

Abstract
Cox17, a copper chaperone for cytochrome c oxidase, is an essential and highly conserved protein. The structure and mechanism of functioning of Cox17 are unknown, and even its metalbinding stoichiometry is elusive. In the present study, we demonstrate, using electrospray ionization–MS, that porcine Cox17 binds co-operatively four Cu+ ions. Cu4Cox17 is stable at pH values above 3 and fluorescence spectra indicate the presence of a solvent-shielded multinuclear Cu(I) cluster. Combining our results with earlier EXAFS results on yeast CuCox17, we suggest that Cu4Cox17 contains a Cu4S6-type cluster. At supramillimolar concentrations, dithiothreitol extracts metals from Cu4Cox17, and an apparent copper dissociation constant KCu=13 fM was calculated from these results. Charge-state distributions of different Cox17 forms suggest that binding of the first Cu+ ion to Cox17 causes a conformational change from an open to a compact state, which may be the rate-limiting step in the formation of Cu4Cox17. Cox17 binds non-co-operatively two Zn2+ ions, but does not bind Ag+ ions, which highlights its extremely high metal-binding specificity. We further demonstrate that porcine Cox17 can also exist in partly oxidized (two disulphide bridges) and fully oxidized (three disulphide bridges) forms. Partly oxidized Cox17 can bind one Cu+ or Zn2+ ion, whereas fully oxidized Cox17 does not bind metals. The metal-binding properties of Cox17 imply that, in contrast with other copper chaperones, Cox17 is designed for the simultaneous transfer of up to four copper ions to partner proteins. Metals can be released from Cox17 by non-oxidative as well as oxidative mechanisms.
Keywords: co-operative metal binding, copper cluster, cytochrome c oxidase copper chaperone (Cox17), mass spectrometry, charge-state distribution analysis
Abbreviations: CCO, cytochrome c oxidase; Cox17, a CCO copper chaperone; DTT, dithiothreitol; ESI, electrospray ionization
INTRODUCTION
Cytochrome c oxidase (CCO) is a key catabolic enzyme, which is localized in the mitochondrial inner membrane and terminates the respiratory chain by transferring electrons from cytochrome c to molecular oxygen [1]. During evolution, CCO has acquired the ability to transfer four electrons and protons that convert molecular oxygen into water and thus avoid the formation of reactive oxygen metabolites. CCO accomplishes this challenging biochemical task by the help of three copper ions and two haems. Copper ions constitute a binuclear CuA site and a mononuclear CuB site on the mitochondrially synthesized CCO subunits Cox2 and Cox1 respectively [2]. Cellular copper metabolism is highly regulated and copper is delivered to key copper proteins, including CCO, by specific metal-transfer proteins, such as copper chaperones [3].
Copper chaperone for CCO, identified in yeast as a factor facilitating the incorporation of copper into CCO, was designated as Cox17 [4]. Cox17 is a low-molecular-mass protein, which is highly conserved from yeast to mammals. Many observations demonstrate that Cox17 is essential to activate CCO in yeast [5] and mammals [6]; however, the mechanism of functioning of Cox17 is still elusive. It was initially suggested that, in yeast, Cox17 might shuttle copper from the cytosol to the mitochondrial intermembrane space [7]; however, recent results indicate that Cox17 functioning may be restricted to the mitochondrial intermembrane space. There is biological evidence that yeast Cox17 functions in concert with the mitochondrial inner-membrane protein Sco1 in the delivery of copper, specifically to the CuA sites of CCO [8,9]. It is suggested that Cox11, another mitochondrial inner-membrane protein, delivers copper to the CuB sites of CCO [10]; however, the details of metal transfer are not known.
The functional properties of Cox17 should rely on its metalbinding properties, which are, however, largely unknown. According to current knowledge, both yeast and mammalian Cox17 may contain two or three copper atoms per molecule of protein, whereas the metal content depends on the way in which the recombinant protein is produced [6,11,12]. Moreover, Cox17 loses metals during purification [7] and tends to oxidize [13,14], which further complicates determination of its metal-binding stoichiometry. An attempt has been made to determine the metalbinding stoichiometry of yeast Cox17 by ESI (electrospray ionization)–MS, which is the method of choice for this purpose. For unknown reasons, ESI–MS measurements did not detect any metallated Cox17 species [12].
Structural information about the metal-binding motif in Cox17 is available only from fluorescence and EXAFS studies of yeast CuCox17 samples. Fluorescence of CuCox17 suggests that copper is bound in the form of Cu+ ions to a solvent-shielded polycopper cluster [11,12]. EXAFS studies of yeast CuCox17 containing two copper atoms/molecule indicated that a binuclear Cu(I)–thiolate cluster may exist in Cox17 [11]. However, similar studies with yeast CuCox17 containing three copper atoms/molecule demonstrated the existence of a more uniform polycopper–thiolate cluster [12], the type and composition of which are still unknown.
The aim of the present study was to uncover the stoichiometry and mechanism of metal binding to Cox17. Using mammalian Cox17 isolated from porcine intestine and a novel design of ESI–MS experiments, we demonstrate that fully reduced Cox17 binds co-operatively four Cu+ ions into an acid-stable cluster, whereas partially reduced Cox17 binds only one metal ion and fully oxidized Cox17 does not bind metals. The fluorescence properties of Cu4Cox17 are similar to the yeast CuCox17 and indicate that copper is bound in the form of Cu+ ions into a solvent-shielded cluster. We further demonstrate that thiol reagents such as DTT (dithiothreitol) and GSH can extract copper from Cu4Cox17 in a concentration-dependent manner and we determine the apparent dissociation constant KCu for the Cu4Cox17 complex. Metal-binding properties of Cox 17 reveal new aspects of its biological functioning and these are also discussed.
