

Abstract
Intrinsic tenase consists of activated Factors IX (IXa) and VIII (VIIIa) assembled on a negatively charged phospholipid surface. In vivo, this surface is mainly provided by activated platelets. In vitro, phosphatidylcholine/phosphatidylserine vesicles are often used to mimic natural pro-coagulant membranes. In the present study, we developed a quantitative mathematical model of Factor X activation by intrinsic tenase. We considered two situations, when complex assembly occurs on either the membrane of phospholipid vesicles or the surface of activated platelets. On the basis of existing experimental evidence, the following mechanism for the complex assembly on activated platelets was suggested: (i) Factors IXa, VIIIa and X bind to their specific platelet receptors; (ii) bound factors form complexes on the membrane: platelet-bound Factor VIIIa provides a high-affinity site for Factor X and platelet-bound Factor IXa provides a high-affinity site for Factor VIIIa; (iii) the enzyme–cofactor–substrate complex is assembled. This mechanism allowed the explanation of co-operative effects in the binding of Factors IXa, VIIIa and X to platelets. The model was reduced to obtain a single equation for the Factor X activation rate as a function of concentrations of Factors IXa, VIIIa, X and phospholipids (or platelets). The equation had a Michaelis–Menten form, where apparent Vmax and Km were functions of the factors’ concentrations and the internal kinetic constants of the system. The equation obtained can be used in both experimental studies of intrinsic tenase and mathematical modelling of the coagulation cascade. The approach of the present study can be applied to research of other membrane-dependent enzymic reactions.
Keywords: blood coagulation, Factor VIII, Factor IX, Factor X, mathematical modelling, platelet
Abbreviations: fVa, Factor Va; fVIII(a), Factor VIII(a); fIX(a), Factor IX(a); fX(a), Factor X(a); PC, phosphatidylcholine; PS, phosphatidylserine; PS/PC, vesicles composed of PS and PC
INTRODUCTION
fX (Factor X) activation during blood coagulation occurs via two pathways: the extrinsic tenase complex formed by Factor VIIa and tissue factor, and the intrinsic tenase complex composed of fIXa (Factor IXa) and fVIIIa (Factor VIIIa) [1]. There is a consensus that physiological coagulation is initiated by extrinsic tenase, while its propagation is provided by positive feedback via intrinsic tenase [1–3]. The deficiencies of the components of intrinsic tenase, fVIIIa and fIXa, lead to severe bleeding disorders, haemophilias A and B respectively [3,4], which emphasizes the importance of this complex.
fIXa is an Mr 44000 two-chain vitamin K-dependent serine protease capable of hydrolysing the Arg194-Ile195 peptide bond in the fX molecule, which leads to its activation [5]. Although this reaction can proceed slowly in solution, it is significantly accelerated in the presence of negatively charged phospholipid surfaces [6]. In vivo, these surfaces are mainly provided by activated platelets and plasma lipoproteins [7]. Erythrocytes have been reported to contribute substantially to providing pro-coagulant surface in blood [8], but this fact is not generally accepted [9,10]. For in vitro studies of fX activation by intrinsic tenase, synthetic phospholipid vesicles [usually composed of PS (phosphatidylserine) and PC (phosphatidylcholine) at a 25:75 molar ratio] are often used to substitute for physiological surfaces.
The rate of the reaction is increased further by the presence of fVIIIa, a non-enzymic cofactor protein. After fIXa, fVIIIa and fX bind to the platelet membrane or to the surface of phospholipid vesicle [12], fIXa forms a complex with fVIIIa on the phospholipid surface (in solution, the complex is also formed, but with dramatically lower affinity [11]), which can activate fX five orders of magnitude more rapidly than fIXa in solution [6]. In turn, fXa forms a homologous complex with fVa, prothrombinase complex, which catalyses the conversion of prothrombin into thrombin [12].
During the last decade, a number of studies reported kinetic data on fX activation by intrinsic tenase in the presence of either phospholipid vesicles or platelets. The apparent kinetic constants of this reaction [6,11,13], the apparent equilibrium constants of fIXa and fVIIIa dissociation [11,14–16], and the parameters of the binding of fIXa, fVIIIa and fX to phospholipids [17,18] or platelets [19–22] have been determined. However, although a substantial amount of empirical information has been obtained, the mechanisms of the tenase complex assembly and function remain poorly understood. For prothrombinase, the first model was suggested two decades ago [23,24]. In contrast, the only theoretical study on intrinsic tenase [25] investigated the external regulation of the tenase depending on pathways of activation and inhibition of fIXa, fVIIIa and fXa, but did not consider interactions between coagulation factors and phospholipid surfaces. Two simple models considering this interaction have been recently proposed as parts of larger models of the blood coagulation system [26,27]. However, these studies do not focus on intrinsic tenase specifically. In the present study, we attempted to combine accumulated experimental data and existing theoretical concepts, and to develop a detailed mathematical model describing quantitatively the kinetics of fX activation by the fIXa–fVIIIa complex on activated platelets or phospholipid vesicles. For activated platelets, we propose a reaction mechanism, according to which fX and fVIIIa bind from plasma to their specific low-affinity high-capacity receptors on the platelet surface to be transferred to high-affinity receptors formed by fVIIIa and fIXa respectively.
Portions of this work were presented at the XIX Congress of the International Society on Thrombosis and Haemostasis, 12–18 July 2003, Birmingham, U.K. [27a].
MATERIALS AND METHODS
Concepts of the model
The model is based on the mechanism depicted in Figure 2. fIXa, fVIIIa and fX bind to the membrane and subsequently interact by means of diffusion. We disregarded the possibility of direct interaction between solution-phase proteins and surface-bound proteins on the basis of previous reports [23,28,29]. We did not consider the formation of the tenase complex in solution because of its high dissociation constant in the absence of either phospholipids or platelets [11]. fX activation by fIXa alone was also ignored, being slow in comparison with activation of fX by the fIXa–fVIIIa complex [13].
Figure 2. Mechanisms of fX activation by intrinsic tenase used in the model.

(A) Assembly of the intrinsic tenase complex and fX activation on the surface of phospholipid vesicles. fIXa, fVIIIa and fX bind to the membrane from solution. Membrane-bound factors form the fIXa–fVIIIa and fVIIIa–fX complexes (reactions 1 and 3), which subsequently bind fX (reaction 2) and fIXa (reaction 4) respectively to form the final fIXa–fVIIIa–fX complex. (B) Assembly of the intrinsic tenase complex and fX activation on the platelet membrane. fIXa, fVIIIa and fX bind to the membrane from solution; in contrast with (A), each factor binds to its specific site (to illustrate the binding specificity, platelet binding sites for fIXa, fVIIIa and fX are represented as shapes complementary to triangles, circles and squares respectively). fVIIIa and fIXa form intrinsic tenase localized on fIXa receptor, which subsequently binds fX. Platelet-bound fVIIIa can also form a receptor for fX [21], which is followed by the binding of fIXa [22] and the assembly of the final enzyme–cofactor–substrate complex.
Two approaches have been suggested to describe surface-mediated reactions. Nesheim et al. [23] proposed a concept of ‘interface shell’ between membrane and solution, where local concentrations of substrate and enzyme are high, and the local increase in concentration accounts for acceleration of the catalysis. The other approach considers two-dimensional reactions on the surface [28]. These approaches are mathematically equivalent, so we do not distinguish between them. It is necessary to point out that the approach used does not consider that at high concentrations of phospholipid vesicles, the bound proteins can segregate to different vesicles.
Area of applicability of Model 1
Model 1 describes the activation of fX by the fIXa–fVIIIa complex assembled on the surface of synthetic PS/PC (phospholipid vesicles composed of PS and PC) at a molar ratio of 25:75. This model is designed to describe the reaction under the following conditions: human proteins, physiological conditions (37 °C, pH 7.2–7.4, 2 mM Ca2+, 150 mM NaCl), fIXa (fVIIIa) concentration within picomolar to low nanomolar range, fX concentration within high nanomolar to low micromolar range; phospholipid at micromolar or low millimolar concentration.
Area of applicability of Model 2
Model 2 describes activation of fX by the fIXa–fVIIIa complex assembled on the membrane of activated platelets. This model is designed to describe the reaction under the following conditions: human proteins and platelets, physiological conditions (37 °C, pH 7.2–7.4, 2 mM Ca2+, 150 mM NaCl), fIXa (fVIIIa) concentration is in picomolar to low nanomolar range, fX concentration is in high nanomolar to low micromolar range; platelet concentration is below approx. 109 ml−1.
