicated in triclosan binding [6]. Consistent with the above observations, the crystal structure of PfENR solved with NAD+ and triclosan demonstrated that the mode of triclosan binding was very similar to that observed in the E. coli–NAD+–triclosan complex [8]. The residues Ala-217, Asn-218, Met-281 and Phe-368 are present close to the triclosan binding site of PfENR. Hence, to characterize the role played by these residues of PfENR in triclosan binding, we generated the following mutants: A217V, A217G, N218A, N218D, M281A, M281T, F368A and F368I.
The mutants were expressed, purified by Ni2+-nitrilotriacetate affinity chromatography and characterized by determining the kinetic constants for their binding to substrates and the inhibitor, triclosan. The study reports that, as in the case of E. coli FabI, substitution of Ala-217 by an amino acid with a bulkier side chain is not tolerated for triclosan binding. The other mutant enzymes also have reduced affinity for triclosan, probably due to abrogation of important contacts between the side chains of the amino acids and triclosan.
MATERIALS AND METHODS
Materials
Media components were obtained from Hi-media (Delhi, India). β-NADH, β-NAD+, crotonoyl-CoA, imidazole and SDS/PAGE reagents were obtained from Sigma. Triclosan was obtained from Kumar organic products (Bangalore, India). His-bind resin and anti-His tag antibody were obtained from Novagen (Madison, U.S.A.). Protein molecular mass marker was obtained from MBI (Fermentas Inc., U.S.A.). Anti-mouse rabbit antibody and prestained molecular mass marker were obtained from Bangalore Genei (Bangalore, India). All other chemicals used were of analytical grade.
Strains and plasmids
E. coli DH5α cells were used during the cloning of the mutants. pET-28a(+) vector (Novagen, Madison, WI, U.S.A.) and BL21 (DE3) cells (Novagen) were used for the expression of mutant PfENRs. Primers for constructing the mutants were obtained from Sigma.
Construction of A217V, A217G, N218A, N218D, F368A and F368I mutants
The single point mutants of A217V, A217G, N218A, N218D, F368A and F368I were generated by the PCR overlap extension method. A pET 28a(+)-derived vector [7] containing the sequence of PfENR (GenBank accession number AF332608) was used as the template plasmid for all the reactions. For making the A217V mutant, the two PCR products with overlapping ends were generated by using the ‘PlasF’ primer together with primers ‘A217VF’ and ‘A217R’ together with ‘PlasR’. The PCR cycles used were: 95 °C for 3 min followed by 25×(95 °C for 1 min, 50 °C for 1 min, 72 °C for 1 min) and 72 °C for 7 min. The combined PCR product was digested with NcoI and BamHI and cloned into pET28a(+) digested with the same enzymes. A217G, N218A, N218D, F368A and F368I were generated in a similar manner using the primers listed in Table 1. The correct sequences of all constructs were verified by DNA sequencing.
Table 1. Overview of the oligonucleotide primers used to generate the PfENR mutants.
The mutation sites are underlined.