EXPERIMENTAL
Purification of porcine Cox17
Porcine Cox17 was isolated from porcine intestine as a fully oxidized apoprotein. Briefly, porcine intestines were processed by the method described in [15] before size-exclusion chromatography. The Cox17-containing fraction from size-exclusion chromatography was further purified on a preparative cation-exchange column at pH 5.2 and thereafter at pH 2.5. Finally, Cox17 was purified to homogeneity by preparative reversed-phase HPLC. The presence of Cox17 in the fractions was monitored by LC (liquid chromatography)/ESI–MS using an Ettan™ ESI–TOF (ESI–time-of-flight) mass spectrometer (accuracy level, 5 ppm) connected on-line to an Ettan LC (both from Amersham Biosciences, Uppsala, Sweden). The fraction containing pure Cox17 was freeze-dried.
MS measurements with Cox17
Freeze-dried apoCox17 was dissolved at a concentration of 75 μM in argon-saturated 20 mM ammonium acetate (pH 7.6), and this stock solution was used for further experiments. Cox17 was reduced at 45 °C with 2 or 4 mM DTT, and the reduction level at various incubation times (2–110 min) was determined by mass measurements. Metal-binding experiments were performed with fully oxidized, partly reduced and fully reduced Cox17 forms.
Reconstitution of various Cox17 forms with copper was performed as follows. First, Cu(II) acetate was dissolved at 150 μM concentration in argon-saturated 20 mM ammonium acetate (pH 7.6) and Cu2+ was reduced to Cu+ by the addition of 2 mM DTT. Freshly prepared metal salt solution (1–6 equivalents) was added to the oxidized or reduced apoCox17, and the mixture was incubated for 1 min at 25 °C. The sample was diluted with 20 mM ammonium acetate (pH 7.6) to a final protein concentration of 1.4 μM (final concentration of DTT in the MS probe was 0.15–0.35 mM) and infused by a syringe pump at 10 μl/min into an Ettan™ ESI–TOF mass spectrometer. Mass spectra were recorded for 2–3 min at a capillary exit voltage of 150 V.
Reconstitution of Cox17 with Zn2+ and Ag+ ions was performed as described above, except that DTT was not added to the metal salt solutions. Cu(II) acetate, zinc acetate and silver nitrate were obtained from Sigma.
Studies of pH dependence were performed as described above with the only exception that the reconstitution mixture (2 μl) was diluted with different buffers and organic acid solutions (50 μl) before MS measurements. The following solutions were used: 20 mM ammonium acetate with pH values between 4.80 and 8.6, 20 mM acetic acid (pH 3.2), 20 mM formic acid (pH 2.7) and 0.1% trifluoroacetic acid (pH 1.9).
Influence of DTT and GSH on copper binding by Cox17 was studied by adding 4 equivalents of Cu+ ions to Cox17, followed by 13.5-fold dilution of the reconstitution mixture with 20 mM ammonium acetate (pH 7.6), which contained 0–10 mM DTT or 0–16 mM GSH. The mixture obtained (Cox17 concentration, 1.8 μM) was used for MS measurements.
UV and fluorescence spectroscopy
UV and fluorescence titration of fully reduced Cox17 (3.0 μM) with Cu+ or Ag+ ions was performed in 20 mM ammonium acetate buffer (pH 7.6) at 25 °C. Cu+ was prepared as described above and the final concentration of DTT in the final sample was 0.3–0.5 mM. UV spectra were recorded on a UV 1601PC spectrophotometer (Shimadzu, Kyoto, Japan) and fluorescence spectra were recorded on a 4500 spectrofluorimeter (Hitachi, Tokyo, Japan) with an excitation wavelength of 300 nm, an excitation slit width of 5 nm and an emission slit width of 20 nm.
RESULTS
Reduction of Cox17
The mass spectrum of the isolated Cox17 showed three major peaks with charges +5, +6 and +7. Deconvolution of the spectrum gave a molecular mass of 6688.97 Da, which agrees well with the theoretical molecular mass of Cox17 containing three disulphide bridges (6688.96 Da). The incubation of fully oxidized Cox17 with 2 mM DTT at 45 °C for 2 min did not change the distribution of ion species, but increased the molecular mass of the protein to 6691.0 Da, which corresponds to that of Cox17 containing two disulphide bridges. Further reduction took more time and it is only after 110 min incubation that the protein displayed a molecular mass of 6694.82 Da, which agrees with the theoretical molecular mass of fully reduced Cox17 (6694.88 Da). Fully reduced Cox17 exposes six ion species (charges from +5 to +9) in the mass spectrum. According to a previous report, a distinct pattern of disulphide bridges between cysteine residues 22–25, 23–54 and 35–44 exists in fully oxidized porcine Cox17 [13]. Our results demonstrate that there is one labile disulphide bond and two more stable core disulphide bonds in fully oxidized porcine Cox17.
Reconstitution of fully reduced Cox17 with Cu+ ions
Mass spectra of fully reduced Cox17, reconstituted at pH 7.6 with increasing concentrations of Cu+ ions, are presented in Figure 1. The addition of 1 equivalent of Cu+ ions to apoCox17 generates small amounts of two metalloforms corresponding to Cu1Cox17 and Cu4Cox17 (Figure 1b). After the addition of 2 equivalents of Cu+ ions, peaks of apoCox17 and Cu4Cox17 are predominant and of equal intensity (Figure 1c). Since only 50% of Cox17 can be saturated with four Cu+ ions under these conditions, this result indicates that the relative intensities of apoCox17 and Cu4Cox17 peaks in the mass spectra are almost equal. By the addition of 4 equivalents of Cu+ ions, the MS spectrum exposes one major peak of Cu4Cox17 together with minor apoCox17, Cu1Cox17 and Cu5Cox17 forms (Figure 1d). Addition of 6 equivalents of Cu+ ions leads to a relative increase of the Cu5Cox17 peak (Figure 1e). Mass analysis of Cu4Cox17 (assuming that four Cu+ ions are bound) indicates that the molecular mass of Cox17 protein in this complex is 6695.14 Da. This value is very close to the theoretical value for fully reduced Cox17 protein (6694.88 Da), which demonstrates that there are no disulphides in Cu4Cox17 and copper is bound exclusively in the form of Cu+ ions.