Assumptions of the model
First, we assumed all platelets (phospholipid vesicles) to be identical and did not consider their statistical size/composition variability. Secondly, for the majority of coagulation factors, the rate constants for association/dissociation with membranes are unknown, and only equilibrium dissociation constants have been reported (Tables 1 and 2). We used the rapid equilibrium assumption, i.e. we assume that the binding of factors to both synthetic and platelet membranes is rapid compared with the rate of catalysis itself [23,24], based on high association rates reported for fVIIIa, fXa and fVa [18,29,30]. Importantly, this assumption may be invalid for small phospholipid vesicles, when the catalytic efficiency of such rapid enzymes as tenase and prothrombinase is likely to be determined by the rate of substrate delivery [31,32]. Thirdly, formation of complexes by membrane-bound proteins is assumed to be rapid. Therefore we used a steady-state approach to determine the concentrations of these complexes (eqns A9–A11 and A48–A50). Although the kinetic constants of these reactions for the components of intrinsic tenase have not been reported, the high rate of the fXa–fVa complex formation [27] suggests high efficiency of surface diffusion for homologous proteins. Fourthly (for phospholipid vesicles only), the phospholipid membrane, unlike the platelet surface, is not specific, and the proteins compete with each other for binding. Taking into consideration typical values of the binding constants and stoichiometries (Table 1) and the typical concentrations (fIXa, approx. 1 nM; fVIIIa, approx. 1 nM; fX approx. 100–1000 nM; and PS/PC, approx. 1–1000 μM), we disregarded the fact that fIXa and fVIIIa occupy binding sites on the phospholipid surface. We assumed that fX is the only factor that can considerably occupy the membrane and displace other factors. Fifthly (for platelets only), platelets in the model have three types of specific binding sites, for fIXa, fVIIIa and fX respectively (Table 2). The pro-coagulant activity of activated platelets depends on the type of activator [21,33]. In the present study, we considered only thrombin-activated platelets. Because the number of binding sites that appears after activation by thrombin does not depend on the thrombin concentration within a wide range [19], we assumed that the number of sites of each type is constant (Table 2) and did not consider competition between the factors for these sites [19–21]. This assumption is not valid for an in vivo situation, since fX competes with prothrombin [20] for the binding sites, and fIXa and fVIIIa share part of their sites with fIX and fVIII respectively [19,21]. Therefore, to adjust our model to plasma conditions, some corrections described in the Discussion should be introduced.
Table 1. Parameters of the model describing intrinsic tenase on phospholipids (Model 1).
| Experimentally determined parameter | Experimental value | Experimental conditionsa | Reference | Relationship between model and experimental parameters | Value used in the model |
|---|---|---|---|---|---|
| kcat (app) | 1740 min−1 | P=25 μM PS/PC | [13] | Eqn A46 | kcatlocal=720 min−1 |
| 136 min−1 | P=25 μM PS/PC | [11] | |||
| 198b min−1 | P=5 μM PS/PC | [6] | |||
| 439b min−1 | P=10 μM PS/PC | [6] | |||
| 436b min−1 | P=25 μM PS/PC | [6] | |||
| Km (app) | 190 nM | P=25 μM PS/PC | [13] | Eqn A39, eqn A47 | KdVIIIa-X local=Kmlocal/k=9.2×10−4 |
| 23c nM | P=25 μM PS/PC | [11] | |||
| 24b nM | P=5 μM PS/PC | [6] | |||
| 45b nM | P=10 μM PS/PC | [6] | |||
| 83b nM | P=25 μM PS/PC | [6] | |||
| Kd (app) | 1.4 nM | P=25 μM PS/PC | [16] | Eqn A42 | KdIXa-VIIIa local/k=7×10−6 |
| 2.3 nM | P=25 μM PS/PC | [11] | |||
| 0.07 nM | P=10 μM PS/PC | [15] | |||
| KdX | 580 nM | – | [43] | – | 40 nM |
| 150 nM | [17] | ||||
| 40b nM | [44] | ||||
| SX | 2×42d | – | [17] | – | 84 |
| KdIXa | 930 nM | – | [43] | – | 930 nM |
| 1000b nM | [45] | ||||
| SIXa | 2×42d,e | – | [17] | – | 84 |
| KdVIIIa | 0.65 nMf | – | [18] | – | 0.65 nM |
| SVIIIa | 89g | – | [46] | – | 360 |
| 170h | [46] | ||||
| 360i | [47] |
a Only the data necessary to obtain the model parameters from the corresponding experimentally determined parameters are presented in this column.
b The value was obtained for bovine proteins.
c The values used in the model are underlined.
d For the PS/1-palmitoyl-2-oleoyl-3-sn-PC (20/80) vesicles.
e We assume that SIXa≈SX on the basis of [48].
f For the PS/PC/phosphatidylethanolamine (4/76/20) vesicles, at pH 6.0.
g For fVIII, PS/PC/phosphatidylethanolamine (30/67.5/2.5) vesicles.
h For fVIII, PS/PC/phosphatidylethanolamine (20/77.5/2.5) vesicles.
i For fVIII, 1,2-dioleoyl-sn-glycero-PS/1,2-dioleoyl-sn-glycero-PC (25:75) vesicles.
Table 2. Parameters of the model describing intrinsic tenase on activated platelets (Model 2).
| Experimentally determined parameter | Experimental value | Experimental conditionsa | Reference | Relationship between model and experimental parameters | Value used in the model |
|---|---|---|---|---|---|
| Vmax | 12.4 nM·min−1 | [IXa]=10 pM, [VIIIa]=5 units·ml−1 n=5×107 platelets·ml−1 | [13] | Eqn A71 | kcatlocal=6350 min−1 |
| 2.5 nM·min−1 | [IXa]=1 nM, [VIIIa]=5 units ml−1n=5×107 platelets·ml−1 | [41] | Eqn A77 | ||
| Km (app) | 160 nM | [IXa]=10 pM, [VIIIa]=5 units·ml−1n=5×107 platelets·ml−1 | [13] | Eqn A71 | Kmlocal/k=1216 molecules/platelet |
| 22.6b nM | [IXa]=1 nM, [VIIIa]=5 units ml−1n=5×107 platelets·ml−1 | [41] | Eqn A76 | ||
| KdIX (app) | 0.56 nM | fVIIIa and fX were in saturation | [19] | Eqn A68 | KdIXa-VIIIa local/k=278 molecules/platelet |
| KdX | 320 nM | – | [20] | – | 320 nM |
| nX | 16000 sites/platelet | – | [20] | – | 16000 sites/platelet |
| KdIXa | 2.57 nM | – | [19] | – | 2.57 nM |
| nIXa | 250 sites/platelet | – | [19] | – | 250 sites/platelet |
| KdVIIIa | 1.5 nM | – | [21] | – | 1.5 nM |
| 1.7 nM | [49] | ||||
| nVIIIa | 750 sites/platelet | – | [21] | – | 750 sites/platelet |
| KdVIIIa-X (app) | 30 nM | – | [21] | Eqn A66 | KdVIIIa-X local/k=1655 molecules/platelet |
a Only the data necessary to obtain the model parameters from the corresponding experimentally determined parameters are presented in this column.
b The values used in the model are underlined.
Fitting of generated curves
The binding curves obtained theoretically were fitted to the one-site (see Figure 5A–5C) or two-site (see Figure 5B, upper curve) standard binding model using the direct non-linear least-squares method implemented in Microcal Origin 6.0 (Microcal Software). The apparent parameters of Michaelis kinetics Vmax and Km, shown in Figures 2 and 3, were obtained with the same method.
Figure 5. Specific binding of each component of the fX-activating complex to platelets depends on the presence of two other components (theoretical curves).

(A) The binding of fIXa (at indicated increasing concentrations) to activated platelets (2×108 per ml) in the presence (○) or in the absence (▪) of 5 nM fVIIIa and 1500 nM fX. The solid line shows the approximation of the curve (○) with the one-site binding model. (B) The binding of fVIIIa (at the indicated increasing concentrations) to activated platelets (2×108 per ml) in the presence (○) or in the absence (▪) of 45 nM fIXa and 1500 nM fX. The broken line represents the approximation of the curve (○) with the one-site binding model. The dotted line shows the approximation of the same curve with the two-site binding model. The inset shows the low-concentration section of the plot. (C) The binding of fX (at indicated increasing concentrations) to activated platelets (2×108 per ml) in the presence (○) or in the absence (▪) of 10 nM fIXa and 10 nM fVIIIa. The solid line shows the approximation of the curve (○) with the two-site binding model. The calculations of the binding of fIXa, fVIIIa and fX were carried out using eqns A63, A62 and A61 respectively. The constants are listed in Table 2. The binding of fIXa to the shared fIXa/fIX sites is not considered in these calculations. The results of approximations are given in Table 3.
Figure 3. Kinetics of intrinsic tenase on phospholipids.

(A) Dependence of the initial rate of fX activation on concentration of PS/PC (25:75) vesicles. Theoretical dependence (solid curve) is obtained using Model 1 (eqns A34, A24, A27, A28 and A29). The values of the constants are listed in Table 1. The conditions of the experiment were: 200 nM fX, 1 nM fIXa, 0.06 nM fVIIIa and increasing concentrations of phospholipids. Data (▪) taken from [18]. (B) Dependence of the apparent maximal velocity Vmax (▪) and the apparent Michaelis constant Km (○) of fX activation by intrinsic tenase on concentration of PS/PC (25:75) vesicles. The calculations were carried out using Model 1 (eqns A34, A24, A27, A28 and A29). The values of the constants are listed in Table 1. The conditions of the simulation were: 1 nM fIXa, 0.06 nM fVIIIa and increasing concentrations of phospholipids. To find Vmax and Km, the rate of fXa production is calculated at 1, 5, 10, 20, 50 and 100 nM of fX, and the dependence of activation rate on fX concentration is approximated with the non-linear least-squares method (see the Materials and methods section). The solid lines are drawn with the help of B-spline interpolation.