| Primer | Sequence |
|---|---|
| PlasF | 5′-ACGTCCCATGGTGCATCATCATCATCATCATAATGAAGATATTTGTTTT ATTGCTGGTATAGG-3′ |
| PlasR | 5′-ATATGGATCCTCAATCATCTGGTAAAAACATTATATTTAATCCGTTATCC ACATATATTGTCTG-3′ |
| A217GF | 5′-CTCGTTCATTCTTTAGGCAACGCTAAAGAAGTTCAAAAAG-3′ |
| A217GR | 5′-CTTTTTGAACTTCTTTAGCGTTGCCTAAAGAATGAACGAG-3′ |
| A217VF | 5′-CTCGTTCATTCTTTAGTTAACGCTAAAGAAGTTCAAAAAG-3′ |
| A217VR | 5′-CTTTTTGAACTTCTTTAGCGTTAACTAAAGAATGAACGAG-3′ |
| N218AF | 5′-CGTTCATTCTTTAGCTGCGGCTAAAGAAGTTC-3′ |
| N218AR | 5′-GAACTTCTTTAGCCGCAGCTAAAGAATGAACG-3′ |
| N218DF | 5′-CGTTCATTCTTTAGCTGATGCTAAAGAAGTTC-3′ |
| N218DR | 5′-GAACTTCTTTAGCATCAGCTAAAGAATGAACG-3′ |
| M281AF | 5′-GGCTATGGTGGAGGTGCGTCGAGCGCTAAAGCTG-3′ |
| M281AR | 5′-CAGCTTTAGCGCTCGACGCACCTCCACCATAGCC-3′ |
| M281TF | 5′-GGCTATGGTGGAGGTACCTCGAGCGCTAAAGCTG-3′ |
| M281TR | 5′-CAGCTTTAGCGCTCGAGGTACCTCCACCATAGCC-3′ |
| F368AF | 5′-GCTAGCCAAAATTATACAGCGATAGATTATGCAATAGAG-3′ |
| F368AR | 5′-CTCTATTGCATAATCTATCGCTGTATAATTTTGGCTAGC-3′ |
| F368IF | 5′-GCTAGCCAAAATTATACAATTATAGATTATGCAATAGAG-3′ |
| F368IR | 5′-CTCTATTGCATAATCTATAATTGTATAATTTTGGCTAGC-3′ |
Construction of M281A and M281T mutants
The M281A mutant was generated by amplification of the wild-type PfENR plasmid using M281AF and M281AR primers. The PCR cycles used were: 95 °C for 3 min, 19×(95 °C for 1 min, 62 °C for 1 min, 72 °C for 6.5 min) and 72 °C, 7 min. The PCR product was digested with DpnI and transformed into E. coli DH5α cells. The colonies were screened by digesting with XhoI, as the primers lead to a silent mutation in PfENR leading to the generation of a XhoI site. The M281T mutant was generated in a similar manner using the primers listed in Table 1. The correct sequences was verified by DNA sequencing.
Overexpression and purification of wild-type and mutant PfENRs
Wild-type and mutant PfENRs were expressed and purified as described previously [7]. Briefly, the plasmid containing PfENR was transformed in BL21(DE3) cells. Cultures were grown at 37 °C for 12 h, pelleted and resuspended in 20 mM Tris/HCl buffer containing 500 mM NaCl and 5 mM imidazole, pH 7.4. The cultures were lysed by sonication followed by purification of the His-tagged PfENR on a Ni2+-nitrilotriacetate–agarose column using an imidazole gradient. PfENR eluted at 400 mM imidazole concentration. The purity of the protein was confirmed by SDS/PAGE (10% gel). The fractions containing pure protein were pooled and concentrated using 10 kDa centripreps (Amicon, Danvers, MA, U.S.A.) for further experiments. The pET 28a(+) constructs harbouring the mutated PfENR sequences were transformed into BL21(DE3) cells, and the mutant proteins were purified using the same protocol as described above for the wild-type PfENR. The molar extinction coefficienct, A280, of mutant ENRs was estimated using the ProtParam tool at http://tw.expasy.org/tools/protparam.html.
Enzyme assay
All experiments were carried out on a Jasco V-530 UV-Vis spectrophotometer. ENR was assayed at 25 °C by monitoring the decrease in A340 due to the oxidation of NADH using crotonoyl-CoA as a substrate [7]. The standard reaction mixture in a total volume of 100 μl contained 20 mM Tris/HCl buffer, pH 7.4, 500 mM NaCl, 100 μM crotonoyl-CoA and 100 μM NADH. The Km for each substrate was determined by varying the concentrations of that substrate while keeping the concentration of the other substrate constant. The kinetic parameters were obtained by fitting initial velocity data to the Michaelis–Menten equation by non-linear regression analysis using GraphPad Prism software.