Figure 1. Binding of Cu+ ions to Cox17.

Mass spectra of Cox17 (1.4 μM) reconstituted with 1–6 equivalents of Cu+ ions in the presence of 20 mM ammonium acetate (pH 7.6) and 0.15–0.35 mM DTT at 25 °C. Charge state +6 ions are presented and numbers on the peaks denote the metal stoichiometry of the complex.
Yeast CuCox17 is oligomeric at high protein concentrations and the dimer/tetramer equilibrium constant is equal to 20 μM [12]. Our MS experiments were performed at a protein concentration of 1.4–1.8 μM and we detected no dimeric or oligomeric species in the mass spectra of porcine CuCox17 forms.
Influence of pH on the binding of Cu+ to Cox17
Mass spectra of Cox17 reconstituted with four Cu+ equivalents and diluted with buffers to different pH values demonstrate that Cu4Cox17 and Cu5Cox17 forms are stable at and above pH 4.8. At pH 3.2, the relative intensity of the peak corresponding to apoCox17 starts to increase, and at pH 1.9, apoCox17 is the major form. At acidic pH values, minute amounts of the intermediate metalloforms Cu2Cox17 and Cu3Cox17 also occur in the mass spectra, which are totally absent at pH 7.6. The presence of the intermediate metalloforms is indicative of the existence of metal exchange between apoCox17 and Cu4Cox17. Yeast CuCox17 starts to decompose also at pH 3 [12] and, therefore, its pH stability is similar to that of porcine Cu4Cox17, which implies similar co-ordination of metals in mammalian and yeast CuCox17 complexes.
Influence of DTT and GSH on the binding of Cu+ to Cox17
Mass spectra of Cox17 reconstituted with four Cu+ equivalents in the presence of increasing concentrations of DTT are shown in Figure 2. At 0.95 mM DTT, the peak corresponding to apoCox17 and Cu1Cox17 starts to increase (Figure 2b); at 3 mM DTT, the apoCox17 and Cu4Cox17 peaks show almost equal intensities (Figure 2c); at 9.55 mM DTT, apoCox17 is the major peak in the mass spectrum together with minor Cu1Cox17 and Cu4Cox17 peaks (Figure 2e). The results obtained demonstrated that, at supramillimolar concentrations, DTT extracts metals from Cu4Cox17, and allowed us to calculate the dissociation constant for the Cu4Cox17 complex. The binding results were analysed according to the following scheme:
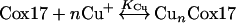 |
(1) |
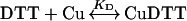 |
(2) |
Here, KCu is the dissociation constant for the CunCox17 complex and Kd the conditional dissociation constant for the Cu+–DTT complex; Kd=6.31×10−12 M at pH=7.4, intensity I=0.1 and T=25 °C [16]. To determine the KCu value, we used the general method described in [17]. First, the concentrations of free Cu+ ions in the presence of various concentrations of DTT were calculated using the Kd value presented above. Furthermore, Y, the fractional content of Cu4Cox17 species, was calculated from the intensities of apoCox17 and Cu4Cox17 peaks in the mass spectra using the relation Y=ICu4Cox17/(ICox17+ICu4Cox17), and the latter parameter was correlated with the concentration of free copper ions (Figure 3). The binding isotherm obtained was non-linearly fitted with a common hyperbolic equation, describing a simple 1:1 binding equilibrium (n=1). The analysis yielded a good fit (R2=0.95) with an apparent dissociation constant KCu=(1.3±0.3)×10−14 M (Figure 3). The binding results were analysed also in Hill coordinates (Figure 3, inset), which yielded a Hill coefficient h=1.35±0.11 and log KCu=14.0 (KCu=1.0×10−14 M). The Hill coefficient being close to unity confirms that binding of Cu+ ions to Cox17 could be reasonably described by the equilibrium eqn (3) where h=1 and, therefore, there is no need to use more complicated binding schemes. The program Origin 6.1 (OriginLab Corp., Northampton, MA, U.S.A.) was used for the fitting.
Figure 2. Binding of Cu+ ions to Cox17 in the presence of DTT.

Mass spectra of Cox17 (2 μM) reconstituted with 4 equivalents of Cu+ ions in the presence of 20 mM ammonium acetate (pH 7.6) and DTT at various concentrations. Concentrations of DTT: (a) 0.3 mM, (b) 0.75 mM, (c) 3.0 mM, (d) 6 mM and (e) 10 mM. Charge state +6 ions are presented and numbers on the peaks denote the metal stoichiometry of the complex.
Figure 3. Determination of the dissociation constant Kd for the Cu4Cox17 complex.

Dependence of the fractional content of metallated Cu4Cox17, Y=ICu4Cox17/(ICox17+ICu4Cox17), on the concentration of free Cu+ ions in the metal competition experiment [20 mM ammonium acetate (pH 7.6); 25 °C]. ——, Curve fitted for hyperbola with KCu=1.3×10−14 M is shown. Inset: the same results in Hill coordinates [Φ=Y/(1−Y)] and a linear fit (——) are shown; h (represented by n in the Figure), Hill coefficient.
