

Abstract
Hofmann coordination polymers (CPs) that couple the well-studied spin transition of the FeII central ion with electron-responsive ligands provide an innovative strategy toward multifunctional metal-organic frameworks (MOFs). Here, we developed a 2D planar network consisting of metal-cyanide-metal sheets in an unusual coordination mode, brought about by infinitely π-stacked redox-active bipyridinium derivatives as axial ligands. The obtained family of materials show vivid thermochromism attributed to electron transfer and/or electronic spin state change processes that can occur either independently or concomitantly. Importantly, the redox activity of the ligands within the structure leads to the quasi-reversible electrochemical reduction reaction on a spin-crossover complex at solid state. These observations have been confirmed via temperature-dependent single-crystal X-ray diffraction, magnetic measurements, Mössbauer, EPR, optical and vibrational spectroscopies as well as quantum chemical calculations.
Subject terms: Metal-organic frameworks, Coordination chemistry
Hofmann-type MOFs with an uncommon structural arrangement show thermochromism based on spin crossover behaviour and/or an intramolecular electron transfer between non-bridged CN units and the employed redox-active cationic bipyridinium ligand.
Introduction
Multi-switchable and readable molecules able to reversibly tune their intrinsic properties in response to one or several external stimuli have received much attention as potential components in a great variety of applications, e.g. memory devices1, sensors2,3, or catalysts4. In general, multifunctional compounds consist of several bistable building blocks, each able to respond to external stimuli (such as light, temperature, electrochemical or magnetic input)5,6. Among the different building blocks, bipyridinium units7–11 are especially promising because of their excellent electrochemical properties due to their electron-deficient character: the one-electron reduction in diluted solutions yields the paramagnetic bipyridinium radical cation, whereas it induces the dimerization of the molecules in a concentrated solution. In order to take advantage of the redox properties, bipyridinium derivatives have been successfully included in a large variety of systems spanning the fields of supramolecular chemistry, coordination chemistry, host-guest compounds, electrochromic materials etc. aiming to induce an inter or intra-molecular electron transfer (ET) by applying an external stimulus (temperature, light irradiation or pressure)12–18. As an example, Coe et al. 19 reported the possibility for pentacyano-iron-pyridinium derivatives to undergo an ET due to the electron donor and acceptor character of the building blocks. Much more common are examples of coordination polymers where the observed piezo, photo, or thermal ET is due to the presence of an electron-deficient viologen derivative, and a carboxylic acid acting as the electron-rich counterpart17,20,21. In such configurations, the ET occurs through hydrogen bonding, the most likely electron donor being the uncoordinated carboxylate oxygen atom. In general, the key structural factors favoring ET pathways are: (i) short distances between donor and acceptor, where the ET transfer will occur through either space or hydrogen bonds and (ii) planar structures to allow for electron delocalization. However, one should note that the structural changes before and after the ET observed in the literature are subtle.
Metal organic frameworks (MOFs) on a bipyridinium basis can be excellent candidates to observe an ET due to their structural arrangement. Recently, Guo et al.12 reported a 2D semiconductor MOF composed by π-stacked redox active N-methylpyridinium cations, sandwiched by cyanide-bridged layers that shows light and temperature-induced color change with the formation of stable radicals even in an ambient atmosphere. In this case, the observed thermo- and photo-activated ET within these materials involves the pyridinium unit as acceptor and the uncoordinated CN- moiety as donor. These MOF structures are closely related to Prussian blue and/or Hofmann clathrate derivatives which offer excellent structural platforms to establish multifunctionality, due to their intrinsic magnetic properties: photomagnetism, magnetic ordering, valence tautomerism and/or a spin crossover (SCO)22–26.
In general, Hofmann clathrates are formed by three major building blocks, first off, a transition metal center, second, a ligand (L) and lastly, a cyanometallate anion [M(CN)x]n- (x = 2 or 4, n = 1 or 2). Usually, this combination affords 2D planar metal−cyanide−metal sheets completed by a mono- or bidentate neutral organic ligand L, most often ammonia27, pyridine28–32, triazole33–37 or pyrazine38–40 derivatives, with a general formula of {Fe(L)x[µ4-M’(CN)4]} (x = 1 or 2; M’= Ni, Pd or Pt) or {Fe(L)x[µ2-M”(CN)2]} (x = 1 or 2; M”= Cu, Ag or Au)28. The modulation of the ligand field around the iron center through the different units leads to the introduction of the SCO properties. To our knowledge, at least three exceptions to this general formula have been described in literature: i) if the ligand is a strong chelate, for example a quinoline derivative (aqin), 1D chains of [Fe(aqin)2(μ2-M(CN)4)] (M = NiII and PtII) are obtained, where [M(CN)4]2- acts as a bridge between the iron sites41; ii) if the ligand is tetradentate, a {[Fe(μ4-bztpy)μ2-Pt(CN)4]·0.5bztpy} structure emerges, where 2D sheets, formed by the ligand and the FeII metal center, are again interconnected by [M(CN)4]2- building blocks42, and iii) very recently, Real et al.43 described two isomorphous structures, where the ligand carries a positive charge due to a spontaneous protonation during the crystallization process. The authors suggested that the positive charge could be responsible for an additional class of Hofmann-clathrates with the general formula {Fe(L)2[µ2-M(CN)4]}. It should be noted that in all of the above cases, the specific properties (electronic, steric, etc.) of the ligands are responsible for the final structure of these modified Hofmann-type clathrates, which, nevertheless, maintain a SCO behavior. Thus, we think that Hofmann-type clathrates offer an excellent structural platform to establish multifunctionality through introduction of a redox-active ligand, such as a bipyridinium-type derivative.
In this work, we explore this idea by synthesizing a large family of non-innocent ligands, which contain three functional building blocks (Fig. 1a; see Supplementary Figs 1–5 for characterization), the most important being a monocationic pyridinium unit that is responsible for the redox activity12,44–47. Attached to it, an aryl group allows for the fine-tuning of the delocalized electron density of the ligand by modifying the substituent R in para position. Lastly, a pyridine group is added to ensure the coordination to the iron center. The pyridinium moiety contains an electropositive, quaternary nitrogen atom that can be reduced to the highly air-sensitive neutral (radical) species (Fig. 1b). As demonstrated through several examples in the literature, the stability of the radical state can be largely improved by e.g. incorporating it in a MOF structure. In the case of the compound reported by Guo et al.12 the stabilization of the radical is due to π−π and cation-π interactions within the network. With this idea in mind, the abovementioned redox-active ligands were used to synthesize a family of Hofmann-type MOFs, which, as we show below, host a wealth of electronic phenomena, including both SCO and ET processes as well as redox activity.
Fig. 1. Redox properties of ligands.

a One-electron redox process on the studied R-pbpyCl ligands (R-pbpy+= 1-(4-R-phenyl)−4,4’-bipyridinium); b Cyclic voltammetry of CH3-pbpy+ (1) showing the reversible reduction of the bipyridinium unit (see Supplementary Fig. 6 for R = Br (2), COOCH3 (3) and NO2 (4)). [C] = 10−4 M in 0.1 M TBAP/CH3CN; reference: Ag+|Ag electrode ([Ag+]=10−2 M in CH3CN); scan rate: 100 mV·s−1.
Results
In the present work, we report the synthesis and characterization of the first redox active spin crossover coordination polymers belonging to this family. These compounds are isostructural and consist of 2D extended sheets with the molecular formula {Fe(R-pbpy+)2[µ2-M(CN)4]2·2H2O} (R being methyl (1, CH3), bromo (2, Br), ester (3, COOCH3) or nitro (4, NO2), R-pbpy+= 1-(4-R-phenyl)−4,4’-bipyridinium and M1 = Ni2+ (a) or M2 = Pt2+ (b)). The {Fe(R-pbpy+)2[µ2-M(CN)4]2·2H2O} Hofmann CPs were synthesized as microcrystalline powders and as single crystals. The direct powder precipitation of the compounds is obtained when K2[M(CN)4]·xH2O (M = Ni2+ or Pt2+) is added to an EtOH/H2O (1:1) solution containing the ligand and Fe(BF4)2·6H2O. The purity of the products has been assessed by elemental analysis (Supplementary Methods) and powder X-ray diffraction (XRD) (Supplementary Figs. 9–16). Single-crystals suitable for XRD analysis were obtained by slow crystallization in an H-tube from the diffuse reaction of Fe(BF4)2·6H2O, the ligand R-pbpy+ (R=CH3, Br or COOCH3) and K2[Pt(CN)4] in a molar ratio of 1:2:2 (see Supplementary Tables 9, 10 & 12 for compounds 1b, 2b & 3b respectively).