Enzyme-inhibition studies
The inhibition of PfENR activity was monitored by the spectro-photometric assay performed as described above, except that triclosan was added prior to the initiation of the reaction by addition of crotonoyl-CoA. The studies were performed in the presence of 1% DMSO used for solubilizing the inhibitors. In order to study the inhibition of ENR by triclosan by a steady-state approach, a modified protocol was followed as described in [17]. ENR was pre-incubated with the respective inhibitor and various concentrations of the coenzymes at 4 °C for 5 h. This was warmed to 25 °C and assay started by the addition of crotonoyl-CoA. The mechanism of inhibition was identified by studying the dependence of the apparent inhibition constant (Ki′) on the substrate concentration. The Ki′ was determined using the equation:
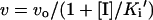 |
(1) |
where v is the rate in the presence of the inhibitor, vo is the uninhibited rate, and I is inhibitor.
The substrate dependence of Ki′ for competitive, uncompetitive and mixed noncompetitive kinetics is defined by Equations 2, 3 and 4 respectively (where Kis is the inhibitor dissociation constant for a competitive inhibitor and Kii is the inhibitor dissociation constant for an uncompetitive inhibitor).
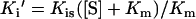 |
(2) |
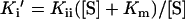 |
(3) |
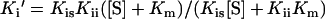 |
(4) |
RESULTS
We have demonstrated previously that the inhibition of PfENR by the well-known antibacterial, triclosan and determined the inhibition constants for the same [1,7]. Here, we have examined the effect of mutations that span the triclosan binding region, on the affinity of triclosan as well as its substrate binding and enzymatic activity.
Expression and purification of wild-type and mutant ENR
Wild-type and mutant ENRs were purified as described previously [7]. The mobility of the mutant proteins as analysed by SDS/PAGE was similar to the wild-type protein (see supplementary online data http://www.BiochemJ.org/bj/381/bj3810735add.htm). The expression of the recombinant His-tagged wild-type and mutant ENR was also analysed using anti-His antibody, and a band corresponding to the position of band in SDS/PAGE was obtained (see supplementary online data). The expected molecular mass of ENR as calculated using the ProtParam tool at http://kr.expasy.org/tools/ was 38166 Da. The molecular mass of purified ENR was further confirmed by MALDI–TOF (matrix-assisted laser-desorption ionization–time-of-flight)-MS to be 38163 (±2) Da (all data are given as means±S.E.M.). The molecular masses of the mutant ENRs were also confirmed by MALDI–TOF-MS and were found to be the expected molecular masses.
Gel filtration and CD analyses of the mutants
Changes in the overall shape or the quaternary structure of the molecule, potentially introduced by mutagenesis, was first probed using size exclusion chromatography. Wild-type PfENR elutes as a single peak at a volume of 7.6 ml on a Superdex-200 gel-filtration column (see supplementary online data as above). The mutants eluted at the same volume as their wild-type counterpart. The elution positions of the wild-type and mutants of PfENR correspond to a relative molecular mass of 150 (±10) kDa indicating the enzymes to be homotetramers and that the mutations did not alter the overall shape or the quaternary structure of PfENR. CD spectroscopy was used to investigate potential perturbations in the secondary and tertiary structure of PfENR mutants, which indicated that the various amino acid substitutions had no effect on the overall folding of the resultant mutant protein (supplementary online data).
Kinetic analysis of the mutants
As can be seen in Table 2, the kinetic constants (Km and kcat) for A217V, A217G, M281A, M281T, F368A and F368I remain unchanged with respect to the wild-type ENR. Thus, the mutations do not affect the catalytic efficiency of the enzyme. Interestingly, N218A and N218D mutants had an increase in the Km for NADH as compared with the wild-type. The Km value of wild-type PfENR for NADH was calculated previously as 33 μM [7]. N218A and N218D had a Km value of 73 μM and 100 μM respectively. Such a change for the E. coli enzyme has not been observed.
Table 2. Kinetic constants for NADH and crotonoyl-CoA with respect to wild-type and mutant PfENR.