Similar to DTT, GSH has an influence on the copper binding to Cox17, but the effects appear at slightly higher concentrations of the reagent (3.7 mM GSH, Y=0.73; 7.41 mM GSH, Y=0.61; 14.8 mM GSH, Y=0.40). Quantitative analysis of these results has not been attempted since the binding of Cu+ to GSH is very complex and conditional binding constants for the Cu+–GSH complex available in the literature are not as reliable as for DTT [16]. However, the results obtained provide a quantitative picture of the distribution of copper between Cox17 and GSH at physiological concentrations of GSH (0.5–5 mM).
Reconstitution of fully reduced Cox17 with Ag+ and Zn2+ ions
After the addition of 4 equivalents of Ag+ ions to fully reduced Cox17 at pH 7.6, apoCox17 still remains the prevalent form and only a minor peak of Ag1Cox17 is observed (Figure 4b). Mass spectra of fully reduced Cox17 reconstituted at pH 7.6 with 4 equivalents of Zn2+ ions expose minor apoCox17 and two major peaks corresponding to Zn1Cox17 and Zn2Cox17 with almost equal intensity (Figure 4c). The addition of 6 equivalents of Zn2+ ions increases the relative intensity of the Zn2Cox17 peak only slightly; however, a substantial amount of apoCox17 and Zn1Cox17 together with minor Zn3Cox17 is still present (results not shown). The titration of fully reduced Cox17 with Cd2+ ions yields a mixture of apoCox17, Cd1Cox17, Cd2Cox17 and minor Cd3Cox17, which is similar to the titration of Cox17 with Zn2+ ions, and the corresponding spectrum is shown in Figure 4(d). By the addition of 4 equivalents of Fe2+ ions to the reduced Cox17, only apoCox17 is detected (Figure 4e).
Figure 4. Binding of Ag+ and bivalent metal ions by Cox17.

Mass spectra of Cox17 (1.4 μM) reconstituted with 4 equivalents of Ag+ (b), Zn2+ (c), Cd2+ (d) and Fe2+ ions (e) in 20 mM ammonium acetate (pH 7.6) and 0.3 mM DTT at 25 °C. Charge state +6 ions are presented and numbers on the peaks denote the metal stoichiometry of the complex.
Reconstitution of fully and partially oxidized Cox17 with Cu+ and Zn2+ ions
The reconstitution of fully oxidized Cox17 was probed with Cu2+ and Zn2+ ions, but metallated Cox17 species were not detected. Partially reduced Cox17 can bind maximally one Cu+ or Zn2+ ion under metal-excess conditions (spectra not presented). Cu1Cox17 formed with partially oxidized Cox17 is stable at neutral pH values. However, it starts to decompose at pH 5.5 itself and will be only a minor form at pH 4.8, which demonstrates that this metalloform is substantially less acid-stable compared with Cu4Cox17. Reasons for the lower pH stability of partially reduced Cu1Cox17 compared with fully reduced Cu4Cox17 are unclear at the moment. However, these results imply that metal-binding sites in these Cox17 forms are composed of different amino acid residues and/or have different solvent accessibilities.
Conformational analysis
Information about the conformational states of Cox17 protein under a variety of conditions can be obtained from the charge-state distribution analysis of ESI mass spectra, which relies on the well-confirmed assumption that in the ESI process, compact protein conformations produce lower-charge-state ions when compared with more open conformations [18]. Our ESI–MS results demonstrate that fully and partially oxidized Cox17 forms show three major peaks in the mass spectrum with charges +5, +6 and +7 (Figure 5a), whereas fully reduced Cox17 exposes five charge states with charges between +5 and +9 (Figure 5b). These results indicate that fully and partially oxidized Cox17 have compact structures, which are opened only after the reduction of all three disulphide bridges. Figure 5(c) demonstrates that binding of one Cu+ ion to fully reduced Cox17 causes a shift in the charge-state distribution from higher to lower charge states, which indicates that the formation of Cu1Cox17 is accompanied by a conformation change from an open to a compact state. Cu4Cox17 has also the charge-state distribution characteristic of the compact conformation (Figure 5d); therefore, there are no substantial conformational changes in the formation of Cu4Cox17 from Cu1Cox17 species. Metalloforms of partially oxidized Cox17 with Cu+ (Figure 5e) and Zn2+ ions (results not shown) have also a charge-state distribution characteristic of the compact conformational state, which indicates that binding of one metal by partially oxidized Cox17 occurs without substantial conformational changes.
Figure 5. Charge-state analysis of different forms of Cox17.

Mass spectra of (a) fully oxidized Cox17, (b) reduced Cox17, (c) reduced Cox17 reconstituted with 1 equivalent of Cu+ ions, (d) reduced Cox17 reconstituted with 4 equivalents of Cu+ ions and (e) partially oxidized Cox17 reconstituted with 4 equivalent of Cu+ ions. Conditions: 1.4 μM Cox17, 20 mM ammonium acetate (pH 7.6) and 0.3 mM DTT at 25 °C. Charge states of ions (underlined) are presented and numbers on the peaks denote the metal stoichiometry of the complex.
UV and fluorescence spectroscopies
UV absorption and fluorescence spectra of Cox17 at increasing concentrations of Cu+ ions are presented in Figure 6. Addition of Cu+ ions leads to the appearance of a broad absorption band between 240 and 320 nm with a tailing to 350 nm, which is similar to yeast CuCox17 and characteristic of Cu(I)–thiolate charge-transfer transitions [11,12]. There is an initial sharp increase in UV absorption after the addition of 1–4 equivalents of Cu+ ions (Figure 6a, inset), which quite probably reflects the formation of Cox17 complexes with Cu+ ions. Further increase in the absorption at Cu+-to-protein stoichiometries higher than 4 is mainly caused by increase in the concentration of the Cu+–DTT complex, which also absorbs in this region.
Figure 6. UV absorption and fluorescence studies.