RESULTS
The mechanism of assembly of the fIXa–fVIIIa–fX complex on PS/PC vesicles
An enzyme–cofactor–substrate ternary complex can be assembled via various pathways. If we assume that coagulation factors have to bind to the membrane before complex assembly (see the Materials and methods section), then the range of possible two-dimensional reactions leading to assembly of the fIXa–fVIIIa–fX complex is limited to those shown in Figure 1. All possible combinations of binary complexes would give seven unique models of a ternary complex assembly [34]. The data of multiple studies strongly suggest that at least one binary complex, composed of fIXa and fVIIIa, actually exists [14,35]. This narrows the number of possible assembly models down to four: (i) fIXa–fVIIIa is the only possible binary complex; (ii) both the fIXa–fVIIIa and fVIIIa–fX complexes exist; (iii) both the fIXa–fVIIIa and fIXa–fX complexes exist; (iv) all three binary complexes shown in Figure 1 exist. The fIXa–fVIIIa interaction does not seem to be affected by the addition of fX [14], which is inconsistent with model ‘a’ (see the Appendix, eqn A40). The existing evidence suggests that both the fIXa–fX [6,13,28] and the fVIIIa–fX [22] binary complexes can be formed during fX activation. However, we have chosen model ‘b’ on the basis of the following evidence: (i) The estimated value of the local dissociation constant KIXa-X locald for fIXa and fX is approx. 0.05 (see the Appendix). This value exceeds the estimated value of the dissociation constant of fIXa and fVIIIa by four orders of magnitude and also exceeds the value of the estimated local Michaelis constant by two orders of magnitude (Table 1). This suggests that formation of the fIXa–fX complex in the presence of fVIII would be unfavourable and unlikely to contribute significantly to fX activation. (ii) During assembly of the homologous prothrombinase complex, the cofactor–substrate fVa–fII (Factor II) binary complex formation is the preferred pathway of prothrombin activation both in the absence [34] and in the presence [36] of phospholipid membrane. (iii) Recent data suggest that formation of the fVIIIa–fX complex is important during fX activation by intrinsic tenase on the surface of activated platelets [22]. Finally, we tested the accuracy of mechanism ‘b’ a posteriori. The model was modified to include formation of the fIXa–fX complex (eqns A35–A36). Substitution of eqns A35–A36 for eqns A27–A28 (KIXa-X locald/k=0.05) did not change the rates of fXa formation (the difference was less than 0.1% under various conditions; results not shown).
Figure 1. Possible pathways of the enzyme–cofactor–substrate complex formation.

The ternary complex of fIXa, fVIIIa, and fX can be assembled on the membrane via several possible pathways. There can be three possible binary complexes: (1) the fIXa–fVIIIa complex, (2) the fIXa–fX complex and (3) the fVIIIa–fX complex. Depending on which complexes do exist, their combination gives seven possible alternative models of the final complex assembly [34]: a single binary complex (1, 2 or 3) is required for assembly of the ternary complex; two complexes contribute to the ternary complex formation (1 and 2, 1 and 3, or 2 and 3); contribution of all three complexes (1, 2 and 3) is significant.
Kinetics of intrinsic tenase on phospholipid vesicles
The theoretical concepts of the model are depicted in Figure 2(A). An important feature of this system is the non-specificity of the binding sites on PS/PC membranes. This implies that a protein in excess can displace other proteins, thus decreasing the efficiency of the reaction (in particular, it can lead to inhibition of the reaction by excess substrate or excess enzyme [23]). The process of the model development is detailed in the Appendix. It gives eqn A34 (see Appendix) for the initial rate of the fXa production by the fIXa–fVIIIa complex assembled on phospholipids (Model 1), where [XM], A, B and C are given by eqns A24, A27, A28 and A29 respectively (see Appendix). The local constants are derived from the reported experimentally determined apparent constants with the help of conversion eqns A39, A42, A46 and A47 (see Table 1). Model 1 predicts a Michaelis-like dependence of fX activation rate on fX concentration, the apparent Michaelis constant being linearly dependent on the phospholipid concentration (see eqn A45 and Figure 3B). Parameters A and B determine the distribution of fVIIIa and fIXa respectively, between the binding sites and volume. For example, if we multiply eqn A27 by (P-[XM]·SX)/KVIIIad·SVIIIa and by the concentration of free fVIIIa, the first term on the right side of the equation would be fVIIIa bound to phospholipid membrane, the second term would be free fVIIIa in solution, and the third term would be fVIIIa bound to fX. Parameter C indicates assembly of the final fIXa–fVIIIa–fX complex with regard to the fIXa–fVIIIa complex: 1/C is the fraction of the fIXa–fVIIIa complex, which is not bound to fX.
Kinetics of the intrinsic tenase on synthetic phospholipid vesicles obtained by simulation is shown in Figure 3. Initial rate of fX activation rapidly reaches saturation (Figure 3A), followed by a slow decrease due to an excess of phospholipid surface. At the saturation point, all factors are quantitatively bound, and further increase of membrane concentration decreases the local factor concentration and the frequency of molecule collisions. Mathematically, this effect is seen as a linear increase of the apparent Michaelis constant Km (app) (see eqn A45) shown in Figure 3(B) and of the Kd (app) of intrinsic tenase (see eqn A42). These effects were shown experimentally in early studies [6,35] using bovine proteins (see Table 1). Although a direct quantitative comparison between these data and our simulations cannot be drawn, the qualitative dependence of kcat (app) and Km (app) on phospholipid concentration [6] (see Table 1) correlates well with the dependences for Vmax and Km (app) respectively, as shown in Figure 3(B).
The mechanism of assembly of the fIXa–fVIIIa–fX complex on activated platelets
The mechanisms of assembly of intrinsic tenase on activated platelets are under intensive investigation at present [37]. The current paradigm for intrinsic tenase assembly on the membrane of activated platelets is the so-called ‘three-receptor model’ suggested by Walsh and colleagues [21,22,38]. According to this model, fIXa, fVIIIa and fX possess individual, highly specific, but interacting, receptors on the surface of activated platelets. These factors bind independently to their receptors from plasma and form complexes on the membrane by means of two-dimensional diffusion [28,29,39].
It was shown that in the presence of fVIIIa and fX, the Kd for fIXa binding to platelets was decreased from 2.57 nM to 0.56 nM, while the number of binding sites for fIXa remained constant [19]. A similar phenomenon was observed for fVIIIa: in the presence of fIXa and fX, the Kd (app) for fVIIIa binding to platelets was decreased from 1.5 nM to 0.8 nM [21]. In contrast with fIXa, the number of binding sites for fVIIIa also changed: from approx. 750 to approx. 1200 sites/platelet in the absence of fIX [21], and from approx. 750 to approx. 1000 sites/platelet, when approx. 300 of approx. 515 sites/platelet for fIXa were blocked by an excess of fIX [22]. A recent study has shown that in the presence of excess prothrombin (which displaced fX from approx. 16000 shared sites) and fIX (which displaced fIXa from approx. 300 out of approx. 515 shared sites), fVIIIa and fX both bound to approx. 1000 sites with Kd values of 5 nM and 0.8 nM respectively [22]. The presence of fIXa and fVIIIa in the absence of fIX induced the appearance of approx. 1200 and not approx. 1000 highly specific binding sites for fX.
We used these data to develop a comprehensive model of interaction of the factors on the platelet membrane. We took into consideration that the number of binding sites for fVIIIa in the presence of fIXa and fX increases by approx. 450 sites/platelet, which is close to the total number of fIXa-binding sites (515) per platelet. Moreover, if approx. 300 sites shared by fIX and fIXa are blocked by excess fIX, the number of fVIIIa sites increases only by approx. 250 sites [22], which is close to the number of sites left for fIXa. We speculate that the enzyme–substrate fIXa–fVIIIa–fX complex is assembled on the receptor for fIXa; i.e. platelet-bound fVIIIa and the fX–fVIIIa complex are ‘passed on’ to the site consisting of fIXa bound to its receptor (Figure 2B). With this assumption, the increase in the number of fVIIIa-binding sites in the presence of fIXa and fX can be explained by the fact that the sites for fIXa are added to the original number of fVIIIa-binding sites. In turn, the mutual increase in affinity may be related to lower dissociation of the factors in the complex from the surface of platelets. Our model also includes another hypothesis [22] that fX bound to its receptor on the platelet surface is subsequently presented to the binding site consisting of fVIIIa bound to its receptor, forming a 1:1 complex. Considerations similar to those for the PS/PC vesicles allow us to exclude other models of the fIXa–fVIIIa–fX complex assembly. The final scheme of intrinsic tenase assembly on the surface of activated platelets is shown in Figure 2(B).