The unit cell of {Fe(CH3-pbpy+)2[µ2-Pt(CN)4]2·2H2O} (1b) (P21/c monoclinic space group) determined by single crystal XRD is shown in Fig. 2a. The FeII ions are coordinated sixfold, interconnected via four pseudo square-planar tetracyano-platinate ions, forming 2D metal-cyanide-metal sheets. Only two CN- groups of each tetracyano-platinate are coordinated to the FeII ions while the two others remain uncoordinated. The coordination sphere of FeII is completed by the N atoms from the two monodentate, redox-active ligands in an axial configuration. The structure shows two molecules of crystal water forming hydrogen bonds with the uncoordinated CN- moieties of two stacked 2D planes. While this can be seen for the entire family of compounds, compound 3b shows a much higher disorder of the water molecules, enabling us to place them only in partial occupation.
Fig. 2. Crystallographic structure of compound 1b.
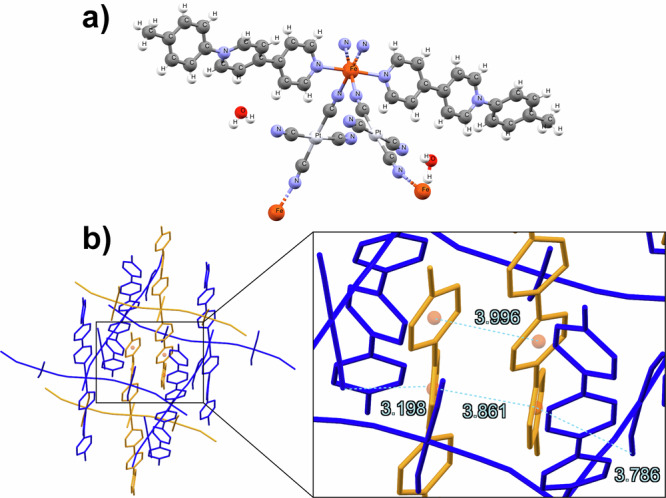
a unit cell of 1b at 260 K (full crystallographic data for all measured samples are provided in Supplementary Tables); b the structure of 1b is made up of 2D metal-cyanide-metal sheets (here in blue), intercalated by the π−π stacked axial ligands of the sheets above and below. Hydrogen atoms and water molecules were omitted for clarity. All distances are shown in Ångström (Å).
The structure of this family of compounds is the same as the one recently reported by the Real group43. The void space in each metal-cyanide-metal sheet (see Fig. 2b, in blue) is filled by the π−π stacked axial ligands of the next sheets to either side (see Fig. 2b, in yellow). Taking into account that the entire series presented in this work is isostructural, we provide here experimental proof for the hypothesis proposed in their work43: the presence of a cationic species is responsible for this Hofmann-type structure, with the two anionic non-coordinated CN- units ensuring the overall neutrality of the compound.
Magnetic susceptibility and optical reflectivity measurements for all compounds were performed over 80–325 K (see Fig. 3 for 1-4a, as well as cross for 1-4b). At room temperature, the compounds are different shades of pink/purple. Upon cooling, a clear and gradual color change to a dark violet/blue is observed in optical reflectivity measurements for all compounds, whereas in magnetic measurements only R = CH3 and Br show a complete transition from the high spin (HS) to the low spin (LS) state. This difference in the magnetic properties may have its origin in the different electron densities on the ligands. The pyridinium moiety is an electron-deficient group, where the positive charge is delocalized over the aromatic rings. The introduction of an electron-withdrawing group, like NO2 or COOCH3, leads to the destabilization of the pyridinium unit, whereas the contrary is true for electron-donating groups. In order to prove this hypothesis, a systematic study of the electron donor/acceptor nature of the substituents vs SCO should be undertaken, this is, however, out of the scope of the present article.
Fig. 3. Magnetic and optical reflectivity measurements of compounds 1-4a.

χaT vs. T (χa stands for the molar magnetic susceptibility) and variable temperature optical reflectivity in cooling and heating modes for samples of 1-4a {Fe(R-pbpy+)2[Ni(CN)4]2·2H2O} (R = CH3, (curve a; red), Br (curve b; blue), COOCH3 (curve c; yellow) and NO2 (curve d; green). The magnetic measurements are shown in the respective colors, the corresponding optical reflectivity curves in grey dots.
Fig. 3: Magnetic and optical reflectivity measurements of compounds 1-4a: χaT vs T (χa stands for the molar magnetic susceptibility) and variable temperature optical reflectivity in cooling and heating modes for samples of 1-4a {Fe(R-pbpy+)2[Ni(CN)4]2·2H2O} (R = CH3, (curve a; red), Br (curve b; blue), COOCH3 (curve c; yellow) and NO2 (curve d; green). The magnetic measurements are shown in the respective colors, the corresponding optical reflectivity curves in grey dots.
As a color change is characteristic for a spin transition, it is expected that those two phenomena occur at comparable temperatures. Here, however, the optical and magnetic properties are not always correlated. Compounds 1a & 2a show a reversible, gradual change of χaT with two steps at 200 K and 145 K for R = CH3 and at 228 K and 183 K for R = Br (Fig. 3 a, b). For both compounds, the reversible optical transition seems to start first, and is only roughly correlated with the magnetic changes, with the first step leading to a pronounced color change, while the second seems more subtle (vide infra). For compound 3a (R = COOMe) (Fig. 3c), the optical change follows the magnetic properties more closely, with a partial SCO at low temperatures. On the contrary, compound 4a (R = NO2) (Fig. 3d) does not show a SCO in the measured temperature range, however, a pronounced color change is observed anyways. Particularly interesting, it does not only show a gradual color change until the lowest measured temperatures, but also an abrupt transition with a hysteresis loop of 58 K (between 232 K and 290 K). While the overall magnetic behavior of the respective Pt compounds (1-4b, see Supplementary Fig. 8) mirrors the Ni counterparts, the spin transition temperatures lie approximately 10 K higher.
Taking into account the literature on viologen/bipyridinium-based compounds and the structure of our materials, one hypothesis for the origin of the thermochromism, other than the SCO, is the radical formation within the MOF structure. Indeed as described in literature, this type of compounds can exhibit photo-, thermo- and/or piezo-chromic phenomena due to an electron transfer process originating from donor groups, such as the iron or the non-coordinated CN− groups, to the organic ligands12. Thermochromic behavior is observed for many viologen/bipyridinium-based compounds, usually occurring at or above room temperature14. In our case, it is possible that the introduction of the bipyridinium unit into a protective/stabilizing framework allows for this phenomenon to occur at lower temperatures. In order to closely examine this hypothesis, we chose two compounds for more detailed analysis: First, {Fe(CH3-pbpy+)2[µ2-M(CN)4]2·2H2O} (1a, b), which allowed us to compare optical and magnetic data with a detailed crystallographic study, and second, {Fe(NO2-pbpy+)2[µ2-Ni(CN)4]2·xH2O} (4a), in order to gain insight into the non-magnetic transition via an in-depth spectroscopic analysis.
The optical properties of 1a and 1b have been carefully checked by UV-vis-NIR spectroscopy and compared to the parent clathrate compound {Fe(py)2[µ4-Pt(CN)4]·2H2O}. At 293 K (HS state), the parent compound presents two main absorption bands centered at 355 and 900 nm, where the near-infrared band is attributed to an MLCT transition (Fig. 4, red curve)48. The compound (1b), however, presents an additional absorption band at 535 nm which is rather unusual for HS SCO compounds - it appears when exchanging the innocent pyridine ligand for the redox active R-pbpy+ unit (Fig. 4, purple curve). In parallel, spectroelectrochemical analysis of the methyl ligand shows that upon reduction at −1.2 V, two new absorption bands emerge at 384 and 545 nm, which are attributed to the reduced, radical form of the ligand (see Supplementary Fig. 7). This indicates, that upon incorporation into the MOF, the ligand might have received an electron leading to the single CH3-pbpy• radical, which could explain the strong absorption at 535 nm. It should be noted that this absorption band has been seen for all {Fe(R-pbpy+)2[µ2-M(CN)4]2·2H2O} compounds. In addition, isostructural Zn derivatives {Zn(R-pbpy+)2[µ2-Ni(CN)4]2·2H2O} were synthesized (see Supplementary Fig. 17 for powder XRD of R = CH3 (1b-Zn) and Supplementary Fig. 18 for crystal structure of R = Br (2b-Zn)), which, even though they contain the respective redox-active ligand, only show a small shoulder around 500 nm (Fig. 4, teal curve), indicating that the FeII ion also plays an important role at the origin of this absorption band.
