

Abstract
T cell-derived cancers are hallmarked by heterogeneity, aggressiveness, and poor clinical outcomes. Available targeted therapies are severely limited due to a lack of target antigens that allow discrimination of malignant from healthy T cells. Here, we report a novel approach for the treatment of T cell diseases based on targeting the clonally rearranged T cell receptor displayed by the cancerous T cell population. As a proof of concept, we identified an antibody with unique specificity toward a distinct T cell receptor (TCR) and developed antibody-drug conjugates, precisely recognizing and eliminating target T cells while preserving overall T cell repertoire integrity and cellular immunity. Our anti-TCR antibody-drug conjugates demonstrated effective receptor-mediated cell internalization, associated with induction of cancer cell death with strong signs of apoptosis. Furthermore, cell proliferation-inhibiting bystander effects observed on target-negative cells may contribute to the molecules’ anti-tumor properties precluding potential tumor escape mechanisms. To our knowledge, this represents the first anti-TCR antibody-drug conjugate designed as custom-tailored immunotherapy for T cell-driven pathologies.
Keywords: antibody-drug conjugate, idiotype, targeted therapy, T cell leukemia, T cell lymphoma, T cell receptor, yeast surface display
Graphical abstract
Harald Kolmar and colleagues report a novel approach for the treatment of the difficult-to-treat T cell lymphoma/leukemia based on targeting the clonally rearranged T cell receptor expressed by the malignant T cell population. The developed antibody-drug conjugates precisely eliminate target T cells while preserving the integrity of the T cell repertoire and cellular immunity.
Introduction
T cell malignancies represent a clinically heterogeneous group of disorders that derive from clonal dysfunctional T cells arising through distinct mechanisms at different stages of development.1,2,3 Lymphoid B cell neoplasms occur more frequently than cancers of T cell origin, which account for only about 10% of non-Hodgkin lymphomas and 15% of acute lymphoblastic leukemias (ALLs).4,5,6 While there are several immunotherapeutic agents available for the treatment of B cell diseases such as monoclonal antibodies (mAbs), bispecific antibodies, antibody-drug conjugates (ADCs), and chimeric antigen receptor (CAR) T cells, patients suffering from T cell malignancies have limited therapeutic options, relying primarily on chemotherapy, which is associated with a poor prognosis.7,8,9,10,11 A prospective cohort study on peripheral T cell lymphoma (TCL) demonstrated that 68% of patients were identified as refractory (47%) or relapsed (21%) within a median time of 8 months after receiving first-line treatment, and out of these patients, 47% died after a median follow up of 38 months.12 In T cell ALL, response rates reach up to 85% in 5-year event-free survival with contemporary chemotherapy, but in relapsed disease, event-free and overall survival rates are less than 25%.13 The concept of successful therapy for B cell malignancies is mainly based on targeting of pan-B cell antigens such as CD19 or CD20 entailing B cell aplasia, which is clinically tolerated and, in most cases, compensated by periodic immunoglobulin infusion.14,15,16,17 However, applying this concept to T cell lymphoma is not feasible since it would lead to a permanent and ultimately fatal loss of healthy T cells.18 Despite advances in understanding T cell disease biology, no antigens that discriminate malignant from healthy T cells have been identified. Recent advances include targeting of antigens with limited expression on healthy T cells and elevated presence on malignant T cells.19 To date, two antibody-based drugs following this concept have received FDA approval for TCL: mogamulizumab, an anti-CCR4 mAb, and brentuximab vedotin, an anti-CD30 ADC; besides, there are several antibody-derived molecules currently undergoing clinical investigation.19,20,21
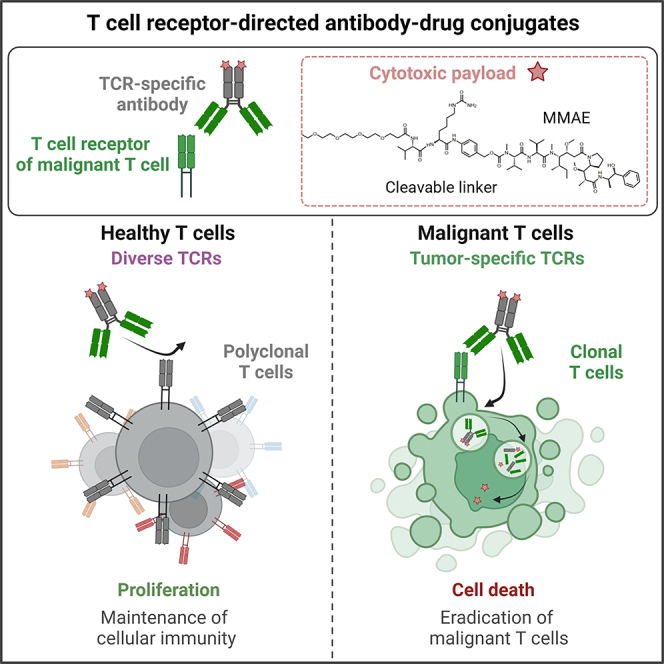
The αβ T cell receptor (TCR) constitutes a key element in the adaptive immune response mediating recognition and discrimination of self and foreign antigenic material. Consisting of disulfide-linked TCR α and TCR β chains imparting peptide-major histocompatibility complex recognition, the TCR is constitutively associated with cluster of differentiation 3 (CD3) dimers responsible for transduction of activation signals. During T cell development, TCR diversity is generated through somatic recombination of multiple variable (V), diversity (D; only for β chain), and joining (J) germline gene segments to the constant (C) region genes.22 This results in distinct TCR rearrangement patterns that establish the antigen binding site with the V gene fragments encoding complementarity-determining regions (CDRs) CDR1 and CDR2 and the junctional V(D)J site coding for CDR3, which is the most varied sequence in the molecule providing the major diversity of the TCR repertoire.23,24 Apart from healthy T lymphocytes, TCR expression is observed in mature T cell cancers including peripheral T cell lymphomas (PTCLs), adult T cell leukemia/lymphoma, and a substantial fraction of T-ALL.25,26,27,28,29 Consequently, healthy T cells display a variety of different TCRs, whereas malignant T cells typically form clonal populations exclusively expressing one unique TCR.30,31 This renders TCRs a highly promising target for cancer therapy offering the opportunity to selectively eradicate malignant T cells while sparing healthy T cells and thus preserving cellular immunity. Previous attempts to target tumor-specific TCRs involving anti-T cell receptor β chain constant domains 1 (TRBC1) as well as anti-TRBV8 and TRBV5 CAR T cells and bispecific antibodies targeting TRBV5-5 or TRBV12 in combination with CD3 have shown encouraging anti-tumor effects and significantly reduced T cell aplasia in preclinical models.29,32,33 Beyond addressing specific TRBC or TRBV elements, it is feasible to address the TCR variable regions carrying unique antigenic determinants, referred to as idiotype.34
Here we describe the generation of TCR idiotype-directed ADCs for the treatment of T cell-derived cancers (Figure 1). To this end, it was necessary to identify and express a TCR, e.g., of a malignant T cell population. Subsequently, a chicken was immunized with the target TCR, followed by isolation of the genetic material and construction of a single-chain variable fragment (scFv) yeast surface display (YSD) library, which was screened by fluorescence-activated cell sorting (FACS) for TCR binders. The isolated TCR binders were tested toward off-target effects using polyclonal T lymphocytes derived from human healthy donor blood. The lead candidate, reformatted as full-length antibody, was further characterized regarding stability, aggregation behavior, affinity, cellular binding, and internalization properties. To ensure cytotoxicity of the molecule, the anti-TCR antibody was conjugated to the cytotoxin monomethyl auristatin E (MMAE) via two different approaches resulting in ADCs with drug-to-antibody ratios (DARs) of 2 and 2–8, respectively. The TCR idiotype-targeting ADCs effectively deplete cancerous T cells in vitro with varying potency depending on their drug load while sparing healthy T lymphocytes.
Figure 1.

Concept overview
Identification and expression of tumor-specific TCR was followed by chicken immunization. Afterward, splenic RNA was isolated, and cDNA was synthesized. Genes encoding the variable antibody domains were amplified and assembled as scFv gene string for yeast surface display and subsequent screening by FACS. TCR-binding scFvs were cloned into bacterial and mammalian vectors for scFv and scFv-Fc expression, respectively. After selection of a lead candidate, the TCR idiotype-specific binder was reformatted as full-length antibody and subsequently characterized. Generation of ADCs by antibody MMAE conjugation was conducted by two different approaches resulting in DARs of 2 and 2–8, respectively. Created with BioRender.com.
Results
Library generation and screening for TCR idiotype-targeting antibodies
TCRs are present in all jawed vertebrates from lower vertebrates to mammals.35 In addition to the diversification in overall structure and amino acid sequence, TCRs differ in their variable regions defined by six CDRs.36 In order to obtain antibodies that target the variable region of a specific TCR, we conducted an animal immunization and subsequent screening campaign. As a proof of concept TCRA6, an αβ TCR specific for the T cell leukemia-associated human T cell lymphotropic virus type 111–19 HTLV-1 peptide (A2-Tax) complexed with the human leukocyte antigen (HLA-A∗0201) was used as target to generate TCR-specific antibodies.37,38,39 Previous attempts involving immunization of chickens revealed high diversities of avian antibodies directed against human proteins.40,41 Therefore, an adult laying hen was immunized with the soluble target TCRA6, which elicited a strong immune response (Figure S1). Based on the genetic material from chicken, an scFv YSD library consisting of 6 × 109 transformants was generated and screened using FACS. For library sorting, yeast cells were stained for scFv surface presentation using an anti-c-myc antibody fluorescein isothiocyanate (FITC) conjugate and antigen binding by DyLight650-labeled TCRA6. Over two consecutive rounds of screening using 500 nM antigen, we were able to enrich a yeast population binding to TCRA6 (Figure 2). In a third round, antigen concentration was reduced to 50 nM, and yeast cells demonstrating strong interaction with TCRA6 were sorted for further investigations.
Figure 2.