Kinetics of intrinsic tenase on activated platelets
The process of model development is detailed in the Appendix. This process gives eqn A60 (see Appendix) for the rate of fX activation by intrinsic tenase assembled on platelets (Model 2), where [XM], [VIIIaM] and [IXaM] are given by eqns A57, A58 and A59 respectively (see Appendix). To avoid complex notation, the symbols denoting the constants and the concentrations are the same in the ‘platelet’ and ‘phospholipid’ sections of this paper, although the values and the meaning of these constants are different (Tables 1 and 2). Definitions of these constants and concentrations are given in the Appendix. Additional eqns A61, A62 and A63 can be used to calculate the total concentrations of bound fX, fVIIIa and fIXa respectively, and thus to obtain the apparent dissociation constants and the numbers of binding sites. The model predicts that at near-physiological conditions, the rate of fX activation depends linearly on platelet concentration (Figure 4), in agreement with the earlier study [33]. It should be noted that the reported values of catalytic efficiency of intrinsic tenase on platelets are somewhat controversial. As follows from Table 2, two studies from the same laboratory determined that Vmax at 10 pM of fIXa [13] is 5-fold higher than Vmax at 1 nM of fIXa [41]. This gives a range of uncertainty in the determination of klocalcat of about two orders of magnitude. For our model, in order to fit the experimental curve in Figure 4(A), we assumed klocalcat=6350 min−1, which is within this range.
Figure 4. Kinetics of intrinsic tenase on activated platelets.

(A) Dependence of the initial rate of fX activation on concentration of thrombin-activated platelets. Theoretical dependence (solid curve) is obtained using Model 2 (eqns A60, A57, A58 and A59). The values of the constants are listed in Table 2. The conditions of the experiment were 500 nM fX, 50 nM fIXa, 0.1 nM fVIIIa and increasing concentrations of platelets. From Rosing, J., van Rijn, J. L., Bevers, E. M., van Dieijen, G., Comfurius, P. and Zwaal, R. F. (1985) The role of activated human platelets in prothrombin and factor X activation. Blood 65, 319–332. Copyright American Society of Haematology, used with permission. (B) Dependence of the apparent Vmax (▪) and the apparent Km (○) of fX activation by the intrinsic tenase on concentration of thrombin-activated platelets. The calculations were carried out using Model 2 (eqns A60, A57, A58 and A59). The values of the constants are listed in Table 2. The conditions of the simulation were 0.1 nM fIXa, 5 nM fVIIIa and increasing concentrations of platelets. To find Vmax and Km, the rate of fXa production is calculated at 5, 10, 20, 50 and 100 nM of fX, and dependence of the activation rate on fX concentration is approximated with the non-linear least-squares method (see the Materials and methods section). The solid lines are drawn with the help of B-spline interpolation.
The dependence of apparent parameters of Michaelis kinetics on concentration of activated platelets is shown in Figure 4(B). Unlike in the presence of phospholipids, the apparent Michaelis constant does not change within this range of platelet concentrations and Vmax increases linearly due to a relatively low binding capacity of platelets. Therefore we used a simplified form of eqn A4, omitting the concentration of binding sites in the denominator to derive further eqns A57, A58 and A59. This assumption is valid for normal plasma concentrations of platelets (see the Materials and methods section). However, in aggregates, where platelet concentration is higher by orders of magnitude, our model in its present form could be incorrect because of the possible effect of inhibition by excess surface.
In Figure 5, we present binding curves for each component of the fX-activating complex and activated platelets in the absence and in the presence of two other components. The curves are obtained using eqns A61, A62 and A63 and are fitted to the one- or two-site binding model (see the Materials and methods section). As follows from Table 3, the presence of fVIIIa and fX causes a 5-fold increase in the fIXa apparent affinity, and the presence of fIXa and fVIIIa induces the appearance of approx. 600 additional high-affinity binding sites for fX. These are expected results as the effects were included in the model a priori (see the Appendix). The binding curve for fVIIIa (see Figure 5B and Table 3) is of major interest, as the Kd (app) and the number of binding sites in the presence of fIXa and fX were not included a priori. When the one-site binding model (Figure 5B, broken line) is used for fitting, the apparent number of fVIIIa-binding sites increases from 750 sites/platelet to 929 sites/platelet, and the Kd (app) decreases from 1.5 nM to 1.08 nM, in good agreement with the experimental data [21,22].
Table 3. Binding of coagulation factors to thrombin-activated platelets: theoretical resultsa.
| Ligand | Two other factors | Number of binding sites/platelet (n) | Kd (app) (nM) |
|---|---|---|---|
| fIXa | Absentb | 250 | 2.57 |
| Presentc | 250 | 0.69 | |
| fVIIIa | Absentb | 750 | 1.5 |
| Presentc | 926 (750 and 193)d | 1.1 (1.5 and 0.34)d | |
| fX | Absentb | 16000 | 320 |
| Presentc | 16002 and 755d | 320 and 28.7d |
a The conditions of calculations are given in the legend to Figure 5.
b Fixed values.
c Obtained values.
d Fitting to the two-site binding model.
Importantly, the form of eqn A62 suggests that fVIIIa in the presence of fIXa and fX has in fact several families of binding sites: its own binding sites on activated platelets and those provided by fIXa. Therefore we also performed the fitting of the upper curve in Figure 5(B) by the two-site model (Figure 5B, dotted line). While visually this curve is nearly indistinguishable from the one-site approximation (enlarged in the inset), it yields a quite different result: it predicts the existence of two types of binding sites, of 750 sites with Kd=1.5 nM and 197 sites with Kd=0.32 nM, with the effectiveness of approximation (in terms of the sum of squares of deviations) of 1.20×10−26, while for the one-site model it is 21.46.
DISCUSSION
In the present study, we have developed a detailed mathematical model of fX activation by intrinsic tenase, which is one of the most important reactions of blood coagulation. We have considered two most frequently occurring situations: (i) the pro-coagulant surface is provided by phospholipid vesicles (the model consists of eqns A34, A24, A27, A28 and A29); (ii) the pro-coagulant surface is provided by activated platelets (the model consists of eqns A60, A57, A58 and A59). The first experimental design is frequently used for in vitro experiments, as phospholipid vesicles represent a standard model of the pro-coagulant surface, while the second situation occurs in vivo. An advantage of this approach is that the final model is a simple equation which has the form:
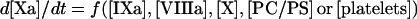 |
(1) |
in contrast with the models of prothrombinase suggested earlier [23,24]. This makes our model a handy tool for experimental design and analysis, although imposing some limitations on the area of applicability. Our approach can also be used to model the kinetics of other membrane-dependent reactions in the coagulation cascade and other biochemical networks, such as activation of prothrombin by prothrombinase, activation of protein C by the complex of thrombin and thrombomodulin, inactivation of fVa by activated protein C, reactions of complement, etc.
FX activation by intrinsic tenase assembled on phospholipid vesicles
The model of intrinsic tenase assembled on phospholipids (Model 1) is in agreement with the experimental data on the rate of fX activation at different phospholipid concentrations (Figure 3A). It predicts a linear dependence of the apparent Michaelis constant and the Kd (app) of fIXa and fVIIIa on phospholipid concentration (Figure 3B and eqn A42); the Vmax value has a maximum, which is also in agreement with the reported experimental data [6,35]. Although the model of intrinsic tenase on synthetic phospholipids describes an artificial system, this model may have applications in vivo. Indeed, as shown, the pro-coagulant surface for tenase assembly can be provided by plasma lipoproteins [7], the surface of which is most likely to be non-specific. If so, eqns A34, A24, A27, A28 and A29, although developed for synthetic PS/PC vesicles, could be used for lipoproteins upon substitution of the values of kinetic constants.
Activation of fX by intrinsic tenase assembled on activated platelets
The existence of highly specific receptors for fIXa, fVIIIa and fX on activated platelets and correlation between the rate of fX activation and the occupancy of these receptors is well established [28,39]. These receptors seem to be complex structures consisting of phospholipids and membrane proteins [21]. The apparent affinity and the apparent number of binding sites for each component of the fX-activating complex (fIXa, fVIIIa and fX) depend in a complex way on the concentration of two other components (for details, see the Results section). Based on these data, we suggest a kinetic scheme of fIXa, fVIIIa and fX interactions with platelets (Figure 2B). Our model is based on the ‘three-receptor model’ proposed by Walsh and colleagues [21,22] and it states that: (i) each of the proteins (fIXa, fVIIIa or fX) independently binds to its specific receptor and subsequently they form complexes on the membrane by means of two-dimensional diffusion [28]; (ii) membrane-bound fX is ‘passed on’ to the specific site consisting of fVIIIa bound to its receptor, and fX–fVIIIa complex subsequently binds fIXa (the hypothesis proposed in [22]); however, the ‘classic’ order of complex assembly (fIXa and fVIIIa form the enzymic complex and then bind fX) is also possible; (iii) by analogy with ‘b’, the enzyme–cofactor complex and the final enzyme–cofactor–substrate complex are formed on the site consisting of fIXa bound to its receptor (the hypothesis of the present study). With this assembly mechanism, fX would bind from solution to a large area containing approx. 10000 low-affinity receptors, and be subsequently transferred to approx. 1000 high-affinity sites provided by fVIIIa [38]; where, again, they would be directed to approx. 250 sites of fIXa, where catalysis occurs.