Fig. 4. UV-vis-NIR absorption spectra at 293 K.
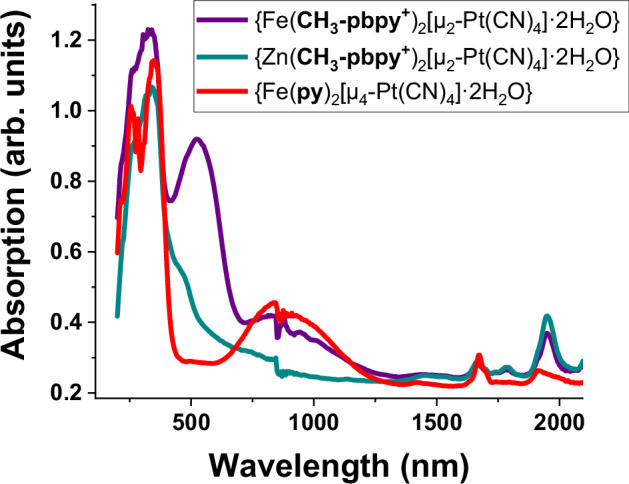
Compound 1b (purple), {Zn(CH3-pbpy+)2[Pt(CN)4]2·2H2O} (teal) and {Fe(py)2[Pt(CN)4]·2H2O} (red).
The evolution of the absorbance spectra during the cooling process has been monitored for the parent compound {Fe(py)2[µ4-Ni(CN)4]·2H2O} (Supplementary Fig. 19), as well as the compounds 1a (see Fig. 5a, b) and 4a (see Fig. 5c, d) (check Supplementary Information for compounds 1b (Supplementary Fig. 20), 2a (Supplementary Fig. 21) and 3a (Supplementary Fig. 22)). For 1a, we observe the disappearance of the absorption bands around 1085 nm and 820 nm, as well as the emergence of a shoulder at 575 nm, which is supported by the well-defined isosbestic point until 200 K at 690 nm (see Fig. 5a). Typically, if only a SCO is observed in the material, as can be seen for the parent compound {Fe(py)2[µ4-Ni(CN)4]·2H2O}, the evolution of absorption band intensities (535, 815 and 1000 nm) follows the SCO phenomenon (see Supplementary Fig. 19b). Fig. 5b shows the variation of the absorption band intensities with respect to the magnetic and optical properties of 1a, explaining the discrepancy between the two measurements. In contrast to the parent compound, only the evolution of the absorption intensity at 820 nm seems to follow the SCO (see also Supplementary Fig. 20 (1b) and Supplementary Fig. 21 (2a)), the absorption band at 575 nm, however, follows the optical transition, while a contribution at 1085 nm decreases right from the start. This indicates that in addition to the SCO, at least one more thermochromic phenomenon is occurring upon changing the temperature.
Fig. 5. Temperature dependent UV-vis-NIR absorption spectra of 1a and 4a.

a compound 1a upon cooling and b comparison of the evolution of χaT and optical reflectivity of 1a and the absorption intensities at selected wavelengths as a function of the temperature; c compound 4a upon cooling and d comparison of the evolution of χaT and optical reflectivity of 4a with the absorption intensity at 600 nm as a function of the temperature upon cooling and heating. The absorption intensity scales for 575 nm (b; compound 1a, in green) and 600 nm (d; compound 4a, in green) have been inverted to facilitate a comparison with the optical behavior. The photographs indicate the color of the samples at selected temperatures, clearly showing the observed hysteresis loop in compound 4a. The peaks marked with * stem from water/humidity.
Even more intriguing is the behavior of compound 4a, where the observed thermochromism is not related to a spin transition (see Fig. 5c, d). Upon cooling, the intensity of the absorption band at 600 nm strongly increases (Fig. 5c), which can be superposed with the optical reflectivity measurements. As there is no magnetic transition, no absorption changes were observed around 700–800 nm. For a side-to-side comparison, showing that the color change is fully reversible for the discussed compounds please see Supplementary Fig. 23.
Based on these findings we conclude that the absorption band observed at 580–605 nm corresponds to the optical transition, while the changes around 700 to 800 nm describe the magnetic behavior, thus indicating that besides the SCO at least one more thermochromic phenomenon occurs in each sample. Since the formation of R-pbpy• radicals gives rise to spectral changes around 500–600 nm as well as around 800–1000 nm (in case of dimer formation)7,49–52, it is tempting to associate these events with an ET between the ligand and some donor species (e.g. the iron center or the non-coordinated CN− groups). To better understand the origin of this SCO-independent thermochromism, TD-DFT calculations and detailed structural analysis (for compound 1b) as well as EPR, Mössbauer, Raman, and Infrared absorption (IR) spectroscopy studies (1a and 4a) have been carried out in the range of 300-80 K.
The electronic excitation calculations were performed within linear-response in time-dependent density-functional theory (LR-TD-DFT) using a simplified model fragment of compound 1b, based on the crystal structure in the LS state (total electronic spin S=0) and in the HS state (S = 2). The low-energy parts of the calculated spectra of LS {Fe(CH3-pbpy+)2[μ2-Pt(CN)4]4}4- and HS {Fe(CH3-pbpy+)2[μ2-Pt(CN)4]4}4- spectra are dominated by an intense Fe → CH3-pbpy+ MLCT transition located at 763 nm and 1185 nm (Fig. 6, blue and red), which correspond to the experimental bands observed at 580 nm and 1065 nm, respectively. The red-shift of the calculated spectra can be ascribed to the approximations underlying the LR-TD-DFT calculations and possibly to the use of a molecular fragment and hence to the neglect of the crystalline environment. The influence of an electron transfer on the optical properties was investigated by performing LR-TD-DFT calculations on the doubly reduced HS fragment (S=3) {Fe(CH3-pbpy•)2[μ2-Pt(CN)4]4}6- only since the self-consistent-field calculations on the mono-reduced HS fragment lead to a degenerate electronic state (see Supplementary Discussion). The calculated spectrum of the doubly reduced HS fragment presents two intense bands (Fig. 6, yellow) that are independent π−π transitions occurring at 796 nm and 663 nm and are centred each on one or the other reduced ligand. The overlap of these intra-ligand absorption bands with the MLCT bands found in the calculated spectra of LS and HS {Fe(CH3-pbpy+)2[μ2-Pt(CN)4]4}4- supports the conclusion drawn from the experiments that the temperature dependence of the absorption intensities is likely also due to the presence of reduced CH3-pbpy• ligands at elevated temperatures.
Fig. 6. Calculated absorption spectra.
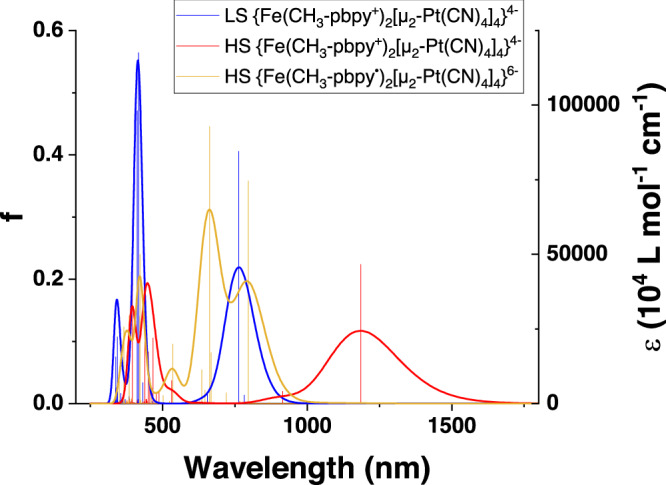
LS (blue) and HS (red) of {Fe(CH3-pbpy+)2[μ2-Pt(CN)4]4}4- as well as the spectrum of HS {Fe(CH3-pbpy•)2[μ2-Pt(CN)4]4}6- (yellow).
EPR does not show a signal characteristic for a radical, which is, however, to be expected in the case of dimer formation7,49–52. This hypothesis is also in agreement with the observation of an absorption band near 800–1000 nm, characteristic of bipyridinium dimer species52, and with the observation of π−π interactions between the ligands (vide infra).