UV absorption spectra (a) and fluorescence spectra (b) of Cox17 after the addition of Cu+ ions. Conditions: 20 mM ammonium acetate (pH 7.6) at 25 °C. (a, b) Insets: absorption at 265 nm (a) or luminescence intensity at 585 nm as a function of the mole equivalents of Cu+ ions added.
In the fluorescence spectra, the addition of Cu+ ions leads to the appearance of a fluorescence emission band centred at 585 nm (Figure 6b). Similar fluorescence has been observed also in yeast CuCox17 [11,12] and other proteins containing multinuclear Cu+ clusters such as metallothioneins [19], transcription factor Ace1 [12] and others. It is generally suggested that emission close to 600 nm arises from multinuclear Cu+ clusters in solvent-shielded environments [11,12,19], and our fluorescence studies point to the presence of such a type of cluster(s) also in porcine CuCox17. The fluorescence titration curve (Figure 6b, inset) is sigmoidal and the steepest increase in fluorescence intensity occurs after the addition of 2–4 equivalents of Cu+. These results indicate that fluorescence arises from CuCox17 species having metal load higher than 2, which according to MS data are Cu4Cox17 and Cu5Cox17. A further slight increase in fluorescence intensity at metal stoichiometries higher than 4 suggests that CuCox17 forms with metal stoichiometry greater than 4, e.g. Cu5Cox17, have slightly higher fluorescence intensities when compared with Cu4Cox17.
Cox17 reconstituted with up to 8 equivalents of Ag+ ions did not show any fluorescence after excitation at 300 nm, which confirms that Cox17 cannot bind Ag+ ions into a multinuclear solvent-shielded complex.
DISCUSSION
The ESI–MS studies reported here allow an in-depth understanding of the interplay between different molecular states and metalloforms of porcine Cox17, and reveal the mechanism of metal binding to mammalian Cox17. In general, our results demonstrate that, depending on the environmental conditions, mammalian Cox17 may exist in three main states, corresponding to fully reduced, partially oxidized (two disulphide bridges) or fully oxidized (three disulphide bridges) protein forms, which differ substantially in their metal-binding properties. Fully reduced Cox 17 is designed for specific binding of four Cu+ ions, partially oxidized Cox17 binds one metal with low metal specificity and fully oxidized Cox17 does not bind metals.
Mechanism of metal binding to fully reduced Cox17
Our results demonstrate clearly that fully reduced Cox17 binds co-operatively four Cu+ ions. The co-operative character of the binding emerges from the fact that Cu4Cox17 is formed already by the addition of 1 and 2 equivalents of Cu+ ions to Cox17 and intermediate metalloforms; Cu2Cox17 and Cu3Cox17 are practically absent from the spectrum throughout the titration. There is a minor Cu1Cox17 metalloform present in the mass spectra throughout the titration and its fractional content is higher at low metal-to-protein ratios, which suggests that Cu1Cox17 is an intermediate metalloform in the formation of Cu4Cox17 species. Conformational analysis indicates that the formation of Cu1Cox17 is accompanied by a conformational change of Cox17 from an open to a more compact state and, therefore, it is reasonable to suggest that this conformational change may be the rate-limiting step in the formation of Cu1Cox17, which binds co-operatively three more Cu+ ions into the Cu4Cox17 complex.
Our results indicate that porcine Cox17 can form a complex also with five Cu+ ions. The peak corresponding to Cu5Cox17 is present in MS spectra after the addition of 2 equivalents of Cu+ ions (Figure 1c) and its fractional content increases with higher metal-to-protein ratios. These results demonstrate that the Cu5Cox17 peak does not arise from the non-specific binding of metal excess; rather, they indicate that there is an equilibrium between Cu4Cox17 and Cu5Cox17 forms in the metal-binding scheme of Cox17. The total metal binding scheme for Cox17 could be presented by three equilibria, as follows:
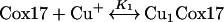 |
(3) |
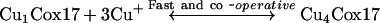 |
(4) |
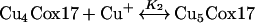 |
(5) |
where K1 and K2 are elementary dissociation constants for the Cu1Cox17 and Cu5Cox17 complexes respectively.
Our results do not agree with results in the literature regarding the metal-binding stoichiometry of Cox17; therefore, critical analysis of previous results should also be provided. So far, the metal-binding stoichiometry of Cox17 has been deduced from quantitative metal analyses, which have demonstrated that the recombinant Cox17 produced from a glutathione S-transferase fusion protein contains approx. two copper atoms per molecule and the CuCox17 produced as untagged protein contains approx. three copper atoms per molecule for both yeast [11,12] and mammalian Cox17 [6]. Our ESI–MS results demonstrate clearly that mammalian Cox17 does not form metalloforms with metal stoichiometries 2 and 3, and the Cu+-binding stoichiometry of the major metalloform is 4. Importantly, our studies demonstrate that DTT can, at supramillimolar concentrations, extract metals from porcine Cu4Cox17. In the light of these results, it can be inferred that the common usage of up to 150 mM concentrations of DTT in the reduction of the Cox17 sample, and lower mM concentrations of DTT in further purification buffers [6,11,12] can easily lead to partial loss of metals from CuCox17 and underestimation of its metal-binding stoichiometry by common methods. We have applied the ESI–MS technique for correct determination of the metal-binding stoichiometry of Cox17. It should be noticed that sample preparation in our MS studies differs substantially from that in earlier MS studies of yeast CuCox17 [12]. In earlier MS studies, sample preparation included extensive dialysis of yeast CuCox17 against buffers containing 1 mM DTT [12], which may lead to metal depletion. We avoided sample dialysis or desalting and have confirmed that fast desalting of porcine CuCox17 samples, by the method described in [20], leads to metal depletion and substantial decrease of all peaks in the mass spectrum.