Our calculations using Model 2 show that the scheme in Figure 2(B) gives results that are consistent with experimentally observed effects (see Figure 5 and Table 3). The model also predicts that the presence of both fIXa and fX leads to the appearance of a new family of high-affinity (approx. 0.3 nM) binding sites for fVIIIa; however, precise experiments are required to test this prediction (Figure 5B and Table 3). Thorough examination of fVIIIa (at low concentrations) binding to activated platelets may provide evidence for or against our model. A more direct approach to detect these newly formed fIXa-dependent fVIIIa-binding sites is to block fVIIIa's own sites. For example, this can be done by an excess of the isolated C2 domain of fVIIIa, which contains the phospholipid-binding site of fVIII. It was shown that the C2 domain competes with fVIIIa for its binding sites on platelets, but does not interact with fIXa [42]. Experimental determination of dependences of the apparent Vmax and Km on platelet concentration and their comparison with model predictions (Figure 4B) can also significantly contribute to the understanding of the processes of complex formation on the phospholipid membrane. Further investigation of this mechanism requires experimental determination of dependence of KVIIIad, nVIIIa on [IXa] and [X], and KIXad, nIXa on [VIIIa] and [X]. Comparison of these dependences with the predictions of different models seems to be a promising approach of investigation of this mechanism.
Possible applications and extensions of the model
In their present forms, Models 1 and 2 can describe only the initial kinetics of fX activation and only in the systems that contain no additional proteins except for fIXa, fVIIIa and fX. It is important, however, to have a model which is valid for the conditions of plasma and for various protein environments. In this section, we summarize the approaches that can considerably extend the area of applicability of the model.
Product inhibition on PS/PC
First, eqns A34, A24, A27, A28 and A29 of Model 1, describing intrinsic tenase on phospholipids, were derived based on the assumption that fX is the only protein to occupy the membrane of vesicles. This assumption is applicable only for analysis of the initial kinetics of the reaction. At later stages, accumulation of fXa may produce the effect of product inhibition: fXa will bind to the membrane and displace fIXa, fVIIIa and fX. The binding of fXa to phospholipids occurs with the same affinity and stoichiometry as the binding of fX [17], and this product competition can be described by Model 1 after substitution of eqn 2 for eqn A23:
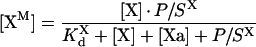 |
(2) |
Intrinsic tenase assembly on activated platelets in plasma
Although activated platelets express highly specific binding sites, a few plasma proteins homologous with fIXa, fVIIIa or fX can compete to a certain extent for the respective binding sites. The most important is prothrombin, which shares with fX the binding sites on activated platelets [20]. When systems containing prothrombin are considered, eqn A57 should be modified according to the standard competition formula:
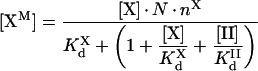 |
(3) |
where KdII is the equilibrium dissociation constant for prothrombin binding to activated platelets. The validity of eqn 3 is supported by its agreement (results not shown) with the experiments of intrinsic tenase competitive inhibition by prothrombin fragment 1 [28]. fVIIIa and fIXa share part of their specific binding sites with their predecessors fVIII and fIX respectively [19,21]. However, plasma fVIII is quantitatively bound to von Willebrand factor, which effectively inhibits fVIII binding to platelets [21]. Therefore correction of the model for possible competition between fVIIIa and fVIII for platelets in plasma is not required. In contrast with fVIII, fIX has been shown not only to affect, but also even to potentiate fX activation by intrinsic tenase, though the mechanism of this effect remains unclear [40]. Further elucidation of this mechanism is required in order to develop an adequate model of fX activation on activated platelets in the presence of fIX.
Acknowledgments
We are grateful to Dr Mikhail V. Ovanesov (National Research Center for Hematology) for careful review and constructive criticism of the manuscript. This study was supported in part by grant from Russian Foundation for Basic Research, project No. 03-04-48338, to F. I. A., and by NATO Collaborative Linkage Grant No. 979210 to E. L. S. and F. I. A.
APPENDIX
Derivation of basic equations
Initially, we derive general equations to describe the binding of a protein from solution to a membrane and association/dissociation of two membrane-bound proteins. First, we consider the binding of some factor F to a membrane. The binding equation is:
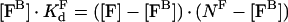 |
(A1) |
where [FB] is the concentration of bound factor F with respect to total volume, [F] is the total factor F concentration, KFd is the equilibrium dissociation constant of factor F and its binding site on the membrane, NF is the concentration of factor F binding sites on the membrane with respect to total volume. From this quadratic equation, the equilibrium concentration of bound factor F is:
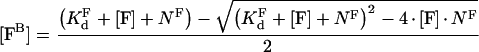 |
(A2) |
Assuming that
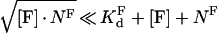 |
(A3) |
we can use the first-order approximation of the square root in eqn A2 to obtain:
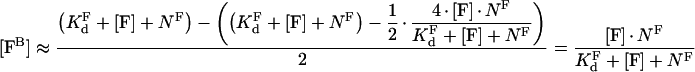 |
(A4) |
Eqn A4, being simpler and more convenient for analysis than eqn A2, has a wider area of applicability than the standard binding model:
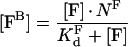 |
(A5) |
Eqn A3 holds for fIXa, fVIIIa, fX and platelet/phospholipid membrane in the area of applicability specified for our model (see the Materials and methods section). To describe the interaction between membrane-bound factors, we introduce the local concentration of factor F, [FB]local. Physically, this local concentration is either the two-dimensional surface density of factor F on the surface of a platelet/vesicle [28] or the three-dimensional concentration of factor F in the thin ‘interface shell’ between the bulk solution and the platelet/vesicle [23,24]. These two approaches are mathematically equivalent, so we do not specify the dimensionality of [FB]local: it can be either nmol/cm2 or nmol/l. By its definition, [FB]local is directly proportional to [FB] and is inversely proportional to the concentration of platelets/vesicles [23,24,28]:
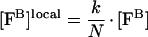 |
(A6) |
where N is the concentration of platelets or phospholipids, and k is the proportionality factor. The dimensionality of k depends on the dimensionality of [FB]local. If N were expressed in phospholipid vesicles per litre, then parameter k would have simple physical interpretation: its dimensionality will be [litre−1] and the value of k will be reciprocal relative to the volume of the ‘interface shell’ of a single vesicle. Next, we consider interaction of two membrane-bound proteins, factors F1 and F2. Let their local equilibrium dissociation constant equal K1–2 locald. Dimensionality of this local constant is identical with that of [FB]local, and the constant has a straightforward definition as the ratio of product of equilibrium local concentrations of complex-free factors to the concentration of the complex. Then the local concentration of the F1–F2 complex by analogy with equations A2, A3, and A4 is defined:
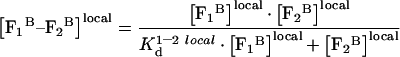 |
(A7) |
Rearranging eqn A7 with the help of eqn A6, we obtain the concentration of the complex with respect to the total volume:
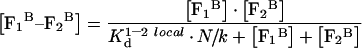 |
(A8) |
By analogy with eqn A4, eqn A8 is valid as long as either [F1B] or [F2B] is much smaller than the other value or as long as both are much smaller than K1–2 locald. The approach used to derive eqns A4 and A8 is frequently used throughout the following computations.