57Fe Mössbauer spectroscopy is a useful technique to determine the spin and oxidation states of iron. Indeed, in case of an ET event from the FeII central ion to the ligand, a characteristic signature of FeIII might be visible via Mössbauer studies. Thus, Mössbauer spectra have been recorded for compounds 1-4a in the range of 300–80 K (see Supplementary Fig. 24–27). In Fig. 7a, b the spectra at 80 K and 323 K are shown respectively for 1a (see Supplementary Fig. 24 for additional temperatures). At 323 K an asymmetric doublet is observed, which can be clearly assigned to a FeIIHS species based on the values of isomer shift (δCS = 1.005(17) mm s−1) and quadrupole splitting (ΔEQ = 0.987(35) mm s−1). Upon cooling, the intensity of this doublet gradually decreases whereas a new doublet emerges, and finally, at 80 K, the sample shows a fully symmetric FeIILS doublet (δCS = 0.4674(40) mm s-1 and ΔEQ = 0.2353(64) mm s−1). These observations are characteristic of a spin state switching between FeIIHS (S = 2) and FeIILS (S = 0), and can be correlated to the magnetic data. The asymmetry of a quadrupole-split Mössbauer line at high temperatures remains, however, intriguing. Such line asymmetry may have different origins, including preferential crystalline orientation (texture), lattice vibrational anisotropy (Goldanskii-Karyagin effect), a residual FeIILS species, or a relaxational line broadening53. Nevertheless, based on the temperature dependence of the spectrum, characterized by a well-defined shoulder at higher temperatures and a symmetrical doublet at the lowest temperatures, the best fit of the spectra is obtained by assuming the existence of a second iron species (δCS = 0.24(10) mm s−1 and ΔEQ = 0.55(17) mm s−1) with a population of ca. 29 ± 8 % at 323 K. This species might be ascribed to a ferric ion, possibly FeIIIHS, and shows a notable temperature dependence, indicating that it does not stem from the accidental oxidation of the samples. The Mössbauer results thus support the UV-visible data, showing that a part of the iron ions participates in an ET towards the ligands when increasing the temperature – in partial overlap with the SCO phenomenon. One shall also note that there is only one SCO active iron site (confirmed both by Mössbauer and XRD measurements). This means that the two-step SCO observed in magnetism (see Fig. 3a, b) is not linked to the presence of two distinct FeII sites, but could arise due to a coupling of different physical phenomena. A plausible hypothesis is a coupling between the SCO and ET events.
Fig. 7. Mössbauer spectra of 1a and 4a.

1a {Fe(CH3-pbpy+)2[Ni(CN)4]2·2H2O} a at 80 K and b at 323 K, in comparison to spectra of 4a {Fe(NO2-pbpy+)2[Ni(CN)4]2·2H2O} c at 80 K and d at 295 K.
Figs 7c, d show the Mössbauer absorption spectra for compound 4a at 80 K and 295 K respectively (see Supplementary Fig. 26 for additional temperatures). Even though we were able to improve spectral quality by enriching sample 4a with 57Fe, it is difficult to determine a unique fit due to the rather large doublet present in the spectra. At ambient temperature, a symmetric doublet is observed, consisting of at least two FeII HS species (FeII HS (1) δCS = 1.114(20) mm s−1 and ΔEQ = 1.493(85) mm s−1 and (2) δCS = 1.1330(55) mm s−1 and ΔEQ = 1.016(13) mm s−1). Upon cooling we see the emergence of a new FeII HS species with a large quadrupole splitting, reaching up to 20 % population at 80 K (FeII HS (3) δCS = 1.2290(88) mm s−1 and ΔEQ = 2.800(37) mm s−1). This indicates that the coordination environment of part of the iron centers presents changes with temperature, which might stem from an electron transfer to the ligand and explain the color change observed for this sample. The asymmetric nature of the spectra at low temperatures is partially attributed to the increase of a fourth Fe species (δCS = 0.5326(76) mm s−1 and ΔEQ = 0.302(11) mm s−1), that might correspond to a small LS fraction or a FeIII species. Since we do not observe a SCO in compound 4a, we believe that the latter is more likely.
As both the SCO and ET phenomena should be reflected by changes in vibrational spectroscopy, variable temperature Raman and Infrared (IR) spectra of the samples were acquired (see Fig. 8). Raman microscopy measurements of 1a (see Fig. 8a) reveal new peaks during the cooling process (e.g. at 685 cm−1) and, in addition, the intensity ratio between peaks located around 1612 cm−1 changes. These peaks are attributed to the stretching and deformation vibrations of the C-H and C-N bonds of the ligand. Moreover, a shift of the CN modes to higher wavenumbers, caused by the SCO, can be observed (see Supplementary Fig. 28). Compound 4a (see Fig. 8b) shows similar behavior with new peaks around 659 and 1275 cm-1 and an intensity ratio change between peaks located around 1620 cm−1. As for compound 1a, it shows two distinct C-N stretching modes, since only two of the four CN− moieties are coordinated to the FeII, however, as expected in this case, no CN mode shift can be detected.
Fig. 8. Vibrational spectroscopy analysis of compounds 1a and 4a.

a Raman spectra (acquired with a 785 nm laser) of 1a recorded at 323 K (red), 193 K (blue), and 93 K (yellow); b Raman spectra (acquired with a 785 nm laser) of 4a recorded at 283 K (red), 243 K (blue) and 133 K (yellow); c temperature dependent Infrared absorption spectrum of compound 1a and d temperature dependent Infrared absorption spectrum of compound 4a.
The analysis of the Raman spectral changes is complicated by the strong thermochromism of the samples, potentially leading to the resonance enhancement of certain Raman modes. IR absorption studies (see Fig. 8c, d), on the other hand, enabled us to follow the temperature-induced changes of vibrational modes with less ambiguity. Importantly, we observed significant changes in the out-of-plane CH bending regions (both fundamental and overtone) of the aromatic ligands both in the SCO active and inactive derivatives (see Supplementary Figs. 29 and 30, for region of 900 to 1200 cm−1 of compound 1a and 4a, respectively). This behavior strongly contrasts with that of the parent clathrate compound {Fe(py)2[µ4-Ni(CN)4]·2H2O}, which displays only slight shifts of vibrational modes associated with the pyridine ligand54. Since the compounds are isostructural at each temperature (no symmetry change) we can thus infer a significant variation in the structure of the R-pbpy+ ligands upon temperature changes. Indeed, the uncoordinated ν(C-N) modes display significant changes as a function of the temperature as well. Compound 1a (Fig. 8c) presents two asymmetric doublets at 2159 and 2135 cm−1 attributed to the two different C-N modes (coordinated and uncoordinated, respectively). When increasing the temperature, a new peak emerges at 2120 cm−1 in the spectral region attributed to the uncoordinated CN- units. For compound 4a a similar behavior is observed with a shoulder emerging in the same position (see Fig. 8d). These observations can be related to an ET phenomenon between the CN− group, which could act as an electron donor, and the bipyridinium ligands. Similar arguments have been proposed in the paper of Guo et al. for the origin of the ET observed in their compound12.
As mentioned in the introduction, the occurrence of ET phenomena in compounds containing bipyridinium derivatives is associated with several subtle structural features. According to the Marcus ET theory, the shorter the distance between an electron donor and acceptor the faster the ET will occur, the maximum distance possible being around 4 Å. The ET is often related to the presence of hydrogen bonds and favored by π−π interactions in the crystal packing. In addition, the transfer is more likely to occur if the interannular torsion angle of the pyridinium units is negligible14. To explore the structural details, a thorough crystallographic study of the single crystal 1b {Fe(CH3-pbpy+)2[µ2-Pt(CN)4]2·2H2O} was undertaken at various temperatures. At 300 K, the average Fe–N lengths for the cyano and viologen units vary between 2.15 and 2.22 Å (see Fig. 9c), which is in close agreement with a FeII ion in the HS state. Interestingly, the axial CH3-pbpy+ ligands are non-equivalent in regards to the interannular angle of the bipyridinium unit: while one shows a torsion angle of around 44°, the other unit is almost fully planar (see Fig. 9a, c). Moreover, very short distances of less than 3 Å can be observed between the uncoordinated CN- units and the bipyridinium units of the axial ligands. In addition, the uncoordinated CN− groups show a clear structural disorder at higher temperature (280 K and 290 K), confirming the observations made in IR spectroscopy. Furthermore, π−π interactions between two CH3-pbpy+ ligands are present, possibly explaining the EPR silent behavior of the compounds. All four phenomena are in line with the aforementioned requirements for an ET at ambient temperature within our structure and would suggest an electron transfer from the uncoordinated CN- groups to the axial ligand, in agreement with the evolution of the IR absorption bands discussed above.
Fig. 9. In-detail crystallographic analysis of compound 1b.

a side view of two π-stacked units of 1b at 290 K, indicating where interactions and structural changes are observed in temperature. Fe-N distances are marked in light blue, the Fe-N-C angle in purple, the π−π stacking and bipyridinium-NC distances in red and green, respectively, and torsion angles in orange. Water molecules are omitted for clarity. b Evolution of the distance Fe1-N1 and the torsion angle of the flat bipyridinium unit (C011-C019-C01B-C00U) upon cooling of 1b compared to its magnetic behavior, error bars designate standard deviation; c selection of metal-to-ligand bond lengths (Å) and torsion angles (°) for 1b at 150 K and 290 K.