From our results, it is reasonable to suggest that the mammalian CuCox17 sample with an average copper content equal to three atoms per molecule contains approx. 75% of Cu4Cox17 together with apo- and low-stoichiometry forms of Cox17. We cannot automatically extend this conclusion to yeast CuCox17, since no complementary MS data are available for yeast Cox17 as yet. However, there are many convincing reasons to believe that yeast and mammalian Cox17 have a similar general mechanism of metal binding. First, yeast and mammalian Cox17 are homologous proteins, which are highly conserved during evolution. Secondly, copper forms of yeast and mammalian Cox17 show similar copper contents after isolation [6,11,12]. Finally, yeast and mammalian CuCox17 forms have similar pH stability and spectroscopic properties.
Structure of the metal-binding motif in CuCox17
The spatial structure of Cu4Cox17 is unknown; however, accumulated spectroscopic data have allowed us to draw a number of conclusions about the structure of the metal-binding motif in CuCox17. Metal binding co-operativity and stoichiometry together with fluorescence properties suggest that a solvent-shielded tetranuclear Cu(I) cluster exists in the mammalian Cox17. The UV absorption spectrum and pH dependence of porcine Cu4Cox17 indicate that thiolates from cysteine residues could be involved in metal binding; however, without EXAFS or structural data, we cannot completely exclude participation of other types of amino acid residues, especially histidine or methionine residue(s), in the ligation of metals by the mammalian Cox17. At the moment, EXAFS data are available only for yeast CuCox17 samples with metal content 2 [11] and 3 [12]. Both spectra demonstrate that there is a polycopper cluster in yeast CuCox17 and copper atoms are trigonally co-ordinated by thiolates exclusively [12]. Moreover, it has been noticed that the X-ray absorption near-edge spectrum of yeast CuCox17 is very similar to the corresponding spectrum of the well-characterized [Cu4(SPh)6]2− model cluster [12]. In the structure of [Cu4(SPh)6]2−, four Cu+ ions form an approximate tetrahedron and metals are trigonally co-ordinated by bridging thiolates above each of the six edges (Figure 7a) [21]. Combining EXAFS results on yeast CuCox17 with our MS data, we can suggest that yeast CuCox17 contains the [Cu4(S)6]2− cluster, which is very similar to the well-known [Cu4(S)6]2− model clusters depicted in Figure 7(a). It is reasonable to assume that all the six conserved cysteine residues of Cox17 (Figure 7b) participate in the ligation of metals by both yeast and mammalian Cox17, which may explain their absolute evolutionary conservation from yeast to mammals. Our model is consistent with in vivo mutation experiments of the yeast Cox17, since it has been demonstrated that single mutations of the N-terminal cysteine residues at positions 23, 24 and 26 of yeast Cox17 [22], as well as C-terminal Cys-36 and Cys-57, lead to respiratory-deficient phenotype [22,23]. In vivo mutation experiments are not conclusive for Cys-47, since its mutation to a serine residue resulted in the functional protein [22], whereas substitution of the same residue by an arginine residue leads to the non-functional protein [23]. Although the model of the copper-binding motif in Cox17 presented in this paper is consistent with our in vitro results on mammalian Cox17 and earlier in vitro and in vivo results on yeast Cox17, it could be finally proven by EXAFS or structural studies of mammalian Cu4Cox17, which are currently in progress.
Figure 7. Proposed structure of the metal-binding motif in Cox17.

(a) Structure of the [Cu4(S)6]2− model cluster proposed for the structure of the copper–thiolate cluster in Cu4Cox17; (b) sequence alignment of yeast and mammalian Cox17.
Reconstitution of Cox17 with different metals demonstrates that fully reduced Cox17 can bind also bivalent Zn2+ ions, but the binding is non-co-operative and the protein can bind maximally two Zn2+ ions. This difference could be explained by the different co-ordination preferences of univalent Cu+ and bivalent Zn2+ ions, and this demonstrates that Cox17 is adapted for the binding of Cu+ ions. However, it is surprising that based on MS and fluorescence analyses, Cox17 could not be reconstituted with Ag+ ions. The co-ordination preferences of Ag+ are similar to those of Cu+ ions, and Ag+ can successfully substitute Cu+ ions in metal–thiolate clusters of the transcription factor Ace1 [24] and in yeast metallothionein Cup1 [25]. At the moment, we can explain the discrimination of Ag+ ions by Cox17 only with their slightly larger ionic radius (RAg+=1.13 Å; 1 Å=0.1 nm) compared with that of Cu+ ions (RCu+=0.98 Å), and we suggest that Ag+ ions may be size-excluded from binding to the multinuclear Ag+ cluster of Cox17. In biological systems, an extremely high specificity of Cox17 for binding of Cu+ ions may be needed to restrict the incorporation of Ag+ ions into CCO.
The biological context
By analysing the metal-binding affinity of Cox17 (Kd=13 fM) in the biological context, we can conclude that the apparent metal-binding affinity of Cox17 is slightly lower compared with that of superoxide dismutase (Kd=6 fM) [26], but is substantially lower compared with that of metallothioneins (Kd<0.05 fM) [27]. Our experimental results demonstrate that micromolar Cox17 can successfully compete with supramillimolar GSH for copper binding, which means that, under physiological conditions, Cox17 can extract copper mainly from the GSH-bound copper pool.