Screening of scFv immune library for TCRA6 binders by FACS
Sorting rounds 1–3 of the chicken-derived yeast surface-displayed scFv library are depicted in green with respective TCRA6 concentrations and percentages of cells in sorting gates. Negative controls without antigen incubation are shown in gray. Surface presentation was detected by anti-c-myc antibody FITC conjugate, and antigen binding was analyzed using DyLight650-labeled TCRA6. Density plots were created by FlowJo v10 Software.
Sequence analysis of four randomly selected scFv-displaying yeast single clones revealed four unique anti-TCRA6 (aTCRA6) scFv candidates (S1, S4, S7, and S10), which were heterogeneously expressed in Escherichia coli. To investigate, whether the scFvs specifically recognize TCRA6 via CDR binding, flow cytometric studies were performed by incubation of the respective scFvs with Jurkat wild-type (WT) T cells, a cell line derived from an acute T cell leukemia patient, which displays a cell-specific TCR differing from the target TCRA6.42 Besides, on-target binding was verified using an engineered Jurkat T cell line exclusively expressing the target TCRA6, referred to as Jurkat TCRA6, which was generated by a successive approach of CRISPR-Cas9-based knockout of the inherent Jurkat αβ TCR and lentiviral transduction with the α and β chain of TCRA6. While two antibody single chains, aTCRA6 S4 and S7, revealed equal interaction with Jurkat TCRA6 target cells and Jurkat WT off-target cells, suggesting binding to constant portions of the TCR, S1 and S10, demonstrated 12- and 27-fold increased binding of target cells compared to off-target cells, respectively, indicating specificity for TCRA6 (Figure S2). The two best-performing scFvs were subsequently scrutinized in a biolayer interferometry (BLI) measurement that validated binding to immobilized TCRA6. Thereby, candidate aTCRA6 S1 showed superior binding properties compared to aTCRA6 S10 (Figure S3A). Affine binding was further demonstrated in cellular binding assays with Jurkat TCRA6 cells (Figure S3B). To exclude interaction with constant and off-target variable TCR regions, binding to polyclonal T lymphocytes derived from human healthy donor blood was investigated by flow cytometry (Figure S3C). The scFv aTCRA6 S1 was selected as the lead candidate due to its remarkable binding to TCRA6 on molecular and cellular level, while not targeting Jurkat WT cells as well as primary T lymphocytes from human blood, thus providing the desired anti-idiotypic properties.
Generation and functional characterization of anti-TCRA6 antibody
The scFv aTCRA6 S1 was reformatted as scFv-Fc fusion and as Fab-Fc full-length, chimeric chicken-human IgG antibody, expressed in Expi293F and purified via protein A affinity chromatography. Integrity, size, and purity of the proteins were initially confirmed using reducing SDS-PAGE (Figure S4A). Size exclusion chromatography (SEC) revealed favorable aggregation properties, with profiles displaying high uniformity and expected retention times of the analytes under native conditions (Figure S4B). Thermal stability, investigated via differential scanning fluorimetry, was determined by melting temperatures of 60°C for both aTCRA6 scFv-Fc and aTCRA6 Fab-Fc (Figure S4C). In order to quantify binding kinetics, BLI measurements were conducted by loading the antibodies onto biosensors and subsequent association of TCRA6 in varying concentrations. Both formats of aTCRA6 showed excellent apparent affinities amounting to 5.0 nM (aTCRA6 scFv-Fc) and 1.6 nM (aTCRA6 Fab-Fc) and exhibited low off-rates, which indicated slow dissociation (Figure 3A). In line with molecular interactions, titration of aTCRA6 antibodies on Jurkat TCRA6 target cells resulted in curves demonstrating strong binding with estimated on-cell affinities of 24 nM (aTCRA6 scFv-Fc) and 6 nM (aTCRA6 Fab-Fc), while no off-target binding was observed on Jurkat WT T cells (Figure 3B). Idiotype specificity of aTCRA6 antibodies was further confirmed by 220-fold (aTCRA6 scFv-Fc) and 660-fold (aTCRA6 Fab-Fc) increased binding of target cells compared to off-target cells using saturating antibody concentrations, respectively. Taken together, the aTCRA6 scFv-Fc/Fab-Fc variants unveiled eminent biophysical properties combined with excellent antigen-binding abilities in molecular and cellular setups. Overall, the full-length aTCRA6 antibody in Fab format outperformed scFv-Fc fusion in terms of binding affinity.
Figure 3.

Binding properties of aTCRA6 scFv-Fc/Fab-Fc
(A) Binding kinetics. BLI measurement of aTCRA6 scFv-Fc (left) and aTCRA6 Fab-Fc (right) loaded onto AHC biosensor tips and associated with 3.9–62.5 nM TCRA6. Kinetic parameters were estimated using Savitzky-Golay filtering and 1:1 Langmuir modeling. (B) Cellular binding. Flow cytometry analysis of Jurkat TCRA6 target cells and Jurkat WT off-target T cells incubated with varying concentrations of aTCRA6 scFv-Fc (1–1,000 nM; left) or aTCRA6 Fab-Fc (0.5–500 nM; right) and stained via anti-human IgG Fc-PE secondary detection antibody. On-cell KDs were determined using variable slope four-parameter fit. Results are shown as mean RFU, and error bars represent standard deviation derived from experimental triplicates. Data are representative of three independent experiments.
Cytotoxicity of anti-TCRA6 ADCs
Engendering cytotoxic effects, we generated ADCs based on the identified aTCRA6 Fab-Fc antibody (sequence provided in Table S1). To this end, aTCRA6 was provided with either 2 or up to 8 (average DAR ∼5) payload units consisting of PEG4 linker, valine-citrulline dipeptide serving as cathepsin substrate, p-aminobenzyl alcohol self-immolative spacer, and the cytotoxin MMAE (Figure 4A). A DAR of 2 was obtained through a site-specific two-step approach of enzyme-mediated azide modification of the heavy chain’s C terminus equipped with a recognition sequence for lipoate-protein ligase A and click chemistry using DBCO-modified payload.40 ADCs armed with approximately 5 cytotoxins on average, referred to as DAR52-8, were generated via partial reduction of endogenous interchain cysteines and subsequent click reaction of thiols with maleimide-conjugated payload. Degree of conjugation was assessed by hydrophobic interaction chromatography (HIC), revealing successful payload attachment without remaining unconjugated fractions for both strategies (Figures S5B and S5C). By integration of signals, the average DAR for the cysteine-coupled aTCRA6 ADC was determined to be 4.9 (Figure S5C).
Figure 4.

Internalization and cytotoxicity of aTCRA6 ADC variants
(A) Schematic representation of aTCRA6 ADCs. In aTCRA6 DAR2 (left), the anti-TCRA6 heavy chains are C-terminally modified with DBCO-PEG4-Val-Cit-PAB-MMAE. In aTCRA6 DAR52-8 (right), the interchain cysteines of anti-TCRA6 mAb are conjugated to maleimide-PEG4-Val-Cit-PAB-MMAE. DBCO, dibenzocyclooctyne; PEG, polyethylene glycol; Val, valine, Cit, citrulline; PAB, p-aminobenzyl alcohol; MMAE, monomethyl auristatin E. (B) Cytotoxicity was assessed by exposition of Jurkat TCRA6 target cells and Jurkat WT off-target cells to aTCRA6 DAR2 (0.06–400 nM) and aTCRA6 DAR52-8 (0.005–30 nM) for 72 h. Cell proliferation was normalized to untreated control cells (0 nM). Internalization was studied by application of pHAb-conjugated aTCRA6 antibody in varying concentrations (0.04–250 nM) to Jurkat TCRA6 target cells and Jurkat WT off-target cells and incubation overnight. Fold internalization was calculated by the ratio of relative fluorescence units (RFU) of the respective antibody sample and the untreated sample without antibody (0 nM). EC50s were determined using variable slope four-parameter fit. Results are shown as mean, and error bars represent standard deviation derived from experimental triplicates. Data are representative of at least two independent experiments.
Since efficient internalization is a prerequisite for ADCs to properly exert their anti-proliferative effects, we first investigated internalization properties of aTCRA6. Therefore, the mAb was labeled with a pH-dependent dye exclusively exerting fluorescence under acidic conditions as given upon internalization and subsequent trafficking into endosomes and lysosomal vesicles.43 Incubation of pH-dye-conjugated aTCRA6 with Jurkat TCRA6 target and Jurkat WT off-target cells was conducted overnight, followed by flow cytometric analysis. The fraction of endocytosed aTCRA6 increased in a concentration-dependent manner for Jurkat TCRA6 cells (Figure 4B). However, internalization in Jurkat WT cells was significantly lower, overall indicating potent and specific receptor-mediated uptake.
In order to evaluate in vitro cytotoxicity of the generated ADCs, Jurkat TCRA6 target cells and Jurkat WT off-target cells were exposed to the DAR2 and DAR52-8 ADCs for 72 h. Both ADCs exerted a robust concentration-dependent anti-proliferative effect on target T cells (Figure 4B). As expected, higher antibody drug load of aTCRA6 resulted in augmented cytotoxicity with the DAR2 ADC revealing an EC50 of 3.6 nM and 93% maximal killing and the DAR52-8 ADC exhibiting an EC50 of 0.1 nM and 97% maximal killing. Surprisingly, the half maximal effective concentration was decreased by a factor of 30 for the higher DAR ADC variant, indicating a markedly more potent drug delivery and intracellular MMAE release. Jurkat WT cells, serving as negative control, remained almost unaffected by ADC treatment. Application of ADCs at concentrations exceeding 100 nM presumably caused death of off-target cells due to unspecific ADC uptake.