Kinetics of intrinsic tenase on phospholipids
Through [IXaM], [VIIIaM] and [XM], we denote the concentrations of fIXa, fVIIIa and fX respectively, which are bound to membrane, but are free (i.e. are not in a complex). The concentration with superscript ‘B’ denotes the total concentrations of a bound factor (i.e. [IXaB]=[IXaM]+[IXaB–VIIIaB]+[IXaB–VIIIaB–XB]). To obtain quasi-steady concentrations of membrane complexes, we use eqns A9, A10 and A11 for the reactions on the membrane in Figure 2(A):
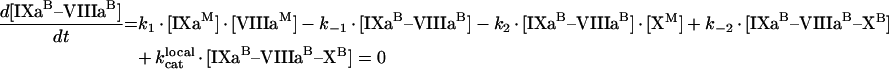 |
(A9) |
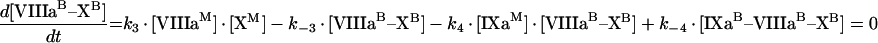 |
(A10) |
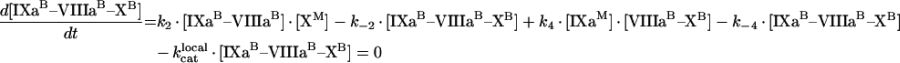 |
(A11) |
where k1, k2, k3 and k4 are effective kinetic association constants of the direct reactions 1, 2, 3 and 4 of Figure 2(A); k−1, k−2, k−3 and k−4 are the rate constants of the respective reverse reactions. According to eqn A8, k1, k2, k3 and k4 are inversely proportional to phospholipid concentration P (see eqns A18, A19 and A20). Solving this system of algebraic equations, we obtain [IXaB–VIIIaB], [VIIIaB–XB] and [IXaB–VIIIaB–XB]:
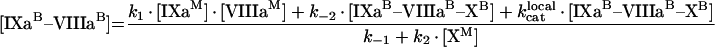 |
(A12) |
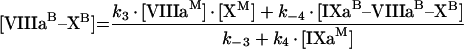 |
(A13) |
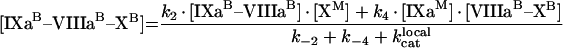 |
(A14) |
The set of eqns A12–A14 is solved after the reduction of high-order terms in eqns A12 and A13 (see [34] for the analysis of a similar system):
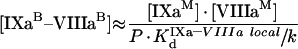 |
(A15) |
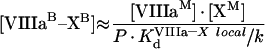 |
(A16) |
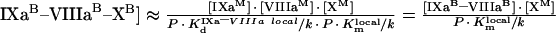 |
(A17) |
where we denote:
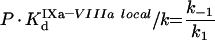 |
(A18) |
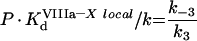 |
(A19) |
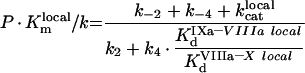 |
(A20) |
The next step is to find connection between the bulk ([IXa], [VIIIa] and [X]) and bound ([IXaM], [VIIIaM] and [XM]) factor concentrations. Binding sites for factors on phospholipid vesicles are non-specific. Therefore, to calculate the concentrations of bound factors, competition between factors has to be taken into account. Moreover, the binding of different factors to phospholipids occurs with different stoichiometry. We assume that the only factor to occupy the membrane significantly is fX (see the Materials and methods section). The equations of equilibrium binding are:
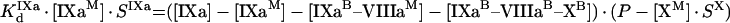 |
(A21) |
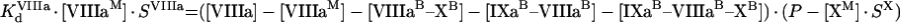 |
(A22) |
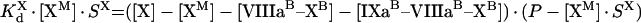 |
(A23) |
where SIXa, SVIIIa and SX are the doubled stoichiometries of binding of fIXa, fVIIIa and fX respectively to phospholipid. The values are doubled because only a part of phospholipid molecules (from half to two thirds, depending on the vesicle size) is located on the outer leaflet of the membrane of the vesicle [17]. Accordingly, the expressions P/SIXa, P/SVIIIa and P/SX give the concentrations of binding sites for the factors with respect to total volume. Solution of eqns A21, A22 and A23 is:
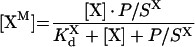 |
(A24) |
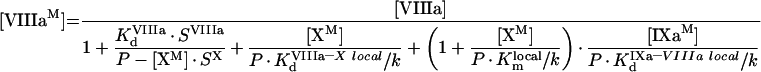 |
(A25) |
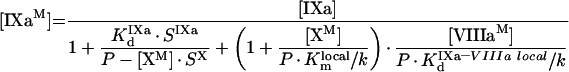 |
(A26) |
To solve this system, we take into consideration that we do not need individual solutions for [VIIIaM] and [IXaM], but only for their product. For better readability of equations, we denote:
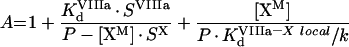 |
(A27) |
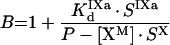 |
(A28) |
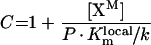 |
(A29) |
After rearrangement, eqns A25 and A26 can be written in the form:
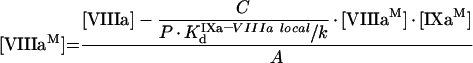 |
(A30) |
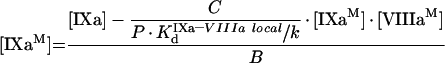 |
(A31) |
Multiplication of these equations and reduction of the fourth-order term ([IXaM]·[VIIIaM])2 gives the following solution:
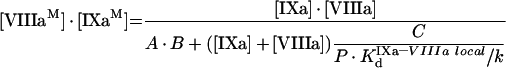 |
(A32) |
 |
(A33) |
The rate of fX activation is defined by the equation obtained from eqns A17 and A33:
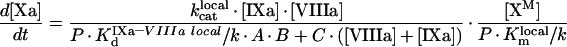 |
(A34) |
If we consider the most general model ‘d’ of the fIXa–fVIIIa–fX complex assembly (all the binary complexes shown in Figure 1 exist), then these parameters will have the form:
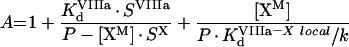 |
(A35) |
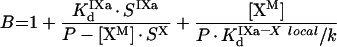 |
(A36) |
The value of KdIXa–X local/k can be estimated from the value of the apparent Michaelis constant for the fX activation by fIXa. Specifically, KdIXa–X local/k is obtained from the ratio of the Michaelis constant and phospholipid concentration (in a manner similar to eqn A47). Estimation on the basis of [13] gives a value of KdIXa–X local/k=0.05.
It is difficult to measure the values of local two-dimensional constants kcatlocal, Kmlocal/k, KdVIIIa–X local/k and KdIXa–VIIIa local/k in direct experiments. However, they can be obtained from measurable parameters of the reaction of fX activation (Table 1). From eqn A27, the Kd (app) of the fIXa–fVIIIa complex is defined as:
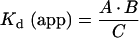 |
(A37) |
By substituting eqns A27, A28 and A29 into eqn A37, rearranging and simplifying (assuming for this experiment that [XM]≪P/SX), we get:
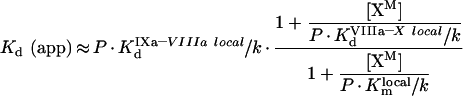 |
(A38) |
This equation provides information about two constants. First, this constant is reported not to be influenced by fX addition [14]. Therefore
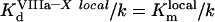 |
(A39) |
Otherwise, the Kd (app) would depend upon fX addition:
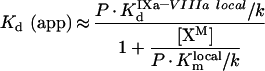 |
(A40) |
From eqns A38 and A39 it follows that:
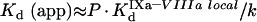 |
(A41) |
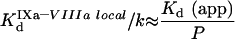 |
(A42) |
Under the conditions used in Table 1 of [11] and in [13], fVIIIa is in excess, and from eqn A34 we obtain:
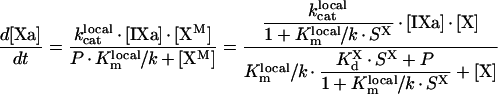 |
(A43) |
The apparent catalytic and Michaelis constants are expressed:
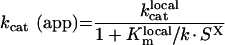 |
(A44) |
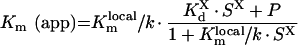 |
(A45) |
Finally, the values of model parameters are:
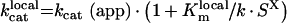 |
(A46) |
 |
(A47) |
Summarizing, the rate of fX activation by intrinsic tenase on phospholipids is determined by eqn A34, into which eqns A24, A27, A28 and A29 are substituted. These equations form Model 1. The parameters, used in these equations, can be obtained from the experimentally determined values with the help of eqns A39, A42, A46 and A47 (Table 1).
Kinetics of intrinsic tenase on activated platelets
The process of intrinsic tenase assembly on the surface of platelet membrane differs from that on the surface of phospholipid vesicles. Coagulation factors bind to their specific sites, and there is no competition for the surface between factors. However, bound factors and their specific receptors interact in a complex way, which complicates the modelling. To avoid complex notation, the symbols denoting the constants and the concentrations are the same both in the ‘platelet’ and ‘phospholipid’ sections of the present paper, although these constants have different values and different meanings (Tables 1 and 2). As for phospholipids, [IXaM], [VIIIaM] and [XM] are the concentrations of fIXa, fVIIIa and fX respectively, which are bound to the membrane, but are free there. To obtain the quasi-steady concentrations of complexes, we use the equations of reactions written in accordance with Figure 2(B), which are identical with eqns A9, A10 and A11. The association constants used in these equations, however, are the apparent constants, which are inversely proportional to the concentration of surface, according to eqn A8. This fact is used below in eqns A51, A52 and A53. Solving this system of algebraic equations, we obtain the values of [IXaB–VIIIaB], [VIIIaB–XB] and [IXaB–VIIIaB–XB], which are given by eqns A12, A13 and A14. By analogy with phospholipids:
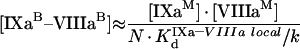 |
(A48) |
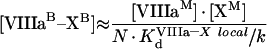 |
(A49) |