Upon cooling, significant changes can be observed in the crystal structure. At 150 K the spin transition is virtually complete as indicated by the typical shortening of the Fe-N distances for both the cyano and pyridine units to 1.945 Å and 2.013 Å, respectively (see Fig. 9b, c). It is important to mention that the bipyridinium unit showing a fully planar interannular angle is strongly affected by temperature, reaching a more twisted state with an angle of 12–14° when cooling to 150 K. As a thermo-induced ET is much more likely in flat bipyridinium units compared to twisted ones, this confirms that the process will more likely take place at (or above) ambient temperature. In addition, it is interesting to notice that the distances between the uncoordinated CN- and bipyridinium units do not show a large variation in temperature, even though we observe a shortening of the Fe-N bonds occurring upon the spin transition. This is another indication for the coupling between the uncoordinated CN- and bipyridinium units. Overall, the planar configuration of the bipyridinium unit at 300 K combined with the short distances to the uncoordinated CN- moiety, which likely acts as an electron donor, is perfectly in line with the hypothesis of an intramolecular ET within the structure.
The multifunctionality of the compounds induced by the presence of a redox-active ligand, has been highlighted through cyclic voltammetry and spectro-electrochemical studies (see Fig. 10). There are several examples in the literature concerning the chemical reduction of Hofmann-type clathrates in order to modulate the SCO properties. Ohba et al.55 conducted a post synthetic oxidative addition of iodine to their compound, which coordinates to the Pt ions, oxidizing them from PtII to PtIV. Depending on the iodine content and due to pore size restrictions, they were able to oxidize up to half of all Pt ions, thereby modulating the transition temperature of their compound in a range of 300 to 400 K, while maintaining the cooperative transition. Similarly, Zuo et al.56 doped a tetra(4-pyridyl)tetrathiafulvalene based FeII MOF with I2, thereby modifying its magnetic behavior and increasing the MOFs conductivity. Tong et al.57 reported a cationic metal organic framework with larger pores, where the chemical oxidation by coordination of iodine or bromine was possible for all AuII sites, thus modifying the SCO behavior from a hysteretic one-step transition to a three- or two-step transition, respectively. They show that after the subsequent reduction of the compound using ascorbic acid, the one-step hysteretic transition can be reinstated, however, with permanent changes in spectroscopic and SCO properties. Coronado et al.58 have reported a SCO active polyoxometalate (POM) compound, that can undergo a chemical or electrochemical reduction due to the redox-active linkers employed. They show this reduction to be reversible, the magnetic properties of the chemically re-oxidized compound are, however, modified. Recently, a review focusing on SCO and valence tautomerism conductors has been published, discussing synthesis strategies, magnetic and conductive properties as well as the influence of the materials inherent bistability on their conductivity59. Interesting concepts are the co-crystallization of a bistable matrix with a conductive unit (often a redox-active, planar molecule) – here, SCO materials offer interesting prospects due to the volume change seen upon the spin transition.
Fig. 10. Cyclic voltammetry and Raman spectroscopy of compound 1a.

a Cyclic voltammetry of 1a versus Ag+|AgCl in the solid state. 5 mg of the target material were dispersed in 500 µL of a 4% PVA solution in water and vigorously sonicated, then 125 µL of a 4% BORAX solution in water were added, forming a gel that was then applied onto a glassy carbon electrode (3 mm diameter). The scan rate was 50 mV s–1 in 1 M KCl; b in-situ Raman spectroscopy measurements of the 1a, showing the pristine clathrate (red) after the reduction (blue) and after re-oxidation (yellow). The dashed lines highlight the new peaks of the reduced compound.
Since the Hofmann clathrate derivatives presented in this work are not soluble, their electrochemical redox properties were studied in the solid state, discussed here for the R=CH3 derivative. 1a shows a reduction potential of E1/2 = −0.765 V vs. Ag+|AgCl (see Fig. 10a), which is ascribed to the one-electron reduction of the pyridinium unit, in line with previous reports in the literature on the electrochemical behavior of bipyridinium derivatives46,47. The quasi-reversible cyclic voltammogram shows a higher current intensity for the anodic wave, which might be related to the dissolution of the compound. In addition, compound 1a was investigated spectro-electrochemically at a negative potential. Fig. 10b shows the Raman spectra of 1a before and after reduction, followed by a re-oxidation. After the reduction at −1.0 V vs. Ag+|AgCl, we remark a complete modification of the Raman spectra with the emergence of several new peaks (e.g. at 995, 1190, and 1537 cm−1) in the region attributed to the stretching vibrations of the ligand. These modifications are ascribed to the one-electron reduction of the bipyridinium ligand within the MOF structure. The re-oxidation shows the reversibility of the system as the spectrum of the pristine compound is fully recovered. This result contrasts with the abovementioned studies carried out on the chemical reduction or oxidation of similar compounds and proves electrochemistry to be a useful method to obtain multifunctional materials. It shows that this family of compounds is able to respond to multiple external stimuli such as temperature or an applied potential in order to attain several stable and reversible states, allowing us to classify these materials as multifunctional.
In conclusion, we have shown that the rational design of a series of non-innocent ligands for Hofmann-type clathrates affords a family of redox-active SCO compounds, {Fe(R-pbpy+)2[µ2-M(CN)4]2·2H2O} (R = CH3, Br, COOCH3 or NO2; M = Ni or Pt), displaying an unusual structural arrangement due to the cationic nature of the ligand. The investigation of the optical properties of the compounds over a temperature range of 100–300 K reveals their thermo-chromic character which cannot be solely ascribed to the SCO phenomenon. Indeed, the combination of spectroscopic, crystallographic, and theoretical findings points to the occurrence of at least one additional electron transfer process and the associated formation of radical dimers within the structure. From Mössbauer and UV visible data, it is clear that the iron ion plays an important role in the ET properties. In addition, vibrational spectroscopies show strong changes in the stretching modes of the uncoordinated CN− group upon cooling. This result, and the observed short distances of 3 Å between the bipyridinium unit and the uncoordinated CN− moieties indicate the latter as the most likely origin of the ET. In addition, the presence of π−π interactions between the bipyridinium ligands, which stabilize the formed dimers, as well as the small inter-annular torsion angle observed for the latter, satisfy the structural requirements for an ET to occur. Importantly, we have shown that this family of compounds is able to undergo a quasi-reversible electrochemical redox process that can be followed via in situ Raman spectroscopy. This last result confirms the multi-stability and multi-addressability of our compounds, thus opening the door to a new class of switchable materials.
Methods
Materials
All purchased chemicals were used as received: Fe(BF4)2·6H2O: Iron(II) tetrafluoroborate, K2[M(CN)4]: Potassium tetracyanometallate (M = Ni or Pt), 4,4’-bipyridine, 2,4-dinitrochlorobenzene, trimethylamine, methyl-4-aminobenzoate, 4-methylaniline, 4-bromoaniline, 4-nitroaniline.
Synthesis
Ligands were synthesized in two steps: First, 20 mmol (3.12 g) 4,4’-bipyridine and 20 mmol (4.05 g) 2,4-dinitrochlorobenzene are dissolved in 50 ml acetone and stirred at reflux for 12 h, cooled, then the precipitate that formed during reaction (di-NO2-pbpyCl) was filtered and washed multiple times with acetone (Yield: 77%). Second, 3 equivalents of a 4-R-aniline and 1 equivalent of di-NO2-pbpyCl were dissolved in 15 ml of ethanol abs. stirred at reflux for 4 h, cooled, and then, 20 ml dist. H2O was added to remove the byproduct as precipitate. All solvent was evaporated and the crude product dissolved in as little methanol as possible, then, the ligand R-pbpyCl was precipitated in diethyl ether and washed multiple times with diethylether to remove impurities. Please check the Supplementary Methods section for a more detailed description and the characterisation (1H-NMR and mass spectrometry results) of all synthesized ligands. Clathrates were synthesized with 0.5 mmol of the respective ligand (L = CH3-pbpyCl, COOCH3-pbpyCl, NO2-pbpyCl or Br-pbpyCl), that are dissolved in 10 mL 50/50 v% EtOH/H2O dist., then 0.25 mmol of Fe(BF4)2 · 6 H2O are added with a small amount of ascorbic acid. Once dissolved, 0.5 mmol of K2[Ni(CN)4]·x H2O or K2[Pt(CN)4] are added, and purple precipitates form immediately. The mixture is stirred for 1 h, filtered and the precipitate is washed several times with EtOH/H2O dist., then dried in the oven. The purity of the microcrystalline powders was investigated via CHN and Inductively Coupled Plasma (ICP) analysis as well as powder X-ray diffraction (please see Supplementary Methods and Supplementary Figs.). Crystals were grown with 0.35 mmol of the respective ligand (L = CH3-pbpyCl, COOCH3-pbpyCl or Br-pbpyCl), that are dissolved in 8 mL 50/50 v% EtOH/H2O dist. and filled into an H-tube, then 0.1 mmol of Fe(BF4)2 · 6 H2O in 0.5 mL 50/50 v% EtOH/H2O dist. are added on one side and 0.2 mmol of K2[Pt(CN)4] in 0.5 mL 50/50 v% EtOH/H2O dist. are added on the other side. After several days/weeks, red crystals are formed and characterized via single-crystal X-ray diffraction.