To further scrutinize the cytotoxic properties of the generated ADCs, we measured induction of apoptosis in TCRA6-expressing target cells compared to Jurkat WT cells. Therefore, cells were exposed to ADCs at concentrations triggering maximal killing for a period of 72 h, stained with annexin V-FITC/propidium iodide (PI), and evaluated in microscopic and flow cytometric analysis. Treatment of target cells with both 300 nM aTCRA6 DAR2 and 30 nM aTCRA6 DAR52-8, resulted in significantly higher proportions of early apoptotic cells, as indicated by annexin V binding to phosphatidylserines exposed on the outer side of the cell membrane, as well as late apoptotic/necrotic cells with completely compromised cell membranes, as suggested by PI staining of cellular DNA (Figures 5 and S7). Compared to untreated controls (0 nM), the aTCRA6 DAR2 and DAR52-8 ADCs provoked 11- and 13-fold increase of overall annexin V positivity in Jurkat TCRA6 cells, respectively (Figure 6). Morphologically, the cells displayed strong apoptotic signs post ADC regimen, which can be attributed to the anti-mitotic agent MMAE delivered by the antibody, which is described to cause cell-cycle arrest in G2/M phase and subsequent induction of apoptosis (Figure S7).44,45 In TCRA6-negative Jurkat WT cells, induction of apoptosis by ADC administration was observed in a minor section for DAR2 (12%) and was barely detectable for DAR52-8 (5%). Consistent with the observed cytotoxic properties, the DAR52-8 ADC demonstrated superior total cell death induction, defined by effective concentration, maximal killing, and off-target effect, compared to the DAR2 ADC. This renders aTCRA6 DAR52-8 an ideal, promising molecule to combat T cell cancer, offering a wide therapeutic window by high tumor selectivity through anti-idiotypic properties and high potency via effective drug load.
Figure 5.

Apoptosis induction of aTCRA6 ADC variants
Jurkat TCRA6 target cells and Jurkat WT off-target cells were exposed to 300 nM aTCRA6 DAR2 and 30 nM aTCRA6 DAR52-8 for 72 h. Cells were stained with annexin V-FITC and propidium iodide (PI) and analyzed by flow cytometry. Percentage of annexin V-FITC+/PI+ and annexin V-FITC+/PI− (apoptotic cells) is depicted in right bar chart. Significant differences (p < 0.05) between ADCs and 0 nM control were determined using an unpaired, two-tailed t test and are depicted by ∗ (0.1234 (ns), 0.0332 (∗), 0.0021 (∗∗), 0.0002 (∗∗∗), and <0.0001 (∗∗∗∗)). Results are shown as mean, and error bars represent standard deviation derived from experimental triplicates. Data are representative of two independent experiments.
Figure 6.

Bystander activity of aTCRA6 ADC variants
(A) Direct killing was determined by exposition of Jurkat TCRA6 target cells to aTCRA6 DAR2 (0.05–300 nM) and aTCRA6 DAR52-8 (0.005–30 nM) for 96 h. (B) Supernatants of pre-treated Jurkat TCRA6 target cells were transferred to freshly seeded Jurkat WT off-target cells in a ratio of 1:1 and incubated for 96 h. Results are shown as mean, and error bars represent standard deviation derived from experimental duplicates. Data are representative of at least two independent experiments.
Bystander effect of anti-TCRA6 ADCs
Depending on the developmental stage of the T cell upon malignant transformation, oligoclonal patterns have been observed in lymphoma cell populations.1 While leukemias arrest at a certain stage of development and mature T cell-derived cancers have completed TCR rearrangement and form monoclonal lymphomas/leukemias, immature lymphoid precursor cells progress through sequential rearrangements that result in a clonotypic heterogeneous pool of lymphoma cells.28,46,47 Moreover, downregulation of TCR expression has been observed in cancer cells.18,29 Circumventing such tumor escape mechanisms, an ADC is capable to mediate bystander effects, (mainly) by internalization and degradation in target cells and subsequent permeation and diffusion of free payload, leading to the death of surrounding cells. Investigating the in vitro bystander activity of our aTCRA6 DAR2 and aTCRA6 DAR52-8 ADCs, direct cell killing was measured using Jurkat TCRA6 target cells, and bystander killing was analyzed by transfer of respective cell culture supernatants to Jurkat WT off-target cells. Consistent with our preliminary cytotoxicity data, direct killing of TCRA6-positive Jurkat cells was potently induced by the ADCs post 96 h (Figure 6A). Subsequently, cell culture supernatants of aTCRA6 ADC- and mAb-treated target cells were applied to TCRA6-negative Jurkat WT cells and incubated for an additional 96 h. Although target-negative Jurkat WT cells alone are sensitive to MMAE, but insensitive to aTCRA6 ADCs, supernatant transfer triggered cell death in a dose-dependent manner (Figures 4, S6, and 6B). Compared to half maximal effective doses for direct killing of target cells, EC50s for bystander killing of target-negative cells are 7- to 14-fold increased, reflecting the effect of released MMAE on surrounding cells. While the primary mechanism of action of aTCRA6 ADCs is targeted delivery of MMAE to TCRA6 idiotype-expressing tumor cells, the cytotoxicity may be augmented by the bystander activity on target-positive as well as target-negative cells located in the tumor microenvironment.48
Discussion
In contrast to the tremendous progress that has been made in the treatment of B cell lymphomas, the development of therapies for T cell-derived cancers remains challenging. Current investigational immunotherapies focus primarily on CAR T cells targeting pan-T cell antigens such as CD4, CD5, and CD7.49,50,51 However, the success in clinical applications is severely hampered by fratricide, T cell aplasia, and product contamination with malignant cells.18,49,52 To overcome the first two hurdles in adopting CAR technology for application in T cell malignancies, antigens with limited expression on normal cells, e.g., CD30, CD37, and CD1a, are addressed.53,54,55 Following this concept, targeting of tumor-specific TCR constant domains has become more popular, ensuring selective eradication of tumor cells. Maciocia et al. made use of the fact that T cells mutually exclusively express the TCR β chain constant domains 1 or 2 (TRBC1 and TRBC2). Thus, TRBC1-based targeting in a CAR T cell setup unveiled compelling in vitro and in vivo anti-tumor effects under preservation of approximately half of the normal T cell population.29 In addition to the issue of product contamination in the case of T cell malignancies, manufacturing of CAR T cells is a logistically time- and resource-intensive process due to the autologous nature of the personalized therapy approach. In the particular case of TRBC1-directed CAR T cells, CAR T cell efficacy may be further hampered by limited persistence due to TCR cross-linking on healthy T lymphocytes and CAR T cell cytolysis. Therefore, alternative strategies have been explored, including bispecific antibodies against distinct variable chains of the TCR β chain, which have proven effective in harnessing the host immune system’s T cells to mediate cytotoxicity via CD3 recruitment in mouse models of human T cell cancers.32 Another appealing antibody-based therapy class is represented by ADCs, which completely exclude bidirectional T cell killing while precisely inflicting chemotherapeutic damage on tumor cells.
In this study, we developed anti-TCR idiotype ADCs for specific elimination of T cell malignancies. Starting with the identification of an anti-idiotypic scFv derived from a chicken immune library, we reformatted the binder as full-length antibody, which was further conjugated to the cytotoxic payload MMAE (via a cleavable linker) under generation of aTCRA6 ADCs with DARs of 2 and 5 on average. Besides the remarkable single-digit nanomolar affinity of the aTCRA6 antibody on molecular and cellular levels, aTCRA6 ADCs demonstrated precise and potent in vitro anti-tumor activity, predominantly based on the induction of apoptotic cell death. Following release of the anti-mitotic agent MMAE, anti-proliferative bystander effects were observed in off-target cells further contributing to aTCRA6 ADCs’ tumoricidal properties.
ADCs share a common architecture including a mAb component covalently bound to a cytotoxic payload via a synthetic linker. However, ADCs based on the same mAb carrier may exhibit different potencies, safeties, and pharmacokinetics due to independently modifiable key parameters such as linker technology, drug properties, as well as stoichiometry and placement of warheads.56 Our aTCRA6 ADCs share mAb, protease-labile linker and payload, but they differ in their bioconjugation method, resulting in distinct antibody-payload linkages and DARs (Figure 4A). While chemoenzymatic payload coupling for generation of the DAR2 variant occurred site specifically at the antibody’s C terminus under formation of a triazole, reaction of the reduced interchain cysteines with maleimide-modified payload results in thio-succinimide linkages and a heterogeneous drug load distribution varying from 2 to 8 entities per antibody. By further optimization of the conjugation conditions, a DAR of up to 8 is reportedly achievable.57,58 Expectedly, comparing our two aTCRA6 ADC variants, the ADC with higher payload outperformed the DAR2 ADC in terms of cytotoxicity and associated (absence of) off-target effects. It is common practice to increase the number of drugs per antibody in order to augment the potency of an ADC in vitro, albeit in vivo studies have revealed that excessive hydrophobic drug loading resulted in decreased efficacy due to reduced tumor exposure as they clear faster from circulation.59,60,61 Previous investigations using anti-CD30 antibody brentuximab (or cAC10) in interchain cysteine conjugation-based MMAE ADCs containing DARs of 2, 4, and 8 determined the highest therapeutic index for DAR4 ADCs in xenograft-bearing immunodeficient mice.62 Encouraging cancer treatment outcomes with cAC10-Val-Cit-PABC-MMAE, known as brentuximab vedotin, which consists of an average of four molecules of MMAE attached to the interchain cysteine residues, led to the first FDA approval of an ADC in 2011, for therapy of relapsed Hodgkin lymphoma and relapsed systemic anaplastic large cell lymphoma (ALCL).63 Since tumors with a low percentage of CD30-positive cells can exhibit a comparable clinical response to brentuximab vedotin as high-CD30-expressing disease, bystander killing mechanisms likely contribute to ADC-mediated anti-tumor activity.64,65 Linker design and drug chemistry dictate the ability of the cytotoxin to diffuse into surrounding cells and exert bystander effects, enabling the elimination of heterogeneous target-expressing populations.59,66 Bystander activity may also be valuable for TCR targeting, as cancerous T cells often downregulate expression of the TCR or form oligoclonal populations in case of transformed lymphoid precursors undergoing TCR rearrangement.1,18,29 However, in PTCL, malignant transformation mostly occurs after TCR rearrangement, resulting in monoclonality and over 95% TCR expression.1,25 In leukemias (e.g., T-ALL), T cells become arrested at a certain stage of development. Consequently, in one-third of leukemia cases where TCRs are displayed, the cancerous populations are of monoclonal nature.5,67 While pioneering approaches of Levy and Stevenson specifically addressing the clonally rearranged cell surface receptor in B cell lymphomas were superseded by pan-B cell targeting immunotherapies with demonstrable safety profiles, this concept may be rational in the context of T cell-derived cancers.68,69 By precise targeting of malignant T cells, the relative T cell repertoire integrity is maintained, circumventing immunodeficiency and T cell aplasia.