Data are given as means±S.E.M.
| kcat (s−1) | Km (μM) (Crotonoyl-CoA) | Km (μM) (NADH) | |
|---|---|---|---|
| Wild-type | 1.62 (±0.06) | 160±5 | 33±3 |
| A217V | 1.2 (±0.02) | 150±4 | 28±4.2 |
| A217G | 0.9 (±0.1) | 173±6 | 35±2.2 |
| N218A | 152±4 | 73±5 | |
| N218D | 1.5 (±0.07) | 165±7 | 100±10 |
| M281A | 168±5 | 39±4 | |
| M281T | 1.1 (±0.07) | 155±3.4 | 25±12 |
| F368A | 152±6 | 31±3 | |
| F368I | 1.05 (±0.15) | 147±2 | 35±14 |
Inhibition constants of wild-type and mutant PfENR
Earlier studies on the E. coli and S. aureus FabI have shown that triclosan forms a ternary complex with NAD+, the oxidized cofactor [16,18–21]. Moreover, a ternary complex formation between ENR from P. falciparum, triclosan and NAD+ was also demonstrated in our earlier studies [1,7]. These observations were confirmed by the crystal structure of PfENR [8]. Both our modelling studies and the crystal structure of the ternary complex of PfENR with triclosan and NAD+, show that the ring A of triclosan, in addition to interacting face-to-face with the nicotinamide ring of NAD+, also makes additional van der Waals interactions with certain PfENR residues including Phe-368 [6,8]. Ring B is located in a pocket bounded by pyrophosphate and nicotinamide moieties of NAD+, by the peptide backbone residues 217–231 and by the side chains of Asn-218, Val-222, Tyr-277 and Met-281 (Figure 1). In order to analyse the contribution of these residues towards triclosan binding, we mutated key residues that interact with rings A and B of triclosan. Also, mutations in these residues have been found to confer resistance of E. coli FabI towards triclosan.
Figure 1. Schematic representation of the triclosan binding pocket of PfENR.

Triclosan and NAD+ are shown in black. The residues mutated in the present study (Ala-217, Asn-218, Met-281, and Phe-368) are shown in dark grey. The figure was generated in WebLab ViewerLite and rendered using POV-Ray.
The value of Ki′ for the inhibition of different mutants by triclosan with respect to NAD+ was estimated in the presence of 250 μM NADH and varying concentrations of NAD+. The curve was fitted to competitive, uncompetitive and noncompetitive inhibition using Equations 2, 3 and 4. The inhibition constants of triclosan with respect to the different mutants are shown in Table 3.
Table 3. Inhibition constants (Ki) for triclosan binding to wild-type and mutant PfENR.
| Enzyme | Inhibition constant (Ki) (nM) |
|---|---|
| Wild-type | 0.03 (±0.001) |
| A217V | 232.0 (±5.0) |
| A217G | 0.57 (±0.05) |
| N218A | 2.10 (±0.10) |
| N218D | 1.50 (±0.04) |
| M281A | 9.34 (±0.81) |
| M281T | 10.0 (±0.89) |
| F368A | 7.20 (±0.90) |
| F368I | 6.30 (±0.56) |
Residues interacting with ring B of triclosan
The Ki of triclosan for A217V [232 (±5) nM] is far greater than the wild-type PfENR [0.03 (±0.001) nM], demonstrating its low affinity for A217V (Figure 2A). The A217G PfENR mutant also showed lower affinity towards triclosan as compared with the wild-type ENR with a Ki of 0.57 (±0.05) nM (Figure 2B). N218A and N218D had a Ki of 2.1 (±0.1) and 1.5 (±0.04) nM respectively. M281A and M281T had a Ki of 9.34 (±0.81) and 10.0 (±0.89) nM respectively. A plot of Ki versus different concentrations of NAD+ is shown as Figures 2(A) and 2(B) for two representative examples, A217V and A217G. In all the cases the curve fits well to uncompetitive kinetics demonstrating the preference of triclosan for binding to PfENR as well as its mutant counterparts in the presence of NAD+.
Figure 2. Inhibition of A217V (A) and A217G (B) PfENR mutants by triclosan.

The plots of Ki′ versus NAD+ concentration fit best to uncompetitive inhibition (solid lines) with a Ki of 232 (±5) nM and 0.57 (±0.05) nM for A217V and A217G respectively. Best fits for competitive (broken lines) and noncompetitive (dotted lines) are also shown.