Measurements
Elemental analyses were performed using a Perkin Elmer 2400 series II Instrument for CHN analysis and ICAP 6300 Thermo RF Generator with CID detector for Inductively Coupled Plasma analysis. Mass Spectrometry was performed on a ThermoScientific LCQ Fleet mass spectrometer with ion trap in electrospray ionization mode. Nuclear Magnetic Resonance (NMR), 1H-NMR spectra were obtained at 298 K in DMSO-d6 as an internal reference and were recorded on a Bruker Avance 300 or Bruker Avance 400. Chemical shifts are reported in parts per million (ppm). Optical reflectivity measurements were carried out on a Linkam THMS 600 heating-cooling stage for microscopy, where a few mg of sample were placed and flattened. The samples were heated to 323 K and the stage was purged under dry N2 flow for 30 min. Then the samples were cooled to 93 K and heated back up to 323 K at a speed of 5 °C/min for two cycles. Pictures were taken every 12 seconds using a Motic stereomicroscope equipped with a color camera and graphically analyzed using the ImageG software. UV-vis-NIR spectra of powders were obtained on a Perkin Elmer LAMBDA 950 Spectrophotometer with an Integrating Sphere and a spectral range of 200 to 2500 nm in diffuse scattering mode. For temperature-dependent measurements, the samples were placed inside a Linkam stage (see above) and thermalized for 2 min at the respective temperature before taking a UV-vis-NIR spectra. Raman spectra were measured on an Xplora (Horiba) Raman micro-spectrometer equipped with an edge filter and a diode laser (785 nm, 0.1 mW). The laser beam was focused on the sample by an ×50 objective (numerical aperture = 0.5), which was also used to collect the scattered photons. For spectral calibration, a pure Si wafer was used. Spectra were recorded with an acquisition time of 6 × 10 s from 50 to 2300 cm−1. The measurements were carried out using a Linkam heating stage for microscopy (see above). Spectra were recorded each 10 °C after the temperature had stabilized. FTIR spectra were recorded on a PerkinElmer Frontier FTIR/FIR instrument equipped with a Spotlight 150 IR microscope in a range of 4000 to 600 cm−1 at a resolution of 0.125 cm−1 and 16 scans. The measurements were carried out using a Linkam heating stage for microscopy (see above) using ZnSe windows. Spectra were recorded each 10 °C after the temperature had stabilized. 57Fe Mössbauer spectra were recorded using a conventional constant-acceleration-type spectrometer equipped with a 57Co source and a liquid nitrogen cryostat. Least-squares fittings of the Mössbauer spectra were carried out with the assumption of Lorentzian line shapes using the Recoil software. The isomer shift values are given with respect to metallic iron at room temperature. Variable-temperature magnetic susceptibility data were obtained at cooling and heating rates of 2 or 4 K min−1 under a field of 1 kOe using a Quantum Design MPMS magnetometer. Electrochemical measurements were conducted in a glovebox with a standard one-compartment, three-electrode electrochemical cell using a BioLogic SP300 potentiostat. Experiments in solution were carried out in anhydrous CH3CN containing tetra-n-butylammonium perchlorate (0.1 M) as the supporting electrolyte. The solution concentrations used for the electrochemical studies were typically 10−4 M. The counter electrode was a platinum wire, and an ALS Co. Ltd. Ag+|Ag electrode ([Ag+] = 10−2 M in CH3CN) served as the reference. This electrode was separated from the solution by a bridging compartment. For CV measurements, the working electrode was a 3 mm diameter vitreous carbon disk, which was polished with 1 μm diamond paste before each recording. UV-visible spectroelectrochemical analyses were carried out using a diode array spectrophotometer equipped with a dip probe (10 mm) connected to the spectrophotometer through an optical fiber. Measurements on solid-state materials were carried out in 1 M KCl (aq), using an aqueous reference electrode ALS Co. Ltd. Ag+|AgCl (3 M NaCl) and a Pt wire as counter electrode. 5 mg of the target material were dispersed in 500 µL of a 4% PVA solution in water and vigorously sonicated, then 125 µL of a 4 % BORAX solution in water were added, forming a gel that was then applied onto a glassy carbon electrode (3 mm diameter). The scan rate was 100 mV s−1.
Powder and Single-crystal X-ray crystallographic studies
The powder X-ray diffraction (XRD) analyzes were carried out at room temperature on a Rigaku Miniflex600 benchtop powder diffractometer in a theta/2 theta geometry using a Cu anode (K-Alpha1 [Å] 1.54060), a fast 1D detector with energy selection, and a 6-position sample changer. Samples were measured in a range of 2theta from 5 to 60°, every 0.02°, with a speed of 5° per minute. For single crystal XRD, the crystals were kept in their mother liquor until they were dipped into perfluoropolyether oil and their structure was determined. The chosen crystals were mounted on a Mitegen micromount. Data were collected at different temperatures (from 100 K to 300 K) on a XtaLAB Synergy (Rigaku) diffractometer using either a Cu-Kα radiation (λ = 1.54184 Å) or a Mo-Kα radiation (λ = 0.71073 Å) micro-source, equipped with an Oxford Cryosystems Cooler Device. The structures have been solved using the new dual-space algorithm program SHELXT60 and refined by means of least-squares procedures using SHELXL-201860 program included either in the software package WinGX61 version 1.639 or the package OLEX262. The Atomic Scattering Factors were taken from International Tables for X-Ray Crystallography63. Hydrogen atoms were placed geometrically and refined using a riding model, except for the hydrogens of water molecules which were located by Fourier Differences. All non-hydrogen atoms were anisotropically refined. Ellipsoid plots in the figures of the crystallography section were generated using the software ORTEP-364. The crystal structures have been deposited at the Cambridge Crystallographic Data Centre and allocated the deposition numbers 2321396-2321412, 2327738, 2327739, and 2355273.
Calculations of electron absorption spectra
All calculations have been performed with the ADF program65,66 using the statistical average of orbital potentials (SAOP) exchange-correlation potential and including scalar-relativistic effects within the zero-order regular approximation (ZORA)67,68. The atoms were described by relativistic all-electron Slater-type basis sets from the ADF program69: the TZP basis set of triple-ζ polarized quality was employed for the Fe and Pt atoms, and the DZP basis set of double-ζ polarized quality for the N, C, and H atoms. The fragments were solvated in water using the conductor-like screening model of solvation (COSMO)70. The electronic absorption properties have been studied by performing electronic excitation calculations within linear response in time-dependent density functional theory (LR-TDDFT)71,72 and the Tamm-Dancoff (TDA) approximation73. The analysis of the predicted transitions was done in terms of transition between occupied and virtual natural transition orbitals (NTO)74. The calculations were run restricted for the LS state and unrestricted for the considered HS state with the projection of the total spin moment S along the z-axis constrained to MS = +S.
Supplementary information
Acknowledgements
This work was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Programme (grant agreement No. 101019522, AB), the University of Toulouse III (CPJ 288890, SC), and the Agence Nationale de la Recherche (Project ANR-23-CPJ1-0112-01, SC). LMLD acknowledges high-performance computing time from the HPC facilities of the University of Geneva. AR acknowledges CNCS - UEFISCDI for the financial support of the HighSensSpin project (Contract no. 77/2022).
Author contributions
LG&SC (experimental work and manuscript), DP&LV (crystallographic studies), LMLD (theoretical calculations), AR (magnetic measurements), and GM&AB (manuscript).