An anthracycline-based treatment has been considered the standard frontline regimen for most types of PTCL, despite the fact that in most cases, results are suboptimal.70,71 We propose an individualized strategy of tumor eradication feasible through TCR sequencing in order to identify the disease-causing T cell clone, providing a novel T cell lymphoma/leukemia therapy.72 Thus, patients newly diagnosed with T cell diseases may receive chemotherapeutic first-line treatment to control tumor burden, and in case of relapse, TCR sequencing could be performed followed by generation of anti-TCR idiotype antibodies as exemplary demonstrated by chicken immunization. However, the anti-idiotype approach necessitates the production of a custom-made antibody for each individual patient. At present, implementation of personalized therapies is not as advanced, since time and resources required to manufacture and obtain regulatory approval frequently exceed therapeutic value. Nevertheless, individualized immunotherapy promises to broaden the responder patient population by highly specific targeting of tumor cells.73 Attempts utilizing patient-tailored antibodies have primarily focused on hematologic cancers, achieving promising results, e.g., in multiple myeloma patients.74,75,76,77,78 However, as an additional technical hurdle, humanization of the avian-derived molecule is imperative to produce a non-immunogenic antibody variant for therapeutic applications, which can be implemented by a straightforward method of chicken CDR grafting onto a human germline framework based on Vernier residue randomization, as previously described by our group.79,80 Regarding drug resistance, patients may experience TCR-negative relapses following TCR-directed therapy, as frequently observed in previous clinical applications of single-antigen targeting biologics.81,82,83 In addition, administration of ADCs for therapeutic purposes is often hampered by dose-limiting toxicities.84,85 Ultimately, preclinical and early clinical studies are warranted to evaluate the feasibility and toxicity of anti-TCR idiotype-targeting ADCs for treatment of T cell malignancies.
In summary, we have demonstrated a unique approach to combat malignant T cells without inducing systemic immunosuppression derived from ablation of the entire T cell subset. With the development of anti-TCR idiotype ADCs, we provide proof of concept for precise tumor targeting by recognizing the clonally rearranged T lymphocyte receptor, potent anti-tumor activity based on the highly cytotoxic payload, and a favorable safety profile due to the exclusion of off-target effects. Our study may contribute to the future lymphoma/leukemia management by delivering a novel targeted immunotherapeutic starting point urgently needed in the setting of T cell-driven pathologies.
Materials and methods
Immunization of chicken and construction of yeast library
Chicken immunization and scFv YSD library construction were performed as reported previously.41,86 Briefly, a pathogen-free adult laying hen (Gallus gallus domesticus) was immunized with TCRA6 (produced in-house) in combination with immune adjuvant AddaVax (InvivoGen, Toulouse, France) on days 1, 14, 28, 35, and 56.37 The animal was sacrificed on day 63 for spleen resection and subsequent isolation of RNA. Additionally, antibody serum titer against TCRA6 was determined by ELISA. The immunization procedure as well as RNA extraction were executed at Davids Biotechnologie (Regensburg, Germany). Ethical approval for animal immunization was granted to Davids Biotechnologie (Regensburg, Germany). The experimental procedures and the care of the animals complied with EU animal welfare laws and regulations. For library generation, cDNA was synthesized from total splenic RNA. Subsequently, genes encoding VH and VL were amplified, randomly combined into scFv gene strings, and transferred into a linearized YSD vector (pCT) via homologous recombination in Saccharomyces cerevisiae strain EBY100 (MATa URA3-52 trp1 leu2Δ1 his3Δ200 pep4:HIS3 prb1Δ1.6R can1 GAL [pIU211:URA3]) (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Library generation in EBY100 cells was conducted according to the yeast transformation protocol published by Benatuil et al.87
Yeast library screening
Induction of scFv expression and surface presentation were accomplished by inoculation of yeast cells in synthetic galactose minimal medium with casein amino acids (SG-CAA) and incubation overnight at 30°C and 180 rpm. For library sorting, cells were harvested by centrifugation and washed once with PBS+0.1% (w/v) BSA (PBS-B). Antigen staining was conducted with DyLight650-labeled TCRA6 conjugated beforehand using 5-fold excess of DyLight650 NHS Ester (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Simultaneously, staining for surface presentation using anti-c-myc antibody FITC conjugate (Miltenyi Biotec, Bergisch Gladbach, Germany; diluted 1:50) was performed for 30 min on ice. After washing three times with PBS-B, the yeast library was screened using BD Influx cell sorter with corresponding BD FACS Software v1.0 (BD, California, USA). During the sorting process collected yeast cells were plated on synthetic dextrose minimal medium with casein amino acids (SD-CAA) agar plates and incubated for 48 h at 30°C. General handling of yeast cells and scFv YSD library screening by FACS are described elsewhere.86
Expression and purification of scFv, scFv-Fc, and Fab-Fc constructs
Reformatting, expression, and purification of scFvs were performed as described previously.88 In brief, isolated yeast vectors were sequenced, and scFv-encoding genes were cloned into a pET30 plasmid using golden gate assembly (GGA), followed by recombinant expression in Escherichia coli SHuffle T7 Express (New England Biolabs, Ipswich, Massachusetts, USA). A two-step affinity purification was conducted including IMAC and Strep-TactinXT purification, followed by buffer exchange against PBS. Production of Fc-fused scFvs and full-length antibodies (Fab-Fc) was performed by transfection of Expi293F cells with pTT5-derived GGA vectors and ExpiFectamine 293 Transfection Kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA). For purification of Fc-containing antibody constructs, cell culture supernatants were collected 5 days after transfection, sterile filtered, and applied to a HiTrap protein An HP column (GE Healthcare, Piscataway, New Jersey, USA) using an ÄKTA pure chromatography system (GE Healthcare, Piscataway, New Jersey, USA). Buffer exchange against PBS was performed using a HiTrap Desalting column (GE Healthcare, Piscataway, New Jersey, USA).
Generation of Jurkat TCRA6 cell line
Engineered Jurkat cells exclusively expressing the TCRA6 were generated in succession by CRISPR-Cas9-based knockout of the individual Jurkat αβ TCR chains, enrichment by FACS, and lentiviral transduction of the respective TCRA6 α and β chains.
To this end, knockout of the endogenous Jurkat αβ TCR chains was performed as described in the Amaxa 4D-Nucleofector protocol by nucleofection of 3 × 105 Jurkat WT cells using the 4D-Nucleofector, the SE Cell Line Kit, and the CK-116 nucleofection program (Lonza, Switzerland). A Cas9/gRNA ribonucleoprotein complex (PNA Bio, Newbury Park, California, USA; 20 pmol) with chemically modified sgRNAs (Synthego Menlo Park, California, USA; 100 pmol) targeting the TRAC locus (5′-AGAGUCUCUCAGCUGGUACA-3′, targeting exon 1) and TRBC1 locus (5′-CUUUCCAGAGGACCUGAACA-3′, targeting exon 1) were utilized to achieve optimal nucleofection efficiency.89 Following nucleofection and 1-week expansion, engineered Jurkat cells were washed with PBS both before and after incubation with an anti-αβ TCR BUV737-conjugated antibody (BD Horizon, Becton Dickinson, New Jersey, USA; diluted 1:40) for 30 min at 4°C to confirm the absence of αβ TCR surface expression using the FACS Celesta Cell Analyzer (BD, California, USA). Subsequently TCR-negative Jurkat cells were enriched using the FACS Aria Fusion (BD, California, USA).
For further engineering of the TCR-negative Jurkat cells, the encoding DNA sequence for TCRA6 α/β chain was cloned into a modified version of the pSLCAR-CD19-28z lentiviral transfer plasmid (Addgene plasmid #135991) generated by Scott McComb and colleagues.90 Production of lentiviral particles with the generated TCRA6 vectors was conducted using a 3rd generation plasmid system as described recently, with minor modifications of the protocol.91 Afterward 1 × 105 TCR-negative Jurkat cells were transduced by spinfection (32°C, 800 × g, 90 min) with a multiplicity of infection of 3–5, expanded for 2 weeks, and enriched for αβ TCR-positive surface expression as described using the FACS Aria Fusion (BD, California, USA).
Cell lines
T cells including Jurkat WT (Clone E6-1, DSMZ ACC 282) and Jurkat TCRA6 cells were cultured at 37°C and 5% CO2. Jurkat WT cells were maintained in RPMI-1640 supplemented with 10% FBS and 1% penicillin-streptomycin. Jurkat TCRA6 cells were maintained in RPMI-1640 supplemented with 20% FBS, 10 mM HEPES, and 1% penicillin-streptomycin. Cells were sub-cultured every 2–3 days. Expi293F cells were cultured in Expi293 Expression Medium (Thermo Fisher Scientific, Waltham, Massachusetts, USA), sub-cultured every 3–4 days, and incubated at 37°C and 8% CO2.
Thermal shift assay
Thermal stability was analyzed by differential scanning fluorimetry using a CFX Connect Real-Time PCR Detection System (Bio-Rad, California, USA) with a temperature gradient from 20°C to 95°C and 0.5°C/10 s. The derivatives of the melt curves were calculated with the corresponding Bio-Rad CFX Maestro software to determine the melt temperatures (Tm). All reactions were performed in PBS in presence of 1 mg/mL protein and SYPRO Orange (Thermo Fisher Scientific, Waltham, Massachusetts, USA; diluted 1:00).
Biolayer interferometry
For biolayer interferometric measurements, the Octet RED96 system (ForteBio, Sartorius, Göttingen, Germany) was used. Therefore, respective biosensor tips were soaked in PBS (pH 7.4) for at least 10 min before start of the assay.
For testing of two chicken-derived scFvs aTCRA6 S1 and S10, High Precision Streptavidin biosensors (SAX; Sartorius, Sartorius, Göttingen, Germany) were loaded with biotinylated TCRA6. All following steps were performed using kinetics buffer (KB; Sartorius, Göttingen, Germany). Association was measured for 200 s with 500 nM of respective scFvs followed by dissociation for 200 s.