Residues interacting with ring A of triclosan
A Ki of 7.2 (±0.9) and 6.3 (±0.56) nM was obtained for F368A and F368I PfENR mutants respectively, while that for its wild-type counterpart was found to be 0.03 nM, implying the importance of this interaction for binding of triclosan to PfENR.
DISCUSSION
Triclosan inhibits ENR (one of the enzymes of the FAS pathway) from E. coli and P. falciparum very potently. However, its affinity for the enzyme from Mycobacterium tuberculosis is 10000-fold lower [22]. Also, another diphenyl ether, 2–2′ dihydroxydiphenyl ether, inhibits the enzyme from E. coli 1000-fold more potently as compared with the P. falciparum enzyme [1,7]. The crystal structures of ENR (FabI) from E. coli [19–21], M. tuberculosis [13] and P. falciparum [8] are now available. As yet, the reasons for such large differences in the binding affinities are not clear, but this stresses the need to understand the structural features that govern such differences in the affinities of the enzyme from different sources. Multiple sequence alignment of ENR from different organisms, comparing the PfENR with its counterparts from other organisms, is shown in Figure 3. In this study, we have taken a mutational approach to study the triclosan binding site of ENR from P. falciparum, which highlights the subtle differences in the binding site of PfENR as compared with ENRs from other sources. Thus, the knowledge of these differences would help in the design of potent inhibitors of ENR.
Figure 3. Multiple sequence alignment of ENR from P. falciparum (P. falci; accession number AF332608) with ENR from B. napus (accession number P80030), E. coli (accession number P29132), M. tuberculosis (M. tuber; accession number P46533).

The residues mutated in the present study are shown by *. Conserved residues are shown in white with a black background, negative residues are indicated by −, positive by +, aliphatic by l, aromatic by a, tiny by t, small by s, big by b, charged by c, polar by p, and hydrophobic by h (all below the alignment on the line labelled Consensus/100%). The figure was generated using CHROMA [24].
The role of a particular amino acid residue of a protein towards interaction with a ligand can be judged by site-directed mutagenesis. Hence, we performed a series of mutations spanning the triclosan-binding site of ENR. The expression of recombinant ENR–His-tagged fusion protein and the purification of the ENR was confirmed by using anti-His antibody. To exclude the possibility that the triclosan binding to mutant ENRs was decreased due to the introduction of gross structural changes, we first analysed the oligomerization status and conformation of wild-type and mutant ENR by gel-filtration and CD spectroscopy (supplementary online data). Thereafter, we performed a thorough analysis of the binding of PfENR mutants to triclosan.
We had demonstrated previously that triclosan acts as a potent inhibitor of PfENR [7]. As the concentration of NAD+ increases during the course of the reaction catalysed by ENR, and since NAD+ potentiates inhibition of ENR by triclosan, a modified protocol was followed. The reaction mixtures containing NAD+ were pre-incubated for 5 h at 4 °C in order to achieve a steady-state before starting the assay (also ensuring that the concentration of NAD+ does not change significantly during the course of assay) [17]. Triclosan demonstrated uncompetitive kinetics with an inhibition constant of 0.03 nM at saturating NAD+ concentration, demonstrating that triclosan binds to the enzyme-NAD+ complex with far greater affinity compared with the enzyme alone [7,25]. Indeed, an increase in the binding constant of triclosan towards PfENR in the presence of NAD+ has been observed in surface plasmon resonance experiments [25].