Peer review
Peer review information
Nature Communications thanks Mohamed Hegazy, Jun Tao, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
The X-ray crystallographic data for samples 1b (from 150 K to 290 K, one structure every 10 K ((CCDC-2321398 to (CCDC-2321412)), 2b (CCDC-2327738 (100 K), CCDC-2327739 (300 K)) and 3b (CCDC-2321396 (100 K), CCDC-2321397 (300 K)), as well as 2b-Zn (CCDC-2355273 (300 K)) generated in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC). These data can be obtained free of charge from The Cambridge Crystallographic Data Center via https://www.ccdc.cam.ac.uk/structures/?. The corresponding tables are provided in the Supplementary Information file. We declare that the data supporting the findings of this study are available within the article and its Supplementary Information files. All relevant source data are available from the corresponding author upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Saioa Cobo, Email: saioa.cobo@lcc-toulouse.fr.
Azzedine Bousseksou, Email: azzedine.bousseksou@lcc-toulouse.fr.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-51385-8.
References
- 1.Balzani, V., Credi, A. & Venturi, M. in Molecular Devices and Machines: Concepts and Perspectives for the Nanoworld 69–106 (John Wiley & Sons, Ltd, 2008). 10.1002/9783527621682.ch4.
- 2.Balamurugan, A. & Lee, H. A visible light responsive on–off polymeric photoswitch for the colorimetric detection of nerve agent mimics in solution and in the vapor phase. Macromolecules49, 2568–2574 (2016). [Google Scholar]
- 3.Lu, T. et al. Hierarchical photonic structured stimuli-responsive materials as high-performance colorimetric sensors. Nanoscale8, 10316–10322 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Natali, M. & Giordani, S. Molecular switches as photocontrollable “smart” receptors. Chem. Soc. Rev.41, 4010–4029 (2012). [DOI] [PubMed] [Google Scholar]
- 5.Tao, C., Li, Y. & Wang, J. The progress of electrochromic materials based on metal–organic frameworks. Coord. Chem. Rev.475, 214891 (2023). [Google Scholar]
- 6.Chatir, E. et al. Photochromic metallopolymer based on dithienylethene as a molecular calculator. Chem. Mater.34, 5912–5918 (2022). [Google Scholar]
- 7.Geraskina, M. R., Dutton, A. S., Juetten, M. J., Wood, S. A. & Winter, A. H. The viologen cation radical Pimer: A case of dispersion-driven bonding. Angew. Chem. Int. Ed.56, 9435–9439 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Trabolsi, A. et al. Radically enhanced molecular recognition. Nat. Chem.2, 42–49 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Wu, Y. et al. Probing distance-dependent charge-transfer character in excimers of extended viologen cyclophanes using femtosecond vibrational spectroscopy. J. Am. Chem. Soc.139, 14265–14276 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Zhan, T.-G. et al. Supramolecular radical polymers self-assembled from the stacking of radical cations of rod-like viologen di- and trimers. Org. Chem. Front.3, 1635–1645 (2016). [Google Scholar]
- 11.Sagara, T. & Tahara, H. Redox of viologen for powering and coloring. Chem. Rec.21, 2375–2388 (2021). [DOI] [PubMed] [Google Scholar]
- 12.Yu, X.-Q., Sun, C., Liu, B.-W., Wang, M.-S. & Guo, G.-C. Directed self-assembly of viologen-based 2D semiconductors with intrinsic UV–SWIR photoresponse after photo/thermo activation. Nat. Commun.11, 1179 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu, J.-X. et al. Achieving large thermal hysteresis in an anthracene-based manganese(II) complex via photo-induced electron transfer. Nat. Commun.13, 2646 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang, N.-N., Han, Y.-F. & Wang, M.-S. Planar Viologen-based crystalline compounds showing heat-induced electron transfer and thermochromism. Cryst. Growth Des.22, 5293–5299 (2022). [Google Scholar]
- 15.Xu, G. et al. Photochromism of a Methyl Viologen Bismuth(III) Chloride: Structural variation before and after UV irradiation. Angew. Chem. Int. Ed.46, 3249–3251 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Cadman, L. K., Mahon, M. F. & Burrows, A. D. Inclusion of viologen cations leads to switchable metal–organic frameworks. Faraday Discuss.225, 414–430 (2021). [DOI] [PubMed] [Google Scholar]
- 17.Sui, Q. et al. Piezochromism and hydrochromism through electron transfer: new stories for viologen materials. Chem. Sci.8, 2758–2768 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sui, Q. et al. Coordination-modulated piezochromism in metal–viologen materials. J. Mater. Chem. C.5, 12400–12408 (2017). [Google Scholar]
- 19.Coe, B. J. et al. Pentacyanoiron(II) as an Electron Donor Group for Nonlinear Optics: Medium-responsive properties and comparisons with related Pentaammineruthenium(II) Complexes. J. Am. Chem. Soc.128, 12192–12204 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Li, H.-Y. et al. Photochromic properties of a Series of Zinc(II)–Viologen complexes with structural regulation by anions. Cryst. Growth Des.17, 6311–6319 (2017). [Google Scholar]
- 21.Yang, D.-D., Zheng, H.-W., Liang, Q.-F. & Zheng, X.-J. Multistimuli-induced structural variations of a Zn(II)−viologen coordination polymer and the two-step photochromism. Dyes Pigments200, 110188 (2022). [Google Scholar]
- 22.Hegazy, M. B. Z., Hassan, F. & Hu, M. Hofmann-Type Cyanide bridged coordination polymers for advanced functional nanomaterials. Small20, 2306709 (2023). [DOI] [PubMed] [Google Scholar]
- 23.Muñoz, M. C. & Real, J. A. Thermo-, piezo-, photo- and chemo-switchable spin crossover iron(II)-metallocyanate based coordination polymers. Coord. Chem. Rev.255, 2068–2093 (2011). [Google Scholar]
- 24.Huang, Y. & Ren, S. Multifunctional Prussian blue analogue magnets: Emerging opportunities. Appl. Mater. Today22, 100886 (2021). [Google Scholar]
- 25.Ohkoshi, S. et al. High proton conductivity in Prussian blue analogues and the interference effect by magnetic ordering. J. Am. Chem. Soc.132, 6620–6621 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Aguilà, D., Prado, Y., Koumousi, E. S., Mathonière, C. & Clérac, R. Switchable Fe/Co Prussian blue networks and molecular analogues. Chem. Soc. Rev.45, 203–224 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Hofmann, K. A. & Küspert, F. Verbindungen von Kohlenwasserstoffen mit Metallsalzen. Z. Anorg. Allg. Chem.15, 204–207 (1897). [Google Scholar]
- 28.Ni, Z.-P. et al. Recent advances in guest effects on spin-crossover behavior in Hofmann-type metal-organic frameworks. Coord. Chem. Rev.335, 28–43 (2017). [Google Scholar]
- 29.Hiiuk, V. M. et al. Two-step spin crossover in Hofmann-type coordination polymers [Fe(2-phenylpyrazine)2{M(CN)2}2] (M = Ag, Au). Inorg. Chem.61, 2093–2104 (2022). [DOI] [PubMed] [Google Scholar]
- 30.Turo-Cortés, R. et al. Bistable Hofmann-type FeIi spin-crossover two-dimensional polymers of 4-Alkyldisulfanylpyridine for prospective grafting of monolayers on metallic surfaces. Inorg. Chem.60, 9040–9049 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agustí, G. et al. Thermal and light-induced spin crossover phenomena in new 3D Hofmann-like microporous metalorganic frameworks produced as bulk materials and nanopatterned thin films. Chem. Mater.20, 6721–6732 (2008). [Google Scholar]
- 32.Bartual-Murgui, C. et al. Enhanced porosity in a new 3D Hofmann-like network exhibiting humidity sensitive cooperative spin transitions at room temperature. J. Mater. Chem.21, 7217–7222 (2011). [Google Scholar]
- 33.Brennan, A. T., Zenere, K. A., Kepert, C. J., Clegg, J. K. & Neville, S. M. Three distinct spin-crossover pathways in halogen-appended 2D Hofmann frameworks. Inorg. Chem.60, 3871–3878 (2021). [DOI] [PubMed] [Google Scholar]