Kinetics measurements were conducted for affinity determination of aTCRA6 scFv-Fc and aTCRA6 Fab-Fc. Anti-human IgG Fc capture biosensors (AHC; Sartorius Sartorius, Göttingen, Germany) were used to immobilize the antibodies. After a quenching step in KB (Sartorius, Göttingen, Germany), an association step using TCRA6 (produced in-house) with concentrations ranging from 3.9–62.5 nM was performed followed by a dissociation step in KB (400 s/step). Association in KB served as reference and was subtracted prior to evaluation steps. Data analysis was performed using ForteBio data analysis software 9.0. Binding kinetics including the equilibrium constant KD were determined using Savitzky-Golay filtering and 1:1 Langmuir model.
Isolation of primary T cells from human healthy donor blood
Peripheral blood mononuclear cells (PBMCs) and subsequently primary T cells were isolated from buffy coats of fresh blood of healthy donors supplied by the German Red Cross Blood Donation Service (DRK-Blutspendedienst Baden-Württemberg-Hessen, Frankfurt, Germany). This study was approved by the Ethics Committee of the Goethe University Frankfurt, Germany (approval no. 329/10). All participants gave written informed consent in accordance with the Declaration of Helsinki.
Briefly, PBMCs were isolated by density gradient centrifugation using Biocoll (Biochrom, Cambridge, United Kingdom), followed by CD3-positive enrichment of primary T cells using the EasySep Human CD3 Positive Selection Kit II (StemCell Technologies, Vancouver, Canada) according to the instructions provided by the manufacturer. Afterward, primary T cells were cultivated in RPMI-1640 supplemented with 10% human serum (DRK-Blutspendedienst Baden-Württemberg-Hessen, Frankfurt, Germany), 25 mM HEPES, 1% penicillin-streptomycin, and 50 IU/mL IL-2 (PROLEUKIN S, Novartis Pharma, Basel, Switzerland).92
Cellular binding assay
Cellular binding of the antibodies was determined by flow cytometry. Engineered Jurkat TCRA6 cells served as target cells. Primary T cells derived from human healthy donor blood or Jurkat WT cells were used as negative controls to analyze unspecific cell binding. To this end, cells (1.5 × 105 cells/well) were washed with PBS-B and subsequently incubated with the respective aTCRA6 antibody constructs in varying concentrations for 30 min on ice. Followed by another PBS-B washing step, anti-human IgG Fc PE-conjugated secondary antibody (Thermo Fisher Scientific, Waltham, Massachusetts, USA; diluted 1:50), anti-his AF647-conjugated secondary antibody (Thermo Fisher Scientific, Waltham, Massachusetts, USA; diluted 1:50), or mouse anti-his secondary antibody (Qiagen, Venlo, Netherlands; diluted 1:50) and anti-mouse IgG APC-conjugated tertiary antibody (Thermo Fisher Scientific, Waltham, Massachusetts, USA; diluted 1:50) were applied for 20 min on ice. After another washing step with PBS-B, flow cytometry was performed using CytoFLEX S System (Beckman Coulter, Minnesota, USA). In case of antibody affinity titration to cells, the mean fluorescence intensity was plotted against the respective logarithmic concentration. The resulting curves were fitted with a variable slope four-parameter fit using GraphPad Prism.
Internalization assay
Investigations toward receptor-mediated antibody internalization were performed using pHAb Amine Reactive dye (Promega, Wisconsin, USA) according to the manufacturer’s instructions. In brief, aTCRA6 Fab-Fc was conjugated with pHAb dye and applied to Jurkat WT and Jurkat TCRA6 T cells (2 × 104 cells/well) in different concentrations (0.04–250 nM) in a 96-well plate. After incubation overnight, cells were washed once with PBS, and internalization was measured using flow cytometry. Fold internalization was calculated by the ratio of relative fluorescence units (RFU) of the respective antibody sample and the untreated sample without antibody (0 nM). The resulting curves were fitted with a variable slope four-parameter fit.
Generation of ADCs
ADCs with a DAR of 2 were generated via a two-step approach of enzymatic modification and click chemistry reaction for conjugation of MMAE to the Fc fragment. Therefore, the C terminus of the antibody heavy chain was genetically fused with a lipoic acid ligase 12 aa acceptor peptide (LAP), which serves as recognition sequence for lipoate-protein ligase A (LplA) from Escherichia coli.93 Lipoic acid ligase reaction was conducted with 0.1 equivalents (eq.) of a mutant lipoic acid ligase A (LplAW37V)94 accepting various carboxylic acid derivatives in the presence of 5 mM ATP, 5 mM Mg(Ac)2, and 10–20 eq. azide-bearing lipoic acid derivative (synthesized in-house) in PBS (pH 7.4) for 1 h at 37°C. Azide-functionalized antibody was loaded onto protein A resin (protein A HP SpinTrap; Cytiva, Massachusetts, USA), and strain-promoted azide-alkyne [3 + 2] cycloaddition with 5 eq. DBCO-PEG4-Val-Cit-PAB-MMAE was performed overnight at 4°C. After acidic elution of ADC from protein A column, the buffer was exchanged to PBS (pH 7.4), and successful conjugation was confirmed by HIC.
ADCs with a DAR of 2–8 were generated via a two-step approach including partial reduction of interchain disulfide bonds and thiol-maleimide Michael addition click reaction for conjugation of MMAE to the endogenous cysteines. Reduction of cystines was achieved by 2 h, 37°C incubation with 10 eq. tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl). Subsequent reaction with 16 eq. maleimide-PEG4-Val-Cit-PAB-MMAE was performed for 2 h at room temperature, followed by quenching of conjugation reaction using 50 eq. N-acetylcysteine for 15 min at 37°C. The ADC was purified from the reaction mixture using protein A spin columns (protein A HP SpinTrap; Cytiva, Massachusetts, USA). After acidic elution of ADC from protein A resin, the buffer was exchanged to PBS (pH 7.4), and conjugation including DAR distribution was analyzed by HIC.
Hydrophobic interaction chromatography
HIC was performed using TSKGel Butyl-NPR column (Tosoh Bioscience, Griesheim, Germany) in combination with 1260 Infinity chromatography system (Agilent Technologies, Waldbronn, Germany) to analyze successful antibody-MMAE conjugation as well degree of conjugation. Separation was obtained using Eluent A (1.5 M (NH4)2SO4, 25 mM Tris [pH 7.5]) and Eluent B (25 mM Tris [pH 7.5]) in a linear gradient of 0%–100% Eluent B over 25 min at a flow rate of 0.9 mL/min. 30 μg of protein samples were injected at a concentration of 0.3 mg/mL, and protein elution was monitored by absorbance at 220 nm. The average DAR was determined by integration of the absorbance peak areas of the different species using the following equation: , where refers to the individual DAR value and refers to the peak area of the respective DAR species.95
Size exclusion chromatography
Analytical SEC using TSKgel SuperSW3000 column (Tosoh Bioscience, Griesheim, Germany) in combination with 1260 Infinity chromatography system (Agilent Technologies, Waldbronn, Germany) was performed to assess aggregation behavior of antibodies and ADCs. Chromatography was run with 30 μg protein (0.3 mg/mL) at a flow rate of 0.35 mL/min with PBS for 20 min, and protein elution was detected by measuring absorbance at 220 nm.
Cytotoxicity assay
Cytotoxic effects of aTCRA6 DAR2 and aTCRA6 DAR52-8 ADCs were estimated by exposing Jurkat WT and Jurkat TCRA6 T cells to different ADC concentrations. Cell viability was analyzed 72 h post ADC addition by a fluorometric method using CellTiter-Blue Cell Viability Assay (Promega, Wisconsin, USA) according to the manufacturer’s instructions. Briefly, cells were seeded in a 96-well plate (1 × 104 cells/well) with the desired ADC concentrations ranging from 0.06–400 nM (for DAR2 ADC) or 0.005–30 nM (for DAR52-8 ADC) in a serial dilution. After 72 h, redox dye (resazurin) was added to the cells, and the plate was incubated for 3–4 h. Fluorescence of resorufin (560Ex/590Em) was recorded using CLARIOstar plus microplate reader (BMG LABTECH, Offenburg, Germany). Cell proliferation was normalized to untreated control cell fluorescence values. The resulting curves were fitted with a variable slope four-parameter fit, and EC50s were calculated using GraphPad Prism.
Apoptosis assay
To determine induction of apoptosis of aTCRA6 DAR2 and aTCRA6 DAR52-8 ADCs in Jurkat WT and Jurkat TCRA6 cells, annexin V/PI staining was conducted. Therefore, cells were seeded in a 24-well plate (5 × 104 cells/well) and incubated with 300 nM (DAR2 ADC) or 30 nM (DAR52-8 ADC) ADCs for 72 h. Annexin V-FITC/PI staining ROTITEST Annexin V (Carl Roth & Co. KG, Karlsruhe, Germany) was applied for apoptosis detection of T cells according to the manufacturer’s instructions. The analysis was performed using CytoFLEX S System (Beckman Coulter, Minnesota, USA) and the fluorescence microscope Zeiss Axio V.A1 with Axio Cam ICM1 and AxioVision 1.0.1.0 software (Carl Zeiss AG, Jena, Germany).
Bystander killing assay
To investigate bystander activity of ADCs, target Jurkat TCRA6 cells were exposed to ADCs for 4 days, and subsequent supernatant was transferred to off-target Jurkat WT cells, and cell viability was determined after 4 days of incubation. Therefore, Jurkat TCRA6 cells were seeded in a 96-well plate (2 × 104 cells/well) with the desired ADC concentrations ranging from 0.05–300 nM (DAR2 ADC) or 0.005–30 nM (DAR52-8 ADC) in a serial dilution. For direct killing, Jurkat TCRA6 cells were seeded at a density of 1 × 104 cells/well. After 96 h, viability readout for direct cell killing was measured using CellTiter-Blue Cell Viability Assay (Promega, Wisconsin, USA). For bystander effect analysis, target-negative Jurkat WT cells were seeded at a density of 1 × 104 cells/well after 96 h of initial ADC incubation. Centrifuged cell culture supernatant of the ADC-treated target-positive Jurkat TCRA6 cells was transferred to the seeded Jurkat WT cells and incubated for 96 h at 37°C with 5% CO2. Cell viability was determined using the CellTiter-Blue Cell Viability Assay (Promega, Wisconsin, USA) and CLARIOstar plus microplate reader (BMG LABTECH, Offenburg, Germany), with the detailed protocol described in section cytotoxicity assay. Cell proliferation was normalized to untreated control cell fluorescence values. The resulting curves were fitted with a variable slope four-parameter fit, and EC50s were calculated using GraphPad Prism.