Single amino acid substitutions (G93V, M159T and F203L) in the sequence of E. coli FabI have been shown to confer resistance to triclosan [9]. The structure of the E. coli–NAD+–triclosan complex revealed that these residues make direct contacts with triclosan. The mutation of Gly-93 to Val specifically led to a 100-fold decrease in the MIC, which can be attributed to the steric contacts between the side chains of Val and triclosan [9,14,22]. This is also the site for G93A/S/C/V E. coli mutants resistant to diazaborine, again through the introduction of adverse steric contacts [15]. Although the M. smegmatis InhA mutant, S94A, leads to triclosan resistance [11], the same mutation does not affect the affinity of triclosan for InhA from M. tuberculosis [22]. Consistent with these observations, the mutation of Ala-217 to Val of PfENR led to a dramatic decrease in the affinity of triclosan, as apparent from the value of Ki (Table 3). The E. coli mutant G93V showed 9000-fold reduced affinity for triclosan as compared with the wild-type FabI [14]. As can be seen in Figure 1, Ala-217 comes close to the 2,4-dichlorophenoxy ring (ring B) of triclosan. Thus, it seems reasonable to conclude that even in the case of PfENR, the mutation of Ala-217 to Val leads to unacceptable steric contacts between the side chain of valine and triclosan, leading to reduced affinity of triclosan for the enzyme. Interestingly enough, the substitution of alanine by the smaller amino acid, glycine, also led to decreased affinity, but to a limited extent. There was a 19-fold decrease in the inhibition constant of A217G mutant with respect to the wild-type. The 2-chloro atom of ring B of triclosan is positioned close to the side chain of Ala-217 [6,17]. The decrease in the affinity of the A217G mutant for triclosan could be due to loss of contacts between triclosan and the side chain of alanine. In contrast, the E. coli enzyme has a glycine at the corresponding position, and its replacement by alanine reduced the affinity considerably for triclosan. Thus, despite having a nearly similar tertiary structure and the binding pocket, the malarial enzyme differs strikingly from its bacterial counterpart.
The mutation of Asn-218, Met-281 and Phe-368 also made the enzyme resistant towards triclosan, albeit to different extents. As illustrated in Figure 1, all three residues are located close to the inhibitor-binding site of the ternary complex of ENR-triclosan and NAD+. The ring B of triclosan was located in a pocket interacting with pyrophosphate and nicotinamide moieties of NAD+, by peptide backbone residues 217–231 and by side chains of Asn-218, Val-222, Tyr-277 and Met-281 [6]. The mutation of Asn-218 to aspartic acid led to a 50-fold decrease in the affinity of the malarial enzyme for triclosan. The mutant M281T had 333-fold reduced affinity for triclosan. This could be because of the loss of van der Waals contacts between the 4-chloro atom of ring B and hydrophobic side chain of Met-281 [6,8]. In order to rule out the possibility that the addition of the β-branched threonine could introduce indirect effects due to the perturbation of the local structure, the M281A mutant was made, that showed Ki similar to M281T.
As observed from the crystal structure of the ternary complex of P. falciparum with triclosan and NAD+, the ring A of triclosan makes van der Waals interactions with the side chain of Phe-368 [6]. Also, the 4-chloro atom of triclosan makes several van der Waals contacts with Phe-368 [6,17]. The mutation of Phe-368 to alanine and isoleucine led to 240- and 210-fold decrease in the affinity of enzyme for the inhibitor highlighting the importance of stacking and the van der Waals interactions between ring A of triclosan and Phe-368 of the enzyme.
Thus, to conclude, the strong affinity of PfENR for triclosan is compromised due to critical mutations at its active site. Because of the subtle but significant differences observed in these studies for the contribution of key residues to the binding affinity of PfENR for triclosan, in contrast to its bacterial counterparts, it should be possible to design better and specific analogues of triclosan as antimalarial agents that spare the bacterial enzyme. Also, several point mutations reduced inhibitory potency of triclosan without affecting the catalytic properties of the enzyme, indicating that resistant strains could arise under the pressure of the biocide triclosan. Our findings, therefore, hint at the need for the design and development of more effective inhibitors, and, more importantly, their usage in combination therapies to ward off the emergence of drug resistance strains [23].
Online data
Acknowledgments
This work was supported by a grant from the Department of Biotechnology, Government of India to N. S. and A. S.