- 34.Murphy, M. J. et al. Guest programmable multistep spin crossover in a Porous 2-D Hofmann-type material. J. Am. Chem. Soc.139, 1330–1335 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Klein, Y. M. et al. Spin crossover intermediate plateau stabilization in a flexible 2-D Hofmann-type coordination polymer. Chem. Commun.50, 3838–3840 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Milin, E. et al. Elastic frustration triggering photoinduced hidden hysteresis and multistability in a two-dimensional photoswitchable Hofmann-like spin-crossover metal–organic framework. Inorg. Chem.55, 11652–11661 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Kuzevanova, I. S. et al. Spin crossover in iron(II) Hofmann clathrates analogues with 1,2,3-triazole. Dalton Trans.50, 9250–9258 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Niel, V., Martinez-Agudo, J. M., Muñoz, M. C., Gaspar, A. B. & Real, J. A. Cooperative spin crossover behavior in Cyanide-Bridged Fe(II)−M(II) bimetallic 3D Hofmann-like Networks (M = Ni, Pd, and Pt). Inorg. Chem.40, 3838–3839 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Cobo, S. et al. Single-laser-shot-induced complete bidirectional spin transition at room temperature in single crystals of (FeII(pyrazine)(Pt(CN)4)). J. Am. Chem. Soc.130, 9019–9024 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Valverde-Muñoz, F. J. et al. Strong cooperative spin crossover in 2D and 3D FeII–MI, II Hofmann-like coordination polymers based on 2-Fluoropyrazine. Inorg. Chem.55, 10654–10665 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Setifi, F. et al. Spin crossover Iron(II) coordination polymer chains: syntheses, structures, and magnetic characterizations of [Fe(aqin)2(μ2-M(CN)4)] (M = Ni(II), Pt(II), aqin = Quinolin-8-amine). Inorg. Chem.53, 97–104 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Yang, J.-H., Zhao, Y.-X., Xue, J.-P., Yao, Z.-S. & Tao, J. Reverse Hofmann-type spin-crossover compound showing a multichannel controllable color change in an ambient environment. Inorg. Chem.60, 7337–7344 (2021). [DOI] [PubMed] [Google Scholar]
- 43.Orellana-Silla, A. et al. Symmetry breaking and cooperative spin crossover in a Hofmann-type coordination polymer based on negatively charged {FeII(μ2-[MII(CN)4])2}n2n– layers (MII = Pd, Pt). Inorg. Chem.62, 12783–12792 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ji, H. et al. Pyridinium-functionalized ionic metal–organic frameworks designed as bifunctional catalysts for CO2 fixation into cyclic carbonates. ACS Appl. Mater. Interfaces12, 24868–24876 (2020). [DOI] [PubMed] [Google Scholar]
- 45.Sun, J.-K., Yang, X.-D., Yang, G.-Y. & Zhang, J. Bipyridinium derivative-based coordination polymers: From synthesis to materials applications. Coord. Chem. Rev.378, 533–560 (2019). [Google Scholar]
- 46.Bakkar, A. et al. A redox- and photo-responsive quadri-state switch based on dimethyldihydropyrene-appended cobalt complexes. J. Mater. Chem. C.4, 1139–1143 (2016). [Google Scholar]
- 47.Bakkar, A. et al. Electrochemical control of the switching process of photochromic dimethyldihydropyrene derivatives. Chem. Commun.53, 9360–9363 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Olejnik, A., Kopec, W., Maskowicz, D. & Sawczak, M. Spin-resolved band structure of Hofmann Clathrate [Fe(pz)2Pt(CN)4] as an essential tool to predict optical spectra of metal–organic frameworks. ACS Appl. Mater. Interfaces15, 15848–15862 (2023). [DOI] [PubMed] [Google Scholar]
- 49.Monk, P. M. S., Hodgkinson, N. M. & Ramzan, S. A. Spin pairing (‘dimerisation’) of the viologen radical cation: kinetics and equilibria. Dyes Pigments43, 207–217 (1999). [Google Scholar]
- 50.Kahlfuss, C. et al. Reversible dimerization of viologen radicals covalently linked to a calixarene platform: Experimental and theoretical aspects. Comptes Rendus Chim.17, 505–511 (2014). [Google Scholar]
- 51.Kahlfuss, C. et al. Chemically and electrochemically triggered assembly of viologen radicals: towards multiaddressable molecular switches. Chem. Eur. J.21, 2090–2106 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Iordache, A. et al. Redox control of rotary motions in ferrocene-based elemental ball bearings. J. Am. Chem. Soc.134, 2653–2671 (2012). [DOI] [PubMed] [Google Scholar]
- 53.Reguera, E., Yee-Madeira, H., Demeshko, S., Eckold, G. & Jimenez-Gallegos, J. Nature of the observed asymmetry in Mössbauer Spectra of Iron (2+) Hexacyanometallates (III). Z. Für. Phys. Chem.223, 701–711 (2009). [Google Scholar]
- 54.Molnár, G. et al. Vibrational Spectroscopy of Cyanide-bridged, Iron(II) spin-crossover coordination polymers: estimation of vibrational contributions to the entropy change associated with the spin transition. J. Phys. Chem. B106, 9701–9707 (2002). [Google Scholar]
- 55.Ohtani, R. et al. Precise control and consecutive modulation of spin transition temperature using chemical migration in porous coordination polymers. J. Am. Chem. Soc.133, 8600–8605 (2011). [DOI] [PubMed] [Google Scholar]
- 56.Wang, H.-Y. et al. Photo- and electronically switchable spin-crossover Iron(II) metal–organic frameworks based on a Tetrathiafulvalene ligand. Angew. Chem. Int. Ed.56, 5465–5470 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Wu, S.-G. et al. Redox-programmable spin-crossover behaviors in a cationic framework. J. Am. Chem. Soc.144, 14888–14896 (2022). [DOI] [PubMed] [Google Scholar]
- 58.Palacios-Corella, M. et al. Redox and guest tunable spin-crossover properties in a polymeric polyoxometalate. Chem. Sci.14, 3048–3055 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang, M., Li, Z.-Y., Ishikawa, R. & Yamashita, M. Spin crossover and valence tautomerism conductors. Coord. Chem. Rev.435, 213819 (2021). [Google Scholar]
- 60.Sheldrick, G. M. SHELXT – Integrated space-group and crystal-structure determination. Act. Cryst. A71, 3–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Farrugia, L. J. WinGX suite for small-molecule single-crystal crystallography. J. Appl. Cryst.32, 837–838 (1999). [Google Scholar]
- 62.Dolomanov, O. V., Bourhis, L. J., Gildea, R. J., Howard, J. A. K. & Puschmann, H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Cryst.42, 339–341 (2009). [Google Scholar]
- 63.INTERNATIONAL Tables for X-Ray Crystallography. IV (Kynoch Press, Birmingham, England,(1974).
- 64.Farrugia, L. J. ORTEP-3 for Windows - a version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Cryst.30, 565–565 (1997). [Google Scholar]
- 65.te Velde, G. et al. Chemistry with ADF. J. Comput. Chem.22, 931–967 (2001). [Google Scholar]
- 66.SCM, ADF (Amsterdam Density Functional), Vrije Universiteit, Amsterdam, The Netherlands, 2021.
- 67.Rosa, A. et al. Electronic Spectra of M(CO)6 (M = Cr, Mo, W) revisited by a relativistic TDDFT approach. J. Am. Chem. Soc.121, 10356–10365 (1999). [Google Scholar]
- 68.Van Lenthe, E., Baerends, E. J. & Snijders, J. G. Relativistic regular two‐component Hamiltonians. J. Chem. Phys.99, 4597–4610 (1993). [Google Scholar]
- 69.Van Lenthe, E. & Baerends, E. J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem.24, 1142–1156 (2003). [DOI] [PubMed] [Google Scholar]
- 70.Pye, C. C. & Ziegler, T. An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package. Theor. Chem. Acc.101, 396–408 (1999). [Google Scholar]
- 71.Casida, M. E. Time-dependent density functional response theory for molecules. in Recent Advances in Density Functional Methods 1 155–192 (World Scientific, 1995).
- 72.van Gisbergen, S. J. A., Snijders, J. G. & Baerends, E. J. Implementation of time-dependent density functional response equations. Comput. Phys. Comm.118, 119–138 (1999). [Google Scholar]
- 73.Hirata, S. & Head-Gordon, M. Time-dependent density functional theory within the Tamm–Dancoff approximation. Chem. Phys. Lett.314, 291–299 (1999). [Google Scholar]
- 74.Martin, R. L. Natural transition orbitals. J. Chem. Phys.118, 4775–4777 (2003). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The X-ray crystallographic data for samples 1b (from 150 K to 290 K, one structure every 10 K ((CCDC-2321398 to (CCDC-2321412)), 2b (CCDC-2327738 (100 K), CCDC-2327739 (300 K)) and 3b (CCDC-2321396 (100 K), CCDC-2321397 (300 K)), as well as 2b-Zn (CCDC-2355273 (300 K)) generated in this study have been deposited at the Cambridge Crystallographic Data Center (CCDC). These data can be obtained free of charge from The Cambridge Crystallographic Data Center via https://www.ccdc.cam.ac.uk/structures/?. The corresponding tables are provided in the Supplementary Information file. We declare that the data supporting the findings of this study are available within the article and its Supplementary Information files. All relevant source data are available from the corresponding author upon request.