Data and code availability
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Acknowledgments
The authors would like to thank Peter Bitsch for synthesis of azide-modified lipoic acid as well as technical support in bioconjugation analytics. We acknowledge support by the German Research Foundation (grant KO1390/14-1) and the Open Access Publishing Fund of Technical University of Darmstadt.
Author contributions
K.S.: conceptualization, investigation, data curation, and writing – original draft. J. Habermann: conceptualization, investigation, and writing – review & editing. P.W.: conceptualization, investigation, and writing – review & editing. J. Harwardt: investigation and writing – review & editing. E.U.: conceptualization and writing - review & editing. H.K.: conceptualization, project administration, and writing – original draft.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omton.2024.200850.
Supplemental information
References
- 1.Iyer A., Hennessey D., Gniadecki R. Clonotype pattern in T-cell lymphomas map the cell of origin to immature lymphoid precursors. Blood Adv. 2022;6:2334–2345. doi: 10.1182/bloodadvances.2021005884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Belver L., Ferrando A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer. 2016;16:494–507. doi: 10.1038/nrc.2016.63. [DOI] [PubMed] [Google Scholar]
- 3.Malcolm T.I.M., Hodson D.J., Macintyre E.A., Turner S.D. Challenging perspectives on the cellular origins of lymphoma. Open Biol. 2016;6 doi: 10.1098/rsob.160232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.The Non-Hodgkin's Lymphoma Classification Project; A Clinical Evaluation of the International Lymphoma Study Group Classification of Non-Hodgkin's Lymphoma. Blood. 1997;89:3909–3918. [PubMed] [Google Scholar]
- 5.Pui C.-H., Robison L.L., Look A.T. Acute lymphoblastic leukaemia. Lancet (London, England) 2008;371:1030–1043. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 6.Bhansali R.S., Barta S.K. SOHO State of the Art Updates and Next Questions | Challenging Cases in Rare T-Cell Lymphomas. Clin. Lymphoma Myeloma Leuk. 2023;23:642–650. doi: 10.1016/j.clml.2023.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asselin B.L., Devidas M., Wang C., Pullen J., Borowitz M.J., Hutchison R., Lipshultz S.E., Camitta B.M. Effectiveness of high-dose methotrexate in T-cell lymphoblastic leukemia and advanced-stage lymphoblastic lymphoma: a randomized study by the Children's Oncology Group (POG 9404) Blood. 2011;118:874–883. doi: 10.1182/blood-2010-06-292615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mak V., Hamm J., Chhanabhai M., Shenkier T., Klasa R., Sehn L.H., Villa D., Gascoyne R.D., Connors J.M., Savage K.J. Survival of patients with peripheral T-cell lymphoma after first relapse or progression: spectrum of disease and rare long-term survivors. J. Clin. Oncol. 2013;31:1970–1976. doi: 10.1200/JCO.2012.44.7524. [DOI] [PubMed] [Google Scholar]
- 9.Schmitz N., Lenz G., Stelljes M. Allogeneic hematopoietic stem cell transplantation for T-cell lymphomas. Blood. 2018;132:245–253. doi: 10.1182/blood-2018-01-791335. [DOI] [PubMed] [Google Scholar]
- 10.Teachey D.T., Pui C.-H. Comparative features and outcomes between paediatric T-cell and B-cell acute lymphoblastic leukaemia. Lancet Oncol. 2019;20:e142–e154. doi: 10.1016/S1470-2045(19)30031-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bock A.M., Nowakowski G.S., Wang Y. Bispecific Antibodies for Non-Hodgkin Lymphoma Treatment. Curr. Treat. Options Oncol. 2022;23:155–170. doi: 10.1007/s11864-021-00925-1. [DOI] [PubMed] [Google Scholar]
- 12.Bellei M., Foss F.M., Shustov A.R., Horwitz S.M., Marcheselli L., Kim W.S., Cabrera M.E., Dlouhy I., Nagler A., Advani R.H., et al. The outcome of peripheral T-cell lymphoma patients failing first-line therapy: a report from the prospective, International T-Cell Project. Haematologica. 2018;103:1191–1197. doi: 10.3324/haematol.2017.186577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raetz E.A., Teachey D.T. T-cell acute lymphoblastic leukemia. Hematology. 2016;2016:580–588. doi: 10.1182/asheducation-2016.1.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maude S.L., Teachey D.T., Porter D.L., Grupp S.A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015;125:4017–4023. doi: 10.1182/blood-2014-12-580068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cappell K.M., Kochenderfer J.N. Long-term outcomes following CAR T cell therapy: what we know so far. Nat. Rev. Clin. Oncol. 2023;20:359–371. doi: 10.1038/s41571-023-00754-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein C., Jamois C., Nielsen T. Anti-CD20 treatment for B-cell malignancies: current status and future directions. Expert Opin. Biol. Ther. 2021;21:161–181. doi: 10.1080/14712598.2020.1822318. [DOI] [PubMed] [Google Scholar]
- 17.Zinzani P.L., Minotti G. Anti-CD19 monoclonal antibodies for the treatment of relapsed or refractory B-cell malignancies: a narrative review with focus on diffuse large B-cell lymphoma. J. Cancer Res. Clin. Oncol. 2022;148:177–190. doi: 10.1007/s00432-021-03833-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fleischer L.C., Spencer H.T., Raikar S.S. Targeting T cell malignancies using CAR-based immunotherapy: challenges and potential solutions. J. Hematol. Oncol. 2019;12:141. doi: 10.1186/s13045-019-0801-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iżykowska K., Rassek K., Korsak D., Przybylski G.K. Novel targeted therapies of T cell lymphomas. J. Hematol. Oncol. 2020;13:176. doi: 10.1186/s13045-020-01006-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makita S., Tobinai K. Mogamulizumab for the treatment of T-cell lymphoma. Expert Opin. Biol. Ther. 2017;17:1145–1153. doi: 10.1080/14712598.2017.1347634. [DOI] [PubMed] [Google Scholar]
- 21.Richardson N.C., Kasamon Y.L., Chen H., Claro R.A. de, Ye J., Blumenthal G.M., Farrell A.T., Pazdur R. FDA Approval Summary: Brentuximab Vedotin in First-Line Treatment of Peripheral T-Cell Lymphoma. Oncologist. 2019;24:e180–e187. doi: 10.1634/theoncologist.2019-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davis M.M., Bjorkman P.J. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334:395–402. doi: 10.1038/334395a0. [DOI] [PubMed] [Google Scholar]
- 23.Robins H.S., Campregher P.V., Srivastava S.K., Wacher A., Turtle C.J., Kahsai O., Riddell S.R., Warren E.H., Carlson C.S. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood. 2009;114:4099–4107. doi: 10.1182/blood-2009-04-217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rudolph M.G., Stanfield R.L., Wilson I.A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006;24:419–466. doi: 10.1146/annurev.immunol.23.021704.115658. [DOI] [PubMed] [Google Scholar]
- 25.Went P., Agostinelli C., Gallamini A., Piccaluga P.P., Ascani S., Sabattini E., Bacci F., Falini B., Motta T., Paulli M., et al. Marker expression in peripheral T-cell lymphoma: a proposed clinical-pathologic prognostic score. J. Clin. Oncol. 2006;24:2472–2479. doi: 10.1200/JCO.2005.03.6327. [DOI] [PubMed] [Google Scholar]
- 26.Jamal S., Picker L.J., Aquino D.B., McKenna R.W., Dawson D.B., Kroft S.H. Immunophenotypic analysis of peripheral T-cell neoplasms. A multiparameter flow cytometric approach. Am. J. Clin. Pathol. 2001;116:512–526. doi: 10.1309/QF6N-VAQW-N74H-4JE2. [DOI] [PubMed] [Google Scholar]
- 27.Foucar K. Mature T-cell leukemias including T-prolymphocytic leukemia, adult T-cell leukemia/lymphoma, and Sézary syndrome. Am. J. Clin. Pathol. 2007;127:496–510. doi: 10.1309/KWJYBCCGTB90B6AE. [DOI] [PubMed] [Google Scholar]
- 28.Asnafi V., Beldjord K., Boulanger E., Comba B., Le Tutour P., Estienne M.-H., Davi F., Landman-Parker J., Quartier P., Buzyn A., et al. Analysis of TCR, pT alpha, and RAG-1 in T-acute lymphoblastic leukemias improves understanding of early human T-lymphoid lineage commitment. Blood. 2003;101:2693–2703. doi: 10.1182/blood-2002-08-2438. [DOI] [PubMed] [Google Scholar]
- 29.Maciocia P.M., Wawrzyniecka P.A., Philip B., Ricciardelli I., Akarca A.U., Onuoha S.C., Legut M., Cole D.K., Sewell A.K., Gritti G., et al. Targeting the T cell receptor β-chain constant region for immunotherapy of T cell malignancies. Nat. Med. 2017;23:1416–1423. doi: 10.1038/nm.4444. [DOI] [PubMed] [Google Scholar]