It has been established that Cox17 participates in the delivery of copper to CCO in both yeast [9] and animal cells [6]. However, the mechanisms of metal transfer by Cox17 as well as distinct protein partners are still not known. The biochemical results presented strongly imply that the biologically active metallated Cox17 form in the cells is Cu4Cox17, and metals are bound to Cox17 and released from Cu4Cox17 in a co-operative manner. Consequently, Cox17 is a multicopper donor suitable for the simultaneous transfer of up to four copper ions to partner proteins. Metal binding capacity and mechanism are substantially different between Cox17 and two other known cellular copper chaperones, CCS (copper chaperone for superoxide dismutase) and Atx1 (copper chaperone for cation-transporting P-type ATPase), both of which bind one copper ion per protein monomer [3]. There is evidence that Cox17 provides copper exclusively to the CuA site of CCO [23], which contains two copper ions. It is known that CCO is a dimer [2], and an instant saturation of both the CuA sites on dimeric apoCCO by Cu4Cox17 would be minimal and represents the most effective biological solution for specific metal transfer from Cox17 to the CuA sites of CCO. Suggestions that Cu4Cox17 delivers metals to Sco1 [8] should be, according to the co-operative metal-release mechanism, considered unrealistic since Sco1 binds only one copper ion per molecule [28,29]. At the same time, the formation of oligomeric metal-transfer complexes with the participation of Sco1, and also Cox11, are theoretically not excluded.
From the results presented, it is evident that metals can be released from Cu4Cox17 by two different metal-release mechanisms. According to the first mechanism, non-oxidative metal-release mechanism, Cu4Cox17 can transfer metals to partners with higher metal-binding affinity, which is facilitated by the kinetic lability of metal–thiolate clusters. According to the second mechanism, oxidative mechanism, metals could be released from the copper–thiolate cluster of Cox17 by oxidation of the cysteine residues. Such an oxidative mechanism operates in the metal release from zinc–thiolate clusters of metallothionein [30]. The oxidative metal-release mechanism is supported by the observation that Cox17 exists in different oxidative states and tends to form a distinct pattern of disulphides [13,14]. Moreover, it is also realistic that CCO may oxidize Cox17 during the metal-transfer act. Oxidative metal-release mechanism implies, however, that oxidized Cox17 should be reduced before the next metal-transfer cycle. According to our results, the full reduction of Cox17 with 2 mM DTT takes more than 1 h at 45 °C, which means that under cellular conditions, this reaction should be catalysed by specific thioreductases. Following this line of hypothetical biological events, it is not excluded that such a thioreductase is the Sco1 protein, which acts downstream of Cox17 in the assembly of functional CCO [23]. Sco proteins contain a thioredoxin fold and it was already suggested that they may play a catalytic role in the assembly of functional CCO [29,31]. So far, it has been suggested that Sco1 may reduce disulphides in the CuA site of CCO [29,31]. Alternatively, Sco1 may reduce also oxidized Cox17 and restore its metal-binding capacity, which is necessary for its functioning as copper chaperone for CCO.
The biological mechanism of metal transfer by Cox17 proposed above is yet to be verified further. However, the biochemical results presented provide an important framework for understanding the mechanism of Cox17 action in biological systems.
Acknowledgments
This work was supported by grants from the Swedish Institute, Estonian Science Foundation project no. 5635, EU 5th Framework Project No. 02435 and Karolinska Institutet.
References
- 1.Ferguson-Miller S., Babcock G. T. Heme/copper terminal oxidases. Chem. Rev. 1996;96:2889–2908. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 2.Tsukihara T., Aoyama H., Yamashita E., Tomizaki T., Yamaguchi H., Shinzawa-Itoh K., Nakashima R., Yaono R., Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 3.Huffman D. L., O'Halloran T. V. Function, structure, and mechanism of intracellular copper trafficking proteins. Annu. Rev. Biochem. 2001;70:677–701. doi: 10.1146/annurev.biochem.70.1.677. [DOI] [PubMed] [Google Scholar]
- 4.Glerum D. M., Shtanko A., Tzagoloff A. Characterization of Cox17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J. Biol. Chem. 1996;271:14504–14509. doi: 10.1074/jbc.271.24.14504. [DOI] [PubMed] [Google Scholar]
- 5.Amaravadi R., Glerum D. M., Tzagoloff A. Isolation of a cDNA encoding the human homolog of Cox17, a yeast gene essential for mitochondrial copper recruitment. Hum. Genet. 1997;99:329–333. doi: 10.1007/s004390050367. [DOI] [PubMed] [Google Scholar]
- 6.Takahashi Y., Kako K., Kashiwabara S., Takehara A., Inada Y., Arai H., Nakada K., Kodama H., Hayashi J., Baba T., et al. Mammalian copper chaperone Cox17p has an essential role in activation of cytochrome c oxidase and embryonic development. Mol. Cell. Biol. 2002;22:7614–7621. doi: 10.1128/MCB.22.21.7614-7621.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beers J., Glerum D. M., Tzagoloff A. Purification, characterization, and localization of yeast Cox17p, a mitochondrial copper shuttle. J. Biol. Chem. 1997;272:33191–33196. doi: 10.1074/jbc.272.52.33191. [DOI] [PubMed] [Google Scholar]