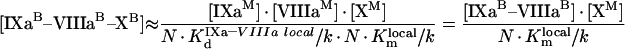 |
(A50) |
where
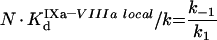 |
(A51) |
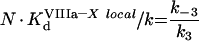 |
(A52) |
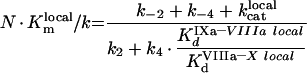 |
(A53) |
The next step is to find a connection between the bulk ([IXa], [VIIIa] and [X]) and the local ([IXaM], [VIIIaM] and [XM]) concentrations. Note that eqn A55 suggests that the fVIIIa–fX complex can dissociate from the platelet membrane with the same rate as fVIIIa [22]. We did not consider this reaction in the model for phospholipids, assuming that [VIIIaM]≫[VIIIaB–XaB]. To simplify the system, we do not consider the reverse reaction (binding of fX to platelet-bound fVIIIa from solution) because its contribution is negligible under normal conditions [22,28]. However, this simplification makes it impossible to use our model to describe experiments in which fX sites are blocked with prothrombin [22]. The binding equations are:
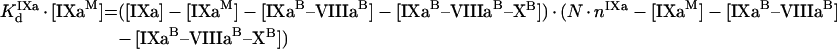 |
(A54) |
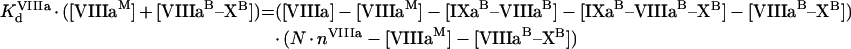 |
(A55) |
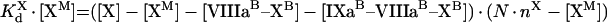 |
(A56) |
Solution with the help of eqns A48, A49 and A50 gives:
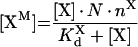 |
(A57) |
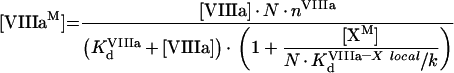 |
(A58) |
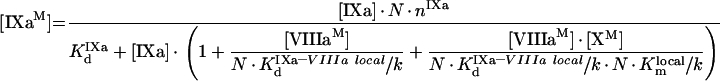 |
(A59) |
To obtain eqns A57–A59, we take into account that [IXa]≫[IXaB]=[IXaM]+[IXaB–VIIIaB]+[IXaB–VIIIaB–XB], [VIIIa]≫[VIIIaB]=[IXaM]+[VIIIaB–XB]+[IXaB–VIIIaB]+[IXaB–VIIIaB–XB], [X]≫[XB]=[XM]+[VIIIaB–XB]+[IXaB–VIIIaB–XB] (see the Materials and methods section). In particular, with these additional assumptions, identical eqns A23 and A56 give eqns A24 and A57 respectively, which have different forms. The formula for the rate of fX activation has the form:
 |
(A60) |
In summary, the rate of fX activation by intrinsic tenase on activated platelets is defined by eqn A60, into which eqns A57, A58 and A59 are substituted. These equations form Model 2. Besides, eqns A57, A58 and A59 allow total concentrations of bound factors to be obtained:
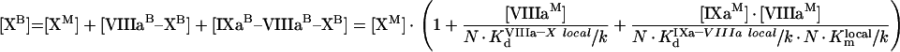 |
(A61) |
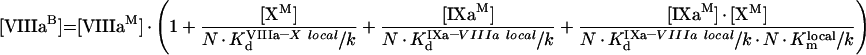 |
(A62) |
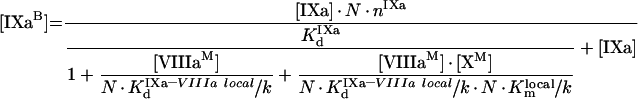 |
(A63) |
It follows from these equations that, for example for fIXa, the number of binding sites at saturating concentrations of fVIIIa and fX does not change (see eqn A63), while the Kd (app) is decreased. As for phospholipids, we should obtain the values of local constants from the experimentally determined parameters (Table 2). The values of KIXa–VIIIa locald/k and KVIIIa–X locald/k can be obtained from the experiments of co-ordinate binding of factors to platelets. fVIIIa is known to form the binding site for fX with a Kd (app) of approx. 30 nM [21]. From eqns A49, A57 and A58, we see that in this case:
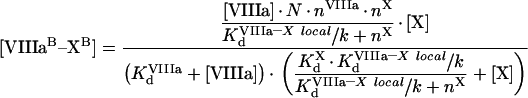 |
(A64) |
and we obtain:
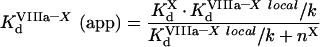 |
(A65) |
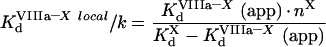 |
(A66) |
To obtain the local dissociation constant of fIXa and fVIIIa, Klocald/k, we use eqn A63. In the presence of excess fVIIIa and fX, the Kd (app) is:
 |
(A67) |
and the value of the local constant is:
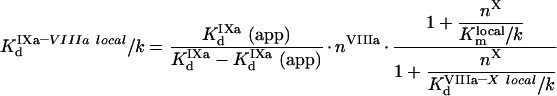 |
(A68) |
Finally, to obtain the values of the local catalytic constant and the local Michaelis constants, we used two different experimental reports [13,41]. Under the conditions of the earlier study [13], when [IXa]=10 pM≪KIXad, we get from eqn A60:
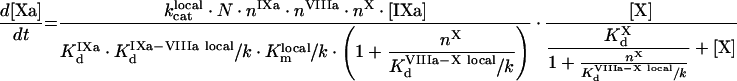 |
(A69) |
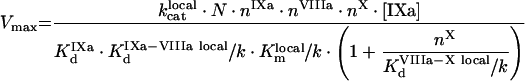 |
(A70) |
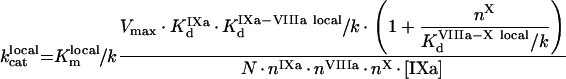 |
(A71) |
In contrast with [13], in [41], the factors saturate their binding sites, and from eqn A60 we get:
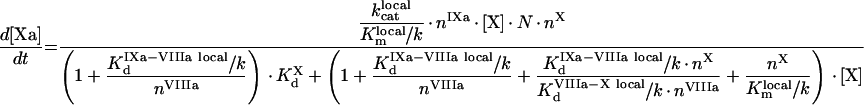 |
(A72) |
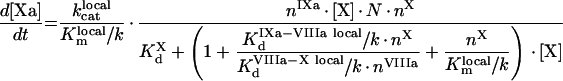 |
(A73) |
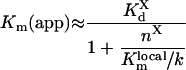 |
(A74) |
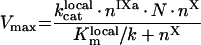 |
(A75) |
Values of local constants are:
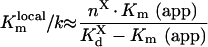 |
(A76) |
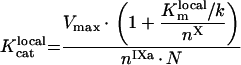 |
(A77) |
References
- 1.Mann K. G. Biochemistry and physiology of blood coagulation. Thromb. Haemostasis. 1999;82:165–174. [PubMed] [Google Scholar]
- 2.Mann K. G., Butenas S., Brummel K. The dynamics of thrombin formation. Arterioscler. Thromb. Vasc. Biol. 2003;23:17–25. doi: 10.1161/01.atv.0000046238.23903.fc. [DOI] [PubMed] [Google Scholar]
- 3.Ovanesov M. V., Krasotkina J. V., Ul'yanova L. I., Abushinova K. V., Plyushch O. P., Domogatskii S. P., Vorob'ev A. I., Ataullakhanov F. I. Hemophilia A and B are associated with abnormal spatial dynamics of clot growth. Biochim. Biophys. Acta. 2002;1572:45–57. doi: 10.1016/s0304-4165(02)00278-7. [DOI] [PubMed] [Google Scholar]
- 4.Thompson A. R. Structure, function, and molecular defects of factor IX. Blood. 1986;67:565–572. [PubMed] [Google Scholar]
- 5.Venkateswarlu D., Perera L., Darden T., Pedersen L. G. Structure and dynamics of zymogen human blood coagulation factor X. Biophys. J. 2002;82:1190–1206. doi: 10.1016/S0006-3495(02)75476-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Dieijin G., Tans G., Rosing J., Hemker H. C. The role of phospholipid and factor VIIIa in the activation of bovine factor X. J. Biol. Chem. 1981;256:3433–3442. [PubMed] [Google Scholar]
- 7.Saenko E. L., Shima M., Sarafanov A. G. Role of activation of the coagulation factor VIII in interaction with vWf, phospholipid, and functioning within the factor Xase complex. Trends Cardiovasc. Med. 1999;9:185–192. doi: 10.1016/s1050-1738(00)00019-0. [DOI] [PubMed] [Google Scholar]
- 8.Peyrou V., Lormeau J. C., Herault J. P., Gaich C., Pfliegger A. M., Herbert J. M. Contribution of erythrocytes to thrombin generation in whole blood. Thromb. Haemostasis. 1999;81:400–406. [PubMed] [Google Scholar]
- 9.Zwaal R. F., Schroit A. J. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89:1121–1132. [PubMed] [Google Scholar]
- 10.Andrews D. A., Low P. S. Role of red blood cells in thrombosis. Curr. Opin. Hematol. 1999;6:76–82. doi: 10.1097/00062752-199903000-00004. [DOI] [PubMed] [Google Scholar]
- 11.Gilbert G. E., Arena A. A. Activation of the factor VIIIa–factor IXa enzyme complex of blood coagulation by membranes containing phosphatidyl-L-serine. J. Biol. Chem. 1996;271:11120–11125. doi: 10.1074/jbc.271.19.11120. [DOI] [PubMed] [Google Scholar]
- 12.Mann K. G., Nesheim M. E., Church W. R., Haley P., Krishnaswamy S. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood. 1990;76:1–16. [PubMed] [Google Scholar]
- 13.Rawala-Sheikh R., Ahmad S. S., Ashby B., Walsh P. N. Kinetics of coagulation factor X activation by platelet-bound factor IXa. Biochemistry. 1990;29:2606–2611. doi: 10.1021/bi00462a025. [DOI] [PubMed] [Google Scholar]
- 14.Duffy E. J., Parker E. T., Mutucumarana V. P., Johnson A. E., Lollar P. Binding of factor VIIIa and factor VIII to factor IXa on phospholipid vesicles. J. Biol. Chem. 1992;267:17006–17011. [PubMed] [Google Scholar]
- 15.Mathur A., Zhong D., Sabharwal A. K., Smith K. J., Bajaj S. P. Interaction of factor IXa with factor VIIIa. Effects of protease domain Ca2+ binding site, proteolysis in the autolysis loop, phospholipid, and factor X. J. Biol. Chem. 1997;272:23418–23426. doi: 10.1074/jbc.272.37.23418. [DOI] [PubMed] [Google Scholar]
- 16.Neuenschwander P., Jesty J. A comparison of phospholipid and platelets in the activation of human factor VIII by thrombin and factor Xa, and in the activation of factor X. Blood. 1988;72:1761–1770. [PubMed] [Google Scholar]
- 17.Cutsforth G. A., Whitaker R. N., Hermans J., Lentz B. R. A new model to describe extrinsic protein binding to phospholipid membranes of varying composition: application to human coagulation proteins. Biochemistry. 1989;28:7453–7461. doi: 10.1021/bi00444a045. [DOI] [PubMed] [Google Scholar]