- 30.Kim H., Kim I.S., Chang C.L., Kong S.Y., Lim Y.T., Kong S.G., Cho E.H., Lee E.Y., Shin H.J., Park H.J., et al. T-Cell Receptor Rearrangements Determined Using Fragment Analysis in Patients With T-Acute Lymphoblastic Leukemia. Ann. Lab. Med. 2019;39:125–132. doi: 10.3343/alm.2019.39.2.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oon M.L., Lim J.Q., Lee B., Leong S.M., Soon G.S.-T., Wong Z.W., Lim E.H., Li Z., Yeoh A.E.J., Chen S., et al. T-Cell Lymphoma Clonality by Copy Number Variation Analysis of T-Cell Receptor Genes. Cancers. 2021;13 doi: 10.3390/cancers13020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paul S., Pearlman A.H., Douglass J., Mog B.J., Hsiue E.H.-C., Hwang M.S., DiNapoli S.R., Konig M.F., Brown P.A., Wright K.M., et al. TCR β chain-directed bispecific antibodies for the treatment of T cell cancers. Sci. Transl. Med. 2021;13 doi: 10.1126/scitranslmed.abd3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li F., Zhang H., Wang W., Yang P., Huang Y., Zhang J., Yan Y., Wang Y., Ding X., Liang J., et al. T cell receptor β-chain-targeting chimeric antigen receptor T cells against T cell malignancies. Nat. Commun. 2022;13:4334. doi: 10.1038/s41467-022-32092-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bona C.A. Idiotypes in Medicine: Autoimmunity, Infection and Cancer. Anti-idiotypes (Elsevier); 1997. [Google Scholar]
- 35.Antonacci R., Massari S., Linguiti G., Caputi Jambrenghi A., Giannico F., Lefranc M.-P., Ciccarese S. Evolution of the T-Cell Receptor (TR) Loci in the Adaptive Immune Response: The Tale of the TRG Locus in Mammals. Genes. 2020;11 doi: 10.3390/genes11060624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong W.K., Leem J., Deane C.M. Comparative Analysis of the CDR Loops of Antigen Receptors. Front. Immunol. 2019;10:2454. doi: 10.3389/fimmu.2019.02454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sádio F., Stadlmayr G., Stadlbauer K., Gräf M., Scharrer A., Rüker F., Wozniak-Knopp G. Stabilization of soluble high-affinity T-cell receptor with de novo disulfide bonds. FEBS Lett. 2020;594:477–490. doi: 10.1002/1873-3468.13616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cole D.K., Sami M., Scott D.R., Rizkallah P.J., Borbulevych O.Y., Todorov P.T., Moysey R.K., Jakobsen B.K., Boulter J.M., Baker B.M., Yi L. Increased Peptide Contacts Govern High Affinity Binding of a Modified TCR Whilst Maintaining a Native pMHC Docking Mode. Front. Immunol. 2013;4:168. doi: 10.3389/fimmu.2013.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furukawa Y., Fujisawa J., Osame M., Toita M., Sonoda S., Kubota R., Ijichi S., Yoshida M. Frequent clonal proliferation of human T-cell leukemia virus type 1 (HTLV-1)-infected T cells in HTLV-1-associated myelopathy (HAM-TSP) Blood. 1992;80:1012–1016. [PubMed] [Google Scholar]
- 40.Schoenfeld K., Harwardt J., Habermann J., Elter A., Kolmar H. Conditional activation of an anti-IgM antibody-drug conjugate for precise B cell lymphoma targeting. Front. Immunol. 2023;14 doi: 10.3389/fimmu.2023.1258700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grzeschik J., Yanakieva D., Roth L., Krah S., Hinz S.C., Elter A., Zollmann T., Schwall G., Zielonka S., Kolmar H. Yeast Surface Display in Combination with Fluorescence-activated Cell Sorting Enables the Rapid Isolation of Antibody Fragments Derived from Immunized Chickens. Biotechnol. J. 2019;14 doi: 10.1002/biot.201800466. [DOI] [PubMed] [Google Scholar]
- 42.Gioia L., Siddique A., Head S.R., Salomon D.R., Su A.I. A genome-wide survey of mutations in the Jurkat cell line. BMC Genom. 2018;19:334. doi: 10.1186/s12864-018-4718-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z., Wang M., Yao X., Li H., Li S., Liu L., Yu D., Li X., Fang J., Huang C. Development of novel anti-CD19 antibody-drug conjugates for B-cell lymphoma treatment. Int. Immunopharmacol. 2018;62:299–308. doi: 10.1016/j.intimp.2018.06.034. [DOI] [PubMed] [Google Scholar]
- 44.Cunningham D., Parajuli K.R., Zhang C., Wang G., Mei J., Zhang Q., Liu S., You Z. Monomethyl Auristatin E Phosphate Inhibits Human Prostate Cancer Growth. Prostate. 2016;76:1420–1430. doi: 10.1002/pros.23226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Best R.L., LaPointe N.E., Azarenko O., Miller H., Genualdi C., Chih S., Shen B.-Q., Jordan M.A., Wilson L., Feinstein S.C., Stagg N.J. Microtubule and tubulin binding and regulation of microtubule dynamics by the antibody drug conjugate (ADC) payload, monomethyl auristatin E (MMAE): Mechanistic insights into MMAE ADC peripheral neuropathy. Toxicol. Appl. Pharmacol. 2021;421 doi: 10.1016/j.taap.2021.115534. [DOI] [PubMed] [Google Scholar]
- 46.Yoneda N., Tatsumi E., Kawano S., Matsuo Y., Minowada J., Yamaguchi N. Human recombination activating gene-1 in leukemia/lymphoma cells: expression depends on stage of lymphoid differentiation defined by phenotype and genotype. Blood. 1993;82:207–216. [PubMed] [Google Scholar]
- 47.Iyer A., Hennessey D., O'Keefe S., Patterson J., Wang W., Wong G.K.-S., Gniadecki R. Branched evolution and genomic intratumor heterogeneity in the pathogenesis of cutaneous T-cell lymphoma. Blood Adv. 2020;4:2489–2500. doi: 10.1182/bloodadvances.2020001441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kovtun Y.V., Audette C.A., Ye Y., Xie H., Ruberti M.F., Phinney S.J., Leece B.A., Chittenden T., Blättler W.A., Goldmacher V.S. Antibody-drug conjugates designed to eradicate tumors with homogeneous and heterogeneous expression of the target antigen. Cancer Res. 2006;66:3214–3221. doi: 10.1158/0008-5472.CAN-05-3973. [DOI] [PubMed] [Google Scholar]
- 49.Raikar S.S., Fleischer L.C., Moot R., Fedanov A., Paik N.Y., Knight K.A., Doering C.B., Spencer H.T. Development of chimeric antigen receptors targeting T-cell malignancies using two structurally different anti-CD5 antigen binding domains in NK and CRISPR-edited T cell lines. OncoImmunology. 2018;7 doi: 10.1080/2162402X.2017.1407898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gomes-Silva D., Srinivasan M., Sharma S., Lee C.M., Wagner D.L., Davis T.H., Rouce R.H., Bao G., Brenner M.K., Mamonkin M. CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood. 2017;130:285–296. doi: 10.1182/blood-2017-01-761320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma G., Shen J., Pinz K., Wada M., Park J., Kim S., Togano T., Tse W. Targeting T Cell Malignancies Using CD4CAR T-Cells and Implementing a Natural Safety Switch. Stem Cell Rev. Rep. 2019;15:443–447. doi: 10.1007/s12015-019-09876-5. [DOI] [PubMed] [Google Scholar]
- 52.Cooper M.L., Choi J., Staser K., Ritchey J.K., Devenport J.M., Eckardt K., Rettig M.P., Wang B., Eissenberg L.G., Ghobadi A., et al. An "off-the-shelf" fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32:1970–1983. doi: 10.1038/s41375-018-0065-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramos C.A., Ballard B., Zhang H., Dakhova O., Gee A.P., Mei Z., Bilgi M., Wu M.-F., Liu H., Grilley B., et al. Clinical and immunological responses after CD30-specific chimeric antigen receptor-redirected lymphocytes. J. Clin. Invest. 2017;127:3462–3471. doi: 10.1172/JCI94306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scarfò I., Ormhøj M., Frigault M.J., Castano A.P., Lorrey S., Bouffard A.A., van Scoyk A., Rodig S.J., Shay A.J., Aster J.C., et al. Anti-CD37 chimeric antigen receptor T cells are active against B- and T-cell lymphomas. Blood. 2018;132:1495–1506. doi: 10.1182/blood-2018-04-842708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sánchez-Martínez D., Baroni M.L., Gutierrez-Agüera F., Roca-Ho H., Blanch-Lombarte O., González-García S., Torrebadell M., Junca J., Ramírez-Orellana M., Velasco-Hernández T., et al. Fratricide-resistant CD1a-specific CAR T cells for the treatment of cortical T-cell acute lymphoblastic leukemia. Blood. 2019;133:2291–2304. doi: 10.1182/blood-2018-10-882944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sochaj A.M., Świderska K.W., Otlewski J. Current methods for the synthesis of homogeneous antibody-drug conjugates. Biotechnol. Adv. 2015;33:775–784. doi: 10.1016/j.biotechadv.2015.05.001. [DOI] [PubMed] [Google Scholar]
- 57.Walsh S.J., Bargh J.D., Dannheim F.M., Hanby A.R., Seki H., Counsell A.J., Ou X., Fowler E., Ashman N., Takada Y., et al. Site-selective modification strategies in antibody-drug conjugates. Chem. Soc. Rev. 2021;50:1305–1353. doi: 10.1039/d0cs00310g. [DOI] [PubMed] [Google Scholar]
- 58.Lyon R.P., Bovee T.D., Doronina S.O., Burke P.J., Hunter J.H., Neff-LaFord H.D., Jonas M., Anderson M.E., Setter J.R., Senter P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015;33:733–735. doi: 10.1038/nbt.3212. [DOI] [PubMed] [Google Scholar]
- 59.Matikonda S.S., McLaughlin R., Shrestha P., Lipshultz C., Schnermann M.J. Structure-Activity Relationships of Antibody-Drug Conjugates: A Systematic Review of Chemistry on the Trastuzumab Scaffold. Bioconjug. Chem. 2022;33:1241–1253. doi: 10.1021/acs.bioconjchem.2c00177. [DOI] [PubMed] [Google Scholar]
- 60.Bryant P., Pabst M., Badescu G., Bird M., McDowell W., Jamieson E., Swierkosz J., Jurlewicz K., Tommasi R., Henseleit K., et al. In Vitro and In Vivo Evaluation of Cysteine Rebridged Trastuzumab-MMAE Antibody Drug Conjugates with Defined Drug-to-Antibody Ratios. Mol. Pharm. 2015;12:1872–1879. doi: 10.1021/acs.molpharmaceut.5b00116. [DOI] [PubMed] [Google Scholar]