- 8.Glerum D. M., Shtanko A., Tzagoloff A. Sco1 and Sco2 act as high copy suppressors of a mitochondrial copper recruitment defect in Saccharomyces cerevisiae. J. Biol. Chem. 1996;271:20531–20535. doi: 10.1074/jbc.271.34.20531. [DOI] [PubMed] [Google Scholar]
- 9.Punter F. A., Adams D. L., Glerum D. M. Characterization and localization of human Cox17, a gene involved in mitochondrial copper transport. Hum. Genet. 2000;107:69–74. doi: 10.1007/s004390000339. [DOI] [PubMed] [Google Scholar]
- 10.Carr H. S., George G. N., Winge D. R. Yeast Cox11, a protein essential for cytochrome c oxidase assembly, is a Cu(I)-binding protein. J. Biol. Chem. 2002;277:31237–31242. doi: 10.1074/jbc.M204854200. [DOI] [PubMed] [Google Scholar]
- 11.Srinivasan C., Posewitz M. C., George G. N., Winge D. R. Characterization of the copper chaperone Cox17 of Saccharomyces cerevisiae. Biochemistry. 1998;37:7572–7577. doi: 10.1021/bi980418y. [DOI] [PubMed] [Google Scholar]
- 12.Heaton D. N., George G. N., Garrison G., Winge D. R. The mitochondrial copper metallochaperone Cox17 exists as an oligomeric, polycopper complex. Biochemistry. 2001;40:743–751. doi: 10.1021/bi002315x. [DOI] [PubMed] [Google Scholar]
- 13.Chen Z. W., Bergman T., Ostenson C. G., Efendic S., Mutt V., Jörnvall H. Characterization of dopuin, a polypeptide with special residue distributions. Eur. J. Biochem. 1997;249:518–522. doi: 10.1111/j.1432-1033.1997.t01-2-00518.x. [DOI] [PubMed] [Google Scholar]
- 14.Takenouchi T., Fujimoto M., Shimamoto A., Munekata E. Isolation and characterization of Cox17p from porcine heart by determining its survival-promoting activity in NIH3T3 cells. Biochim. Biophys. Acta. 1999;1472:498–508. doi: 10.1016/s0304-4165(99)00158-0. [DOI] [PubMed] [Google Scholar]
- 15.Eriste E., Norberg A., Bonetto V., Nepomuceno D., Lovenberg T. W., Sillard R., Jörnvall H. A C-terminally elongated form of PHI from porcine intestine. Cell. Mol. Life Sci. 1999;56:709–713. doi: 10.1007/s000180050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krezel A., Lesniak W., Jezowska-Bojczuk M., Mlynarz P., Brasun J., Kozlowski H., Bal W. Coordination of heavy metals by dithiothreitol, a commonly used thiol group protectant. J. Inorg. Biochem. 2001;84:77–88. doi: 10.1016/s0162-0134(00)00212-9. [DOI] [PubMed] [Google Scholar]
- 17.Hitomi Y., Outten C. E., O'Halloran T. V. Extreme zinc-binding thermodynamics of the metal sensor/regulator protein, ZntR. J. Am. Chem. Soc. 2001;123:8614–8615. doi: 10.1021/ja016146v. [DOI] [PubMed] [Google Scholar]
- 18.Kaltashov I. A., Eyles S. J. Studies of biomacromolecular conformations and conformational dynamics by mass spectrometry. Mass Spectrom. Rev. 2002;21:31–71. doi: 10.1002/mas.10017. [DOI] [PubMed] [Google Scholar]
- 19.Stillman M. J. Metallothioneins. Coord. Chem. Rev. 1995;144:461–511. [Google Scholar]
- 20.Palumaa P., Eriste E., Njunkova O., Pokras L., Jörnvall H., Sillard R. Brain-specific metallothionein-3 has higher metal-binding capacity than ubiquitous metallothioneins and binds metals noncooperatively. Biochemistry. 2002;41:6158–6163. doi: 10.1021/bi025664v. [DOI] [PubMed] [Google Scholar]
- 21.Coucouvanis D., Murray C. N., Kanodia S. K. Metal-mercaptide chemistry. Synthesis and structural characterization of the [Cu(SC6H5)]2− anion. Rational synthesis and the structure of the [Cu4(SC6H5)6] cluster. Inorg. Chem. 1980;19:2993–2998. [Google Scholar]
- 22.Heaton D., Nittis T., Srinivasan C., Winge D. R. Mutational analysis of the mitochondrial copper metallochaperone Cox17. J. Biol. Chem. 2000;275:37582–37587. doi: 10.1074/jbc.M006639200. [DOI] [PubMed] [Google Scholar]
- 23.Punter F. A., Glerum D. M. Mutagenesis reveals a specific role for Cox17p in copper transport to cytochrome oxidase. J. Biol. Chem. 2003;278:30875–30880. doi: 10.1074/jbc.M302358200. [DOI] [PubMed] [Google Scholar]
- 24.Dameron C. T., Winge D. R., George G. N., Sansone M., Hu S., Hamer D. A copper–thiolate polynuclear cluster in the ACE1 transcription factor. Proc. Natl. Acad. Sci. U.S.A. 1991;88:6127–6131. doi: 10.1073/pnas.88.14.6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peterson C. W., Narula S. S., Armitage I. M. 3D solution structure of copper and silver-substituted yeast metallothioneins. FEBS Lett. 1996;379:85–93. doi: 10.1016/0014-5793(95)01492-6. [DOI] [PubMed] [Google Scholar]
- 26.Rae T. D., Schmidt P. J., Pufahl R. A., Culotta V. C., O'Halloran T. V. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- 27.Vasak M., Kägi J. H. R. Metallothioneins. In: King R. B., editor. Encyclopedia of Inorganic Chemistry. New York: John Wiley & Sons; 1994. pp. 2229–2241. [Google Scholar]
- 28.Beers J., Glerum D. M., Tzagoloff A. Purification and characterization of yeast Sco1p, a mitochondrial copper protein. J. Biol. Chem. 2002;277:22185–22190. doi: 10.1074/jbc.M202545200. [DOI] [PubMed] [Google Scholar]
- 29.Balatri E., Banci L., Bertini I., Cantini F., Ciofi-Baffoni S. Solution structure of Sco1: a thioredoxin-like protein involved in cytochrome c oxidase assembly. Structure (Cambridge) 2003;11:1431–1443. doi: 10.1016/j.str.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 30.Maret W. Cellular zinc and redox states converge in the metallothionein/thionein pair. J. Nutr. 2003;133:1460S–1462S. doi: 10.1093/jn/133.5.1460S. [DOI] [PubMed] [Google Scholar]
- 31.Chinenov Y. V. Cytochrome c oxidase assembly factors with a thioredoxin fold are conserved among prokaryotes and eukaryotes. J. Mol. Med. 2000;78:239–242. doi: 10.1007/s001090000110. [DOI] [PubMed] [Google Scholar]