References
- 1.Surolia N., Surolia A. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nature Med. 2001;7:167–173. doi: 10.1038/84612. [DOI] [PubMed] [Google Scholar]
- 2.Waller R. F., Keeling P. J., Donald R. G. K., Striepen B., Handman E., Lang-Unnasch N., Cowman A. F., Besra G. S., Roos D. S., McFadden G. I. Nuclear-encoded proteins target to the plastid in Toxoplasma gondii and Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 1998;95:12352–12357. doi: 10.1073/pnas.95.21.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rao S. P., Surolia A., Surolia N. Triclosan: A shot in the arm for antimalarial chemotherapy. Mol. Cell Biochem. 2003;253:55–63. doi: 10.1023/a:1026049217966. [DOI] [PubMed] [Google Scholar]
- 4.Gornicki P. Apicoplast fatty acid biosynthesis as a target for medical intervention in apicomplexan parasites. Int. J. Parasitol. 2003;33:885–896. doi: 10.1016/s0020-7519(03)00133-4. [DOI] [PubMed] [Google Scholar]
- 5.Muralidharan J., Suguna K., Surolia A., Surolia N. Exploring the interaction energies for the binding of hydroxydiphenyl ethers to enoyl-acyl carrier protein reductases. J. Biomol. Struct. Dyn. 2003;20:589–594. doi: 10.1080/07391102.2003.10506875. [DOI] [PubMed] [Google Scholar]
- 6.Suguna K., Surolia A., Surolia N. Structural basis for triclosan and NAD binding to enoyl-ACP reductase of Plasmodium falciparum. Biochem. Biophys. Res. Commun. 2001;283:224–228. doi: 10.1006/bbrc.2001.4747. [DOI] [PubMed] [Google Scholar]
- 7.Kapoor M., Dar M. J., Surolia A., Surolia N. Kinetic determinants of the interaction of enoyl-ACP reductase from Plasmodium falciparum with its substrates and inhibitors. Biochem. Biophys. Res. Comm. 2001;289:832–837. doi: 10.1006/bbrc.2001.6061. [DOI] [PubMed] [Google Scholar]
- 8.Perozzo R., Kuo M., Sidhu A. S., Valiyaveettil J. T., Bittman R., Jacobs W. R., Jr, Fidock D. A., Sacchettini J. C. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J. Biol. Chem. 2002;277:13106–13114. doi: 10.1074/jbc.M112000200. [DOI] [PubMed] [Google Scholar]
- 9.McMurray L. M., Oethinger M., Levy S. B. Triclosan targets lipid synthesis. Nature (London) 1998;394:531–532. doi: 10.1038/28970. [DOI] [PubMed] [Google Scholar]
- 10.Fan F., Yan K., Wallis N. G., Reed S., Moore T. D., Rittenhouse S. F., DeWolf W. E., Jr, Huang J., McDevitt D., Miller W. H., et al. Defining and combating the mechanisms of triclosan resistance in clinical isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 2002;46:3343–3347. doi: 10.1128/AAC.46.11.3343-3347.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McMurry L. M., McDermott P. F., Levy S. B. Genetic evidence that InhA of Mycobacterium smegmatis is a target for triclosan. Antimicrob. Agents Chemother. 1999;43:711–713. doi: 10.1128/aac.43.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergler H., Wallner P., Ebeling A., Leitinger B., Fuchsbichler S., Aschauer H., Kollenz G., Högenauer G., Turnowsky F. Protein EnvM is the NADH-dependent enoyl-ACP reductase (FabI) of Escherichia coli. J. Biol. Chem. 1994;269:5493–5496. [PubMed] [Google Scholar]
- 13.Rafferty J. B., Simon J. W., Baldock C., Artymiuk P. J., Baker P. J., Stuitje A. R., Slabas A. R., Rice D. W. Common themes in redox chemistry emerge from the X-ray structure of oilseed rape (Brassica napus) enoyl acyl carrier protein reductase. Structure. 1995;3:927–938. doi: 10.1016/S0969-2126(01)00227-1. [DOI] [PubMed] [Google Scholar]