- 61.Adem Y.T., Schwarz K.A., Duenas E., Patapoff T.W., Galush W.J., Esue O. Auristatin antibody drug conjugate physical instability and the role of drug payload. Bioconjug. Chem. 2014;25:656–664. doi: 10.1021/bc400439x. [DOI] [PubMed] [Google Scholar]
- 62.Hamblett K.J., Senter P.D., Chace D.F., Sun M.M.C., Lenox J., Cerveny C.G., Kissler K.M., Bernhardt S.X., Kopcha A.K., Zabinski R.F., et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004;10:7063–7070. doi: 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- 63.Senter P.D., Sievers E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012;30:631–637. doi: 10.1038/nbt.2289. [DOI] [PubMed] [Google Scholar]
- 64.Brown M.P., Staudacher A.H. Could bystander killing contribute significantly to the antitumor activity of brentuximab vedotin given with standard first-line chemotherapy for Hodgkin lymphoma? Immunotherapy. 2014;6:371–375. doi: 10.2217/imt.14.13. [DOI] [PubMed] [Google Scholar]
- 65.Masuda S., Miyagawa S., Sougawa N., Sawa Y. CD30-targeting immunoconjugates and bystander effects. Nat. Rev. Clin. Oncol. 2015;12:245. doi: 10.1038/nrclinonc.2014.159-c1. [DOI] [PubMed] [Google Scholar]
- 66.Staudacher A.H., Brown M.P. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br. J. Cancer. 2017;117:1736–1742. doi: 10.1038/bjc.2017.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Campana D., van Dongen J.J., Mehta A., Coustan-Smith E., Wolvers-Tettero I.L., Ganeshaguru K., Janossy G. Stages of T-cell receptor protein expression in T-cell acute lymphoblastic leukemia. Blood. 1991;77:1546–1554. [PubMed] [Google Scholar]
- 68.Hamblin T.J., Cattan A.R., Glennie M.J., Stevenson G.T., Stevenson F.K., Watts H.F., Stevenson G.T. Initial experience in treating human lymphoma with a chimeric univalent derivative of monoclonal anti-idiotype antibody. Blood. 1987;69:790–797. [PubMed] [Google Scholar]
- 69.Miller R.A., Maloney D.G., Warnke R., Levy R. Treatment of B-cell lymphoma with monoclonal anti-idiotype antibody. N. Engl. J. Med. 1982;306:517–522. doi: 10.1056/NEJM198203043060906. [DOI] [PubMed] [Google Scholar]
- 70.Armitage J.O. The aggressive peripheral T-cell lymphomas: 2017. Am. J. Hematol. 2017;92:706–715. doi: 10.1002/ajh.24791. [DOI] [PubMed] [Google Scholar]
- 71.Sibon D. Peripheral T-Cell Lymphomas: Therapeutic Approaches. Cancers. 2022;14 doi: 10.3390/cancers14092332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Simone M. de, Rossetti G., Pagani M. Single Cell T Cell Receptor Sequencing: Techniques and Future Challenges. Front. Immunol. 2018;9:1638. doi: 10.3389/fimmu.2018.01638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Scheetz L., Park K.S., Li Q., Lowenstein P.R., Castro M.G., Schwendeman A., Moon J.J. Engineering patient-specific cancer immunotherapies. Nat. Biomed. Eng. 2019;3:768–782. doi: 10.1038/s41551-019-0436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Meeker T.C., Lowder J., Maloney D.G., Miller R.A., Thielemans K., Warnke R., Levy R. A clinical trial of anti-idiotype therapy for B cell malignancy. Blood. 1985;65:1349–1363. [PubMed] [Google Scholar]
- 75.Santos C.F., Chen I., States D.J., Darwech I., McCord A. PRE-CLINICAL ANTI-TUMOR ACTIVITY OF A RAPIDLY-SYNTHESIZED MONOCLONAL ANTIBODY TARGETING B-CELL RECEPTOR POSITIVE LYMPHOMA. Hematol. Oncol. 2019;37:515–517. [Google Scholar]
- 76.Ott P.A., Hu Z., Keskin D.B., Shukla S.A., Sun J., Bozym D.J., Zhang W., Luoma A., Giobbie-Hurder A., Peter L., et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pawlyn C., Davies F.E. Toward personalized treatment in multiple myeloma based on molecular characteristics. Blood. 2019;133:660–675. doi: 10.1182/blood-2018-09-825331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Puttemans J., Stijlemans B., Keyaerts M., Vander Meeren S., Renmans W., Fostier K., Debie P., Hanssens H., Rodak M., Pruszynski M., et al. The Road to Personalized Myeloma Medicine: Patient-specific Single-domain Antibodies for Anti-idiotypic Radionuclide Therapy. Mol. Cancer Ther. 2022;21:159–169. doi: 10.1158/1535-7163.MCT-21-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bogen J.P., Elter A., Grzeschik J., Hock B., Kolmar H. Humanization of Chicken-Derived Antibodies by Yeast Surface Display. Methods Mol. Biol. 2022;2491:335–360. doi: 10.1007/978-1-0716-2285-8_18. [DOI] [PubMed] [Google Scholar]
- 80.Elter A., Bogen J.P., Hinz S.C., Fiebig D., Macarrón Palacios A., Grzeschik J., Hock B., Kolmar H. Humanization of Chicken-Derived scFv Using Yeast Surface Display and NGS Data Mining. Biotechnol. J. 2021;16 doi: 10.1002/biot.202000231. [DOI] [PubMed] [Google Scholar]
- 81.Charette M. de, Houot R. Hide or defend, the two strategies of lymphoma immune evasion: potential implications for immunotherapy. Haematologica. 2018;103:1256–1268. doi: 10.3324/haematol.2017.184192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaimi Y., Takahashi Y., Taniguchi H., Ochi T., Makino H., Makita S., Iwaki N., Fukuhara S., Munakata W., Ogawa C., et al. Loss of or decrease in CD30 expression in four patients with anaplastic large cell lymphoma after brentuximab vedotin-containing therapy. Virchows Arch. 2024;43:1161–1166. doi: 10.1007/s00428-024-03764-1. [DOI] [PubMed] [Google Scholar]
- 83.Schoenfeld K., Harwardt J., Kolmar H. Better safe than sorry: dual targeting antibodies for cancer immunotherapy. Biol. Chem. 2024;405:443–459. doi: 10.1515/hsz-2023-0329. [DOI] [PubMed] [Google Scholar]
- 84.Connors J.M., Jurczak W., Straus D.J., Ansell S.M., Kim W.S., Gallamini A., Younes A., Alekseev S., Illés Á., Picardi M., et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin's Lymphoma. N. Engl. J. Med. 2018;378:331–344. doi: 10.1056/NEJMoa1708984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wadleigh M., Richardson P.G., Zahrieh D., Lee S.J., Cutler C., Ho V., Alyea E.P., Antin J.H., Stone R.M., Soiffer R.J., DeAngelo D.J. Prior gemtuzumab ozogamicin exposure significantly increases the risk of veno-occlusive disease in patients who undergo myeloablative allogeneic stem cell transplantation. Blood. 2003;102:1578–1582. doi: 10.1182/blood-2003-01-0255. [DOI] [PubMed] [Google Scholar]
- 86.Bogen J.P., Grzeschik J., Krah S., Zielonka S., Kolmar H. Rapid Generation of Chicken Immune Libraries for Yeast Surface Display. Methods Mol. Biol. 2020;2070:289–302. doi: 10.1007/978-1-4939-9853-1_16. [DOI] [PubMed] [Google Scholar]
- 87.Benatuil L., Perez J.M., Belk J., Hsieh C.-M. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng. Des. Sel. 2010;23:155–159. doi: 10.1093/protein/gzq002. [DOI] [PubMed] [Google Scholar]
- 88.Hinz S.C., Elter A., Rammo O., Schwämmle A., Ali A., Zielonka S., Herget T., Kolmar H. A Generic Procedure for the Isolation of pH- and Magnesium-Responsive Chicken scFvs for Downstream Purification of Human Antibodies. Front. Bioeng. Biotechnol. 2020;8:688. doi: 10.3389/fbioe.2020.00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hendel A., Bak R.O., Clark J.T., Kennedy A.B., Ryan D.E., Roy S., Steinfeld I., Lunstad B.D., Kaiser R.J., Wilkens A.B., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bloemberg D., Nguyen T., MacLean S., Zafer A., Gadoury C., Gurnani K., Chattopadhyay A., Ash J., Lippens J., Harcus D., et al. A High-Throughput Method for Characterizing Novel Chimeric Antigen Receptors in Jurkat Cells. Mol. Ther. Methods Clin. Dev. 2020;16:238–254. doi: 10.1016/j.omtm.2020.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Müller S., Bexte T., Gebel V., Kalensee F., Stolzenberg E., Hartmann J., Koehl U., Schambach A., Wels W.S., Modlich U., Ullrich E. High Cytotoxic Efficiency of Lentivirally and Alpharetrovirally Engineered CD19-Specific Chimeric Antigen Receptor Natural Killer Cells Against Acute Lymphoblastic Leukemia. Front. Immunol. 2019;10:3123. doi: 10.3389/fimmu.2019.03123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Prommersberger S., Hudecek M., Nerreter T. Antibody-Based CAR T Cells Produced by Lentiviral Transduction. Curr. Protoc. Immunol. 2020;128 doi: 10.1002/cpim.93. [DOI] [PubMed] [Google Scholar]
- 93.Puthenveetil S., Liu D.S., White K.A., Thompson S., Ting A.Y. Yeast display evolution of a kinetically efficient 13-amino acid substrate for lipoic acid ligase. J. Am. Chem. Soc. 2009;131:16430–16438. doi: 10.1021/ja904596f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baruah H., Puthenveetil S., Choi Y.-A., Shah S., Ting A.Y. An engineered aryl azide ligase for site-specific mapping of protein-protein interactions through photo-cross-linking. Angew. Chem. 2008;47:7018–7021. doi: 10.1002/anie.200802088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Singh A.P., Sharma S., Shah D.K. Quantitative characterization of in vitro bystander effect of antibody-drug conjugates. J. Pharmacokinet. Pharmacodyn. 2016;43:567–582. doi: 10.1007/s10928-016-9495-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.