- 18.Saenko E. L., Scandella D., Yakhyaev A. V., Greco N. J. Activation of factor VIII by thrombin increases its affinity for binding to synthetic phospholipid membranes and activated platelets. J. Biol. Chem. 1998;273:27918–27926. doi: 10.1074/jbc.273.43.27918. [DOI] [PubMed] [Google Scholar]
- 19.Ahmad S. S., Rawala-Sheikh R., Walsh P. N. Comparative interactions of factor IX and factor IXa with human platelets. J. Biol. Chem. 1989;264:3244–3251. [PubMed] [Google Scholar]
- 20.Scandura J. M., Ahmad S. S., Walsh P. N. A binding site expressed on the surface of activated platelets is shared by factor X and prothrombin. Biochemistry. 1996;35:8890–8902. doi: 10.1021/bi9525029. [DOI] [PubMed] [Google Scholar]
- 21.Ahmad S. S., Scandura J. M., Walsh P. N. Structural and functional characterization of platelet receptor-mediated factor VIII binding. J. Biol. Chem. 2000;275:13071–13081. doi: 10.1074/jbc.275.17.13071. [DOI] [PubMed] [Google Scholar]
- 22.Ahmad S. S., Walsh P. N. Coordinate binding studies of the substrate (factor X) with the cofactor (factor VIII) in the assembly of the factor X activating complex on the activated platelet surface. Biochemistry. 2002;41:11269–11276. doi: 10.1021/bi025785v. [DOI] [PubMed] [Google Scholar]
- 23.Nesheim M. E., Tracy R. P., Mann K. G. “Clotspeed,” a mathematical simulation of the functional properties of prothrombinase. J. Biol. Chem. 1984;259:1447–1453. [PubMed] [Google Scholar]
- 24.Nesheim M. E., Tracy R. P., Tracy P. B., Boskovic D. S., Mann K. G. Mathematical simulation of prothrombinase. Methods Enzymol. 1992;215:316–328. doi: 10.1016/0076-6879(92)15074-m. [DOI] [PubMed] [Google Scholar]
- 25.Jesty J. Interaction of feedback control and product inhibition in the activation of factor X by factors IXa and VIII. Haemostasis. 1991;21:208–218. doi: 10.1159/000216230. [DOI] [PubMed] [Google Scholar]
- 26.Bungay S. D., Gentry P. A., Gentry R. D. A mathematical model of lipid-mediated thrombin generation. Math. Med. Biol. 2003;20:105–129. doi: 10.1093/imammb/20.1.105. [DOI] [PubMed] [Google Scholar]
- 27.Kuharsky A. L., Fogelson A. L. Surface-mediated control of blood coagulation: the role of binding site densities and platelet deposition. Biophys. J. 2001;80:1050–1074. doi: 10.1016/S0006-3495(01)76085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27a.Panteleev M., Saenko E. L., Ataullakhanov F. I. Kinetics of factor X activation by membrane-bound complex of factor IXa and factor VIIIa. J. Thromb. Haemostasis. 2003;1(suppl. 1):P1066. doi: 10.1042/BJ20031748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scandura J. M., Walsh P. N. Factor X bound to the surface of activated human platelets is preferentially activated by platelet-bound factor IXa. Biochemistry. 1996;35:8903–8913. doi: 10.1021/bi9525031. [DOI] [PubMed] [Google Scholar]
- 29.Krishnaswamy S., Jones K. C., Mann K. G. Prothrombinase complex assembly: kinetic mechanism of enzyme assembly on phospholipid vesicles. J. Biol. Chem. 1988;263:3823–3834. [PubMed] [Google Scholar]
- 30.Bardelle C., Furie B., Furie B. C., Gilbert G. E. Membrane binding kinetics of factor VIII indicate a complex binding process. J. Biol. Chem. 1993;268:8815–8824. [PubMed] [Google Scholar]
- 31.Giesen P. L., Willems G. M., Hemker H. C., Hermens W. T. Membrane-mediated assembly of the prothrombinase complex. J. Biol. Chem. 1991;266:18720–18725. [PubMed] [Google Scholar]
- 32.Giesen P. L., Willems G. M., Hermens W. T. Production of thrombin by prothrombinase complex is regulated by membrane-mediated transport of prothrombin. J. Biol. Chem. 1991;266:1379–1382. [PubMed] [Google Scholar]
- 33.Rosing J., van Rijn J. L., Bevers E. M., van Dieijen G., Comfurius P., Zwaal R. F. The role of activated human platelets in prothrombin and factor X activation. Blood. 1985;65:319–332. [PubMed] [Google Scholar]
- 34.Boskovic D. S., Giles A. R., Nesheim M. E. Studies of the role of factor Va in the factor Xa-catalyzed activation of prothrombin, fragment 1.2-prethrombin-2 and dansyl-L-glutamyl-glycyl-L-arginine-meizothrombin in the absence of phospholipid. J. Biol. Chem. 1990;265:10497–10505. [PubMed] [Google Scholar]
- 35.van Dieijen G., van Rijn J. L., Govers-Riemslag J. W., Hemker H. C., Rosing J. Assembly of the intrinsic factor X activating complex – interactions between factor IXa, factor VIIIa and phospholipid. Thromb. Haemostasis. 1985;53:396–400. [PubMed] [Google Scholar]
- 36.van de Waart P., Hemker H. C., Lindhout T. Interaction of prothrombin with factor Va–phospholipid complexes. Biochemistry. 1984;23:2838–2842. doi: 10.1021/bi00307a047. [DOI] [PubMed] [Google Scholar]
- 37.Nesheim M. E., Furmaniak-Kazmierczak E., Henin C., Côté G. On the existence of platelet receptors for factor V(a) and factor VIII(a) Thromb. Haemostasis. 1993;70:80–86. [PubMed] [Google Scholar]
- 38.Ahmad S. S., London F. S., Walsh P. N. The assembly of the factor X-activating complex on activated human platelets. J. Thromb. Haemostasis. 2003;1:48–59. doi: 10.1046/j.1538-7836.2003.00020.x. [DOI] [PubMed] [Google Scholar]
- 39.Ahmad S. S., Rawala-Sheikh R., Walsh P. N. Platelet receptor occupancy with factor IXa promotes factor X activation. J. Biol. Chem. 1989;264:20012–20016. [PubMed] [Google Scholar]
- 40.London F. S., Walsh P. N. Zymogen factor IX potentiates factor IXa-catalyzed factor X activation. Biochemistry. 2000;39:9850–9858. doi: 10.1021/bi000245o. [DOI] [PubMed] [Google Scholar]
- 41.Wilkinson F. H., Ahmad S. S., Walsh P. N. The factor IXa second epidermal growth factor (EGF2) domain mediates platelet binding and assembly of the factor X activating complex. J. Biol. Chem. 2002;277:5734–5741. doi: 10.1074/jbc.M107753200. [DOI] [PubMed] [Google Scholar]
- 42.Ahmad S. S., Walsh P. N. The role of C2 domain of factor VIII in the assembly of factor X-activating complex on platelet membrane. Blood. 2001;98:706a. doi: 10.1021/bi0511033. [DOI] [PubMed] [Google Scholar]
- 43.Burri B. J., Edgington T. S., Fair D. S. Molecular interactions of the intrinsic activation complex of coagulation: binding of native and activated human factors IX and X to defined phospholipid vesicles. Biochim. Biophys. Acta. 1987;923:176–186. doi: 10.1016/0304-4165(87)90002-x. [DOI] [PubMed] [Google Scholar]
- 44.Erb E. M., Stenflo J., Drakenberg T. Interaction of bovine coagulation factor X and its glutamic-acid-containing fragments with phospholipid membranes: a surface plasmon resonance study. Eur. J. Biochem. 2002;269:3041–3046. doi: 10.1046/j.1432-1033.2002.02981.x. [DOI] [PubMed] [Google Scholar]
- 45.Beals J. M., Castellino F. J. The interaction of bovine factor IX, its activation intermediate, factor IX alpha, and its activation products, factor IXa alpha and factor IXa beta, with acidic phospholipid vesicles of various compositions. Biochem. J. 1986;236:861–869. doi: 10.1042/bj2360861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilbert G. E., Furie B. C., Furie B. Binding of human factor VIII to phospholipid vesicles. J. Biol. Chem. 1990;265:815–822. [PubMed] [Google Scholar]
- 47.Spaargaren J., Giesen P. L., Janssen M. P., Voorberg J., Willems G. M., van Mourik J. A. Binding of blood coagulation factor VIII and its light chain to phosphatidylserine/phosphatidylcholine bilayers as measured by ellipsometry. Biochem. J. 1995;310:539–545. doi: 10.1042/bj3100539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mertens K., Cupers R., Van Wijngaarden A., Bertina R. M. Binding of human blood-coagulation Factors IXa and X to phospholipid membranes. Biochem. J. 1984;223:599–605. doi: 10.1042/bj2230599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X., Gabriel D. A. The physical exchange of factor VIII (FVIII) between von Willebrand factor and activated platelets and the effect of the FVIII B-domain on platelet binding. Biochemistry. 1997;36:10760–10767. doi: 10.1021/bi970052+. [DOI] [PubMed] [Google Scholar]