- 14.Sivaraman S., Zwahlen J., Bell A. F., Hedstrom L., Tonge P. J. Structure–activity studies of the inhibition of FabI, the enoyl reductase from Escherichia coli, by triclosan: kinetic analysis of mutant FabIs. Biochemistry. 2003;42:4406–4413. doi: 10.1021/bi0300229. [DOI] [PubMed] [Google Scholar]
- 15.Baldock C., Rafferty J. B., Stuitje A. R., Slabas A. R., Rice D. W. The X-ray structure of Escherichia coli enoyl reductase with bound NAD+ at 2.1 Å resolution. J. Mol. Biol. 1998;284:1529–1546. doi: 10.1006/jmbi.1998.2271. [DOI] [PubMed] [Google Scholar]
- 16.Dessen A., Quemard A., Blanchard J. S., Jacobs W. R., Jr, Sacchettini J. C. Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science (Washington, D.C.) 1995;267:1638–1641. doi: 10.1126/science.7886450. [DOI] [PubMed] [Google Scholar]
- 17.Ward W. H. J., Holdgate G. A., Rowsell S., Mclean E. G., Pauptit R. A., Clayton E., Nichols W. W., Colls J. G., Minshull C. A., Jude D. A., et al. Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochemistry. 1999;38:12514–12525. doi: 10.1021/bi9907779. [DOI] [PubMed] [Google Scholar]
- 18.Heath R. J., Li J., Roland G. E., Rock C. O. Inhibition of the Staphylococcus aureus NADPH-dependent enoyl-acyl carrier protein reductase by triclosan and hexachlorophene. J. Biol. Chem. 2000;275:4654–4659. doi: 10.1074/jbc.275.7.4654. [DOI] [PubMed] [Google Scholar]
- 19.Stewart M. J., Parikh S., Xiao G., Tonge P. J., Kisker C. Structural basis and mechanism of enoyl reductase inhibition by triclosan. J. Mol. Biol. 1999;23:859–865. doi: 10.1006/jmbi.1999.2907. [DOI] [PubMed] [Google Scholar]
- 20.Roujeinikova A., Levy C. W., Rowsell S., Sedelnikova S., Baker P. J., Minshull C. A., Mistry A., Colls J. G., Camble R., Stuitje A. R., et al. Crystallographic analysis of triclosan bound to enoyl reductase. J. Mol. Biol. 1999;294:527–535. doi: 10.1006/jmbi.1999.3240. [DOI] [PubMed] [Google Scholar]
- 21.Levy C. W., Roujeinikova A., Sedelnikova S., Baker P. J., Stuitje A. R., Slabas A. R., Rice D. W., Rafferty J. B. Molecular basis of triclosan activity. Nature (London) 1999;398:383–384. doi: 10.1038/18803. [DOI] [PubMed] [Google Scholar]
- 22.Parikh S. L., Xiao G., Tonge P. J. Inhibition of InhA, the enoyl reductase from Mycobacterium tuberculosis, by triclosan and isoniazid. Biochemistry. 2000;39:7645–7650. doi: 10.1021/bi0008940. [DOI] [PubMed] [Google Scholar]
- 23.Ndyomugyenyi R., Magnussen P., Clarke S. The efficacy of chloroquine, sulfadoxine-pyrimethamine and a combination of both for the treatment of uncomplicated Plasmodium falciparum malaria in an area of low transmission in western Uganda. Trop. Med. Int. Health. 2004;9:47–52. doi: 10.1046/j.1365-3156.2003.01167.x. [DOI] [PubMed] [Google Scholar]
- 24.Goodstadt L., Ponting C. P. CHROMA: consensus-based colouring of multiple alignments for publication. Bioinformatics. 2001;17:845–846. doi: 10.1093/bioinformatics/17.9.845. [DOI] [PubMed] [Google Scholar]
- 25.Kapoor M., Gopalakrishnapai J., Surolia N., Surolia A. Kinetic and structural analysis of the increased affinity of enoyl-ACP reductase for triclosan in the presence of NAD+ Biochem. J. (2004) 2004;381:725–733. doi: 10.1042/BJ20040228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.